 Junxia Huang
Junxia Huang Xinzhi Xu
Xinzhi Xu Ji Yang
Ji Yang- Department of Dermatology, Zhongshan Hospital, Fudan University, Shanghai, China
T helper 17 (Th17) cells are characterized by the secretion of the IL-17 cytokine and are essential for the immune response against bacterial and fungal infections. Despite the beneficial roles of Th17 cells, unrestrained IL-17 production can contribute to immunopathology and inflammatory autoimmune diseases, including multiple sclerosis, rheumatoid arthritis, and inflammatory bowel disease. Although these diverse outcomes are directed by the activation of Th17 cells, the regulation of Th17 cells is incompletely understood. The discovery that microRNAs (miRNAs) are involved in the regulation of Th17 cell differentiation and function has greatly improved our understanding of Th17 cells in immune response and disease. Here, we provide an overview of the biogenesis and function of miRNA and summarize the role of miRNAs in Th17 cell differentiation and function. Finally, we focus on recent advances in miRNA-mediated dysregulation of Th17 cell fate in autoimmune diseases.
Introduction
Activated CD4+ T helper (Th) cells are critical mediators of immune response, and the dysregulation of Th cell activation and differentiation is associated with inflammatory disease. Activated Th cells can be divided into functionally distinct subsets based on their cytokine production and transcription factor expression. These subsets include Th1, Th2, Th17, and Foxp3+ T regulatory cells as well as the recently described T follicular helper, Th9, and Th22 cells. Specific effector functions have been described for each Th cell subset but have been challenged by the identification of Th17 cells, which secrete IL-17. Under physiological conditions, Th17 cells accumulate at the mucosal surfaces of the gut, skin, and lung where they exert a protective immune function (1). Elimination of Th17 cells, but not Th1 cells, prevents the development of experimental autoimmune encephalomyelitis (EAE), an autoimmune disease of the central nervous system (2). We now know that Th17 cells are involved in nearly all major autoimmune diseases, including multiple sclerosis (MS), rheumatoid arthritis (RA), inflammatory bowel disease (IBD), and systemic lupus erythematosus (SLE) (3). Due to the strong association of Th17 cells with autoimmune diseases, there is a substantial interest in understanding the divergent functions of Th17 cells in homeostatic and disease states. Th17 cells are recruited to sites of inflammation by chemokine receptors, such as chemokine receptor 6 (CCR6). At the inflammation sites, Th17 cells recruit neutrophils and macrophages via secretion of cytokines, including interleukin-17A (IL-17A)/IL-17F and granulocyte-macrophage colony-stimulating factor (GM-CSF). Pro-inflammatory recruitment of neutrophils and macrophages ultimately results in tissue damage. Moreover, Th17 cells retain a degree of plasticity that enables them to co-express alternate effector cytokines, such as interferons γ (IFN-γ) or IL-10, expanding the pro-inflammatory effects of Th17 cells or change the “pathogenic” Th17 cells to “protective” Th17 cell (4–6). In addition to their role in inflammation, Th17 cells promote the maturation of B cells and play a critical role in the formation of ectopic lymphoid follicles in target organs (7, 8). The balance between Th17 cells and regulatory T cells (Treg) also emerged as an important factor in regulating autoimmunity (9). Given the strong association of Th17 cells with multiple inflammatory and autoimmune disorders, it is crucial to investigate the underlying mechanisms that balance homeostatic and pro-inflammatory Th17 cells in disease states. Improving our understanding of these mechanisms will contribute to clarify the immunologic mechanism of an autoimmune disease.
MicroRNAs (miRNAs) are a group of short, single-stranded, non-coding RNAs (10). The first miRNA was discovered in 1993 in the nematode Caenorhabditis elegans. These RNA molecules were reported to control the temporal development of C. elegans via an antisense RNA-RNA interaction (11). Today, extensive studies have elucidated the molecular functions of miRNAs and have demonstrated many key roles for miRNA in physiological and pathological processes, including metabolism, differentiation, inflammation, cancer, and immunity (12). Interference in miRNA biogenesis or deletion of specific miRNA results in a range of disease states. For example, T cell-specific deletion of Dicer, an enzyme required for microRNAs generation, leads to the reduction of CD4+ T cells and defective effector function of them (13). The absence of miRNAs in CD4+ T cells hinders the development of Treg cells. Also, CD4Cre dicerΔ/Δ mouse can develop an inflammatory bowel disease (14). In addition, global miRNA-deficiency in CD4+ T cells results in a reduced IL-17 production by cytokine-stimulated Th17 cells (14, 15), implying the existence of miRNAs that specifically regulate IL-17 expression. Moreover, several miRNAs are dysregulated in an autoimmune disease and contribute to the pathogenesis of autoimmune inflammation by interfering with the differentiation and function of Th17 cells. Thus, understanding how miRNAs regulate the differentiation and effector function of Th17 cells is critical to the development of novel therapeutic strategies for inflammatory autoimmune diseases and for miRNA-based therapeutics.
In this review, we first outline the biogenesis and function of miRNA. Then, we discuss the mechanisms by which miRNAs participate in Th17 cell differentiation and function. Finally, we focus on how the dysregulation of miRNAs in Th17 cells contributes to the development of an inflammatory autoimmune disease.
The Biogenesis and Function of miRNAs
The biogenesis of miRNA begins with the generation of primary miRNAs (pri-miRNAs), which are typically transcribed by RNA polymerase II. The pri-miRNA transcripts are cleaved in the nucleus to form hairpin pre-miRNAs that are exported to the cytoplasm for additional processing to become ~22-nucleotide miRNA duplexes. The resulting duplexes are segregated, and individual single-strand miRNA are incorporated into the multiprotein RNA-induced silencing complex (RISC) (16–18). The functional miRNA will guide the RISC to complementary mRNA sequences, which are located primarily in the 3′ untranslated region (3′ UTR), to repress mRNA expression. In some cases, miRNAs repress target gene expression by triggering the degradation of target mRNAs and, in other cases, miRNAs repress mRNA translation at initiation or elongation stages. The biogenesis and function of miRNA are widely accepted and have been recently reviewed (19, 20). An overview of miRNA biogenesis and function is depicted in Figure 1. Importantly, individual miRNA can target multiple mRNAs, and each mRNA transcript can be recognized by multiple miRNAs. Thus, the regulation of mRNA by miRNA is extremely complex and is not fully understood. In addition to their intracellular function, miRNAs can also contribute a cell-to-cell communication via vesicle or protein carrier transportation (21–23). Secreted miRNAs can be detected in the circulation and in other bodily fluids, highlighting their potential as disease biomarkers. Moreover, miRNAs are stable in stored samples, even after periods of years, making them attractive for clinical use (24). Indeed, significant correlations have been observed between the serum or tissue miRNA profiles and disease stage or activity. For example, several miRNAs have been associated with a specific disease or treatment response after miRNA profiling in whole blood, peripheral blood mononuclear cell (PBMC), brain lesions, and synovial fluid of MS, RA, and SLE patients compared to healthy donors (25–27). Unfortunately, results have been inconsistent between studies. The source of this variation may be due to relatively small sample sizes and lack of validation in independent cohorts. Thus, large-scale studies using standardized protocols are essential to enhance the clinical development of miRNA biomarkers.
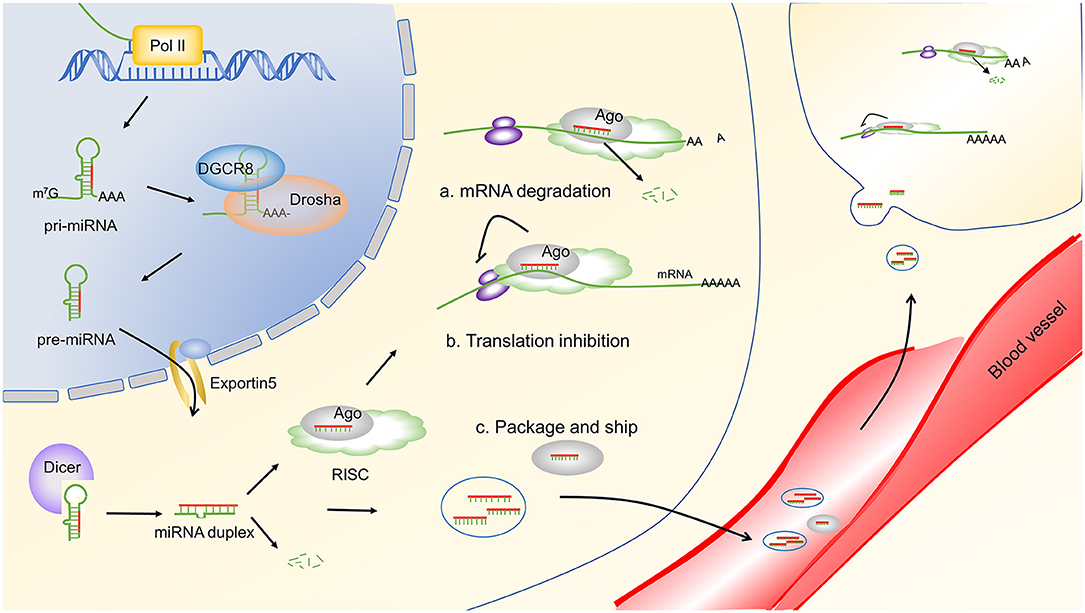
Figure 1. miRNA biogenesis and function. miRNA genes are transcribed by RNA polymerase II (Pol II) to generate pri-miRNAs. Then, the pri-miRNAs were cleaved to be mature miRNAs through a serious of steps. The first step is mediated by the DGCR8 complex, which generates the pre-miRNA. Then, the pre-miRNAs are exported to the cytoplasm by exportin 5. In the cytoplasm, pre-miRNAs are cleaved by Dicer to produce ~22 nucleotides of duplex miRNAs. One strand of the resulting duplex is bound by Ago proteins to form a RISC, and the other strand is commonly degraded. Finally, the mature miRNA guides the RISC to target mRNAs through complementary base pairing and represses target gene expression by either destabilizing target mRNAs (A) or repressing their translation (B). Alternatively, miRNAs can be packaged into vesicles and transported to other cells to exert their function (C). Arrow heads indicate a downstream response, and blunt lines indicate inhibition. Pol II, RNA polymerase II; m7G, 7-methylguanosine-cap; AAA, poly(A) tail; pri-miRNA, primary miRNAs; pre-miRNA, precursor miRNA; DGCR8, DiGeorge syndrome critical region gene 8; Ago, argonaute; RISC, RNA-induced silencing complex.
miRNA in Th17 Cell Differentiation
miRNAs are deeply involved in both the positive and negative regulation of Th17 cell differentiation. Here, we focus on four main pathways that influence Th17 cell differentiation and are regulated by miRNA: TGF-β, janus kinase-signal transducer and activator of transcription 3 (JAK-STAT3), nuclear factor kappa-B (NF-κB), and phosphoinositide 3-kinase-protein kinase B- mammalian target of rapamycin (PI3K-AKT-mTOR).
TGF-β-mediated phosphorylation of Smad2/3 is critical in the differentiation of Th17 cells. Activated Smad2/3 associate with Smad4 and translocate to the nucleus to activate the transcription (28). miR-18 inhibits Th17 cell differentiation by directly targeting Smad4 (29), whereas miR-301a enhances Th17 cell differentiation by targeting Smad nuclear interacting protein 1 (SNIP1), which competes with Smad4 to bind the coactivator CBP/P300 (30). Similarly, miR-21 (31) and miR-181c (32) induce Th17 cell differentiation via the inhibition of Smad7. Smad7 is an inhibitory SMAD protein that competitively binds to TGF-β receptor I, preventing the formation of the Smad2/3 complex and inhibiting the interaction with Smad4 (28). An overview of these mechanisms is shown in Figure 2. IL-6, IL-21, and IL-23 are cytokines that positively regulate Th17 differentiation via JAK-STAT3 signaling. Depletion of miR-29 or miR-10 in dendritic cell (DCs) promotes Th17 cell differentiation by releasing the inhibition of IL-23p40 and enhancing the production of IL-23 (33–35). Cytokine receptors on T cells activate JAK-STAT signaling to promote Th17 cell differentiation. These receptors are targeted by miRNA, ultimately influencing the differentiation of Th17 cells. Specifically, miR-34a, miR-1299, and miR-124 can target IL-6R (37, 39, 40); miR-30a targets IL-21R (36); and miR-34a and miR-326 target IL-23R (37, 38). Additionally, miR-30a-5p and miR-1246 target gp130 (39, 41), a component of the functional IL-6 family receptor complex. Cytokine receptors activate JAK, which subsequently phosphorylates STAT3. miR-29a-3p, miR-21-5p, miR-93-5p, and miR-20b impair Th17 cell differentiation by directly targeting STAT3 (45–47). In contrast, miR-155 (42), miR-384 (43), and miR-301a (44) promote the differentiation of Th17 cells through the targeted inhibition of suppressor of cytokine signaling 1 (SOCS1), suppressor of cytokine signaling 3 (SOCS3), and protein inhibitor of activated STAT3 (PIAS3), respectively. SOCS1, SOCS3, and PIAS3 are negative regulators of the JAK-STAT3 signaling pathway (49, 50). An overview of these mechanisms is shown in Figure 2.
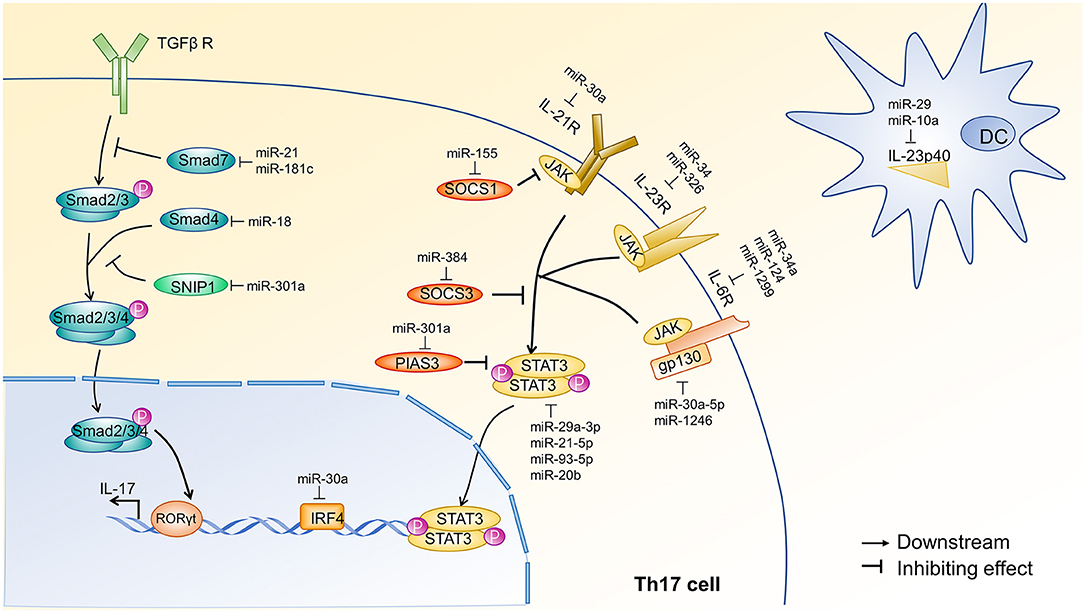
Figure 2. miRNA regulation of TGF-β and JAK-STAT3 signal transduction pathways during Th17 cell differentiation. The initial activation of Th17 cell differentiation is driven by the synergistic effect of JAK-STAT3 and transforming growth factor-β (TGF-β) signaling, which induce RORγt expression in naïve CD4+ T cells via two distinct mechanisms. JAK-STAT3 signaling is initiated by IL-6, IL-21, and IL-23. Dendritic cells (DCs) are the primary source of IL-23, and DC production of IL-23 is inhibited by miR-29 (33, 34) and miR-10 (35). Cytokine receptors on Th17 cells can also be inhibited by miRNAs. For example, miR-30a (36) inhibits IL-21R; miR-34a (37) and miR-326 (38) inhibit IL-23R; and miR-34a (37), miR-1299 (39), and miR-124 (40) inhibit IL-6R. Additionally, miR-30a-5p (39) and miR-1246 (41) target gp130, a component of the functional IL-6 family receptor complex. JAK-mediated phosphorylation of STAT3 occurs after cytokine receptor activation and is inhibited at various steps by SOCS1, SOCS3, and PIAS3. miR-155 (42), miR-384 (43), and miR-301a (44) target SOCS1, SOCS3, and PIAS3, respectively. Finally, STAT3 is directly inhibited by miR-29a-3p, miR-93-5p (45), miR-21-5p (46), and miR-20b (47). The TGF-β signal transduction pathway involves the sequential phosphorylation of Smad 2, 3, and 4, which ultimately induce IL-17 transcription. Smad7 and SNIP1 negatively regulate TGF-β signal transduction and are targeted by miRNA. Specifically, miR-21 (31) and miR-181c (32) inhibit Smad7, and miR-301a (30) inhibits SNIP1. Finally, miR-18 (29) can target Smad4. Of note, miR-30a (48) targets IRF4, another transcription factor required for the induction of ROR and IL-17A. Each of these pathways affects the expression of IL-17 in Th17 cells. Arrow heads indicate a downstream response, and blunt lines indicate inhibition. TGF-βR, transforming growth factor-β receptor; Smad, drosophila mothers against decapentaplegic protein; SNIP1, SMAD nuclear interacting protein1; ROR, Retinoid-related orphan receptor; IRF4, interferon regulatory factor4; IL-21R, Interleukin 21 receptor; IL-23R, interleukin 23 receptor; IL-6R, interleukin 6 receptor; JAK, Janus kinase; gp130, glycoprotein130; SOCS1, suppressor of cytokine signaling1; SOCS3, suppressor of cytokine signaling3; PIAS3, protein inhibitor of activated STAT3; STAT3, signal transducer and activator of transcription3; P, phosphate; DC, dendritic cell; IL-23p40, IL-12 subunit p40.
NF-κB activation in response to TCR engagement is important for Th17 cell activation and differentiation by regulating the retinoic acid-related orphan receptor-γt (RORγt) (51), an important transcriptional regulator of Th17 cell differentiation (52). Tumor necrosis factor receptor-associated factor 6 (TRAF6) and interleukin 1 Receptor associated kinase 1 (IRAK1) jointly regulate NF-κB signaling in autoreactive CD4+ T cells. miR-146a targets TRAF6/IRAK1, blocks the NF-κB-mediated secretion of IL-6/IL-21, and inhibits the development of pathogenic Th17 cells (53). Similarly, miR-15b, an inhibitor of O-GlcNAc transferase (OGT), suppresses the differentiation of Th17 cells through an impaired O-linked N-acetylglucosamine glycosylation of NF-κB (54). Finally, miR-301a promotes Th17 cell differentiation through the downregulation of SNIP1, which suppresses the activity of NF-κB (30). An overview of these mechanisms is shown in Figure 3. Hypoxia-inducible factor 1 (Hif-1α) increases the glycolytic activity of T cells, which is crucial for rapid Th17 cell expansion (62, 63). Studies have found that miR-210 and miR-18 negatively regulate Th17 differentiation by targeting Hif-1α (29, 58). Hif-1α is activated through PI3K-AKT-mTOR signal transduction, which is targeted at several stages by miRNA. For example, miR-183, miR-99a, and miR-150 suppress Th17 cells expansion by downregulating mTOR, which is an upstream activator of Hif-1α (56, 57). In contrast, the miR-17-92 cluster represses the expression of phosphatase and tensin homology (PTEN), which antagonizes PI3K, thereby activating the PI3K-AKT-mTOR axis and facilitate Th17 cell differentiation (55, 64). An overview of these mechanisms is shown in Figure 3.
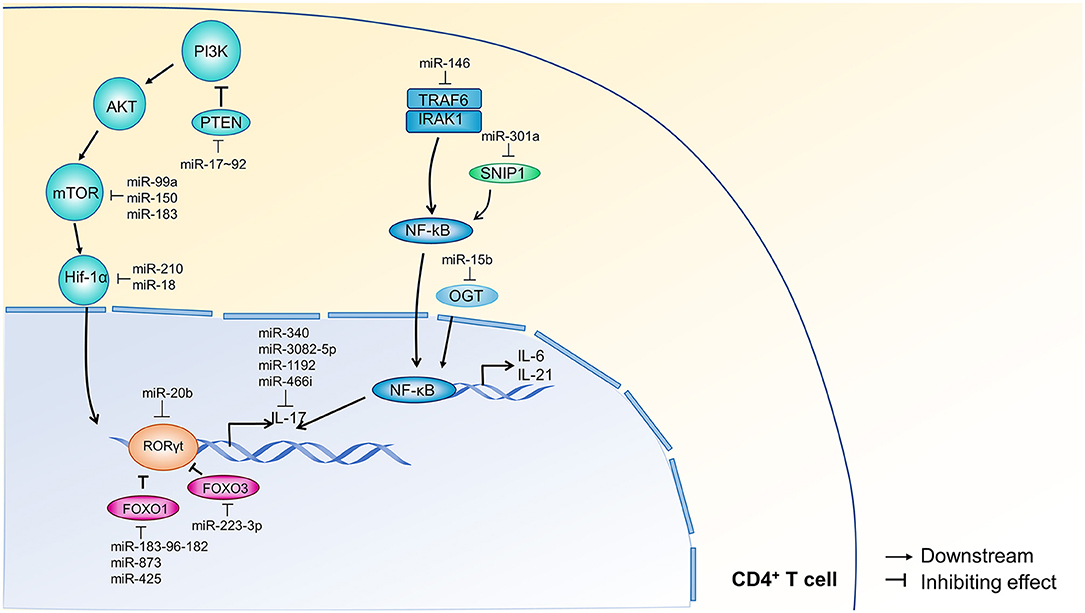
Figure 3. miRNA regulation of NF-κB and PI3K signal transduction pathways during Th17 cell differentiation. PI3K-AKT-mTOR-Hif-1α signaling is required for Th17 cell expansion, and NF-κB signaling contributes to the autocrine production of IL-6 and IL-21 in T cells. PI3K signals through AKT and mTOR to activate Hif-1α and downstream transcription. PTEN is a negative inhibitor of PI3K and is targeted by miR-17~92 (55). Additionally, miR-183 (56), miR-99a, and miR-150 (57) inhibit mTOR; miR-210 and miR-18 (29, 58) inhibit Hif-1α. TRAF6/IRAK1 and SNIP1 are upstream regulators of NF-κB, which contributes to the transcription of IL-6, IL-21, and IL-17. TRAF6 and IRAK1 are inhibited by miR-146 (53), and SNIP1 is inhibited by miR-301a (30). OGT also contributes to NF-κB-mediated transcription and is targeted by miR-15b (54). Within the nucleus, miR-20b (47) targets ROR, which is also inhibited by FOXO1 and FOXO3. FOXO1 is targeted by miR-183-96-182 (15), miR-873 (59), and miR-425 (60), and FOXO3 is targeted by miR-223-3p (61). IL-17 itself is inhibited by miR-340, miR-3082-5p, miR-1192, and miR466i. Arrow heads indicate a downstream response, and blunt lines indicate inhibition. PI3K, phosphoinositide 3-kinase; PTEN, phosphatase and tensin homolog deleted on chromosome ten; AKT, Ser/Thr kinase; mTOR, mammalian target of rapamycin; Hif-1α, hypoxia inducible factor 1α; ROR, retinoic acid-related orphan receptor; FOXO1, factor forkhead box protein O1; FOXO3, factor forkhead box protein O3; TRAF6, tumor necrosis factor receptor-associated factor 6; IRAK1, interleukin-1 receptor-associated kinase; SNIP1, SMAD nuclear interacting protein 1; NF-κB, nuclear factor κB; OGT, O-linked N-acetylglucosamine transferase; PCR2, polycomb repressive complex 2; JARID2, Jumonji and AT-rich interaction domain-containing 2.
In addition to influencing these well-known Th17 cell differentiation pathways, miRNA can also directly target IL-17 and its transcriptional regulators: RORγt, Forkhead box O1 (FOXO1), and Forkhead box O3 (FOXO3). miR-1192 (65), miR-3082-5p (66), miR-466i (67), and miR-340 (68) directly bind to the 3′ UTR of IL-17A mRNA and decrease the expression of IL-17A, which impedes Th17 cell differentiation. Similarly, Th17 cell differentiation is impaired by miR-20b (47) inhibition of RORγt. FOXO1 and FOXO3 are negative regulators of the ROR transcription factors and are targeted by several miRNAs to promote Th17 differentiation (15, 59, 60). An overview of these mechanisms is shown in Figure 3.
miRNA-Mediated Th17 Cell Abnormality in Autoimmune Diseases
As discussed above, miRNAs modulate the expression of many regulatory proteins that are required for the normal development and function of Th17 cells. Moreover, aberrant expression of miRNAs contributes to the pathology of autoimmune disease through various mechanisms (25, 69, 70). In this section, we focus on how miRNAs regulate Th17 cell differentiation to influence autoimmune diseases, including MS, RA, IBD, and SLE.
miRNAs in MS
Multiple sclerosis (MS) is a chronic, inflammatory, demyelinating, and neurodegenerative disease of the central nervous system (CNS) in young adults. Evidence points to the IL-23-Th17 cell axis as a major inducer of CNS inflammation (71). Indeed, IL-23p19−/− mice, IL-17A−/− mice, and mice treated with anti-IL-17A are resistant to the induction of experimental autoimmune encephalomyelitis (EAE), a mouse model of MS (72, 73). As IL-17 and IL-22 disrupt tight junction proteins in CNS endothelial cells (74, 75), activated Th17 cells freely migrate through the blood-brain barrier into MS lesions (76). Moreover, infiltrating Th17 cells secrete GM-CSF, which recruits DCs and macrophages to promote and sustain tissue inflammation (3, 77).
The first miRNAs implicated in Th17 cell differentiation and MS development were miR-326 and miR-155. Elevated miR-326 levels are associated with IL-17A expression in CD4+ T cells from MS patients. Moreover, miR-326 upregulation promotes the generation of Th17 cells, but not Th1 cells, by targeting E26 avian erythroblastosis virus transcription factor-1 (Ets-1) (78). O'Connell's group reported that miR-155 is required for Th1 and Th17 differentiation during EAE development. Consistent with their findings, another study has demonstrated that miR-155 is enriched in brain-infiltrating myelin-autoreactive CD4+ T cells and promotes Th17 development by targeting two Hsp40 genes, DnaJ homolog subfamily member A2 (DNAJA2) and DnaJ heat shock protein family member B1 (DNAJB1) (79). Beyond its role in T cells, miR-155 is also necessary for the secretion of Th17-related cytokines in DCs (80). Other miRNAs also promote Th17 cell differentiation and exacerbate inflammatory response in EAE. For instance, miR-21 (31) and miR-181c (32) are drastically elevated in cytokine-induced-Th17 cells in vitro, and their upregulation promotes Th17 cell differentiation by targeting Smad7 (Figure 2). Mice deficient for miR-21 or miR-181c have a defective Th17 cell differentiation and slow EAE progression. Of note, these studies were conducted by two independent groups that used different cytokine protocols to induce Th17 cell differentiation, and the differentiation efficiency varied between the two groups. Therefore, additional studies are needed to verify that the miRNA levels are associated with Th17 cell differentiation and not with the cytokine protocol used. Moreover, whether miR-21 and miR-181c are synergistic or competitive in the promotion of Th17 cell differentiation and EAE progression remains unknown. The miR-183-96-182 (15) cluster and miR-301a are also overexpressed in T cells infiltrating the CNS in EAE. Mechanistic studies found that miR-301a activates STAT3 signaling by targeting protein inhibitors of activated STAT3 (PIAS3), an inhibitor of STAT3, ultimately promoting Th17 cell differentiation (44). Interestingly, IL-6-mediatated activation of STAT3 strongly induces miR-183-96-182 expression, which in turn inhibits FOXO1 in pathogenic Th17 cells. Similarly, STAT3 activation mediates the induction of miR-384 in CD4+ T cells, which reduces the expression of SOCS3, elevates the Th17/Treg ratio, and aggravates inflammation in EAE (43, 81). One final example of a positive regulation of Th17 cell differentiation by miRNA is the miR-132/212 cluster. The miR-132/212 cluster is induced by aryl hydrocarbon receptor (AHR) activation and amplifies AHR-mediated induction of Th17 cells by targeting B-cell lymphoma 6 (Bcl6) (82).
Astrocytes contribute to CNS inflammation in MS and respond to IL-17 by releasing chemokines and pro-inflammatory cytokines that recruit Th17 cells to the site of inflammation. Liu's group reported that miR-409-3p, miR-1896, and miR-873 are upregulated in IL-17 stimulated astrocytes and target SOCS3 and A20. Upregulation of these miRNAs further induces the production of inflammatory cytokines and chemokines, such as IL-1β, IL-6, IP-10, and MCP-1, to recruit pathogenic CD4+ T cells to the CNS, ultimately aggravating EAE development (83, 84).
miRNAs also negatively regulate Th17 cell differentiation and protect against inflammatory responses in MS. For example, miR-146a is an important negative regulator of the inflammatory response. Mice deficient for miR-146a develop a more severe EAE featuring an exaggerated Th17 cell response to autoantigens due to an enhancement of the IL-6/IL-21 pathway in CD4+ T cells. Although miR-146a is a negative feedback regulator of NF-κB signaling, expression of miR-146a is induced by NF-κB. In turn, miR-146a targets TRAF6 and IRAK1 to repress NF-κB signaling (53). Additionally, miR-30a and miR-20b are downregulated in differentiated Th17 cells and in CD4+ T cells from MS patients and EAE mice. Mice infected with lentivirus-mediated-miR-30a overexpression or lentivirus-mediated-miR-20b overexpression exhibit defective Th17 cell differentiation and slow EAE progression. Disulfiram, diphenhydramine hydrochloride, and toll like receptor 4 (TLR4) activation increase miR-30a expression, which in turn targets IRF4 and IL-21R, resulting in a suppressed Th17 cell differentiation (36, 48, 85). miR-20b also suppresses Th17 cell differentiation by directly inhibiting STAT3 and ROR (47). As miRNAs that promote or inhibit Th17 cell differentiation in MS can exist in the same cell type or even within the same cell, the coordination and regulation of miRNA signaling is a crucial question. Future research should address the network regulation of miRNAs in the same cells and how this regulation contributes to the pathogenesis of MS. A systematic analysis using genomics and deep-sequencing approaches is necessary to clarify this complex regulatory network.
miRNAs in RA
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterized by a chronic inflammation of the synovial tissues in joints associated with bone and cartilage damage. In RA patients, IL-17 levels are elevated in the synovial fluid and synovial tissue. Th17 cells predominantly express CC chemokine receptor 6 (CCR6) and migrate to joints in response to CC chemokine ligand 20 (CCL20), the ligand of CCR6, which is expressed in synoviocytes (86, 87). The aggregation of Th17 cells is an important early event in joint inflammation and is followed by a series of inflammatory amplification events, including fibroblast-like synoviocyte activation, synovial-resident innate lymphoid cell expansion, osteoclast differentiation, and neutrophil infiltration (88–92).
Similar to MS, miR-155 is upregulated in synovial fluid (SF) macrophages of RA patients. Overexpression of miR-155 increases the level of pro-inflammatory cytokines and drives Th17 cell differentiation and function by directly targeting Src homolog 2-containing inositol phosphatase-1 (SHIP-1). In vivo, miR-155 knockout results in the profound suppression of antigen-specific Th17 cells and relieves collagen-induced arthritis (93).
Additionally, the imbalance between Th17 and Treg cell populations is known to be involved in RA pathogenesis. RA patients have elevated levels of Th17 cells and reduced levels of Treg cells in PBMC, which results in a high Th17/Treg ratio. Although miR-21 levels are negatively correlated with the Th17/Treg ratio in PBMC from RA patients (94), the function of miR-21 in T cells has not been studied. In contrast, miR-34 elevates the Th17/Treg ratio (95, 96) and is regulated by GM-CSF and TLR ligands in blood DCs from patients with RA. Moreover, miR-34 drives DC activation and inflammatory cytokine production by targeting AXL, a critical molecular switch that inhibits TLR ligand-induced DC activation (95). In CD4+ T cells from RA patients, miR-34a expression is induced by IL-6 and TNF-α through the NF-κB signaling pathway. Furthermore, upregulation of miR-34a disrupts the Th17/Treg balance by targeting FOXP3 and contributes to the progress of collagen-induced arthritis in mice (96). Interestingly, IL-6 and TNF-α can also induce miR-10b-5p expression, which dampens IL-17 production by targeting MAP3K7 (97). Similar to MS, there is a complex regulatory network that connects inflammatory cytokines and miRNAs in RA.
miRNAs in IBD
Inflammatory Bowel Disease (IBD) is described as chronic inflammatory disorders of the gastrointestinal tract, including Crohn's disease (CD) and ulcerative colitis (UC). Although Th17 cells provide a protective effect in the gut under steady states, they are also a chief pathogenic contributor in IBD (67, 98). Most Th17 cells reside in mucosal surfaces to stop the invasion of pathogens and to facilitate intestinal barrier integrity (99, 100). However, pathogenic Th17 profiles have been found in patients with IBD (98, 101, 102). Indeed, transferring the microbiota of IBD patients into germ-free mice increases intestinal Th17 cells and exacerbates colitis (103).
Individual miRNA can exert similar or opposing effects depending on the inflammatory disorder. For instance, expression of miR-301a is elevated in MS as well as in the inflamed mucosa and PBMC of CD and UC patients. In IBD, pro-inflammatory cytokines, like TNF-α, IL-23, and IL-6, enhance miR-301a expression in CD4+ T cells, which in turn facilitate Th17 cell differentiation and TNF-α production by inhibiting SNIP1. Downregulation of miR-301a in vivo alleviates 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis in mice (30). In contrast, miR-34a has a potent pro-inflammatory role in RA but is a safeguard for the inflammatory stem cell niche and reparative regeneration in Citrobacter rodentium-induced colitis in mice. Wang's group demonstrated that miR-34a inhibits Th17 cell differentiation, expansion, and recruitment by targeting IL-6R, IL-23R, and CCL22. Moreover, they found that miR-34a restrains inflammation-induced stem cell proliferation by targeting interleukin-17 receptor D (IL-17RD) (37). It is important to note that these studies were performed in mouse and Jurkat cells, and validation of the role of miR-34a in human inflammatory mucosa is still necessary.
Data from a mouse model of IBD suggest that the development of disease requires the transition of a subset of Th17 cells to Th1-like cells that express IFN-γ (4, 104). miR-340 facilitates the pathogenic “Th1-like” differentiation of a subset of Th17 cells in human CD4+ T cells (105) whereas miR-210 inhibits this conversion (58). Wang et al. demonstrated that miR-210 inhibits Th17 cell differentiation during reoxygenation by targeting Hif-1α. Specifically, mice with miR210−/− T cells have elevated levels of Th17 cells, enhanced conversion of Th17 cells to “Th1-like” cells, and more severe disease in an adoptive transfer colitis model (58). These studies suggest that the pathogenic transformation of Th17 cells may contribute to IBD.
Dendritic cells are potent supporters of Th17 cells differentiation because they secrete many of the cytokines required for Th17 polarization. miR-29 and miR-10a are markedly downregulated in DCs from IBD patients due to nucleotide-binding oligomerization domain 2 (NOD2) polymorphisms and elevated TNF-α, respectively. Both miR-29 and miR-10a inhibit IL-23-driven Th17 and IL-12-driven Th1 cells responses by targeting IL-12/IL-23p40, which downregulates mucosal inflammation. Moreover, miR-10a also targets NOD2 in DCs. Therefore, the inter-regulation of miR-10a and miR-29 needs to be further explored (34, 35). Finally, miR-125 and miR-219 are downregulated in CD4+ T cells from the peripheral blood and inflamed mucosa of IBD patients. Lentivirus-mediated upregulation of miR-125 or miR-219a-5p in CD4+T cells inhibits both IL-17A and IFN-γ production (106, 107). The pathogenicity of mucosal Th17 cells is not only defined by IL-17 secretion but also their plastic nature in IBD. The role of miRNAs in the conversion of Th17 to Th1-like cell needs further investigation to better understand IBD pathogenesis.
miRNAs in SLE
Systemic lupus erythematosus (SLE) is an autoimmune disease that affects almost every organ in the body and is characterized by protean clinical manifestations. Similar to other inflammatory autoimmune diseases, pathogenic functions of Th17 cells have been reported in SLE. Th17 cells and IL-17 levels are elevated in PBMC and various tissues from patients with active SLE compared to healthy donors (108–111). Consistent with the observations in RA, the percentage of Tregs is reduced in patients with active SLE, which elevates the Th17/Treg ratio. This skewed balance between Th17 cells and Treg cells contributes to the pathogenesis of SLE (108, 112, 113). Additionally, IL-17 promotes the activation of B cells and autoantibody production in SLE (114–116). Our group has demonstrated that IL-17 increases the expression of adhesion molecules and induces the adherence of T cells to human umbilical vein endothelial cells (HUVECs) (108). Another group found that Th17 cells contribute to atherosclerosis in SLE (117, 118). Together, these studies suggest that IL-17 and Th17 cells may contribute to a vascular injury in an active SLE.
miR-155 is involved in several inflammatory autoimmune diseases and has also been explored in SLE using miR-155-deficient mice. In MRL/lpr or pristane-induced lupus mice, miR-155 deficiency reduces IL-17, autoantibody production, and renal inflammation (119, 120). These effects are similar to those observed after the modulation of miR-155 in other inflammatory autoimmune diseases. Additionally, miR-873 is elevated in Th17 cells and in PBMC from SLE patients and facilitates the differentiation of Th17 cells through FOXO1 inhibition (59). Contrary to miR-155 and miR-873, miR-101-3p and miR-183 are reduced in patients with SLE. miR-101-3p blocks Th17 cell polarization by downregulating histone deacetylase 9 (HDAC9) in SLE (121), and miR-183 reduces Th17 cell polarization and SLE progression by targeting mTOR in MRL/lpr mice (56). Overall, the miRNAs' role in regulating Th17 cells to affect SLE development is least studied. More researches are required to explore the new miRNAs and related regulation mechanisms.
miRNAs in Other Autoimmune Diseases
Psoriasis is a chronic, immune-mediated, inflammatory cutaneous disease. Skin biopsies taken from patients with psoriasis show a high expression of IL-17 and IL-22 (122). Furthermore, the numbers of Th17 and Th1 cells in blood and skin lesions of psoriasis patients are elevated and are positively correlated with disease activity (123, 124). Wu's group reported that an enhanced expression of miR-210 induces Th17 and Th1 cell generation in peripheral blood and skin lesions of psoriasis patients or psoriasis-like mouse models. Silencing miR-210 by systemic or topical administrating of anti-miR-210 ameliorates dermatitis and decreases the percentage of Th17 and Th1 cells in splenic cells of imiquimod (IMQ)-treated mice (125, 126). Contrary to miR-210, miR-340 is reduced in psoriasis-like mouse models. Treatment with agomir-miR-340 alleviates the severity of IMQ-induced psoriasis in mice by directly targeting IL-17A (68). The combination treatment of miR-210-antigomir and miR-340-agomir in psoriasis deserves further study.
In mice with experimental autoimmune uveitis (EAU), miR-21-5p levels increase in accordance with the proportion of Th17 cells. miR-21-5p participates in the progression of EAU by affecting the Th17/Treg ratio via the regulation of IL-10. Inhibition of miR-21-5p reduces inflammatory injuries and retinal cell apoptosis (127). miR-223-3p promotes pathogenic Th17 cell response via suppression of FOXO3. MiR-233 knockdown decreases pathogenic function of Th17 cells and ameliorates the development of EAU (61). Additionally, miR-155−/− mice is also resistant to EAU due to the defective induction of Th17 and Th1 cell expansion (128).
Hashimoto's thyroiditis (HT) is a common autoimmune disease characterized by lymphocyte infiltration and thyroid follicular tissue lesion. Similar to the observations in MS patients, miR-326 is elevated in the PBMC of HT patients compared to healthy donors as well as in an iodine-induced thyroiditis NOD.H-2h4 mouse model. miR-326 promotes Th17 cell differentiation by targeting A disintegrin and metalloproteinase 17 (ADAM17) and Ets-1 in PBMC from HT patients and in a thyroiditis mouse model, respectively (38, 129).
Based on the studies discussed here, several miRNAs appear to be closely related to Th17 cell differentiation in various autoimmune diseases, such as miR-155, miR-326, and miR-21. However, the mechanisms by which they regulate Th17 cells are inconsistent. These inconsistences are likely due to inherent differences in the disease models and cells. A clear picture of miRNA-target genes has yet to be resolved and is complicated by the multiplicity of miRNA targets. A single miRNA can fine-tune the expression of hundreds of mRNA transcripts, and each mRNA can be targeted by hundreds of miRNAs. Therefore, future research should focus on a systematic analysis to identify key miRNA-target genes in various Th17 cell-dependent autoimmune diseases. Functions of miRNAs and their target genes in Th17 cells and autoimmune diseases are shown in Supplementary Table 1.
The Clinical Utilization of miRNA in Autoimmune Diseases
The application of miRNAs in disease diagnosis and treatment is under active investigation. Recent studies have shown that various autoimmune diseases have distinct miRNAs expression profiles. A microarray analysis plus real-time quantitative PCR identified that miR-126 was specifically enriched only in the blood of the SLE patients instead of healthy donors and RA patients (130). In another study, TaqMan Low Density Arrays were used to detect 365 miRNAs in PBMC of SLE patients and healthy controls. Results showed that miR-21 was upregulated and strongly correlated with SLE disease activity (131). MiR-103a-3p elevated both in whole blood and PBMC of RA patients and asymptomatic first-degree relatives (FDRs) compared with healthy controls, and the altered miR-103a-3p expression in at risk FDRs prior to RA onset (132). In patients with MS, Keren et al. observed that a decreased expression of miR-337-3p is correlated with an increased T1/T2 lesion load, which suggested that miR-337-3p is a potential biomarker for MS disease progression. In addition, miR-199a downregulation is negatively correlated with patients with disability, and the dysregulation of miR-199a can differentiate MS from RA, several types of neurologic disorders, and healthy controls (133). These miRNAs may be potential biomarkers for autoimmune disease diagnosis. However, to date, no miRNAs have met both the high sensitivity and specificity requirements for the clinical application in an autoimmune disease. A larger sample size and other disease control groups are necessary for further verification.
Several miRNA therapies have entered human clinical trials. So far, however, no miRNA drug candidates have entered the phase III of clinical trials. An immunity related example is Cobomarsen (MRG-106), a locked nucleic acid-modified oligonucleotide inhibitor of miR-155, currently being evaluated in a phase II clinical study of patients with cutaneous T cell lymphoma (clinical trial number: NCT03837457). Here, we have detailed the influence of miR-155 in Th17 cell differentiation and in the pathogenesis of various autoimmune diseases. Based on these studies, MRG-106 may also have beneficial therapeutic applications in Th17 cell-dependent autoimmune diseases. However, several challenges must be overcome before miRNA drugs can be considered for the clinical treatment of Th17-dependent autoimmune diseases. The biggest challenge is to identify the optimal miRNA candidates in each disease type. The miRNAs identified in individual studies may be influenced by heterogeneity, confounding inclusion of additional cell types in clinical samples, and the absolute quantity of Th17 cells purified from clinical samples. Although Th17 cell-specific miRNAs have been identified in cytokine-induced-Th17 cells in vitro, these Th17 cells are not perfect replications of the in vivo Th17 cells. The induced-Th17 cell differentiation and miRNA profiles are influenced by the cytokine profiles used to polarize Th17 cells. Moreover, the identification of miRNA targets in each autoimmune disease is complicated. Fortunately, technological advances, such as single cell sequencing, may help to systematically analyze miRNA-target networks and select key miRNA target candidates. Additional challenges include designing optimal miRNA delivery vehicles that enable tissue-specific or Th17 cell-specific targeting, limiting the off-target effects and immunogenicity to a safe degree.
Conclusion
In summary, miRNAs are important regulators in Th17 cell differentiation and contribute to the etiopathology and progression of autoimmune diseases. Regulating the expression of miRNAs may reduce or inhibit pathological Th17 cells, which is a promising therapeutic direction for autoimmune diseases. Although miRNA-based therapies are still in the early stages of investigation, numerous studies have advanced our understanding of the effects of miRNA at the pathophysiological level. Further investigation of miRNAs and their molecular mechanisms in Th17 cell-dependent autoimmune diseases will help miRNA-based therapies become a long-term clinical reality.
Author Contributions
All authors conducted the literature review for the article and substantially contributed to the discussion of content, writing of the manuscript, and editing prior to submission.
Funding
This work was supported by grants from the Medical Guide Project from Science and Technology Commission of Shanghai Municipality (CN) (20ZR1411500, 19411962500); National Natural Science Foundation of China (82073436); Outstanding Youth Medical Talents of Shanghai (Rising Stars of Medical Talent) Youth Development Program; Zhuoxue Plan of Fudan University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.593473/full#supplementary-material
References
1. Khader SA, Gaffen SL, Kolls JK. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. (2009) 2:403–11. doi: 10.1038/mi.2009.100
2. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
3. Yasuda K, Takeuchi Y, Hirota K. The pathogenicity of Th17 cells in autoimmune diseases. Semin Immunopathol. (2019) 41:283–97. doi: 10.1007/s00281-019-00733-8
4. Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci U S A. (2015) 112:7061–6. doi: 10.1073/pnas.1415675112
5. Paroni M, Maltese V, De Simone M, Ranzani V, Larghi P, Fenoglio C, et al. Recognition of viral and self-antigens by TH1 and TH1/TH17 central memory cells in patients with multiple sclerosis reveals distinct roles in immune surveillance and relapses. J Allergy Clin Immunol. (2017) 140:797–808. doi: 10.1016/j.jaci.2016.11.045
6. Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. (2009) 30:646–55. doi: 10.1016/j.immuni.2009.05.001
7. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. (2011) 35:986–96. doi: 10.1016/j.immuni.2011.10.015
8. Mitsdoerffer M, Lee Y, Jager A, Kim HJ, Korn T, Kolls JK, et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci U S A. (2010) 107:14292–7. doi: 10.1073/pnas.1009234107
9. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev. (2014) 13:668–77. doi: 10.1016/j.autrev.2013.12.004
10. Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev. (2015) 87:3–14. doi: 10.1016/j.addr.2015.05.001
11. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. (1993) 75:843–54. doi: 10.1016/0092-8674(93)90529-y
12. Kabekkodu SP, Shukla V, Varghese VK, J DS, Chakrabarty S, Satyamoorthy K. Clustered miRNAs and their role in biological functions and diseases. Biol Rev Camb Philos Soc. (2018) 93:1955–86. doi: 10.1111/brv.12428
13. Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. (2005) 202:261–9. doi: 10.1084/jem.20050678
14. Cobb BS, Hertweck A, Smith J, O'Connor E, Graf D, Cook T, et al. A role for Dicer in immune regulation. J Exp Med. (2006) 203:2519–27. doi: 10.1084/jem.20061692
15. Ichiyama K, Gonzalez-Martin A, Kim BS, Jin HY, Jin W, Xu W, et al. The microRNA-183-96-182 cluster promotes T helper 17 cell pathogenicity by negatively regulating transcription factor foxo1 expression. Immunity. (2016) 44:1284–98. doi: 10.1016/j.immuni.2016.05.015
16. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. (2014) 15:509–24. doi: 10.1038/nrm3838
17. Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol. (2019) 20:5–20. doi: 10.1038/s41580-018-0059-1
18. Zlotorynski E. Insights into the kinetics of microRNA biogenesis and turnover. Nat Rev Mol Cell Biol. (2019) 20:511. doi: 10.1038/s41580-019-0164-9
19. Iwakawa HO, Tomari Y. The functions of microRNAs: mRNA decay and translational repression. Trends Cell Biol. (2015) 25:651–65. doi: 10.1016/j.tcb.2015.07.011
20. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. (2015) 16:421–33. doi: 10.1038/nrg3965
21. Chen X, Liang H, Zhang J, Zen K, Zhang CY. Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol. (2012) 22:125–32. doi: 10.1016/j.tcb.2011.12.001
22. Duan W, Zhang W, Jia J, Lu Q, Eric Gershwin M. Exosomal microRNA in autoimmunity. Cell Mol Immunol. (2019) 16:932–4. doi: 10.1038/s41423-019-0319-9
23. Mori MA, Ludwig RG, Garcia-Martin R, Brandao BB, Kahn CR. Extracellular miRNAs: from biomarkers to mediators of physiology and disease. Cell Metab. (2019) 30:656–73. doi: 10.1016/j.cmet.2019.07.011
24. Grasedieck S, Schöler N, Bommer M, Niess JH, Tumani H, Rouhi A, et al. Impact of serum storage conditions on microRNA stability. Leukemia. (2012) 26:2414–6. doi: 10.1038/leu.2012.106
25. Long H, Wang X, Chen Y, Wang L, Zhao M, Lu Q. Dysregulation of microRNAs in autoimmune diseases: pathogenesis, biomarkers and potential therapeutic targets. Cancer Lett. (2018) 428:90–103. doi: 10.1016/j.canlet.2018.04.016
26. Evangelatos G, Fragoulis GE, Koulouri V, Lambrou GI. MicroRNAs in rheumatoid arthritis: from pathogenesis to clinical impact. Autoimmun Rev. (2019) 18:102391. doi: 10.1016/j.autrev.2019.102391
27. D'Ambrosio A, Pontecorvo S, Colasanti T, Zamboni S, Francia A, Margutti P. Peripheral blood biomarkers in multiple sclerosis. Autoimmun Rev. (2015) 14:1097–110. doi: 10.1016/j.autrev.2015.07.014
28. Konkel JE, Chen W. Balancing acts: the role of TGF-beta in the mucosal immune system. Trends Mol Med. (2011) 17:668–76. doi: 10.1016/j.molmed.2011.07.002
29. Montoya MM, Maul J, Singh PB, Pua HH, Dahlstrom F, Wu N, et al. A distinct inhibitory function for miR-18a in Th17 cell differentiation. J Immunol. (2017) 199:559–69. doi: 10.4049/jimmunol.1700170
30. He C, Shi Y, Wu R, Sun M, Fang L, Wu W, et al. miR-301a promotes intestinal mucosal inflammation through induction of IL-17A and TNF-alpha in IBD. Gut. (2016) 65:1938–50. doi: 10.1136/gutjnl-2015-309389
31. Murugaiyan G, da Cunha AP, Ajay AK, Joller N, Garo LP, Kumaradevan S, et al. MicroRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest. (2015) 125:1069–80. doi: 10.1172/JCI74347
32. Zhang Z, Xue Z, Liu Y, Liu H, Guo X, Li Y, et al. MicroRNA-181c promotes Th17 cell differentiation and mediates experimental autoimmune encephalomyelitis. Brain Behav Immun. (2018) 70:305–14. doi: 10.1016/j.bbi.2018.03.011
33. Botta C, Cuce M, Pitari MR, Caracciolo D, Gulla A, Morelli E, et al. MiR-29b antagonizes the pro-inflammatory tumor-promoting activity of multiple myeloma-educated dendritic cells. Leukemia. (2018) 32:1003–15. doi: 10.1038/leu.2017.336
34. Brain O, Owens BM, Pichulik T, Allan P, Khatamzas E, Leslie A, et al. The intracellular sensor NOD2 induces microRNA-29 expression in human dendritic cells to limit IL-23 release. Immunity. (2013) 39:521–36. doi: 10.1016/j.immuni.2013.08.035
35. Wu W, He C, Liu C, Cao AT, Xue X, Evans-Marin HL, et al. miR-10a inhibits dendritic cell activation and Th1/Th17 cell immune responses in IBD. Gut. (2015) 64:1755–64. doi: 10.1136/gutjnl-2014-307980
36. Qu X, Zhou J, Wang T, Han J, Ma L, Yu H, et al. MiR-30a inhibits Th17 differentiation and demyelination of EAE mice by targeting the IL-21R. Brain Behav Immun. (2016) 57:193–9. doi: 10.1016/j.bbi.2016.03.016
37. Wang L, Wang E, Wang Y, Mines R, Xiang K, Sun Z, et al. miR-34a is a microRNA safeguard for Citrobacter-induced inflammatory colon oncogenesis. Elife. (2018) 7:e39479. doi: 10.7554/eLife.39479
38. Liu Y, Cui X, Wang S, Liu J, Zhao N, Huang M, et al. Elevated microRNA-326 levels regulate the IL-23/IL-23R/Th17 cell axis in Hashimoto's thyroiditis by targeting a disintegrin and metalloprotease 17. Thyroid. (2020) 30:1327–37. doi: 10.1089/thy.2019.0552
39. Schiavinato J, Haddad R, Saldanha-Araujo F, Baiochi J, Araujo AG, Santos Scheucher P, et al. TGF-beta/atRA-induced Tregs express a selected set of microRNAs involved in the repression of transcripts related to Th17 differentiation. Sci Rep. (2017) 7:3627. doi: 10.1038/s41598-017-03456-8
40. Zhou L, Wang J, Li J, Li T, Chen Y, June RR, et al. 1,25-Dihydroxyvitamin D3 ameliorates collagen-induced arthritis via suppression of Th17 cells through miR-124 mediated inhibition of IL-6 signaling. Front Immunol. (2019). 10:178. doi: 10.3389/fimmu.2019.00178
41. Xie K, Liu L, Chen J, Liu F. Exosomal miR-1246 derived from human umbilical cord blood mesenchymal stem cells attenuates hepatic ischemia reperfusion injury by modulating T helper 17/regulatory T balance. IUBMB Life. (2019) 71:2020–30. doi: 10.1002/iub.2147
42. Li J, Zhang J, Guo H, Yang S, Fan W, Ye N, et al. Critical role of alternative M2 skewing in miR-155 deletion-mediated protection of colitis. Front Immunol. (2018) 9:904. doi: 10.3389/fimmu.2018.00904
43. Qu X, Han J, Zhang Y, Wang Y, Zhou J, Fan H, et al. MiR-384 regulates the Th17/Treg ratio during experimental autoimmune encephalomyelitis pathogenesis. Front Cell Neurosci. (2017) 11:88. doi: 10.3389/fncel.2017.00088
44. Mycko MP, Cichalewska M, Machlanska A, Cwiklinska H, Mariasiewicz M, Selmaj KW. MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc Natl Acad Sci U S A. (2012) 109:E1248–57. doi: 10.1073/pnas.1114325109
45. Zhu D, Tian J, Wu X, Li M, Tang X, Rui K, et al. G-MDSC-derived exosomes attenuate collagen-induced arthritis by impairing Th1 and Th17 cell responses. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:165540. doi: 10.1016/j.bbadis.2019.165540
46. Zhou J, Li X, Wu X, Zhang T, Zhu Q, Wang X, et al. Exosomes released from tumor-associated macrophages transfer miRNAs that induce a Treg/Th17 cell imbalance in epithelial ovarian cancer. Cancer Immunol Res. (2018) 6:1578–92. doi: 10.1158/2326-6066.CIR-17-0479
47. Zhu E, Wang X, Zheng B, Wang Q, Hao J, Chen S, et al. miR-20b suppresses Th17 differentiation and the pathogenesis of experimental autoimmune encephalomyelitis by targeting RORgammat and STAT3. J Immunol. (2014) 192:5599–609. doi: 10.4049/jimmunol.1303488
48. Zhao M, Sun D, Guan Y, Wang Z, Sang D, Liu M, et al. Disulfiram and diphenhydramine hydrochloride upregulate miR-30a to suppress IL-17-associated autoimmune inflammation. J Neurosci. (2016) 36:9253–66. doi: 10.1523/JNEUROSCI.4587-15.2016
49. Liang Y, Xu WD, Peng H, Pan HF, Ye DQ. SOCS signaling in autoimmune diseases: molecular mechanisms and therapeutic implications. Eur J Immunol. (2014) 44:1265–75. doi: 10.1002/eji.201344369
50. Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science. (1997) 278:1803–5. doi: 10.1126/science.278.5344.1803
51. Oh H, Ghosh S. NF-κB: roles and regulation in different CD4(+) T-cell subsets. Immunol Rev. (2013) 252:41–51. doi: 10.1111/imr.12033
52. Stockinger B, Omenetti S. The dichotomous nature of T helper 17 cells. Nat Rev Immunol. (2017) 17:535–44. doi: 10.1038/nri.2017.50
53. Li B, Wang X, Choi IY, Wang YC, Liu S, Pham AT, et al. miR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J Clin Invest. (2017) 127:3702–16. doi: 10.1172/JCI94012
54. Liu R, Ma X, Chen L, Yang Y, Zeng Y, Gao J, et al. MicroRNA-15b suppresses Th17 differentiation and is associated with pathogenesis of multiple sclerosis by targeting O-GlcNAc transferase. J Immunol. (2017) 198:2626–39. doi: 10.4049/jimmunol.1601727
55. Liu SQ, Jiang S, Li C, Zhang B, Li QJ. miR-17-92 cluster targets phosphatase and tensin homology and Ikaros Family Zinc Finger 4 to promote TH17-mediated inflammation. J Biol Chem. (2014) 289:12446–56. doi: 10.1074/jbc.M114.550723
56. Li X, Luo F, Li J, Luo C. MiR-183 delivery attenuates murine lupus nephritis-related injuries via targeting mTOR. Scand J Immunol. (2019) 90:e12810. doi: 10.1111/sji.12810
57. Warth SC, Hoefig KP, Hiekel A, Schallenberg S, Jovanovic K, Klein L, et al. Induced miR-99a expression represses Mtor cooperatively with miR-150 to promote regulatory T-cell differentiation. Embo j. (2015) 34:1195–213. doi: 10.15252/embj.201489589
58. Wang H, Flach H, Onizawa M, Wei L, McManus MT, Weiss A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat Immunol. (2014) 15:393–401. doi: 10.1038/ni.2846
59. Liu L, Liu Y, Yuan M, Xu L, Sun H. Elevated expression of microRNA-873 facilitates Th17 differentiation by targeting forkhead box O1 (Foxo1) in the pathogenesis of systemic lupus erythematosus. Biochem Biophys Res Commun. (2017) 492:453–60. doi: 10.1016/j.bbrc.2017.08.075
60. Yang X, He Q, Guo Z, Xiong F, Li Y, Pan Y, et al. MicroRNA-425 facilitates pathogenic Th17 cell differentiation by targeting forkhead box O1 (Foxo1) and is associated with inflammatory bowel disease. Biochem Biophys Res Commun. (2018) 496:352–8. doi: 10.1016/j.bbrc.2018.01.055
61. Wei Y, Chen S, Sun D, Li X, Wei R, Li X, et al. miR-223-3p promotes autoreactive Th17 cell responses in experimental autoimmune uveitis (EAU) by inhibiting transcription factor FOXO3 expression. Faseb J. (2019) 33:13951–65. doi: 10.1096/fj.201901446R
62. Rostamzadeh D, Yousefi M, Haghshenas MR, Ahmadi M, Dolati S, Babaloo Z. mTOR Signaling pathway as a master regulator of memory CD8(+) T-cells, Th17, and NK cells development and their functional properties. J Cell Physiol. (2019) 234:12353–68. doi: 10.1002/jcp.28042
63. Nagai S, Kurebayashi Y, Koyasu S. Role of PI3K/Akt and mTOR complexes in Th17 cell differentiation. Ann N Y Acad Sci. (2013) 1280:30–4. doi: 10.1111/nyas.12059
64. Essig K, Hu D, Guimaraes JC, Alterauge D, Edelmann S, Raj T, et al. Roquin suppresses the PI3K-mTOR signaling pathway to inhibit T helper cell differentiation and conversion of Treg to Tfr cells. Immunity. (2017) 47:1067–82.e12. doi: 10.1016/j.immuni.2017.11.008
65. Singh NP, Singh UP, Rouse M, Zhang J, Chatterjee S, Nagarkatti PS, et al. Dietary indoles suppress delayed-type hypersensitivity by inducing a switch from proinflammatory Th17 cells to anti-inflammatory regulatory T cells through regulation of microRNA. J Immunol. (2016) 196:1108–22. doi: 10.4049/jimmunol.1501727
66. Al-Ghezi ZZ, Singh N, Mehrpouya-Bahrami P, Busbee PB, Nagarkatti M, Nagarkatti PS. AhR activation by TCDD (2,3,7,8-Tetrachlorodibenzo-p-dioxin) attenuates pertussis toxin-induced inflammatory responses by differential regulation of tregs and Th17 cells through specific targeting by microRNA. Front Microbiol. (2019) 10:2349. doi: 10.3389/fmicb.2019.02349
67. Chen J, Adamiak W, Huang G, Atasoy U, Rostami A, Yu S. Interaction of RNA-binding protein HuR and miR-466i regulates GM-CSF expression. Sci Rep. (2017) 7:17233. doi: 10.1038/s41598-017-17371-5
68. Bian J, Liu R, Fan T, Liao L, Wang S, Geng W, et al. miR-340 alleviates psoriasis in mice through direct targeting of IL-17A. J Immunol. (2018) 201:1412–20. doi: 10.4049/jimmunol.1800189
69. Garo LP, Murugaiyan G. Contribution of microRNAs to autoimmune diseases. Cell Mol Life Sci. (2016) 73:2041–51. doi: 10.1007/s00018-016-2167-4
70. Chen JQ, Papp G, Szodoray P, Zeher M. The role of microRNAs in the pathogenesis of autoimmune diseases. Autoimmun Rev. (2016) 15:1171–80. doi: 10.1016/j.autrev.2016.09.003
71. Hiltensperger M, Korn T. The interleukin (IL)-23/T helper (Th)17 axis in experimental autoimmune encephalomyelitis and multiple sclerosis. Cold Spring Harb Perspect Med. (2018) 8:a029637. doi: 10.1101/cshperspect.a029637
72. McGinley AM, Sutton CE, Edwards SC, Leane CM, DeCourcey J, Teijeiro A, et al. Interleukin-17A serves a priming role in autoimmunity by recruiting IL-1β-producing myeloid cells that promote pathogenic T cells. Immunity. (2020) 52:342–56.e6. doi: 10.1016/j.immuni.2020.01.002
73. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. (2003) 421:744–8. doi: 10.1038/nature01355
74. Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. Faseb J. (2010) 24:1023–34. doi: 10.1096/fj.09-141978
75. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. (2007) 13:1173–5. doi: 10.1038/nm1651
76. Dos Passos GR, Sato DK, Becker J, Fujihara K. Th17 cells pathways in multiple sclerosis and neuromyelitis optica spectrum disorders: pathophysiological and therapeutic implications. Mediators Inflamm. (2016) 2016:5314541. doi: 10.1155/2016/5314541
77. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. (2011) 12:560–7. doi: 10.1038/ni.2027
78. Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. (2009) 10:1252–9. doi: 10.1038/ni.1798
79. Mycko MP, Cichalewska M, Cwiklinska H, Selmaj KW. miR-155-3p Drives the Development of autoimmune demyelination by regulation of heat shock protein 40. J Neurosci. (2015) 35:16504–15. doi: 10.1523/JNEUROSCI.2830-15.2015
80. O'Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. (2010) 33:607–19. doi: 10.1016/j.immuni.2010.09.009
81. Han J, Liu Y, Zhen F, Yuan W, Zhang W, Song X, et al. STAT3 Regulates miR-384 transcription during Th17 polarization. Front Cell Dev Biol. (2019) 7:253. doi: 10.3389/fcell.2019.00253
82. Nakahama T, Hanieh H, Nguyen NT, Chinen I, Ripley B, Millrine D, et al. Aryl hydrocarbon receptor-mediated induction of the microRNA-132/212 cluster promotes interleukin-17-producing T-helper cell differentiation. Proc Natl Acad Sci U S A. (2013) 110:11964–9. doi: 10.1073/pnas.1311087110
83. Liu X, Zhou F, Yang Y, Wang W, Niu L, Zuo D, et al. MiR-409-3p and MiR-1896 co-operatively participate in IL-17-induced inflammatory cytokine production in astrocytes and pathogenesis of EAE mice via targeting SOCS3/STAT3 signaling. Glia. (2019) 67:101–12. doi: 10.1002/glia.23530
84. Liu X, He F, Pang R, Zhao D, Qiu W, Shan K, et al. Interleukin-17 (IL-17)-induced microRNA 873 (miR-873) contributes to the pathogenesis of experimental autoimmune encephalomyelitis by targeting A20 ubiquitin-editing enzyme. J Biol Chem. (2014) 289:28971–86. doi: 10.1074/jbc.M114.577429
85. Qu X, Han J, Zhang Y, Wang X, Fan H, Hua F, et al. TLR4-RelA-miR-30a signal pathway regulates Th17 differentiation during experimental autoimmune encephalomyelitis development. J Neuroinflammation. (2019) 16:183. doi: 10.1186/s12974-019-1579-0
86. Kaneko S, Kondo Y, Yokosawa M, Furuyama K, Segawa S, Tsuboi H, et al. The RORgammat-CCR6-CCL20 axis augments Th17 cells invasion into the synovia of rheumatoid arthritis patients. Mod Rheumatol. (2018) 28:814–25. doi: 10.1080/14397595.2017.1416923
87. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. (2007) 204:2803–12. doi: 10.1084/jem.20071397
88. Hirota K, Hashimoto M, Ito Y, Matsuura M, Ito H, Tanaka M, et al. Autoimmune Th17 cells induced synovial stromal and innate lymphoid cell secretion of the cytokine GM-CSF to initiate and augment autoimmune arthritis. Immunity. (2018) 48:1220–32.e5. doi: 10.1016/j.immuni.2018.04.009
89. Piper C, Pesenacker AM, Bending D, Thirugnanabalan B, Varsani H, Wedderburn LR, et al. T cell expression of granulocyte-macrophage colony-stimulating factor in juvenile arthritis is contingent upon Th17 plasticity. Arthritis Rheumatol. (2014) 66:1955–60. doi: 10.1002/art.38647
90. Kim KW, Kim HR, Kim BM, Cho ML, Lee SH. Th17 cytokines regulate osteoclastogenesis in rheumatoid arthritis. Am J Pathol. (2015) 185:3011–24. doi: 10.1016/j.ajpath.2015.07.017
91. Takeuchi Y, Hirota K, Sakaguchi S. Impaired T cell receptor signaling and development of T cell-mediated autoimmune arthritis. Immunol Rev. (2020) 294:164–76. doi: 10.1111/imr.12841
92. Lubberts E. The IL-23-IL-17 axis in inflammatory arthritis. Nat Rev Rheumatol. (2015) 11:562. doi: 10.1038/nrrheum.2015.128
93. Kurowska-Stolarska M, Alivernini S, Ballantine LE, Asquith DL, Millar NL, Gilchrist DS, et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc Natl Acad Sci U S A. (2011) 108:11193–8. doi: 10.1073/pnas.1019536108
94. Dong L, Wang X, Tan J, Li H, Qian W, Chen J, et al. Decreased expression of microRNA-21 correlates with the imbalance of Th17 and Treg cells in patients with rheumatoid arthritis. J Cell Mol Med. (2014) 18:2213–24. doi: 10.1111/jcmm.12353
95. Kurowska-Stolarska M, Alivernini S, Melchor EG, Elmesmari A, Tolusso B, Tange C, et al. MicroRNA-34a dependent regulation of AXL controls the activation of dendritic cells in inflammatory arthritis. Nat Commun. (2017) 8:15877. doi: 10.1038/ncomms15877
96. Xie M, Wang J, Gong W, Xu H, Pan X, Chen Y, et al. NF-κB-driven miR-34a impairs Treg/Th17 balance via targeting Foxp3. J Autoimmun. (2019) 102:96–113. doi: 10.1016/j.jaut.2019.04.018
97. Chen L, Al-Mossawi MH, Ridley A, Sekine T, Hammitzsch A, de Wit J, et al. miR-10b-5p is a novel Th17 regulator present in Th17 cells from ankylosing spondylitis. Ann Rheum Dis. (2017) 76:620–5. doi: 10.1136/annrheumdis-2016-210175
98. Calderon-Gomez E, Bassolas-Molina H, Mora-Buch R, Dotti I, Planell N, Esteller M, et al. Commensal-specific CD4(+) cells from patients with Crohn's disease have a T-helper 17 inflammatory profile. Gastroenterology. (2016) 151:489–500.e3. doi: 10.1053/j.gastro.2016.05.050
99. Song X, Dai D, He X, Zhu S, Yao Y, Gao H, et al. Growth factor FGF2 cooperates with interleukin-17 to repair intestinal epithelial damage. Immunity. (2015) 43:488–501. doi: 10.1016/j.immuni.2015.06.024
100. Hegazy AN, West NR, Stubbington MJT, Wendt E, Suijker KIM, Datsi A, et al. Circulating and tissue-resident CD4(+) T cells with reactivity to intestinal microbiota are abundant in healthy individuals and function is altered during inflammation. Gastroenterology. (2017) 153:1320–37.e16. doi: 10.1053/j.gastro.2017.07.047
101. Dragasevic S, Stankovic B, Sokic-Milutinovic A, Milosavljevic T, Milovanovic T, Lukic S, et al. Importance of TLR9-IL23-IL17 axis in inflammatory bowel disease development: Gene expression profiling study. Clin Immunol. (2018) 197:86–95. doi: 10.1016/j.clim.2018.09.001
102. Bsat M, Chapuy L, Rubio M, Wassef R, Richard C, Schwenter F, et al. Differential pathogenic Th17 profile in mesenteric lymph nodes of Crohn's disease and ulcerative colitis patients. Front Immunol. (2019) 10:1177. doi: 10.3389/fimmu.2019.01177
103. Britton GJ, Contijoch EJ, Mogno I, Vennaro OH, Llewellyn SR, Ng R, et al. Microbiotas from humans with inflammatory bowel disease alter the balance of gut Th17 and RORγt(+) regulatory T cells and exacerbate colitis in mice. Immunity. (2019) 50:212–24.e4. doi: 10.1016/j.immuni.2018.12.015
104. Ueno A, Jeffery L, Kobayashi T, Hibi T, Ghosh S, Jijon H. Th17 plasticity and its relevance to inflammatory bowel disease. J Autoimmun. (2018) 87:38–49. doi: 10.1016/j.jaut.2017.12.004
105. Chen Q, He Q, Xiu W, Chen Y, Guo Z. miR-340 affects sauchinone inhibition of Th17 cell differentiation and promotes intestinal inflammation in inflammatory bowel disease. Biochem Biophys Res Commun. (2020) 526:1157–63. doi: 10.1016/j.bbrc.2020.04.038
106. Ge Y, Sun M, Wu W, Ma C, Zhang C, He C, et al. MicroRNA-125a suppresses intestinal mucosal inflammation through targeting ETS-1 in patients with inflammatory bowel diseases. J Autoimmun. (2019) 101:109–20. doi: 10.1016/j.jaut.2019.04.014
107. Shi Y, Dai S, Qiu C, Wang T, Zhou Y, Xue C, et al. MicroRNA-219a-5p suppresses intestinal inflammation through inhibiting Th1/Th17-mediated immune responses in inflammatory bowel disease. Mucosal Immunol. (2020) 13:303–12. doi: 10.1038/s41385-019-0216-7
108. Yang J, Chu Y, Yang X, Gao D, Zhu L, Yang X, et al. Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum. (2009) 60:1472–83. doi: 10.1002/art.24499
109. Vincent FB, Northcott M, Hoi A, Mackay F, Morand EF. Clinical associations of serum interleukin-17 in systemic lupus erythematosus. Arthritis Res Ther. (2013) 15:R97. doi: 10.1186/ar4277
110. Larosa M, Zen M, Gatto M, Jesus D, Zanatta E, Iaccarino L, et al. IL-12 and IL-23/Th17 axis in systemic lupus erythematosus. Exp Biol Med (Maywood). (2019) 244:42–51. doi: 10.1177/1535370218824547
111. Zickert A, Amoudruz P, Sundstrom Y, Ronnelid J, Malmstrom V, Gunnarsson I. IL-17 and IL-23 in lupus nephritis - association to histopathology and response to treatment. BMC Immunol. (2015) 16:7. doi: 10.1186/s12865-015-0070-7
112. Alvarez-Rodriguez L, Martinez-Taboada V, Calvo-Alen J, Beares I, Villa I, Lopez-Hoyos M. Altered Th17/Treg ratio in peripheral blood of systemic lupus erythematosus but not primary antiphospholipid syndrome. Front Immunol. (2019) 10:391. doi: 10.3389/fimmu.2019.00391
113. Rother N, van der Vlag J. Disturbed T cell signaling and altered Th17 and regulatory T cell subsets in the pathogenesis of systemic lupus erythematosus. Front Immunol. (2015) 6:610. doi: 10.3389/fimmu.2015.00610
114. Lee SY, Lee SH, Seo HB, Ryu JG, Jung K, Choi JW, et al. Inhibition of IL-17 ameliorates systemic lupus erythematosus in Roquin(san/san) mice through regulating the balance of TFH cells, GC B cells, Treg and Breg. Sci Rep. (2019) 9:5227. doi: 10.1038/s41598-019-41534-1
115. Ferretti E, Ponzoni M, Doglioni C, Pistoia V. IL-17 superfamily cytokines modulate normal germinal center B cell migration. J Leukoc Biol. (2016) 100:913–8. doi: 10.1189/jlb.1VMR0216-096RR
116. Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. (2008) 9:166–75. doi: 10.1038/ni1552
117. Lopez P, Rodriguez-Carrio J, Martinez-Zapico A, Perez-Alvarez AI, Benavente L, Caminal-Montero L, et al. IgM anti-phosphorylcholine antibodies associate with senescent and IL-17+ T cells in SLE patients with a pro-inflammatory lipid profile. Rheumatology (Oxford). (2020) 59:407–17. doi: 10.1093/rheumatology/kez264
118. Lopez P, Rodriguez-Carrio J, Martinez-Zapico A, Perez-Alvarez AI, Suarez-Diaz S, Mozo L, et al. Low-density granulocytes and monocytes as biomarkers of cardiovascular risk in systemic lupus erythematosus. Rheumatology (Oxford). (2020) 59:1795. doi: 10.1093/rheumatology/keaa225
119. Xin Q, Li J, Dang J, Bian X, Shan S, Yuan J, et al. miR-155 Deficiency ameliorates autoimmune inflammation of systemic lupus erythematosus by targeting S1pr1 in Faslpr/lpr mice. J Immunol. (2015) 194:5437–45. doi: 10.4049/jimmunol.1403028
120. Leiss H, Salzberger W, Jacobs B, Gessl I, Kozakowski N, Bluml S, et al. MicroRNA 155-deficiency leads to decreased autoantibody levels and reduced severity of nephritis and pneumonitis in pristane-induced lupus. PLoS One. (2017) 12:e0181015. doi: 10.1371/journal.pone.0181015
121. Sun H, Guo F, Xu L. Downregulation of microRNA-101-3p participates in systemic lupus erythematosus progression via negatively regulating HDAC9. J Cell Biochem. (2020) 121:4310–20. doi: 10.1002/jcb.29624
122. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. (2007) 445:648–51. doi: 10.1038/nature05505
123. Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. (2008) 128:1207–11. doi: 10.1038/sj.jid.5701213
124. Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, Skorcheva I, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. (2009) 129:2175–83. doi: 10.1038/jid.2009.65
125. Wu R, Zeng J, Yuan J, Deng X, Huang Y, Chen L, et al. MicroRNA-210 overexpression promotes psoriasis-like inflammation by inducing Th1 and Th17 cell differentiation. J Clin Invest. (2018) 128:2551–68. doi: 10.1172/JCI97426
126. Feng H, Wu R, Zhang S, Kong Y, Liu Z, Wu H, et al. Topical administration of nanocarrier miRNA-210 antisense ameliorates imiquimod-induced psoriasis-like dermatitis in mice. J Dermatol. (2020) 47:147–54. doi: 10.1111/1346-8138.15149
127. Shi L, Guo H, Li Z, Wang Y, Wang Y, Cui Y. Adenovirus-mediated down-regulation of miR-21-5p alleviates experimental autoimmune uveoretinitis in mice. Int Immunopharmacol. (2019) 74:105698. doi: 10.1016/j.intimp.2019.105698
128. Escobar T, Yu CR, Muljo SA, Egwuagu CE. STAT3 activates miR-155 in Th17 cells and acts in concert to promote experimental autoimmune uveitis. Invest Ophthalmol Vis Sci. (2013) 54:4017–25. doi: 10.1167/iovs.13-11937
129. Zhao N, Zou H, Qin J, Fan C, Liu Y, Wang S, et al. MicroRNA-326 contributes to autoimmune thyroiditis by targeting the Ets-1 protein. Endocrine. (2018) 59:120–9. doi: 10.1007/s12020-017-1465-4
130. Wang H, Peng W, Ouyang X, Li W, Dai Y. Circulating microRNAs as candidate biomarkers in patients with systemic lupus erythematosus. Transl Res. (2012) 160:198–206. doi: 10.1016/j.trsl.2012.04.002
131. Stagakis E, Bertsias G, Verginis P, Nakou M, Hatziapostolou M, Kritikos H, et al. Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann Rheum Dis. (2011) 70:1496–506. doi: 10.1136/ard.2010.139857
132. Anaparti V, Smolik I, Meng X, Spicer V, Mookherjee N, El-Gabalawy H. Whole blood microRNA expression pattern differentiates patients with rheumatoid arthritis, their seropositive first-degree relatives, and healthy unrelated control subjects. Arthritis Res Ther. (2017) 19:249. doi: 10.1186/s13075-017-1459-x
Keywords: miRNAs, T helper 17 cell, systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease, autoimmune disease
Citation: Huang J, Xu X and Yang J (2021) miRNAs Alter T Helper 17 Cell Fate in the Pathogenesis of Autoimmune Diseases. Front. Immunol. 12:593473. doi: 10.3389/fimmu.2021.593473
Received: 10 August 2020; Accepted: 18 February 2021;
Published: 21 April 2021.
Edited by:
Linda L. Kusner, George Washington University, United StatesReviewed by:
Murugaiyan Gopal, Harvard Medical School, United StatesGábor Papp, University of Debrecen, Hungary
Copyright © 2021 Huang, Xu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji Yang, eWFuZy5odWFAeWVhaC5uZXQ=
†These authors have contributed equally to this work