 Jennifer Wittwer
Jennifer Wittwer David Bradley
David Bradley
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 25 February 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.612496
This article is part of the Research TopicThe Immunomodulatory Roles of AdipocytesView all 11 articles
The cardiometabolic syndrome involves a clustering of metabolic and cardiovascular factors which increase the risk of patients developing both Type 2 Diabetes Mellitus and cardio/cerebrovascular disease. Although the mechanistic underpinnings of this link remain uncertain, key factors include insulin resistance, excess visceral adiposity, atherogenic dyslipidemia, and endothelial dysfunction. Of these, a state of resistance to insulin action in overweight/obese patients appears to be central to the pathophysiologic process. Given the increasing prevalence of obesity-related Type 2 Diabetes, coupled with the fact that cardiovascular disease is the number one cause of mortality in this patient population, a more thorough understanding of the cardiometabolic syndrome and potential options to mitigate its risk is imperative. Inherent in the pathogenesis of insulin resistance is an underlying state of chronic inflammation, at least partly in response to excess adiposity. Within obese adipose tissue, an immunomodulatory shift occurs, involving a preponderance of pro-inflammatory immune cells and cytokines/adipokines, along with antigen presentation by adipocytes. Therefore, various adipokines differentially expressed by obese adipocytes may have a significant effect on cardiometabolism. Clusterin is a molecular chaperone that is widely produced by many tissues throughout the body, but is also preferentially overexpressed by obese compared lean adipocytes and relates strongly to multiple components of the cardiometabolic syndrome. Herein, we summarize the known and potential roles of circulating and adipocyte-specific clusterin in cardiometabolism and discuss potential further investigations to determine if clusterin is a viable target to attenuate both metabolic and cardiovascular disease.
Although the exact diagnostic criteria varies (1–3), the metabolic syndrome involves a clustering of abnormalities including obesity, insulin resistance, hypertension, and dyslipidemia. These in turn heighten the risk of cardio- and cerebrovascular disease (CVD) [elevated risk of primary and recurrent stroke (4) and myocardial infarction (5)], Type 2 Diabetes Mellitus (T2D) (6, 7), and non-alcoholic fatty liver disease/steatohepatitis (NAFLD/NASH) (8). Initially termed the metabolic syndrome, Reaven's syndrome, or Syndrome X, among others (9, 10), the ramifications of metabolic disease on CVD risk have subsequently led to a broadening of terminology (i.e., the cardiometabolic syndrome). Although the criteria are the same (Table 1), the term cardiometabolic syndrome has gained more widespread acceptance due to the intersection of risk factors that contribute to both CVD and metabolic disease and involve similar pathophysiologic processes.
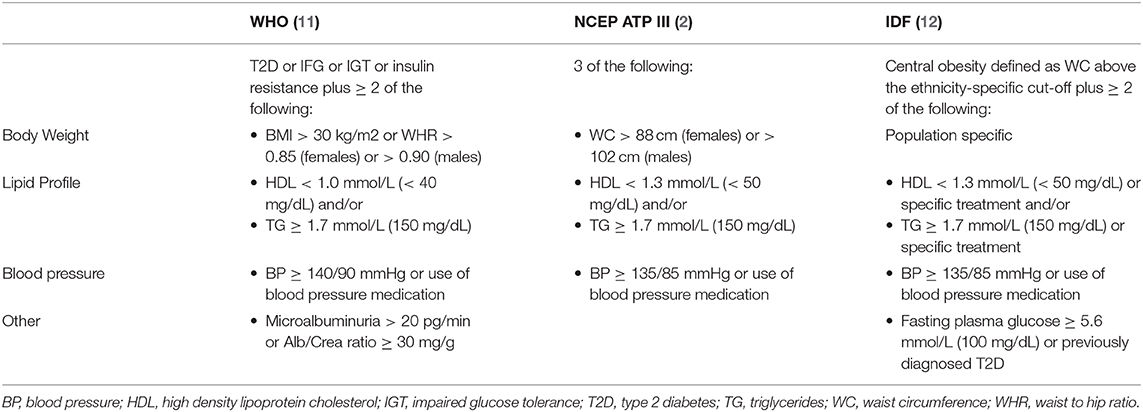
Table 1. Clinical definitions of the cardiometabolic syndrome based on the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults Adult Treatment Panel III, the International Diabetes Federation (IDF), and the World Health Organization (WHO).
The cardiometabolic syndrome is highly prevalent, affecting over 30% of the adult population in the United States (U.S.) and rising, with especially high prevalence rates (>40%) in patients older than 60 years old (13, 14). Compared to the general population, the relative risk for developing CVD with coexistent cardiometabolic syndrome is doubled (15), with 3-fold the risk of T2D (13). In addition, all-cause mortality is higher in those with the cardiometabolic syndrome. Importantly, factors related to ethnicity/race, gender, and socio-economics affect risk, with the highest rates occurring in non-Hispanic white men and black women (14). In addition, socio-economic factors such as low education level and advanced age are independently associated with a higher risk of the cardiometabolic syndrome. The reasons for these differences are incompletely understood and likely multifactorial, but remain a critical focus of future research with significant public health ramifications (16–19).
Over 35% of the adult US population is obese (20), and excess adiposity contributes to multiple complications including T2D and accelerated rates of CVD (21). In fact, CVD is the number one cause of mortality in diabetic patients, with a 2–3-fold higher risk of clinical atherosclerosis (22), illustrating a close association between metabolic disease and CV risk. As such, underlying the dysfunction in cardiometabolic disease are four interrelated central features: insulin resistance, excess visceral adiposity, atherogenic dyslipidemia, and endothelial dysfunction (23). Of these, obesity-related insulin resistance appears to the most important trigger. Among all the cardiometabolic risk factors, the relationship between insulin resistance and hypertension is the best established, and end-organ insulin resistance is a central tenet in its pathophysiology (24). Various mechanisms have been put forth to explain this connection including a decrease in insulin-mediated renal artery vasodilatation and uncompensated sodium reabsorption, with a resultant increase in blood pressure. Systemic and vascular insulin resistance occurs in conjunction with inappropriate activation of the renin–angiotensin–aldosterone system (RAAS) (25). Hyperinsulinemia also increases sympathetic nervous system activity (26), contributing further to the development of hypertension, a prominent component of the cardiometabolic syndrome.
Obesity and its associated comorbidities (including T2D and CVD) are associated with a state of chronic low-grade inflammation (27) that is well-recognized as a major cause of decreased insulin sensitivity (28–30). Inflammatory pathway activation has been observed in all classical insulin target tissues, indicating the key role of inflammation in driving the pathogenesis of systemic insulin resistance. Particularly, in adipose tissue (AT), macrophages play a central role (28, 31, 32); however, recent studies have highlighted the importance of several other key immune cells in maintaining lean AT, including immunosuppressive regulatory T (Treg) cells, which contribute to a “Type 2” anti-inflammatory immunoenvironment (33, 34). In obesity, this immunologic milieu is shifted to a more pro-inflammatory state, in which the normal architecture, energy storage, and endocrine activities of adipocytes are profoundly altered. Activation of a proinflammatory pathway in AT leads to the secretion of numerous cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (Il-6) and interleukin-1β (IL-1β) (35) that activate toll-like receptors (TLR2 and TLR4) and impair glucose uptake (36). Cytokines also impair suppression of AT lipolysis, with resultant free fatty acid (FFA) release into the circulation (37–39), which hinders the ability of insulin to stimulate muscle glucose uptake (40) and suppress hepatic glucose production (41), the two major factors in the pathogenesis of insulin resistance. Therefore, disruption in AT fatty acid metabolism is likely an underlying factor in cardiometabolic disease, by promoting both hyperglycemia and dyslipidemia.
Obesity, the cardiometabolic syndrome, and T2D have also long been associated with higher risk of cerebrovascular disease and cognitive decline (42–52). One potential reason for this connection is that insulin has direct effects on neurotransmission and neuropathology in the brain (53–56), including alterations in the production, degradation and clearance of β-amyloid (Aβ) that lead to plaque deposition in Alzheimer's disease (57). Various murine models of obesity and diabetes (including after high-fat diet feeding) (58–61) have indicated a relationship between peripheral and “central” insulin resistance, and in humans altered metabolic brain activity occurs in peripherally insulin-resistant subjects (62–64), with dysregulation in CNS insulin signaling (65–67). In fact, intravenous insulin infusion (57, 68, 69), inhaled insulin (69, 70), the insulin-sensitizing agent pioglitazone (70, 71), metformin (72, 73), and weight-loss interventions, including bariatric surgery, have demonstrated beneficial effects on memory (74–77). Cerebrovascular disease (78–80) and vascular dementia (81, 82) are also strongly related to insulin resistance, even independent of frank diabetes, and the Insulin Resistance Intervention after Stroke (IRIS) trial established that improving insulin sensitivity can prevent cerebrovascular events (83).
The human clusterin (CLU) gene (encodes the protein clusterin/apolipoprotein J) was first identified by Blaschuk et al. (84). This highly conserved gene consists of nine exons located on chromosome 8 that encode different isoforms resulting from alternative splicing and post-translational modifications (glycosylation, disulfide bond cleavage, etc.) (85, 86). The CLU gene promoter is highly conserved among species, with numerous identified regulatory elements including TGF-β inhibitory element, activator protein-1 and−2, and nuclear factor, but is also responsive to many environmental and cytokines that vary depending on the involved tissue (87–89). Although expressed by nearly every tissue in the human body, clusterin is predominantly made by epithelial tissues during embryonic development and in the testis, ovary, adrenal gland, liver, heart and brain of adults (85, 86). Its identified receptors are varied and often tissue-specific and include the HDL cholesterol receptor, low density lipoprotein-related protein 2 (LRP/megalin) (90), ApoER2 (91), and very low density lipoprotein receptor (VLDLR), many of which are critical to cardiovascular health.
There are two major forms of clusterin: a stress-induced, non-glycosylated, nucleocytostolic 55kDa variant (nCLU) consisting of parallel α and β chains, and a secreted or cytosolic variant (sCLU) that is proteolytically cleaved, connected by five disulfide bonds, and released from cells in an antiparallel fashion (92). Heterodimeric sCLU circulates mainly as a component of high-density lipoprotein (HDL) cholesterol, but has also been found to be bound to apolipoprotein (Apo) A1, various lipids, paroxanase, beta (β)-amyloid protein, and complement proteins, among others [summarized in Trougakos and Gonos (93)]. In healthy subjects, a higher prevalence of sCLU is bound to cardioprotective HDL cholesterol, suggesting that secreted clusterin may play a role in preventing progression of vascular disease (94). In contrast, nCLU predominantly promotes ionizing radiation-induced death of cells and triggers apoptosis in a BAX-dependent mechanism, and has yet to be linked with cardiometabolic pathology (95). Therefore, the remainder of this review will focus on the relationship of CVD and metabolic disease with sCLU.
One of the major roles of clusterin is to act as a molecular chaperone that assists folding of secreted proteins (87). Clusterin may also serve as a sensor of oxidative stress and is reduced upon exposure to acute stress (96). As a result of its ubiquitous nature, it has been implicated in a wide range of pathologic processes including cancer development and progression, complement regulation, and sperm maturation (93, 97, 98). CLU gene transcription and protein expression is upregulated in breast cancer (99), ovarian cancer (100), and prostate cancer (101), and inhibition of CLU expression protects the cell from apoptosis induced by chemotherapy, radiotherapy, and androgen/estrogen depletion (102–104). Clusterin is also involved in CNS lipid trafficking (105, 106) and is widely expressed in the brain (107). Accordingly, clusterin has clinical associations with Alzheimer's disease (AD) (108, 109) and has been proposed as a biomarker of AD (110). In fact, risk variants in CLU are strongly associated with AD (108). In patients with both mild cognitive impairment and AD, clusterin levels are elevated in the brain, cerebrospinal fluid, and blood (111–114), and accordingly CLU gene expression is elevated in these pathologic conditions (107).
There are numerous identified mechanisms by which circulating clusterin could impact the risk of metabolic disease. Leptin resistance has been demonstrated in both murine models and human obesity, with reduced transport across the blood-brain-barrier (BB) (115). In turn, sCLU affects the transport of leptin across the BBB via LDL cholesterol (116), and through its binding to the receptor LRP2 can sensitize leptin receptors in the hypothalamus (117). This suggests that clusterin may play a role in modulating appetite and contributing to obesity (117). Clusterin can also directly affect insulin signaling and inflammation, two factors that can lead to insulin resistance, via its actions on macrophage phosphoinositide 3-kinase (PI3K; a mediator of insulin signaling) and NFκB (a major pro-inflammatory pathway in insulin resistance) (118). Clusterin induces directional migration of macrophages acting as a chemoattractant (119). This stimulates the expression and secretion of TNF-α and various chemotactic cytokines allowing clusterin to serve as a link between inflammation and remodeling of tissues by directing immune cells (120). Therefore, clusterin plays a significant role in inflammation and immune responses through its molecular interactions with complement factors, immunoglobulins, and inflammatory pathways (121).
In support of these identified mechanistic processes, both murine and human studies have demonstrated a significant link between circulating clusterin and features of the metabolic syndrome. Skeletal muscle and hepatic gene expression of CLU increase following high-fat diet feeding in mice, and whole body clusterin knockout mice are insulin sensitive compared to wild-type mice (122). Obese patients without diabetes following a 2 week very low calorie diet have reduced plasma clusterin levels (123), and in obese compared to lean subjects, plasma clusterin levels are elevated and positively relate to body mass index, waist circumference, markers of inflammation (hsCRP and retinol-binding protein-4) (124), and insulin resistance (125). In addition, polymorphisms in CLU have been linked to insulin resistance [by the homeostasis model of insulin resistance [HOMA-IR] and impaired insulin secretion [HOMA-β]] (126). In contrast to these deleterious metabolic effects, clusterin has been shown to reduce hepatic fibrosis via stellate cell downregulation of the Smad3 signaling pathway (127).
The mechanistic effects of circulating clusterin on CVD are controversial, due to seemingly paradoxical effects in the existing literature, and the mechanisms behind such a link remain unclear. Clusterin is found in a subset of dense HDL cholesterol particles and has wide-ranging effects on lipid transport (121, 128). In plasma, clusterin forms HDL particles with ApoA-I and ApoE and aids in the transfer of HDL cholesterol from peripheral tissues to the liver, diverting lipoproteins away from atherosclerotic lesions (129, 130). In contrast, clusterin may have a deleterious effect on the antioxidant activity of paroxanase-1 (PON1), whose deficiency enhances atherosclerosis by increasing the accumulation of oxidized phospholipids in atherosclerotic plaques (131).
There are multiple lines of evidence suggesting that human clusterin may have a significant clinical association with multiple facets of cardiovascular risk. Circulating plasma clusterin (sCLU) levels are strongly associated with the pro-inflammatory factor C-reactive protein (CRP) (124), various lipid markers of heightened cardiovascular risk, and increasing systolic and diastolic blood pressure (90, 132). Circulating clusterin is also negatively associated with leptin in obesity-related CVD (133). In addition, clusterin bound to HDL cholesterol is reduced in obese males and is associated with lower levels of HDL cholesterol, higher TGs (134) and low-density lipoprotein (LDL) cholesterol levels, and accelerated atherogenesis (135), and may confer higher cardiovascular risk during the aging process (135). Interestingly, proteomic analysis has shown that higher levels of clusterin are found in carotid atherosclerotic compared to non-atherosclerotic plaques (136). Not all studies, however, have confirmed a beneficial role for clusterin in CVD. A recent study showed that lower serum clusterin was associated with higher rates of mortality in heart failure patients (137), indicating some uncertainty on the importance of circulating clusterin in the CVD process.
The adipocyte is no longer viewed as simply a storage depot for lipids, but is now recognized as an important determinant of an obesity-related proinflammatory environment, instigating inflammation in expanding AT (138). Despite significant progress in our understanding of the role of the adipocyte as an immumodulator, and evidence that circulating plasma and HDL cholesterol bound clusterin may be involved in the metabolic syndrome, insulin resistance, atherogenesis, and CV risk, the importance of adipocyte-derived clusterin in human cardiometabolic disease remains largely unknown. In whole human AT, CLU gene expression is higher in obese compared to lean subjects, and is decreased following weight loss induced by VLCD or bariatric surgery (123). We have recently shown that clusterin derived specifically from the adipocyte may play an important role in cardiometabolic disease (90). In obese compared to lean human subjects, adipocyte gene expression and protein levels of clusterin were higher and responsive to (FFA) palmitate stimulation (a major component of a high fat diet enriched in fatty acids) (139). In addition, we found strong associations of adipocyte clusterin with systemic insulin resistance, multiple components of the metabolic syndrome (HDL cholesterol, the ratio of HDL cholesterol to total cholesterol, and TGs, and both systolic and diastolic blood pressure), and overall CVD risk and mortality. In this same study, clusterin treatment of human liver cells reduced insulin signaling by lowering Akt phosphorylation and promoting key genes involved in gluconeogenesis; yet hepatic expression of the major regulator of hepatic de novo lipogenesis [sterol regulatory element-binding protein-1 [SREBP-1]] and APOA1 were decreased in response to clusterin binding to LRP2. These results suggest that the liver receptor LRP2 may be a key target for the potential cardiometabolic role of clusterin. Knockdown of SREBP-1 can perpetuate hyperglycemia via enhanced gluconeogenesis and reduced glycolysis and glycogen synthesis (140). APOA1 is a major protein associated with HDL cholesterol particles in plasma which facilitates efflux of cholesterol from cells, notably from macrophages within atherosclerotic plaques, to the liver for excretion. Low plasma APOA1 levels are also a strong predictor of CVD (141). In a mouse model prone to non-alcoholic steatohepatitis (NASH) adipocyte CLU expression also paralleled an increase in liver fat, hepatic fibrosis, and steatohepatitis (90).
Although these results suggest several mechanisms by which clusterin could link insulin resistance, metabolic disease, and CVD (Figure 1), further investigation is needed to fully elucidate the cardiometabolic role of AT clusterin, and specifically clusterin derived from the adipocyte. Although treatment with the FFA palmitate stimulates clusterin release in vitro, other potential triggers for clusterin expression are possible. These include AT hypoxia, which has previously been shown to increase clusterin expression in other cell types outside of AT (142). In addition, the effects of adipocyte-derived clusterin on the AT immunoenvironment and the skewed balance of pro- and anti-inflammatory cytokines observed in human obesity is also unknown.
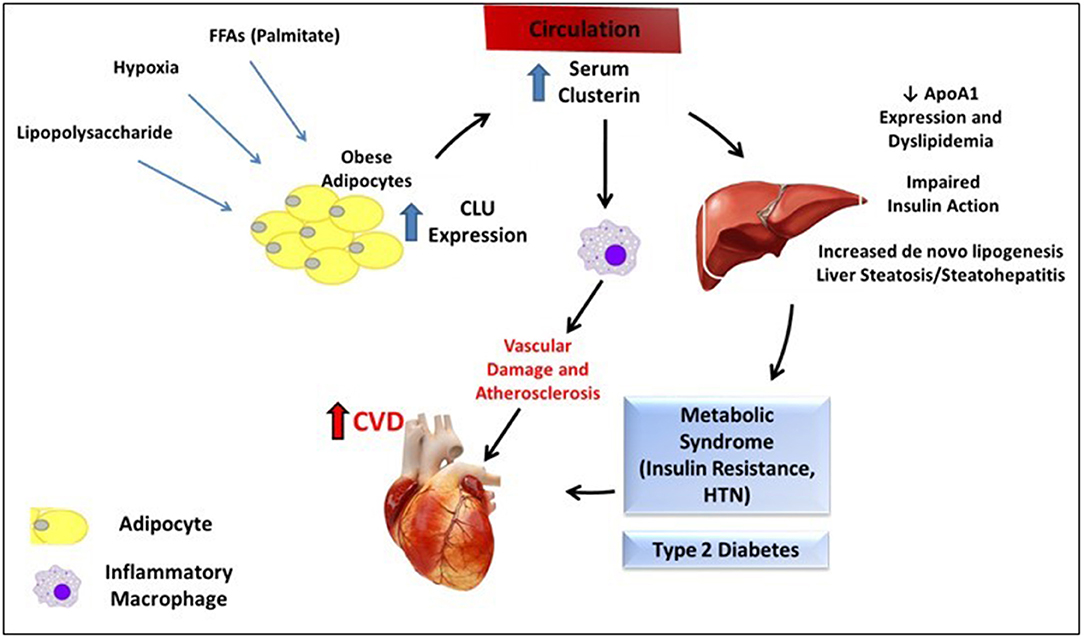
Figure 1. Summary of proposed mechanism for clusterin-mediated cardiometabolic disease. Various stimuli may increase adipocyte expression of CLU from adipocytes in the setting of obesity. Circulating clusterin subsequently has multiple effects on the liver (reduction in ApoA1 expression, dyslipidemia, impaired insulin signaling, and potentially increased steatosis and inflammation) and on macrophages, which may contribute to the cardiometabolic syndrome, and increase CVD risk.
The cardiometabolic syndrome is a clustering of metabolic and cardiovascular abnormalities that increase the risk of CVD, T2D, and all-cause mortality. The rising prevalence of the cardiometabolic syndrome, both in the U.S. and worldwide, make a more thorough understanding of its pathophysiologic underpinnings imperative. Although likely multifactorial, the presence of obesity-related insulin resistance appears to be a central, if not instigating factor. Systemic and tissue-specific insulin resistance not only affect endothelial function and leads to atherogenic dyslipidemia, but propagate a pro-inflammatory environment that includes excess release of detrimental FFAs into the circulation. Clusterin is a ubiquitous protein secreted by many organs/tissues throughout the body. Although studies have implicated circulating clusterin in multiple metabolic and cardio/cerebrovascular abnormalities, a unifying mechanism remains elusive, and the current literature is inconsistent and inconclusive. In particular, the importance of AT derived clusterin, strongly associated with many metabolic and CVD risk factors, requires further investigation. This includes understanding the exact mechanistic processes by which it acts locally within AT and systemically in the liver, endothelial cells, and the vasculature. Isolating its effects, potentially through the development of adipocyte-specific clusterin knockout and overexpression models, will be instrumental in determining if it is a viable target to attenuate features of the cardiometabolic syndrome.
JW and DB co-wrote the manuscript. Both authors contributed to the article and approved the submitted version.
This study was supported by grants from the American Diabetes Association 1-16-ICTS-049), The National Institutes of Health KL2 Scholar Award KL2TR001068, and The Ohio State University College of Medicine Office of Research Bridge Funding Program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National heart, lung, and blood institute scientific statement: executive summary. Crit Pathw Cardiol. (2005) 4:198–203. doi: 10.1161/CIRCULATIONAHA.105.169405
2. E. National Cholesterol Education Program Expert Panel on Detection, A. Treatment of high blood cholesterol in, third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. (2002) 106:3143–421. doi: 10.1161/circ.106.25.3143
3. Zimmet P, Magliano D, Matsuzawa Y, Alberti G, Shaw J. The metabolic syndrome: a global public health problem and a new definition. J Atherosc Thromb. (2005) 12:295–300. doi: 10.5551/jat.12.295
4. Li X, Li X, Lin H, Fu X, Lin W, Li M, et al. Metabolic syndrome and stroke: a meta-analysis of prospective cohort studies. J Clin Neurosci. (2017) 40:34–8. doi: 10.1016/j.jocn.2017.01.018
5. Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation. (2003) 107:391–7. doi: 10.1161/01.CIR.0000055014.62083.05
6. Adams LA, Waters OR, Knuiman MW, Elliott RR, Olynyk JK. NAFLD as a risk factor for the development of diabetes and the metabolic syndrome: an eleven-year follow-up study. Am J Gastroenterol. (2009) 104:861–7. doi: 10.1038/ajg.2009.67
7. Defronzo RA, Tripathy D, Schwenke DC, Banerji M, Bray GA, Buchanan TA, et al. Prediction of diabetes based on baseline metabolic characteristics in individuals at high risk. Diabetes Care. (2013) 36:3607–12. doi: 10.2337/dc13-0520
8. Golabi P, Otgonsuren M, de Avila L, Sayiner M, Rafiq N, Younossi ZM. Components of metabolic syndrome increase the risk of mortality in nonalcoholic fatty liver disease (NAFLD). Medicine. (2018) 97:e0214. doi: 10.1097/MD.0000000000010214
9. Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. (1988) 37:1595–607. doi: 10.2337/diab.37.12.1595
10. Haller H. [Epidermiology and associated risk factors of hyperlipoproteinemia]. Z Gesamte Inn Med. (1977) 32:124–8.
11. Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. (1998) 15:539–53. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S
12. Alberti KG, Zimmet P, Shaw JI, D.F.E.T.Group FC. The metabolic syndrome–a new worldwide definition. Lancet. (2005) 366:1059–62. doi: 10.1016/S0140-6736(05)67402-8
13. Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third national health and nutrition examination survey. JAMA. (2002) 287:356–9. doi: 10.1001/jama.287.3.356
14. Moore JX, Chaudhary N, Akinyemiju T. Metabolic syndrome prevalence by race/ethnicity and sex in the united states, national health and nutrition examination survey, 1988-2012. Prev Chronic Dis. (2017) 14:E24. doi: 10.5888/pcd14.160287
15. Galassi, Reynolds K, He J. Metabolic syndrome and risk of cardiovascular disease: a meta-analysis. Am J Med. (2006) 119:812–9. doi: 10.1016/j.amjmed.2006.02.031
16. Strath S, Swartz A, Parker S, Miller N, Cieslik L. Walking and metabolic syndrome in older adults. J Phys Act Health. (2007) 4:397–410. doi: 10.1123/jpah.4.4.398
17. Mankowski RT, Aubertin-Leheudre M, Beavers DP, Botoseneanu A, Buford TW, Church T, et al. Sedentary time is associated with the metabolic syndrome in older adults with mobility limitations–The LIFE study. Exp Gerontol. (2015) 70:32–6. doi: 10.1016/j.exger.2015.06.018
18. Denys K, Cankurtaran M, Janssens W, Petrovic M. Metabolic syndrome in the elderly: an overview of the evidence. Acta Clin Belg. (2009) 64:23–34. doi: 10.1179/acb.2009.006
19. Akinyemiju T, Jha M, Moore JX, Pisu M. Disparities in the prevalence of comorbidities among US adults by state medicaid expansion status. Prev Med. (2016) 88:196–202. doi: 10.1016/j.ypmed.2016.04.009
20. Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010. JAMA. (2012) 307:491–7. doi: 10.1001/jama.2012.39
21. Bray GA. Complications of obesity. Ann Intern Med. (1985) 103:1052–62. doi: 10.7326/0003-4819-103-6-1052
22. Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA. (1979) 241:2035–8. doi: 10.1001/jama.241.19.2035
23. Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech. (2009) 2:231–7. doi: 10.1242/dmm.001180
24. Kirk EP, Klein S. Pathogenesis and pathophysiology of the cardiometabolic syndrome. J Clin Hypertens. (2009) 11:761–5. doi: 10.1111/j.1559-4572.2009.00054.x
25. Manrique C, Lastra G, Sowers JR. New insights into insulin action and resistance in the vasculature. Ann N Y Acad Sci. (2014) 1311:138–50. doi: 10.1111/nyas.12395
26. Modan M, Halkin H. Hyperinsulinemia or increased sympathetic drive as links for obesity and hypertension. Diabetes Care. (1991) 14:470–87. doi: 10.2337/diacare.14.6.470
27. Romeo GR, Lee J, Shoelson SE. Metabolic syndrome, insulin resistance, and roles of inflammation–mechanisms and therapeutic targets. Arterioscler Thromb Vasc Biol. (2012) 32:1771–6. doi: 10.1161/ATVBAHA.111.241869
28. Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des. (2008) 14:1225–30. doi: 10.2174/138161208784246153
29. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. (1995) 95:2409–15. doi: 10.1172/JCI117936
30. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. (1993) 259:87–91. doi: 10.1126/science.7678183
31. Blaszczak AM, Jalilvand A, Liu J, Wright VP, Suzo A, Needleman B, et al. Human visceral adipose tissue macrophages are not adequately defined by standard methods of characterization. J Diabetes Res. (2019) 2019:8124563. doi: 10.1155/2019/8124563
32. Brestoff JR, Artis D. Immune regulation of metabolic homeostasis in health and disease. Cell. (2015) 161:146–60. doi: 10.1016/j.cell.2015.02.022
33. Cipolletta D. Adipose tissue-resident regulatory T cells: phenotypic specialization, functions and therapeutic potential. Immunology. (2014) 142:517–25. doi: 10.1111/imm.12262
34. Deiuliis J, Shah Z, Shah N, Needleman B, Mikami D, Narula V, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS ONE. (2011) 6:e16376. doi: 10.1371/journal.pone.0016376
35. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. (2010) 72:219–46. doi: 10.1146/annurev-physiol-021909-135846
36. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. (2012) 18:363–74. doi: 10.1038/nm.2627
37. Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. (2011) 121:1858–70. doi: 10.1172/JCI43378
38. Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. (2000) 106:171–6. doi: 10.1172/JCI10583
39. Boden G. Fatty acid-induced inflammation and insulin resistance in skeletal muscle and liver. Curr Diabetes Rep. (2006) 6:177–81. doi: 10.1007/s11892-006-0031-x
40. Kelley DE, Mokan M, Simoneau JA, Mandarino LJ. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J Clin Invest. (1993) 92:91–8. doi: 10.1172/JCI116603
41. Ferrannini E, Barrett EJ, Bevilacqua S, DeFronzo RA. Effect of fatty acids on glucose production and utilization in man. J Clin Invest. (1983) 72:1737–47. doi: 10.1172/JCI111133
42. Cheng D, Noble J, Tang MX, Schupf N, Mayeux R, Luchsinger JA. Type 2 diabetes and late-onset Alzheimer's disease. Dement Geriatr Cogn Disord. (2011) 31:424–30. doi: 10.1159/000324134
43. Panza F, Frisardi V, Capurso C, Imbimbo BP, Vendemiale G, Santamato A, et al. Metabolic syndrome and cognitive impairment: current epidemiology and possible underlying mechanisms. J Alzheimers Dis. (2010) 21:691–724. doi: 10.3233/JAD-2010-091669
44. Ott, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: the rotterdam study. Neurology. (1999) 53:1937–42. doi: 10.1212/WNL.53.9.1937
45. Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O'Brien PC, et al. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. (1997) 145:301–8. doi: 10.1093/oxfordjournals.aje.a009106
46. Arvanitakis Z, Wilson RS, Bennett DA. Diabetes mellitus, dementia, and cognitive function in older persons. J Nutr Health Aging. (2006) 10:287–91.
47. Wu JH, Haan MN, Liang J, Ghosh D, Gonzalez HM, Herman WH. Impact of antidiabetic medications on physical and cognitive functioning of older Mexican Americans with diabetes mellitus: a population-based cohort study. Ann Epidemiol. (2003) 13:369–76. doi: 10.1016/S1047-2797(02)00464-7
48. Wu JH, Haan MN, Liang J, Ghosh D, Gonzalez HM, Herman WH. Impact of diabetes on cognitive function among older Latinos: a population-based cohort study. J Clin Epidemiol. (2003) 56:686–93. doi: 10.1016/S0895-4356(03)00077-5
49. Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, Winblad B, et al. Diabetes, Alzheimer disease, and vascular dementia: a population-based neuropathologic study. Neurology. (2010) 75:1195–202. doi: 10.1212/WNL.0b013e3181f4d7f8
50. Yaffe K, Blackwell T, Kanaya AM, Davidowitz N, Barrett-Connor E, Krueger K. Diabetes, impaired fasting glucose, and development of cognitive impairment in older women. Neurology. (2004) 63:658–63. doi: 10.1212/01.WNL.0000134666.64593.BA
51. Stoeckel LE, Arvanitakis Z, Gandy S, Small D, Kahn CR, Pascual-Leone A, et al. Complex mechanisms linking neurocognitive dysfunction to insulin resistance and other metabolic dysfunction. F1000Res. (2016) 5:353. doi: 10.12688/f1000research.8300.2
52. Logroscino G, Kang JH, Grodstein F. Prospective study of type 2 diabetes and cognitive decline in women aged 70-81 years. BMJ. (2004) 328:548. doi: 10.1136/bmj.37977.495729.EE
53. Clarke DW, Boyd FT Jr, Kappy MS, Raizada MK. Insulin binds to specific receptors and stimulates 2-deoxy-D-glucose uptake in cultured glial cells from rat brain. J Biol Chem. (1984) 259:11672–5. doi: 10.1016/S0021-9258(20)71260-3
54. Raizada MK, Shemer J, Judkins JH, Clarke DW, Masters BA, LeRoith D. Insulin receptors in the brain: structural and physiological characterization. Neurochem Res. (1988) 13:297–303. doi: 10.1007/BF00972477
55. Smythe GA, Bradshaw JE, Nicholson MV, Grunstein HS, Storlien LH. Rapid bidirectional effects of insulin on hypothalamic noradrenergic and serotoninergic neuronal activity in the rat: role in glucose homeostasis. Endocrinology. (1985) 117:1590–7. doi: 10.1210/endo-117-4-1590
56. Uemura E, Greenlee HW. Insulin regulates neuronal glucose uptake by promoting translocation of glucose transporter GLUT3. Exp Neurol. (2006) 198:48–53. doi: 10.1016/j.expneurol.2005.10.035
57. Craft S, Asthana S, Cook DG, Baker LD, Cherrier M, Purganan K, et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer's disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology. (2003) 28:809–22. doi: 10.1016/S0306-4530(02)00087-2
58. Ramos-Rodriguez JJ, Ortiz O, Jimenez-Palomares M, Kay KR, Berrocoso E, Murillo-Carretero MI, et al. Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice. Psychoneuroendocrinology. (2013) 38:2462–75. doi: 10.1016/j.psyneuen.2013.05.010
59. Arnold SE, Lucki I, Brookshire BR, Carlson GC, Browne CA, Kazi H, et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol Dis. (2014) 67:79–87. doi: 10.1016/j.nbd.2014.03.011
60. Liu Z, Patil IY, Jiang T, Sancheti H, Walsh JP, Stiles BL, et al. High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PLoS ONE. (2015) 10:e0128274. doi: 10.1145/2818302
61. Martins IV, Rivers-Auty J, Allan SM, Lawrence CB. Mitochondrial abnormalities and synaptic loss underlie memory deficits seen in mouse models of obesity and Alzheimer's disease. J Alzheimers Dis. (2017) 55:915–32. doi: 10.3233/JAD-160640
62. Tschritter O, Preissl H, Hennige AM, Stumvoll M, Porubska K, Frost R, et al. The cerebrocortical response to hyperinsulinemia is reduced in overweight humans: a magnetoencephalographic study. Proc Natl Acad Sci USA. (2006) 103:12103–8. doi: 10.1073/pnas.0604404103
63. Tschritter O, Preissl H, Yokoyama Y, Machicao F, Haring HU, Fritsche A. Variation in the FTO gene locus is associated with cerebrocortical insulin resistance in humans. Diabetologia. (2007) 50:2602–3. doi: 10.1007/s00125-007-0839-1
64. Anthony K, Reed LJ, Dunn JT, Bingham E, Hopkins D, Marsden PK, et al. Attenuation of insulin-evoked responses in brain networks controlling appetite and reward in insulin resistance: the cerebral basis for impaired control of food intake in metabolic syndrome? Diabetes. (2006) 55:2986–92. doi: 10.2337/db06-0376
65. Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. (2012) 122:1316–38. doi: 10.1172/JCI59903
66. Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease–is this type 3 diabetes? J Alzheimers Dis. (2005) 7:63–80. doi: 10.3233/JAD-2005-7107
67. Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease- associated Abeta oligomers. J Clin Invest. (2012) 122:1339–53. doi: 10.1172/JCI57256
68. Kern W, Peters A, Fruehwald-Schultes B, Deininger E, Born J, Fehm HL. Improving influence of insulin on cognitive functions in humans. Neuroendocrinology. (2001) 74:270–80. doi: 10.1159/000054694
69. Craft S, Newcomer J, Kanne S, Dagogo-Jack S, Cryer P, Sheline Y, et al. Memory improvement following induced hyperinsulinemia in Alzheimer's disease. Neurobiol Aging. (1996) 17:123–30. doi: 10.1016/0197-4580(95)02002-0
70. Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging. (2011) 32:1626–33. doi: 10.1016/j.neurobiolaging.2009.10.009
71. Hanyu H, Sato T, Kiuchi A, Sakurai H, Iwamoto T. Pioglitazone improved cognition in a pilot study on patients with Alzheimer's disease and mild cognitive impairment with diabetes mellitus. J Am Geriatr Soc. (2009) 57:177–9. doi: 10.1111/j.1532-5415.2009.02067.x
72. Luchsinger JA, Perez T, Chang H, Mehta P, Steffener J, Pradabhan G, et al. Metformin in amnestic mild cognitive impairment: results of a pilot randomized placebo controlled clinical trial. J Alzheimers Dis. (2016) 51:501–14. doi: 10.3233/JAD-150493
73. Koenig AM, Mechanic-Hamilton D, Xie SX, Combs MF, Cappola AR, Xie L, et al. Effects of the insulin sensitizer metformin in Alzheimer disease: pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis Assoc Disord. (2017) 31:107–13. doi: 10.1097/WAD.0000000000000202
74. Siervo M, Arnold R, Wells JC, Tagliabue A, Colantuoni A, Albanese E, et al. Intentional weight loss in overweight and obese individuals and cognitive function: a systematic review and meta-analysis. Obes Rev. (2011) 12:968–83. doi: 10.1111/j.1467-789X.2011.00903.x
75. Handley JD, Williams DM, Caplin S, Stephens JW, Barry J. Changes in cognitive function following bariatric surgery: a systematic review. Obes Surg. (2016) 26:2530–7. doi: 10.1007/s11695-016-2312-z
76. Gunstad J, Strain G, Devlin MJ, Wing R, Cohen RA, Paul RH, et al. Improved memory function 12 weeks after bariatric surgery. Surg Obes Relat Dis. (2011) 7:465–72. doi: 10.1016/j.soard.2010.09.015
77. Alosco ML, Galioto R, Spitznagel MB, Strain G, Devlin M, Cohen R, et al. Cognitive function after bariatric surgery: evidence for improvement 3 years after surgery. Am J Surg. (2014) 207:870–6. doi: 10.1016/j.amjsurg.2013.05.018
78. Kernan WN, Inzucchi SE, Viscoli CM, Brass LM, Bravata DM, Horwitz RI. Insulin resistance and risk for stroke. Neurology. (2002) 59:809–15. doi: 10.1212/WNL.59.6.809
79. Kernan WN, Inzucchi SE, Viscoli CM, Brass LM, Bravata DM, Shulman GI, et al. Impaired insulin sensitivity among nondiabetic patients with a recent TIA or ischemic stroke. Neurology. (2003) 60:1447–51. doi: 10.1212/01.WNL.0000063318.66140.A3
80. Bravata DM, Wells CK, Kernan WN, Concato J, Brass LM, Gulanski BI. Association between impaired insulin sensitivity and stroke. Neuroepidemiology. (2005) 25:69–74. doi: 10.1159/000086286
81. Craft S. Insulin resistance and cognitive impairment: a view through the prism of epidemiology. Arch Neurol. (2005) 62:1043–4. doi: 10.1001/archneur.62.7.1043-a
82. Willette AA, Xu G, Johnson SC, Birdsill AC, Jonaitis EM, Sager MA, et al. Insulin resistance, brain atrophy, and cognitive performance in late middle-aged adults. Diabetes Care. (2013) 36:443–9. doi: 10.2337/dc12-0922
83. Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, et al. Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med. (2016) 374:1321–31. doi: 10.1056/NEJMoa1506930
84. Fritz IB, Burdzy K, Setchell B, Blaschuk O. Ram rete testis fluid contains a protein (clusterin) which influences cell-cell interactions in vitro. Biol Reprod. (1983) 28:1173–88. doi: 10.1095/biolreprod28.5.1173
85. Wong P, Pineault J, Lakins J, Taillefer D, Leger J, Wang C, et al. Genomic organization and expression of the rat TRPM-2 (clusterin) gene, a gene implicated in apoptosis. J Biol Chem. (1993) 268:5021–31. doi: 10.1016/S0021-9258(18)53497-9
86. Wong P, Taillefer D, Lakins J, Pineault J, Chader G, Tenniswood M. Molecular characterization of human TRPM-2/clusterin, a gene associated with sperm maturation, apoptosis and neurodegeneration. Eur J Biochem. (1994) 221:917–25. doi: 10.1111/j.1432-1033.1994.tb18807.x
87. Park S, Mathis KW, Lee IK. The physiological roles of apolipoprotein J/clusterin in metabolic and cardiovascular diseases. Rev Endocr Metab Disord. (2014) 15:45–53. doi: 10.1007/s11154-013-9275-3
88. Wilson MR, Easterbrook-Smith SB. Clusterin is a secreted mammalian chaperone. Trends Biochem Sci. (2000) 25:95–8. doi: 10.1016/S0968-0004(99)01534-0
89. Michel D, Chatelain G, Herault Y, Brun G. The expression of the avian clusterin gene can be driven by two alternative promoters with distinct regulatory elements. Eur J Biochem. (1995) 229:215–23. doi: 10.1111/j.1432-1033.1995.0215l.x
90. Bradley D, Blaszczak A, Yin Z, Liu J, Joseph JJ, Wright V, et al. Clusterin impairs hepatic insulin sensitivity and adipocyte clusterin associates with cardiometabolic risk. Diabetes Care. (2019) 42:466–75. doi: 10.2337/dc18-0870
91. Leeb C, Eresheim C, Nimpf J. Clusterin is a ligand for apolipoprotein E receptor 2 (ApoER2) and very low density lipoprotein receptor (VLDLR) and signals via the Reelin-signaling pathway. J Biol Chem. (2014) 289:4161–72. doi: 10.1074/jbc.M113.529271
92. Jones SE, Jomary C. Clusterin. Int J Biochem Cell Biol. (2002) 34:427–31. doi: 10.1016/S1357-2725(01)00155-8
93. Trougakos IP, Gonos ES. Clusterin/apolipoprotein J in human aging and cancer. Int J Biochem Cell Biol. (2002) 34:1430–48. doi: 10.1016/S1357-2725(02)00041-9
94. Riwanto M, Rohrer L, Roschitzki B, Besler C, Mocharla P, Mueller M, et al. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation. (2013) 127:891–904. doi: 10.1161/CIRCULATIONAHA.112.108753
95. Leskov KS, Araki S, Lavik JP, Gomez JA, Gama V, Gonos ES, et al. CRM1 protein-mediated regulation of nuclear clusterin (nCLU), an ionizing radiation-stimulated, Bax-dependent pro-death factor. J Biol Chem. (2011) 286:40083–90. doi: 10.1074/jbc.M111.252957
96. Antonelou MH, Kriebardis AG, Stamoulis KE, Trougakos IP, Papassideri IS. Apolipoprotein J/Clusterin is a novel structural component of human erythrocytes and a biomarker of cellular stress and senescence. PLoS ONE. (2011) 6:e26032. doi: 10.1371/journal.pone.0026032
97. Trougakos IP, Djeu JY, Gonos ES, Boothman DA. Advances and challenges in basic and translational research on clusterin. Cancer Res. (2009) 69:403–6. doi: 10.1158/0008-5472.CAN-08-2912
98. Trougakos IP, Gonos ES. Regulation of clusterin/apolipoprotein J, a functional homologue to the small heat shock proteins, by oxidative stress in ageing and age-related diseases. Free Radic Res. (2006) 40:1324–34. doi: 10.1080/10715760600902310
99. Yom CK, Woo HY, Min SY, Kang SY, Kim HS. Clusterin overexpression and relapse-free survival in breast cancer. Anticancer Res. (2009) 29:3909–12.
100. Wei L, Xue T, Wang J, Chen B, Lei Y, Huang Y, et al. Roles of clusterin in progression, chemoresistance and metastasis of human ovarian cancer. Int J Cancer. (2009) 125:791–806. doi: 10.1002/ijc.24316
101. July LV, Akbari M, Zellweger T, Jones EC, Goldenberg SL, Gleave ME. Clusterin expression is significantly enhanced in prostate cancer cells following androgen withdrawal therapy. Prostate. (2002) 50:179–88. doi: 10.1002/pros.10047
102. Liu T, Liu PY, Tee AE, Haber M, Norris MD, Gleave ME, et al. Over-expression of clusterin is a resistance factor to the anti-cancer effect of histone deacetylase inhibitors. Eur J Cancer. (2009) 45:1846–54. doi: 10.1016/j.ejca.2009.03.002
103. Saad F, Hotte S, North S, Eigl B, Chi K, Czaykowski P, et al. Canadian Uro-Oncology, randomized phase II trial of Custirsen (OGX-011) in combination with docetaxel or mitoxantrone as second-line therapy in patients with metastatic castrate-resistant prostate cancer progressing after first-line docetaxel: CUOG trial P-06c. Clin Cancer Res. (2011) 17:5765–73. doi: 10.1158/1078-0432.CCR-11-0859
104. Laskin JJ, Nicholas G, Lee C, Gitlitz B, Vincent M, Cormier Y, et al. Phase I/II trial of custirsen (OGX-011), an inhibitor of clusterin, in combination with a gemcitabine and platinum regimen in patients with previously untreated advanced non-small cell lung cancer. J Thorac Oncol. (2012) 7:579–86. doi: 10.1097/JTO.0b013e31823f459c
105. Humphreys DT, Carver JA, Easterbrook-Smith SB, Wilson MR. Clusterin has chaperone-like activity similar to that of small heat shock proteins. J Biol Chem. (1999) 274:6875–81. doi: 10.1074/jbc.274.11.6875
106. Wyatt, Yerbury J, Poon S, Dabbs R, Wilson M. Chapter 6: the chaperone action of Clusterin and its putative role in quality control of extracellular protein folding. Adv Cancer Res. (2009) 104:89–114. doi: 10.1016/S0065-230X(09)04006-8
107. Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM. Expression of novel Alzheimer's disease risk genes in control and Alzheimer's disease brains. PLoS ONE. (2012) 7:e50976. doi: 10.1371/journal.pone.0050976
108. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. (2009) 41:1088–93. doi: 10.1038/ng.440
109. Schrijvers EM, Koudstaal PJ, Hofman A, Breteler MM. Plasma clusterin and the risk of Alzheimer disease. JAMA. (2011) 305:1322–6. doi: 10.1001/jama.2011.381
110. Li X, Ma Y, Wei X, Li Y, Wu H, Zhuang J, et al. Clusterin in Alzheimer's disease: a player in the biological behavior of amyloid-beta. Neurosci Bull. (2014) 30:162–8. doi: 10.1007/s12264-013-1391-2
111. Nilselid AM, Davidsson P, Nagga K, Andreasen N, Fredman P, Blennow K. Clusterin in cerebrospinal fluid: analysis of carbohydrates and quantification of native and glycosylated forms. Neurochem Int. (2006) 48:718–28. doi: 10.1016/j.neuint.2005.12.005
112. Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y, et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry. (2010) 67:739–48. doi: 10.1001/archgenpsychiatry.2010.78
113. Yang C, Wang H, Li C, Niu H, Luo S, Guo X. Association between clusterin concentration and dementia: a systematic review and meta-analysis. Metab Brain Dis. (2018) 34:129–40. doi: 10.1007/s11011-018-0325-0
114. Morgan AR, Touchard S, O'Hagan C, Sims R, Majounie E, Escott-Price V, et al. The correlation between inflammatory biomarkers and polygenic risk score in Alzheimer's disease. J Alzheimers Dis. (2017) 56:25–36. doi: 10.3233/JAD-160889
115. Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, et al. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet. (1996) 348:159–61. doi: 10.1016/S0140-6736(96)03173-X
116. Bajari TM, Strasser V, Nimpf J, Schneider WJ. A model for modulation of leptin activity by association with clusterin. FASEB J. (2003) 17:1505–7. doi: 10.1096/fj.02-1106fje
117. Gil SY, Youn BS, Byun K, Huang H, Namkoong C, Jang PG, et al. Clusterin and LRP2 are critical components of the hypothalamic feeding regulatory pathway. Nat Commun. (2013) 4:1862. doi: 10.1038/ncomms2896
118. Shim YJ, Kang BH, Jeon HS, Park IS, Lee KU, Lee IK. Clusterin induces matrix metalloproteinase-9 expression via ERK1/2 and PI3K/Akt/NF-kappaB pathways in monocytes/macrophages. J Leukoc Biol. (2011) 90:761–9. doi: 10.1189/jlb.0311110
119. Kang BH, Shim YJ, Tae YK, Song JA, Choi BK, Park IS. Clusterin stimulates the chemotactic migration of macrophages through a pertussis toxin sensitive G-protein-coupled receptor and Gbetagamma-dependent pathways. Biochem Biophys Res Commun. (2014) 445:645–50. doi: 10.1016/j.bbrc.2014.02.071
120. Shim YJ, Kang BH, Choi BK, Park IS, Min BH. Clusterin induces the secretion of TNF-alpha and the chemotactic migration of macrophages. Biochem Biophys Res Commun. (2012) 422:200–5. doi: 10.1016/j.bbrc.2012.04.162
121. Falgarone G, Chiocchia G. Chapter 8: clusterin: a multifacet protein at the crossroad of inflammation and autoimmunity. Adv Cancer Res. (2009) 104:139–70. doi: 10.1016/S0065-230X(09)04008-1
122. Kwon MJ, Ju TJ, Heo JY, Kim YW, Kim JY, Won KC, et al. Deficiency of clusterin exacerbates high-fat diet-induced insulin resistance in male mice. Endocrinology. (2014) 155:2089–101. doi: 10.1210/en.2013-1870
123. Klouckova J, Lacinova Z, Kavalkova P, Trachta P, Kasalicky M, Haluzikova D, et al. Plasma concentrations and subcutaneous adipose tissue mRNA expression of clusterin in obesity and type 2 diabetes mellitus: the effect of short-term hyperinsulinemia, very-low-calorie diet and bariatric surgery. Physiol Res. (2016) 65:481–92. doi: 10.33549/physiolres.933121
124. Won JC, Park CY, Oh SW, Lee ES, Youn BS, Kim MS. Plasma clusterin (ApoJ) levels are associated with adiposity and systemic inflammation. PLoS ONE. (2014) 9:e103351. doi: 10.1371/journal.pone.0103351
125. Flehmig G, Scholz M, Kloting N, Fasshauer M, Tonjes A, Stumvoll M, et al. Identification of adipokine clusters related to parameters of fat mass, insulin sensitivity and inflammation. PLoS ONE. (2014) 9:e99785. doi: 10.1371/journal.pone.0099785
126. Daimon M, Oizumi T, Karasawa S, Kaino W, Takase K, Tada K, et al. Association of the clusterin gene polymorphisms with type 2 diabetes mellitus. Metabolism. (2011) 60:815–22. doi: 10.1016/j.metabol.2010.07.033
127. Seo HY, Lee SH, Lee JH, Kang YN, Choi YK, Hwang JS, et al. Clusterin attenuates hepatic fibrosis by inhibiting hepatic stellate cell activation and downregulating the Smad3 signaling pathway. Cells. (2019) 8:1442. doi: 10.3390/cells8111442
128. Bergmeier C, Siekmeier R, Gross W. Distribution spectrum of paraoxonase activity in HDL fractions. Clin Chem. (2004) 50:2309–15. doi: 10.1373/clinchem.2004.034439
129. Bettuzzi S. Conclusions and perspectives. Adv Cancer Res. (2009) 105:133–50. doi: 10.1016/S0065-230X(09)05008-8
130. Ishikawa Y, Akasaka Y, Ishii T, Komiyama K, Masuda S, Asuwa N, et al. Distribution and synthesis of apolipoprotein J in the atherosclerotic aorta. Arterioscler Thromb Vasc Biol. (1998) 18:665–72. doi: 10.1161/01.ATV.18.4.665
131. Navab M, Hama-Levy S, Van Lenten BJ, Fonarow GC, Cardinez CJ, Castellani LW, et al. Mildly oxidized LDL induces an increased apolipoprotein J/paraoxonase ratio. J Clin Invest. (1997) 99:2005–19. doi: 10.1172/JCI119369
132. Aronis KN, Vamvini MT, Chamberland JP, Mantzoros CS. Circulating clusterin (apolipoprotein J) levels do not have any day/night variability and are positively associated with total and LDL cholesterol levels in young healthy individuals. J Clin Endocrinol Metab. (2011) 96:E1871–5. doi: 10.1210/jc.2011-1555
133. Poulakou MV, Paraskevas KI, Wilson MR, Iliopoulos DC, Tsigris C, Mikhailidis DP, et al. Apolipoprotein J and leptin levels in patients with coronary heart disease. In vivo. (2008) 22:537–42.
134. Hoofnagle AN, Wu M, Gosmanova AK, Becker JO, Wijsman EM, Brunzell JD, et al. Low clusterin levels in high-density lipoprotein associate with insulin resistance, obesity, and dyslipoproteinemia. Arterioscler Thromb Vasc Biol. (2010) 30:2528–34. doi: 10.1161/ATVBAHA.110.212894
135. Baralla, Sotgiu E, Deiana M, Pasella S, Pinna S, Mannu A, et al. Plasma clusterin and lipid profile: a link with aging and cardiovascular diseases in a population with a consistent number of centenarians. PLoS ONE. (2015) 10:e0128029. doi: 10.1371/journal.pone.0128029
136. Aragones G, Auguet T, Guiu-Jurado E, Berlanga A, Curriu M, Martinez S, et al. Proteomic profile of unstable atheroma plaque: increased neutrophil defensin 1, clusterin, and apolipoprotein e levels in carotid secretome. J Proteome Res. (2016) 15:933–44. doi: 10.1021/acs.jproteome.5b00936
137. Koller L, Richter B, Winter MP, Sulzgruber P, Potolidis C, Liebhart F, et al. Clusterin/apolipoprotein J is independently associated with survival in patients with chronic heart failure. J Clin Lipidol. (2017) 11:178–84. doi: 10.1016/j.jacl.2016.11.009
138. Grassmann S, Wirsching J, Eichelmann F, Aleksandrova K. Association between peripheral adipokines and inflammation markers: a systematic review and meta-analysis. Obesity. (2017) 25:1776–85. doi: 10.1002/oby.21945
139. Sears B, Perry M. The role of fatty acids in insulin resistance. Lipids Health Dis. (2015) 14:121. doi: 10.1186/s12944-015-0123-1
140. Ruiz R, Jideonwo V, Ahn M, Surendran S, Tagliabracci VS, Hou Y, et al. Sterol regulatory element-binding protein-1 (SREBP-1) is required to regulate glycogen synthesis and gluconeogenic gene expression in mouse liver. J Biol Chem. (2014) 289:5510–7. doi: 10.1074/jbc.M113.541110
141. McQueen MJ, Hawken S, Wang X, Ounpuu S, Sniderman A, Probstfield J, et al. Lipids, lipoproteins, and apolipoproteins as risk markers of myocardial infarction in 52 countries (the INTERHEART study): a case-control study. Lancet. (2008) 372:224–33. doi: 10.1016/S0140-6736(08)61076-4
Keywords: adipocyte, clusterin, cardiometabolic disease, type 2 ddiabetes mellitus, inflammation
Citation: Wittwer J and Bradley D (2021) Clusterin and Its Role in Insulin Resistance and the Cardiometabolic Syndrome. Front. Immunol. 12:612496. doi: 10.3389/fimmu.2021.612496
Received: 30 September 2020; Accepted: 04 February 2021;
Published: 25 February 2021.
Edited by:
Emira Ayroldi, University of Perugia, ItalyReviewed by:
Gareth S. D. Purvis, University of Oxford, United KingdomCopyright © 2021 Wittwer and Bradley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Bradley, ZGF2aWQuYnJhZGxleUBvc3VtYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.