
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Immunol., 15 April 2021
Sec. Primary Immunodeficiencies
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.606099
This article is part of the Research TopicInterstitial Lung Disease in Primary ImmunodeficienciesView all 12 articles
 Olivia A. C. Lamers1†
Olivia A. C. Lamers1† Bas M. Smits1,2†
Bas M. Smits1,2† Helen Louisa Leavis2
Helen Louisa Leavis2 Godelieve J. de Bree3
Godelieve J. de Bree3 Charlotte Cunningham-Rundles4
Charlotte Cunningham-Rundles4 Virgil A. S. H. Dalm5
Virgil A. S. H. Dalm5 Hsi-en Ho4
Hsi-en Ho4 John R. Hurst6
John R. Hurst6 Hanna IJspeert5Sabine M. P. J. Prevaes7
Hanna IJspeert5Sabine M. P. J. Prevaes7 Alex Robinson6Astrid C. van Stigt5Suzanne Terheggen-Lagro8
Alex Robinson6Astrid C. van Stigt5Suzanne Terheggen-Lagro8 Annick A. J. M. van de Ven9
Annick A. J. M. van de Ven9 Klaus Warnatz10,11Janneke H. H. M. van de Wijgert12
Klaus Warnatz10,11Janneke H. H. M. van de Wijgert12 Joris van Montfrans1*
Joris van Montfrans1*Introduction: Besides recurrent infections, a proportion of patients with Common Variable Immunodeficiency Disorders (CVID) may suffer from immune dysregulation such as granulomatous-lymphocytic interstitial lung disease (GLILD). The optimal treatment of this complication is currently unknown. Experienced-based expert opinions have been produced, but a systematic review of published treatment studies is lacking.
Goals: To summarize and synthesize the published literature on the efficacy of treatments for GLILD in CVID.
Methods: We performed a systematic review using the PRISMA guidelines. Papers describing treatment and outcomes in CVID patients with radiographic and/or histologic evidence of GLILD were included. Treatment regimens and outcomes of treatment were summarized.
Results: 6124 papers were identified and 42, reporting information about 233 patients in total, were included for review. These papers described case series or small, uncontrolled studies of monotherapy with glucocorticoids or other immunosuppressants, rituximab monotherapy or rituximab plus azathioprine, abatacept, or hematopoietic stem cell transplantation (HSCT). Treatment response rates varied widely. Cross-study comparisons were complicated because different treatment regimens, follow-up periods, and outcome measures were used. There was a trend towards more frequent GLILD relapses in patients treated with corticosteroid monotherapy when compared to rituximab-containing treatment regimens based on qualitative endpoints. HSCT is a promising alternative to pharmacological treatment of GLILD, because it has the potential to not only contain symptoms, but also to resolve the underlying pathology. However, mortality, especially among immunocompromised patients, is high.
Conclusions: We could not draw definitive conclusions regarding optimal pharmacological treatment for GLILD in CVID from the current literature since quantitative, well-controlled evidence was lacking. While HSCT might be considered a treatment option for GLILD in CVID, the risks related to the procedure are high. Our findings highlight the need for further research with uniform, objective and quantifiable endpoints. This should include international registries with standardized data collection including regular pulmonary function tests (with carbon monoxide-diffusion), uniform high-resolution chest CT radiographic scoring, and uniform treatment regimens, to facilitate comparison of treatment outcomes and ultimately randomized clinical trials.
Common variable immunodeficiency disorders (CVID) are the most common symptomatic primary immunodeficiencies, with an estimated incidence between 1:10.000 and 1:50.000 (1). Patients typically suffer from recurrent respiratory tract infections, such as bronchitis, sinusitis, otitis media and pneumonia. Moreover, they are often affected by immune dysregulation, a term which encompasses auto-immune manifestations, auto-inflammatory disease and lymphoproliferation, and by malignancy (2). Infection risk in CVID can be minimized by means of antimicrobial prophylaxis and immunoglobulin replacement therapy (IgRT). In contrast, immune dysregulation is much more difficult to prevent and treat, and remains a major cause of morbidity and mortality (3–6).
Granulomatous lymphocytic interstitial lung disease (GLILD) is one of the complications of CVID and is considered the pulmonary manifestation of multi-system immune dysregulation. GLILD occurs in approximately 10-20% of patients with CVID and was reported to be responsible for a reduction in life expectancy of more than 50% after diagnosis in adult patients, from a median of 28.9 to 13.7 years (6, 7). GLILD may be asymptomatic, or may present with non-specific symptoms such as cough and dyspnea on exertion (4). Small or large nodules, consolidations and ground glass abnormalities in the lower regions of the lung on high-resolution CT-scan are highly suggestive of GLILD (8). The diagnosis can be confirmed by biopsy (via video-assisted thoracoscopic surgery, transbronchial or percutaneous intervention) and FDG-PET-CT may be used for the identification of active inflammatory lesions elsewhere (4, 9). The combination of routine chest CT-scans and pulmonary function tests, including specifically diffusing capacity of carbon monoxide, should be used to identify GLILD in CVID and monitor disease progression (9).
The etiology of GLILD is still poorly understood. Maglione and colleagues pointed out that patients with X-linked agammaglobulinemia (XLA) have severe antibody deficiency that is even more pronounced than CVID but only rarely develop GLILD (10). Patients with XLA lack mature B-cells, whereas patients with CVID have peripheral B-cells, although often with impaired function, suggesting that B-lymphocytes may play a causative role in GLILD development. Indeed, lymphocytic (but not the granulomatous) progression has been associated with an increased production of B-cell activating factor (BAFF), which in turn leads to activation of the anti-apoptotic factor Bcl-2, thereby promoting B-cell survival as well as an increase of IgM producing CD21 low B-cells (10). Unger et al. linked the expansion of CD21low B-cells with disproportionally high numbers of Th1 cells and increased interferon-γ production, probably reflecting the aberrant combined T-B interaction in the pathogenesis of interstitial lung disease in CVID (11). It has also been suggested that viral infections may trigger GLILD, as Wheat et al. identified a correlation between human herpesvirus 8 (HHV8) infection and the disease (12). However, since the publication of the original article describing this correlation, no further evidence has been provided for this hypothesis. Finally, an association between interstitial lung disease and an increased relative abundance of Streptococcus in the oropharyngeal microbiome in CVID was recently identified (13).
The treatment of GLILD mostly consists of immunosuppressive medication, in addition to IgRT and other supportive measures such as physiotherapy. According to the British Lung Foundation/United Kingdom Primary Immunodeficiency Network Consensus Statement, glucocorticoids are the first line of therapy for GLILD (9). Most clinicians agree that azathioprine, mycophenolate mofetil (MMF) and rituximab are second-line choices when glucocorticoids are not effective or when attempting to spare their use (9). Although alternative medication may also be prescribed, there is no consensus about the use of other biologic therapies or disease-modifying anti-rheumatic drugs (DMARDs) (9).
Current GLILD treatment guidelines are based on expert opinion rather than on robust scientific evidence. An objective review of the existing evidence is needed to minimize potential biases associated with expert opinion, and to identify knowledge gaps. Therefore, our aim was to systematically review the existing literature on treatment of GLILD in CVID patients. To the best of our knowledge, this is the first systematic review on that topic.
We searched PubMed and EMBASE for publications on treatment of GLILD in CVID patients (last search on March 27th 2020, see Appendix for Search String). Articles describing patients with CVID and GLILD who were treated with pharmacological therapy and/or a hematopoietic stem cell transplantation (HSCT) were included. Improvement of disease activity parameters (symptoms, pulmonary function tests and radiological findings) and mortality served as outcomes.
We focused our search on patients with CVID and GLILD. Studies describing patients with monogenetic diseases causing a CVID-like phenotype (such as CTLA-4 haploinsufficiency and LRBA deficiency) were included.
The consensus GLILD definition of the British Lung Foundation/United Kingdom Primary Immunodeficiency Network was used: “GLILD is a distinct clinic-radio-pathological interstitial lung disease occurring in patients with CVID, associated with a lymphocytic infiltrate and/or granuloma in the lung, and in whom other conditions have been considered and where possible excluded” (9). Only articles that reported radiological findings on a CT-scan or histological analysis of biopsies compliant with this definition of GLILD were included.
All non-English articles were excluded for purposes of practicality. Conference abstracts, while read and taken into consideration, were excluded from the review as they were not peer-reviewed.
Two independent investigators (O.L. and B.S.) selected articles on the basis of title and abstract. Blinding of the investigators was achieved by inserting all articles in a common online database (Rayyan), which has a blinding feature and allows each researcher to select articles independently of the other. Ultimately, the selection of articles of each researcher was compared to the other. If there were any selection discrepancies, the articles were discussed until a unanimous decision about in- or exclusion could be made. Data were extracted from the eligible full-text articles using a standardized data extraction sheet. The extracted data were summarized descriptively and reported in tables. We could not conduct meta-analyses because the selected articles contained insufficient quantitative data.
If the use of multiple treatment regimens in one patient was reported, the effect of the treatment regimens was evaluated separately. When escalation or switching of treatment was deemed necessary by the authors, the previous regimen was deemed insufficient. To evaluate the effect of treatment regimens, both qualitative and quantitative assessments of GLILD activity were analyzed. Descriptive improvement of pulmonary function tests, radiological findings and symptoms (e.g. “shortness of breath”, “coughing”) were used for the qualitative evaluation of disease activity. Significant improvement was defined as a relapse-free improvement of at least one of these parameters and no deterioration of the other parameters. Pre- and post-treatment pulmonary function test results were used for the quantitative evaluation of disease activity, and significant improvement of pulmonary function was here defined as a 10% increase in at least one pulmonary function test parameter.
Overall risk of bias of each study was assessed by means of a self-designed tool based on the PRISMA guidelines (14). This tool took into account the quality of the studies (based on the number of patients and controls, and on descriptions of outcomes, medication dosages and follow-up procedures) and possible confounders (smoking, age, comorbidity, and results of genetic testing). Each study was assigned a rating for each of these categories, ‘good’ (+) if the highest quality standard was attained with clear quantitative outcomes, ‘intermediate’ (+/-) if some information was reported but quantitative measures were lacking, and ‘insufficient’ (-) if the information was not reported at all. The overall risk of bias was determined as follows: ‘high risk of bias’ if the study had four or more insufficient or eight or more intermediate judgments; ‘intermediate risk of bias’ if the study was marked insufficient on two to four items or intermediate on four to eight items; and ‘low risk of bias’ if the study had only one insufficient judgment or a maximum of three intermediate judgments.
The level of evidence for each study and the degree of recommendation in clinical practice were determined following the criteria formulated by the Centre for Evidence Based Medicine (15).
The search identified 6124 articles on PubMed and EMBASE and seven additional papers via snowballing (Figure 1). After removal of duplicates, 5304 articles were screened, 65 full-text papers were read, and 42 articles were deemed eligible. 233 patients were described in total. The findings are summarized below, sorted by treatment modality. Qualitative and quantitative lung function findings are shown in Figure 2.
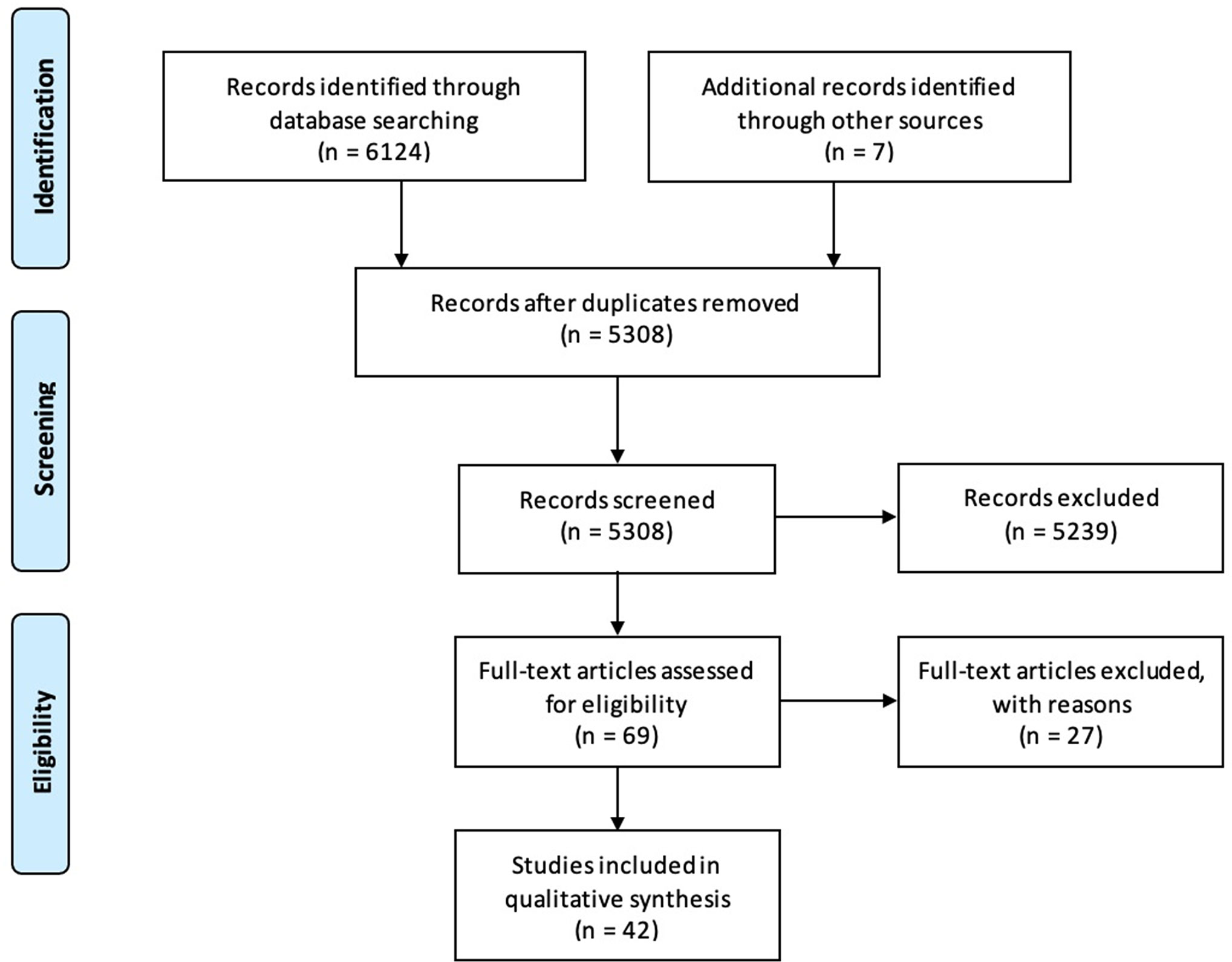
Figure 1 PRISMA flow chart for article inclusion.
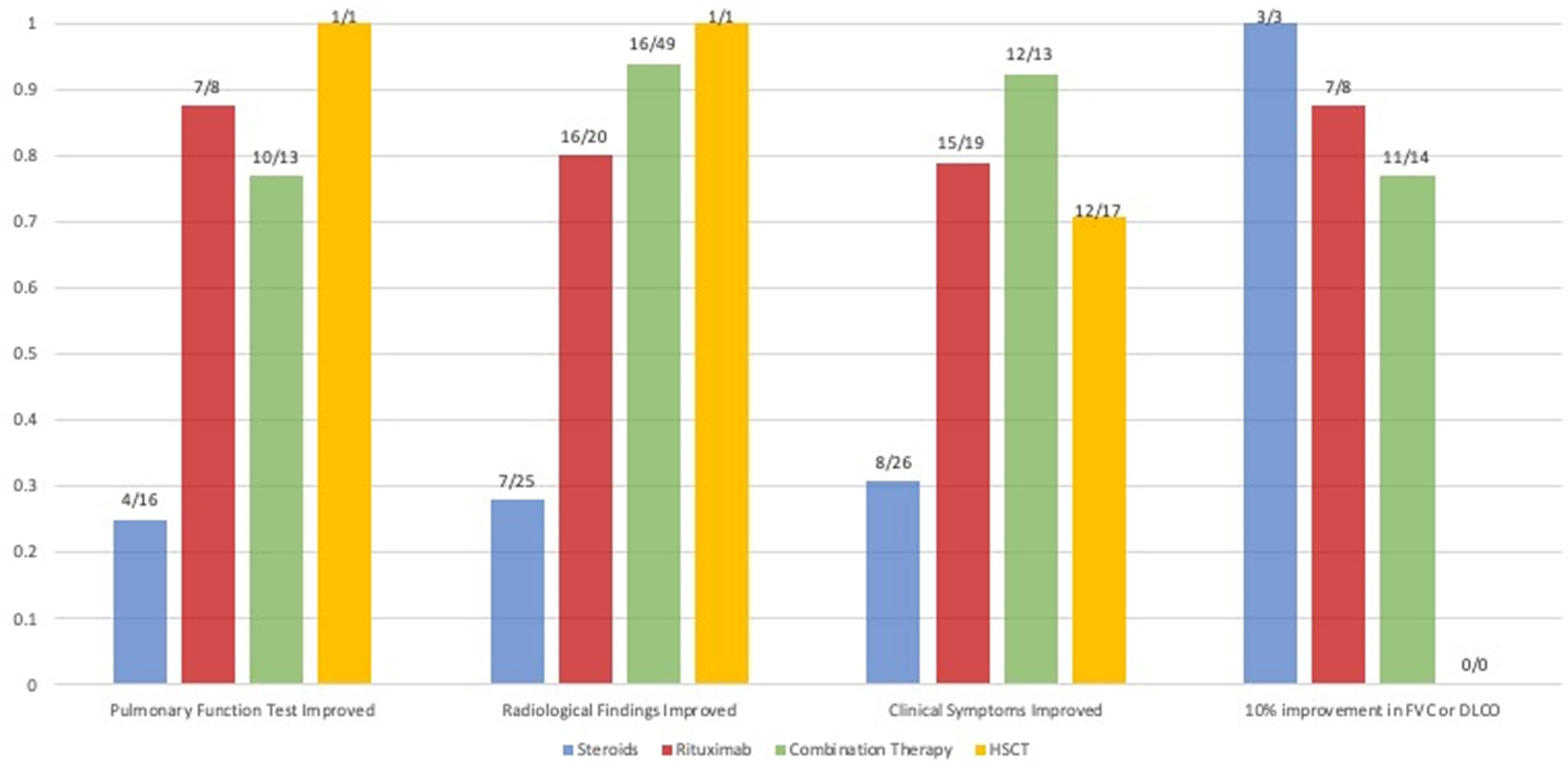
Figure 2 Comparison of the available qualitative and quantitative outcomes of studies that reported on patients (N) treated with steroids, rituximab monotherapy and rituximab combination therapy. The proportion of patients that had a qualitatively reported improvement of pulmonary function tests, radiological findings and the proportion that had a quantitative improvement of their forced vital capacity (FVC) or diffusion capacity of the lung for carbon monoxide (DLCO) of 10% after therapy is shown. Due to a lack of quantitative data, statistics could not be performed.
There were three papers describing GLILD in patients with B lymphocyte related primary antibody deficiency other than CVID (such as IgA or IgG subclass deficiency, or selective antibody deficiency for polysaccharide antigens). These articles are listed in the Supplementary Material (Table S1).
Glucocorticoids have been identified as the first line treatment for GLILD by the British Lung Foundation/United Kingdom Primary Immunodeficiency Network (2017) (9).
Six articles specifically reported on the use of glucocorticoids for the treatment of GLILD in patients with CVID, as shown in Table 1. The first report dates back to 1982 and describes the case of a woman who was treated with high-dose prednisone for six weeks. Symptoms initially subsided but relapsed when the medication was tapered (18). Ten additional studies included glucocorticoid treatment as one of several therapies (Tables 2 and 3). Five of these reported no effect of glucocorticoids (26, 27, 31, 36–38), one reported relapse after initial remission (29) and four reported treatment success (16, 17, 20, 21). The article by Kanathur et al. is particularly interesting as it describes a case in which glucocorticoids initially failed to have any effect at all but were associated with the resolution of symptoms when paired with splenectomy (19).
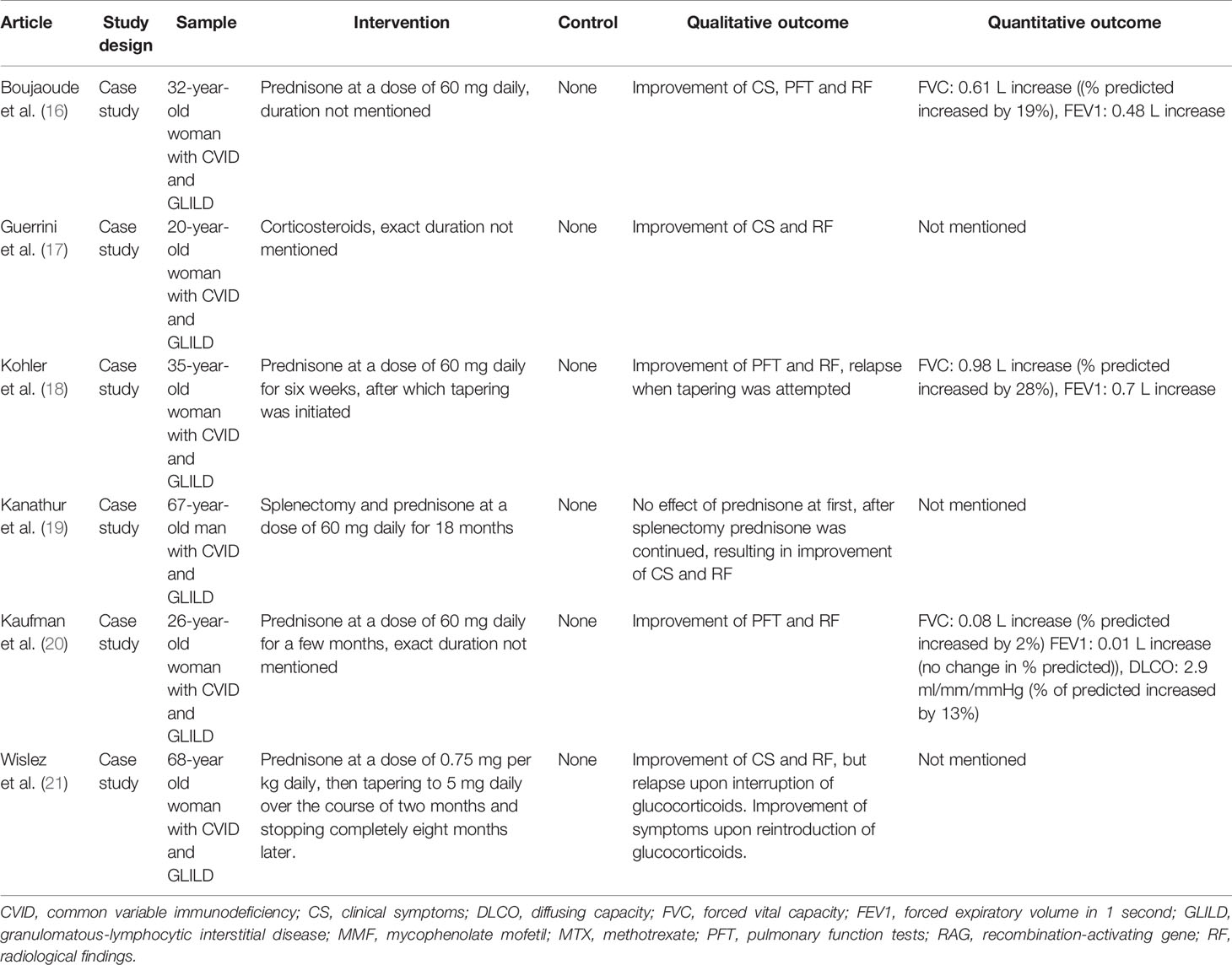
Table 1 Studies reporting treatment of GLILD in PID with corticosteroids.
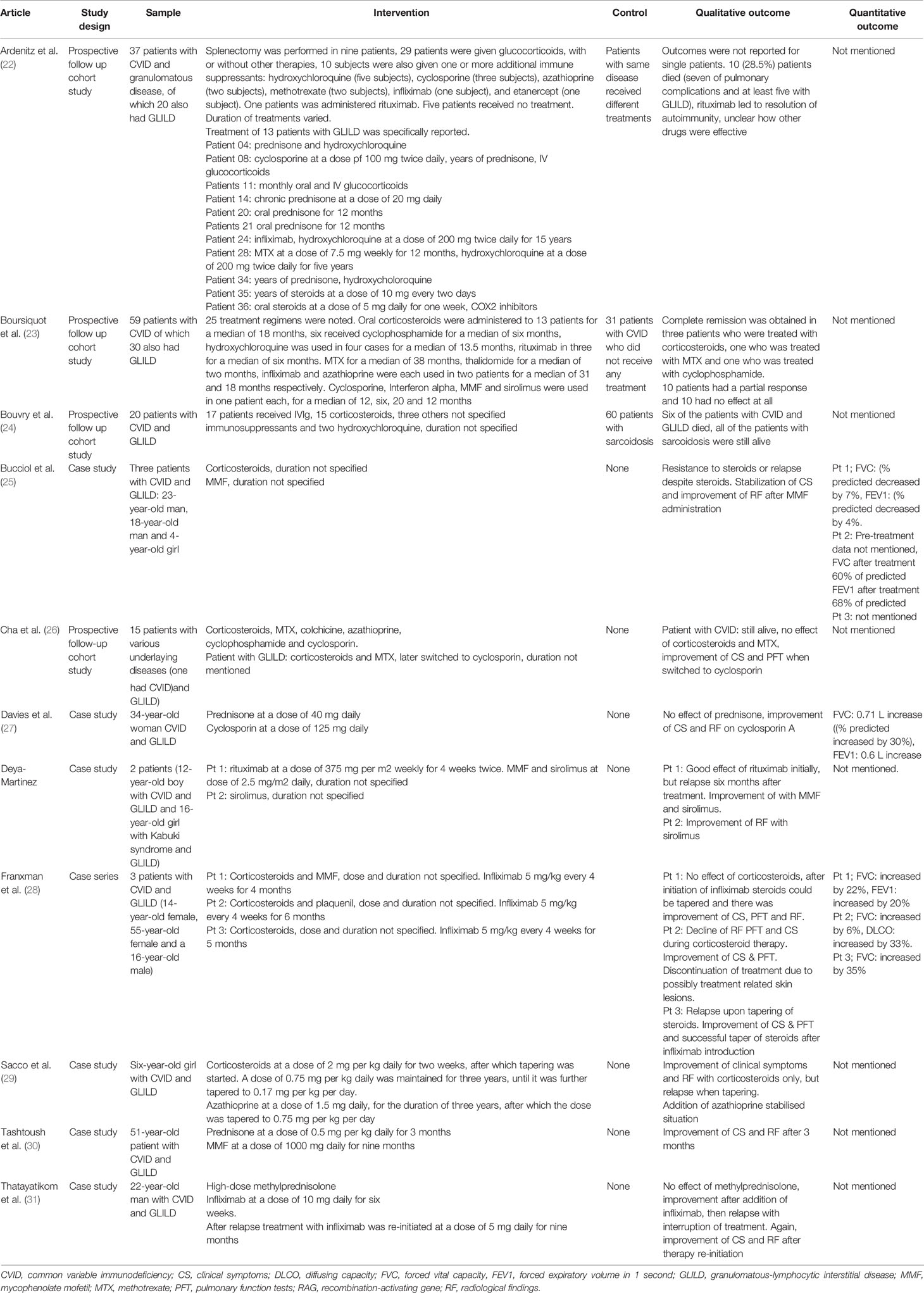
Table 2 Studies reporting treatment of GLILD in antibody deficiencies with various immunosuppressants.
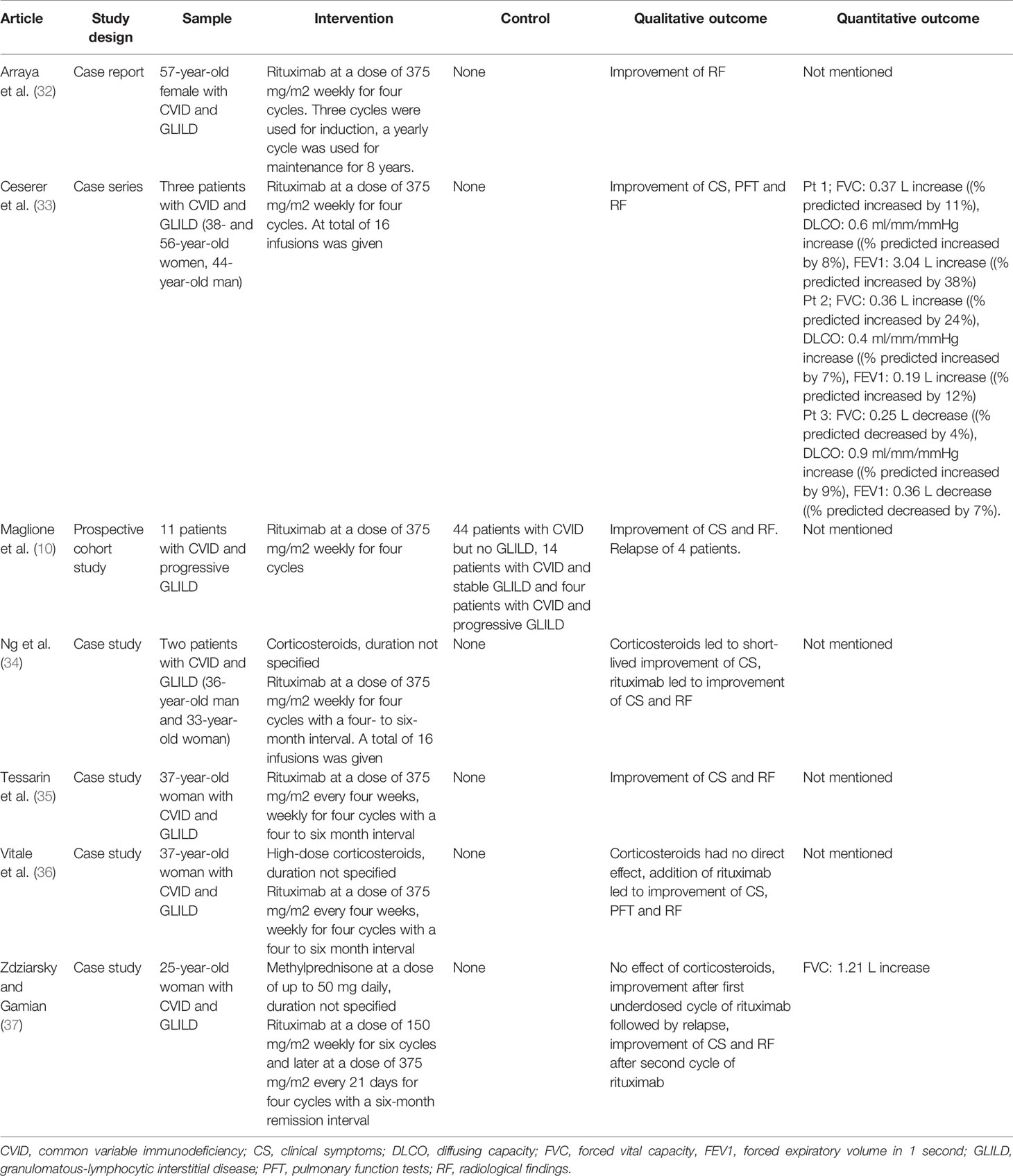
Table 3 Studies reporting treatment of GLILD in PID with rituximab.
Besides glucocorticoids, other immunosuppressants for the treatment of GLILD have been evaluated (Table 2). Examples encountered in the literature included methotrexate (MTX), cyclophosphamide, mycophenolate (MMF), azathioprine, cyclosporin, hydroxychloroquine, tacrolimus and sirolimus.
Boursiquot et al. assessed the efficacy of both MTX and cyclophosphamide in the treatment of GLILD. The researchers prospectively followed 59 patients with CVID, of whom 30 had GLILD. Different treatment regimens were initiated in 25 patients with CVID and GLILD (Table 2). Complete remission was obtained in three (out of 13) patients who were treated with glucocorticoids, one (out of one) who was treated with MTX and one (out of five) who was treated with cyclophosphamide. Ten patients had a partial response and the remainder showed no effect at all (23).
Other articles reported the use of MMF for the treatment of GLILD. Bucciol et al. described three patients with GLILD. Glucocorticoids were ineffective, but a switch to MMF resulted in stabilization of symptoms and improvement of clinical and radiologic findings in all three cases (25). More evidence was provided by Tashtoush et al., who published a case report about a 51-year old woman with CVID and GLILD. This patient achieved remission after induction therapy with glucocorticoids for 3 months and MMF maintenance therapy for 9 months (30).
As emerged from the Delphi Study of the British Lung Foundation/United Kingdom Primary Immunodeficiency Network, azathioprine is another drug that is often used for the treatment of GLILD. An article dating back to 1996 by Sacco et al. reported the case of a six-year-old girl with CVID and severe GLILD. The patient was treated with glucocorticoids with good effect, but tapering of the medication resulted in disease relapse. This prompted the physicians to add azathioprine, which halted disease progression. The combination of prednisone and azathioprine was maintained for three years, after which they were tapered to 5 mg every other day and 0.75 mg per kg daily, respectively (29).
Albeit less frequently reported, several articles describe the use of cyclosporine for the treatment of GLILD. Davies et al. reported the case of a 34-year old woman with CVID and GLILD who responded well to glucocorticoid therapy, but had recurrent relapses after tapering. The patient was eventually treated with cyclosporine, with good effect (27). Similar results were observed by Cha et al.: a patient with CVID and concomitant GLILD was initially treated with glucocorticoids, but achieved disease remission only when therapy was switched to cyclosporin (26).
Deya-Martinez et al. showed that the immunosuppressant sirolimus can be useful in the treatment of GLILD. A boy with CVID and GLILD, who had been previously treated with rituximab and who had relapsed, was switched to sirolimus monotherapy and achieved remission of symptoms (39).
Two articles reported the use of DMARDs for the treatment of GLILD in relatively large patient series. Both papers described variable regimens of multiple drugs, without mentioning the outcomes.
Ardeniz described the long-term follow up of a group of 37 patients with CVID and granulomatous disease, of which 20 patients had GLILD. Patients were treated with a different combination of drugs, including glucocorticoids, cyclosporine, hydroxychloroquine, infliximab, etanercept and rituximab. Outcomes were not clearly reported. Over the follow-up period of 25 years, 10 of the 37 patients included in the study died. Of those, at least five had GLILD (22).
Bouvry compared outcomes of CVID patients with GLILD with those of patients with sarcoidosis. Patients were treated with different immunosuppressants over the course of the study. Results were not clearly reported, the main difference between the two groups was that patients with CVID and GLILD had worse outcomes than those with sarcoidosis (24).
Biologicals, also known as biological medicinal products, are drugs which are (partially) produced by living organisms by means of recombinant DNA technologies (40). For GLILD specifically, infliximab, rituximab and abatacept have been used.
Infliximab is a monoclonal antibody that binds to TNFα and blocks signaling, thus interfering with a central mechanism of inflammation (41). Thatayatikom et al. reported a 22-year-old man with CVID and life-threatening GLILD, who was first unsuccessfully treated with glucocorticoids, but achieved remission after treatment with infliximab for nine months (31). Additionally, Franxman, Howe & Baker described three patients who all showed remission of GLILD on CT scan and pulmonary function tests, after 4 months, 8 months and 5 months of treatment, respectively (28).
Rituximab is a monoclonal antibody that depletes B-cells, by binding to CD20 molecules on their surface (42). Seven studies focused on rituximab monotherapy for GLILD (Table 3). Arraya, Cereser, Ng and Tessarin all reported cases of patients with CVID and GLILD who were successfully treated with rituximab monotherapy (at a dose of 375 mg/m2 weekly for four weeks) (32–35). Maglione et al. followed 73 patients for 18 months: 44 patients had CVID only, 14 had concomitant stable GLILD, and 15 had concomitant progressive GLILD. 11 of the 15 patients with progressive GLILD were treated with rituximab at a dose of 375 mg/m2 weekly for four weeks: all experienced stabilization or improvement of disease activity, however four relapsed 18 months after completion of therapy (10).
Of particular interest is the study by Zdziarsky and Gamian’s, describing a 25-year old woman with CVID and GLILD who was treated with rituximab monotherapy at a relatively low dose of 150 mg/m2 weekly for six weeks because of risk of infection (37). This resulted in incomplete remission of clinical symptoms, and the patient relapsed six months later. Treatment with rituximab was repeated, this time at a dose of 375 mg/m2, resulting in complete remission for a period of 30 months.
Eight studies evaluated combination chemotherapy with rituximab and azathioprine (Table 4). The rationale behind this combination chemotherapy is that B- and T-lymphocytes are targeted simultaneously (38). Chase and colleagues were the first ones to pioneer this approach. They performed a longitudinal prospective cohort study in which they followed seven patients with CVID and GLILD, who were treated with intravenous rituximab and oral azathioprine for 18 months. All patients experienced some degree of improvement in radiological findings (38). These results were confirmed by Pathria, Routes, Limsuwat and Tillman, who reported successful treatment of patients with CVID and GLILD with combination chemotherapy (44–46, 49). Vitale et al., reported successful addition of combination therapy with rituximab to glucocorticoid treatment in a 17-year old patient with CVID and GLILD after initial unresponsiveness to glucocorticoid monotherapy (36). Jolles’ and Sood’s articles showed that azathioprine can be replaced by other drugs with similar mechanisms of action. For example, Jolles et al. described a 51-year old woman with CVID and GLILD treated with a combination of rituximab and MMF, because of intolerance of azathioprine. Five months into treatment, the patient experienced an improvement of symptoms, alongside better pulmonary function and radiologic results (43). Sood et al. reported an improvement of GLILD related symptoms in the case of a 16-year old boy with 22q.11 deletion syndrome who was treated with rituximab and 6-mercaptopurine (48). One additional article by Verbsky et al. was added to the review despite its publishing date (June 2020) being after the last literature search (March 2020). We choose to mention this article, because the planned publication of the paper was known to the authors at the time of the literature search and, most importantly, because its results are highly relevant for this systematic review. The authors performed a retrospective chart review of 39 patients with CVID and GLILD who were treated with a combination of rituximab and azathioprine or rituximab and MMF. The median follow-up period was four years. 37 patients were included in the final analysis and of those 34 (92%) experienced an improvement of GLILD-related parameters. 27 patients (73%) experienced sustained remission, whereas nine patients (24%) relapsed after a median of 3.2 months. Of those relapsing, two patients died of septicemia and respiratory failure, respectively (47).
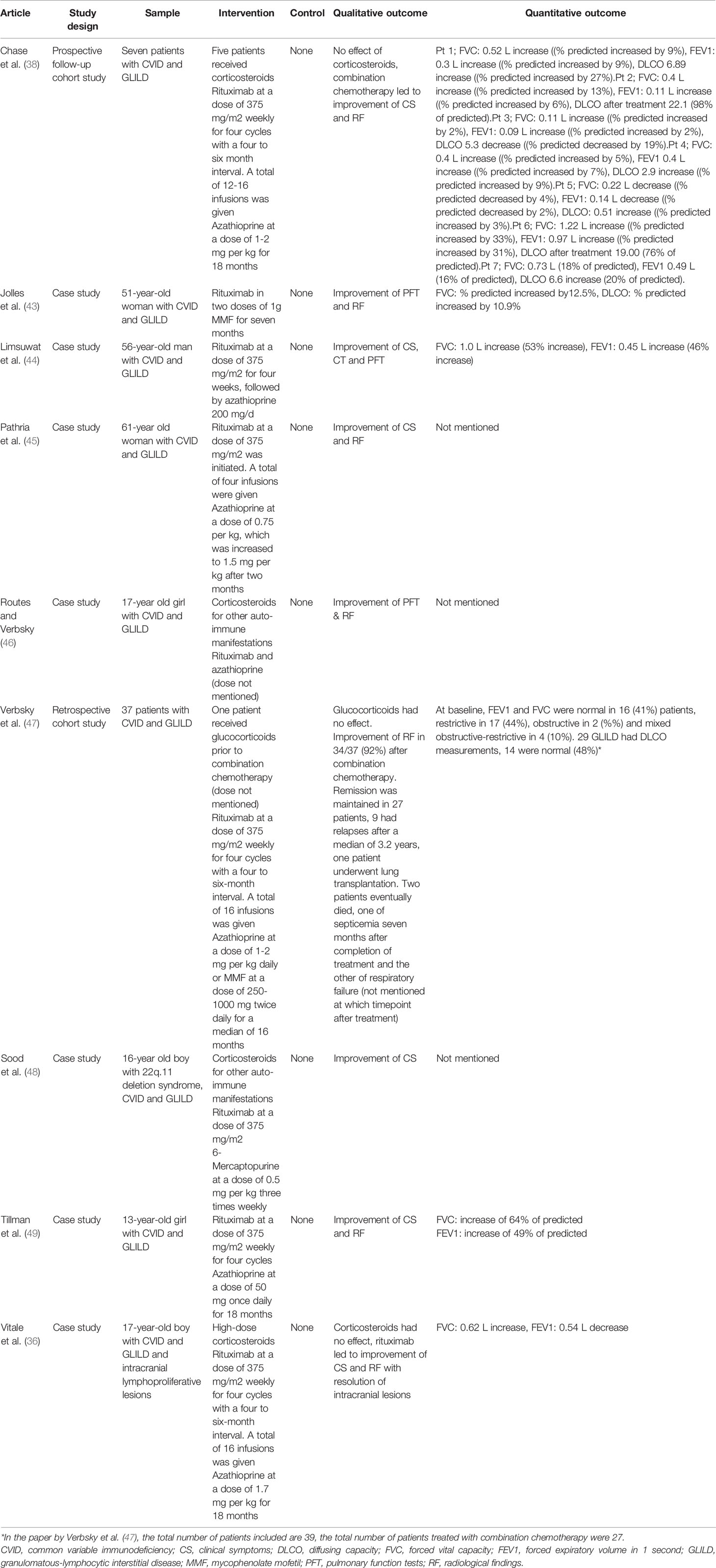
Table 4 Studies reporting treatment of GLILD in antibody deficiencies with combination chemotherapy.
CTLA-4 haploinsufficiency and LRBA deficiency result in a phenotype similar to CVID with severe immunodeficiency, lymphoproliferation and autoimmunity. In the physiological state, T lymphocyte responses are regulated by binding of the B7 ligand to CTLA-4 thus blocking T-cell activation, whereas LRBA is involved in intracellular trafficking and, among others, preserves CTLA-4 from degradation (50, 51), causing excessive immune activation. Abatacept consists of the Fc region of immunoglobulin IgG1 fused to CTLA-4 (52) and thus serves as a CTLA-4 fusion protein preventing excessive T lymphocyte proliferation in patients with CTLA-4 haploinsufficiency and LRBA deficiency.
A total of three articles described the use of abatacept for the treatment of GLILD (Table 5). Schwab and colleagues performed a longitudinal prospective cohort study in which they followed 133 patients with CTLA-4 haploinsufficiency. Of these, two patients who presented with GLILD treated with abatacept experienced improvement of both clinical symptoms and radiologic findings (51).
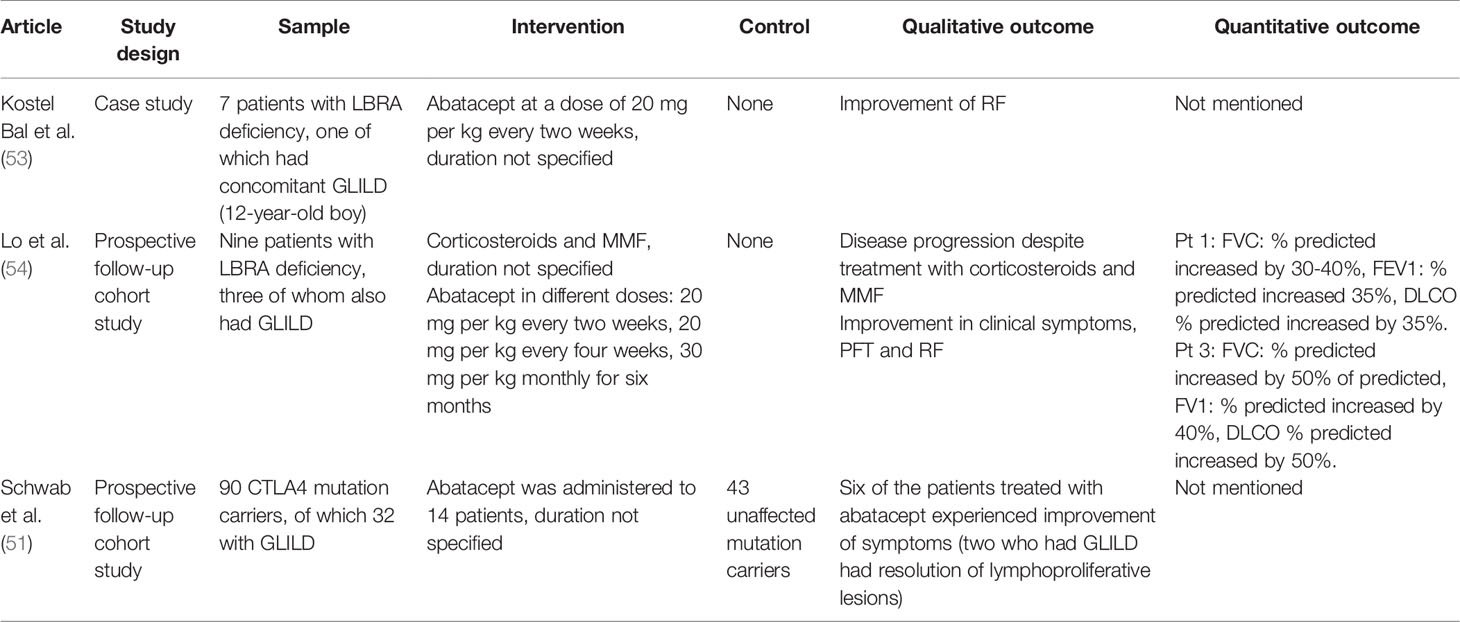
Table 5 Studies reporting treatment of GLILD in PID with abatacept.
Lo and colleagues reported three patients with LRBA deficiency and GLILD, who experienced significant improvements in lung function and radiological findings after treatment with abatacept (54). Bal replicated these results, findings abatacept to be useful in the treatment of GLILD in a 12-year old boy with LRBA deficiency (53).
HSCT holds the promise of being a definitive treatment for GLILD as it can correct the underlying immunodeficiency and the associated GLILD instead of just alleviating GLILD related symptoms. However, it is associated with considerable risks, including Graft versus Host Disease (GvHD) and serious infections, both associated with considerable morbidity. This risk is likely higher in those with established structural lung disease.
Five studies reported on HSCT for CVID patients with associated GLILD (Table 6). Wehr followed 25 patients with CVID who underwent HSCT. Five patients had GLILD: four experienced an improvement of the CVID-related complications; one died 104 days after transplantation due to acute GvHD and infectious complications (60). Wehr’s papers also includes four patients which were discussed in Rizzi’s publication in 2011 (56). Hartono published the case of a 23- year-old woman who presented with a CVID-like phenotype due to a STAT1 gain-of-function mutation and GLILD: after HSCT there was an improvement of radiologic findings (55). Mixed outcomes were reported by both Seidel and Tesc. Seidel and colleagues performed an international survey and collected information about 12 patients with CVID-like disease due to underlying LRBA deficiency (seven of whom also had GLILD), who underwent HSCT. Four patients went into partial remission, whereas three of them died (57). Tesch published a prospective follow-up study of 76 patients with LRBA deficiency, of which 24 underwent HSCT. Of these 24 patients, 17 of the 24 patients survived and all of the seven patients with concomitant GLILD experienced an improvement of GLILD related symptoms. Two patients who did not have GLILD before HSCT, developed the disease after the procedure (59).

Table 6 Studies reporting treatment of GLILD in antibody deficiencies with HSCT.
All studies had an overall intermediate or high risk of bias (Table 7). This was largely due to the small sample sizes and lack of controls. Outcomes were mostly reported qualitatively, with few data about pulmonary function tests and a lack of standardized CT evaluation. The duration of follow-up was typically limited, meaning that long-term outcomes of patients remained uncertain. As far as confounders are concerned, smoking status was not always reported. Finally, genetic testing for CTLA-4 haploinsufficiency and LRBA deficiency only became available as of 2012, meaning that older articles could not make this additional distinction.
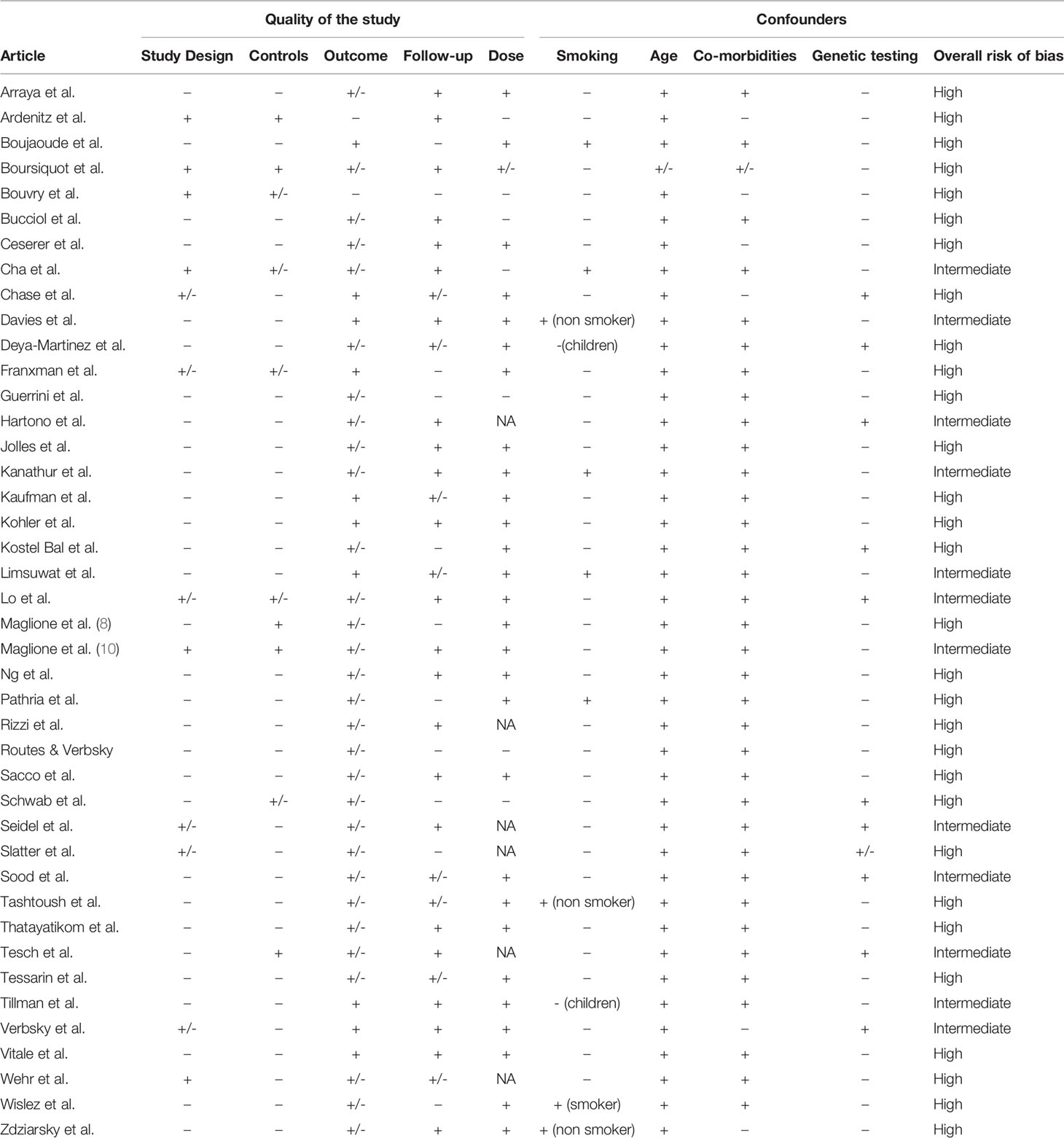
Table 7 Quality of studies analyzing treatment for GLILD in primary antibody deficiencies.
In 27 studies the level of evidence was 4, and in 12 studies the level of evidence of 3. The associated level of practice recommendations was weak in both groups.
To our knowledge, this is the most comprehensive systematic review analyzing treatment efficacy for GLILD in CVID. We show that there is still much uncertainty about the optimal treatment for GLILD and that more basic scientific and clinical research is needed in order to establish the best standard of care.
There are many factors influencing the choice of treatment. Apart from efficacy, risk-to-benefit ratio and patient preference, drug availability and cost may also play a role. Several studies reported that the efficacy of glucocorticoid monotherapy is limited. Other immunosuppressants were often used as second-line therapy with varying results. Rituximab monotherapy and combination chemotherapy with rituximab and azathioprine emerged as promising second-line treatments. Abatacept has been used in patients with CTLA-4 and LRBA mutations, but has not been routinely used in other patient populations as of yet. Finally, HSCT may be an option when other treatments have failed, but reported survival after HSCT in CVID has been poor.
Our findings suggest that glucocorticoids, although widely used as first line therapy, failed to induce remission in 57% (17 individuals) of patients using glucocorticoids (18, 23, 26, 27, 31, 36–38). Treatment with glucocorticoids led to a partial response in 13% (four individuals) and failed to maintain remission in 7% (two individuals) of patients (18, 29). There are, however, also literature reports about the positive effects of glucocorticoids (16, 17, 20, 21). 23% (seven individuals) of all patients using glucocorticoids had resolution of symptoms. It is currently unclear how much reporting bias has occurred in the reports describing the use of for example glucocorticoids for treatment of GLILD. Based on current knowledge, it remains unclear how the benefits of glucocorticoids in some patients may weigh against the side-effects of long-term treatment.
With respect to the category of the (biological) DMARDs, MMF, azathioprine, cyclosporine, sirolimus and infliximab have demonstrated efficacy in single case reports. Yet, because of the anecdotal nature of the studies and the relatively small patient populations they were described in, there is insufficient evidence to make definitive statements. While a previous survey has shown that most physicians agree on the implementation of azathioprine and MMF, there is no consensus as far as other (biological) DMARDs are concerned (9).
We found that rituximab monotherapy was effective in treating GLILD in most cases, although relapses did occur after B cell reconstitution (10, 39). Combination chemotherapy with rituximab and azathioprine is another potential treatment regimen in patients with CVID and GLILD. Our collected data show that this combination of drugs was effective at inducing remission in all cases, even where other therapies had failed (36–38). However, there are also indications that upon prolonged follow-up, relapses may occur (10, 47). The findings on rituximab are in line with published literature which indicates both rituximab and rituximab-based chemotherapy are effective treatments for GLILD in CVID (9). The current literature does not allow to determine whether rituximab monotherapy is superior, equally effective or inferior to rituximab-based combination chemotherapy.
Abatacept is often implemented in the treatment of GLILD in patients with CTLA-4 haploinsufficiency and LRBA deficiency. Results were promising as the drug was effective in most reported cases. Although abatacept is mostly implemented for the treatment of patients with CTLA-4 or LRBA related diseases, it would be interesting to see whether it could be of benefit in other GLILD patient populations as well.
HSCT is a potentially curative treatment for immunodeficiencies and GLILD, yet is associated with the risk of serious complications. Our results show that when successfully carried out, HSCT does indeed lead to resolution of GLILD symptoms in most cases. One exception was two patients in the study by Tesch et al., who developed GLILD after HSCT (59). On the other hand, the reported mortality rate was still relatively high compared to overall survival of patients transplanted for other types of PID. While for patients with CVID and GLILD the survival after HSCT varied between 48% and 70%, in PIDs in general it approaches 90% (61). Furthermore, the procedure of HSCT encompasses immunosuppression as a result of the conditioning and replacement of hematopoietic stem cells, and it is as yet not fully proven which of these two components is responsible for the reduction of GLILD activity after HSCT. There are many factors influencing transplantation outcome, including HLA matching, severity of pre-existing lung disease, infections and the presence of active inflammation in other organs which can make transplant more hazardous. Bone-marrow microenvironment, that is, the complex interplay of local and systemic factors driving and influencing stem cell development, has recently emerged as a potential contributor to the success or failure of HSCT. As pointed out by Troilo and colleagues, approximately half of patients with CVID undergoing HSCT experience incomplete B-cell reconstitution. By studying development and maturation of B-cells of immunodeficient patients with different genetic mutations in vitro, the researchers found that patients with a non-supportive bone-marrow niche may not allow for adequate immune cell reconstitution and may have worse outcomes (62). These findings may help in in the prediction of which CVID patients with GLILD could benefit from HSCT.
Furthermore, our study did not find clear differences in treatment responses between children (27 individuals) and adults (228) with GLILD. While mortality is higher in patients with pediatric-onset disease (63) almost all literature reports of children with GLILD showed a positive response to treatment. However, in order to make a clear statement about the prognosis of pediatric-onset GLILD, long-term follow-up data would be required.
This is the first review that comprehensively summarizes all peer-reviewed data about the treatment of GLILD in CVID. A systematic approach was implemented according to the internationally recognized PRISMA guidelines that aimed at identifying all existing literature on the treatment of GLILD in CVID. Two databases were searched and, in order to reduce the risk of bias, the screening process was carried out by two independent blinded researchers.
Despite efforts to minimize weaknesses, several limitations need discussion. First of all, there might be bias intrinsic to the published studies. Glucocorticoids are considered first-line treatment for GLILD (9), which could mean that their efficacy is taken for granted and successfully treated patients are under-reported.
Further, the definition of GLILD used throughout this paper may have some limitations. Even though we strictly adhered to the internationally recognized definition of GLILD used by the British Lung Foundation/United Kingdom Primary Immunodeficiency Network, we must acknowledge that GLILD is a spectrum of symptoms and manifestations and that the impact on daily life and response to treatment may differ accordingly. Hence, there is a certain degree of interindividual variation that is difficult to quantify in the absence of detailed and objective information, such as standard radiological scores and pulmonary function tests.
Moreover, we excluded several case reports describing patients with CVID and granulomatous disease, often classified as sarcoidosis, not fulfilling the current GLILD criteria. However, some of these patients may have suffered from GLILD. Indeed, there are several case reports describing patients who were misdiagnosed with sarcoidosis and who were frequently unresponsive to glucocorticoid monotherapy, similarly to the results described in this review (64–66).
Moreover, treatment regimens were strictly defined to enable comparison of the effects of different types of monotherapy. In addition, strict criteria for evaluation of remission of GLILD were formulated. Because of this, small positive effects of treatment might have been underreported in this study.
Finally, long-term effects of medication are seldom mentioned, including the risk of infection linked to the prolonged use of immunosuppressants. This could either mean that the added effect of immunosuppressants in already immunocompromised individuals is negligible or that there is some degree of reporting bias at play. Similarly, little to no side-effects were mentioned in the analyzed literature. However, glucocorticoids are unsuitable long-term therapy candidates because of detrimental effects on metabolism, bone density, growth and behavior. As mentioned previously, the quality of the evidence was relatively low, because none of the included studies had an experimental set-up. The choice of outcome measures was heterogeneous, and often only qualitative assessments were made, thus preventing meta-analysis. Possible confounders were rarely mentioned in the reviewed literature. Hence, it was difficult to make any final recommendations for clinical practice based on the available literature.
Understanding the cause of GLILD is critical in finding a cure for this disease. About 10-20% of patients with CVID develop GLILD, which suggests that the complication is brought on by a combination of (epi-) genetic and/or environmental factors rather than a single cause (7). It could be postulated that individuals with GLILD are a specific subset of the patient population with CVID, with a susceptibility for lymphoproliferation. Reverse thinking by translating from the bench back to hypothesis formulation can help assemble a workable theoretical framework. If, as is currently thought, GLILD is a form of immune dysregulation, there are potentially two important players, namely T-cells and B-cells (67).
The efficacy of second-line immunosuppressants that selectively target T-cells suggest they have an important role in the pathogenesis of GLILD. On the other hand, the successful use of rituximab in the treatment of the disease supports the idea that B-cells may be important effector cells, either initiating or maintaining inflammation in GLILD. A combined role of T- and B-lymphocytes has also been suggested: superior efficacy of the combination of azathioprine and rituximab compared to rituximab monotherapy would plead in favor of this hypothesis (38).
However, fundamental research into the pathophysiology of GLILD is needed to corroborate any of the above-mentioned hypotheses. In patients in whom monogenetic defects are identified, personalized medicine with individualized treatment strategies could be devised. Histopathological analysis, where available, may support this. Abatacept in CTLA-4 haploinsufficiency and LRBA deficiency is a good example of how personalized medicine is already being implemented in clinical practice.
In order to improve patient care and treatment of GLILD, it is important to screen for the condition, and define the best standard of treatment (9). RCTs are still lacking, because, due to the low incidence of GLILD, it is difficult to recruit sufficient numbers of participants. However, a combined effort by international consortium of medical centers, could allow for standardized data collection on a much larger scale, including pulmonary function tests and a uniform radiographic high-resolution CT scan score. Indeed, studies such as STILPAD are on-going and will inform on this. Until then, uniform standardized reporting on GLILD is crucial. Based on previous literature, this should at least include information on how the GLILD diagnosis was made, dosage and interval of the intervention, treatment-associated side effects (both short- and long-term), pre- and post-treatment CT scores using a universal scoring method, pulmonary function tests including carbon-monoxide diffusion and lymphocyte phenotyping data, ideally using validated tools. Results could provide scientific backup for current treatment strategies and help create new, evidence-based treatment protocols.
Based on this systematic review of the current literature, which was often of low quality with a high risk of bias, it is impossible to define which therapeutic option is optimal in treating GLILD in CVID.
Corticosteroid monotherapy seems suboptimal for many patients, rituximab monotherapy and combination chemotherapy with rituximab and azathioprine were effective in most reported cases. The use of abatacept has so far been only implemented as therapy for patients with pathogenic CTLA-4 and LRBA mutations. HSCT is the only curative treatment for GLILD, yet not free of risks. While much is left open and uncertain, what has become most evident throughout this review is that there remain many critical knowledge gaps concerning treatment of GLILD. Etiology and optimal treatment for the disease are questions that require urgent answers, as they may lead to better and more specific treatment regimens. In the future, larger well-designed studies evaluating therapeutic strategies should be carried out, with uniform quantitative outcomes.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
OL and BS created the search string, selected the articles included in the review, wrote the paper, and created the tables. JM chose the review topic, and guided the research and writing process. JW gave advice about the methodology and reviewed the final text. CC-R and H-eH provided additional raw data which was included in the review. VD, GB, JH, H-eH, HI, HL, ST-L, SP, AR, AS, AV, and KW gave advice during the synthesis of the results, commented on the draft papers, and reviewed the final text. All authors contributed to the article and approved the submitted version.
Financial support for this publication was provided by the Louise Vehmeijer Foundation, “e-GLILDnet” and the European Respiratory Society.
JH and KW co-chair the European Respiratory Society-funded e-GLILDnet Clinical Research Collaboration which is a collaboration with ESID (the European Society for Immunodeficiencies).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer EK declared a past co-authorship with one of the authors CC-R to the handling editor.
Publication of this paper was made possible through financial support of “e-GLILDnet” and the European Respiratory Society.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.606099/full#supplementary-material
1. Salzer U, Warnatz K, Peter HH. Common variable immunodeficiency - an update. Arthritis Res Ther (2012) 14(5):1–11. doi: 10.1186/ar4032
2. Cinetto F, Scarpa R, Pulvirenti F, Quinti I, Agostini C, Milito C. Appropriate lung management in patients with primary antibody deficiencies. Expert Rev Respir Med (2019) 13:823–38. doi: 10.1080/17476348.2019.1641085
3. Baumann U, Miescher S, Vonarburg C. Immunoglobulin replacement therapy in antibody deficiency syndromes: are we really doing enough? Clin Exp Immunol (2014) 178:83–5. doi: 10.1111/cei.12521
4. Baumann U, Routes JM, Soler-Palacin P, Jolles S. The Lung in Primary Immunodeficiencies: New Concepts in Infection and Inflammation. Front Immunol (2018) 9:1–15. doi: 10.3389/fimmu.2018.01837
5. Pulvirenti F, Pecoraro A, Cinetto F, Milito C, Valente M, Santangeli E, et al. Gastric Cancer Is the Leading Cause of Death in Italian Adult Patients With Common Variable Immunodeficiency. Front Immunol (2018) 9:1–9. doi: 10.3389/fimmu.2018.02546
6. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood (2012) 119:1650–7. doi: 10.1182/blood-2011-09-377945
7. Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol (2004) 114:415–21. doi: 10.1016/j.jaci.2004.05.057
8. Maglione PJ, Overbey JR, Radigan L, Bagiella E, Cunningham-Rundles C. Pulmonary radiologic findings in common variable immunodeficiency: clinical and immunological correlations. Ann Allergy Asthma Immunol (2014) 113:452–9. doi: 10.1016/j.anai.2014.04.024
9. Hurst JR, Verma N, Lowe D, Baxendale HE, Jolles S, Kelleher P, et al. British Lung Foundation/United Kingdom Primary Immunodeficiency Network Consensus Statement on the Definition, Diagnosis, and Management of Granulomatous-Lymphocytic Interstitial Lung Disease in Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Practice (2017) 5:938–45. doi: 10.1016/j.jaip.2017.01.021
10. Maglione PJ, Gyimesi G, Cols M, Radigan L, Ko HBM, Weinberger T, et al. BAFF-driven B cell hyperplasia underlies lung disease in common variable immunodeficiency. JCI Insight (2019) 4(5):1–15. doi: 10.1172/jci.insight.122728
11. Unger S, Seidl M, van Schouwenburg P, Rakhmanov M, Bulashevska A, Frede N, et al. The TH1 phenotype of follicular helper T cells indicates an IFN-gamma-associated immune dysregulation in patients with CD21low common variable immunodeficiency. J Allergy Clin Immunol (2018) 141:730–40. doi: 10.1016/j.jaci.2017.04.041
12. Wheat WH, Cool CD, Morimoto Y, Rai PR, Kirkpatrick CH, Lindenbaum BA, et al. Possible role of human herpesvirus 8 in the lymphoproliferative disorders in common variable immunodeficiency. J Exp Med (2005) 202:479–84. doi: 10.1084/jem.20050381
13. Berbers RM, Mohamed Hoesein FAA, Ellerbroek PM, van Montfrans JM, Dalm V, van Hagen PM, et al. Low IgA Associated With Oropharyngeal Microbiota Changes and Lung Disease in Primary Antibody Deficiency. Front Immunol (2020) 11:1245. doi: 10.3389/fimmu.2020.01245
14. Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, Ioannidis JPA, et al. The PRISMA Statement for Reporting Systematic Reviews and Meta-Analyses of Studies That Evaluate Health Care Interventions: Explanation and Elaboration. Ann Internal Med (2009) 151:W65–94. doi: 10.7326/0003-4819-151-4-200908180-00136
15. CEBM. Levels of Evidence (2011). Available from: https://www.cebm.net/wp-content/uploads/2014/06/CEBM-Levels-of-Evidence-2.1.pdf.
16. Boujaoude Z, Arya R, Rafferty W, Dammert P. Organising pneumonia in common variable immunodeficiency. BMJ Case Rep (2013) 2013:1–3. doi: 10.1136/bcr-2013-008905
17. Guerrini S, Squitieri NC, Marignetti Q, Puliti A, Pieraccini M, Grechi M, et al. Granulomatous-lymphocytic interstitial lung disease at the emergency department: Think about it! Lung India (2018) 35:360–2. doi: 10.4103/lungindia.lungindia_461_17
18. Kohler PF, Cook RD, Brown WR, Manguso RL. Common Variable Hypogammaglobulinemia with T-Cell Nodular Lymphoid Interstitial Pneumonitis and B-Cell Nodular Lymphoid Hyperplasia - Different Lymphocyte Populations with a Similar Response to Prednisone Therapy. J Allergy Clin Immunol (1982) 70:299–305. doi: 10.1016/0091-6749(82)90066-5
19. Kanathur N, Byrd RP, Fields CL, Roy TM. Noncaseating granulomatous disease in common variable immunodeficiency. South Med J (2000) 93:631–3. doi: 10.1097/00007611-200093060-00023
20. Kaufman J, Komorowski R. Bronchiolitis Obliterans Organizing Pneumonia in Common Variable Immunodeficiency Syndrome. Chest (1991) 100:552–3. doi: 10.1378/chest.100.2.552
21. Wislez M, Sibony M, Naccache JM, Liote H, Carette MF, Oksenhendler E, et al. Organizing pneumonia related to common variable immunodeficiency. Respiration (2000) 67:467–70. doi: 10.1159/000029552
22. Ardeniz O, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Clin Immunol (2009) 133(2):198–207. doi: 10.1016/j.clim.2009.05.001
23. Boursiquot JN, Gerard L, Malphettes M, Fieschi C, Galicier L, Boutboul D, et al. Granulomatous Disease in CVID: Retrospective Analysis of Clinical Characteristics and Treatment Efficacy in a Cohort of 59 Patients. J Clin Immunol (2013) 33:84–95. doi: 10.1007/s10875-012-9778-9
24. Bouvry D, Mouthon L, Brillet PY, Kambouchner M, Ducroix JP, Cottin V, et al. Granulomatosis-associated common variable immunodeficiency disorder: a case control study versus sarcoidosis. Eur Respir J (2013) 41:115–22. doi: 10.1183/09031936.00189011
25. Bucciol G, Petrone A, Putti MC. Efficacy of mycophenolate on lung disease and autoimmunity in children with immunodeficiency. Pediatr Pulmonol (2017) 52(10):E73–E6. doi: 10.1002/ppul.23757
26. Cha SI, Fessler MB, Cool CD, Schwarz ML, Brown KK. Lymphoid interstitial pneumonia: clinical features, associations and prognosis. Eur Respir J (2006) 28:364–9. doi: 10.1183/09031936.06.00076705
27. Davies CWH, Juniper MC, Gray W, Gleeson FV, Chapel HM, Davies RJO. Lymphoid interstitial pneumonitis associated with common variable hypogammaglobulinaemia treated with cyclosporin A. Thorax (2000) 55:88–90. doi: 10.1136/thorax.55.1.88
28. Franxman TJ, Howe LE, Baker JR. Infliximab for Treatment of Granulomatous Disease in Patients with Common Variable Immunodeficiency. J Clin Immunol (2014) 34:820–7. doi: 10.1007/s10875-014-0079-3
29. Sacco O, Fregonese B, Picco P, Faraci M, Facchetti P, Pistoia V, et al. Common Variable immunodeficiency presenting in a girl as lung infiltrates and mediastinal adenopathies leading to severe ‘‘superior vena caval’’ syndrome. Eur Respir J (1996) 9:1958–61. doi: 10.1183/09031936.96.09091958
30. Tashtoush B, Memarpour R, Ramirez J, Bejarano P, Mehta J. Granulomatous-lymphocytic interstitial lung disease as the first manifestation of common variable immunodeficiency. Clin Respir J (2018) 12:337–43. doi: 10.1111/crj.12511
31. Thatayatikom A, Thatayatikom S, White AJ. Infliximab treatment for severe granulomatous disease in common variable immunodeficiency: a case report and review of the literature. Ann Allergy Asthma Immunol (2005) 95:293–300. doi: 10.1016/S1081-1206(10)61228-8
32. Arraya M, Castro Y, Navarro J, Sarmiento E, Fernández-Cruz E, Carbone JC. Rituximab for granulomatous lymphocytic interstitial lung disease in a patient with common variable immunodeficiency. Is single therapy enough? Int J Clin Reumatol (2018) 13(1):38–42. doi: 10.4172/1758-4272.1000159
33. Cereser L, De Carli R, Girometti R, De Pellegrin A, Reccardini F, Frossi B, et al. Efficacy of rituximab as a single-agent therapy for the treatment of granulomatous and lymphocytic interstitial lung disease in patients with common variable immunodeficiency. J Allergy Clin Immunol Practice (2019) 7:1055–+. doi: 10.1016/j.jaip.2018.10.041
34. Ng J, Wright K, Alvarez M, Hunninghake GM, Wesemann DR. Rituximab Monotherapy for Common Variable Immune Deficiency-Associated Granulomatous-Lymphocytic Interstitial Lung Disease. Chest (2019) 155:E117–E21. doi: 10.1016/j.chest.2019.01.034
35. Tessarin G, Bondioni MP, Rossi S, Palumbo L, Soresina A, Badolato R, et al. Rituximab as a Single Agent for Granulomatous Lymphocytic Interstitial Lung Disease in Common Variable Immune Deficiency. J Investigational Allergol Clin Immunol (2019) 29:470–1. doi: 10.18176/jiaci.0450
36. Vitale J, Convers KD, Goretzke S, Guzman M, Noyes B, Parkar N, et al. Serum IL-12 and soluble IL-2 receptor levels as possible biomarkers of granulomatous and lymphocytic interstitial lung disease in common variable immunodeficiency: A case report. J Allergy Clin Immunol Practice (2015) 3:273–6. doi: 10.1016/j.jaip.2014.09.019
37. Zdziarski P, Gamian A. Lymphoid Interstitial Pneumonia in Common Variable Immune Deficiency - Case Report With Disease Monitoring in Various Therapeutic Options: Pleiotropic Effects of Rituximab Regimens. Front Pharmacol (2019) 9:1–8. doi: 10.3389/fphar.2018.01559
38. Chase NM, Verbsky JW, Hintermeyer MK, Waukau JK, Tomita-Mitchell A, Casper JT, et al. Use of Combination Chemotherapy for Treatment of Granulomatous and Lymphocytic Interstitial Lung Disease (GLILD) in Patients with Common Variable Immunodeficiency (CVID). J Clin Immunol (2013) 33:30–9. doi: 10.1007/s10875-012-9755-3
39. Deya-Martinez A, Esteve-Sole A, Velez-Tirado N, Celis V, Costa J, Cols M, et al. Sirolimus as an alternative treatment in patients with granulomatous-lymphocytic lung disease and humoral immunodeficiency with impaired regulatory T cells. Pediatr Allergy Immunol (2018) 29:425–32. doi: 10.1111/pai.12890
40. Declerck PJ. Biologicals and biosimilars: a review of the science and its implications. Gabi J Generics Biosimilars Initiative J (2012) 1:13–6. doi: 10.5639/gabij.2012.0101.005
41. Levin AD, Wildenberg ME, van den Brink GR. Mechanism of Action of Anti-TNF Therapy in Inflammatory Bowel Disease. J Crohns Colitis (2016) 10:989–97. doi: 10.1093/ecco-jcc/jjw053
42. Gurcan HM, Keskin DB, Stern JNH, Nitzberg MA, Shekhani H, Ahmed AR. A review of the current use of rituximab in autoimmune diseases. Int Immunopharmacol (2009) 9:10–25. doi: 10.1016/j.intimp.2008.10.004
43. Jolles S, Carne E, Brouns M, El-Shanawany T, Williams P, Marshall C, et al. FDG PET-CT imaging of therapeutic response in granulomatous lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Exp Immunol (2017) 187:138–45. doi: 10.1111/cei.12856
44. Limsuwat C, Daroca PJ, Lasky JA. A 56-Year-Old-Man With Common Variable Immunodeficiency and Worsening Dyspnea Common variable immunodeficiency with granulomatous and lymphocytic interstitial lung disease. Chest (2018) 13(1):154:E27–30. doi: 10.1016/j.chest.2017.11.034
45. Pathria M, Urbine D, Zumberg MS, Guarderas J. Management of granulomatous lymphocytic interstitial lung disease in a patient with common variable immune deficiency. BMJ Case Rep (2016) 2016:1–5. doi: 10.1136/bcr-2016-215624
46. Routes JM, Verbsky JW. Immunodeficiency Presenting as an Undiagnosed Disease. Pediatr Clinics North A (2017) 64:27–+. doi: 10.1016/j.pcl.2016.08.007
47. Verbsky JW, Hintermeyer MK, Simpson PM, Feng M, Barbeau J, Rao N, et al. Rituximab and antimetabolite treatment of granulomatous and lymphocytic interstitial lung disease in common variable immunodeficiency. J Allergy Clin Immunol (2020) 147(2):704–12. doi: 10.1016/j.jaci.2020.07.021.
48. Sood AK, Funkhouser W, Handly B, Weston B, Wu EY. Granulomatous-Lymphocytic Interstitial Lung Disease in 22q11.2 Deletion Syndrome: a Case Report and Literature Review. Curr Allergy Asthma Rep (2018) 18:14. doi: 10.1007/s11882-018-0769-7
49. Tillman R, Guillerman RP, Trojan T, Silva-Carmona M, Chinn IK. Treatment-Responsive Granulomatous-Lymphocytic Interstitial Lung Disease in a Pediatric Case of Common Variable Immunodeficiency. Front Pediatr (2019) 7:105. doi: 10.3389/fped.2019.00105
50. Gamez-Diaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol (2016) 137:223–30. doi: 10.1016/j.jaci.2015.09.025
51. Schwab C, Gabrysch A, Olbrich P, Patino V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol (2018) 142:1932–46. doi: 10.1016/j.jaci.2018.02.055
52. Blair HA, Deeks ED. Abatacept: A Review in Rheumatoid Arthritis. Drugs (2017) 77:1221–33. doi: 10.1007/s40265-017-0775-4
53. Bal SK, Haskologlu S, Serwas NK, Islamoglu C, Aytekin C, Kendirli T, et al. Multiple Presentations of LRBA Deficiency: a Single-Center Experience. J Clin Immunol (2017) 37:790–800. doi: 10.1007/s10875-017-0446-y
54. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (2015) 349(6246):436–40. doi: 10.1126/science.aaa1663
55. Hartono SP, Vargas-Hernandez A, Ponsford MJ, Chinn IK, Jolles S, Wilson K, et al. Novel STAT1 Gain-of-Function Mutation Presenting as Combined Immunodeficiency. J Clin Immunol (2018) 38:753–6. doi: 10.1007/s10875-018-0554-3
56. Rizzi M, Neumann C, Fielding AK, Marks R, Goldacker S, Thaventhiran J, et al. Outcome of allogeneic stem cell transplantation in adults with common variable immunodeficiency. J Allergy Clin Immunol (2011) 128:1371–4.e2. doi: 10.1016/j.jaci.2011.07.055
57. Seidel MG, Bohm K, Dogu F, Worth A, Thrasher A, Florkin B, et al. Treatment of severe forms of LPS-responsive beige-like anchor protein deficiency with allogeneic hematopoietic stem cell transplantation. J Allergy Clin Immunol (2018) 141:770–5.e1. doi: 10.1016/j.jaci.2017.04.023
58. Slatter MA, Engelhardt KR, Burroughs LM, Arkwright PD, Nademi Z, Skoda-Smith S, et al. Hematopoietic stem cell transplantation for CTLA4 deficiency. J Allergy Clin Immun (2016) 138(2):615–9. doi: 10.1016/j.jaci.2016.01.045
59. Tesch VK, Abolhassani H, Shadur B, Zobel J, Mareika Y, Sharapova S, et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol (2020) 145:1452–63. doi: 10.1016/j.jaci.2019.12.896
60. Wehr C, Gennery AR, Lindemans C, Schulz A, Hoenig M, Marks R, et al. Multicenter experience in hematopoietic stem cell transplantation for serious complications of common variable immunodeficiency. J Allergy Clin Immunol (2015) 135:988–97.e6. doi: 10.1016/j.jaci.2014.11.029
61. Laberko A, Gennery AR. Clinical considerations in the hematopoietic stem cell transplant management of primary immunodeficiencies. Expert Rev Clin Immunol (2018) 14:297–306. doi: 10.1080/1744666X.2018.1459189
62. Troilo A, Wehr C, Janowska I, Venhoff N, Thiel J, Rawluk J, et al. Nonpermissive bone marrow environment impairs early B-cell development in common variable immunodeficiency. Blood (2020) 135:1452–7. doi: 10.1182/blood.2019003855
63. Baloh C, Reddy A, Henson M, Prince K, Buckley R, Lugar P. 30-Year Review of Pediatric- and Adult-Onset CVID: Clinical Correlates and Prognostic Indicators. J Clin Immunol (2019) 39:678–87. doi: 10.1007/s10875-019-00674-9
64. Allaoui A, Moudatir M, Echchilal K, Alaoui FZ, Elkabli H. A Misleading Diagnosis of Sarcoidosis in an Older Woman. Eur J Case Rep Intern Med (2017) 4:000463. doi: 10.12890/2017_000463
65. Fasano MB, Sullivan KE, Sarpong SB, Wood RA, Jones SM, Johns CJ, et al. Sarcoidosis and common variable immunodeficiency. Report of 8 cases and review of the literature. Med (Baltimore) (1996) 75:251–61. doi: 10.1097/00005792-199609000-00002
66. Sutor G, Fabel H. Sarcoidosis and common variable immunodeficiency. A case of a malignant course of sarcoidosis in conjunction with severe impairment of the cellular and humoral immune system. Respiration (2000) 67:204–8. doi: 10.1159/000029488
67. Park JH, Levinson AI. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Immunol (2010) 134:97–103. doi: 10.1016/j.clim.2009.10.002
Population: patients with PID and GLILD
Intervention: treatment (pharmacological and/or stem cell transplantation)
Control: no therapy or placebo
Outcome: clinical symptoms, pulmonary function tests, radiologic findings, mortality
“common variable immunodeficiency”[MeSH] OR CVID [Title/Abstract] OR common variable immunodeficiency [Title/Abstract] OR primary immunodeficiency [Title/Abstract] OR GLILD [Title/Abstract] OR antibody deficiency [Title/Abstract] OR granulomatous lymphocytic interstitial lung disease [Title/Abstract] OR granulomatous disease[Title/Abstract] OR interstitial lung disease [Title/Abstract] OR ILD [Title/Abstract] OR granulomatous lung disease [Title/Abstract] OR lymphocytic interstitial pneumonitis [Title/Abstract] OR lymphoid interstitial pneumonitis [Title/Abstract] OR LIP [Title/Abstract]
AND “hematopoietic stem cell transplantation”[MeSH] OR hematopoietic stem cell transplantation[Title/Abstract] OR HSCT[Title/Abstract] OR stem cell transplantation[Title/Abstract] OR SCT[Title/Abstract] OR “abatacept”[MeSH] OR abatacept[Title/Abstract] OR corticosteroid*[Title/Abstract] OR prednisone[Title/Abstract] OR methotrexate[Title/Abstract] OR “mycophenolic acid”[MeSH] OR “mycophenolic acid” [Title/Abstract] OR mycophenolate mofetil[Title/Abstract] OR rituximab[Title/Abstract] OR “azathioprine”[MeSH] OR azathioprine[Title/Abstract] OR immunosuppressant[Title/Abstract] OR immunomodulator[Title/Abstract]
‘common variable immunodeficiency’/exp OR ‘common variable immodeficiency’:ab,ti,kw OR CVID:ab,ti,kw OR ‘primary immunodeficienc*’:ab,ti,kw OR ‘antibody deficiency’:ab,ti,kw OR GLILD:ab,ti,kw OR ‘granulomatous lymphocytic interstitial lung disease’/exp OR ‘granulomatous lymphocytic interstitial lung disease’:ab,ti,kw OR ILD:ab,ti,kw OR ‘granulomatous lung disease’:ti,ab,kw OR ‘interstitial lung disease’:ab,ti,kw OR ‘lymphocytic interstitial pneumonia’:ti,ab,kw OR ‘lymphocytic interstitial pneumonitis’:ti,ab,kw OR ‘lymphoid interstitial pneumonitis’:ti,ab,kw
AND ‘stem cell transplantation’/exp OR ‘stem cell transplantation’:ti,ab,kw OR ‘hematopoietic stem cell transplantation’:ti,ab,kw OR abatacept/exp OR abatacept:ab,ti,kw OR corticosteroid/exp OR corticosteroid:ab,ti,kw OR prednisone:ab,ti,kw OR ‘mycophenolic acid’/exp OR ‘mycophenolic acid’:ti,ab,kw OR ‘mycophenolate mofetil’/exp OR ‘mycophenolate mofetil’:ti,ab,kw OR methotrexate/exp OR methotrexate:ab,ti,kw OR immunosuppressant:ti,ab,kw OR immunomodulator:ab,ti,kw
Keywords: systematic review, immunodeficiency, common variable immunodeficiency, CVID, granulomatous lymphocytic interstitial lung disease, GLILD, treatment
Citation: Lamers OAC, Smits BM, Leavis HL, de Bree GJ, Cunningham-Rundles C, Dalm VASH, Ho H-e, Hurst JR, IJspeert H, Prevaes SMPJ, Robinson A, van Stigt AC, Terheggen-Lagro S, van de Ven AAJM, Warnatz K, van de Wijgert JHHM and van Montfrans J (2021) Treatment Strategies for GLILD in Common Variable Immunodeficiency: A Systematic Review. Front. Immunol. 12:606099. doi: 10.3389/fimmu.2021.606099
Received: 14 September 2020; Accepted: 24 March 2021;
Published: 15 April 2021.
Edited by:
Michael Jordan, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Erinn Kellner, Cincinnati Children’s Hospital Medical Center, United StatesCopyright © 2021 Lamers, Smits, Leavis, de Bree, Cunningham-Rundles, Dalm, Ho, Hurst, IJspeert, Prevaes, Robinson, van Stigt, Terheggen-Lagro, van de Ven, Warnatz, van de Wijgert and van Montfrans. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joris van Montfrans, Si52YW5Nb250ZnJhbnNAdW1jdXRyZWNodC5ubA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.