 Shirley Pollack1,2*
Shirley Pollack1,2* Israel Eisenstein1
Israel Eisenstein1 Adi Mory3Tamar Paperna3Ayala Ofir3Hagit Baris-Feldman3Karin Weiss3
Adi Mory3Tamar Paperna3Ayala Ofir3Hagit Baris-Feldman3Karin Weiss3 Nóra Veszeli4
Nóra Veszeli4 Dorottya Csuka4Revital Shemer5
Dorottya Csuka4Revital Shemer5 Fabian Glaser6
Fabian Glaser6 Zoltán Prohászka4Daniella Magen1,5
Zoltán Prohászka4Daniella Magen1,5- 1Pediatric Nephrology Institute, Ruth Children’s Hospital, Haifa, Israel
- 2Rappaport Faculty of Medicine, Technion - Israel Institute of Technology, Haifa, Israel
- 3Genetic Institute, Haifa, Israel
- 4Research Laboratory, Department of Internal Medicine and Haematology, and MTA-SE Research Group of Immunology and Hematology, Hungarian Academy of Sciences and Semmelweis University, Budapest, Hungary
- 5Laboratory of Molecular Medicine, Rappaport Faculty of Medicine, Technion - Israel Institute of Technology, Haifa, Israel
- 6Bioinformatics Knowledge Unit, The Lorry I. Lokey Interdisciplinary Center for Life Sciences and Engineering, Technion-Israel Institute of Technology, Haifa, Israel
Background and Objectives: Atypical hemolytic uremic syndrome (aHUS) is mostly attributed to dysregulation of the alternative complement pathway (ACP) secondary to disease-causing variants in complement components or regulatory proteins. Hereditary aHUS due to C3 disruption is rare, usually caused by heterozygous activating mutations in the C3 gene, and transmitted as autosomal dominant traits. We studied the molecular basis of early-onset aHUS, associated with an unusual finding of a novel homozygous activating deletion in C3.
Design, Setting, Participants, & Measurements: A male neonate with eculizumab-responsive fulminant aHUS and C3 hypocomplementemia, and six of his healthy close relatives were investigated. Genetic analysis on genomic DNA was performed by exome sequencing of the patient, followed by targeted Sanger sequencing for variant detection in his close relatives. Complement components analysis using specific immunoassays was performed on frozen plasma samples from the patient and mother.
Results: Exome sequencing revealed a novel homozygous variant in exon 26 of C3 (c.3322_3333del, p.Ile1108_Lys1111del), within the highly conserved thioester-containing domain (TED), fully segregating with the familial disease phenotype, as compatible with autosomal recessive inheritance. Complement profiling of the patient showed decreased C3 and FB levels, with elevated levels of the terminal membrane attack complex, while his healthy heterozygous mother showed intermediate levels of C3 consumption.
Conclusions: Our findings represent the first description of aHUS secondary to a novel homozygous deletion in C3 with ensuing unbalanced C3 over-activation, highlighting a critical role for the disrupted C3-TED domain in the disease mechanism.
Introduction
Atypical hemolytic uremic syndrome (aHUS) is a heterogeneous group of acquired and hereditary thrombotic microangiopathy (TMA), manifested by microangiopathic hemolytic anemia, thrombocytopenia and acute kidney injury, which are usually not preceded by the diarrheal prodromal phase of Shiga Toxin-related HUS. Most forms of aHUS are associated with dysregulation of the alternative complement pathway (ACP), resulting in complement-mediated endothelial cell injury, with ensuing end-organ tissue damage (1, 2).
To date, about two thirds of patients with aHUS carry identifiable mutations or likely-pathogenic risk variants in genes encoding complement pathway proteins (2–5). While loss-of-function mutations are commonly implicated in genes encoding regulatory complement components, including complement Factor H (CFH), complement Factor I (CFI) and membrane cofactor protein (MCP, CD46), gain-of-function mutations are usually associated with complement Factor B (CFB) and C3 (6–8). In conjunction with genetic predisposition, acquired autoantibodies against Factor H (FH) have been implicated in the pathogenesis of aHUS in approximately 10% of cases, and are mostly attributed to genomic rearrangements or deletions in CFH/CFHR1/CFHR3/CFHR4 genes within the regulators of complement activation gene cluster (9, 10). In addition to proteins directly related to the complement cascade, increasing evidence highlights the role of coagulation factors in the pathogenesis of aHUS, including thrombomodulin (THBD), which regulates Factor I-mediated C3b inactivation, and plasminogen (PLG), which enhances plasmin-mediated fibrinolysis and thrombi degradation (11, 12). Finally, mutations in DGKE, encoding diacylglycerol kinase-ϵ (DGKE) have been linked with infantile-onset aHUS, accompanied by nephrotic syndrome and chronic kidney disease (CKD), through an as yet unresolved mechanism (8, 13).
Given the central role of ACP over-activation in the pathogenesis of aHUS, a favorable response to targeted blockade of the complement cascade is highly conceivable. Eculizumab is a monoclonal antibody directed against the human C5 complement component, causing inhibition of C5a release, thereby preventing downstream generation of the membrane attack complex (MAC), C5b-9. As such, it is indicated in acute episodes of aHUS, and as prophylaxis in susceptible patients (3, 14).
In this report we describe a male neonate with a life-threatening presentation of aHUS, associated with a novel homozygous in-frame deletion in the C3 gene. Complement profile analysis suggests unbalanced activation of the ACP, which is further supported by a favorable response to eculizumab therapy. To the best of our knowledge, this is the first report of a homozygous deletion in C3 associated with severe neonatal presentation of aHUS, secondary to C3 over-activation.
Clinical Presentation and Workup
The patient is the 5th child of healthy, 1st-degree cousins of Arab-Muslim origin, born at term after normal pregnancy and delivery. Detailed family history was negative for renal, hematological, autoimmune or recurrent infectious disorders. He was admitted to a local hospital at the age of 3 days for evaluation of extensive jaundice, anemia, thrombocytopenia, and impaired kidney function. Following fulminant renal failure and acute respiratory insufficiency, he was urgently transferred to our care for further evaluation and management. On admission, the infant displayed clinical signs of encephalopathy, accompanied by fluid overload and pulmonary congestion secondary to anuric renal failure, which necessitated mechanical ventilation and renal replacement therapy.
Initial laboratory investigation was compatible with TMA, as evidenced by normocytic anemia, severe thrombocytopenia, significant schistocytosis on peripheral blood smear, a negative Coombs test, and extremely elevated lactate dehydrogenase (LDH) levels. A thorough microbiological workup was negative for an identifiable infectious trigger. Plasma ADAMTS-13 activity was normal, and anti-ADAMTS-13 antibodies were undetected, thereby excluding thrombotic thrombocytopenic purpura (TTP). Coagulation tests excluded diffuse intravascular coagulation (DIC). Metabolic screen showed normal blood levels of homocysteine, methionine and cobalamin C, with no evidence of organic aciduria, thereby excluding cobalamin C deficiency. Serum C3 levels were repeatedly extremely low, with normal C4 levels. A connective tissue disorder panel (collagenogram) was negative. Imaging studies, including renal echo-Doppler, brain ultrasonography and echocardiography were unremarkable.
With a working diagnosis of aHUS, the patient was managed with eculizumab therapy, in conjunction with mechanical ventilation, daily hemodialysis, red blood cell transfusions and prophylactic penicillin therapy. Within several days of eculizumb initiation, the infant showed dramatic clinical and laboratory improvement, followed by rapid neurological, hematological, respiratory and renal recovery. Currently, at the age of 2 years, the child is in sustained remission from aHUS under prophylactic eculizumab therapy, with a history of two reversible aHUS relapses following non-adherence to prescribed eculizumab prophylaxis. Each relapse appeared after 10-14 days delay in scheduled eculizumab therapy, manifesting with thrombotic microangiopathy and acute renal failure. Sequelae of his fulminant presenting episode of aHUS include arterial hypertension, non-nephrotic range proteinuria with normal glomerular filtration rate (GFR), and gross motor developmental delay. His serum C3 levels are constantly low. He does not exhibit increased susceptibility to pyogenic bacterial infections, as reported in patients with homozygous loss-of-function variants in C3 which cause C3 deficiency (15, 16), although infections may have been prevented by prophylactic antibiotic treatment.
Materials and Methods
Genetic Analysis
Genomic DNA was extracted from the patient’s peripheral lymphocytes for clinical exome sequencing, using the TruSight One Sequencing panel (Illumina, Inc., San Diego, CA, USA), according to manufacturer’s instructions. Direct Sanger sequencing was performed on the coding regions and intron-exon boundaries of CFH (exons 1-9 & 11-23), CFI (exons 1-13), MCP (CD46, exons 1-14), C3 (exons 1-41), and CFB (exons 1-18); THBD (exon 1).
Multiplex Ligation-Dependent Probe Amplification (MLPA) analysis was performed using a commercially available assay (P236-A3 ARMD probemix, MRC-Holland).
All available family members were Sanger sequenced for the C3 variant and for additional variations and risk polymorphisms identified in the patient.
Pathogenicity of identified sequence variants was classified according to joint consensus recommendations by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (17).
Complement Profile Analysis
Levels of complement components, regulators and FH autoantibodies were determined on frozen plasma samples of the patient and his mother, according to previously published methods (18). Fresh plasma samples for complement analysis on additional family members were unavaliable.
C3b-FH Complex Modelling
Structural analysis of C3b-FH complex was based on the PDB 3OXU complex structure (19). Interfacial amino acids of C3b were defined as those within a distance < 5 Å of any atom of Factor H. The energy of the C3b-FH complex was computed with pyDock bindEy module for protein structure prediction (20), both for the wild-type and the inferred mutant C3 protein structure. Molecular graphics and analyses were performed with the UCSF ChimeraX program, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, SF, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases (21).
Results
Genetic Analysis
Sequence analysis of the patient revealed a homozygous in-frame deletion of 12bp in exon 26 of the C3 gene (c.3322_3333del, p.Ile1108_Lys1111del) resulting in deletion of highly conserved four amino acids (ILEK) from the inferred protein sequence. The four deleted residues are predicted to be located in the α-chain of the encoded C3 protein, within its highly conserved thioester-containing domain (TED) (NCBI conserved domains). The p.Ile1108_Lys1111del-C3 variant was absent from public variation data bases [dbSNP; 1000 Genomes Projects; gnomAD; The Greater Middle East Variom Project (GME)], and was predicted harmful by mutation prediction software (Mutation Taster).
Several additional common polymorphisms and variants of unknown significance were identified in complement encoding genes of the patient (Figure 1). In detail, he was found to be heterozygous for a substitution in the CFH gene (c.3050C>T, p.Thr1017Ile). This variant has been previously classified as likely benign, based on comparable allele frequencies between healthy individuals and aHUS patients, combined with ambiguous prediction of pathogenicity, according to in-silico prediction software analysis (22). Further identified heterozygous polymorphisms in the patient included the CFH Y402H risk factor for dense deposit disease, as well as the CD46 rs2796267, rs2796268 and rs1962149 polymorphisms, referred as part of the MCPggaac risk haplotype for aHUS (Figure 1).
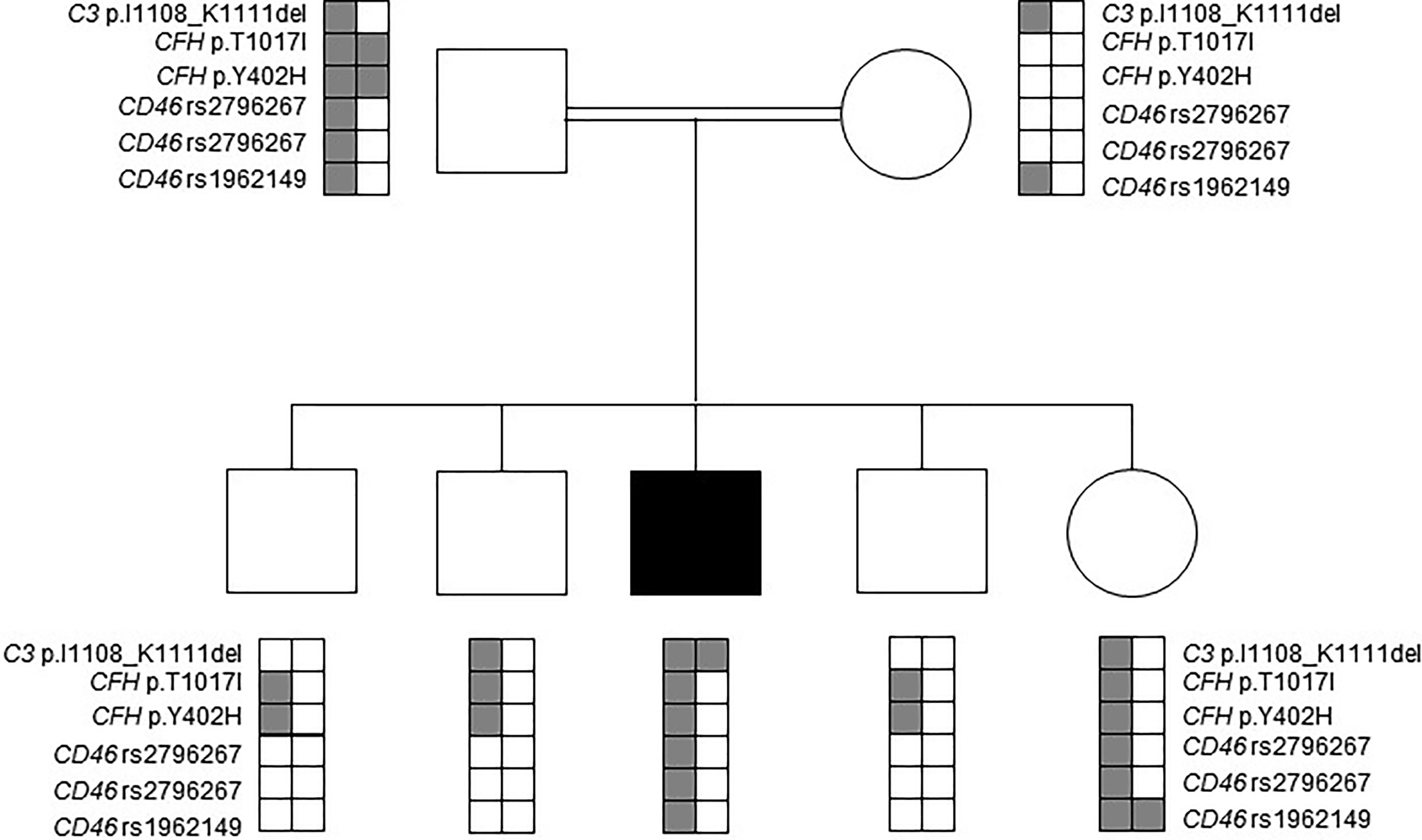
Figure 1 Mutation analysis in the patient’s nuclear family. Affected individual is marked by a black filled square. Rare alleles of the rs2796267, rs2796268 and rs1962149 polymorphisms and their haplotype referred as MCPggaac were reported to be a risk factor for developing aHUS.
MLPA analysis excluded deletions or duplications within the Regulators of Complement Activation (RCA) gene cluster (CFH, CFHR1, CFHR2, CFHR3 and CFHR5) on chromosome 1q31.3, as underlying the disease.
Targeted sequencing of all healthy close relatives excluded homozygosity for the p.Ile1108_Lys1111del-C3 variant. Heterozygosity for the MCPggaac risk haplotype was identified in the healthy father and sister. The rare allele of the CFH Y402H risk polymorphism was identified in the healthy father, sister and brothers, but not in the healthy mother (Figure 1).
Complement Profile Analysis
As shown in Table 1, complement analysis of the patient indicated critically low C3 level, deficient activity of all three complement pathways, decreased complement factor B (FB) level, and increased level of the terminal pathway activation marker. C1q, C4, FI and FH levels were within or slightly below the reference range. Anti-FH antibody titer was negative, below the cut-off.
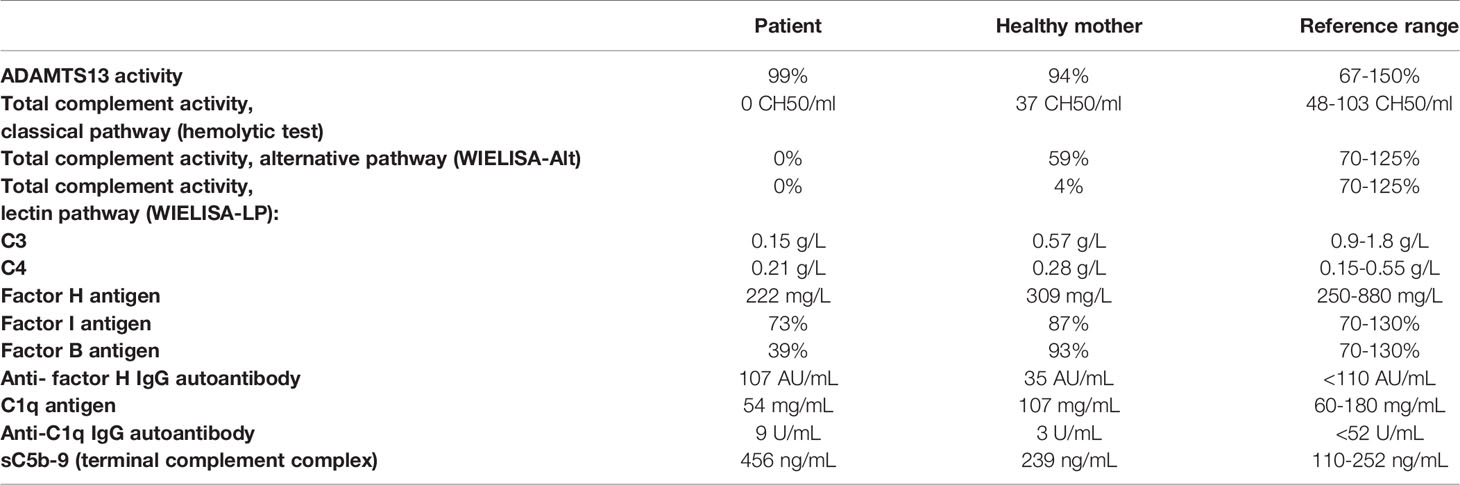
Table 1 Complement profile of the patient and his mother.
Taken together, complement profile analysis of the patient suggests spontaneous activation of the p.lle1108_Lys111de-C3 protein by FB, resulting in ongoing consumption of C3 and FB, and increased generation of the MAC.
C3b-FH Complex Modeling
Structural analysis of C3b-FH complex demonstrates strategic location of the four deleted amino acids (ILEK) within the TED domain, at the C3b-FH interface (Figure 2C). The energy difference between wild-type and mutant C3b-FH complex structure (ΔG-wild-type – ΔG mutant) is estimated to be ΔΔG ~ -5Kcal, suggesting a catastrophic effect exerted by the deletion on interface stability. Hence, the ILEK deletion in mutant C3 is highly likely to disrupt C3b-FH interaction, thereby disabling the inhibitory effect of FH on downstream activation of C3b.
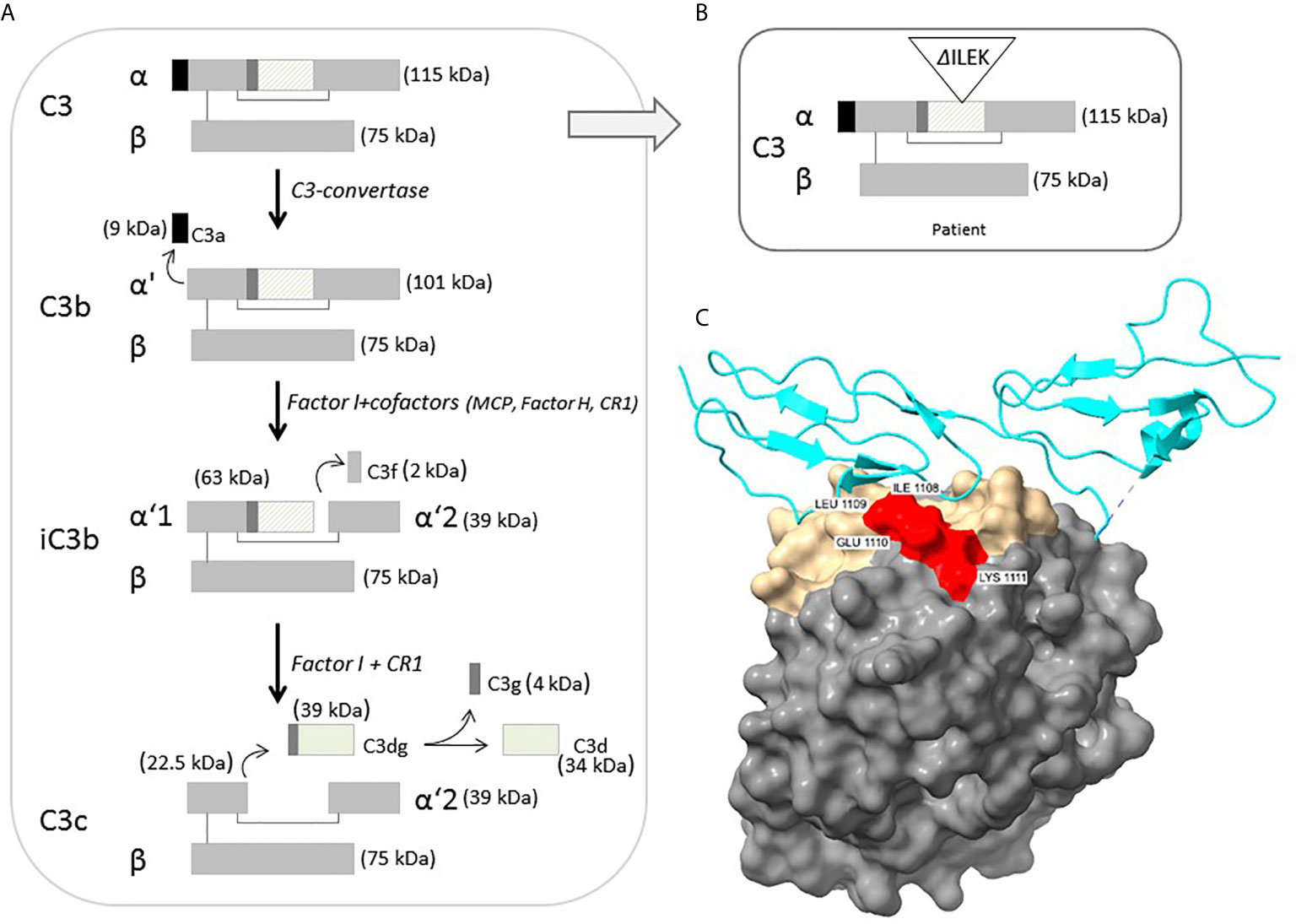
Figure 2 (A) Schematic representation of intact C3 structure, processing and regulation. The diagonal-line filled rectangle represents the TED of C3. (B) Schematic representation of the position of four-residue deleted sequence (ΔILEK) within the TED domain of the patient’s C3 alpha chain. (C) Modelling of the C3b (TED domain)-Factor H complex (from PDB 3OXU) (19), using the UCSF ChimeraX tool. C3b is shown in solvent-excluded molecular surface representation, and Factor H (P08603) in cyan ribbons. All C3b interfacial amino acids are colored gold, while the p.1008-1111 (ILEK) mutant region is colored red. As clearly demonstrated, amino acids ILEK are an integral part of the C3b-Factor H interface.
Discussion
We herein described the clinical and molecular investigation in a male infant from a consanguineous family, with neonatal presentation of fulminant aHUS and multi-organ failure, accompanied by extremely low serum levels of C3, a favorable systemic response to eculizumab therapy, and intolerance to eculizumab cessation. Mutation analysis revealed a small homozygous in-frame deletion of 12 bp in exon 26 of the C3 gene, resulting in four amino acids deletion (ILEK) from the inferred TED of C3, in association with apparent C3 deficiency. However, complement profile analysis indicated unbalanced activation of the ACP, as evidenced by a reduced level of FB denoting its consumption, combined with excessive generation of the terminal MAC. Moreover, structural modelling of C3b-FH complex suggests a disruptive effect of ILEK-deletion on mutant C3b binding to FH, thereby supporting the hypothesis of unbalanced degradation of mutant C3 as underlying the mechanism of aHUS.
C3 is the most abundant complement component protein, playing a crucial role in activation of all three complement pathways. In its mature, unprocessed form, the C3 protein is composed of one α and one β chain linked by a disulfide bond, and consists of 13 domains, harboring intrinsic functional and binding capacity to various effector ligands. Intact C3 is an inert molecule, exhibiting its biological activity only after proteolytic cleavage to functional degradation product (Figure 2A). Under physiological condition, C3b is constantly produced at a very low rate, by spontaneous hydrolysis to C3(H2O). The C3b factor is able to attach to pathogens and host cell surfaces, where it binds FB, which in turn is cleaved by complement factor D (FD). The resulting C3bBb or C3 convertase further cleaves and activates C3, thereby leading to an amplification loop of the alternative complement cascade, culminating in MAC-related tissue damage (23, 24).
aHUS secondary to C3 disruption is relatively rare, especially during the pediatric age group, with an overall reported prevalence ranging between 4.5%-11.4 of aHUS cases (3, 4, 25). C3-associated aHUS is mostly attributed to heterozygous gain-of-function variants in the C3 gene, leading to unbalanced activation of the complement cascade (8, 26–28). Furthermore, the presentation of aHUS in carriers of C3 gain-of-function mutations may be influenced by the presence of the MCPggaac risk haplotype, as previously described (29). In contrast, homozygous loss-of-function variants in C3 are usually implicated in C3 deficiency, manifested by increased susceptibility to recurrent bacterial infections and autoimmunity, unrelated to aHUS (15, 16). To the best of our knowledge, the constellation of aHUS secondary to a small homozygous deletion in C3 associated with unbalanced activation of the ACP has not been previously reported (OMIM databases).
According to the crystallographic structure of C3, the TED, located within residues 963-1268, contains a hidden thioester bond, which is exposed upon C3 cleavage, thereby facilitating covalent binding of C3b to target surface ligands (30, 31). Moreover, conformational displacement of the TED domain has been previously shown to disrupt the interaction of C3b with the ACP regulators, i.e., FH and MCP (32, 33). Hence, the inferred deletion of four highly conserved residues from the TED of the p.Ile1108_Lys1111del-C3 protein (Figures 2B, C), may impose a conformational change of functional significance on the TED, rendering mutant C3b resistant to inactivation, through impaired binding to FH. This hypothesis is further supported by previous reports on the functional significance of various aHUS-related C3 missense variants within the TED, (i.e., p.L1109V, p.P1114L, p.D1115N, p.G1116R, I1157T), residing in close proximity to the location of p.Ile1108_Lys1111del-C3, with accumulating evidence for their disrupted cell surface interaction with FH, or resistance to inactivation by MCP, due to C3b altered conformation (4, 5, 26, 34, 35).
Of note, hereditary aHUS, in general, is well-known for its partial penetrance, resulting in phenotypic variability among carriers of a shared disease-causing variant. Incomplete penetrance is commonly attributed to the harmful effect of background risk variants within ACP regulatory genes, including CFH and MCP risk haplotypes (5, 36, 37). The fact that all heterozygous carriers of the p.lle1108_Lys1111del-C3 variant within the studied family are disease-free, suggests that heterozygosity for this likely-pathogenic activating variant, either in the absence or presence of additional risk variants, is insufficient for the development of full-blown disease, albeit intermediated C3 consumption and ACP activation, as evidenced by the complement profile of the patient’s mother.
In conclusion, we have identified a likely-pathogenic homozygous deletion in C3, associated with life-threatening aHUS secondary to over-activation of the ACP. Complement profile analysis suggests a damaging conformational effect of this deletion on the TED of the C3 protein, rendering it susceptible to unbalanced activation due to impaired interaction with regulatory FH. Our findings enable prenatal diagnosis and early treatment initiation in newly identified subjects at risk, while reinforcing the significance of an intact C3-TED for normal function and regulation of the ACP. Yet, the exact mechanism whereby the identified 4-residue deletion in C3 may affect molecular interactions of the C3 protein with additional regulators of the ACP, with ensuing downstream over-activation of terminal complement cascade, warrants further investigation.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, 2384952.
Ethics Statement
The studies involving human participants were reviewed and approved by the Rambam Health Care Campus Institutional ethics committee. The patients/participants provided their written informed consent to participate in this study. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
SP: Writing – original draft – Preparation, creation and/or presentation of the published work, specifically writing the initial draft (including substantive translation). DM and ZP: Writing – review & editing – Preparation, creation and/or presentation of the published work by those from the original research group, specifically critical review, commentary or revision – including pre- or post-publication stages. IE, AM, TP, AO, HB-F, KW, NV, RS, FG, and DC: research activity - performed the clinical and laboratory analysis, reviewed and edited the paper. All authors contributed to the article and approved the submitted version.
Funding
The study was supported by the Premium Postdoctoral Fellowship Program of the Hungarian Academy of Sciences (PPD2018-016/2018), and by the Kaylie Kidney Research Center of Excellence at Rambam Medical Center.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Noris M, Remuzzi G, Noris M. Atypical Hemolytic-Uremic Syndrome. N Engl J Med (2009) 361(17):1676–87. doi: 10.1056/NEJMra0902814
2. Schaefer F, Ardissino G, Ariceta G, Fakhouri F, Scully M, Isbel N, et al. Clinical and Genetic Predictors of Atypical Hemolytic Uremic Syndrome Phenotype and Outcome. Kidney Int (2018) 94(2):408–18. doi: 10.1016/j.kint.2018.02.029
3. Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, et al. An International Consensus Approach to the Management of Atypical Hemolytic Uremic Syndrome in Children. Pediatr Nephrol (2016) 31(1):15–39. doi: 10.1007/s00467-015-3076-8
4. Schramm EC, Roumenina LT, Rybkine T, Chauvet S, Vieira-Martins P, Hue C, et al. Mapping Interactions Between Complement C3 and Regulators Using Mutations in Atypical Hemolytic Uremic Syndrome. Blood (2015) 125(15):2359–69. doi: 10.1182/blood-2014-10-609073
5. Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, et al. Combined Complement Gene Mutations in Atypical Hemolytic Uremic Syndrome Influence Clinical Phenotype. J Am Soc Nephrol (2013) 24(3):475–86. doi: 10.1681/ASN.2012090884
6. Rodriguez E, Rallapalli PM, Osborne AJ, Perkins SJ, Rodriguez E. New Functional and Structural Insights From Updated Mutational Databases for Complement Factor H, Factor I, Membrane Cofactor Protein and C3. Biosci Rep (2014) 34(5):e00146. doi: 10.1042/BSR20140117
7. Feitz WJC, van de Kar NCAJ, Orth-Höller D, van den Heuvel LPJW, Licht C. The Genetics of Atypical Hemolytic Uremic Syndrome. Med Genet (2018) 30(4):400–9. doi: 10.1007/s11825-018-0216-0
8. Yoshida Y, Kato H, Ikeda Y, Nangaku M, Yoshida Y. Pathogenesis of Atypical Hemolytic Uremic Syndrome. J Atheroscler Thromb (2019) 26(2):99–110. doi: 10.5551/jat.RV17026
9. Moore I, Strain L, Pappworth I, Kavanagh D, Barlow PN, Herbert AP, et al. Association of Factor H Autoantibodies With Deletions of CFHR1, Cfhr3, CFHR4, and With Mutations in CFH, Cfi, CD46, and C3 in Patients With Atypical Hemolytic Uremic Syndrome. Blood (2010) 115(2):379–87. doi: 10.1182/blood-2009-05-221549
10. Valoti E, Alberti M, Iatropoulos P, Piras R, Mele C, Breno M, et al. Rare Functional Variants in Complement Genes and Anti-FH Autoantibodies-Associated Ahus. Front Immunol (2019) 10:853. doi: 10.3389/fimmu.2019.00853
11. Dzik S. Complement and Coagulation: Cross Talk Through Time. Transfus Med Rev (2019) 33(4):199–206. doi: 10.1016/j.tmrv.2019.08.004
12. Bu F, Maga T, Meyer NC, Wang K, Thomas CP, Nester CM, et al. Comprehensive Genetic Analysis of Complement and Coagulation Genes in Atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol (2014) 25(1):55–64. doi: 10.1681/ASN.2013050453
13. Lemaire M, Frémeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, et al. Recessive Mutations in DGKE Cause Atypical Hemolytic-Uremic Syndrome. Nat Genet (2013) 45(5):531–6. doi: 10.1038/ng.2590
14. Fakhouri F, Loirat C. Anticomplement Treatment in Atypical and Typical Hemolytic Uremic Syndrome. Semin Hematol (2018) 55(3):150–8. doi: 10.1053/j.seminhematol.2018.04.009
15. Reis E S, Falcão DA, Isaac L. Clinical Aspects and Molecular Basis of Primary Deficiencies of Complement Component C3 and Its Regulatory Proteins Factor I and Factor H. Scand J Immunol (2006) 63(3):155–68. doi: 10.1111/j.1365-3083.2006.01729.x
16. Okura Y, Kobayashi I, Yamada M, Sasaki S, Yamada Y, Kamioka I, et al. Clinical Characteristics and Genotype-Phenotype Correlations in C3 Deficiency. J Allergy Clin Immunol (2016) 137(2):640–44.e1. doi: 10.1016/j.jaci.2015.08.017
17. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–24. doi: 10.1038/gim.2015.30
18. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
19. Morgan HP, Schmidt CQ, Guariento M, Blaum BS, Gillespie D, Herbert AP, et al. Structural Basis for Engagement by Complement Factor H of C3b on a Self Surface. Nat Struct Mol Biol (2011) 18(4):463–70. doi: 10.1038/nsmb.2018
20. Rosell M, Rodríguez-Lumbreras LA, Fernández-Recio M. Modeling of Protein Complexes and Molecular Assemblies With Pydock. Methods Mol Biol (2020) 2165:175–98. doi: 10.1007/978-1-0716-0708-4_10
21. Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, et al. Ucsf ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci (2021) 30(1):70–82. doi: 10.1002/pro.3943
22. Osborne AJ, Breno M, Borsa NG, Bu F, Frémeaux-Bacchi V, Gale DP, et al. Statistical Validation of Rare Complement Variants Provides Insights Into the Molecular Basis of Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. J Immunol (2018) 200(7):2464–78. doi: 10.4049/jimmunol.1701695
23. de Bruijn MH, Fey GH. Human Complement Component C3: cDNA Coding Sequence and Derived Primary Structure. Proc Natl Acad Sci USA (1985) 82(3):708–12. doi: 10.1073/pnas.82.3.708
24. Janssen BJ, Gros P, Janssen BJC. Structural Insights Into the Central Complement Component C3. Mol Immunol (2007) 44(1-3):3–10. doi: 10.1016/j.molimm.2006.06.017
25. Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaimé F, Dragon-Durey MA, Ngo S, et al. Genetics and Outcome of Atypical Hemolytic Uremic Syndrome: A Nationwide French Series Comparing Children and Adults. Clin J Am Soc Nephrol (2013) 8(4):554–62. doi: 10.2215/CJN.04760512
26. Frémeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J, Brown AL, et al. Mutations in Complement C3 Predispose to Development of Atypical Hemolytic Uremic Syndrome. Blood (2008) 112(13):4948–52. doi: 10.1182/blood-2008-01-133702
27. Lhotta K, Janecke AR, Scheiring J, Petzlberger B, Giner T, Fally V, et al. A Large Family With a Gain-of-Function Mutation of Complement C3 Predisposing to Atypical Hemolytic Uremic Syndrome, Microhematuria, Hypertension and Chronic Renal Failure. Clin J Am Soc Nephrol (2009) 4(8):1356–62. doi: 10.2215/CJN.06281208
28. Sartz L, Olin AI, Kristoffersson AC, Ståhl AL, Johansson ME, Westman K, et al. A Novel C3 Mutation Causing Increased Formation of the C3 Convertase in Familial Atypical Hemolytic Uremic Syndrome. J Immunol (2012) 188(4):2030–7. doi: 10.4049/jimmunol.1100319
29. Lumbreras J, Subias M, Espinosa N, Ferrer JM, Arjona E, Rodríguez de Córdoba S, et al. The Relevance of the MCP Risk Polymorphism to the Outcome of Ahus Associated With C3 Mutations. A Case Report. Front Immunol (2020) 11:1348. doi: 10.3389/fimmu.2020.01348
30. Janssen BJ, Huizinga EG, Raaijmakers HC, Roos A, Daha MR, Nilsson-Ekdahl K, et al. Structures of Complement Component C3 Provide Insights Into the Function and Evolution of Immunity. Nature (2005) 437(7058):505–11. doi: 10.1038/nature04005
31. Janssen BJ, Christodoulidou A, McCarthy A, Lambris JD, Gros P. Structure of C3b Reveals Conformational Changes That Underlie Complement Activity. Nature (2006) 444(7116):213–6. doi: 10.1038/nature05172
32. Wu J, Wu Y-Q, Ricklin D, Janssen BJC, Lambris JD, Grosl P. Structure of Complement Fragment C3b-Factor H and Implications for Host Protection by Complement Regulators. Nat Immunol (2009) 10(7):728–33 doi: 10.1038/ni.1755
33. Wu J, Wu YQ, Ricklin D, Janssen BJ, Lambris JD, Gros P.Wu J, et al. Structure of Complement Fragment C3b–Factor H and Implications for Host Protection by Complement Regulators. Nat Immunol (2009) 10(7):728–33. doi: 10.1038/ni.1755
34. Persson BD, Schmitz NB, Santiago C, Zocher G, Larvie M, Scheu U, et al. Structure of the Extracellular Portion of CD46 Provides Insights Into Its Interactions With Complement Proteins and Pathogens. PloS Pathog (2010) 6(9):e1001122. doi: 10.1371/journal.ppat.1001122
35. Martínez-Barricarte R, Heurich M, López-Perrote A, Tortajada A, Pinto S, López-Trascasa M, et al. The Molecular and Structural Bases for the Association of Complement C3 Mutations With Atypical Hemolytic Uremic Syndrome. Mol Immunol (2015) 66(2):263–73. doi: 10.1016/j.molimm.2015.03.248
36. Sansbury FH, Cordell HJ, Bingham C, Bromilow G, Nicholls A, Powell R, et al. Factors Determining Penetrance in Familial Atypical Haemolytic Uraemic Syndrome. J Med Genet (2014) 51(11):756–64. doi: 10.1136/jmedgenet-2014-102498
Keywords: atypical hemolytic uremic syndrome (aHUS), complement factor C3, eculizumab, alternative complement pathway, homozygous deletion
Citation: Pollack S, Eisenstein I, Mory A, Paperna T, Ofir A, Baris-Feldman H, Weiss K, Veszeli N, Csuka D, Shemer R, Glaser F, Prohászka Z and Magen D (2021) A Novel Homozygous In-Frame Deletion in Complement Factor 3 Underlies Early-Onset Autosomal Recessive Atypical Hemolytic Uremic Syndrome - Case Report. Front. Immunol. 12:608604. doi: 10.3389/fimmu.2021.608604
Received: 21 September 2020; Accepted: 24 May 2021;
Published: 24 June 2021.
Edited by:
Cordula M. Stover, University of Leicester, United KingdomReviewed by:
Kevin James Marchbank, Newcastle University, United KingdomMichal Malina, Newcastle Hospitals, United Kingdom
Elisabetta Valoti, Istituto di Ricerche Farmacologiche Mario Negri (IRCCS), Italy
Copyright © 2021 Pollack, Eisenstein, Mory, Paperna, Ofir, Baris-Feldman, Weiss, Veszeli, Csuka, Shemer, Glaser, Prohászka and Magen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shirley Pollack, c19wb2xsYWNrQHJtYy5nb3YuaWw=