 Pan Liu
Pan Liu Zhengdong Zhang
Zhengdong Zhang Yao Li
Yao Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 February 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.603416
Diabetic kidney disease (DKD) is a major cause of chronic kidney disease (CKD) in many developed and developing countries. Pyroptosis is a recently discovered form of programmed cell death (PCD). With progress in research on DKD, researchers have become increasingly interested in elucidating the role of pyroptosis in DKD pathogenesis. This review focuses on the three pathways of pyroptosis generation: the canonical inflammasome, non-canonical inflammasome, and caspase-3-mediated inflammasome pathways. The molecular and pathophysiological mechanisms of the pyroptosis-related inflammasome pathway in the development of DKD are summarized. Activation of the diabetes-mediated pyroptosis-related inflammasomes, such as nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3), Toll-like receptor 4 (TLR4), caspase-1, interleukin (IL)-1β, and the IL-18 axis, plays an essential role in DKD lesions. By inhibiting activation of the TLR4 and NLRP3 inflammasomes, the production of caspase-1, IL-1β, and IL-18 is inhibited, thereby improving the pathological changes associated with DKD. Studies using high-glucose–induced cell models, high-fat diet/streptozotocin-induced DKD animal models, and human biopsies will help determine the spatial and temporal expression of DKD inflammatory components. Recent studies have confirmed the relationship between the pyroptosis-related inflammasome pathway and kidney disease. However, these studies are relatively superficial at present, and the mechanism needs further elucidation. Linking these findings with disease activity and prognosis would provide new ideas for DKD research.
Cell death includes pyroptosis, apoptosis, necroptosis, necrosis, and autophagy, depending on different biochemical mechanisms and signal transduction pathways (1–5). In 2001, Cookson et al. (6) described a form of cell death in macrophages that depends on caspase-1, which was accompanied by the release of many pro-inflammatory factors. The term “pyroptosis” comes from the Greek “pyro” meaning fire or fever and “ptosis” meaning falling, to describe the pro-inflammatory properties of this cell death process and its relationship with the release of mature interleukin (IL)-1β and IL-18. Pyroptosis is a form of inflammation that is activated by bacteria, pathogens, or their endotoxins, leading to the subsequent activation of caspase-1, accompanied by cell swelling, cell membrane pore formation, cell membrane rupture, cell-permeable dissolution, DNA lysis, inflammasome activation, as well as the release of cell contents and inflammatory mediators, resulting in a robust inflammatory response. This response leads to programmed cell death (PCD) (7, 8). The immune response of the innate immune system after pathogen invasion plays a vital role in maintaining tissue homeostasis and the immune response. Pyroptosis is involved in the innate immune response and protects the body from infection by pathogenic microorganisms (9–11); however, excessive pyroptosis can lead to a variety of autoinflammatory and immune diseases such as massive cell death, tissue damage, organ failure, and even septic shock (12, 13). Recent studies have shown that scorch death plays a vital role in diseases such as liver disease (14), atherosclerosis (15), diabetes mellitus (16), gout (17), epilepsy (18), and tumors (19, 20).
Diabetes is a major global public health problem. The number of people with diabetes increased from 108 million in 1980 to 422 million in 2014 (21), and diabetes is among the leading causes of kidney failure. Diabetic kidney disease (DKD) causes glomerular hypertrophy, basal membrane thickening, glomerular sclerosis, Kimmelstiel-Wilson nodules, an increased glomerular filtration rate (GFR), clinical proteinuria, hypertension, and edema, and is often associated with diabetic retinopathy (22, 23). DKD leads to a decline in the quality of life and shortening of the survival time of patients, thus leading to heavy social and economic burdens. Fernández-Real (24) reported that innate immunity is related to the production of inflammatory cells, and development of obesity, insulin resistance, and other diabetic complications. Microinflammation and extracellular matrix amplification are common pathways for the progression of DKD. Various molecules associated with the inflammatory pathway in DKD include proinflammatory cytokines, chemokines, and Toll-like receptors (TLRs). As an essential innate immune response in the body, pyroptosis is closely related to the progression of DKD owing to the involvement of various pro-inflammatory factors in its activation pathway.
This review focuses on the three pathways of pyroptosis generation. The molecular and pathophysiological mechanisms of pyroptosis-related inflammasomes pathway in the development of DKD are summarized. With this review, we attempted to provide new insights for researchers regarding the development of potential therapies for DKD.
Caspase is a family of cysteine proteases with a primary function in mediating cell death, including apoptosis and pyroptosis (25). Caspase plays a vital role in embryonic development and in maintaining adult tissue balance. The sub-members of this family include caspase-1, caspase-11, and caspase-12 from mouse sources; and caspase-1, caspase-4, caspase-5, and caspase-12 of human origin. Their common features are control of the inflammatory response of host cells to pathogen invasion and the stimulation of damage by the host cell cytoplasm. Pro-caspases are inactive monomers that are not activated until they are subjected to specific stimuli (26). Pro-caspases become activated once they are absorbed into the multi-protein complex of the inflammasome, and then cleave the inactive pro-IL-1β and pro-IL-18 into active IL-1β and IL-18 during the pyroptosis process (27, 28). Caspase-11 can be directly activated by sensing lipopolysaccharides (LPS) in cells infected by various Gram-negative bacteria, thereby inducing pyroptosis in macrophages (7, 29). Similar to caspase-11, caspase-4 and caspase-5 both induce pyroptosis during LPS sensing (30). Caspase-8 has always been considered an apoptosis-related caspase. Subsequent studies showed that caspase-8 can directly regulate the cleavage and activation of gasdermin proteins under specific conditions to induce pyroptosis (31, 32). However, caspase-8 has significantly weaker processing power on gasdermin D (GSDMD) than caspase-1 (33). Recent studies have found that caspase-8 may be an important molecular switch that controls apoptosis, necroptosis, and pyrolysis, and prevents tissue damage (34, 35). Caspase-12 is also an inflammatory caspase, although its function is unknown (36).
Six members of the gasdermin family have been identified in humans: gasdermin A (GSDMA), gasdermin B (GSDMB), gasdermin C (GSDMC), GSDMD, gasdermin E (GSDME, also known as DFNA5), and pejva-kin (PJVK, also known as DFNB59). Seven members of the gasdermin family have been identified in mice: GSDMAs (GSDMA1–3) and GSDMCs (GSDMC1–4) (37–39). The entire gasdermin family has a common membrane-targeting mechanism (40). Among them, GSDMD is currently the gasdermin protein that has been most strongly associated with pyroptosis. The N-terminal and C-terminal domains are formed after cleavage by caspase. The N-terminal domain can be connected to phosphatidylinositol, phosphatidic acid, and phosphatidylserine on the cell membrane, resulting in their aggregation and insertion into the cell membrane to form membrane-spanning pores. GSDM pores are large, non-selective pores with an external diameter of 32 nm and an internal diameter of 10–20 nm (41, 42). This pore size is sufficient to allow for the inflow of H2O and Ca2+ to cause cell swelling. When the cell swells to a certain degree, it disintegrates. The cell contents such as K+, IL-1β, IL-18 (about 4–8 nm in diameter), and other small cytosolic proteins flow out, ultimately resulting in pyroptosis (43–49). The C-terminal domain (GSDMD-CT) is removed, and it is hypothesized to fold back on the N-terminal domain of gasdermin (GSDMD-NT) to inhibit N-terminal function, thereby inhibiting the formation of cell membrane pores and blocking the process of pyroptosis (42, 43). The pore-forming activity of GSDMD plays a vital role in the downstream pathway of pyroptosis mediated by inflammatory caspase (50). GSDMD proteins have been reported to independently modulate inflammatory mediators such as IL-1β release and cell membrane breakdown (48). Given the above characteristics, GSDMD is known as the executor of cell pyroptosis. In addition to GSDMD, mutations in the hydrophobic core of the C-terminal of GSDMA, GSDMA3, GSDMC, and GSDME can cause pyroptosis (50). For example, caspase-3 releases the N-terminal through proteolytic cleavage of GSDME, forming a hole in the membrane, and then converts apoptosis induced by tumor necrosis factor (TNF) or chemotherapy into pyrolysis (51–53). Gasdermin family members are widely expressed in different cells and tissues, but are mainly found in the gastrointestinal tract, skin, and immune cells, indicating that they play an essential role in the physical and mucosal barrier system, and actively eliminate infected cells through pyroptosis (38, 54). Recent studies have suggested that GSDMD pores formed on the plasma membrane can enable Ca2+ influx and activate the endosomal sorting complexes required for transport (ESCRT) mechanism to initiate the repair of membrane pores (55, 56). However, the specific detailed mechanism requires further study and clarification.
The PRR family includes TLRs, C-type lectin receptors (CLRs), retinoic acid-induced gene protein I (retinoic acid-induced gene)-like receptors (RLRs), and nucleotide-binding oligomerization domain-like receptors (NLRs). Some PRRs can form oligomeric protein structures called inflammasomes, promote the protein maturation of IL-1 family cytokines (i.e., IL-1 and IL-18), and mediate pyroptosis in inflammatory forms, accompanied by the final secretion of downstream mediators of inflammation, thereby mobilizing the recruitment of a large number of host immune cells that have different immune outcomes and promote acute inflammatory processes (40, 57). We summarize two representative receptors in this section: TLRs and nucleotide oligomerization domain (NOD)-like receptors (NLRs).
TLRs are the first family of innate immune receptors, described as type I transmembrane proteins anchored to the plasma membrane or endolysin membrane. The primary role of TLRs is to recognize the PRR-mediated activation of pathogen-associated molecular patterns (PAMPs) and host damage-associated molecular patterns (DAMPs), and to induce pyroptosis as a response. More than 10 TLRs have been identified in humans and 12 TLRs have been identified in mice (58). TLRs on the cell surface can recognize extracellular pathogens, whereas microbial nucleic acids are sensed by TLRs located in lysosomes. The changes in 1eucine-richrepeats (LRRs) of different TLRs provide specificity for the ligands that they can recognize. Ligand binding causes most TLRs to form dimers. In addition to forming homodimers, some TLRs can also recognize other additional ligands by forming heterodimers, co-receptors, or accessory proteins. In the presence of ligands, TLR dimers undergo conformational changes such that the intracellular TLR domain can join the downstream signal transduction pathway for adaptor initiation (59, 60).
NLRs are the largest family of cytoplasmic receptors and have a common central NOD. The NLR family is divided into four subfamilies: NLRAs, NLRBs, NLRCs (including NOD1, NOD2, NLRC3, NLRC4, and NLRC5), and NLRPs (including NLRP1–14). NLRs have a variety of immune functions, including response to infection and the formation of inflammasomes (NLRP1, NLRP3, and NLRC4) (61, 62), regulation of antigen presentation (NLRC5, CIITA) (63, 64), regulation of homeostasis in microbial clusters (NLRP6), and a regulatory role in the responses of nuclear factor kappa B (NF-кB) (NLRP6, NLRP12, and NLRC3), MAVS (NLRX1), and STING (NLRC3, NLRX1) (65–68). NAIP/NLRC4 oligomeric polymers rely on the CARD-CARD interaction to recruit apoptosis-associated speck-like protein containing CARD (ASC), and then activate caspase-1 and downstream effector functions. PRR activation (especially those in the cytoplasm), in addition to the induction of cytokine transcription, can also induce pyroptosis and stimulate inflammatory responses. Intracellular bacteria are killed directly by destroying the replicative niche of pathogens and intracellular traps induced by holes, thereby enhancing the immune defense function of pyroptosis (9, 11). NLRP3 is a key member of the NOD-like receptor family, which recognizes microbial and non-microbial risk signals, and induces aseptic inflammation under different conditions (69). In the pyroptosis pathway, NLRP3 can be combined with ASC to recruit pro-caspase-1 to form inflammasomes, which are converted into caspase-1 by hydrolysis. Previous studies have suggested that NOD2, TLR2, TLR4, and NLRP3 inflammasome-mediated inflammation participate in the persistence of DKD inflammation (70, 71).
Cells are stimulated by signals from bacteria and viruses, and different cytoplasmic sensor proteins trigger responses to pathogens and inflammatory factors. Dimerization occurs through combination of the adaptor protein ASC or NLRC4 with the pro-caspase-1 monomer, which activates pro-caspase-1 to become mature caspase-1. Meanwhile, caspase-1 cleaves GSDMD and activates the inactive precursor IL-1β into mature IL-1β. After GSDMD is cleaved, the domains at both ends of the NC are separated, and GSDMD-NT is released. The released GSDMD-NT forms a pore in the cell membrane by recognizing and binding phospholipid molecules on the cell membrane. The formation of pores destroys cell potential energy penetration, leading to cell swelling and eventually cell pyroptosis. IL-1β is also released from cells through the pores, causing a robust inflammatory response (43, 72, 73).
In 2011, Kayagaki et al. (74) discovered non-canonical pyroptotic pathways. In contrast to canonical pyroptotic pathways, the cell wall LPS of Gram-negative bacteria bypasses TLR4 and directly combines with the pro-caspase (-4 and -5 in humans and -11 in murine) to form activated caspase-4/5/11. The activated caspase-4/5/11 cleaves GSDMD and promotes the activation of pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18. Similarly, GSDMD-NT forms a hole in the cell membrane, which causes the release of IL-1β and IL-18 in the cell and induces pyroptosis (29, 38, 75). This pathway does not involve caspase-1; in the absence of caspase-1, human caspase-4/5 and murine caspase-11 can also induce pyroptosis with all associated morphological characteristics (40).
From the perspective of these two inflammasome pathways associated with pyroptosis, in the canonical inflammasome pathway, inflammation sensors detect different microbial signals and activate caspase-1 through ASC or NLRC adaptors. In contrast, the non-canonical inflammasome pathway is activated by caspase-4, caspase-5, and caspase-11, which are directly combined with LPS (76). In addition, recent studies have shown that caspase-11 directly binds to Leishmania lipophosphoglycan (LPG) (77) and oxidized phospholipids (oxPAPC) (78). Although the activation pathways are different, the downstream signaling pathways are all activated caspases that cleave GSDMD and release the N-terminal domain to form membrane pores, eventually leading to pyroptosis. In other words, GSDMD is a necessary downstream component of both the canonical and non-canonical inflammasome pathways associated with pyroptosis (41–43, 46, 47).
In addition, a new pyroptosis pathway was recently discovered. Caspase-3 is well-known as an important effector associated with apoptosis (79). Previous studies have suggested that caspase-3 is not involved in pyroptosis (80). However, researchers recently found that GSDME can convert caspase-3 induced apoptosis into pyroptosis through TNF-α and some chemotherapeutic drugs (52). Various death stimuli or viral infections can lead to an increase in the permeability of the outer mitochondrial membrane, causing the release of cytochrome C and binding to apaf-1, thereby enabling the assembly of apaf-1 apoptotic bodies and activation of caspase-9. The active caspase-9 then cleaves pro-caspase-3 to generate the active caspase-3 heterodimer. In addition, caspase-3 can be activated through the death receptor pathway, which is itself activated by the death receptor ligand on the cell membrane, and then pro-caspase-8 is activated to caspase-8. Active caspase-8 cleaves pro-caspase-3 to generate the active caspase-3 heterodimer (51). Caspase-3 cleaves GSDME into the N-terminal fragment of GSDME (GSDME-NT) and the C-terminal fragment of GSDME (GSDME-CT). GSDME-NT forms membrane pores on the cell membrane and induces pyroptosis (20, 52). In the course of this pathway, although the apoptosis-related proteins in the cells are activated at nearly the same time, the process of cell pyroptosis is faster; therefore, the cells eventually appear as pyroptotic (52). For a more intuitive understanding, we have summarized the three kinds of inflammasome pathways associated with pyroptosis along with graphical interpretations in Figure 1.
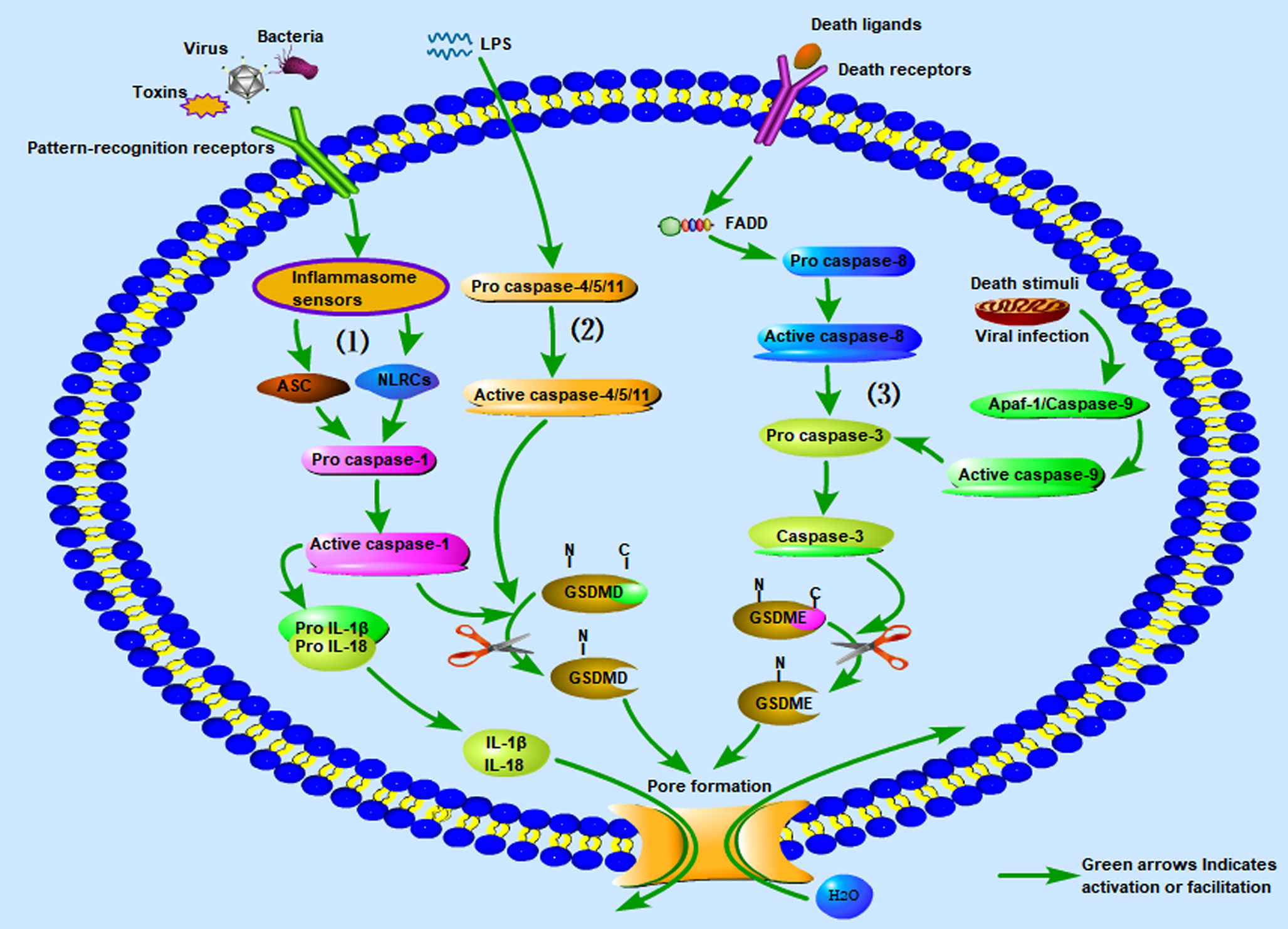
Figure 1 The three kinds of inflammasome pathways associated with pyroptosis. (1) Canonical inflammasome pathway associated with pyroptosis; (2) non-canonical inflammasome pathway associated with pyroptosis; (3) caspase-3 mediated inflammasome pathway associated with pyroptosis. LPS, Lipopolysaccharide.
Some recent studies have demonstrated that IL-1β, an inflammatory factor released by cells during pyroptosis, plays an important role in the pathogenesis of type 2 diabetes mellitus (T2DM) (81). Increased IL-1β has emerged as an essential factor for predicting the occurrence of T2DM. DKD is a common clinical complication in patients with diabetes, and is the leading cause of chronic kidney disease (CKD) in many developed and developing regions. Persistent aseptic inflammatory reactions in the kidney tissue are the pathophysiological basis of diabetic nephropathy (DN) that lead to glomerular capillary damage. The clinical features of DN are a gradual decline in renal function, abnormal levels of albumin (microalbuminuria) in the urine (30 mg/day or 20 g/min), and subsequent proteinuria and end-stage renal disease (ESRD) (82, 83). Once ESRD develops, the mortality rate is high, representing a critical clinical issue (84, 85).
With the expansion of research on DKD, there has been more interest on the potential role of pyroptosis in DKD pathogenesis. The hyperglycemia associated with DN is due to insufficient insulin secretion or insulin resistance, which produces hypoxia and causes excessive production of inflammatory cytokines. If oxidative stress reactions persist, many inflammatory cells are immersed in the matrix (86). The excessive activation of inflammatory cytokines can promote the progression of renal fibrosis (87, 88). Numerous studies have shown that activation of inflammatory factors caused by hyperglycemia plays a crucial role in the progression of DKD (89–91).
The NLRP3 inflammasome is involved in the pathogenesis of various kidney diseases, including acute kidney injury, CKD, DKD, and crystal-related nephropathy (92, 93). The NLRP3 inflammasome promotes disease occurrence and progression in DKD in a high-glucose environment. The production of mitochondrial ROS has been shown to initiate the activation of NLRP3 inflammasomes under diabetic conditions (94–96), further establishing a causal relationship between NLRP3 inflammasome activation and DKD. Inhibition of NLRP3 in the kidney (via silencing of NLRP3 or the studies in NLRP3 knockout mice) may improve renal function, and attenuate glomerular hypertrophy, glomerulosclerosis, mesangial expansion, interstitial fibrosis, inflammation, and expression of TGF-β1 and connective tissue growth factor (CTGF). Inhibition of NLRP3 or caspase-1 inflammasome activation, thereby inhibiting renal inflammation and fibrosis (at least in part), via suppression of oxidative stress in DN imparts protective effects on the kidney (97–99). By inhibiting NLRP3 upstream of the pyrolysis-related inflammasome pathway, the downstream expression of caspase-1, IL-1, and IL-18 can be progressively inhibited. Notably, the NLRP3 inflammasome can be activated via the canonical and non-canonical inflammasome pathways associated with pyroptosis (100). These findings provide a solid theoretical basis for how the NLRP3 inflammasome can be activated by inducing caspase-1 as an essential mediator of pro-inflammatory cytokine production (101, 102).
Clinical studies have found that compared with diabetic and non-diabetic patients without proteinuria, diabetic patients with proteinuria have significantly higher expression levels of IL-1β, IL-18, NLRP3, and serum IL-1β and IL-18 levels, with a proteinuria-positive correlation (95, 102, 103). Clinical research results also show that inhibiting IL-1β can prevent the progression of T2DM (104). ASC and caspase-1 were found to be highly expressed in a streptozotocin (STZ)-induced DN rat model, accompanied by hyperuricemia and hyperlipidemia, and IL-1β and IL-18 levels were elevated. The NLRP3 inflammasome-caspase-l-IL-1β/IL-18 axis is considered to play a critical role in DKD. Likewise, animal experiments have found that uric acid-lowering drugs (such as allopurinol and quercetin) can reduce uric acid and blood lipid levels, inhibit the activation of NLRP3 inflammasomes, and prevent kidney damage caused by STZ (105). Another study found that significant inhibition of NF-κB, and the decrease in IL-1β and TNF-α levels in diabetic rats were related to reductions in TXNIP and NLRP3 expression levels in diabetic kidneys (106). A clinical study showed that IL-18 levels in the serum and urine of patients with T2DM were positively correlated with the degree of proteinuria during follow-up, indicating that IL-18 may also be a risk factor for DKD (107). Insulin secretion disorders caused by pancreatic β-cell dysfunction and impaired insulin action caused by enhanced insulin resistance lead to hyperglycemia in T2DM (108). The combination of guava (Psidium guajava), with demonstrated antioxidant and anti-inflammatory effects, and trehalose on protecting the kidney and pancreas from damage was explored in a rat model of T2DM. The results showed that guava juice and trehalose could inhibit the secretion of IL-1β in the pancreas and kidneys caused by diabetes, and could prevent apoptosis and pyroptosis (109). An et al. (110) administered punicalagin to a mouse model of DN induced by a high-fat diet (HFD) and STZ. After punicalagin intervention, blood urea nitrogen (BUN), serum creatinine (CREA), and urine albumin-creatinine ratios (UACR) were significantly reduced, and the glomerular interstitial hyperplasia and glomerular hypertrophy scores were reduced. This treatment also reduced the expression levels of IL-1β, caspase-1, GSDMD, and NLRP3. The authors also found that punicalagin reduced the high-glucose–mediated protein expression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4) and reduced mitochondrial damage. Thus, by regulating activation of the NLRP inflammasome, the expression of caspase-1 increases, promoting the maturation and release of IL-1β and IL-18, thereby continuing to produce inflammation that leads to kidney damage (Figure 2).
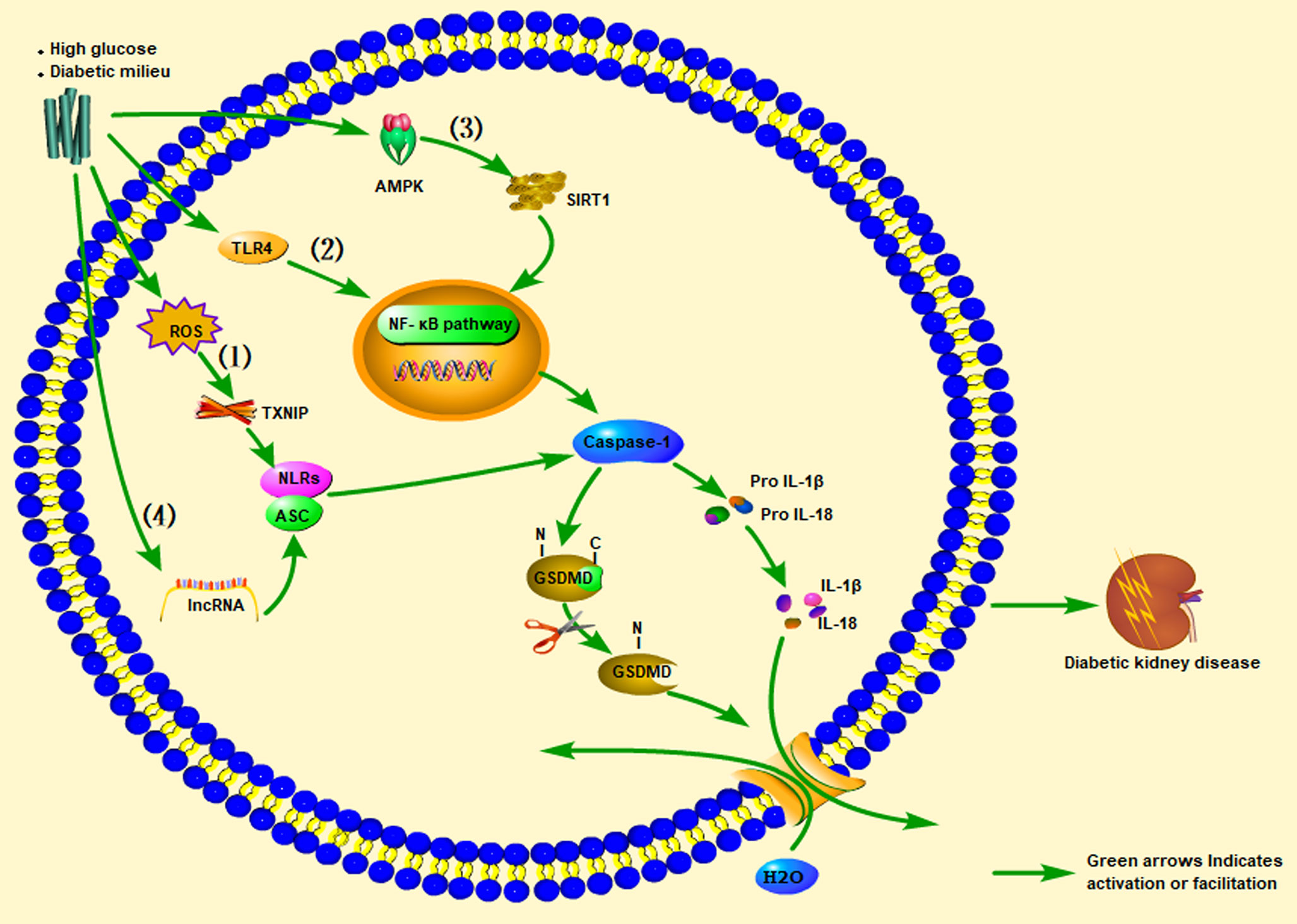
Figure 2 Promotion of inflammasome pathways associated with pyroptosis of cells in the kidney (glomerular endothelial cells, tubular epithelial cells, podocytes, tubular epithelial cells) under high-sugar or diabetes conditions, leading to diabetic kidney disease (DKD). In renal cells stimulated by high glucose (HG): (1) ROS/TXNIP/NLRP3 inflammasome signaling pathway, (2) TLR4/NF-κB inflammasome signaling pathway, (3) AMPK/SIRT1/NF-κB inflammasome signaling pathway, or (4) lncRNA)-related signaling pathways, they all activate pro-caspase-1 to become mature caspase-1. Caspase-1 cleaves GSDMD and activates the inactive pro IL-1β and pro-IL-18 to become mature IL-1β and IL-18, and the released GSDMD-NT forms a pore in the cell membrane, ultimately leading to DKD. ROS, Reactive oxygen species; TXNIP, Thioredoxin-interacting protein; AMPK, Adenosine 5’-monophosphate (AMP)-activated protein kinase; SIRT1, Silent information regulation 2 homolog1; NF-κB, Nuclear factor kappa-B.
Previous studies have found that the A1 adenosine receptor (A1AR) is widely distributed in the renal peritubular capillaries (PTCs) and glomerular afferent arterioles. A1AR is considered an essential regulator of renal tubular-glomerular feedback (TGF) (111). Knockout of A1AR in mice aggravated proteinuria and glomerular damage (112). Moreover, a recent study showed that the A1AR agonist 2-chloro-N6 cyclopentyladenosine (CCPA) plays a protective role in albuminuria related to the loss of megalin in the proximal renal tubules by inhibiting caspase-1/IL-18 signaling in DKD (113).
TLR4 usually signals through its downstream partner MyD88 to activate the NF-κB pathway, leading to ROS and cytokine production. In podocytes stimulated by high glucose, TLR4 activates NF-κB, and increases the release of pro-inflammatory cytokines and chemokines (Figure 2) (114, 115). Wang et al. (116) found that renal tubular damage in DKD patients upregulated the expression of TLR4 and GSDMD in the kidney tissue. In the paraffin-embedded sections of human DN tissues, immunohistochemical staining showed that the expression of GSDMD and TLR4 was positively correlated with albuminuria, interstitial fibrosis, and tubular atrophy scores, and was negatively correlated with the estimated glomerular filtration rate. Injection of the TLR4 inhibitor TAK-242 to db/db mice improved the brush border peeling and atrophy of the kidney tubules, along with interstitial fibrosis. Simultaneously, TLR4 inhibitors could reduce the expression levels of GSDMD and IL-18 in the renal tubular cells of db/db mice, the protein levels of caspase-1 and GSDMD-NT in the renal cortex tissue, and the level of IL-1β in renal homogenates. In addition, human renal tubular epithelial (HK-2) cells treated with high glucose and TAK-242 or parthenolide (an inhibitor of NF-κB) yielded similar results in western blotting, enzyme-linked immunosorbent assay, and flow cytometry. Studies with a diabetic mouse model and HK-2 cell experiments showed that inhibiting TLR4/NF-κB signaling can reverse the increase in GSDMD-NT expression in a high-glucose environment while inhibiting the release of IL-1β. This finding indicates that TLR4 inhibitors significantly inhibited GSDMD-related pyroptosis and reduced kidney damage in db/db mice. In addition, the proteinuria, renal insufficiency, inflammation, and renal fibrosis of STZ-induced diabetic mice with TLR4 knockout were protected, and TLR4 inhibition prevented renal tubular damage and reduced the loss of podocytes in DKD (117, 118). Therefore, the TLR4/NF-κB signaling pathway is involved in the expression of GSDMD in DKD (116).
Li et al. (119) found that geniposide can alleviate renal dysfunction in DN mice induced by an HFD and STZ treatment, which is manifested by reduced serum creatinine SCr) BUN, TNF-α, IL-6, and IL-1β levels. Histological examinations showed that geniposide could reduce glomerular basement membrane thickening and inflammatory cell infiltration. Geniposide also reversed the significant decrease in AMPK, p-AMPK, and SIRT1 levels in a podocyte model induced by high glucose. Geniposide effectively blocked oxidative stress and inflammation, thereby inhibiting DN development. The mechanism was suggested to involve the APMK/SIRT1/NF-κB pathway. Similarly, Chen et al. (120) reported that catalpol could effectively inhibit oxidative stress and inflammation in HFD/STZ-induced DN mice and in a high-glucose–induced podocyte model, and that the mechanism may be related to the AMPK/SIRT1/NF-κB pathway, indicating that catalpol has potential value in the treatment of DN.
With recent research on lncRNAs, exploration of the roles of non-coding RNAs in DN has intensified. Based on circRNA microarray analysis in glucose-stressed HK-2 cells, circACTR2 was found to regulate the pyroptosis and fibrosis of proximal renal tubular cells induced by high glucose. Knockout of circACTR2 significantly inhibited high-glucose–induced IL-1β release, and the production of collagen IV and fibronectin in HK-2 cells. By clarifying the role of circRNAs in the renal tubular cell pyroptosis-related inflammasome, new insights into the pathogenesis and treatment strategies of DKD may be attained (121). Li et al. (122) discovered that miR-23c, as a target of metastasis-associated lung adenocarcinoma tran 1 (MALAT1), directly inhibits the expression of embryonic lethal, abnormal vision, Drosophila-like 1 (ELAVL1), thereby reducing the expression of the downstream factors NLRP3, caspase-1, and IL-1β. In addition, silencing KCNQ1OT1 was shown to inhibit high-glucose–induced inflammation, oxidative stress, and pyroptosis in HK-2 cells by upregulating the expression of miR-506-3p (123). Considering that miR-452-5p is a potential target of GAS5, overexpression of GAS5 could downregulate the expression of mir-452-5p, thereby inhibiting NLRP3, caspase-1, IL-1β, and GSDMD-NT expression in high-glucose–induced HK-2 cells (Figure 2) (124).
Overall, the studies highlighted above found that upregulation of pyroptosis-related inflammatory factors was associated with DKD. Although angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin II receptor blockers (ARBs) is often used to treat DKD clinically, it cannot reverse the condition of DKD. Therefore, more and more molecular studies on DKD are being conducted to seek for better methods to treat DKD. These researches are summarized in Table 1. However, the evidence obtained to date is not sufficient to clarify whether there is a critical relationship between DKD and cell death, and the underlying mechanism of this association. Thus, the key discovery of the possible underlying mechanism linking inflammatory factors and pyroptosis in DKD will provide new insights to clarify and control the incidence and progression of DKD.

Table 1 Strategies to inhibit the inflammasome pathways associated with pyroptosis of diabetic kidney disease.
In this review, we have summarized the mechanism of pyroptosis, including the canonical, non-canonical, and caspase-3-mediated inflammasome pathways associated with pyroptosis. Research on the mechanism of pyroptosis regulation in DKD is rapidly emerging in the field of renal disease and immunology. The activation of diabetes-mediated related factors such as TLR4, NLRP3, caspase-1, IL-1β, IL-18, and GSDMD-NT plays a vital role in the pathophysiology of DKD. By inhibiting the activation of TLR4 or the NLRP3 inflammasome and related pathways, caspase-1, IL-1β, IL-18, and GSDMD-NT are inhibited, leading to renal lesions associated with DKD. The inflammasome is involved in the processes of activation, including the activation of caspase-1, which finally orders the terminal core protein GSDMD to perform its perforation effect that causes the release of IL-1B/IL-18. These are essential links in the process of pyroptosis. Recent studies have confirmed the relationship between the pyroptosis-related inflammasome and kidney disease. However, these studies are relatively superficial at present, they do not prove that pyroptosis is necessarily related to DKD and the mechanism needs further elucidation.
The detailed mechanism underlying the function of GSDMD in DKD in the downstream pathway of pyroptosis remains unclear. Small-molecule inhibitors targeting TLR4, NLRP3, and other inflammatory components are potential therapeutic options for DKD. However, there are still many unknown pathways and targets, and corresponding inhibitors, related to the occurrence and development of DKD related to pyroptosis awaiting further exploration. Studies using high-glucose–induced cell models, HFD/STZ-induced DKD animal models, and human DKD patient kidney tissue biopsies will help determine the spatial and temporal expression of DKD inflammatory components, and link these findings with the activity and prognosis of the disease. These insights may provide research ideas for developing new mechanisms, drugs, and technologies for DKD. Based on the current summary, we propose the following research targets.
First, according to molecular mechanisms related to inflammasomes that have been discovered to date, molecular biology methods could be used to further explore the specific mechanisms of caspase, GSDMD, IL-1β, and IL-18 that contribute to DKD, so as to clarify the pathways underlying the role and relationship of pyroptosis-related inflammasomes in DKD.
Second, based on the known signal activation of ROS, TLR4, NLRP3, and lncRNAs, efforts should be made to discover more small molecules or targeted drugs that can regulate the pathways of DKD-related inflammasomes, thereby bringing new methods and hope for the treatment of DKD.
Finally, more attention should be paid to the pathophysiology of DKD, and to understand the possible potential pathways of the pyroptosis-related inflammasome, which can offer new methods and technologies for the clinical treatment of DKD.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the Sichuan Provincial Health and Family Planning Commission (Project No. 18PJ346).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature (2012) 481:278–86. doi: 10.1038/nature10759
2. Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, et al. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science (1999) 284:156–9. doi: 10.1126/science.284.5411.156
3. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature (2015) 517:311–20. doi: 10.1038/nature14191
4. Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science (2016) 352:aaf2154. doi: 10.1126/science.aaf2154
5. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469:323–35. doi: 10.1038/nature09782
6. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol (2001) 9:113–4. doi: 10.1016/S0966-842X(00)01936-3
7. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev (2017) 277:61–75. doi: 10.1111/imr.12534
8. Wang J, Sahoo M, Lantier L, Warawa J, Cordero H, Deobald K, et al. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog (2018) 14:e1007105. doi: 10.1371/journal.ppat.1007105
9. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol (2010) 11:1136–42. doi: 10.1038/ni.1960
10. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 protects against bacteria that escape the vacuole. Science (2013) 339:975–8. doi: 10.1126/science.1230751
11. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med (2016) 213:2113–28. doi: 10.1084/jem.20151613
12. Aglietti RA, Dueber EC. Recent Insights into the Molecular Mechanisms Underlying Pyroptosis and Gasdermin Family Functions. Trends Immunol (2017) 38:261–71. doi: 10.1016/j.it.2017.01.003
13. Aziz M, Jacob A, Yang WL, Matsuda A, Wang P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J Leukoc Biol (2013) 93:329–42. doi: 10.1189/jlb.0912437
14. Gaul S, Leszczynska A, Alegre F, Kaufmann B, Johnson CD, Adams LA, et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol (2020) 1:156–67. doi: 10.1016/j.jhep.2020.07.041
15. Wu X, Zhang H, Qi W, Zhang Y, Li J, Li Z, et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis (2018) 9:171. doi: 10.1038/s41419-017-0257-3
16. Qiu Z, Lei S, Zhao B, Wu Y, Su W, Liu M, et al. NLRP3 Inflammasome Activation-Mediated Pyroptosis Aggravates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats. Oxid Med Cell Longev (2017) 2017:9743280. doi: 10.1155/2017/9743280
17. Greaney AJ, Maier NK, Leppla SH, Moayeri M. Sulforaphane inhibits multiple inflammasomes through an Nrf2-independent mechanism. J Leukoc Biol (2016) 99:189–99. doi: 10.1189/jlb.3A0415-155RR
18. Tan CC, Zhang JG, Tan MS, Chen H, Meng DW, Jiang T, et al. NLRP1 inflammasome is activated in patients with medial temporal lobe epilepsy and contributes to neuronal pyroptosis in amygdala kindling-induced rat model. J Neuroinflamm (2015) 12:18. doi: 10.1186/s12974-014-0233-0
19. Fan JX, Deng RH, Wang H, Liu XH, Wang XN, Qin R, et al. Epigenetics-Based Tumor Cells Pyroptosis for Enhancing the Immunological Effect of Chemotherapeutic Nanocarriers. Nano Lett (2019) 19:8049–58. doi: 10.1021/acs.nanolett.9b03245
20. Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: A new frontier in cancer. BioMed Pharmacother (2020) 121:109595. doi: 10.1016/j.biopha.2019.109595
21. WHO. Diabetes. (2020). https://www.who.int/news-room/fact-sheets/detail/diabetes.
22. Yang Z, Feng L, Huang Y, Xia N. A Differential Diagnosis Model For Diabetic Nephropathy And Non-Diabetic Renal Disease In Patients With Type 2 Diabetes Complicated With Chronic Kidney Disease. Diabetes Metab Syndr Obes (2019) 12:1963–72. doi: 10.2147/DMSO.S223144
23. Liu S, Guo Q, Han H, Cui P, Liu X, Miao L, et al. Clinicopathological characteristics of non-diabetic renal disease in patients with type 2 diabetes mellitus in a northeastern Chinese medical center: a retrospective analysis of 273 cases. Int Urol Nephrol (2016) 48:1691–8. doi: 10.1007/s11255-016-1331-y
24. Fernandez-Real JM, Pickup JC. Innate immunity, insulin resistance and type 2 diabetes. Diabetologia (2012) 55:273–8. doi: 10.1007/s00125-011-2387-y
25. Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ (2015) 22:526–39. doi: 10.1038/cdd.2014.216
26. Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem (1999) 68:383–424. doi: 10.1146/annurev.biochem.68.1.383
27. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
28. Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature (1997) 386:619–23. doi: 10.1038/386619a0
29. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514:187–92. doi: 10.1038/nature13683
30. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
31. Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci USA (2018) 115:E10888–97. doi: 10.1073/pnas.1809548115
32. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science (2018) 362:1064–9. doi: 10.1126/science.aau2818
33. Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J (2019) 38:e101638. doi: 10.15252/embj.2019101638
34. Newton K, Wickliffe KE, Maltzman A, Dugger DL, Reja R, Zhang Y, et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature (2019) 575:679–82. doi: 10.1038/s41586-019-1752-8
35. Fritsch M, Gunther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature (2019) 575:683–7. doi: 10.1038/s41586-019-1770-6
36. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu Rev Immunol (2020) 38:567–95. doi: 10.1146/annurev-immunol-073119-095439
37. Fujii T, Tamura M, Tanaka S, Kato Y, Yamamoto H, Mizushina Y, et al. (Gsdmd) is dispensable for mouse intestinal epithelium development. Genesis (2008) 46:418–23. doi: 10.1002/dvg.20412
38. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
39. Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol (2017) 27:673–84. doi: 10.1016/j.tcb.2017.05.005
40. Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol (2020) 20:143–57. doi: 10.1038/s41577-019-0228-2
41. Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J (2016) 35:1766–78. doi: 10.15252/embj.201694696
42. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA (2016) 113:7858–63. doi: 10.1073/pnas.1607769113
43. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
44. Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol (2018) 48:584–92. doi: 10.1002/eji.201747404
45. de Torre-Minguela C, Barbera-Cremades M, Gomez AI, Martin-Sanchez F, Pelegrin P. Macrophage activation and polarization modify P2X7 receptor secretome influencing the inflammatory process. Sci Rep (2016) 6:22586. doi: 10.1038/srep22586
46. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526:666–71. doi: 10.1038/nature15541
47. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
48. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res (2015) 25:1285–98. doi: 10.1038/cr.2015.139
49. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity (2018) 48:35–44 e6. doi: 10.1016/j.immuni.2017.11.013
50. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535:111–6. doi: 10.1038/nature18590
51. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun (2017) 8:14128. doi: 10.1038/ncomms14128
52. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547:99–103. doi: 10.1038/nature22393
53. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature (2020) 579:415–20. doi: 10.1038/s41586-020-2071-9
54. Feng S, Fox D, Man SM. Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J Mol Biol (2018) 430:3068–80. doi: 10.1016/j.jmb.2018.07.002
55. Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science (2018) 362:956–60. doi: 10.1126/science.aar7607
56. Vietri M, Radulovic M, Stenmark H. The many functions of ESCRTs. Nat Rev Mol Cell Biol (2020) 21:25–42. doi: 10.1038/s41580-019-0177-4
57. Platnich JM, Muruve DA. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys (2019) 670:4–14. doi: 10.1016/j.abb.2019.02.008
58. De Nardo D. Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine (2015) 74:181–9. doi: 10.1016/j.cyto.2015.02.025
59. Vijay K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int Immunopharmacol (2018) 59:391–412. doi: 10.1016/j.intimp.2018.03.002
60. Lim KH, Staudt LM. Toll-like receptor signaling. Cold Spring Harb Perspect Biol (2013) 5:a011247. doi: 10.1101/cshperspect.a011247
61. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi: 10.1038/nri3452
62. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol (2006) 7:576–82. doi: 10.1038/ni1346
63. Yao Y, Qian Y. Expression regulation and function of NLRC5. Protein Cell (2013) 4:168–75. doi: 10.1007/s13238-012-2109-3
64. Yao Y, Wang Y, Chen F, Huang Y, Zhu S, Leng Q, et al. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res (2012) 22:836–47. doi: 10.1038/cr.2012.56
65. Guo H, Konig R, Deng M, Riess M, Mo J, Zhang L, et al. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell Host Microbe (2016) 19:515–28. doi: 10.1016/j.chom.2016.03.001
66. Lupfer C, Kanneganti TD. Unsolved Mysteries in NLR Biology. Front Immunol (2013) 4:285. doi: 10.3389/fimmu.2013.00285
67. Feng H, Lenarcic EM, Yamane D, Wauthier E, Mo J, Guo H, et al. NLRX1 promotes immediate IRF1-directed antiviral responses by limiting dsRNA-activated translational inhibition mediated by PKR. Nat Immunol (2017) 18:1299–309. doi: 10.1038/ni.3853
68. Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity (2014) 40:329–41. doi: 10.1016/j.immuni.2014.01.010
69. Wang S, Yuan YH, Chen NH, Wang HB. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int Immunopharmacol (2019) 67:458–64. doi: 10.1016/j.intimp.2018.12.019
70. Panchapakesan U, Pollock C. The role of toll-like receptors in diabetic kidney disease. Curr Opin Nephrol Hypertens (2018) 27:30–4. doi: 10.1097/MNH.0000000000000377
71. Wada J, Makino H. Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol (2016) 12:13–26. doi: 10.1038/nrneph.2015.175
72. Liang H, Liu Y. Gasdermins pore cell membrane to pyroptosis. Sci China Life Sci (2016) 59:1090–2. doi: 10.1007/s11427-016-0243-4
73. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol (2009) 7:99–109. doi: 10.1038/nrmicro2070
74. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479:117–21. doi: 10.1038/nature10558
75. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science (2013) 341:1250–3. doi: 10.1126/science.1240988
76. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341:1246–9. doi: 10.1126/science.1240248
77. de Carvalho RVH, Andrade WA, Lima-Junior DS, Dilucca M, de Oliveira CV, Wang K, et al. Leishmania Lipophosphoglycan Triggers Caspase-11 and the Non-canonical Activation of the NLRP3 Inflammasome. Cell Rep (2019) 26:429–437 e5. doi: 10.1016/j.celrep.2018.12.047
78. Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science (2016) 352:1232–6. doi: 10.1126/science.aaf3036
79. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol (2010) 11:621–32. doi: 10.1038/nrm2952
80. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ (2009) 16:3–11. doi: 10.1038/cdd.2008.150
81. Dinarello CA, Donath MY, Mandrup-Poulsen T. Role of IL-1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes (2010) 17:314–21. doi: 10.1097/MED.0b013e32833bf6dc
82. Molitch ME, DeFronzo RA, Franz MJ, Keane WF, Mogensen CE, Parving HH, et al. Nephropathy in diabetes. Diabetes Care (2004) 27(Suppl 1):S79–83. doi: 10.2337/diacare.27.2007.S79
83. Belur Nagaraj S, Pena MJ, Ju W, Heerspink HL, B.E.-D. consortium. Machine Learning based Early Prediction of End-stage Renal Disease in Patients with Diabetic Kidney Disease using Clinical Trials Data. Diabetes Obes Metab (2020) 12:2479–86. doi: 10.1111/dom.14178
84. Flyvbjerg A. The role of the complement system in diabetic nephropathy. Nat Rev Nephrol (2017) 13:311–8. doi: 10.1038/nrneph.2017.31
85. Gerritsen KG, Leeuwis JW, Koeners MP, Bakker SJ, van Oeveren W, Aten J, et al. Elevated Urinary Connective Tissue Growth Factor in Diabetic Nephropathy Is Caused by Local Production and Tubular Dysfunction. J Diabetes Res (2015) 2015:539787. doi: 10.1155/2015/539787
86. Zhang L, Long J, Jiang W, Shi Y, He X, Zhou Z, et al. Trends in Chronic Kidney Disease in China. N Engl J Med (2016) 375:905–6. doi: 10.1056/NEJMc1602469
87. Yang X. Design and optimization of crocetin loaded PLGA nanoparticles against diabetic nephropathy via suppression of inflammatory biomarkers: a formulation approach to preclinical study. Drug Deliv (2019) 26:849–59. doi: 10.1080/10717544.2019.1642417
88. Black LM, Lever JM, Agarwal A. Renal Inflammation and Fibrosis: A Double-edged Sword. J Histochem Cytochem (2019) 67:663–81. doi: 10.1369/0022155419852932
89. Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond) (2013) 124:139–52. doi: 10.1042/CS20120198
90. Perez-Gomez MV, Sanchez-Nino MD, Sanz AB, Zheng B, Martin-Cleary C, Ruiz-Ortega M, et al. Targeting inflammation in diabetic kidney disease: early clinical trials. Expert Opin Investig Drugs (2016) 25:1045–58. doi: 10.1080/13543784.2016.1196184
91. da Silva Cristino Cordeiro V, de Bem GF, da Costa CA, Santos IB, de Carvalho L, Ognibene DT, et al. Euterpe oleracea Mart. seed extract protects against renal injury in diabetic and spontaneously hypertensive rats: role of inflammation and oxidative stress. Eur J Nutr (2018) 57:817–32. doi: 10.1007/s00394-016-1371-1
92. Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol (2020) 21:30–41. doi: 10.1038/s41590-019-0548-1
93. Scarpioni R, Obici L. Renal involvement in autoinflammatory diseases and inflammasome-mediated chronic kidney damage. Clin Exp Rheumatol (2018) 36(Suppl 110):54–60.
94. Han Y, Xu X, Tang C, Gao P, Chen X, Xiong X, et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol (2018) 16:32–46. doi: 10.1016/j.redox.2018.02.013
95. Shahzad K, Bock F, Dong W, Wang H, Kopf S, Kohli S, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int (2015) 87:74–84. doi: 10.1038/ki.2014.271
96. Ding T, Wang S, Zhang X, Zai W, Fan J, Chen W, et al. Kidney protection effects of dihydroquercetin on diabetic nephropathy through suppressing ROS and NLRP3 inflammasome. Phytomedicine (2018) 41:45–53. doi: 10.1016/j.phymed.2018.01.026
97. Wu M, Han W, Song S, Du Y, Liu C, Chen N, et al. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol Cell Endocrinol (2018) 478:115–25. doi: 10.1016/j.mce.2018.08.002
98. Wang S, Li Y, Fan J, Zhang X, Luan J, Bian Q, et al. Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis (2017) 8:e2937. doi: 10.1038/cddis.2017.292
99. Ge C, Xu M, Qin Y, Gu T, Lou D, Li Q, et al. Fisetin supplementation prevents high fat diet-induced diabetic nephropathy by repressing insulin resistance and RIP3-regulated inflammation. Food Funct (2019) 10:2970–85. doi: 10.1039/C8FO01653D
100. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
101. Qiu YY, Tang LQ. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic nephropathy. Pharmacol Res (2016) 114:251–64. doi: 10.1016/j.phrs.2016.11.004
102. Ram C, Jha AK, Ghosh A, Gairola S, Syed AM, Murty US, et al. Targeting NLRP3 inflammasome as a promising approach for treatment of diabetic nephropathy: Preclinical evidences with therapeutic approaches. Eur J Pharmacol (2020) 885:173503. doi: 10.1016/j.ejphar.2020.173503
103. Nakamura A, Shikata K, Hiramatsu M, Nakatou T, Kitamura T, Wada J, et al. Serum interleukin-18 levels are associated with nephropathy and atherosclerosis in Japanese patients with type 2 diabetes. Diabetes Care (2005) 28:2890–5. doi: 10.2337/diacare.28.12.2890
104. Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med (2007) 356:1517–26. doi: 10.1056/NEJMoa065213
105. Wang C, Pan Y, Zhang QY, Wang FM, Kong LD. Quercetin and allopurinol ameliorate kidney injury in STZ-treated rats with regulation of renal NLRP3 inflammasome activation and lipid accumulation. PLoS One (2012) 7:e38285. doi: 10.1371/journal.pone.0038285
106. Samra YA, Said HS, Elsherbiny NM, Liou GI, El-Shishtawy MM, Eissa LA. Cepharanthine and Piperine ameliorate diabetic nephropathy in rats: role of NF-kappaB and NLRP3 inflammasome. Life Sci (2016) 157:187–99. doi: 10.1016/j.lfs.2016.06.002
107. Araki S, Haneda M, Koya D, Sugimoto T, Isshiki K, Chin-Kanasaki M, et al. Predictive impact of elevated serum level of IL-18 for early renal dysfunction in type 2 diabetes: an observational follow-up study. Diabetologia (2007) 50:867–73. doi: 10.1007/s00125-006-0586-8
108. Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet (2005) 365:1333–46. doi: 10.1016/S0140-6736(05)61032-X
109. Lin CF, Kuo YT, Chen TY, Chien CT. Quercetin-Rich Guava (Psidium guajava) Juice in Combination with Trehalose Reduces Autophagy, Apoptosis and Pyroptosis Formation in the Kidney and Pancreas of Type II Diabetic Rats. Molecules (2016) 21:334. doi: 10.3390/molecules21030334
110. An X, Zhang Y, Cao Y, Chen J, Qin H, Yang L. Punicalagin Protects Diabetic Nephropathy by Inhibiting Pyroptosis Based on TXNIP/NLRP3 Pathway. Nutrients (2020) 12(5):1516. doi: 10.3390/nu12051516
111. Rabadi MM, Lee HT. Adenosine receptors and renal ischaemia reperfusion injury. Acta Physiol (Oxf) (2015) 213:222–31. doi: 10.1111/apha.12402
112. Faulhaber-Walter R, Chen L, Oppermann M, Kim SM, Huang Y, Hiramatsu N, et al. Lack of A1 adenosine receptors augments diabetic hyperfiltration and glomerular injury. J Am Soc Nephrol (2008) 19:722–30. doi: 10.1681/ASN.2007060721
113. Tian D, Shi X, Zhao Y, Peng X, Zou L, Xu L, et al. The effect of A1 adenosine receptor in diabetic megalin loss with caspase-1/IL18 signaling. Diabetes Metab Syndr Obes (2019) 12:1583–96. doi: 10.2147/DMSO.S215531
114. Wei M, Li Z, Xiao L, Yang Z. Effects of ROS-relative NF-kappaB signaling on high glucose-induced TLR4 and MCP-1 expression in podocyte injury. Mol Immunol (2015) 68:261–71. doi: 10.1016/j.molimm.2015.09.002
115. Yang S, Zhang J, Wang S, Zhao X, Shi J. SOCS2 overexpression alleviates diabetic nephropathy in rats by inhibiting the TLR4/NF-kappaB pathway. Oncotarget (2017) 8:91185–98. doi: 10.18632/oncotarget.20434
116. Wang Y, Zhu X, Yuan S, Wen S, Liu X, Wang C, et al. TLR4/NF-kappaB Signaling Induces GSDMD-Related Pyroptosis in Tubular Cells in Diabetic Kidney Disease. Front Endocrinol (Lausanne) (2019) 10:603. doi: 10.3389/fendo.2019.00603
117. Ma J, Chadban SJ, Zhao CY, Chen X, Kwan T, Panchapakesan U, et al. TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PLoS One (2014) 9:e97985. doi: 10.1371/journal.pone.0097985
118. Jialal I, Major AM, Devaraj S. Global Toll-like receptor 4 knockout results in decreased renal inflammation, fibrosis and podocytopathy. J Diabetes Complications (2014) 28:755–61. doi: 10.1016/j.jdiacomp.2014.07.003
119. Li F, Chen Y, Li Y, Huang M, Zhao W. Geniposide alleviates diabetic nephropathy of mice through AMPK/SIRT1/NF-kappaB pathway. Eur J Pharmacol (2020) 886:173449. doi: 10.1016/j.ejphar.2020.173449
120. Chen J, Yang Y, Lv Z, Shu A, Du Q, Wang W, et al. Study on the inhibitive effect of Catalpol on diabetic nephropathy. Life Sci (2020) 257:118120. doi: 10.1016/j.lfs.2020.118120
121. Wen S, Li S, Li L, Fan Q. circACTR2: A Novel Mechanism Regulating High Glucose-Induced Fibrosis in Renal Tubular Cells via Pyroptosis. Biol Pharm Bull (2020) 43:558–64. doi: 10.1248/bpb.b19-00901
122. Li X, Zeng L, Cao C, Lu C, Lian W, Han J, et al. Long noncoding RNA MALAT1 regulates renal tubular epithelial pyroptosis by modulated miR-23c targeting of ELAVL1 in diabetic nephropathy. Exp Cell Res (2017) 350:327–35. doi: 10.1016/j.yexcr.2016.12.006
123. Zhu B, Cheng X, Jiang Y, Cheng M, Chen L, Bao J, et al. Silencing of KCNQ1OT1 Decreases Oxidative Stress and Pyroptosis of Renal Tubular Epithelial Cells. Diabetes Metab Syndr Obes (2020) 13:365–75. doi: 10.2147/DMSO.S225791
Keywords: pyroptosis-related, inflammasome pathway, pathogenesis, diabetic kidney disease, targeted inhibition
Citation: Liu P, Zhang Z and Li Y (2021) Relevance of the Pyroptosis-Related Inflammasome Pathway in the Pathogenesis of Diabetic Kidney Disease. Front. Immunol. 12:603416. doi: 10.3389/fimmu.2021.603416
Received: 06 September 2020; Accepted: 12 January 2021;
Published: 22 February 2021.
Edited by:
Zhiguang Zhou, Central South University, ChinaReviewed by:
Robert W. Keane, University of Miami, United StatesCopyright © 2021 Liu, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yao Li, eWFvbGlwaHNAdG9tLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.