 Chao Hong
Chao Hong Rong Jin
Rong Jin Xiaoming Gao
Xiaoming Gao- Institutes of Biology and Medical Sciences, Soochow University, Suzhou, China
Chronic graft-versus-host disease (cGVHD) is one of the most common reasons of late non-relapse morbidity and mortality of patients with allogeneic hematopoietic stem cell transplantation (allo-HSCT). While acute GVHD is considered driven by a pathogenic T cell dominant mechanism, the pathogenesis of cGVHD is much complicated and involves participation of a variety of immune cells other than pathogenic T cells. Existing studies have revealed that antigen presenting cells (APCs) play crucial roles in the pathophysiology of cGVHD. APCs could not only present auto- and alloantigens to prime and activate pathogenic T cells, but also directly mediate the pathogenesis of cGVHD via multiple mechanisms including infiltration into tissues/organs, production of inflammatory cytokines as well as auto- and alloantibodies. The studies of this field have led to several therapies targeting different APCs with promising results. This review will focus on the important roles of APCs and their contributions in the pathophysiology of cGVHD after allo-HSCT.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a widely used life-saving procedure for patients with hematopoietic malignancies including leukemia, lymphoma as well as other non-malignant diseases related with bone marrow failure. However, its success is markedly compromised by the development of graft-versus-host disease (GVHD) after transplantation due to the histoincompatibility between donors and recipients. Donor alloreactive T cells are first primed through recognition of host alloantigens presented by host antigen presenting cells (APCs), and less often, by donor APCs. Upon preparative conditioning (including high dose chemotherapy and/or total body irradiation) caused gastrointestinal tract or tissue damage, the released pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) stimulate the upregulation of costimulatory molecules and production of inflammatory cytokines expressed in APCs. Such APCs subsequently drive the activation and differentiation of donor alloreactive T cells into effector T cells which contribute to GVHD in target organs (1–3). According to the time of onset and pathological mechanisms, GVHD can be divided into acute GVHD (aGVHD) and chronic GVHD (cGVHD). aGVHD usually starts within the first 100 days after allo-HSCT and is mediated mainly by infused donor alloreactive T cells in the grafts. Accompanied with the process of aGVHD, donor hematopoietic stem cells (HSCs) engraft in host bone marrow and develop into various immune cell lineages. Unfortunately, such donor-derived immune cells could be dysfunctional and autoreactive due to the altered microenvironment unable to support their normal development. Many aGVHD survivors could further develop into subsequent cGVHD which usually begins at a later stage (100 days to 2 years after allo-HSCT), though earlier onset (termed overlap cGVHD when concurrent with aGVHD) is also possible (4, 5).
cGVHD is a life-threatening complication which affects 30%–70% patients who have received allo-HSCT (6–8), with prior episode of aGVHD as the most potent risk factor. It remains a leading cause of late non-relapse morbidity and mortality of patients following allo-HSCT (9). The incidence of cGVHD has been increasing in the past two decades attributed to increased use of old age donors and unrelated/mismatched donors, reduced intensity conditioning regimen and G-CSF mobilized peripheral blood stem cells (G-PBSCs) instead of unmanipulated bone marrow grafts (8, 10, 11). Several curative therapies against aGVHD, such as corticosteroids and calcineurin inhibitors and other immune inhibition drugs, have been successfully developed (12). However, therapies for cGVHD are still challenging due to our poor understanding on its much complex and obscure pathogenesis (13). Conventional treatments for cGVHD are glucocorticoids and immunosuppressive drugs which only achieve disease remission in part of the patients (14, 15). Moreover, systemic glucocorticoids often bring long-term complications which increase morbidity and mortality in patients with cGVHD (12, 16). In recent years, ruxolitinib (a selective JAK1/2 inhibitor) has been used in patients with steroid-refractory cGVHD which showed promising clinical results (17). Other cell based therapy such as extracorporeal photopheresis has also been found to benefit the treatment of cGVHD although the immunological mechanism remains elusive (18).
Overview of cGVHD in Patients and Mouse Models
GVHD is a complex immunological process involving both innate and adaptive immune responses. cGVHD and aGVHD have distinct pathogenesis albeit they share some common clinical manifestations (19). Unlike aGVHD in which T cells play dominant pathogenic roles (20), the pathogenesis of cGVHD is comprehensive and involves the infiltration of various inflammatory cells as well as the production of auto- and alloantibodies. The complexity of cGVHD immunopathology also indicates a dysfunction of immune tolerance in the hosts after allo-HSCT, which may be part of the reasons for the unresponsiveness of cGVHD patients to the commonly used immunosuppressive agents (21). Tissue and organ damage caused by donor T cell-mediated aGVHD is crucial for initiating cGVHD. Depletion or inhibition of donor T cells in the grafts by anti-lymphocyte antibodies and high-dose cyclophosphamide in the early post-transplantation period could not only prevent aGVHD but also delay the onset of cGVHD (22–25). cGVHD affects not only epithelial tissues (gastrointestinal tract, lung, liver and skin), mostly targeted in aGVHD, but also many other tissues/organs including oral, esophageal, musculoskeletal, fascial, ocular, joint, and even genital tissues (4, 26–29). Attributed to the introduction of National Institute of Health (NIH) consensus criteria, the diagnosis and scoring for cGVHD have been greatly improved in the last two decades. Fibrosis is the most frequently observed characteristic of cGVHD with cutaneous and pulmonary fibrosis (tissue fibrosis manifesting as scleroderma and bronchiolitis obliterans) as the definitive clinical manifestations (4, 30).
Since human cGVHD is very difficult to study mechanistically, various mouse models of cGVHD have been developed in the last decades (31–36). To recapitulate the natural evolution of clinical cGVHD in human allo-HSCT patients, mouse models have been designed with a more precise imitation of clinic procedures including preparative conditioning (total body irradiation), donor and recipient strain combinations (use semiallogeneic F1 mice or minor histoincompatible mice as recipients), and in some models, use of G-CSF-mobilized splenocytes or peripheral blood grafts instead of conventional bone marrow transplantation (BMT) plus purified splenic T cells to induce cGVHD (37, 38). These aspects permit recipients to survive aGVHD and give time for auto- and alloreactive T cells and B cells to develop and cause cGVHD. Inappropriate BMT conditions such as high dose total body irradiation, or high T cell number in grafts, or use of fully MHC-mismatched donors often correlate with an early mortality (within a couple of weeks) after BMT as a result of severe gastrointestinal aGVHD (20, 39). By adjusting to an optimal BMT condition, an autoimmune-mediated pathology could be induced 4-8 weeks after BMT attributable to chronic autoreactive T cell activation and subsequent autoantibody production (40, 41). Considering of the different kinetics with clinical symptoms observed in patients, the disease occurrence in mouse cGVHD models is often absence or only happens at late stage after BMT. In a mouse model of mixed hematopoietic chimerism, the persistence of host B cells and high levels of circulating IgG autoantibodies were found to be associated with the appearance of sclerodermatous cGVHD-like lesions which were observed 7-9 months after BMT (42). In recent years, CD34+-stem-cell-humanized NSG mice were found to develop cGVHD late after transplantation (more than 24 weeks). These mice reproduce the full spectrum of pleiotropism of human cGVHD in the absence of prior aGVHD which may serve as a great model for cGVHD related research (43).
In cGVHD, donor T cells developed from engrafted HSCs could be both auto- and alloreactive capable of inducing similar disease when adoptively transferred into secondary allogeneic or syngeneic recipients (44, 45). In these mouse models, pathogenic Th17 cells have been implicated to be causative to cGVHD as well as their roles in aGVHD (46–48). Specific antibody-mediated suppression of IL-17 producing cells reduces histopathological damage of skin, salivary gland and liver in cGVHD (47). In addition, T follicular helper (Tfh) cells play a part in cGVHD as well through interaction with auto- and alloreactive germinal center (GC) B cells via expression of both cell surface molecules and IL-21 (41). The pathogenesis of cGVHD is also found to be closely related with deficient development of regulatory cell subsets such as regulatory T cells (Tregs) and regulatory B cells (Bregs) (49, 50). In addition to the contributions of dysfunctional lymphocytes, pathogenic macrophages play important roles in the development of cGVHD, indicating a mutlifactorial pathogenesis of the disease (51, 52). Based on the studies of mouse models, the pathophysiological and immunological evolution of cGVHD should include at least 4 major mechanisms: distorted T cell negative selection in injured host thymus, lack of regulatory cell populations, macrophage-mediated multi-organ fibrosis and loss of B cell tolerance (50–53). cGVHD is a result of immune imbalance between inflammatory immune responses and inhibitory immune mechanisms that maintain immune tolerance. Given that APCs play critical roles in initiation of auto- and alloreactive T cell responses, development/maintenance of central/peripheral immune tolerance, production of profibrotic cytokines as well as auto- and alloantibodies, they are likely important contributors to the development of cGVHD. Below, we review the existing literatures of the functions and contributions of APCs in the pathogenesis of cGVHD (Table 1).

Table 1 Distinct origins and functions of antigen presenting cells (APCs) in chronic graft-versus-host disease.
Dysregulation of Central and Peripheral T Cell Tolerance by Dendritic Cells in cGVHD
Dendritic cells (DCs) at steady state play dual roles in the induction of T cell-mediated adaptive immune response and maintenance of immune tolerance (72, 73). In cGVHD settings after allo-HSCT, DCs are crucial for initiating pathogenic T cell activation in periphery. Their dysfunction also causes failure of autoreactive T cell education in host thymus and loss of T cell peripheral tolerance which contribute to the pathogenesis of cGVHD.
Preclinical Data
During normal thymopoietic development, autoreactive T cells are depleted in the thymus as a result of negative selection which is mediated by the medullary thymic epithelia cells (mTECs) and the presence of intrathymic autoantigen presenting DCs (73–76). However, in allogeneic BMT scenario, preparative conditioning regimen and donor T cell-mediated aGVHD could damage host thymus and impair thymopoiesis, resulting in dysfunction of negative selection and subsequent release of auto- and alloreactive T cells into periphery (77–79). Allogeneic BMT recipient animals of MHC class II deficient bone marrow grafts developed cGVHD which can be prevented by prior thymectomy (44), indicating a regulatory role of donor DCs in T cell central tolerance during cGVHD. Donor T cells escaped from the thymus of recipient of MHC class II deficient bone marrow grafts are autoreactive and pathogenic owing to the dysfunction of DCs and can cause cGVHD when transferred into secondary recipient mice (44). Interestingly, even host T cells become pathogenic in the absence of DC-mediated central tolerance. Unlike radioresistant tissue-resident macrophages, host DCs are radiosensitive and replaced by donor cells shortly after transplantation. A study reported that host T cells derived from radioresistant intrathymic T cell precursors escaped negative selection in mice lack of host intrathymic DCs and caused dermal fibrosis in mouse cGVHD model (80). After escaping from dysfunctional thymus, auto- and alloreactive T cells further differentiate into effector T cells in periphery. DCs are well known as the most potent professional APCs in eliciting peripheral naïve T cell activation. While host DCs are rapidly eliminated early after allo-HSCT, donor DCs predominate in peripheral tissues and contribute to the development of cGVHD by presenting both host and donor antigens to activate donor T cells via indirectly antigen presentation (81, 82).
Clinical Data
Although the appearance of donor DCs occurs early after allo-HSCT, their reconstitution is impaired and requires a long period of time to complete. Conventional DCs (cDCs) and plasmacytoid DCs (pDCs) are two major DC subsets both of which contribute to the induction of donor T cell tolerance against host organs after allo-HSCT (73, 74, 83). A study of pediatric allo-HSCT revealed that cDC numbers returned to normal level within 300-400 days after transplantation while pDC numbers recovered very slowly in these pediatric patients and were always lower than their age-matched healthy controls up to 7 years after transplantation (54). Another study reported that allo-HSCT patients with sooner or higher pDC recovery profile correlated with improved overall survival, indicating pDC count in peripheral blood of allo-HSCT patients is a significant predictor of long-term outcome after allo-HSCT (55).
Pathophysiologic Interpretation and Therapeutic Implications
DCs maintain T cell immune tolerance in both thymus and periphery. Peripheral T cell tolerance can be induced via direct interaction of inhibitory signaling molecules PD-L1/PD-1 and (CD80/86)/CTLA4 expressed on the surface of DCs and T cells, respectively (84–86). Besides, DCs could also promote donor T cell tolerance via expansion of Tregs. In addition to IL-2 dependency, Tregs require costimulatory signals from DCs for their optimal activation and proliferation. Tregs play important roles in the control of pathogenic T cell response and dysfunctional Treg development could cause various autoimmune diseases (87, 88). Decreased numbers of circulating Tregs were found to be correlated with cGVHD in both preclinical and clinical studies (40, 89–91), and adoptive transfer of Tregs could effectively ameliorate cGVHD (92, 93). DCs are important for their role in the induction and maintenance of Tregs and this function is mediated through a MHC class II-dependent interaction (94). It was found that an inflammatory cytokine milieu dominated by TNF during GVHD impairs the MHC class II antigen presentation pathway of cDCs, while MHC class I presentation remains largely intact, and leads to a failure in Treg development which results in a loss of immune tolerance in cGVHD (50, 95). Promoting Treg expansion is a promising approach to prevent cGVHD. Low-dose subcutaneous injection of IL-2 has shown to effectively expand Tregs in vivo and ameliorate cGVHD (96–99). A recent study reported that GM-CSF treatment increased CD4+CD8- DC number and promoted DC-dependent Treg expansion, thus protected mice against the development of skin cGVHD (56), validating an indirect strategy to prevent cGVHD via strengthening DC and Treg interaction.
Activation and Infiltration of Donor Macrophages Contribute to cGVHD
Macrophages are remarkably plastic innate immune cells which can be found in all tissues and exhibit a vast functional diversity in development, maintenance of microenvironment homeostasis, tissue damage repair as well as innate immunity and adaptive immunity (100–102). Tissue-resident macrophages differ from monocyte-derived macrophages in terms of origin, which has been widely investigated in the last decade as immune sentinels in immune defense and resolution of inflammation (103). They are of embryonic origin and found to reside in majority peripheral tissues and organs, replenished by self-renewal independent of bone marrow monocyte replacement at steady state. However, after allo-HSCT, tissue-resident macrophages can be replaced by donor monocyte-derived macrophages which contribute to the pathogenesis of cGVHD.
Preclinical Data
In mouse models, accumulating studies support the concept that donor-derived macrophages could facilitate and intensify the pathophysiology of cGVHD (37, 51, 67, 104). It has been revealed that inhibition of donor macrophage infiltration in tissues and organs could ameliorate mouse cGVHD (65). CSF-1 axis controls macrophage development, differentiation and survival and is critical for monocyte-derived macrophage reconstitution after allo-HSCT. In IL-17-dependent cGVHD models of scleroderma and bronchiolitis obliterans, donor bone marrow-derived macrophages were found infiltrating the skin and lung in a CSF-1/CSF-1R-, but not CCL2/CCR2- or GM-CSF/GM-CSFR-, dependent manner and contribute to the pathogenesis of cGVHD. These macrophages express CD206 and TGF-β but not iNOS, identifying them as M2 macrophages (51). Administration of CSF-1R blocking antibodies significantly reduced HSP47+ myofibroblasts in the skin, indicating a macrophage-dependent accumulation of myofibroblasts in cGVHD (66). The origin of macrophages is important for their profibrotic gene expression as evidenced by a finding that monocyte-derived alveolar macrophages differ significantly from tissue-resident alveolar macrophages and drive lung fibrosis after BMT (105).
Clinical Data
In allo-HSCT scenario, host derived tissue-resident macrophages are eliminated and replaced by donor monocyte differentiated tissue resident macrophages with M2 phenotype which are found associated with the development of cGVHD. CD163, a scavenger receptor with immunoregulatory properties, is expressed mainly on M2 macrophages. Examination of biopsy specimens from patients with skin GVHD showed that increased infiltration of CD163+ M2 macrophages was a significant predictor for refractory GVHD and poor prognosis (106). Soluble CD163 (sCD163) accumulates in the blood of hosts under oxidative stress or severe inflammatory conditions, as a result of direct secretion by activated macrophages or cleavage of membrane-bound CD163 from cell surface by matrix metalloproteinases (107–110). Intriguingly, plasma sCD163 in allo-HSCT patients is a high risk predictor of cGVHD, indicating a role of M2 macrophage activation and oxidative stress in the pathogenesis of cGVHD (69). Macrophage-derived chemokine and CC chemokine receptor 4 were also found to be closely associated with strong T cell infiltration in the buccal mucosa and labial salivary glands in cGVHD patients (68).
Pathophysiologic Interpretation and Therapeutic Implications
Activated donor-derived macrophages could mediate tissue fibrosis via production of profibrotic cytokine TGF-β, which induces the differentiation of fibroblasts into collagen-producing myofibroblasts capable of promoting collagen synthesis and deposition in cGVHD (65, 66, 111, 112). Pirfenidone, approved by U.S. Food and Drug Administration (FDA) for idiopathic pulmonary fibrosis, can also ameliorate cGVHD by inhibiting macrophage infiltration and TGF-β production (65). A recent study found that type 2 cannabinoid receptor expressed on macrophages played a critical role in the regulation of cGVHD and therapeutic targeting of this receptor by agonist showed beneficial effect in a sclerodermatous cGVHD model (113). Additionally, macrophages could contribute to the pathogenesis of cGVHD via interaction with T cells. In cGVHD, alloreactive T cells activate and differentiate into Th1/Tc1, Th17/Tc17, and Tfh cell paradigms in the presence of inflammatory cytokines such as IL-6 and IL-12, while Th17/Tc17 cells play a central role in cGVHD pathophysiology (46–48). IL-17 is a key mediator of pathology in cGVHD and it controls the infiltration of F4/80+ macrophages into skin which facilitate the development of scleroderma (51). It should be noted that both pathogenic macrophages and T cells share some common cytokine requirement. IL-6 is a multifunctional inflammatory cytokine which can activate macrophages and also drive the differentiation of pathogenic Th17 cells. By using a humanized cGVHD mouse model through engraftment of human hematopoietic stem/progenitor cells into hIL-6 transgenic recipient mice, Rintaro et al. reported that co-activation of macrophages and T cells were found in lung and liver and contribute to the pathogenesis of cGVHD (67). IL-6 gene polymorphism is closely associated with the pathogenesis of cGVHD and anti-IL-6R monoclonal antibody (tocilizumab) has been reported to ameliorate cGVHD in some allo-HSCT patients (114, 115).
Loss of B Cell Tolerance in cGVHD
At steady state, B cells develop in bone marrow and undergo negative selection which leads to a state of B cell central tolerance to avoid production and release of autoreactive B cells into periphery. Loss of B cell tolerance and aberrant activation of peripheral B cells contribute to the development of cGVHD (116–118).
Preclinical Data
An intact bone marrow microenvironment is critical for normal B cell lymphopoiesis. Osteoblasts, which could form bone marrow stromal niche for HSCs and B cell progenitors, are targeted by donor pathogenic T cells in GVHD (119, 120). Interestingly, protection of osteoblasts from T cell-mediated damage, by a Treg-expanded graft infusion, could maintain the bone marrow niche for early B cell progenitors and increase the number of pro-B, pre-B and immature B cells in bone marrow and ameliorate cGVHD (121). Aberrant B cell negative selection in host bone marrow causes release of auto- and alloreactive B cells into periphery. These B cells migrate into secondary lymphoid organs and encounter auto- and alloantigens, become activated and then differentiate into plasmablasts or memory B cells via interaction with Tfh cells. Through their expression of cell surface molecules and IL-21, Tfh cells promote mature B cell proliferation, differentiation and secretion of auto- and alloantibodies in cGVHD (41, 62, 122). Both Tfh cells and GC B cells are involved in cGVHD and their functions are mutually dependent. Depletion of B cells could suppress Tfh cells in addition to GC formation in cGVHD (63). These data indicate that T-B cell interaction is an important contributor to the pathogenesis of cGVHD. Interestingly, it was reported that donor B cells in transplants, activated by donor T cells, are also efficient APCs to augment the initial clonal expansion and survival of donor autoreactive T cells which are capable of mediating autoimmune-like cGVHD (61). Recently, a study by Deng et al. has reported that extrafollicular CD4+ T and B cell interactions are more important and sufficient for inducing cGVHD, while GC formation is dispensable (123). They identified PSGL-1lowCD4+ pre-Tfh-like extrafollicular T cells that were critical for the pathogenesis of cGVHD owing to their interaction with B cells, indicating a much complex mechanism of T-B cell interaction in the pathogenesis of cGVHD.
Clinical Data
It was originally found in a case report that a cGVHD patient who developed refractory immune-mediated thrombocytopenia after allo-HSCT responded to B cell depletion therapy (124). This finding provided evidence of B cell dysfunction in the immunopathology of cGVHD and suggested a potential way of cGVHD prevention by B cell depletion. B cell development deficiency is often observed in cGVHD patients, indicating an aberrant bone marrow microenvironment failed to support normal B cell lymphopoiesis and selection during cGVHD (125, 126). Insufficient B lymphopoiesis causes post-transplantational B cell deficiency with decreased bone marrow B cell precursors which has been reported in both aGVHD and cGVHD patients after allo-HSCT (127, 128). In addition, there is increasing evidence showing that aberrant peripheral B cell expansion is a feature of cGVHD owing to their dysfunctional regulation of activation and proliferation. For instance, B cells from patients with active cGVHD are in a heightened metabolic state and resistant to apoptosis due to deficient expression of proapoptotic molecule Bim (129). B cell activating factor of the tumor necrosis family (BAFF), which is produced by macrophages, monocytes, DCs, T cells and stromal cells, plays important roles in B cell metabolism, survival and maintaining autoreactive B cell clones (130–132). In cGVHD patients, increased BAFF concentrations and higher BAFF/B-cell ratios correlate with increased numbers of circulating pre-GC B cells and post-GC plasmablast-like cells (64). These circulating pathogenic B cells are capable of autoantibody production without requiring additional antigen stimulation. Besides, other molecules regulating B cell activation and proliferation could also contribute to B cell-mediated pathogenesis in cGVHD. Increased NOTCH2 activation was found to be closely related with robust BCR responsiveness to alloantigens in B cells from cGVHD patients and suppression of BCR-NOTCH hyperactivation by all-trans retinoic acid could reduce NOTCH2 signaling and prevent B cell proliferation while maintaining functional B cell responses (133).
Pathophysiologic Interpretation and Therapeutic Implications
Production of multiple auto- and alloantibodies is a hallmark of cGVHD, and a variety of auto- and alloantibodies have been found to be associated with the severity of cGVHD (134–137). In mouse cGVHD models of scleroderma and bronchiolitis obliterans, these auto- and alloantibodies are found not only the outcome of dysfunctional B cell activation during cGVHD, but also could be causative to cGVHD pathogenesis (57, 58). Alloantibodies against H-Y minor histocompatibility antigens are significantly associated with cGVHD and disease remission (59). Autoantibodies against platelet-derived growth factor receptor have been found to play a role in the development of skin and lung fibrosis in cGVHD via stimulating type I collagen gene expression through the Ha-Ras-ERK1/2-ROS signaling pathway (60). It has been reported that microRNA-17-92 expression is required for alloantibody production and IgG deposition in the skin in cGVHD (138). A recent study found that checkpoint regulator SLAMF3 could modulate the activation thresholds of B cell subsets and SLAMF3 blockade markedly enhanced autoantibody production in cGVHD, thereby revealing a role of SLAMF3 in the negative regulation of cGVHD via preventing the expansion of autoreactive B cells (139). Since aberrant activation of B cells contributes to the pathogenesis of cGVHD, approaches directly targeting the key downstream kinases of B cell activation have been developed for cGVHD treatment with promising results. Ibrutinib was designed as a selective inhibitor of Bruton’s tyrosine kinase (BTK) and became the first FDA-approved drug for the treatment of steroid-refractory cGVHD in 2017 (140). A small molecule inhibitor of Syk has been found effective in the therapy of cGVHD in mouse models (32, 141). Fostamatinib, a Syk inhibitor drug approved by FDA for the treatment of immune thrombocytopenia, is now under clinical evaluation in patients with cGVHD.
Functions of Other APCs in cGVHD
Among the non-hematopoietic APCs (e.g., epithelial or stromal cells), mTECs play important roles in the induction of T lymphocyte central tolerance and the pathogenesis of cGVHD. Damage of recipient mTECs caused by alloreactive T cells in the donor grafts leads to defective negative selection of donor T cells and release of autoreactive CD4+ T cells into periphery which contribute to the development of cGVHD (71, 77, 142). A recent study has found that transplantation of donor-derived TEC progenitors into cGVHD recipients could restore immune tolerance and ameliorate cGVHD (70). In periphery, non-hematopoietic APCs initiate the initial priming of alloreactive T cells independent of hematopoietic APCs while the latter contribute to the intensification of GVHD (143–145), although most of these studies are based on mouse models of aGVHD. Considering the chronic inflammation and continuing existence of alloreactive T cells in cGVHD, detailed investigation on the role of peripheral non-hematopoietic APCs in pathophysiology of cGVHD is merited.
Concluding Remarks
While traditional treatments of cGVHD with corticosteroids and other immune suppressive agents are facing more and more challenges, it is of great interest to discover key cellular targets to interfere the pathogenesis of cGVHD. Detailed investigation on APCs in the pathophysiology of cGVHD will provide insights into new potential therapeutic treatments, especially for patients with steroid-refractory cGVHD. Attributed to the broad investigations based on mouse cGVHD models, the functional contributions of different APCs to the pathogenesis of cGVHD have been uncovered which were considered to be promising targets for cGVHD treatment (Figure 1). These findings in mouse cGVHD models have been translated into the development of clinical medicines some of which have already showed beneficial results in clinical trials to treat patients with cGVHD (32, 65, 140, 141). However, challenges still remain due to the differences of pathogenesis and kinetics of disease occurrence between mouse models and patients with cGVHD. In addition, there is still lack of effective guidance for selection of optimal therapies for individual patients and none of the drugs available in clinic is effective for all patients with cGVHD. Considering the complexity of cGVHD pathophysiology, comprehensive strategies aiming at multiple APC targets may prove to be more promising in the future.
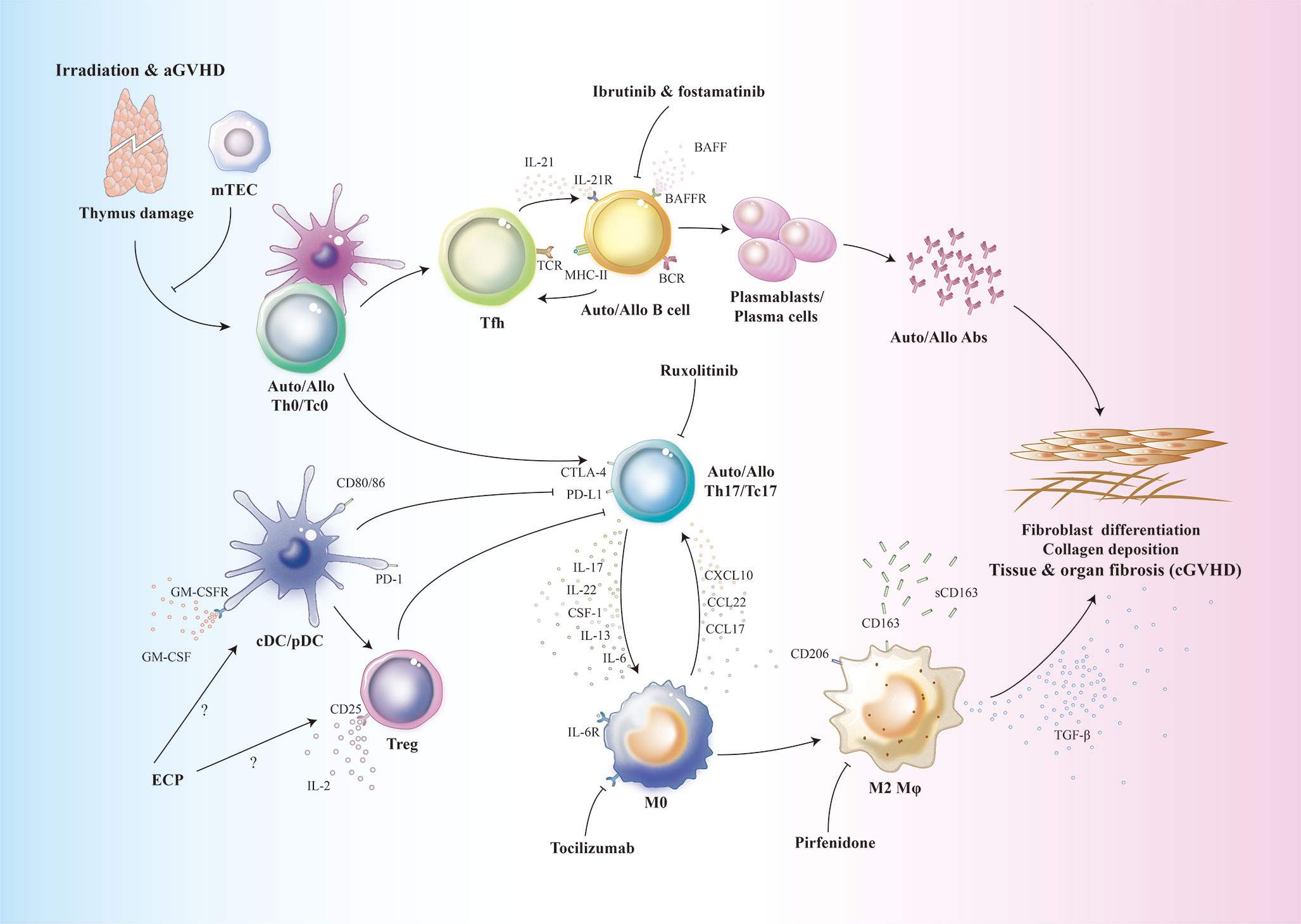
Figure 1 Schematic overview of the functional contributions of APCs to cGVHD. Conditioning regimens such as irradiation, chemotherapy as well as aGVHD cause host thymus damage. Thymic dysfunction contributes to the defective T cell negative selection and release of auto-/alloreactive T cells into periphery. These Th0/Tc0 cells are activated by host or donor DCs and differentiate into auto-/alloreactive Th17/Tc17 and T-follicular helper (Tfh) cells. In germinal center, Tfh cells produce IL-21 which results in activation and expansion of allo-/autoreactive B cells. Elevated levels of BAFF could also contribute to the aberrant B cell expansion. These auto-/alloreactive B cells differentiate into plasmablasts or plasma cells which produce auto-/alloantibodies. Host tissue resident macrophages are eliminated and replaced by donor monocyte derived tissue resident macrophages. These macrophages recruit auto-/alloreactive Th17/Tc17 cells via production of chemokines. After migration into target organs, auto-/alloreactive Th17/Tc17 cells further secrete IL-17 to induce more macrophage infiltration. Under the influence of multiple cytokines such as CSF-1, IL-13 and IL-6, donor monocyte derived macrophages are polarized into TGF-β-producing M2 macrophages. The profibrotic cytokine TGF-β, together with auto-/alloantibodies, contribute to the pathogenesis of cGVHD via inducing fibroblast differentiation into myofibroblasts which promote collagen synthesis and deposition in target organs and tissues. ECP, extracorporeal photopheresis; Fostamatinib, a Syk inhibitor; Ibrutinib, Bruton’s tyrosine kinase inhibitor; Pirfenidone, an anti-fibrotic drug; Ruxolitinib, a selective JAK1/2 inhibitor; Tocilizumab, anti-IL-6R monoclonal antibody.
Author Contributions
CH, RJ, XD, and XG collected all the literatures for reviewing and wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grant from National Key Research and Development Program of China (2017YFA0104502).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Toubai T, Mathewson ND, Magenau J, Reddy P. Danger Signals and Graft-versus-host Disease: Current Understanding and Future Perspectives. Front Immunol (2016) 7:539. doi: 10.3389/fimmu.2016.00539
2. Choi SW, Levine JE, Ferrara JL. Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin North Am (2010) 30:75–101. doi: 10.1016/j.iac.2009.10.001
3. Reddy P. Pathophysiology of acute graft-versus-host disease. Hematol Oncol (2003) 21:149–61. doi: 10.1002/hon.716
4. Jagasia MH, Greinix HT, Arora M, Williams KM, Wolff D, Cowen EW, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant (2015) 21:389–401.e1. doi: 10.1016/j.bbmt.2014.12.001
5. Lee SJ. Classification systems for chronic graft-versus-host disease. Blood (2017) 129:30–7. doi: 10.1182/blood-2016-07-686642
6. Arai S, Arora M, Wang T, Spellman SR, He W, Couriel DR, et al. Increasing incidence of chronic graft-versus-host disease in allogeneic transplantation: a report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant (2015) 21:266–74. doi: 10.1016/j.bbmt.2014.10.021
7. Lee SJ, Flowers ME. Recognizing and managing chronic graft-versus-host disease. Hematol Am Soc Hematol Educ Program (2008) 1:134–41. doi: 10.1182/asheducation-2008.1.134
8. Anasetti C, Logan BR, Lee SJ, Waller EK, Weisdorf DJ, Wingard JR, et al. Peripheral-blood stem cells versus bone marrow from unrelated donors. N Engl J Med (2012) 367:1487–96. doi: 10.1056/NEJMoa1203517
9. Grube M, Holler E, Weber D, Holler B, Herr W, Wolff D. Risk Factors and Outcome of Chronic Graft-versus-Host Disease after Allogeneic Stem Cell Transplantation-Results from a Single-Center Observational Study. Biol Blood Marrow Transplant (2016) 22:1781–91. doi: 10.1016/j.bbmt.2016.06.020
10. Flowers ME, Inamoto Y, Carpenter PA, Lee SJ, Kiem HP, Petersdorf EW, et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood (2011) 117:3214–9. doi: 10.1182/blood-2010-08-302109
11. Stem Cell Trialists’ Collaborative G. Allogeneic peripheral blood stem-cell compared with bone marrow transplantation in the management of hematologic malignancies: an individual patient data meta-analysis of nine randomized trials. J Clin Oncol (2005) 23:5074–87. doi: 10.1200/JCO.2005.09.020
12. Garnett C, Apperley JF, Pavlu J. Treatment and management of graft-versus-host disease: improving response and survival. Ther Adv Hematol (2013) 4:366–78. doi: 10.1177/2040620713489842
13. Macdonald KPA, Betts BC, Couriel D. Emerging Therapeutics for the Control of Chronic Graft-versus-Host Disease. Biol Blood Marrow Transplant (2018) 24:19–26. doi: 10.1016/j.bbmt.2017.10.006
14. Flowers ME, Storer B, Carpenter P, Rezvani AR, Vigorito AC, Campregher PV, et al. Treatment change as a predictor of outcome among patients with classic chronic graft-versus-host disease. Biol Blood Marrow Transplant (2008) 14:1380–4. doi: 10.1016/j.bbmt.2008.09.017
15. Wolff D, Schleuning M, Von Harsdorf S, Bacher U, Gerbitz A, Stadler M, et al. Consensus Conference on Clinical Practice in Chronic GVHD: Second-Line Treatment of Chronic Graft-versus-Host Disease. Biol Blood Marrow Transplant (2011) 17:1–17. doi: 10.1016/j.bbmt.2010.05.011
16. Hayakawa J, Miyamura D, Kimura SI, Gomyo A, Tamaki M, Akahoshi Y, et al. Negative impact of chronic graft-versus-host disease and glucocorticoid on the recovery of physical function after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant (2019) 54:994–1003. doi: 10.1038/s41409-018-0365-4
17. Modi B, Hernandez-Henderson M, Yang D, Klein J, Dadwal S, Kopp E, et al. Ruxolitinib as Salvage Therapy for Chronic Graft-versus-Host Disease. Biol Blood Marrow Transplant (2019) 25:265–9. doi: 10.1016/j.bbmt.2018.09.003
18. Mankarious M, Matthews NC, Snowden JA, Alfred A. Extracorporeal Photopheresis (ECP) and the Potential of Novel Biomarkers in Optimizing Management of Acute and Chronic Graft vs. Host Disease (GvHD). Front Immunol (2020) 11:81. doi: 10.3389/fimmu.2020.00081
19. Cooke KR, Luznik L, Sarantopoulos S, Hakim FT, Jagasia M, Fowler DH, et al. The Biology of Chronic Graft-versus-Host Disease: A Task Force Report from the National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease. Biol Blood Marrow Transplant (2017) 23:211–34. doi: 10.1016/j.bbmt.2016.09.023
20. Markey KA, Macdonald KP, Hill GR. The biology of graft-versus-host disease: experimental systems instructing clinical practice. Blood (2014) 124:354–62. doi: 10.1182/blood-2014-02-514745
21. Flowers ME, Martin PJ. How we treat chronic graft-versus-host disease. Blood (2015) 125:606–15. doi: 10.1182/blood-2014-08-551994
22. Kroger N, Solano C, Wolschke C, Bandini G, Patriarca F, Pini M, et al. Antilymphocyte Globulin for Prevention of Chronic Graft-versus-Host Disease. N Engl J Med (2016) 374:43–53. doi: 10.1056/NEJMoa1506002
23. Kanakry CG, Tsai HL, Bolanos-Meade J, Smith BD, Gojo I, Kanakry JA, et al. Single-agent GVHD prophylaxis with posttransplantation cyclophosphamide after myeloablative, HLA-matched BMT for AML, ALL, and MDS. Blood (2014) 124:3817–27. doi: 10.1182/blood-2014-07-587477
24. Socie G, Schmoor C, Bethge WA, Ottinger HD, Stelljes M, Zander AR, et al. Chronic graft-versus-host disease: long-term results from a randomized trial on graft-versus-host disease prophylaxis with or without anti-T-cell globulin ATG-Fresenius. Blood (2011) 117:6375–82. doi: 10.1182/blood-2011-01-329821
25. Luznik L, Bolanos-Meade J, Zahurak M, Chen AR, Smith BD, Brodsky R, et al. High-dose cyclophosphamide as single-agent, short-course prophylaxis of graft-versus-host disease. Blood (2010) 115:3224–30. doi: 10.1182/blood-2009-11-251595
26. Zeiser R, Blazar BR. Pathophysiology of Chronic Graft-versus-Host Disease and Therapeutic Targets. N Engl J Med (2017) 377:2565–79. doi: 10.1056/NEJMra1703472
27. Anderson BE, Mcniff JM, Jain D, Blazar BR, Shlomchik WD, Shlomchik MJ. Distinct roles for donor- and host-derived antigen-presenting cells and costimulatory molecules in murine chronic graft-versus-host disease: requirements depend on target organ. Blood (2005) 105:2227–34. doi: 10.1182/blood-2004-08-3032
28. Anderson BE, Mcniff JM, Matte C, Athanasiadis I, Shlomchik WD, Shlomchik MJ. Recipient CD4+ T cells that survive irradiation regulate chronic graft-versus-host disease. Blood (2004) 104:1565–73. doi: 10.1182/blood-2004-01-0328
29. Baird K, Pavletic SZ. Chronic graft versus host disease. Curr Opin Hematol (2006) 13:426–35. doi: 10.1097/01.moh.0000245689.47333.ff
30. Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant (2005) 11:945–56. doi: 10.1016/j.bbmt.2005.09.004
31. Schroeder MA, Dipersio JF. Mouse models of graft-versus-host disease: advances and limitations. Dis Model Mech (2011) 4:318–33. doi: 10.1242/dmm.006668
32. Dubovsky JA, Flynn R, Du J, Harrington BK, Zhong Y, Kaffenberger B, et al. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J Clin Invest (2014) 124:4867–76. doi: 10.1172/JCI75328
33. Reddy P, Negrin R, Hill GR. Mouse models of bone marrow transplantation. Biol Blood Marrow Transplant (2008) 14:129–35. doi: 10.1016/j.bbmt.2007.10.021
34. Morris SC, Cheek RL, Cohen PL, Eisenberg RA. Allotype-specific immunoregulation of autoantibody production by host B cells in chronic graft-versus host disease. J Immunol (1990) 144:916–22.
35. Morris SC, Cheek RL, Cohen PL, Eisenberg RA. Autoantibodies in chronic graft versus host result from cognate T-B interactions. J Exp Med (1990) 171:503–17. doi: 10.1084/jem.171.2.503
36. Chu YW, Gress RE. Murine models of chronic graft-versus-host disease: insights and unresolved issues. Biol Blood Marrow Transplant (2008) 14:365–78. doi: 10.1016/j.bbmt.2007.12.002
37. Hill GR, Olver SD, Kuns RD, Varelias A, Raffelt NC, Don AL, et al. Stem cell mobilization with G-CSF induces type 17 differentiation and promotes scleroderma. Blood (2010) 116:819–28. doi: 10.1182/blood-2009-11-256495
38. Macdonald KP, Rowe V, Filippich C, Johnson D, Morris ES, Clouston AD, et al. Chronic graft-versus-host disease after granulocyte colony-stimulating factor-mobilized allogeneic stem cell transplantation: the role of donor T-cell dose and differentiation. Biol Blood Marrow Transplant (2004) 10:373–85. doi: 10.1016/j.bbmt.2004.02.002
39. Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JL. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood (1997) 90:3204–13. doi: 10.1182/blood.V90.8.3204
40. Chen X, Vodanovic-Jankovic S, Johnson B, Keller M, Komorowski R, Drobyski WR. Absence of regulatory T-cell control of TH1 and TH17 cells is responsible for the autoimmune-mediated pathology in chronic graft-versus-host disease. Blood (2007) 110:3804–13. doi: 10.1182/blood-2007-05-091074
41. Flynn R, Du J, Veenstra RG, Reichenbach DK, Panoskaltsis-Mortari A, Taylor PA, et al. Increased T follicular helper cells and germinal center B cells are required for cGVHD and bronchiolitis obliterans. Blood (2014) 123:3988–98. doi: 10.1182/blood-2014-03-562231
42. Perruche S, Marandin A, Kleinclauss F, Angonin R, Fresnay S, Baron MH, et al. Association of mixed hematopoietic chimerism with elevated circulating autoantibodies and chronic graft-versus-host disease occurrence. Transplantation (2006) 81:573–82. doi: 10.1097/01.tp.0000183878.53367.77
43. Sonntag K, Eckert F, Welker C, Muller H, Muller F, Zips D, et al. Chronic graft-versus-host-disease in CD34(+)-humanized NSG mice is associated with human susceptibility HLA haplotypes for autoimmune disease. J Autoimmun (2015) 62:55–66. doi: 10.1016/j.jaut.2015.06.006
44. Sakoda Y, Hashimoto D, Asakura S, Takeuchi K, Harada M, Tanimoto M, et al. Donor-derived thymic-dependent T cells cause chronic graft-versus-host disease. Blood (2007) 109:1756–64. doi: 10.1182/blood-2006-08-042853
45. Zhang Y, Hexner E, Frank D, Emerson SG. CD4+ T cells generated de novo from donor hemopoietic stem cells mediate the evolution from acute to chronic graft-versus-host disease. J Immunol (2007) 179:3305–14. doi: 10.4049/jimmunol.179.5.3305
46. Forcade E, Paz K, Flynn R, Griesenauer B, Amet T, Li W, et al. An activated Th17-prone T cell subset involved in chronic graft-versus-host disease sensitive to pharmacological inhibition. JCI Insight (2017) 2:e92111. doi: 10.1172/jci.insight.92111
47. Okamoto S, Fujiwara H, Nishimori H, Matsuoka K, Fujii N, Kondo E, et al. Anti-IL-12/23 p40 antibody attenuates experimental chronic graft-versus-host disease via suppression of IFN-gamma/IL-17-producing cells. J Immunol (2015) 194:1357–63. doi: 10.4049/jimmunol.1400973
48. Fujiwara H, Maeda Y, Kobayashi K, Nishimori H, Matsuoka K, Fujii N, et al. Programmed death-1 pathway in host tissues ameliorates Th17/Th1-mediated experimental chronic graft-versus-host disease. J Immunol (2014) 193:2565–73. doi: 10.4049/jimmunol.1400954
49. Le Huu D, Matsushita T, Jin G, Hamaguchi Y, Hasegawa M, Takehara K, et al. Donor-derived regulatory B cells are important for suppression of murine sclerodermatous chronic graft-versus-host disease. Blood (2013) 121:3274–83. doi: 10.1182/blood-2012-11-465658
50. Leveque-El Mouttie L, Koyama M, Le Texier L, Markey KA, Cheong M, Kuns RD, et al. Corruption of dendritic cell antigen presentation during acute GVHD leads to regulatory T-cell failure and chronic GVHD. Blood (2016) 128:794–804. doi: 10.1182/blood-2015-11-680876
51. Alexander KA, Flynn R, Lineburg KE, Kuns RD, Teal BE, Olver SD, et al. CSF-1-dependant donor-derived macrophages mediate chronic graft-versus-host disease. J Clin Invest (2014) 124:4266–80. doi: 10.1172/JCI75935
52. Macdonald KP, Blazar BR, Hill GR. Cytokine mediators of chronic graft-versus-host disease. J Clin Invest (2017) 127:2452–63. doi: 10.1172/JCI90593
53. Macdonald KP, Hill GR, Blazar BR. Chronic graft-versus-host disease: biological insights from preclinical and clinical studies. Blood (2017) 129:13–21. doi: 10.1182/blood-2016-06-686618
54. Vakkila J, Thomson AW, Hovi L, Vettenranta K, Saarinen-Pihkala UM. Circulating dendritic cell subset levels after allogeneic stem cell transplantation in children correlate with time post transplant and severity of acute graft-versus-host disease. Bone Marrow Transplant (2005) 35:501–7. doi: 10.1038/sj.bmt.1704827
55. Mohty M, Blaise D, Faucher C, Bardou VJ, Gastaut JA, Viens P, et al. Impact of plasmacytoid dendritic cells on outcome after reduced-intensity conditioning allogeneic stem cell transplantation. Leukemia (2005) 19:1–6. doi: 10.1038/sj.leu.2403558
56. Hotta M, Yoshimura H, Satake A, Tsubokura Y, Ito T, Nomura S. GM-CSF therapy inhibits chronic graft-versus-host disease via expansion of regulatory T cells. Eur J Immunol (2019) 49:179–91. doi: 10.1002/eji.201847684
57. Jin H, Ni X, Deng R, Song Q, Young J, Cassady K, et al. Antibodies from donor B cells perpetuate cutaneous chronic graft-versus-host disease in mice. Blood (2016) 127:2249–60. doi: 10.1182/blood-2015-09-668145
58. Srinivasan M, Flynn R, Price A, Ranger A, Browning JL, Taylor PA, et al. Donor B-cell alloantibody deposition and germinal center formation are required for the development of murine chronic GVHD and bronchiolitis obliterans. Blood (2012) 119:1570–80. doi: 10.1182/blood-2011-07-364414
59. Miklos DB, Kim HT, Miller KH, Guo L, Zorn E, Lee SJ, et al. Antibody responses to H-Y minor histocompatibility antigens correlate with chronic graft-versus-host disease and disease remission. Blood (2005) 105:2973–8. doi: 10.1182/blood-2004-09-3660
60. Svegliati S, Olivieri A, Campelli N, Luchetti M, Poloni A, Trappolini S, et al. Stimulatory autoantibodies to PDGF receptor in patients with extensive chronic graft-versus-host disease. Blood (2007) 110:237–41. doi: 10.1182/blood-2007-01-071043
61. Young JS, Wu T, Chen Y, Zhao D, Liu H, Yi T, et al. Donor B cells in transplants augment clonal expansion and survival of pathogenic CD4+ T cells that mediate autoimmune-like chronic graft-versus-host disease. J Immunol (2012) 189:222–33. doi: 10.4049/jimmunol.1200677
62. Nguyen V, Luzina I, Rus H, Tegla C, Chen C, Rus V. IL-21 promotes lupus-like disease in chronic graft-versus-host disease through both CD4 T cell- and B cell-intrinsic mechanisms. J Immunol (2012) 189:1081–93. doi: 10.4049/jimmunol.1200318
63. Shao L, Lie AK, Zhang Y, Wong CH, Kwong YL. Aberrant germinal center formation, follicular T-helper cells, and germinal center B-cells were involved in chronic graft-versus-host disease. Ann Hematol (2015) 94:1493–504. doi: 10.1007/s00277-015-2394-z
64. Sarantopoulos S, Stevenson KE, Kim HT, Cutler CS, Bhuiya NS, Schowalter M, et al. Altered B-cell homeostasis and excess BAFF in human chronic graft-versus-host disease. Blood (2009) 113:3865–74. doi: 10.1182/blood-2008-09-177840
65. Du J, Paz K, Flynn R, Vulic A, Robinson TM, Lineburg KE, et al. Pirfenidone ameliorates murine chronic GVHD through inhibition of macrophage infiltration and TGF-beta production. Blood (2017) 129:2570–80. doi: 10.1182/blood-2017-01-758854
66. Yamakawa T, Ohigashi H, Hashimoto D, Hayase E, Takahashi S, Miyazaki M, et al. Vitamin A-coupled liposomes containing siRNA against HSP47 ameliorate skin fibrosis in chronic graft-versus-host disease. Blood (2018) 131:1476–85. doi: 10.1182/blood-2017-04-779934
67. Ono R, Watanabe T, Kawakami E, Iwasaki M, Tomizawa-Murasawa M, Matsuda M, et al. Co-activation of macrophages and T cells contribute to chronic GVHD in human IL-6 transgenic humanised mouse model. EBioMedicine (2019) 41:584–96. doi: 10.1016/j.ebiom.2019.02.001
68. Hayashida JN, Nakamura S, Toyoshima T, Moriyama M, Sasaki M, Kawamura E, et al. Possible involvement of cytokines, chemokines and chemokine receptors in the initiation and progression of chronic GVHD. Bone Marrow Transplant (2013) 48:115–23. doi: 10.1038/bmt.2012.100
69. Inamoto Y, Martin PJ, Paczesny S, Tabellini L, Momin AA, Mumaw CL, et al. Association of Plasma CD163 Concentration with De Novo-Onset Chronic Graft-versus-Host Disease. Biol Blood Marrow Transplant (2017) 23:1250–6. doi: 10.1016/j.bbmt.2017.04.019
70. Hu R, Liu Y, Su M, Song Y, Rood D, Lai L. Transplantation of Donor-Origin Mouse Embryonic Stem Cell-Derived Thymic Epithelial Progenitors Prevents the Development of Chronic Graft-versus-Host Disease in Mice. Stem Cells Transl Med (2017) 6:121–30. doi: 10.5966/sctm.2016-0012
71. Muller AMS, Min D, Wernig G, Levy RB, Perez VL, Herretes S, et al. Modeling Chronic Graft-versus-Host Disease in MHC-Matched Mouse Strains: Genetics, Graft Composition, and Tissue Targets. Biol Blood Marrow Transplant (2019) 25:2338–49. doi: 10.1016/j.bbmt.2019.08.001
72. Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol (2019) 19:89–103. doi: 10.1038/s41577-018-0088-1
73. Audiger C, Rahman MJ, Yun TJ, Tarbell KV, Lesage S. The Importance of Dendritic Cells in Maintaining Immune Tolerance. J Immunol (2017) 198:2223–31. doi: 10.4049/jimmunol.1601629
74. Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol (2013) 13:566–77. doi: 10.1038/nri3477
75. Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol (2003) 21:139–76. doi: 10.1146/annurev.immunol.21.120601.141107
76. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol (2014) 14:377–91. doi: 10.1038/nri3667
77. Wu T, Young JS, Johnston H, Ni X, Deng R, Racine J, et al. Thymic damage, impaired negative selection, and development of chronic graft-versus-host disease caused by donor CD4+ and CD8+ T cells. J Immunol (2013) 191:488–99. doi: 10.4049/jimmunol.1300657
78. Van Den Brink MR, Moore E, Ferrara JL, Burakoff SJ. Graft-versus-host-disease-associated thymic damage results in the appearance of T cell clones with anti-host reactivity. Transplantation (2000) 69:446–9. doi: 10.1097/00007890-200002150-00026
79. Teshima T, Reddy P, Liu C, Williams D, Cooke KR, Ferrara JL. Impaired thymic negative selection causes autoimmune graft-versus-host disease. Blood (2003) 102:429–35. doi: 10.1182/blood-2003-01-0266
80. Lee YJ, Min HS, Kang EH, Park HJ, Jeon YK, Kim JH, et al. Sclerodermatous chronic graft-versus-host disease induced by host T-cell-mediated autoimmunity. Immunol Cell Biol (2012) 90:358–67. doi: 10.1038/icb.2011.46
81. Yu H, Tian Y, Wang Y, Mineishi S, Zhang Y. Dendritic Cell Regulation of Graft-Vs.-Host Disease: Immunostimulation and Tolerance. Front Immunol (2019) 10:93. doi: 10.3389/fimmu.2019.00093
82. Stenger EO, Turnquist HR, Mapara MY, Thomson AW. Dendritic cells and regulation of graft-versus-host disease and graft-versus-leukemia activity. Blood (2012) 119:5088–103. doi: 10.1182/blood-2011-11-364091
83. Chung CY, Ysebaert D, Berneman ZN, Cools N. Dendritic cells: cellular mediators for immunological tolerance. Clin Dev Immunol (2013) 2013:972865. doi: 10.1155/2013/972865
84. Probst HC, Mccoy K, Okazaki T, Honjo T, Van Den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol (2005) 6:280–6. doi: 10.1038/ni1165
85. Vanasek TL, Khoruts A, Zell T, Mueller DL. Antagonistic roles for CTLA-4 and the mammalian target of rapamycin in the regulation of clonal anergy: enhanced cell cycle progression promotes recall antigen responsiveness. J Immunol (2001) 167:5636–44. doi: 10.4049/jimmunol.167.10.5636
86. Blazar BR, Carreno BM, Panoskaltsis-Mortari A, Carter L, Iwai Y, Yagita H, et al. Blockade of programmed death-1 engagement accelerates graft-versus-host disease lethality by an IFN-gamma-dependent mechanism. J Immunol (2003) 171:1272–7. doi: 10.4049/jimmunol.171.3.1272
87. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
88. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol (2018) 19:665–73. doi: 10.1038/s41590-018-0120-4
89. Zorn E, Kim HT, Lee SJ, Floyd BH, Litsa D, Arumugarajah S, et al. Reduced frequency of FOXP3+ CD4+CD25+ regulatory T cells in patients with chronic graft-versus-host disease. Blood (2005) 106:2903–11. doi: 10.1182/blood-2005-03-1257
90. Rieger K, Loddenkemper C, Maul J, Fietz T, Wolff D, Terpe H, et al. Mucosal FOXP3+ regulatory T cells are numerically deficient in acute and chronic GvHD. Blood (2006) 107:1717–23. doi: 10.1182/blood-2005-06-2529
91. Matsuoka K, Kim HT, Mcdonough S, Bascug G, Warshauer B, Koreth J, et al. Altered regulatory T cell homeostasis in patients with CD4+ lymphopenia following allogeneic hematopoietic stem cell transplantation. J Clin Invest (2010) 120:1479–93. doi: 10.1172/JCI41072
92. Elias S, Rudensky AY. Therapeutic use of regulatory T cells for graft-versus-host disease. Br J Haematol (2019) 187:25–38. doi: 10.1111/bjh.16157
93. Mcdonald-Hyman C, Flynn R, Panoskaltsis-Mortari A, Peterson N, Macdonald KP, Hill GR, et al. Therapeutic regulatory T-cell adoptive transfer ameliorates established murine chronic GVHD in a CXCR5-dependent manner. Blood (2016) 128:1013–7. doi: 10.1182/blood-2016-05-715896
94. Zou T, Caton AJ, Koretzky GA, Kambayashi T. Dendritic cells induce regulatory T cell proliferation through antigen-dependent and -independent interactions. J Immunol (2010) 185:2790–9. doi: 10.4049/jimmunol.0903740
95. Markey KA, Koyama M, Kuns RD, Lineburg KE, Wilson YA, Olver SD, et al. Immune insufficiency during GVHD is due to defective antigen presentation within dendritic cell subsets. Blood (2012) 119:5918–30. doi: 10.1182/blood-2011-12-398164
96. Matsuoka K, Koreth J, Kim HT, Bascug G, Mcdonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med (2013) 5:179ra43. doi: 10.1126/scitranslmed.3005265
97. Koreth J, Kim HT, Jones KT, Lange PB, Reynolds CG, Chammas MJ, et al. Efficacy, durability, and response predictors of low-dose interleukin-2 therapy for chronic graft-versus-host disease. Blood (2016) 128:130–7. doi: 10.1182/blood-2016-02-702852
98. Kim N, Jeon YW, Nam YS, Lim JY, Im KI, Lee ES, et al. Therapeutic potential of low-dose IL-2 in a chronic GVHD patient by in vivo expansion of regulatory T cells. Cytokine (2016) 78:22–6. doi: 10.1016/j.cyto.2015.11.020
99. Koreth J, Matsuoka K, Kim HT, Mcdonough SM, Bindra B, Alyea EP, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med (2011) 365:2055–66. doi: 10.1056/NEJMoa1108188
100. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature (2013) 496:445–55. doi: 10.1038/nature12034
101. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol (2015) 33:643–75. doi: 10.1146/annurev-immunol-032414-112220
102. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest (2019) 129:2619–28. doi: 10.1172/JCI124615
103. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol (2013) 14:986–95. doi: 10.1038/ni.2705
104. Zhang Y, Mccormick LL, Desai SR, Wu C, Gilliam AC. Murine sclerodermatous graft-versus-host disease, a model for human scleroderma: cutaneous cytokines, chemokines, and immune cell activation. J Immunol (2002) 168:3088–98. doi: 10.4049/jimmunol.168.6.3088
105. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, Mcquattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med (2017) 214:2387–404. doi: 10.1084/jem.20162152
106. Nishiwaki S, Terakura S, Ito M, Goto T, Seto A, Watanabe K, et al. Impact of macrophage infiltration of skin lesions on survival after allogeneic stem cell transplantation: a clue to refractory graft-versus-host disease. Blood (2009) 114:3113–6. doi: 10.1182/blood-2009-03-209635
107. Fabriek BO, Moller HJ, Vloet RP, Van Winsen LM, Hanemaaijer R, Teunissen CE, et al. Proteolytic shedding of the macrophage scavenger receptor CD163 in multiple sclerosis. J Neuroimmunol (2007) 187:179–86. doi: 10.1016/j.jneuroim.2007.04.016
108. Timmermann M, Hogger P. Oxidative stress and 8-iso-prostaglandin F(2alpha) induce ectodomain shedding of CD163 and release of tumor necrosis factor-alpha from human monocytes. Free Radic Biol Med (2005) 39:98–107. doi: 10.1016/j.freeradbiomed.2005.02.031
109. Shimizu K, Ogawa F, Yoshizaki A, Akiyama Y, Kuwatsuka Y, Okazaki S, et al. Increased serum levels of soluble CD163 in patients with scleroderma. Clin Rheumatol (2012) 31:1059–64. doi: 10.1007/s10067-012-1972-x
110. Buechler C, Eisinger K, Krautbauer S. Diagnostic and prognostic potential of the macrophage specific receptor CD163 in inflammatory diseases. Inflammation Allergy Drug Targets (2013) 12:391–402. doi: 10.2174/18715281113126660060
111. Arai M, Ikawa Y, Chujo S, Hamaguchi Y, Ishida W, Shirasaki F, et al. Chemokine receptors CCR2 and CX3CR1 regulate skin fibrosis in the mouse model of cytokine-induced systemic sclerosis. J Dermatol Sci (2013) 69:250–8. doi: 10.1016/j.jdermsci.2012.10.010
112. Barron L, Wynn TA. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am J Physiol Gastrointest Liver Physiol (2011) 300:G723–8. doi: 10.1152/ajpgi.00414.2010
113. Yuan CY, Zhou V, Sauber G, Stollenwerk TM, Komorowski R, Lopez A, et al. Signaling Through the Type 2 Cannabinoid Receptor Regulates the Severity of Acute and Chronic Graft versus Host Disease. Blood (2020). doi: 10.1182/blood.2020004871
114. Cavet J, Dickinson AM, Norden J, Taylor PR, Jackson GH, Middleton PG. Interferon-gamma and interleukin-6 gene polymorphisms associate with graft-versus-host disease in HLA-matched sibling bone marrow transplantation. Blood (2001) 98:1594–600. doi: 10.1182/blood.v98.5.1594
115. Drobyski WR, Pasquini M, Kovatovic K, Palmer J, Douglas Rizzo J, Saad A, et al. Tocilizumab for the treatment of steroid refractory graft-versus-host disease. Biol Blood Marrow Transplant (2011) 17:1862–8. doi: 10.1016/j.bbmt.2011.07.001
116. Mcmanigle W, Youssef A, Sarantopoulos S. B cells in chronic graft-versus-host disease. Hum Immunol (2019) 80:393–9. doi: 10.1016/j.humimm.2019.03.003
117. Li X, Gao Q, Feng Y, Zhang X. Developing role of B cells in the pathogenesis and treatment of chronic GVHD. Br J Haematol (2019) 184:323–36. doi: 10.1111/bjh.15719
118. Yehudai-Ofir D, Henig I, Zuckerman T. Aberrant B cells, autoimmunity and the benefit of targeting B cells in chronic graft-versus-host disease. Autoimmun Rev (2020) 4:102493. doi: 10.1016/j.autrev.2020.102493
119. Shono Y, Ueha S, Wang Y, Abe J, Kurachi M, Matsuno Y, et al. Bone marrow graft-versus-host disease: early destruction of hematopoietic niche after MHC-mismatched hematopoietic stem cell transplantation. Blood (2010) 115:5401–11. doi: 10.1182/blood-2009-11-253559
120. Shono Y, Shiratori S, Kosugi-Kanaya M, Ueha S, Sugita J, Shigematsu A, et al. Bone marrow graft-versus-host disease: evaluation of its clinical impact on disrupted hematopoiesis after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2014) 20:495–500. doi: 10.1016/j.bbmt.2013.12.568
121. Kolupaev OV, Dant TA, Bommiasamy H, Bruce DW, Fowler KA, Tilley SL, et al. Impaired bone marrow B-cell development in mice with a bronchiolitis obliterans model of cGVHD. Blood Adv (2018) 2:2307–19. doi: 10.1182/bloodadvances.2017014977
122. Zeiser R, Sarantopoulos S, Blazar BR. B-cell targeting in chronic graft-versus-host disease. Blood (2018) 131:1399–405. doi: 10.1182/blood-2017-11-784017
123. Deng R, Hurtz C, Song Q, Yue C, Xiao G, Yu H, et al. Extrafollicular CD4(+) T-B interactions are sufficient for inducing autoimmune-like chronic graft-versus-host disease. Nat Commun (2017) 8:978. doi: 10.1038/s41467-017-00880-2
124. Ratanatharathorn V, Carson E, Reynolds C, Ayash LJ, Levine J, Yanik G, et al. Anti-CD20 chimeric monoclonal antibody treatment of refractory immune-mediated thrombocytopenia in a patient with chronic graft-versus-host disease. Ann Intern Med (2000) 133:275–9. doi: 10.7326/0003-4819-133-4-200008150-00011
125. Abdel-Azim H, Elshoury A, Mahadeo KM, Parkman R, Kapoor N. Humoral Immune Reconstitution Kinetics after Allogeneic Hematopoietic Stem Cell Transplantation in Children: A Maturation Block of IgM Memory B Cells May Lead to Impaired Antibody Immune Reconstitution. Biol Blood Marrow Transplant (2017) 23:1437–46. doi: 10.1016/j.bbmt.2017.05.005
126. Bohmann EM, Fehn U, Holler B, Weber D, Holler E, Herr W, et al. Altered immune reconstitution of B and T cells precedes the onset of clinical symptoms of chronic graft-versus-host disease and is influenced by the type of onset. Ann Hematol (2017) 96:299–310. doi: 10.1007/s00277-016-2881-x
127. Sanchez-Garcia J, Serrano J, Gomez P, Martinez F, Martin C, Roman-Gomez J, et al. The impact of acute and chronic graft-versus-host disease on normal and malignant B-lymphoid precursors after allogeneic stem cell transplantation for B-lineage acute lymphoblastic leukemia. Haematologica (2006) 91:340–7.
128. Storek J, Wells D, Dawson MA, Storer B, Maloney DG. Factors influencing B lymphopoiesis after allogeneic hematopoietic cell transplantation. Blood (2001) 98:489–91. doi: 10.1182/blood.v98.2.489
129. Allen JL, Fore MS, Wooten J, Roehrs PA, Bhuiya NS, Hoffert T, et al. B cells from patients with chronic GVHD are activated and primed for survival via BAFF-mediated pathways. Blood (2012) 120:2529–36. doi: 10.1182/blood-2012-06-438911
130. Ng LG, Sutherland AP, Newton R, Qian F, Cachero TG, Scott ML, et al. B cell-activating factor belonging to the TNF family (BAFF)-R is the principal BAFF receptor facilitating BAFF costimulation of circulating T and B cells. J Immunol (2004) 173:807–17. doi: 10.4049/jimmunol.173.2.807
131. Mackay F, Browning JL. BAFF: a fundamental survival factor for B cells. Nat Rev Immunol (2002) 2:465–75. doi: 10.1038/nri844
132. Schneider P, Mackay F, Steiner V, Hofmann K, Bodmer JL, Holler N, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med (1999) 189:1747–56. doi: 10.1084/jem.189.11.1747
133. Poe JC, Jia W, Su H, Anand S, Rose JJ, Tata PV, et al. An aberrant NOTCH2-BCR signaling axis in B cells from patients with chronic GVHD. Blood (2017) 130:2131–45. doi: 10.1182/blood-2017-05-782466
134. Patriarca F, Skert C, Sperotto A, Zaja F, Falleti E, Mestroni R, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol (2006) 34:389–96. doi: 10.1016/j.exphem.2005.12.011
135. Nakasone H, Tian L, Sahaf B, Kawase T, Schoenrock K, Perloff S, et al. Allogeneic HY antibodies detected 3 months after female-to-male HCT predict chronic GVHD and nonrelapse mortality in humans. Blood (2015) 125:3193–201. doi: 10.1182/blood-2014-11-613323
136. Wechalekar A, Cranfield T, Sinclair D, Ganzckowski M. Occurrence of autoantibodies in chronic graft vs. host disease after allogeneic stem cell transplantation. Clin Lab Haematol (2005) 27:247–9. doi: 10.1111/j.1365-2257.2005.00699.x
137. Kuzmina Z, Gounden V, Curtis L, Avila D, Rnp TT, Baruffaldi J, et al. Clinical significance of autoantibodies in a large cohort of patients with chronic graft-versus-host disease defined by NIH criteria. Am J Hematol (2015) 90:114–9. doi: 10.1002/ajh.23885
138. Wu Y, Schutt S, Paz K, Zhang M, Flynn RP, Bastian D, et al. MicroRNA-17-92 is required for T-cell and B-cell pathogenicity in chronic graft-versus-host disease in mice. Blood (2018) 131:1974–86. doi: 10.1182/blood-2017-06-789321
139. Wang N, Yigit B, Van Der Poel CE, Cuenca M, Carroll MC, Herzog RW, et al. The Checkpoint Regulator SLAMF3 Preferentially Prevents Expansion of Auto-Reactive B Cells Generated by Graft-vs.-Host Disease. Front Immunol (2019) 10:831. doi: 10.3389/fimmu.2019.00831
140. Jaglowski SM, Blazar BR. How ibrutinib, a B-cell malignancy drug, became an FDA-approved second-line therapy for steroid-resistant chronic GVHD. Blood Adv (2018) 2:2012–9. doi: 10.1182/bloodadvances.2018013060
141. Flynn R, Allen JL, Luznik L, Macdonald KP, Paz K, Alexander KA, et al. Targeting Syk-activated B cells in murine and human chronic graft-versus-host disease. Blood (2015) 125:4085–94. doi: 10.1182/blood-2014-08-595470
142. Hassan MN, Waller EK. GVHD clears the Aire in thymic selection. Blood (2015) 125:2593–5. doi: 10.1182/blood-2015-03-630871
143. Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, et al. Donor APCs are required for maximal GVHD but not for GVL. Nat Med (2004) 10:987–92. doi: 10.1038/nm1089
144. Reddy P, Maeda Y, Liu C, Krijanovski OI, Korngold R, Ferrara JL. A crucial role for antigen-presenting cells and alloantigen expression in graft-versus-leukemia responses. Nat Med (2005) 11:1244–9. doi: 10.1038/nm1309
Keywords: chronic graft versus host disease, allogeneic hematopoietic stem cell transplantation, antigen presenting cells, immune tolerance, immune regulation
Citation: Hong C, Jin R, Dai X and Gao X (2021) Functional Contributions of Antigen Presenting Cells in Chronic Graft-Versus-Host Disease. Front. Immunol. 12:614183. doi: 10.3389/fimmu.2021.614183
Received: 05 October 2020; Accepted: 11 January 2021;
Published: 24 February 2021.
Edited by:
Ranjit Kumar Sahoo, All India Institute of Medical Sciences, IndiaReviewed by:
Reem Al-Daccak, Institut National de la Santé et de la Recherche Médicale (INSERM), FrancePhilippe Saas, INSERM U1098 Interactions Hôte-Greffon-Tumeur & Ingénierie Cellulaire et Génique, France
Uday Prakash Kulkarni, Christian Medical College & Hospital, India
Copyright © 2021 Hong, Jin, Dai and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chao Hong, Y2hhb2hvbmdAc3VkYS5lZHUuY24=; Xiaoming Gao, eG1nYW9Ac3VkYS5lZHUuY24=