 Tizibt Ashine Bogale1Gaia Faustini1Francesca Longhena1Stefania Mitola2,3Marina Pizzi1Arianna Bellucci1,3*
Tizibt Ashine Bogale1Gaia Faustini1Francesca Longhena1Stefania Mitola2,3Marina Pizzi1Arianna Bellucci1,3*- 1Division of Pharmacology, Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy
- 2Biotechnology Division, Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy
- 3Laboratory for Preventive and Personalized Medicine, Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy
Misfolded proteins, inflammation, and vascular alterations are common pathological hallmarks of neurodegenerative diseases. Alpha-synuclein is a small synaptic protein that was identified as a major component of Lewy bodies and Lewy neurites in the brain of patients affected by Parkinson's disease (PD), Lewy body dementia (LBD), and other synucleinopathies. It is mainly involved in the regulation of synaptic vesicle trafficking but can also control mitochondrial/endoplasmic reticulum (ER) homeostasis, lysosome/phagosome function, and cytoskeleton organization. Recent evidence supports that the pathological forms of α-synuclein can also reduce the release of vasoactive and inflammatory mediators from endothelial cells (ECs) and modulates the expression of tight junction (TJ) proteins important for maintaining the blood–brain barrier (BBB). This hints that α-synuclein deposition can affect BBB integrity. Border associated macrophages (BAMs) are brain resident macrophages found in association with the vasculature (PVMs), meninges (MAMs), and choroid plexus (CPMs). Recent findings indicate that these cells play distinct roles in stroke and neurodegenerative disorders. Although many studies have addressed how α-synuclein may modulate microglia, its effect on BAMs has been scarcely investigated. This review aims at summarizing the main findings supporting how α-synuclein can affect ECs and/or BAMs function as well as their interplay and effect on other cells in the brain perivascular environment in physiological and pathological conditions. Gaps of knowledge and new perspectives on how this protein can contribute to neurodegeneration by inducing BBB homeostatic changes in different neurological conditions are highlighted.
Introduction
Neurodegenerative diseases represent a relevant health burden, especially considering the growing population of elderly subjects. Cerebrovascular disorders such as stroke are considered among the major predisposing factors for the development of neurodegenerative diseases, including Alzheimer's disease (AD) and Parkinson's disease (PD) (1, 2). In particular, PD is the second most common neurodegenerative disorder, affecting 2–3% of the population over the age of 65 years (3). The lack of knowledge on the molecular underpinnings of PD still limits the development of efficient therapies.
Protein aggregates enriched in insoluble α-synuclein fibrils and loss of dopaminergic neurons in the nigrostriatal system are key pathological features of this disorder (4, 5). Of note, the pathological deposition of insoluble α-synuclein at synapses is believed to act as the primum movens for neuronal degeneration in PD, as by hindering neurotransmitter release, it can trigger synaptic failure (6–8). This event can then negatively impinge on axonal projections, thus slowly flowing in a retrograde neurodegenerative process culminating in neuronal cell death (6–8). Additionally, α-synuclein-related neuroinflammation, microglia activation, and vascular degeneration (9–12) have been described as important players in disease pathogenesis. This notwithstanding, whether α-synuclein communicates with other neurovascular components such as border-associated macrophages (BAMs) and vascular endothelial cells (ECs), which are involved in the early phases of ischemic brain damage (13–16), remains to be explored.
Alpha-synuclein is a 14 kDa protein owning an undefined structure in aqueous solutions (17). In neurons, the protein regulates various processes including synaptic function, mitochondrial homeostasis, autophagy/lysosomal functions, and cytoskeletal reorganization (8, 18–24). The diverse domains of α-synuclein and its conformational plasticity allow the interaction with a plethora of other proteins and lipid membranes (20). Alpha-synuclein can also undergo post-translational modifications as amino-terminal and carboxy-terminal nitration and phosphorylation [e.g., Ser129 phosphorylation; (25–27)], which in turn can impact its conformation and can lead to the formation of toxic oligomers and fibrils (20). While oligomers can affect membrane permeability as well as neuronal excitability and engulf protein degradation systems (28–30), fibrils can disrupt the integrity of intracellular organelles and induce chronic inflammation (28, 31). In the brain, α-synuclein is expressed not only in the neuronal cells, but at lower levels also in astrocytes, macrophages, and the microglia (32, 33). In the periphery, the protein is expressed in red blood cells (34, 35), platelets (36), and in other immune cells, such as T cells, B cells, natural killer (NK) cells, and monocytes (32). It has been found that α-synuclein can bind microglia cell surface receptors, thus activating intracellular pathways mediating the release of cytokines and upregulating of proinflammatory genes (10, 37). The protein can also regulate ECs function by blocking the exocytosis of Weibel-Palade bodies (WPBs) (38) and by downregulating the expression of tight junction (TJ) proteins (39).
The deposition of α-synuclein insoluble aggregates named Lewy bodies (LB) or glial cytoplasmic inclusions (GCI) characterizes the brain of patients affected by PD and dementia with LB (DLB) or multiple system atrophy (MSA), respectively (5, 40). For this reason, these disorders are commonly referred to as synucleinopathies. Certain pathological strains of α-synuclein, by moving between the brain cells and across the blood–brain barrier (BBB) interfaces and acting as imprinting templates for the pathological conformational shift of other α-synuclein molecules, are believed to mediate the propagation of pathological aggregates within the brain, from the periphery to the brain, or from the brain to the periphery, with a prion-like fashion (41, 42).
This review focuses on how α-synuclein impacts vascular ECs and BAMs regulation and crosstalk. Current gaps and future perspectives in the context of neurological disorders are also presented.
Alpha-Synuclein Functions in the Central Nervous System (CNS)
To date, the physiological function of α-synuclein has not been fully disclosed, but we know that it controls neurotransmitter release and synaptic plasticity, particularly inhibiting dopamine overflow and modulating synaptic vesicles storage (20, 43, 44).
The full-length α-synuclein isoform consists of 140 amino acids and its structure can be divided into three main regions. The N-terminal part is essential for membrane binding (45–47) and includes the sites of main familial PD mutations, A30P, A53T, and E46K (18, 20), as well as for several post-translational modifications (48). The central domain, called non-amyloid component (NAC), is hydrophobic and highly aggregation-prone (49), and is necessary and sufficient for α-synuclein fibrillation (50). Finally, the C-terminal region is enriched in negative charges (51) and can interact with the N-terminal domain to form a compact aggregation-resistant structure (52).
Alpha-synuclein is described as an intrinsically disordered protein as it can be found in monomeric form (53) or in a stable tetramer (54) when purified at neutral pH. Rapid environmental changes can induce the formation of partially folded intermediates or kinetically trapped transition states (55). Along aging, the high plasticity of α-synuclein, coupled with post-translational modifications and protein enrichment at synaptic sites, can promote in concert the formation of high molecular weight soluble or insoluble aggregates, such as oligomers, protofibrils, or fibrils (20, 21). In PD, α-synuclein deposition is thought to play a pathogenic role in triggering both central and peripheral neurons degeneration, thus underlying the onset of motor and non-motor symptoms, respectively (56, 57). Interestingly, both monomeric and aggregated α-synuclein can be transferred from cell-to-cell (neuron-neuron, neuron-glia), and also across the BBB, thus contributing to neuropathology spreading (58). Endocytosis, carrier-mediated transports, and tunneling nanotubes are described as the main mechanisms for these exchanges (59, 60). In addition, impairment in glymphatic transport and lymphatic drainage pathways results in the accumulation of α-synuclein in the brain parenchyma and the progression of PD-like pathology in transgenic mouse models (61). This is in line with evidence supporting that general systemic circulation would act as a route for long-distance transmission of endogenous α-synuclein (62).
Alpha-synuclein can also exert a physiological regulatory action on intracellular organelles, including mitochondria (19), endoplasmic reticulum (ER), mitochondria-ER associated membranes (63), Golgi apparatus (64), and nuclei (65). Although the nuclear localization of α-synuclein was the first to be reported, its involvement in DNA repair mechanisms has been described quite recently (66). Recent findings, showing reduced nucleus to cytoplasmic transport in induced pluripotent stem cell (iPSC)-derived neurons from familial patients with PD bearing A53T mutation or multiplication of the α-synuclein gene locus SNCA (67), support that the protein may also play a role in maintaining nuclear membrane functions. Interestingly, reduced α-synuclein DNA binding associates with transcription deregulation through inhibition of cell cycle-related genes and the nuclear localization of α-synuclein is modulated by its phosphorylation at Serine 129 (68).
The interplay between mitochondria and α-synuclein during the progression of PD still constitutes an issue to be solved, as the exact contribution of mitochondrial deficits and α-synuclein aggregation to dopaminergic neurons degeneration has yet to be clearly elucidated (69, 70). Indeed, the aggregation of α-synuclein induces neural deficits, but it is also evident that mitochondrial dysfunctions are crucial events in the pathogenesis of PD (71, 72). Notably, the observation that α-synuclein is increased following a stroke, and that its induction is involved in the response to post-stroke brain damage, reinforces the idea that the protein can act as a pivotal regulator of neuronal resilience to injury (35, 73–75).
Numerous studies have shown that α-synuclein accumulation and aggregation can activate neuroinflammation (76–79), in agreement with the evidence showing increased levels of tumor necrosis factor alpha (TNFα), interleukin-1β (IL-1β), and IL-6 in the brains of patients with PD (80–82). In particular, reactive microglia have been found in PD brains (83, 84) and in transgenic mouse models of PD, and can be activated by α-synuclein pathological deposition (85, 86). The main mechanisms involved in α-synuclein aggregates-related microglia response are the activation of nod-like receptor (NLR) pyrin domain containing 3 (NLRP3) or caspase 1 inflammasome and nuclear factor-κB (NF-κB) signaling (87). Moreover, the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) pathway has been found to modulate the migration of microglial cells exposed to the protein (88, 89). Of note, lipopolysaccharide (LPS) and IL-1β increase the expression of α-synuclein in human macrophages (90, 91), while murine macrophages are activated by full length α-synuclein in vitro and in vivo (92, 93). Finally, the expression of α-synuclein in peripheral blood mononuclear cells (PBMCs) (94) and its modulation in PD brains support that α-synuclein may be implicated in the modulation of systemic inflammatory responses, even though its exact contribution is to be further investigated (32).
Alpha-Synuclein in ECs
Endothelial cells constitute a distinct cell population coating the innermost lining of blood and lymphatic vessels (95, 96). These cells are known to exhibit differential gene expression, morphology, and function across the vascular tree and organs of the body (97, 98). However, to which extent such heterogeneity impacts the endothelial dysfunctions in neurodegenerative diseases such as PD remains unclear. Cerebral ECs exert multiple functions, including the formation of the BBB, the regulation of immune cells trafficking and vascular hemostasis, and the control of cell migration and proliferation (95). In the CNS, ECs organize and maintain the BBB through anatomical and molecular interactions with neurons, pericytes, astrocytes, microglia, and perivascular macrophages in the neurovascular unit (NVU) [(99–103); Figure 1]. Moreover, ECs and astrocytes secrete and deposit basement membranes (BM) that provide additional barrier functions [(104); Figure 1]. Interestingly, studies in stroke and PD models showed that BBB disruption leads to enhanced neuroinflammation and accumulation of toxic forms of α-synuclein, which in turn could promote the progression of neuronal loss by impacting on diverse components of the BBB [(73, 105); Figure 1].
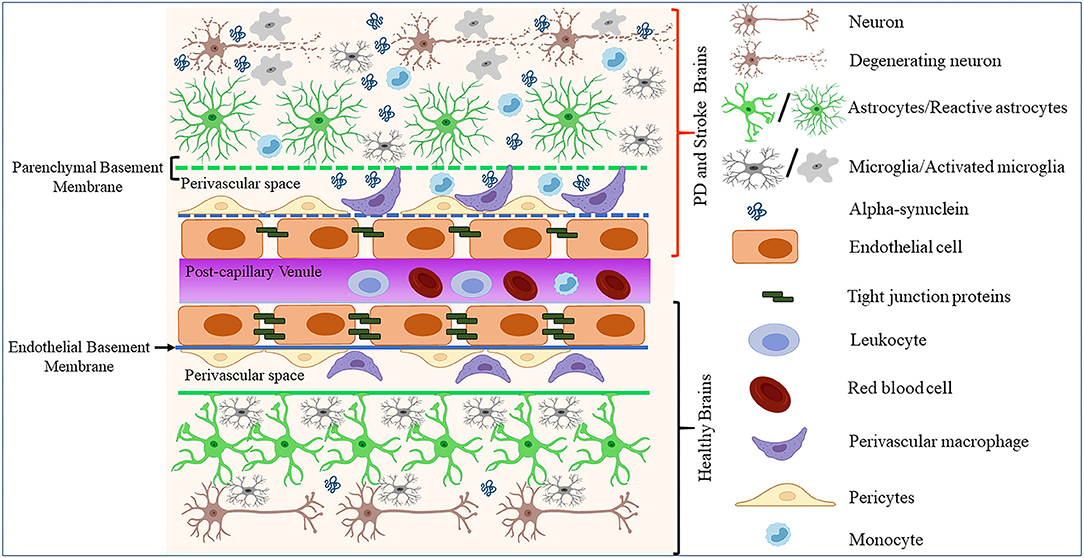
Figure 1. Schematic representation of how pathological α-synuclein-accumulation occurring in PD or stroke can disrupt the physiological homeostasis of the BBB by affecting its diverse components. The BBB is composed of microvascular endothelial cells, pericytes, astrocytes, and BM components deposited by ECs (endothelial BM) and astrocytes (parenchymal BM). More recently, perivascular macrophages and vessel-associated microglia were found to play a role in the maintenance and repair of BBB whose disruption is detected in various neurological disorders including stroke and PD. This could result in BM damage (dotted lines), downregulation of TJ proteins, abnormal accumulation, and spreading of toxic forms of proteins such as α-synuclein, activation of glial cells and PVMs, and infiltration of peripheral leukocytes and monocytes, leading to neuronal degeneration.
The expression of α-synuclein in vascular ECs supplying the brain and peripheral organs has been known for a long time (96). In the normal human brain, a gradient distribution appears to exist, where α-synuclein is present in higher levels in ECs of leptomeningeal vessels, while intra-parenchymal and capillary ECs show lower and no expressions, respectively (106). Nonetheless, the existence of such graded expression in PD, its functional relevance, and regulation have not been elucidated yet. Conversely, ECs lines, including those derived from cerebral micro-vessels, exhibit low endogenous α-synuclein levels when compared to neurons (38, 39, 106).
Interestingly, transmitted-electron microscopy studies addressing subcellular localization in ECs, identified α-synuclein near WPBs, elongated intracellular granules that contain chemokines, cytokines, and adhesive molecules which are rapidly released into the extracellular space by agonists and modulate ECs response to stimuli (38). Pathological conditions such as hypoxia, ischemia, inflammation, and oxidative stress increase α-synuclein levels, its aggregation in neurons, and to some extent in non-neuronal cells in vivo and in vitro (35, 73, 74, 90, 107–109). However, similar stimuli failed to upregulate α-synuclein levels in ECs (106, 110), supporting the need for a better understanding of the mechanisms regulating its expression in these cells. Interestingly, wild-type and mutant α-synuclein inhibit the agonist-induced-release of von Willebrand factor (vWF) and P-selectin translocation from WPBs in ECs (38). These processes enable ECs to control vascular homeostasis during inflammatory response and thrombosis [(111, 112); Figure 2]. Indeed, agonists such as thrombin, vascular endothelial growth factors (VEGF), histamine, and superoxide can induce an increase in intracellular calcium levels. Subsequently, calcium binds and activates calmodulin which then triggers the translocation of Ral specific guanine exchange factor (RalGDS), from cytosol to plasma membrane and activates membrane-bound RalA (a small GTPase and substrate for RalGDS) by exchanging GDP with GTP [(112); Figure 2A]. Afterward, the RalA-GTP interacts with and assembles exocyst, a multi-protein complex important in targeting vesicles to membranes (113), to promote the exocytosis of WPBs (Figure 2A). In parallel, forskolin, or epinephrine can increase cyclic adenosine monophosphate (cAMP) levels thus inducing protein kinase A activation, which also causes RalGDS membrane translocation [(38, 112); Figure 2A]. Upon activation of these pathways, α-synuclein binds to both RalGDS and β-arrestin, thus enhancing their interaction and inhibiting their dissociation and translocation to the plasma membrane, thereby preventing WPBs exocytosis [(38); Figure 2B]. Moreover, it may be also feasible to foresee that exocytosis might also be prevented by inactivation of phospholipase D (PLD) due to α-synuclein overexpression-related ER stress (114) or enhanced polymerization of actin filaments by α-synuclein [(18, 115, 116); Figure 2B], that by immobilizing WPBs would avoid their release.
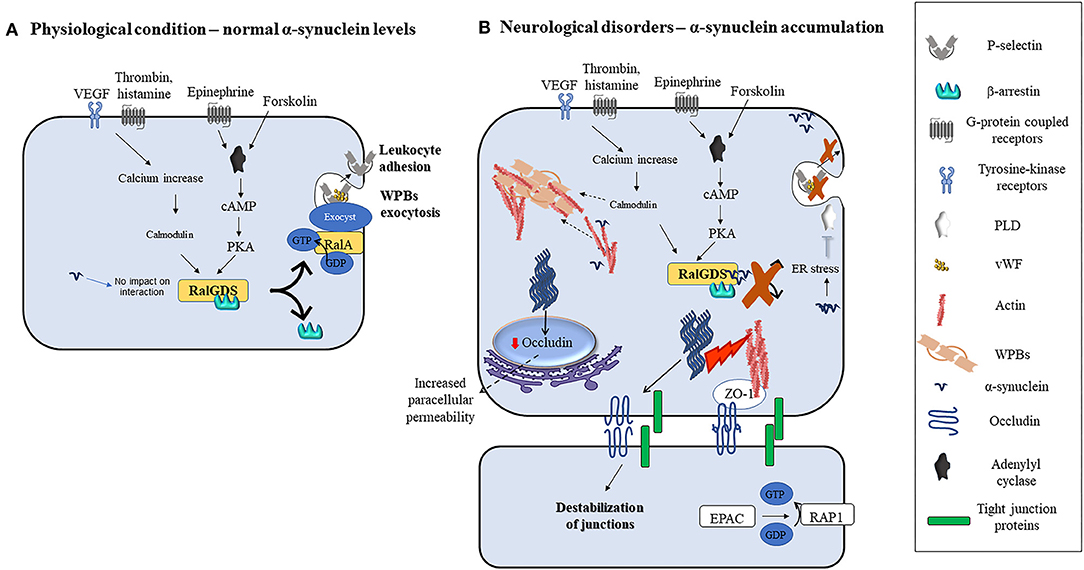
Figure 2. Modulation of ECs function by α-synuclein in physiological or pathological conditions. (A) Various agonists bind to endothelial cells G-protein coupled receptors (thrombin, superoxide, histamine, and epinephrine) or tyrosine kinase receptors (VEGF) to activate different intracellular pathways that lead to the release of contents of WPBs and translocation of P-selectin leading to leukocyte adhesion. Thrombin, VEGF, histamine, and superoxide lead to intracellular calcium increase and calmodulin activation, while forskolin and epinephrine increase intracellular cAMP and activate PKA. Calmodulin and PKA then trigger the translocation of RalGDS from cytosol to plasma membrane that activates membrane-bound RalA by exchanging GDP with GTP. This results in WPBs exocytosis. (B) Alpha-synuclein inhibits the release of contents of WPBs in vitro by various putative mechanisms which include binding to RalGDS and β-arrestin to prevent their dissociation, and hence exchange of RalA, blocking PLD activity, and immobilization of WPBs through enhanced actin polymerization. Fibrillary α-synuclein destabilizes TJ thereby affecting ECs paracellular permeability. This may occur through the downregulation of occludin and ZO-1.
Alpha-synuclein is present in the cerebrospinal fluid (CSF) and the blood and transported across the BBB (59, 117–120). In particular, α-synuclein can be transferred by multiple transport mechanisms including carriers such as lipoprotein receptor-related protein-1 (LRP-1) (59) or extracellular vesicles (EVs) (121). Exosome-derived α-synuclein induces oligomerization of endogenous soluble protein in recipient cells and contributes to intercellular propagation of pathology. The CSF of patients with PD show α-synuclein containing exosomes derived from various cells including microglia and exert different functions (122, 123). In line with this, erythrocyte-derived exosomes containing α-synuclein from patients with PD induce microglial activation in vivo and in vitro, thus suggesting that erythrocyte-derived extravasated α-synuclein may play a role in disease pathogenesis (121). These evidences support that further studies are needed to understand how exosome-associated physiological or pathological forms of the protein may impact on brain immune cells and ECs function and thus on BBB integrity.
Alpha-synuclein-induced inflammation might contribute first to the stimulation of rapid ECs response, which by driving the contraction of ECs, leads to the formation of gaps between them. This reshaping of ECs alters the continuous ECs layer mediating the improvement of its paracellular permeability and induces the activation of ECs. Consequently, the induction of proinflammatory molecules production and release from ECs increases the local blood flow. These events, in conjunction with the ECs layer alteration, prompt BBB dysfunction, leading to the extravasation of protein-rich exudates as well as to the recruitment and activation of circulating leukocytes, that further promote neuroinflammation (124). In particular, it may be feasible that the chronic upregulation of TNF-α and IL-1β associated with α-synuclein deposition, observed in patients with PD and animal models (82), might induce sustained activation of ECs. The consequent activation of NF-κB and activator protein 1 (AP-1) and the production of vascular cell adhesion molecule 1 (VCAM 1) and intercellular adhesion molecule 1 (ICAM 1) (124), would thus set the stage for enhanced neuroinflammation, BBB injury, and neurodegeneration (Figure 2). On this line, the mechanisms linking α-synuclein deposition to endothelial injury warrants further investigation.
Evidence supports that α-synuclein preformed fibrils (pffs) downregulate the expression of occludin and of zonula occludens 1 (ZO-1) (Figure 2B). As a consequence, the transport across intercellular junctions between ECs could be improved (39). However, α-synuclein pffs do not trigger endothelial dysfunction or release of proinflammatory cytokines from ECs in culture (39), supporting that these cells are less vulnerable to α-synuclein toxicity. On the other hand, activation of Ras homologous guanosine triphosphate phosphatase (RhoGTPases) leads to distinct effects on the ECs' barrier function depending on the type of GTPase activated (125, 126). For instance, excess activation of RhoA by thrombin or VEGF induces the formation of stress fibers which destabilizes intercellular junctions and downregulates the expression of eNOS, thereby promoting paracellular permeability and endothelial dysfunction [(126); Figure 2]. A recent study on a human brain-chip modeling the substantia nigra (SN) showed that α-synuclein fibrils can induce increased paracellular permeability (127). Interestingly, transcriptomic analysis of the ECs in the brain-chip revealed the upregulation of genes involved in inflammation, oxidative stress, autophagy, efflux system, and extracellular matrix deposition and the downregulation of genes that encode for TJ proteins (127). Conversely, the overexpression of A30P mutated α-synuclein has been found to upregulate collagen IV α2 chain (COL4A2), a major constituent of BMs in vivo and in vitro (22), further supporting that α-synuclein changes may impact the BBB integrity also by affecting this component. However, whether and how α-synuclein influences these pathways to regulate ECs functions at the BBB or the secretion and assembly of other BM elements, their degrading enzymes, or interaction with receptor proteins and other neighboring cells still needs to be addressed. Likewise, since endothelial dysfunction might, in turn, alter the transport of α-synuclein between the brain and vasculature, thus promoting its accumulation and the progression of α-synuclein pathology, studies addressing whether and how BBB dysfunction may impact PD progression could bring new insights into our basic understanding of the pathophysiology of this disorder.
Indeed, brains of patients with PD show evidence of endothelial degeneration, downregulation of TJ proteins, and even angiogenesis (39, 105, 128, 129). These changes were observed mostly in the SN, locus coeruleus (LC), and caudate putamen (CP), brain regions where α-synuclein-induced degeneration is prominent, and to a lesser extent in the cerebral cortex (105, 128, 129). Moreover, pathological alterations in the capillary BM including collagen deposition and thickening are evident in PD brains (12, 128, 130). It is thus plausible to speculate that such changes may reduce the efficiency of the exchange of molecules between the brain and vasculature, rendering neurons vulnerable to oxidative stress and accumulating cellular waste products.
Angiogenesis is a well-recognized adaptive response to cerebral hypoxia or ischemia and is regulated by BM proteins and their integrin receptors (131). Interestingly, the integrin receptor αvβ is upregulated in angiogenic vessels (131, 132) and in cerebral vessels of patients with PD and incidental LB disease (iLBD) (129), suggesting that the immature nascent vessels generated in PD brains, could contribute to neuroinflammation by facilitating the infiltration of peripheral immune cells and inflammatory or toxic factors (129). Consistently, co-localization of areas of leakage of an intravascular tracer with β3 integrin-expressing new vessels, indicating the presence of both angiogenesis and compromised BBB, has been observed in toxin-induced animal model of PD (132). Based on Braak's PD staging (56), patients with iLBD may represent an early disease stage where LB is restricted to LC and SN (129). Therefore, the presence of angiogenesis in patients with iLBD and PD supports that α-synuclein related vascular dysfunction might precede or/and contribute to the progression of neuroinflammation and neurodegeneration. This is further substantiated by findings showing that α-synuclein-related angiogenesis and downregulation of TJ proteins are not necessarily related to inflammation (39, 129). Indeed, recent findings indicate that dysfunction in BBB accompanied by pathological activation of pericytes precedes the onset of neuronal degeneration in a mouse model of PD (133), thus supporting that vascular dysfunction may be an early pathogenic events leading to neuronal damage. Furthermore, VEGF plays a protective role in PD through a direct effect on dopaminergic neurons (134) or via the canonical VEGF receptor (VEGFR2) pathway (135). VEGF released by activated astrocytes and microglia acts in a paracrine fashion to modulate ECs structure and function both in PD and DLB patients and in animal models of PD (136). This notwithstanding, whether α-synuclein is involved in the upregulation of VEGF in ECs remains to be investigated.
Interestingly, in postmortem sections of the trans-entorhinal (TEC) cortex (Figures 3A–C) and CP (not shown) from sporadic patients with PD, we observed perivascular accumulation of α-synuclein immunoreactivity (in blue) in correspondence of some vessels. Conversely, the brains of healthy or negative controls did not exhibit this feature (Figures 3D–F). By double immunolabeling of laminin α2 (brown) and α-synuclein (violet), we found that in the PD brains, α-synuclein-positive perivascular staining could be identified either in the outer (Figure 3G) and inner (Figure 3H) sides of the perivascular basement membrane, while in control brains, it did not show α-synuclein positivity in the proximity of laminin α2 staining (Figure 3I).
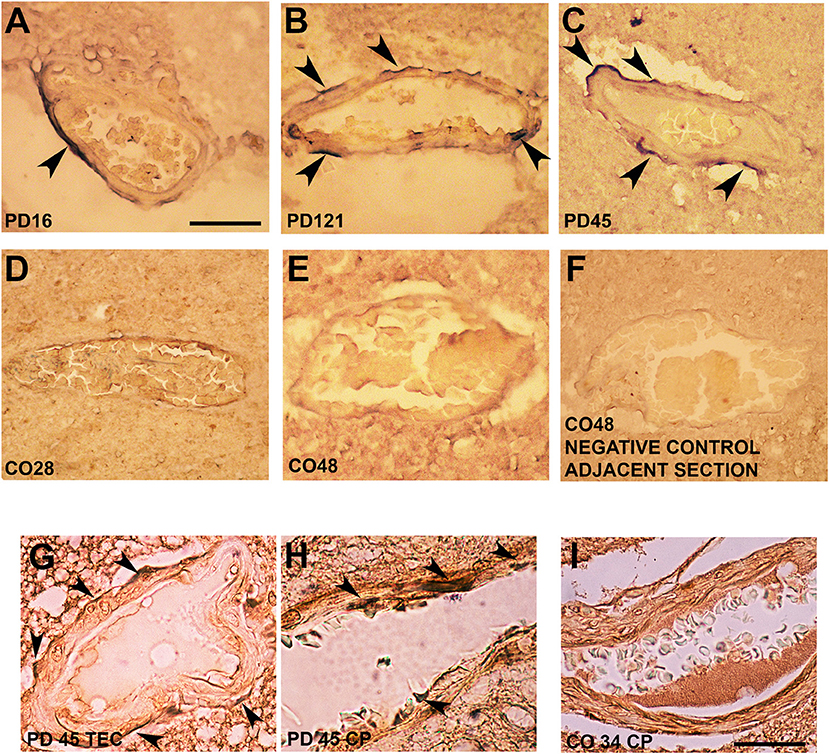
Figure 3. Alpha-synuclein perivascular immunoreactivity in postmortem sections from sporadic patients with PD. For these experiments, sections from three patients with PD (PD16, disease duration 18 years; PD45, disease duration 19 years; and PD121, disease duration 4 years) and three healthy controls (CO28, CO34, and CO48), kindly supplied by the Parkinson's UK Brain Bank, were analyzed. Briefly, sections were treated for antigen retrieval with 10 mM sodium citrate (20′ at 95°C) and 10% formic acid (15′ at RT). After 1 h incubation at room temperature (RT) with blocking solution (2% w/vol bovine serum albumin, 3% vol/vol normal goat serum, 0.3% Triton X-100 diluted in PBS 0.1 M pH 7.4) the 5-μm slices were subjected to either single α-synuclein (Sin211 MA5-12272 Thermo Fisher Scientific, Waltham, USA; dilution 1:500) or double laminin α2 (4H8-2, abcam ab11576; dilution 1:100)/α-synuclein (Sin211, MA5-12272 Thermo Fisher; dilution 1:500) immunolabeling according to previously described protocols (137, 138). Single α-synuclein immunopositive signal was revealed by Blue Alkaline Phosphatase (Vector Laboratories, Burlingame, CA) acquired by using a 40X objective, while for double immunolabeling laminin α2 was revealed by brown 3,3-diaminobenzidine (DAB) and α-synuclein by violet (Nickel supplemented) DAB (Vector laboratories) and acquired by a 100X objective. All the images were acquired by using an inverted light microscope (Olympus BX41; Olympus, Milan, Italy). (A–C) Representative images of perivascular α-synuclein immunolabeling (blue, arrows) in the TEC of three sporadic PD cases (PD 16, PD 121, and PD 45). (D–F) Images from the TEC of two of the healthy controls analyzed (CO28 and CO48). (D,E) The absence of α-synuclein immunolabeling in control brains, while (F) shows a representative image from a negative control for the immunostaining performed without the addition of the primary antibody on an adjacent section of CO48. (G,H) Representative images showing the presence of α-synuclein violet immunolabeling at the outer (G, arrows) and inner (H, arrows) side of laminin α2-positive perivascular BM in the brain of a sporadic patient with PD (PD45). Images are representative of the TEC (G) and CP (H). (I) Representative image showing the absence of α-synuclein accumulation around laminin α2-immunolabeling in the proximity of a vessel of the CP of a healthy control (CO34). Scale bar: (A–F) 40 μm; (G–I) 25 μm.
Although further studies are needed to corroborate whether and how α-synuclein deposition affects these cells, these findings, when coupled to the aforementioned noxious effects exerted by pathological α-synuclein on ECs, support that perivascular accumulation of the protein, by inducing ECs activation may compromise brain vessels integrity exacerbating astrocyte and microglia activation thus promoting neurodegeneration and BBB disruption (Figure 4).
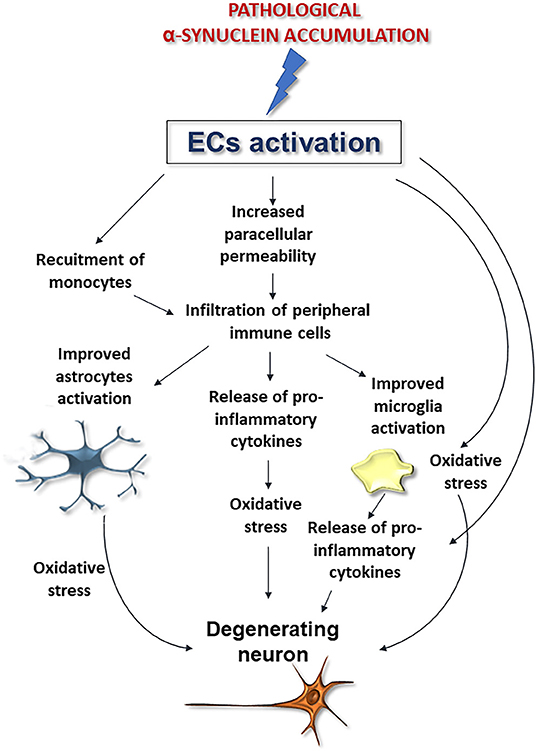
Figure 4. Possible downstream effects of pathological α-synuclein-induced activation of ECs on microglia, astrocytes, and neurons. Alpha-synuclein accumulation-induced ECs activation could promote BBB disruption and peripheral immune cell infiltration, improve microglia and astrocytes activation and exacerbate oxidative stress, thus promoting neuronal degeneration.
Alpha-Synuclein in BAMs and Other Perivascular Cells
Border-associated macrophages are a subset of CNS myeloid cells (macrophages) that like microglia originate prenatally in the yolk sac (139), invade the brain during the early prenatal period, and localize in the choroid plexus, perivascular, and leptomeningeal spaces. BAMs form stable populations with the sole exception of the choroid plexus macrophages that exchange with peripheral monocytes (139, 140). Indeed, BAMs can be anatomically distinguished into perivascular macrophages (PVM), meningeal macrophages (MAM), and choroid plexus associated macrophages (CPM) (139, 140). In healthy brains, PVMs are involved in the regulation of BBB permeability and phagocytosis of pathogens but can also promote the entrance of peripheral immune cells into the brain (102). Under pathological conditions such as cerebral amyloid angiopathy (CAA), AD (141) and PD (141), PVMs can participate in the clearance of toxic amyloid-β and α-synuclein. Consistently, the depletion of PVMs using clodronate-containing liposomes in a mouse model of PD resulted in the increased expression of VCAM 1, the infiltration of T cells, and the propagation of α-synuclein pathology (142). Moreover, in rats that underwent transient ischemia followed by reperfusion injury, BAMs were involved in promoting peripheral immune cell infiltration and vascular permeability without impacting the extent of ischemic damage, thus suggesting that additional studies are needed to fully understand the modulatory role of these cells in cerebrovascular dysfunctions and neuroinflammation (13).
Border associated macrophages are involved in immune surveillance and support the entrance of peripheral immune cells into the CNS under pathological conditions (143, 144). In rodents, monkeys, and humans, PVMs express the mannose receptor CD206 (145) and the scavenger receptor CD163, which under physiological conditions is expressed on tissue macrophages, with the exception of microglia and some monocytes (146–148). Recent research reports, dissecting the molecular signature of brain macrophages in mice at the single-cell level, reported a clear segregation of BAMs from microglia, identified a BAMs core gene signature, and even showed heterogeneity within BAMs (149–152). In stroke animal models and patients, a unique transcriptional signature of BAMs, their local proliferation and migration in the brain parenchyma, have been detected (14).
It is now believed that BAMs play a role in immune function, BBB integrity, and lymphatic clearance (139, 140). Currently, the identification of BAMs mainly relies on the use of anatomical studies aimed at disclosing their localization in the brain thanks to the use of few reliable molecular markers (139, 140). However, this approach is not applicable in the presence of inflammatory conditions or tissue injury when peripheral monocytes/macrophages enter the brain and reside in the same location and express similar molecular markers (139). Despite these limitations, remarkable progresses have been made to fully characterize and understand their role in the normal and diseased brain.
Alpha-synuclein is expressed by microglia and peripheral monocytes/macrophages in a lower amount compared to neurons (32), but the expression in BAMs has not been described yet. Since BAMs share similar ontogeny and molecular and immunologic characteristics with microglia (139, 140), they might exhibit analogous changes and activation states to α-synuclein stimulation (153, 154). Indeed, it has been described that BAMs and microglia display multiple similarities such as the expression of myeloid-specific markers. Among them, ionized calcium-binding adaptor molecule 1(Iba1), F4/80 (mouse) or EMR1 (human), chemokine receptors, scavenger receptors, receptor tyrosine kinases, Integrins, pattern recognition receptors (PRRs), and cytokines receptors (155). α-synuclein is known to induce inflammatory response and migration of microglia (32, 89, 156). For instance, previous studies showed that α-synuclein induces NOX2 activation in microglia by binding to toll-like receptor 2 (TLR-2) and CD11b leading to microglia-mediated neuronal toxicity (157). Similarly, α-synuclein binds to TLR-4 and activates NF-kB signaling which then induces a selective autophagy pathway named synucleinphagy and release of exosomes containing the protein, thus contributing to the intercellular spread of α-synuclein pathology (154).
Interestingly, monomeric α-synuclein can also impact microglia polarization by conferring an anti-inflammatory profile to these cells through the interaction with extracellular signal-regulated kinase (ERK) and the recruitment of the ERK/NF-κB, and peroxisome proliferator-activated receptor γ (PPARγ) pathways (158). It may thus be feasible that these pathways may also be activated upon exposure of BAMs to α-synuclein.
It is worth mentioning that pericytes can also play a role in the formation, maintenance, and regulation of BBB (159). Additionally, pericytes (60) and astrocytes (160) can mediate the transfer of α-synuclein between cells of the NVU, suggesting a possible role of non-neuronal cells in α-synuclein pathology spreading in PD (161). In vitro studies also showed that α-synuclein activates pericytes which in turn release proinflammatory mediators that can mediate BBB dysfunction (162). Early pericyte activation associated with BBB leakage has been recently described in a human α-synuclein overexpression-based mouse model of PD (133), thus further supporting that vascular pathology can constitute a relevant pathophysiological aspect of PD.
Peripheral immune cells such as lymphocytes have also been involved in the pathogenesis of PD. Indeed, studies in the brains of patients with PD and animal models showed that T cells with upregulated expression of the ICAM 1 receptor lymphocyte function-associated antigen-1 (LFA1) can promote leukocyte infiltration (163). Alpha-synuclein-specific T-cell reactivity has been found to be higher in early PD while decreasing in patients with late-stage disease (164). When considering the increase of α-synuclein in animal models of stroke (73), where Treg cells interact with ICAM1 on inflamed microvessels and platelets promoting vascular dysfunction (165), this evidence suggests that the accumulation of α-synuclein occurring following brain ischemia could very well-boost these pathogenic processes.
Several studies showed that α-synuclein aggregates can be detected in reactive astrocytes in the brains of patients with PD and animal models (33) suggesting a role of these cells in the clearance and /or spreading of α-synuclein toxicity in the brain parenchyma and NVU. Even though the role of endogenous α-synuclein in astrocytes remains to be fully explored, in disease states, α-synuclein activates astrocytes by interacting with PRRs such as TLR-4 (33). Activated astrocytes can in turn uptake and degrade the protein via the endosomal-lysosomal pathway and contribute to non-cell autonomous degeneration (166, 167).
Recent evidence suggests that α-synuclein can be removed from the brain via extracellular space drainage pathway which includes glymphatic transport and meningeal lymphatic system (61), whose reduction lead to the accumulation of toxic forms of amyloid-β in the brain parenchyma of AD rodent models (168–170). Similarly, a recent study in a transgenic PD mouse model overexpressing human A53T mutated α-synuclein showed that blockage of the deep cervical lymph node reduces glymphatic transport of an intraventricular tracer and promotes the accumulation of α-synuclein and its aggregation in SN, thus leading to the progression of α-synuclein pathology (61).
Taken together, impairment in these systems results in the accumulation of toxic proteins in the brain and contributes to the progression of neurodegenerative diseases. The close association of BAMs to perivascular and lymphatic drainage systems in the brain when coupled to the detection of α-synuclein aggregates in the perivascular space of a PD mouse model (61) supports that understanding of whether and how these cells contribute to the clearance of α-synuclein along these pathways deserves ad-hoc investigation.
In addition to this, it is plausible that the increase in α-synuclein levels observed following ischemia and spinal cord injury (35, 73, 74, 171) could result in a chemotactic gradient for microglia migration and activation (89) contributing to brain damage. Consistently, inhibition of α-synuclein induction following ischemia or spinal cord injury reduces secondary neuronal injury, inflammatory response, and improves neurological outcomes (171, 172). Although, it is known that juxta vascular microglia play a divergent role in repairing vascular injuries following an insult or systemic inflammation (173–175), whether α-synuclein modulates these cells or BAMs remains to be clarified.
Alpha-Synuclein Role in Modulating BBB Cells Interaction
In the normal and diseased brain, ECs communicate with neurons, microglia, pericytes, and astrocytes to regulate vascular function (176). More importantly, the interaction of microglia with ECs can exert divergent roles in regulating BBB integrity (175). In co-cultures of ECs and neurons, α-synuclein fibrils resulted in endothelial dysfunction, but this effect was not observed in ECs monocultures (39), supporting that neuronal-ECs crosstalk at the NVU may be perturbed by pathological α-synuclein.
CD200, a transmembrane protein found to be expressed in neurons, astrocytes, oligodendrocytes, and ECs, transduces signal via its receptor (CD200R), expressed on myeloid cells including microglial and BAMs (177). CD200-CD200R and C-X3-C motif chemokine ligand 1 (CX3CL1)-C-X3-C motif chemokine receptor 1 (CX3CR1) signaling between neurons and microglia helps to maintain microglia in the resting state (178). Inactivation of the transmembrane glycoprotein CD200R in microglia of a toxin-induced PD mouse model results in increased activation of these cells, release of proinflammatory cytokines, loss of dopaminergic neurons in the SN, and behavioral deficits (179). Similarly, monocyte-derived macrophages (MDMs) from patients with PD show dysregulation in CD200R signaling (180).
On the other hand, M2 macrophages express pro-angiogenic factors such as VEGF and fibroblast growth factor 2 (FGF2), that by activating their receptors (VEGFR2 and FGFR) promote angiogenesis and neuronal survival (181). In vitro studies have shown that microglia maintains ECs in a resting state by secreting transforming growth factor-beta (TGF-β), an anti-inflammatory cytokine, while the proinflammatory TNF-α induces ECs proliferation (182). However, whether this kind of communication occurs also between ECs and BAMs or is influenced by α-synuclein still remains to be elucidated.
Secreted toxic species of α-synuclein are known to bind to various cell surface receptors in adjacent cells and activate several intracellular pathways leading to synaptic dysfunction, neurodegeneration, and inflammation (183). On this line, α-synuclein binds to TLR-2, TLR-4, and CD11β integrin to activate NF-kB signaling and assembly of NLRP3 inflammasome in microglia (37, 154, 157). It is thus plausible that various receptors for α-synuclein might exist in different cells, including ECs, and that the protein may regulate their intracellular activities or their crosstalk within neighboring cells such as BAMs.
Current Gaps and Future Perspectives
While α-synuclein associated vascular dysfunction is evident in PD (11, 128), most of the studies aimed at understanding the physiological role of the protein in ECs have been performed by overexpressing the protein through transgene expression (38, 39). Therefore, the role of extracellularly released α-synuclein on these cells still needs to be extensively explored. Furthermore, it is not yet established whether ECs respond to various pathogenic stimuli by regulating α-synuclein level or its transport across the BBB. Future studies exploiting improved models might overcome these limitations.
It is now clear that α-synuclein-associated inflammation contributes to the pathophysiology of PD (10) but research on whether or how α-synuclein modulates the function of macrophages in the brain has been mostly focused on microglia. As a result, our knowledge on the role of α-synuclein in other brain resident macrophages such as BAMs or other perivascular cells is poor. Similarly, despite the presence of studies indicating interplay between ECs and microglia (175), the interplay between perivascular cells and ECs has been scarcely studied. For instance, since recent evidence showed that BAMs and microglia can acquire distinct genetic and molecular phenotypes early in development (152), studying whether α-synuclein plays a role in modulating the signaling pathways mediating the crosstalk between BAMs and ECs could bring novel and significant insights for understanding the biological basis of neurological disorders such as PD or stroke. Similarly, though α-synuclein transfer between cells and across BBB interfaces has been established (59, 154), whether and how BAMs are involved in this event also deserves further investigations.
A deeper understanding of the role of physiological and pathological forms of α-synuclein in the modulation of BAMs and ECs or their interplay may also greatly aid the identification of novel therapeutic targets for stroke or neurodegenerative synucleinopathies.
Author Contributions
TB and GF wrote and revised the article and prepared the figures. TB performed immunostaining. FL revised the article and was involved in image acquisition. AB, MP, and SM did a critical revision of the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 813294.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Tissue samples and associated clinical and neuropathological data were supplied by the Parkinson's UK Brain Bank, funded by Parkinson's UK, a charity registered in England and Wales (258197) and in Scotland (SC037554). Icons of cells used in Figure 1 were created using Biorender.
References
1. Feigin VL, Nichols E, Alam T, Bannick MS, Beghi E, Blake N, et al. Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2019) 18:459–80. doi: 10.1016/S1474-4422(18)30499-X
2. Mattson MP, Duan W, Pedersen WA, Culmsee C. Neurodegenerative disorders and ischemic brain diseases. Apoptosis. (2001) 6:69–81. doi: 10.1023/A:1009676112184
3. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Prim. (2017) 3:1–21. doi: 10.1038/nrdp.2017.13
4. Goedert M, Jakes R, Spillantini MG. The synucleinopathies: twenty years on. J Parkinsons Dis. (2017) 7:S53–71. doi: 10.3233/JPD-179005
5. Spillantini MG, Goedert M. The alpha-synucleinopathies: Parkinson's disease, dementia with Lewy bodies, and multiple. Ann N Y Acad Sci. (2002) 920:16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x
6. Bellucci A, Antonini A, Pizzi M, Spano PF. The end is the beginning: Parkinson's disease in the light of brain imaging. Front Aging Neurosci. (2017) 9:1–5. doi: 10.3389/fnagi.2017.00330
7. Bellucci A, Mercuri NB, Venneri A, Faustini G, Longhena F, Pizzi M, et al. Review: Parkinson's disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol. (2016) 42:77–94. doi: 10.1111/nan.12297
8. Calo L, Wegrzynowicz M, Santivañez-Perez J, Grazia Spillantini M. Synaptic failure and α-synuclein. Movement Disord. (2016) 31:169–77. doi: 10.1002/mds.26479
9. Janda E, Boi L, Carta AR. Microglial phagocytosis and its regulation: a therapeutic target in parkinson's disease? Front Mol Neurosci. (2018) 11:1–8. doi: 10.3389/fnmol.2018.00144
10. Ho MS. Microglia in Parkinson's disease. Adv Exp Med Biol. (2019) 1175:335–53. doi: 10.1007/978-981-13-9913-8_13
11. Yang P, Min X-L, Mohammadi M, Turner C, Faull R, Waldvogel H, et al. Endothelial degeneration of Parkinson's disease is related to alpha-synuclein aggregation. J Alzheimers Dis Parkinsonism. (2017) 7:370. doi: 10.4172/2161-0460.1000370
12. Yang P, Pavlovic D, Waldvogel H, Dragunow M, Synek B, Turner C, et al. String vessel formation is increased in the brain of Parkinson disease. J Parkinsons Dis. (2015) 5:821–36. doi: 10.3233/JPD-140454
13. Pedragosa J, Salas-Perdomo A, Gallizioli M, Cugota R, Miró-Mur F, Briansó F, et al. CNS-border associated macrophages respond to acute ischemic stroke attracting granulocytes and promoting vascular leakage. Acta Neuropathol Commun. (2018) 6:76. doi: 10.1186/s40478-018-0581-6
14. Rajan WD, Wojtas B, Gielniewski B, Miró-Mur F, Pedragosa J, Zawadzka M, et al. Defining molecular identity and fates of CNS-border associated macrophages after ischemic stroke in rodents and humans. Neurobiol Dis. (2020) 137:104722. doi: 10.1016/j.nbd.2019.104722
15. Zille M, Ikhsan M, Jiang Y, Lampe J, Wenzel J, Schwaninger M. The impact of endothelial cell death in the brain and its role after stroke: a systematic review. Cell Stress. (2019) 3:330–47. doi: 10.15698/cst2019.11.203
16. Andjelkovic AV, Xiang J, Stamatovic SM, Hua Y, Xi G, Wang MM, et al. Endothelial targets in stroke: translating animal models to human. Arteriosc Thromb Vasc Biol. (2019) 39:2240–7. doi: 10.1161/ATVBAHA.119.312816
17. Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. (1996) 35:13709–15. doi: 10.1021/bi961799n
18. Bellucci A, Zaltieri M, Navarria L, Grigoletto J, Missale C, Spano P. From alpha-synuclein to synaptic dysfunctions: new insights into the pathophysiology of Parkinson's disease. Brain Res. (2012) 1476:183–202. doi: 10.1016/j.brainres.2012.04.014
19. Faustini G, Marchesan E, Zonta L, Bono F, Bottani E, Longhena F, et al. Alpha-synuclein preserves mitochondrial fusion and function in neuronal cells. Oxidative Med Cell Longevity. (2019) 2019:4246350. doi: 10.1155/2019/4246350
20. Longhena F, Faustini G, Spillantini MG, Bellucci A. Living in promiscuity: the multiple partners of alpha-synuclein at the synapse in physiology and pathology. Int J Mol Sci. (2019) 20:1–24. doi: 10.3390/ijms20010141
21. Cheng F, Vivacqua G, Yu S. The role of alpha-synuclein in neurotransmission and synaptic plasticity. J Chem Neuroanat. (2011) 42:242–8. doi: 10.1016/j.jchemneu.2010.12.001
22. Paiva I, Jain G, Lázaro DF, Jerčić KG, Hentrich T, Kerimoglu C, et al. Alpha-synuclein deregulates the expression of COL4A2 and impairs ER-Golgi function. Neurobiol Dis. (2018) 119:121–35. doi: 10.1016/j.nbd.2018.08.001
23. Villar-Piqué A, Lopes da Fonseca T, Outeiro TF. Structure, function and toxicity of alpha-synuclein: the Bermuda triangle in synucleinopathies. J Neurochem. (2016) 139:240–55. doi: 10.1111/jnc.13249
24. Spillantini MG, Goedert M. Neurodegeneration and the ordered assembly of α-synuclein. Cell Tissue Res. (2018) 373:137–48. doi: 10.1007/s00441-017-2706-9
25. Burai R, Ait-Bouziad N, Chiki A, Lashuel HA. Elucidating the role of site-specific nitration of α-synuclein in the pathogenesis of Parkinson's disease via protein semisynthesis and mutagenesis. J Am Chem Soc. (2015) 137:5041–52. doi: 10.1021/ja5131726
26. Souza JM, Giasson BI, Lee VM-Y, Ischiropoulos H. Chaperone-like activity of synucleins. FEBS Lett. (2000). 474:116–9. doi: 10.1016/S0014-5793(00)01563-5
27. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. (2002) 4:160–4. doi: 10.1038/ncb748
28. Alam P, Bousset L, Melki R, Otzen DA-O. α-Synuclein oligomers and fibrils: a spectrum of species, a spectrum of toxicities. J Neurochem. (2019) 150:522–34. doi: 10.1111/jnc.14808
29. Danzer KM, Schnack C, Sutcliffe A, Hengerer B, Gillardon F. Functional protein kinase arrays reveal inhibition of p-21-activated kinase 4 by alpha-synuclein oligomers. J Neurochem. (2007) 103:2401–7. doi: 10.1111/j.1471-4159.2007.04933.x
30. Choi B-K, Choi M-G, Kim J-Y, Yang Y, Lai Y, Kweon D-H, et al. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci. (2013) 110:4087. doi: 10.1073/pnas.1218424110
31. Flavin WP, Bousset L, Green ZC, Chu Y, Skarpathiotis S, Chaney MJ, et al. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. (2017) 134:629–53. doi: 10.1007/s00401-017-1722-x
32. Pei Y, Maitta RW. Alpha synuclein in hematopoiesis and immunity. Heliyon. (2019) 5:e02590. doi: 10.1016/j.heliyon.2019.e02590
33. Sorrentino ZA, Giasson BI, Chakrabarty PA-O. α-Synuclein and astrocytes: tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol. (2019) 138:1–21. doi: 10.1007/s00401-019-01977-2
34. Barbour R, Kling K, Anderson JP, Banducci K, Cole T, Diep L, et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegener Dis. (2008) 5:55–9. doi: 10.1159/000112832
35. Wu Z, Li X, Zeng M, Qiu H, Feng H, Xu X, et al. Alpha-synuclein alterations in red blood cells of peripheral blood after acute ischemic stroke. Int J Clin Exp Pathol. (2019) 12:1757–63.
36. Hashimoto M, Yoshimoto M, Sisk A, Hsu LJ, Sundsmo M, Kittel A, et al. NACP, a synaptic protein involved in alzheimer's disease, is differentially regulated during megakaryocyte differentiation. Biochem Biophys Res Commun. (1997) 237:611–6. doi: 10.1006/bbrc.1997.6978
37. Ferreira SA, Romero-Ramos M. Microglia response during Parkinson's disease: alpha-synuclein intervention. Front Cell Neurosci. (2018) 12:1–17. doi: 10.3389/fncel.2018.00247
38. Kim KS, Park JY, Jou I, Park SM. Regulation of Weibel-Palade body exocytosis by α-synuclein in endothelial cells. J Biol Chem. (2010) 285:21416–25. doi: 10.1074/jbc.M110.103499
39. Kuan W-L, Bennett N, He X, Skepper JN, Martynyuk N, Wijeyekoon R, et al. α-Synuclein pre-formed fibrils impair tight junction protein expression without affecting cerebral endothelial cell function. Exp Neurol. (2016). 285:72–81. doi: 10.1016/j.expneurol.2016.09.003
40. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. (1998). 95:6469–73. doi: 10.1073/pnas.95.11.6469
41. Herva ME, Spillantini MG. Parkinson's disease as a member of Prion-like disorders. Virus Res. (2015) 207:38–46. doi: 10.1016/j.virusres.2014.10.016
42. Guo M, Wang J, Zhao Y, Feng Y, Han S, Dong Q, et al. OUP accepted manuscript. Brain. (2020) 143:1476–97. doi: 10.1093/brain/awaa090
43. Chadchankar H, Ihalainen J, Tanila H, Yavich L. Decreased reuptake of dopamine in the dorsal striatum in the absence of alpha-synuclein. Brain Res. (2011) 1382:37–44. doi: 10.1016/j.brainres.2011.01.064
44. Yavich L, Tanila H, Vepsäläinen S, Jäkälä P. Role of α-synuclein in presynaptic dopamine recruitment. J Neurosci. (2004) 24:11165–70. doi: 10.1523/JNEUROSCI.2559-04.2004
45. Vamvaca K, Volles MJ, Lansbury PT. The first N-terminal amino acids of α-synuclein are essential for α-helical structure formation in vitro and membrane binding in yeast. J Mol Biol. (2009) 389:413–24. doi: 10.1016/j.jmb.2009.03.021
46. Bartels T, Ahlstrom LS, Leftin A, Kamp F, Haass C, Brown MF, et al. The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys J. (2010) 99:2116–24. doi: 10.1016/j.bpj.2010.06.035
47. Runfola M, De Simone A, Vendruscolo M, Dobson CM, Fusco G. The N-terminal acetylation of α-synuclein changes the affinity for lipid membranes but not the structural properties of the bound state. Sci Rep. (2020) 10:204. doi: 10.1038/s41598-019-57023-4
48. Barrett PJ, Timothy Greenamyre J. Post-translational modification of α-synuclein in Parkinson's disease. Brain Research. (2015) 1628:247–53. doi: 10.1016/j.brainres.2015.06.002
49. Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA. (1993) 90:11282–6. doi: 10.1073/pnas.90.23.11282
50. Giasson BI, Murray IVJ, Trojanowski JQ, Lee VMY. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J Biol Chem. (2001) 276:2380–6. doi: 10.1074/jbc.M008919200
51. Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human α-synuclein. J Biol Chem. (2005) 280:9595–603. doi: 10.1074/jbc.M411805200
52. Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J Am Chem Soc. (2005) 127:476–7. doi: 10.1021/ja044834j
53. Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat Med. (1998) 4:1318–20. doi: 10.1038/3311
54. Gurry T, Ullman O, Fisher CK, Perovic I, Pochapsky T, Stultz CM. The dynamic structure of α-synuclein multimers. J Am Chem Soc. (2013) 135:3865–72. doi: 10.1021/ja310518p
55. Peelaerts W, Baekelandt V. α-Synuclein strains and the variable pathologies of synucleinopathies. J Neurochem. (2016) 139:256–74. doi: 10.1111/jnc.13595
56. Braak H, Del Tredici K, Rüb U, De Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003) 24:197–211. doi: 10.1016/S0197-4580(02)00065-9
57. Engelender S, Isacson O. The threshold theory for Parkinson's disease. Trends Neurosci. (2017) 40:4–14. doi: 10.1016/j.tins.2016.10.008
58. Longhena F, Spano P, Faustini G, Bellucci, Arianna, Missale C, et al. The contribution of α-synuclein spreading to Parkinson's disease synaptopathyr. Neural Plasticity. (2017) 2017:5012129. doi: 10.1155/2017/5012129
59. Sui Y-T, Bullock KM, Erickson MA, Zhang J, Banks WA. Alpha synuclein is transported into and out of the brain by the blood–brain barrier. Peptides. (2014) 62:197–202. doi: 10.1016/j.peptides.2014.09.018
60. Dieriks BV, Park TIH, Fourie C, Faull RLM, Dragunow M, Curtis MA. α-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson's disease patients. Sci Rep. (2017). 7:42984. doi: 10.1038/srep42984
61. Zou W, Pu T, Feng W, Lu M, Zheng Y, Du R, et al. Blocking meningeal lymphatic drainage aggravates Parkinson's disease-like pathology in mice overexpressing mutated α-synuclein. Transl Neurodegener. (2019) 8:1–17. doi: 10.1186/s40035-019-0147-y
62. Arotcarena ML, Dovero S, Prigent A, Bourdenx M, Camus S, Porras G, et al. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain. (2020) 143:1462–75. doi: 10.1093/brain/awaa096
63. Guardia-Laguarta C, Area-Gomez E, Schon EA, Przedborski S. Novel subcellular localization for α-synuclein: possible functional consequences. Front Neuroanat. (2015) 9:17. doi: 10.3389/fnana.2015.00017
64. Thayanidhi N, Helm JR, Nycz DC, Bentley M, Liang Y, Hay JC. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol Biol Cell. (2010) 21:1850–63. doi: 10.1091/mbc.e09-09-0801
65. Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. (2006) 15:3012–23. doi: 10.1093/hmg/ddl243
66. Schaser AJ, Osterberg VR, Dent SE, Stackhouse TL, Wakeham CM, Boutros SW, et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci Rep. (2019) 9:10919. doi: 10.1038/s41598-019-47227-z
67. Chen V, Moncalvo M, Tringali D, Tagliafierro L, Shriskanda A, Ilich E, et al. The mechanistic role of alpha-synuclein in the nucleus: impaired nuclear function caused by familial Parkinson's disease SNCA mutations. Hum Mol Genet. (2020) 29:3107–21. doi: 10.1093/hmg/ddaa183
68. Pinho R, Paiva I, Jercic KG, Fonseca-Ornelas L, Gerhardt E, Fahlbusch C, et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum Mol Genet. (2019) 28:31–50. doi: 10.1093/hmg/ddy326
69. Faustini G, Bono F, Valerio A, Pizzi M, Spano P, Bellucci A. Mitochondria and α-synuclein: friends or foes in the pathogenesis of Parkinson's disease? Genes. (2017) 8:377. doi: 10.3390/genes8120377
70. Zaltieri M, Longhena F, Pizzi M, Missale C, Spano P, Bellucci A. Mitochondrial dysfunction and α-synuclein synaptic pathology in Parkinson's disease: who's on first? Parkinsons Dis. (2015) 2015:108029. doi: 10.1155/2015/108029
71. Pozo Devoto VM, Falzone TL. Mitochondrial dynamics in Parkinson's disease: a role for α-synuclein? Dis Mod Mech. (2017) 10:1075–87. doi: 10.1242/dmm.026294
72. Gao F, Yang J, Wang D, Li C, Fu Y, Wang H, et al. Mitophagy in Parkinson's disease: pathogenic and therapeutic implications. Front Neurol. (2017) 8:527. doi: 10.3389/fneur.2017.00527
73. Kim TH, Mehta SL, Kaimal B, Lyons K, Dempsey RJ, Vemuganti R. Poststroke induction of α-synuclein mediates ischemic brain damage. J Neurosci. (2016) 36:7055–65. doi: 10.1523/JNEUROSCI.1241-16.2016
74. Unal-Cevik I, Gursoy-Ozdemir Y, Yemisci M, Lule S, Gurer G, Can A, et al. Alpha-synuclein aggregation induced by brief ischemia negatively impacts neuronal survival in vivo: a study in A30Palpha-synuclein transgenic mouse. J Cereb Blood Flow Metab. (2011) 31:913–23. doi: 10.1038/jcbfm.2010.170
75. Zhao HQ, Li FF, Wang Z, Wang XM, Feng T. A comparative study of the amount of alpha-synuclein in ischemic stroke and Parkinson's disease. Neurol Sci. (2016) 37:749–54. doi: 10.1007/s10072-016-2485-1
76. Jackson-Lewis V, Jakowec M, Burke RE, Przedborski S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration. (1995) 4:257–69. doi: 10.1016/1055-8330(95)90015-2
77. McGeer PL, Schwab C, Parent A, Doudet D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann Neurol. (2003) 54:599–604. doi: 10.1002/ana.10728
78. Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson's disease. Neurobiol Aging. (2008) 29:1690–701. doi: 10.1016/j.neurobiolaging.2007.04.006
79. Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, et al. Aggregated α-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J. (2005) 19:533–42. doi: 10.1096/fj.04-2751com
80. Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. (1994) 180:147–50. doi: 10.1016/0304-3940(94)90508-8
81. Mogi M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. (1994) 165:208–10. doi: 10.1016/0304-3940(94)90746-3
82. Nagatsu T, Mogi M, Ichinose H, Togari A. Cytokines in Parkinson's disease. J Neural Transm Suppl. (2000) 143–51. doi: 10.1007/978-3-7091-6284-2_12
83. Bartels AL, Willemsen AT, Doorduin J, de Vries EF, Dierckx RA, Leenders KL. [11C]-PK11195 PET: quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson's disease? Parkinsonism Relat Disord. (2010) 16:57–9. doi: 10.1016/j.parkreldis.2009.05.005
84. Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis. (2006) 21:404–12. doi: 10.1016/j.nbd.2005.08.002
85. Béraud D, Hathaway HA, Trecki J, Chasovskikh S, Johnson DA, Johnson JA, et al. Microglial activation and antioxidant responses induced by the Parkinson's disease protein α-synuclein. J Neuroimmune Pharmacol. (2013) 8:94–117. doi: 10.1007/s11481-012-9401-0
86. Hoenen C, Gustin A, Birck C, Kirchmeyer M, Beaume N, Felten P, et al. Alpha-synuclein proteins promote pro-inflammatory cascades in microglia: stronger effects of the A53T mutant. PLoS ONE. (2016) 11:e0162717–e. doi: 10.1371/journal.pone.0162717
87. Bellucci A, Bubacco L, Longhena F, Parrella E, Faustini G, Porrini V, et al. Nuclear factor-κB dysregulation and α-synuclein pathology: critical interplay in the pathogenesis of Parkinson's disease. Front Aging Neurosci. (2020) 12:68. doi: 10.3389/fnagi.2020.00068
88. Wang S, Chu C-H, Stewart T, Ginghina C, Wang Y, Nie H, et al. α-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc Natl Acad Sci USA. (2015). 112:E1926–35. doi: 10.1073/pnas.1417883112
89. Li Y, Niu M, Zhao A, Kang W, Chen Z, Luo N, et al. CXCL12 is involved in α-synuclein-triggered neuroinflammation of Parkinson's disease. J Neuroinflamm. (2019) 16:263. doi: 10.1186/s12974-019-1646-6
90. Tanji K, Mori F, Imaizumi T, Yoshida H, Matsumiya T, Tamo W, et al. Upregulation of α-synuclein by lipopolysaccharide and interleukin-1 in human macrophages. Pathol Int. (2002) 52:572–7. doi: 10.1046/j.1440-1827.2002.01385.x
91. Prigent A, Lionnet A, Durieu E, Chapelet G, Bourreille A, Neunlist M, et al. Enteric alpha-synuclein expression is increased in Crohn's disease. Acta Neuropathol. (2019) 137:359–61. doi: 10.1007/s00401-018-1943-7
92. Lee S-b, Park SM, Ahn KJ, Chung KC, Paik SR, Kim J. Identification of the amino acid sequence motif of alpha-synuclein responsible for macrophage activation. Biochem Biophys Res Commun. (2009) 381:39–43. doi: 10.1016/j.bbrc.2009.02.002
93. Qin H, Buckley JA, Li X, Liu Y, Fox TH, Meares GP, et al. Inhibition of the JAK/STAT pathway protects against α-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J Neurosci. (2016) 36:5144–59. doi: 10.1523/JNEUROSCI.4658-15.2016
94. Shin EC, Cho SE, Lee DK, Hur MW, Paik SR, Park JH, et al. Expression patterns of alpha-synuclein in human hematopoietic cells and in Drosophila at different developmental stages. Mol Cells. (2000) 10:65–70. doi: 10.1007/s10059-000-0065-x
95. Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. (2007) 100:158–73. doi: 10.1161/01.RES.0000255691.76142.4a
97. Aird WC. Endothelial cell heterogeneity. Cold Spring Harbor Perspect Med. (2012) 2:1–13. doi: 10.1101/cshperspect.a006429
98. Hupe M, Li MX, Kneitz S, Davydova D, Yokota C, Kele-Olovsson J, et al. Gene expression profiles of brain endothelial cells during embryonic development at bulk and single-cell levels. Sci Signal. (2017) 10:eaag2476. doi: 10.1126/scisignal.aag2476
99. Profaci CP, Munji RN, Pulido RS, Daneman R. The blood-brain barrier in health and disease: important unanswered questions. J Exp Med. (2020) 217:e20190062. doi: 10.1084/jem.20190062
100. Liebner SA-O, Dijkhuizen RM, Reiss Y, Plate KH, Agalliu D, Constantin G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. (2018) 135:311–36. doi: 10.1007/s00401-018-1815-1
101. Engelhardt B, Sorokin L. The blood–brain and the blood–cerebrospinal fluid barriers: function and dysfunction. Semi Immunopathol. (2009) 31:497–511. doi: 10.1007/s00281-009-0177-0
102. Lapenna A, De Palma M, Lewis CE. Perivascular macrophages in health and disease. Nat Rev Immunol. (2018) 18:689–702. doi: 10.1038/s41577-018-0056-9
103. Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. (2015) 7:a020412. doi: 10.1101/cshperspect.a020412
104. Hannocks M-J, Pizzo ME, Huppert J, Deshpande T, Abbott NJ, Thorne RG, et al. Molecular characterization of perivascular drainage pathways in the murine brain. J Cereb Blood Flow Metab. (2017) 38:669–86. doi: 10.1177/0271678X17749689
105. Gray MT, Woulfe JM. Striatal blood-brain barrier permeability in Parkinson's disease. J Cereb Blood Flow Metab. (2015) 35:747–50. doi: 10.1038/jcbfm.2015.32
106. Tamo W, Imaizumi T, Tanji K, Yoshida H, Mori F, Fukuda I, et al. Expression of α-synuclein in vascular endothelial and smooth muscle cells. Int Congr Ser. (2003) 1251:173–9. doi: 10.1016/S0531-5131(03)00120-1
107. Sun HL, Sun BL, Chen DW, Chen Y, Li WW, Xu MY, et al. Plasma α-synuclein levels are increased in patients with obstructive sleep apnea syndrome. Ann Clin Transl Neurol. (2019) 6:788–94. doi: 10.1002/acn3.756
108. Sharma N, Nehru B. Curcumin affords neuroprotection and inhibits α-synuclein aggregation in lipopolysaccharide-induced Parkinson's disease model. Inflammopharmacology. (2018) 26:349–60. doi: 10.1007/s10787-017-0402-8
109. Prasad K. Oxidative stress, pro-inflammatory cytokines, and antioxidants regulate expression levels of microRNAs in Parkinson's disease. Curr Aging Sci. (2017) 10: 177–84. doi: 10.2174/1874609810666170102144233
110. Tamo W, Imaizumi T, Tanji K, Yoshida H, Mori F, Yoshimoto M, et al. Expression of α-synuclein, the precursor of non-amyloid β component of Alzheimer's disease amyloid, in human cerebral blood vessels. Neuroscience Lett. (2002) 326:5–8. doi: 10.1016/S0304-3940(02)00297-5
111. Lowenstein CJ, Morrell CN, Yamakuchi M. Regulation of Weibel-Palade body exocytosis. Trends Cardiovasc Med. (2005) 15:302–8. doi: 10.1016/j.tcm.2005.09.005
112. Rondaij MG, Bierings R, Kragt A, Van Mourik JA, Voorberg J. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arteriosc Thromb Vasc Biol. (2006) 26:1002–7. doi: 10.1161/01.ATV.0000209501.56852.6c
113. Moskalenko S, Tong C, Rosse C, Mirey G, Formstecher E, Daviet L, et al. Ral GTPases regulate exocyst assembly through dual subunit interactions. J Biol Chem. (2003) 278:51743–8. doi: 10.1074/jbc.M308702200
114. Rappley I, Gitler AD, Selvy PE, LaVoie MJ, Levy BD, Brown HA, et al. Evidence that α-synuclein does not inhibit phospholipase D. Biochemistry. (2009) 48:1077–83. doi: 10.1021/bi801871h
115. Alim MA, Ma Q-L, Takeda K, Aizawa T, Matsubara M, Nakamura M, et al. Demonstration of a role for α-synuclein as a functional microtubule-associated protein. J Alzheimers Dis. (2004) 6:435–42. doi: 10.3233/JAD-2004-6412
116. Lee HJ, Khoshaghideh F, Lee S, Lee SJ. Impairment of microtubule-dependent trafficking by overexpression of α-synuclein. Eur J Neurosc. (2006) 24:3153–62. doi: 10.1111/j.1460-9568.2006.05210.x
117. Borghi R, Marchese R, Negro A, Marinelli L, Forloni G, Zaccheo D, et al. Full length α-synuclein is present in cerebrospinal fluid from Parkinson's disease and normal subjects. Neurosci Lett. (2000) 287:65–7. doi: 10.1016/S0304-3940(00)01153-8
118. Hansson O, Hall S, Öhrfelt A, Zetterberg H, Blennow K, Minthon L, et al. Levels of cerebrospinal fluid α-synuclein oligomers are increased in Parkinson's disease with dementia and dementia with Lewy bodies compared to Alzheimer's disease. Alzheimers Res Ther. (2014) 6:4–9. doi: 10.1186/alzrt255
119. Bates CA, Zheng W. Brain disposition of α-Synuclein: roles of brain barrier systems and implications for Parkinson's disease. Fluids Barr CNS. (2014) 11:1–9. doi: 10.1186/2045-8118-11-17
120. Peelaerts W, Bousset L, Van Der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. (2015). 522:340–4. doi: 10.1038/nature14547
121. Matsumoto J, Stewart T, Sheng L, Li N, Bullock K, Song N, et al. Transmission of α-synuclein-containing erythrocyte-derived extracellular vesicles across the blood-brain barrier via adsorptive mediated transcytosis: another mechanism for initiation and progression of Parkinson's disease? Acta Neuropathol Commun. (2017) 5:71. doi: 10.1186/s40478-017-0470-4
122. Budnik V, Ruiz-Cañada C, Wendler F. Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci. (2016) 17:160–72. doi: 10.1038/nrn.2015.29
123. Stuendl A, Kunadt M, Kruse N, Bartels C, Moebius W, Danzer KM, et al. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson's disease and dementia with Lewy bodies. Brain. (2016) 139:481–94. doi: 10.1093/brain/awv346
124. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. (2007) 7:803–15. doi: 10.1038/nri2171
125. Beckers CML, van Hinsbergh VWM, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemos. (2010) 103:40–55. doi: 10.1160/TH09-06-0403
126. Spindler V, Schlegel N, Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc Res. (2010) 87:243–53. doi: 10.1093/cvr/cvq086
127. Pediaditakis I, Kodella KR, Manatakis DV, Hinojosa CD, Manolakos ES, Rubin LL, et al. Modeling alpha-synuclein pathology in a human brain-chip to assess blood-brain barrier disruption in Parkinson's disease. bioRxiv. (2020). doi: 10.1101/2020.07.22.207340
128. Guan J, Pavlovic D, Dalkie N, Waldvogel HJ, O'Carroll SJ, Green CR, et al. Vascular degeneration in parkinsons disease. Brain Pathology. (2013) 23:154–64. doi: 10.1111/j.1750-3639.2012.00628.x
129. Bradaric BD, Patel A, Schneider JA, Carvey PM, Hendey B. Evidence for angiogenesis in Parkinson's disease, incidental Lewy body disease, and progressive supranuclear palsy. J Neural Transm. (2012) 119:59–71. doi: 10.1007/s00702-011-0684-8
130. Farkas E, De Jong GI, De Vos RAI, Jansen Steur ENH, Luiten PGM. Pathological features of cerebral cortical capillaries are doubled in Alzheimer's disease and Parkinson's disease. Acta Neuropathol. (2000) 100:395–402. doi: 10.1007/s004010000195
131. Li L, Welser JV, Milner R. Absence of the αvβ3 integrin dictates the time-course of angiogenesis in the hypoxic central nervous system: accelerated endothelial proliferation correlates with compensatory increases in α5β1 integrin expression. J Cereb Blood Flow Metab. (2010) 30:1031–43. doi: 10.1038/jcbfm.2009.276
132. Carvey PM, Zhao CH, Hendey B, Lum H, Trachtenberg J, Desai BS, et al. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur J Neurosc. (2005). 22:1158–68. doi: 10.1111/j.1460-9568.2005.04281.x
133. Elabi O, Gaceb A, Carlsson R, Padel T, Soylu-Kucharz R, Cortijo I, et al. Human α-synuclein overexpression in a mouse model of Parkinson's disease leads to vascular pathology, blood brain barrier leakage and pericyte activation. Sci Rep. (2021) 11:1120. doi: 10.1038/s41598-020-80889-8
134. Yasuhara T, Shingo T, Kobayashi K, Takeuchi A, Yano A, Muraoka K, et al. Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson's disease. Eur J Neurosc. (2004) 19:1494–504. doi: 10.1111/j.1460-9568.2004.03254.x
135. Lange C, Storkebaum E, De Almodóvar CR, Dewerchin M, Carmeliet P. Vascular endothelial growth factor: a neurovascular target in neurological diseases. Nat Rev Neurol. (2016) 12:439–54. doi: 10.1038/nrneurol.2016.88
136. Shim JW, Madsen JR. VEGF signaling in neurological disorders. Int J Mol Sci. (2018) 19:1–22. doi: 10.3390/ijms19010275
137. Bellucci A, Bugiani O, Ghetti B, Spillantini MG. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener Dis. (2011) 8:221–9. doi: 10.1159/000322228
138. Longhena F, Faustini G, Varanita T, Zaltieri M, Porrini V, Tessari I, et al. Synapsin III is a key component of α-synuclein fibrils in Lewy bodies of PD brains. Brain Pathol. (2018) 28:875–88. doi: 10.1111/bpa.12587
139. Goldmann T, Wieghofer P, Jordão MJC, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. (2016) 17:797–805. doi: 10.1038/ni.3423
140. Yang T, Guo R, Zhang F. Brain perivascular macrophages: Recent advances and implications in health and diseases. CNS Neurosci Therap. (2019) 25:1318–28. doi: 10.1111/cns.13263
141. Koizumi T, Kerkhofs D, Mizuno T, Steinbusch HWM, Foulquier S. Vessel-associated immune cells in cerebrovascular diseases: from perivascular macrophages to vessel-associated microglia. Front Neurosci. (2019) 13:1291. doi: 10.3389/fnins.2019.01291
142. Orr CF, Rowe DB, Halliday GM. An inflammatory review of Parkinson's disease. Prog Neurobiol. (2002) 68:325–40.
143. Gupta P, Lai SM, Sheng J, Tetlak P, Balachander A, Claser C, et al. Tissue-resident CD169+ macrophages form a crucial front line against plasmodium infection. Cell Rep. (2016) 16:1749–61. doi: 10.1016/j.celrep.2016.07.010
144. Bechmann I, Priller J, Kovac A, Böntert M, Wehner T, Klett FF, et al. Immune surveillance of mouse brain perivascular spaces by blood-borne macrophages. Eur J Neurosci. (2001) 14:1651–8. doi: 10.1046/j.0953-816x.2001.01793.x
145. Galea I, Palin K, Newman TA, Van Rooijen N, Perry VH, Boche D. Mannose receptor expression specifically reveals perivascular macrophages in normal, injured, and diseased mouse brain. Glia. (2005) 49:375–84. doi: 10.1002/glia.20124
146. Fabriek BO, Van Haastert ES, Galea I, Polfliet MMJ, Döpp ED, Van Den Heuvel MM, et al. CD163-positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia. (2005) 51:297–305. doi: 10.1002/glia.20208
147. Kim WK, Alvarez X, Fisher J, Bronfin B, Westmoreland S, McLaurin J, et al. CD163 identifies perivascular macrophages in normal and viral encephalitic brains and potential precursors to perivascular macrophages in blood. Am J Pathol. (2006) 168:822–34. doi: 10.2353/ajpath.2006.050215
148. Zhang Z, Zhang ZY, Schittenhelm J, Wu Y, Meyermann R, Schluesener HJ. Parenchymal accumulation of CD163+ macrophages/microglia in multiple sclerosis brains. J Neuroimmunol. (2011) 237:73–9. doi: 10.1016/j.jneuroim.2011.06.006
149. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity. (2018) 48:380–95.e6. doi: 10.1016/j.immuni.2018.01.011
150. Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. (2019) 22:1021–35. doi: 10.1038/s41593-019-0393-4
151. Jordão MJC, Sankowski R, Brendecke SM, Sagar, Locatelli G, Tai YH, et al. Neuroimmunology: single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. (2019) 363:6425. doi: 10.1126/science.aat7554
152. Utz SG, See P, Mildenberger W, Thion MS, Silvin A, Lutz M, et al. Early fate defines microglia and non-parenchymal brain macrophage development. Cell. (2020) 181:557–73.e18. doi: 10.1016/j.cell.2020.03.021
153. Chang C, Lang H, Geng N, Wang J, Li N, Wang X. Exosomes of BV-2 cells induced by alpha-synuclein: important mediator of neurodegeneration in PD. Neurosci Lett. (2013) 548:190–5. doi: 10.1016/j.neulet.2013.06.009
154. Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. (2020) 11:1386. doi: 10.1038/s41467-020-15119-w
155. Yin J, Valin KL, Dixon ML, Leavenworth JW. The role of microglia and macrophages in CNS homeostasis, autoimmunity, and cancer. J Immunol Res. (2017) 2017:5150678. doi: 10.1155/2017/5150678
156. Kierdorf K, Prinz M. Factors regulating microglia activation. Front Cell Neurosci. (2013) 7:1–8. doi: 10.3389/fncel.2013.00044
157. Hou L, Bao X, Zang C, Yang H, Sun F, Che Y, et al. Integrin CD11b mediates α-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. (2018) 14:600–8. doi: 10.1016/j.redox.2017.11.010
158. Li N, Stewart T, Sheng L, Shi M, Cilento EM, Wu Y, et al. Immunoregulation of microglial polarization: an unrecognized physiological function of alpha-synuclein. J Neuroinflammation. (2020) 17:272. doi: 10.1186/s12974-020-01940-z
159. Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci. (2016) 19:771–83. doi: 10.1038/nn.4288
160. Loria F, Vargas JY, Bousset L, Syan S, Salles A, Melki R, et al. α-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. (2017). 134:789–808. doi: 10.1007/s00401-017-1746-2
161. Stevenson TJ, Murray HC, Turner C, Faull RLM, Dieriks BV, Curtis MA. α-synuclein inclusions are abundant in non-neuronal cells in the anterior olfactory nucleus of the Parkinson's disease olfactory bulb. Sci Rep. (2020). 10:6682. doi: 10.1038/s41598-020-63412-x
162. Dohgu S, Takata F, Matsumoto J, Kimura I, Yamauchi A, Kataoka Y. Monomeric α-synuclein induces blood–brain barrier dysfunction through activated brain pericytes releasing inflammatory mediators in vitro. Microvasc Res. (2019) 124:61–6. doi: 10.1016/j.mvr.2019.03.005
163. Li W, Chen S, Luo Y, Xia Y, Ma Q, Yao Q, et al. Interaction between ICAM1 in endothelial cells and LFA1 in T cells during the pathogenesis of experimental Parkinson's disease. Exp Ther Med. (2020) 20:1021–9. doi: 10.3892/etm.2020.8758
164. Lindestam Arlehamn CS, Dhanwani R, Pham J, Kuan R, Frazier A, Rezende Dutra J, et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson's disease. Nat Commun. (2020). 11:1875. doi: 10.1038/s41467-020-15626-w
165. Lopes Pinheiro MA, Kooij G, Mizee MR, Kamermans A, Enzmann G, Lyck R, et al. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim Biophys Acta. (2016) 1862:461–71. doi: 10.1016/j.bbadis.2015.10.018
166. Filippini A, Mutti V, Faustini G, Longhena F, Ramazzina I, Rizzi F, et al. Extracellular clusterin limits the uptake of alpha-synuclein fibrils by murine and human astrocytes. Glia. (2021) 69:681–96. doi: 10.1002/glia.23920
167. Tsunemi T, Ishiguro Y, Yoroisaka A, Valdez C, Miyamoto K, Ishikawa K, et al. Astrocytes protect human dopaminergic neurons from alpha-synuclein accumulation and propagation. J Neurosci. (2020) 40:8618–28. doi: 10.1523/JNEUROSCI.0954-20.2020
168. Peng W, Achariyar TM, Li B, Liao Y, Mestre H, Hitomi E, et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer's disease HHS Public Access Graphical abstract. Neurobiol Dis. (2016) 93:215–25. doi: 10.1016/j.nbd.2016.05.015
169. Mesquita SD, Louveau A, Vaccari A, Smirnov I, Cornelison C, Kingsmore KM, et al. Alzheimer's disease. Nature. (2019). 560:185–91. doi: 10.1038/s41586-018-0368-8
170. Wang L, Zhang Y, Zhao Y, Marshall C, Wu T, Xiao M. Deep cervical lymph node ligation aggravates AD-like pathology of APP/PS1 mice. Brain Pathol. (2019) 29:176–92. doi: 10.1111/bpa.12656
171. Zeng H, Liu N, Yang YY, Xing HY, Liu XX, Li F, et al. Lentivirus-mediated downregulation of α-synuclein reduces neuroinflammation and promotes functional recovery in rats with spinal cord injury. J Neuroinflammation. (2019) 16:1–19. doi: 10.1186/s12974-019-1658-2
172. Kim TH, Mehta SL, Morris-Blanco KC, Chokkalla AK, Chelluboina B, Lopez M, et al. The microRNA miR-7a-5p ameliorates ischemic brain damage by repressing-synuclein. Sci Signal. (2018) 11:1–12. doi: 10.1126/scisignal.aat4285
173. Lou N, Takano T, Pei Y, Xavier AL, Goldman SA, Nedergaard M. Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc Natl Acad Sci USA. (2016) 113:1074–9. doi: 10.1073/pnas.1520398113
174. Liu C, Wu C, Yang Q, Gao J, Li L, Yang D, et al. Macrophages mediate the repair of brain vascular rupture through direct physical adhesion and mechanical traction. Immunity. (2016) 44:1162–76. doi: 10.1016/j.immuni.2016.03.008
175. Haruwaka K, Ikegami A, Tachibana Y, Ohno N, Konishi H, Hashimoto A, et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun. (2019) 10:5816. doi: 10.1038/s41467-019-13812-z
176. Lok J, Gupta P, Guo S, Kim WJ, Whalen MJ, van Leyen K, et al. Cell–cell signaling in the neurovascular unit. Neurochem Res. (2007) 32:2032–45. doi: 10.1007/s11064-007-9342-9
177. Koning N, Swaab DF, Hoek RM, Huitinga I. Distribution of the immune inhibitory molecules CD200 and CD200R in the normal central nervous system and multiple sclerosis lesions suggests neuron-glia and glia-glia interactions. J Neuropathol Exp Neurol. (2009) 68:159–67. doi: 10.1097/NEN.0b013e3181964113
178. Wang L, Liu Y, Yan S, Du T, Fu X, Gong X, et al. Disease progression-dependent expression of CD200R1 and CX3CR1 in mouse models of parkinson's disease. Aging Dis. (2020) 11:254–68. doi: 10.14336/AD.2019.0615
179. Zhang S, Wang XJ, Tian LP, Pan J, Lu GQ, Zhang YJ, et al. CD200-CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson's disease. J Neuroinflammation. (2011) 8:154. doi: 10.1186/1742-2094-8-154
180. Luo XG, Zhang JJ, Zhang CD, Liu R, Zheng L, Wang XJ, et al. Altered regulation of CD200 receptor in monocyte-derived macrophages from individuals with Parkinson's disease. Neurochem Res. (2010) 35:540–7. doi: 10.1007/s11064-009-0094-6
181. Katoh M, Katoh M. CD157 and CD200 at the crossroads of endothelial remodeling and immune regulation. Stem Cell Investig. (2019) 6:1–7. doi: 10.21037/sci.2019.04.01
182. Welser JV, Li L, Milner R. Microglial activation state exerts a biphasic influence on brain endothelial cell proliferation by regulating the balance of TNF and TGF-β1. J Neuroinflammation. (2010) 7:89. doi: 10.1186/1742-2094-7-89
183. Surguchev AA, Emamzadeh FN, Surguchov A. Cell responses to extracellular α-synuclein. Molecules. (2019) 24:1–7. doi: 10.3390/molecules24020305
Glossary
AD, Alzheimer's disease; AP-1, activator protein 1; BAMs, border associated macrophages; BBB, blood–brain barrier; BM, basement membrane; CAA, cerebral amyloid angiopathy; cAMP, cyclic adenosine monophosphate; CD200R, CD200 receptor; CNS, central nervous system; COL4A2, collagen IV alpha 2 chain; CP, caudate putamen; CPM, choroid plexus associated macrophages; CSF, cerebrospinal fluid; CX3CL1, C-X3-C motif chemokine ligand 1; CX3CR1, C-X3-C motif chemokine receptor 1; DAB, 3,3-diaminobenzidine; DLB, dementia with LB; ECs, endothelial cells; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; EVs, extracellular vesicles; FGF2, fibroblast growth factor 2; FGFR, FGF2 receptor; GCI, glial cytoplasmic inclusions; Iba1, ionized calcium-binding adaptor molecule 1; ICAM 1, intercellular adhesion molecule 1; iLBD, incidental LB disease; IL, interleukin; iPSC, induced pluripotent stem cell; LB, Lewy bodies; LC, locus coeruleus; LFA, lymphocyte function-associated antigen-1; LPS, lipopolysaccharide; LRP-1, lipoprotein receptor-related protein-1; MAM, meningeal macrophages; MDMs, monocyte-derived macrophages; MSA, multiple system atrophy; NAC, non-amyloid component; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor-κB; NK, natural killer; NLR, nod-like receptor; NLRP3, NLR pyrin domain containing 3; NOX, NADPH oxidase; NVU, neurovascular unit; PBMCs, peripheral blood mononuclear cells; PBS, phosphate buffer saline; PD, Parkinson's disease; pffs, preformed fibrils; PLD, phospholipase D; PPARγ, peroxisome proliferator-activated receptor γ; PRRs, pattern recognition receptors; PVM, perivascular macrophages; RalGDS, Ral specific guanine exchange factor; RhoGTPases, Ras homologous guanosine triphosphate phosphatase; RT, room temperature; SN, substantia nigra; TEC, trans-entorhinal cortex; TGF-β, transforming growth factor-beta; TJ, tight junction; TLR-2, toll-like receptor 2; TLR-4, toll-like receptor 4; TNFα, tumor necrosis factor alpha; VCAM 1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factors; VEGFR2, VEGF receptor; vWF, Willebrand factor; WPBs, Weibel-Palade bodies; ZO-1, zonula occludens 1.
Keywords: Parkinson's disease, α-synuclein, endothelial cells, blood brain barrier, border-associated macrophages, perivascular cells
Citation: Bogale TA, Faustini G, Longhena F, Mitola S, Pizzi M and Bellucci A (2021) Alpha-Synuclein in the Regulation of Brain Endothelial and Perivascular Cells: Gaps and Future Perspectives. Front. Immunol. 12:611761. doi: 10.3389/fimmu.2021.611761
Received: 29 September 2020; Accepted: 27 January 2021;
Published: 19 February 2021.
Edited by:
Giuseppe Locatelli, University of Bern, SwitzerlandReviewed by:
Marina Romero-Ramos, Aarhus University, DenmarkAshley Harms, University of Alabama at Birmingham, United States
Elisa Greggio, University of Padua, Italy
Copyright © 2021 Bogale, Faustini, Longhena, Mitola, Pizzi and Bellucci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arianna Bellucci, YXJpYW5uYS5iZWxsdWNjaUB1bmlicy5pdA==