
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 18 May 2021
Sec. Cytokines and Soluble Mediators in Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.601842
This article is part of the Research Topic New Antigen-Specific Strategies for Prophylaxis and Treatment of Immune-Mediated Diseases View all 9 articles
 Ekaterina O. Gubernatorova1,2*
Ekaterina O. Gubernatorova1,2* Olga A. Namakanova1,2
Olga A. Namakanova1,2 Ekaterina. A. Gorshkova2,3
Ekaterina. A. Gorshkova2,3 Alexandra D. Medvedovskaya1,2
Alexandra D. Medvedovskaya1,2 Sergei A. Nedospasov1,2,3,4
Sergei A. Nedospasov1,2,3,4 Marina S. Drutskaya1,3*
Marina S. Drutskaya1,3*Asthma is a heterogeneous inflammatory disease characterized by airflow obstruction, wheezing, eosinophilia and neutrophilia of the airways. Identification of distinct inflammatory patterns characterizing asthma endotypes led to the development of novel therapeutic approaches. Cytokine or cytokine receptor targeting by therapeutic antibodies, such as anti-IL-4 and anti-IL-5, is now approved for severe asthma treatment. However, the complexity of cytokine networks in asthma should not be underestimated. Inhibition of one pro-inflammatory cytokine may lead to perturbed expression of another pro-inflammatory cytokine. Without understanding of the underlying mechanisms and defining the molecular predictors it may be difficult to control cytokine release that accompanies certain disease manifestations. Accumulating evidence suggests that in some cases a combined pharmacological inhibition of pathogenic cytokines, such as simultaneous blockade of IL-4 and IL-13 signaling, or blockade of upstream cytokines, such as TSLP, are more effective than single cytokine targeting. IL-6 and TNF are the important inflammatory mediators in the pathogenesis of asthma. Preliminary data suggests that combined pharmacological inhibition of TNF and IL-6 during asthma may be more efficient as compared to individual neutralization of these cytokines. Here we summarize recent findings in the field of anti-cytokine therapy of asthma and discuss immunological mechanisms by which simultaneous targeting of multiple cytokines as opposed to targeting of a single cytokine may improve disease outcomes.
Asthma is a heterogeneous disease characterized by chronic airway inflammation affecting almost 20% of population in some countries (1). Asthma is now considered as an umbrella term for diagnosis of distinct mechanistic pathways and clinical manifestations (2). Typically, patients with asthma develop various respiratory symptoms: shortness of breath, cough and chest tightness, but underlying molecular events driving the pathogenesis, as well as disease severity, may vary significantly.
Severe asthma is defined as a condition when adequate control of exacerbations cannot be achieved by treatment with high-dose corticosteroids and/or by standard therapy or if symptoms flare up when the treatment is reduced (3, 4). At the cellular level the thickening of basal membrane of respiratory epithelium and high granulocyte counts in sputum represent hallmarks of severe asthma (5). The disease progression is triggered by the activation of mucosal innate immune system. Resident antigen-presenting cells become activated in response to allergens and epithelial alarmins that are released due to the epithelial barrier disruption (6). The downstream mechanisms include activation of Th2/mast cell/eosinophil-mediated pathology, Th1/Th17/neutrophil-mediated pathology, chronic innate immune responses and irreversible airway obstruction. Studies in mice revealed that cytokines play an exceptional role in orchestrating each stage of severe asthma progression (Figure 1) (7). Th17- and Th2-mediated inflammatory pathways are regulated reciprocally during asthma (8), thus, inhibition of type 2 cytokines, such as IL-4 or IL-5, or administration of corticosteroids leads to reorganization of immune response from Th2-mediated eosinophilic to Th17-mediated neutrophilic disease (9, 10). The earliest signature cytokines of the epithelial damage, IL-33, TSLP (thymic stromal lymphopoietin) and IL-25, promote activation and migration of APCs (antigen-presenting cells), which, in turn, produce pro-inflammatory mediators with a broad spectrum of functions (ex. IL-6 and TNF). During the maturation of adaptive immunity, IL-4, IL-5, IL-13 and eotaxin are indispensable for the type 2 immune responses and are associated with differentiation of Th2 and ILC2 (type 2 innate lymphoid cell) subsets as well as with eosinophil migration (11). IL-17 and IL-6 are known regulators of Th17 immune responses and neutrophilia (12). Macrophage-expressed CXCL8 (chemokine ligand 8) fuels neutrophil accumulation and oxidative damage mediated by these cells (9). TGFβ (transforming growth factor beta), IL-22, IL-6 and TNF also contribute to tissue remodeling and fibrosis that are associated with the late stages of asthma progression (13–16). Repeated cycles of epithelial injury and inadequate immune activation lead to chronic inflammation in the lungs.
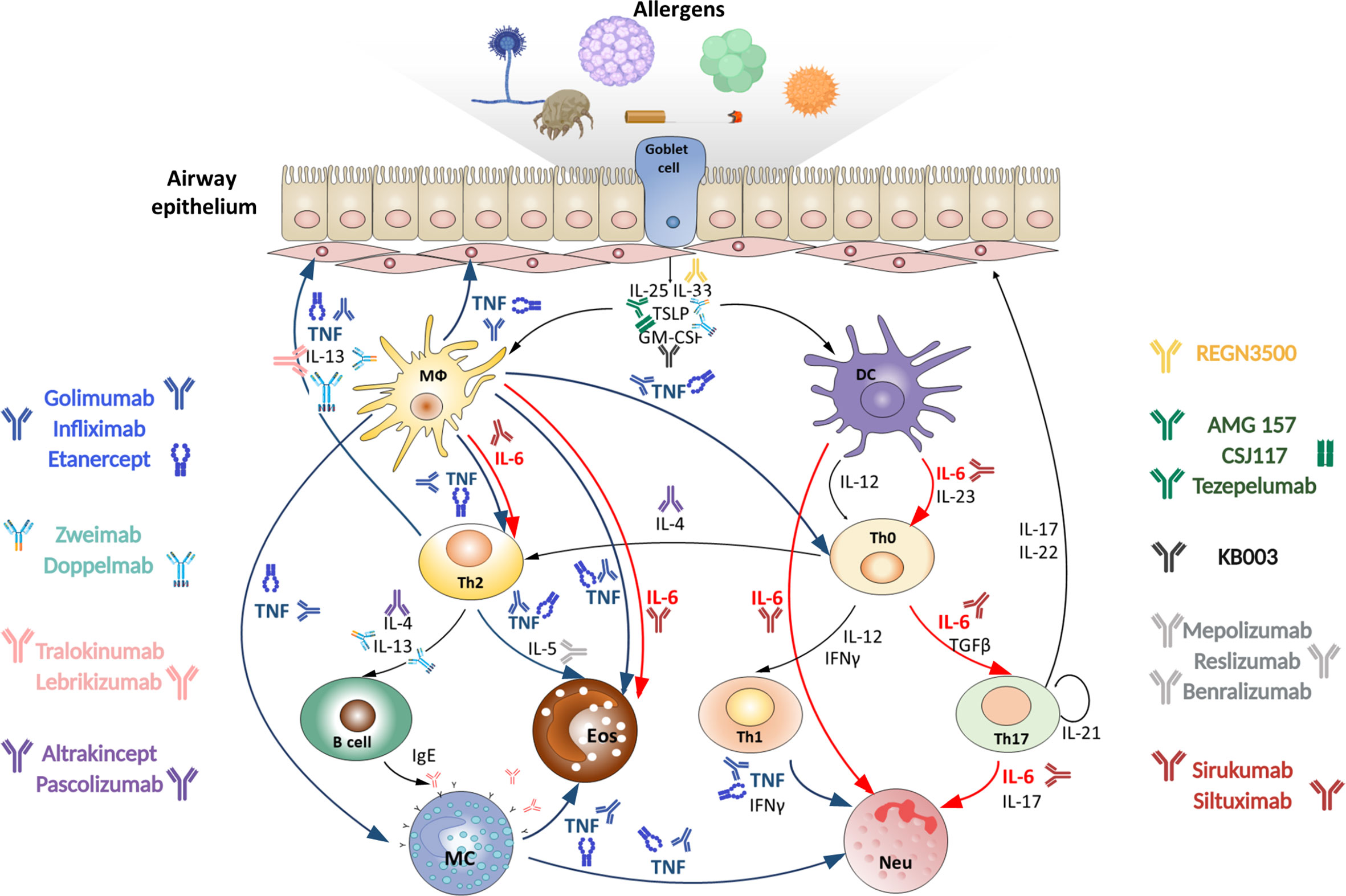
Figure 1 Targeting TNF, IL-6 and other cytokines in acute severe asthma. Severe asthma pathogenesis: epithelial cells exposed to pro‐inflammatory and allergic stimuli release mediators such as TSLP, IL-33 and IL-25, which activate dendritic cells. Within the airway lumen allergens can be captured by dendritic cells, which process antigenic molecules and present them to naïve (Th0) T helper cells. The consequent activation of allergen-specific Th2 cells is responsible for the production of IL-4 and IL-13 that promote B-cell operated synthesis of IgE antibodies and IL-5, which induces eosinophil maturation and survival. Eosinophils activated by IL-5 contribute to oxidative stress. On the other hand, in the presence of IL-23 and IL-6 upon dendritic cell activation Th0 cells differentiate into Th17 cells or into Th1 cells in the presence of IL-12. Th1 and Th17 cells stimulate and induce neutrophilic inflammation, characteristic of severe asthma. Combined pharmacological inhibition of TNF and IL-6 in acute allergic asthma may more effectively reduce the severity of inflammatory response in the lungs as compared to inhibition of these cytokines individually, since it blocks multiple pathogenic mechanisms. MФ, macrophage; DC, dendritic cell; Th, T helper; MC, mast cell; Eos, eosinophil; Neu, neutrophil.
Irreversible bronchoalveolar tissue remodeling in severe asthma patients causes a decrease in respiratory functions and may lead to live-threatening consequences. Сorticosteroids are ineffective in severe asthma treatment, furthermore, they can prevent apoptosis of neutrophils and promote extracellular matrix production, thereby, exacerbating airway remodeling (17). Accumulating evidence suggests that pro-inflammatory cytokines and their receptors may represent promising targets for monoclonal antibody-based therapy. However, pathological mechanisms typically involve multiple cytokines with partly overlapping functions. The idea of combined anti-cytokine inhibition in severe asthma is currently drawing considerable attention. In the next section, we review the most promising clinical studies in this area and provide some perspective for this therapeutic approach.
Airway epithelial cells represent the first line of defense on the host’s mucosal surfaces. The interaction between airway epithelium and the environment is crucial for asthma development. Allergen-induced epithelial damage causes the release of epithelial alarmins - endogenous, constitutively expressed, chemotactic and immune activating proteins (18–20). TSLP, IL-33, GM-CSF (granulocyte-macrophage colony-stimulating factor) and IL-25 are the key airway epithelium-derived cytokines, acting like alarmins, which are released in response to the loss of epithelial cell integrity induced by allergens, proteases or viruses.
Release of TSLP, IL-33, IL-25 and GM-CSF is obviously not restricted to allergy; however, these cytokines mostly contribute to type 2 immune response through specific activation of dendritic cells. In particular, GM-CSF (21) and TSLP (22) overexpression in the lungs may induce spontaneous Th2 response to inhaled ovalbumin. IL-33 was shown to drives IL-1β-dependent Th2 inflammation in mice sensitized and challenged intranasally with ovalbumin and chitin (23), while IL-25 enhances eosinophil recruitment to the airways as well as goblet cell hyperplasia (24). Since epithelial alarmins constitute the first wave of signaling molecules released in response to allergen exposure, the idea of using them as potential therapeutic targets appears attractive.
The efficacy of anti-TSLP, anti-IL-33 and anti-IL-25 as monotherapies for treatment of allergic diseases was addressed in a number of studies. For example, in a mouse model of ovalbumin-induced asthma administration of anti-IL-33 antibodies was shown to decrease eosinophil infiltration, IgE production and Th2 cytokine release (25), as well as airway hyperreactivity (AHR) (26). Specific targeting ST2 (IL1RL1, Interleukin 1 receptor-like 1), an IL-33 receptor, demonstrated similar effects in ovalbumin-challenged mice (27). In a mouse model of persistent house dust mite (HDM)-induced asthma characterized by mixed granulocytic influx in the lungs the anti-IL-33 treatment was shown to prevent airway remodeling (28). Importantly, phase 2a randomized placebo-controlled study of anti-IL-33 in peanut-induced allergy suggests that a single dose of Etokimab, IL-33 blocker, could reduce atopy-related adverse effecs. Therefore, IL-33 targeting is a promising therapeutic strategy for allergic asthma (29) (Table 1).
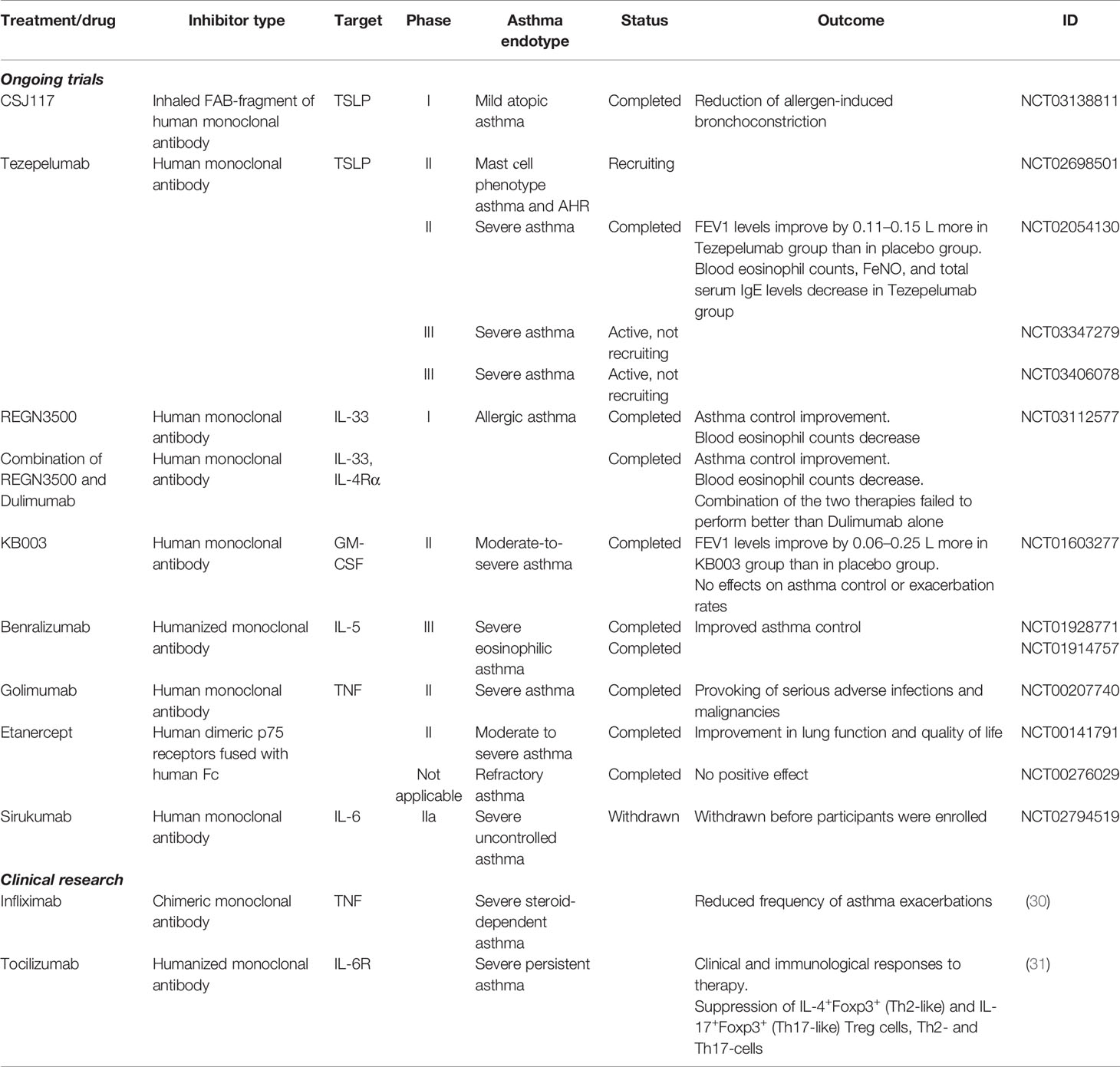
Table 1 Ongoing clinical trials and clinical research of anti-cytokine therapeutic inhibitors.
TSLP is a master-regulator of type 2 response at mucosal surfaces. In a mouse model of chronic HDM-induced allergic asthma (32) and in an ovalbumin-induced asthma both in rats (33) and in mice (34) neutralization of TSLP with monoclonal antibodies decreased Th2 cytokine levels and prevented structural alterations in the airways. Importantly, the efficacy of fully human anti-TSLP monoclonal antibody, AMG 157, was assessed in a small group of patients with mild atopic asthma (35). This double-blind placebo-controlled study revealed that three monthly doses of AMG 157 could reduce both allergen-induced bronchoconstriction and indexes of airway inflammation. Moreover, in a phase 2 trial addition of Tezepelumab, a TSLP blocking agent, resulted in attenuated asthma exacerbation rates in severely affected patients with uncontrolled asthma (36). Currently, two phase 3 trials for Tezepelumab are underway: the NCT03347279 trial will evaluate Tazepelumab in patients with uncontrolled asthma and the NCT03406078 trial will examine whether Tezepelumab treatment can reduce the required daily doses of oral corticosteroids in patients with severe asthma (Table 1). Finally, the development of inhalable high-affinity version of anti-TSLP blocking antibody (37) and the evaluation of its effectiveness (38) is under investigation.
IL-25, or IL-17E, is a member of the IL-17 family of cytokines, regulating type 2 immunity (39, 40). Administration of anti-IL-25 antibodies in mice with ovalbumin- (41) or HDM- and adenoviral smad2–overexpression-induced asthma (42) significantly decreased the Th2 immune responses and attenuated AHR and airway tissue remodeling. Clinical trials of anti-IL-25 efficacy for asthma treatment are yet to be conducted.
Targeting of alarmins proved beneficial as an add-on therapy or in combination with other cytokine inhibitors. Blockade of TSLP together with CRTH2, a chemoattractant receptor homologue expressed on Th2 memory cells, attenuated eosinophilic inflammation and AHR in ovalbumin-challenged mice (43). In line with this, bispecific anti-TSLP, anti-IL-13 reagent was recently developed in a form of monovalent antibody called Zweimab, and bivalent bispecific antibody scaffold – Doppelmab (44). However, further in vivo studies are required to evaluate their efficacy in asthma models.
Despite the substantial role of IL-33 in driving Th2-mediated responses, ST2 deficient mice are not resistant to allergic asthma (45), suggesting that inhibition of IL-33 alone may not be sufficient for preventing asthma development. The detailed analysis of allergic responses in ST2 knockout mice revealed the compensatory increase of TSLP production in response to allergen challenge (46). To test whether anti-IL-33 or anti-TSLP antibodies may attenuate inflammation, RAG1-/- (recombination activating gene 1 knockout) mice were intravenously sensitized by adoptively transferred ILC2 cells from immunocompetent mice and then intranasally challenged with eosinophil extracellular traps. It was found that anti-IL-33, but not anti-TSLP, reduced IL-5 and IL-13 production, while AHR was decreased only in anti-TSLP treated mice (47). The effect of broad neutralization of alarmin-mediated signaling in asthma was further evaluated in ST2-/- mice. Administration of anti-IL-25 and anti-TSLP antibodies during ovalbumin-induced asthma could reduce the infiltration of inflammatory cells into airways, as well as local expression of IL-4, IL-5 and IL-13. Moreover, attenuated airway tissue remodeling and histopathological features related to inflammation were observed under these experimental conditions (48). Interestingly, in a mouse model of influenza-induced exacerbation of allergic asthma only IL-33-specific neutralization resulted in improved AHR, prevented body weight loss and accumulation of inflammatory cells in the lungs, whereas combined administration of anti-TSLP and anti-IL-33 did not provide any additional benefits as compared to anti-IL-33 monotherapy (49). Nevertheless, taking into account the impressive results with anti-TSLP therapy in severe asthma, one may expect that in such case combined anti-IL-33 and anti-TSLP therapy could be even more beneficial. Taken together, these findings suggest that simultaneous targeting of multiple alarmins may provide synergistic therapeutic effects in asthma treatment.
After the initial steps in allergic response and alarmin release, the effector cytokines typically come into play. IL-4, IL-5 and IL-13 are the central Th2 cytokines responsible for eosinophilic inflammation in asthma. IL-5 is required for the maturation and release of eosinophils from the bone marrow and for their tissue accumulation and activation, and, therefore, represents a relevant target for treatment of eosinophilic inflammation. Up to date, two anti-IL-5 antibodies Mepolizumab (50) and Reslizumab (51) are approved for severe eosinophilic asthma, and the third antibody, Benralizumab, has passed phase 3 trials. Post hoc analysis of pooled phase III SIROCCO (NCT01928771) and CALIMA (NCT01914757) data for patients with severe eosinophilic asthma confirmed that Benralizumab improved asthma control (52). Taking together, anti-IL-5 therapy is a valuable treatment option for patients with severe eosinophilic asthma.
Both IL-4 and IL-13 are Th2 cytokines, the aberrant production of which has long been associated with allergic disorders. IL-4 and IL-13 are encoded by adjacent genes (located on chr 5q in humans and chr11 in mice) that share a common regulatory element (GATA-3) and can transmit signals through a shared functional receptor complex IL-4Rα/IL-13Rα1, thus, not surprisingly, these two cytokines have many common functions (53). However, some of their activities appear to be non-redundant. For example, IL-4 predominantly contributes to eosinophilia and Th2 activation (53) but has no impact on airway remodeling and AHR, while IL-13 upregulation is sufficient to induce mucus production, bronchoconstriction and AHR (54). Neutralizing these two cytokines by a monoclonal antibody targeting their common receptor, represents the most impressive example of a multiple cytokine inhibition in asthma. While both anti-IL-13 antibodies, Tralokinumab (55, 56) and Lebrikizumab (57) and both anti-IL-4 antibodies, Altrakincept (58) and Pascolizumab (59), failed to provide beneficial effects for patients with asthma, blocking signal transduction through IL-4- and IL-13-shared receptor complex with Dupilumab, a fully human monoclonal antibody against IL-4Rα, is efficient and is now approved for treatment of severe asthma patients (60–62).
Serum TNF represents an important biomarker for severe asthma (63). TNF-TNFR1 signaling in airway smooth muscle cells causes AHR (64, 65), supports chronic inflammation, as well as lymphocyte and granulocyte infiltration in the lungs (66, 67). Proliferation and transdifferentiation of fibroblasts are also dependent on TNF-induced expression of TGFβ (68). Therefore, blocking this cytokine in severe asthma initially appeared promising, and administration of anti-TNF reagents demonstrated encouraging results in animal asthma models. For example, in acute model of asthma, administration of monoclonal anti-TNF antibodies was associated with reduced inflammatory cell infiltration, airway goblet cell metaplasia and AHR (69, 70).
Anti-TNF biologics as therapeutics for severe asthma were tested in a number of clinical studies. However, Golimumab, a human monoclonal antibody, binding both soluble and transmembrane forms of TNF failed to demonstrate therapeutic effects in severe asthma; furthermore, patients experienced serious side effects (71). Systemic anti-TNF therapy is known to be associated with increased risk of infections and neoplasms (72, 73), and may affect the integrity of granulomas, which control Mycobacterium tuberculosis (74). Moreover, TNF blockade may lead to accumulation of Th17 cells and high levels of IL-17A production (75). On the other hand, Etanercept, a recombinant fusion protein based on TNF receptor 2, significantly improved lung function in patients with severe asthma and high TNF levels in the sputum (76, 77). In line with this, Infliximab, a chimeric monoclonal antibody, significantly decreased asthma exacerbations rate in patients with moderate and corticosteroid-resistant refractory asthma (30, 76, 78). Taken together, these findings suggest that TNF inhibitors remain promising therapeutic reagents for severe asthma, but inhibition of TNF alone may not be sufficient to fully control the disease pathology and also may lead to serious side effects, which are mediated by overexpression of other proinflammatory cytokines.
IL-6 is a pleiotropic cytokine involved both in regulation and induction of chronic inflammation (79, 80) with a wide range of cellular sources and complex receptor system (80–83). IL-6 determines the direction of CD4+ T cell differentiation (84, 85), suppresses the Treg cells, attracts myeloid cells to the site of inflammation and promotes fibrosis (86) in the absence of IL-13.
Increased levels of IL-6 were detected in serum, sputum, and bronchoalveolar lavage fluid (BALF) of asthmatic patients and correlated with disease severity (87, 88). The role of IL-6 from different cellular sources in the respiratory allergic pathology is being actively investigated. It was shown that IL-6 may shift differentiation of macrophages towards alternatively activated macrophages (M2), which play a key role in eosinophilic inflammation in HDM-induced asthma (89). Moreover, mice with IL-6 deficiency in macrophages showed significantly decreased eosinophilic inflammation in the lungs and reduced production of Th2-associated cytokines and IgE (89, 90), while IL-6 deficiency in dendritic cells resulted in decreased Th17-, but not Th2-inflammatory response (90). Thus, IL-6 produced by dendritic cells may contribute to the development of severe neutrophilic asthma. Taken together, IL-6 is implicated in progression, severity and duration of asthma and should be considered as a target for anti-cytokine asthma therapy.
Selective blockade of distinct modes of IL-6 signaling has different outcomes (91, 92). The IL-6 receptor complex includes IL-6R and gp130. The classical pathway is initiated by the interaction of IL-6 to membrane-bound IL-6R, then gp130, localized on the surface of the same cell, joins the complex. The trans-signaling pathway is mediated by a soluble IL-6R (sIL-6R), which may form as the result of alternative splicing or proteolytic shedding. Membrane bound IL-6R is present on the surface of a limited number of cell types, including leukocytes, whereas expression of gp130 is almost ubiquitous providing sensitivity to IL-6 to a broad range of cells upon IL-6/sIL6-R interaction (91). This type of signaling has pathogenic effects in several severe asthma endotypes (93). Both anti-IL-6 antibodies (Sirukumab, Siltuximab) and anti–IL-6R antibodies (Tocilizumab, Sarilumab) are able to block classical and trans-signaling pathways. The newly described third mode of IL-6 signaling – so called trans-presentation - which is involved in formation of pathogenic Th17 cells in EAE (experimental autoimmune encephalomyelitis) mouse model (94) can be inhibited by IL-6R blockers only (91), although the significance of that type of signal transduction in asthma pathogenesis is not yet established.
A clinical trial of Sirukumab, a human monoclonal antibody against IL-6, in severe asthma was withdrawn after FDA had disapproved Sirukumab (NCT02794519) for treatment of rheumatoid arthritis, due to increased number of deaths and malignancies among patients (95). On the contrary, Tocilizumab, a humanized anti-IL-6R monoclonal antibody, is being successfully used for treatment of chronic inflammatory diseases such as rheumatoid arthritis, systemic juvenile idiopathic arthritis and Castleman’s disease (96, 97). Recent case report study on severe pediatric persistent asthma showed clinical and immunological responses to Tocilizumab as an additional therapy without any adverse effects (31). Within 10 months on anti-IL-6R therapy patients showed a decrease in Th2 and Th17 cellular responses, however, peripheral eosinophilia in these patients was not impacted (31). Another clinical study in mild allergic asthma found no evidence that Tocilizumab is able to prevent allergen‐induced bronchoconstriction (98).
Thus, TNF neutralization may have a marked potential to improve lung functions, but demonstrates serious side effects, while blocking of IL-6 does not provide significant therapeutic effect in severe asthma. Interestingly, the simultaneous administration of both inhibitors prevented the increase of eosinophilic and neutrophilic infiltrate in BAL fluid in mouse model of acute HDM-induced asthma (unpublished data). Thus, our preliminary results indicate that combined pharmacological inhibition of TNF and IL-6 in acute allergic asthma may be more effective in reducing the severity of the inflammatory response in the lungs as compared to inhibition of these cytokines individually. More studies are needed to define minimally required doses of the two biologics and to address the issue of possible adverse effects under these conditions.
Effective and safe therapy of severe asthma remains an unresolved problem for modern medicine. Severe eosinophilic asthma is manageable due to FDA approved IL-4Rα blocker – Dupilumab, which affects the two main cytokines of Th2 immune response (IL-4 and IL-13) simultaneously (99). In addition, recent studies in mice revealed therapeutic potential of combined blockade of early markers of epithelial barrier damage – IL-33 and TSLP (44). The strategy for combined anti-cytokine therapy is aimed to overcome functional redundancy of pro-inflammatory cytokines. In this review we discussed the perspective of using combination of blockers to reduce both redundancy and the side effects of a monotherapy. IL-6 and TNF are key pro-inflammatory cytokines implicated in neutrophilia, extracellular matrix production and chronic inflammation during severe asthma. In spite of possible side effects, TNF neutralization is a golden standard in treating rheumatoid arthritis, psoriasis and inflammatory bowel disease therapy (100). Anti-IL-6 drugs are also approved in RA (101). In mice HDM-induced severe asthma a combination of TNF and IL-6 inhibitors prevented IL-6-dependent eosinophilia as IL-6 neutralization helped to suppress the pathological side effects associated with systemic TNF neutralization. Multiple cytokine targeting has demonstrated its efficacy in severe eosinophilic asthma. We propose that similar approach may help to find long-awaited treatment for severe neutrophilic asthma.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
EGu, ON, EGo, AM, MD, and SN discussed the concept and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by RSF grant №19-75-30032.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention (2020). Available at: www.ginasthma.org.
2. Kuruvilla ME, Lee FE, Lee GB. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin Rev Allergy Immunol (2019) 56:219–33. doi: 10.1007/s12016-018-8712-1
3. Lommatzsch M, Virchow JC. Severe Asthma: Definition, Diagnosis and Treatment. Dtsch Arztebl Int (2014) 111:847–55. doi: 10.3238/arztebl.2014.0847
4. Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, Sterk PJ, et al. International ERS/ATS Guidelines on Definition, Evaluation and Treatment of Severe Asthma. Eur Respir J (2014) 43:343–73. doi: 10.1183/09031936.00202013
5. Kim S, Lee CH, Jin KN, Cho SH, Kang HR. Severe Asthma Phenotypes Classified by Site of Airway Involvement and Remodeling Via Chest CT Scan. J Investig Allergol Clin Immunol (2018) 28:312–20. doi: 10.18176/jiaci.0265
6. Wang W, Li Y, Lv Z, Chen Y, Li Y, Huang K, et al. Bronchial Allergen Challenge of Patients With Atopic Asthma Triggers an Alarmin (IL-33, TSLP, and IL-25) Response in the Airways Epithelium and Submucosa. J Immunol (2018) 201:2221–31. doi: 10.4049/jimmunol.1800709
7. Gubernatorova EO, Namakanova OA, Tumanov AV, Drutskaya MS, Nedospasov SA. Mouse Models of Severe Asthma for Evaluation of Therapeutic Cytokine Targeting. Immunol Lett (2019) 207:73–83. doi: 10.1016/j.imlet.2018.11.012
8. Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, et al. TH2 and TH17 Inflammatory Pathways are Reciprocally Regulated in Asthma. Sci Trans Med (2015) 7:301ra129. doi: 10.1126/scitranslmed.aab3142
9. Massoud AH, Charbonnier LM, Lopez D, Pellegrini M, Phipatanakul W, Chatila TA. An Asthma-Associated IL4R Variant Exacerbates Airway Inflammation by Promoting Conversion of Regulatory T Cells to TH17-like Cells. Nat Med (2016) 22:1013–22. doi: 10.1038/nm.4147
10. Menson KE, Mank MM, Reed LF, Walton CJ, Van Der Vliet KE, Ather JL, et al. Therapeutic Efficacy of IL-17A Neutralization With Corticosteroid Treatment in a Model of Antigen-Driven Mixed-Granulocytic Asthma. Am J Physiol Lung Cell Mol Physiol (2020) 319(4):L693–709. doi: 10.1152/ajplung.00204.2020
11. Beckert H, Meyer-Martin H, Buhl R, Taube C, Reuter S. Single and Synergistic Effects of Type 2 Cytokines on Eosinophils and Asthma Hallmarks. J Immunol (2020) 204:550–8. doi: 10.4049/jimmunol.1901116
12. Halwani R, Sultana A, Vazquez-Tello A, Jamhawi A, Al-Masri AA, Al-Muhsen S. Th-17 Regulatory Cytokines IL-21, IL-23, and IL-6 Enhance Neutrophil Production of IL-17 Cytokines During Asthma. J Asthma (2017) 54:893–904. doi: 10.1080/02770903.2017.1283696
13. Zhang Y, Tang H, Yuan X, Ran Q, Wang X, Song Q, et al. TGF-beta3 Promotes MUC5AC Hyper-Expression by Modulating Autophagy Pathway in Airway Epithelium. EBioMedicine (2018) 33:242–52. doi: 10.1016/j.ebiom.2018.06.032
14. Johnson JR, Nishioka M, Chakir J, Risse PA, Almaghlouth I, Bazarbashi AN, et al. IL-22 Contributes to TGF-beta1-mediated Epithelial-Mesenchymal Transition in Asthmatic Bronchial Epithelial Cells. Respir Res (2013) 14:118. doi: 10.1186/1465-9921-14-118
15. Jude JA, Dileepan M, Subramanian S, Solway J, Panettieri RA Jr., Walseth TF, et al. miR-140-3p Regulation of TNF-alpha-induced CD38 Expression in Human Airway Smooth Muscle Cells. Am J Physiol Lung Cell Mol Physiol (2012) 303:L460–8. doi: 10.1152/ajplung.00041.2012
16. Wu Y, Zou F, Lu Y, Li X, Li F, Feng X, et al. SETD7 Promotes TNF-alpha-induced Proliferation and Migration of Airway Smooth Muscle Cells In Vitro Through Enhancing NF-kappaB/CD38 Signaling. Int Immunopharmacol (2019) 72:459–66. doi: 10.1016/j.intimp.2019.04.043
17. Wang M, Gao P, Wu X, Chen Y, Feng Y, Yang Q, et al. Impaired Anti-Inflammatory Action of Glucocorticoid in Neutrophil From Patients With Steroid-Resistant Asthma. Respir Res (2016) 17:153. doi: 10.1186/s12931-016-0462-0
18. Roan F, Obata-Ninomiya K, Ziegler SF. Epithelial Cell-Derived Cytokines: More Than Just Signaling the Alarm. J Clin Invest (2019) 129:1441–51. doi: 10.1172/JCI124606
19. Hammad H, Lambrecht BN. Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity (2015) 43:29–40. doi: 10.1016/j.immuni.2015.07.007
20. Porsbjerg CM, Sverrild A, Lloyd CM, Menzies-Gow AN, Bel EH. Anti-Alarmins in Asthma: Targeting the Airway Epithelium With Next-Generation Biologics. Eur Respir J (2020) 56:2000260. doi: 10.1183/13993003.00260-2020
21. Stampfli MR, Wiley RE, Neigh GS, Gajewska BU, Lei XF, Snider DP, et al. GM-CSF Transgene Expression in the Airway Allows Aerosolized Ovalbumin to Induce Allergic Sensitization in Mice. J Clin Invest (1998) 102:1704–14. doi: 10.1172/JCI4160
22. Zhou B, Comeau MR, De Smedt T, Liggitt HD, Dahl ME, Lewis DB, et al. Thymic Stromal Lymphopoietin as a Key Initiator of Allergic Airway Inflammation in Mice. Nat Immunol (2005) 6:1047–53. doi: 10.1038/ni1247
23. Arae K, Morita H, Unno H, Motomura K, Toyama S, Okada N, et al. Chitin Promotes Antigen-Specific Th2 Cell-Mediated Murine Asthma Through Induction of IL-33-mediated IL-1beta Production by Dcs. Sci Rep (2018) 8:11721. doi: 10.1038/s41598-018-30259-2
24. Tamachi T, Maezawa Y, Ikeda K, Kagami S, Hatano M, Seto Y, et al. IL-25 Enhances Allergic Airway Inflammation by Amplifying a TH2 Cell-Dependent Pathway in Mice. J Allergy Clin Immunol (2006) 118:606–14. doi: 10.1016/j.jaci.2006.04.051
25. Liu X, Li M, Wu Y, Zhou Y, Zeng L, Huang T. Anti-IL-33 Antibody Treatment Inhibits Airway Inflammation in a Murine Model of Allergic Asthma. Biochem Biophys Res Commun (2009) 386:181–5. doi: 10.1016/j.bbrc.2009.06.008
26. Kim YH, Park CS, Lim DH, Ahn SH, Son BK, Kim JH, et al. Beneficial Effect of Anti-interleukin-33 on the Murine Model of Allergic Inflammation of the Lower Airway. J Asthma (2012) 49:738–43. doi: 10.3109/02770903.2012.702841
27. Lee HY, Rhee CK, Kang JY, Byun JH, Choi JY, Kim SJ, et al. Blockade of IL-33/ST2 Ameliorates Airway Inflammation in a Murine Model of Allergic Asthma. Exp Lung Res (2014) 40:66–76. doi: 10.3109/01902148.2013.870261
28. Allinne J, Scott G, Lim WK, Birchard D, Erjefalt JS, Sanden C, et al. IL-33 Blockade Affects Mediators of Persistence and Exacerbation in a Model of Chronic Airway Inflammation. J Allergy Clin Immunol (2019) 144:1624–37.e10. doi: 10.1016/j.jaci.2019.08.039
29. Chinthrajah S, Cao S, Liu C, Lyu SC, Sindher SB, Long A, et al. Phase 2a Randomized, Placebo-Controlled Study of anti-IL-33 in Peanut Allergy. JCI Insight (2019) 4:e131347. doi: 10.1172/jci.insight.131347
30. Taille C, Poulet C, Marchand-Adam S, Borie R, Dombret MC, Crestani B, et al. Monoclonal Anti-TNF-alpha Antibodies for Severe Steroid-Dependent Asthma: A Case Series. Open Respir Med J (2013) 7:21–5. doi: 10.2174/1874306401307010021
31. Esty B, Harb H, Bartnikas LM, Charbonnier LM, Massoud AH, Leon-Astudillo C, et al. Treatment of Severe Persistent Asthma With IL-6 Receptor Blockade. J Allergy Clin Immunol Pract (2019) 7:1639–42.e4. doi: 10.1016/j.jaip.2019.02.043
32. Chen ZG, Zhang TT, Li HT, Chen FH, Zou XL, Ji JZ, et al. Neutralization of TSLP Inhibits Airway Remodeling in a Murine Model of Allergic Asthma Induced by Chronic Exposure to House Dust Mite. PloS One (2013) 8:e51268. doi: 10.1371/journal.pone.0051268
33. Cheng Z, Wang X, Dai LL, Jia LQ, Jing XG, Liu Y, et al. Thymic Stromal Lymphopoietin Signaling Pathway Inhibition Attenuates Airway Inflammation and Remodeling in Rats With Asthma. Cell Physiol Biochem (2018) 47:1482–96. doi: 10.1159/000490865
34. Lin SC, Chou HC, Chen CM, Chiang BL. Anti-Thymic Stromal Lymphopoietin Antibody Suppresses Airway Remodeling in Asthma Through Reduction of MMP and CTGF. Pediatr Res (2019) 86:181–7. doi: 10.1038/s41390-018-0239-x
35. Gauvreau GM, O’Byrne PM, Boulet LP, Wang Y, Cockcroft D, Bigler J, et al. Effects of an Anti-TSLP Antibody on Allergen-Induced Asthmatic Responses. N Engl J Med (2014) 370:2102–10. doi: 10.1056/NEJMoa1402895
36. Corren J, Parnes JR, Wang L, Mo M, Roseti SL, Griffiths JM, et al. Tezepelumab in Adults With Uncontrolled Asthma. N Engl J Med (2017) 377:936–46. doi: 10.1056/NEJMoa1704064
37. Chen Q, Xian D, Xu W, Nian S, Yu H, Wu Y, et al. Affinity Improvement of the Fully Human antiTSLP Recombinant Antibody. Mol Med Rep (2020) 21:759–67. doi: 10.3892/mmr.2019.10880
38. Gauvreau GM, Hohlfeld JM, Grant S, Jain M, Cabanski M, Pertel P, et al. Efficacy and Safety of an Inhaled Anti-TSLP Antibody Fragment in Adults With Mild Atopic Asthma, in: American Journal of Respiratory and Critical Care Medicine. Philadelphia, PA (2020). pp. A4207–7. doi: 10.1164/ajrccm-conference.2020.201.1_MeetingAbstracts.A4207
39. Hong JY, Bentley JK, Chung Y, Lei J, Steenrod JM, Chen Q, et al. Neonatal Rhinovirus Induces Mucous Metaplasia and Airways Hyperresponsiveness Through IL-25 and Type 2 Innate Lymphoid Cells. J Allergy Clin Immunol (2014) 134:429–39. doi: 10.1016/j.jaci.2014.04.020
40. Xu M, Dong C. IL-25 in Allergic Inflammation. Immunol Rev (2017) 278:185–91. doi: 10.1111/imr.12558
41. Ballantyne SJ, Barlow JL, Jolin HE, Nath P, Williams AS, Chung KF, et al. Blocking IL-25 Prevents Airway Hyperresponsiveness in Allergic Asthma. J Allergy Clin Immunol (2007) 120:1324–31. doi: 10.1016/j.jaci.2007.07.051
42. Gregory LG, Jones CP, Walker SA, Sawant D, Gowers KH, Campbell GA, et al. IL-25 Drives Remodelling in Allergic Airways Disease Induced by House Dust Mite. Thorax (2013) 68:82–90. doi: 10.1136/thoraxjnl-2012-202003
43. Lee HY, Lee HY, Hur J, Kang HS, Choi JY, Rhee CK, et al. Blockade of Thymic Stromal Lymphopoietin and CRTH2 Attenuates Airway Inflammation in a Murine Model of Allergic Asthma. Korean J Intern Med (2020) 35:619–29. doi: 10.3904/kjim.2018.248
44. Venkataramani S, Low S, Weigle B, Dutcher D, Jerath K, Menzenski M, et al. Design and Characterization of Zweimab and Doppelmab, High Affinity Dual Antagonistic anti-TSLP/IL13 Bispecific Antibodies. Biochem Biophys Res Commun (2018) 504:19–24. doi: 10.1016/j.bbrc.2018.08.064
45. Zoltowska AM, Lei Y, Fuchs B, Rask C, Adner M, Nilsson GP. The Interleukin-33 Receptor ST2 is Important for the Development of Peripheral Airway Hyperresponsiveness and Inflammation in a House Dust Mite Mouse Model of Asthma. Clin Exp Allergy (2016) 46:479–90. doi: 10.1111/cea.12683
46. Verma M, Liu S, Michalec L, Sripada A, Gorska MM, Alam R. Experimental Asthma Persists in IL-33 Receptor Knockout Mice Because of the Emergence of Thymic Stromal Lymphopoietin-Driven IL-9(+) and IL-13(+) Type 2 Innate Lymphoid Cell Subpopulations. J Allergy Clin Immunol (2018) 142:793–803.e8. doi: 10.1016/j.jaci.2017.10.020
47. Choi Y, Kim YM, Lee HR, Mun J, Sim S, Lee DH, et al. Eosinophil Extracellular Traps Activate Type 2 Innate Lymphoid Cells Through Stimulating Airway Epithelium in Severe Asthma. Allergy (2020) 75:95–103. doi: 10.1111/all.13997
48. An G, Wang W, Zhang X, Huang Q, Li Q, Chen S, et al. Combined Blockade of IL-25, IL-33 and TSLP Mediates Amplified Inhibition of Airway Inflammation and Remodelling in a Murine Model of Asthma. Respirology (2020) 25:603–12. doi: 10.1111/resp.13711
49. Ravanetti L, Dijkhuis A, Dekker T, Sabogal Pineros YS, Ravi A, Dierdorp BS, et al. IL-33 Drives Influenza-Induced Asthma Exacerbations by Halting Innate and Adaptive Antiviral Immunity. J Allergy Clin Immunol (2019) 143:1355–1370 e16. doi: 10.1016/j.jaci.2018.08.051
50. Albers FC, Papi A, Taille C, Bratton DJ, Bradford ES, Yancey SW, et al. Mepolizumab Reduces Exacerbations in Patients With Severe Eosinophilic Asthma, Irrespective of Body Weight/Body Mass Index: Meta-Analysis of MENSA and MUSCA. Respir Res (2019) 20:169. doi: 10.1186/s12931-019-1134-7
51. Bjermer L, Lemiere C, Maspero J, Weiss S, Zangrilli J, Germinaro M. Reslizumab for Inadequately Controlled Asthma With Elevated Blood Eosinophil Levels: A Randomized Phase 3 Study. Chest (2016) 150:789–98. doi: 10.1016/j.chest.2016.03.032
52. Chipps BE, Hirsch I, Trudo F, Alacqua M, Zangrilli JG. Benralizumab Efficacy for Patients With Fixed Airflow Obstruction and Severe, Uncontrolled Eosinophilic Asthma. Ann Allergy Asthma Immunol (2020) 124:79–86. doi: 10.1016/j.anai.2019.10.006
53. Coyle AJ, Le Gros G, Bertrand C, Tsuyuki S, Heusser CH, Kopf M, et al. Interleukin-4 is Required for the Induction of Lung Th2 Mucosal Immunity. Am J Respir Cell Mol Biol (1995) 13:54–9. doi: 10.1165/ajrcmb.13.1.7598937
54. Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, et al. Interleukin-13: Central Mediator of Allergic Asthma. Science (1998) 282:2258–61. doi: 10.1126/science.282.5397.2258
55. Panettieri RA Jr., Sjobring U, Peterffy A, Wessman P, Bowen K, Piper E, et al. Tralokinumab for Severe, Uncontrolled Asthma (STRATOS 1 and STRATOS 2): Two Randomised, Double-Blind, Placebo-Controlled, Phase 3 Clinical Trials. Lancet Respir Med (2018) 6:511–25. doi: 10.1016/S2213-2600(18)30184-X
56. Busse WW, Brusselle GG, Korn S, Kuna P, Magnan A, Cohen D, et al. Tralokinumab did Not Demonstrate Oral Corticosteroid-Sparing Effects in Severe Asthma. Eur Respir J (2019) 53:1800948. doi: 10.1183/13993003.00948-2018
57. Hanania NA, Korenblat P, Chapman KR, Bateman ED, Kopecky P, Paggiaro P, et al. Efficacy and Safety of Lebrikizumab in Patients With Uncontrolled Asthma (LAVOLTA I and LAVOLTA II): Replicate, Phase 3, Randomised, Double-Blind, Placebo-Controlled Trials. Lancet Respir Med (2016) 4:781–96. doi: 10.1016/S2213-2600(16)30265-X
58. Steinke JW. Anti-Interleukin-4 Therapy. Immunol Allergy Clin North Am (2004) 24:599–614, vi. doi: 10.1016/j.iac.2004.06.008
59. Walker BL, Leigh R. Use of Biologicals as Immunotherapy in Asthma and Related Diseases. Expert Rev Clin Immunol (2008) 4:743–56. doi: 10.1586/1744666X.4.6.743
60. Busse WW, Maspero JF, Rabe KF, Papi A, Wenzel SE, Ford LB, et al. Liberty Asthma Quest: Phase 3 Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study to Evaluate Dupilumab Efficacy/Safety in Patients With Uncontrolled, Moderate-to-Severe Asthma. Adv Ther (2018) 35:737–48. doi: 10.1007/s12325-018-0702-4
61. Jonstam K, Swanson BN, Mannent LP, Cardell LO, Tian N, Wang Y, et al. Dupilumab Reduces Local Type 2 Pro-Inflammatory Biomarkers in Chronic Rhinosinusitis With Nasal Polyposis. Allergy (2019) 74:743–52. doi: 10.1111/all.13685
62. Wenzel S, Ford L, Pearlman D, Spector S, Sher L, Skobieranda F, et al. Dupilumab in Persistent Asthma With Elevated Eosinophil Levels. N Engl J Med (2013) 368:2455–66. doi: 10.1056/NEJMoa1304048
63. Russo C, Polosa R. TNF-alpha as a Promising Therapeutic Target in Chronic Asthma: A Lesson From Rheumatoid Arthritis. Clin Sci (Lond) (2005) 109:135–42. doi: 10.1042/CS20050038
64. Amrani Y, Chen H, Panettieri RA Jr. Activation of Tumor Necrosis Factor Receptor 1 in Airway Smooth Muscle: A Potential Pathway That Modulates Bronchial Hyper-Responsiveness in Asthma? Respir Res (2000) 1:49–53. doi: 10.1186/rr12
65. Peters-Golden M, Gleason MM, Togias A. Cysteinyl Leukotrienes: Multi-Functional Mediators in Allergic Rhinitis. Clin Exp Allergy (2006) 36:689–703. doi: 10.1111/j.1365-2222.2006.02498.x
66. Whitehead GS, Thomas SY, Shalaby KH, Nakano K, Moran TP, Ward JM, et al. TNF is Required for TLR Ligand-Mediated But Not Protease-Mediated Allergic Airway Inflammation. J Clin Invest (2017) 127:3313–26. doi: 10.1172/JCI90890
67. Lukacs NW, Strieter RM, Chensue SW, Widmer M, Kunkel SL. TNF-Alpha Mediates Recruitment of Neutrophils and Eosinophils During Airway Inflammation. J Immunol (1995) 154:5411–7.
68. Sullivan DE, Ferris M, Pociask D, Brody AR. Tumor Necrosis Factor-Alpha Induces Transforming Growth Factor-Beta1 Expression in Lung Fibroblasts Through the Extracellular Signal-Regulated Kinase Pathway. Am J Respir Cell Mol Biol (2005) 32:342–9. doi: 10.1165/rcmb.2004-0288OC
69. Kim J, McKinley L, Natarajan S, Bolgos GL, Siddiqui J, Copeland S, et al. Anti-Tumor Necrosis Factor-Alpha Antibody Treatment Reduces Pulmonary Inflammation and Methacholine Hyper-Responsiveness in a Murine Asthma Model Induced by House Dust. Clin Exp Allergy (2006) 36:122–32. doi: 10.1111/j.1365-2222.2005.02407.x
70. Busse PJ, Zhang TF, Schofield B, Kilaru S, Patil S, Li XM. Decrease in Airway Mucous Gene Expression Caused by Treatment With Anti-Tumor Necrosis Factor Alpha in a Murine Model of Allergic Asthma. Ann Allergy Asthma Immunol (2009) 103:295–303. doi: 10.1016/S1081-1206(10)60528-5
71. Wenzel SE, Barnes PJ, Bleecker ER, Bousquet J, Busse W, Dahlen SE, et al. A Randomized, Double-Blind, Placebo-Controlled Study of Tumor Necrosis Factor-Alpha Blockade in Severe Persistent Asthma. Am J Respir Crit Care Med (2009) 179:549–58. doi: 10.1164/rccm.200809-1512OC
72. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis Associated With Infliximab, a Tumor Necrosis Factor Alpha-Neutralizing Agent. N Engl J Med (2001) 345:1098–104. doi: 10.1056/NEJMoa011110
73. Wolfe F, Michaud K. Lymphoma in Rheumatoid Arthritis: The Effect of Methotrexate and Anti-Tumor Necrosis Factor Therapy in 18,572 Patients. Arthritis Rheum (2004) 50:1740–51. doi: 10.1002/art.20311
74. Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, et al. Tumor Necrosis Factor-Alpha is Required in the Protective Immune Response Against Mycobacterium Tuberculosis in Mice. Immunity (1995) 2:561–72. doi: 10.1016/1074-7613(95)90001-2
75. Urbano PCM, Aguirre-Gamboa R, Ashikov A, van Heeswijk B, Krippner-Heidenreich A, Tijssen H, et al. TNF-alpha-induced Protein 3 (TNFAIP3)/A20 Acts as a Master Switch in TNF-alpha Blockade-Driven IL-17A Expression. J Allergy Clin Immunol (2018) 142:517–29. doi: 10.1016/j.jaci.2017.11.024
76. Berry MA, Hargadon B, Shelley M, Parker D, Shaw DE, Green RH, et al. Evidence of a Role of Tumor Necrosis Factor Alpha in Refractory Asthma. N Engl J Med (2006) 354:697–708. doi: 10.1056/NEJMoa050580
77. Holgate ST, Wenzel S, Postma DS, Weiss ST, Renz H, Sly PD. Asthma. Nat Rev Dis Primers (2015) 1:15025. doi: 10.1038/nrdp.2015.25
78. Erin EM, Leaker BR, Nicholson GC, Tan AJ, Green LM, Neighbour H, et al. The Effects of a Monoclonal Antibody Directed Against Tumor Necrosis Factor-alpha in Asthma. Am J Respir Crit Care Med (2006) 174:753–62. doi: 10.1164/rccm.200601-072OC
79. Peters M, Muller AM, Rose-John S. Interleukin-6 and Soluble Interleukin-6 Receptor: Direct Stimulation of gp130 and Hematopoiesis. Blood (1998) 92:3495–504. doi: 10.1182/blood.V92.10.3495.422k47_3495_3504
80. Rose-John S. IL-6 Trans-Signaling Via the Soluble IL-6 Receptor: Importance for the Pro-Inflammatory Activities of IL-6. Int J Biol Sci (2012) 8:1237–47. doi: 10.7150/ijbs.4989
81. Rose-John S. Interleukin-6 Family Cytokines. Cold Spring Harb Perspect Biol (2018) 10:a028415. doi: 10.1101/cshperspect.a028415
82. Schaper F, Rose-John S. Interleukin-6: Biology, Signaling and Strategies of Blockade. Cytokine Growth Factor Rev (2015) 26:475–87. doi: 10.1016/j.cytogfr.2015.07.004
83. Garbers C, Aparicio-Siegmund S, Rose-John S. The IL-6/gp130/STAT3 Signaling Axis: Recent Advances Towards Specific Inhibition. Curr Opin Immunol (2015) 34:75–82. doi: 10.1016/j.coi.2015.02.008
84. Dienz O, Rincon M. The Effects of IL-6 on CD4 T Cell Responses. Clin Immunol (2009) 130:27–33. doi: 10.1016/j.clim.2008.08.018
85. Palmer DC, Restifo NP. Suppressors of Cytokine Signaling (SOCS) in T Cell Differentiation, Maturation, and Function. Trends Immunol (2009) 30:592–602. doi: 10.1016/j.it.2009.09.009
86. Allard JB, Poynter ME, Marr KA, Cohn L, Rincon M, Whittaker LA. Aspergillus Fumigatus Generates an Enhanced Th2-biased Immune Response in Mice With Defective Cystic Fibrosis Transmembrane Conductance Regulator. J Immunol (2006) 177:5186–94. doi: 10.4049/jimmunol.177.8.5186
87. Yokoyama A, Kohno N, Fujino S, Hamada H, Inoue Y, Fujioka S, et al. Circulating Interleukin-6 Levels in Patients With Bronchial Asthma. Am J Respir Crit Care Med (1995) 151:1354–8. doi: 10.1164/ajrccm.151.5.7735584
88. Peters MC, McGrath KW, Hawkins GA, Hastie AT, Levy BD, Israel E, et al. Plasma Interleukin-6 Concentrations, Metabolic Dysfunction, and Asthma Severity: A Cross-Sectional Analysis of Two Cohorts. Lancet Respir Med (2016) 4:574–84. doi: 10.1016/S2213-2600(16)30048-0
89. Draijer C, Robbe P, Boorsma CE, Hylkema MN, Melgert BN. Dual Role of YM1+ M2 Macrophages in Allergic Lung Inflammation. Sci Rep (2018) 8:5105. doi: 10.1038/s41598-018-23269-7
90. Gubernatorova EO, Gorshkova EA, Namakanova OA, Zvartsev RV, Hidalgo J, Drutskaya MS, et al. Non-Redundant Functions of IL-6 Produced by Macrophages and Dendritic Cells in Allergic Airway Inflammation. Front Immunol (2018) 9:2718. doi: 10.3389/fimmu.2018.02718
91. Garbers C, Heink S, Korn T, Rose-John S. Interleukin-6: Designing Specific Therapeutics for a Complex Cytokine. Nat Rev Drug Discov (2018) 17:395–412. doi: 10.1038/nrd.2018.45
92. Kang S, Tanaka T, Narazaki M, Kishimoto T. Targeting Interleukin-6 Signaling in Clinic. Immunity (2019) 50:1007–23. doi: 10.1016/j.immuni.2019.03.026
93. Jevnikar Z, Ostling J, Ax E, Calven J, Thorn K, Israelsson E, et al. Epithelial IL-6 Trans-Signaling Defines a New Asthma Phenotype With Increased Airway Inflammation. J Allergy Clin Immunol (2019) 143:577–90. doi: 10.1016/j.jaci.2018.05.026
94. Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C, et al. Trans-Presentation of IL-6 by Dendritic Cells is Required for the Priming of Pathogenic TH17 Cells. Nat Immunol (2017) 18:74–85. doi: 10.1038/ni.3632
95. Avci AB, Feist E, Burmester GR. Targeting IL-6 or IL-6 Receptor in Rheumatoid Arthritis: What’s the Difference? BioDrugs (2018) 32:531–46. doi: 10.1007/s40259-018-0320-3
96. Tanaka T, Narazaki M, Kishimoto T. Therapeutic Targeting of the Interleukin-6 Receptor. Annu Rev Pharmacol Toxicol (2012) 52:199–219. doi: 10.1146/annurev-pharmtox-010611-134715
97. Kang S, Tanaka T, Kishimoto T. Therapeutic Uses of Anti-Interleukin-6 Receptor Antibody. Int Immunol (2015) 27:21–9. doi: 10.1093/intimm/dxu081
98. Revez JA, Bain LM, Watson RM, Towers M, Collins T, Killian KJ, et al. Effects of Interleukin-6 Receptor Blockade on Allergen-Induced Airway Responses in Mild Asthmatics. Clin Transl Immunol (2019) 8:e1044. doi: 10.1002/cti2.1044
99. Rabe KF, Nair P, Brusselle G, Maspero JF, Castro M, Sher L, et al. Efficacy and Safety of Dupilumab in Glucocorticoid-Dependent Severe Asthma. N Engl J Med (2018) 378:2475–85. doi: 10.1056/NEJMoa1804093
100. Zrubka Z, Gulacsi L, Brodszky V, Rencz F, Alten R, Szekanecz Z, et al. Long-Term Efficacy and Cost-Effectiveness of Infliximab as First-Line Treatment in Rheumatoid Arthritis: Systematic Review and Meta-Analysis. Expert Rev Pharmacoecon Outcomes Res (2019) 19:537–49. doi: 10.1080/14737167.2019.1647104
Keywords: anti-IL-6, anti-TNF, anticytokine therapy, severe asthma, combined cytokine targeting, alarmins in asthma
Citation: Gubernatorova EO, Namakanova OA, Gorshkova EA, Medvedovskaya AD, Nedospasov SA and Drutskaya MS (2021) Novel Anti-Cytokine Strategies for Prevention and Treatment of Respiratory Allergic Diseases. Front. Immunol. 12:601842. doi: 10.3389/fimmu.2021.601842
Received: 09 September 2020; Accepted: 26 April 2021;
Published: 18 May 2021.
Edited by:
Musa R. Khaitov, Institute of Immunology, RussiaReviewed by:
Vanessa Pinho, Federal University of Minas Gerais, BrazilCopyright © 2021 Gubernatorova, Namakanova, Gorshkova, Medvedovskaya, Nedospasov and Drutskaya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ekaterina O. Gubernatorova, ZWthdGVyaW5hLmd1YmVybmF0b3JvdmE0MTJAZ21haWwuY29t; Marina S. Drutskaya, bWFyaW5hZHJ1QGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.