 Yun-feng Wang1†
Yun-feng Wang1† Xue-wu Liu
Xue-wu Liu Sheng-jun Wang
Sheng-jun Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 11 June 2021
Sec. Multiple Sclerosis and Neuroimmunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.582768
This article is part of the Research Topic Antibody-mediated autoimmune diseases of the CNS: Challenges and approaches to diagnosis and management View all 40 articles
Background: The presence of fluid attenuated inversion recovery (FLAIR)-hyperintense lesions in anti-myelin oligodendrocyte glycoprotein (MOG) antibody-associated cerebral cortical encephalitis with seizures (FLAMCES) was recently reported. However, the clinical characteristics and outcome of this rare clinico-radiographic syndrome remain unclear.
Methods: The present study reported two new cases. In addition, cases in the literature were systematically reviewed to investigate the clinical symptoms, magnetic resonance imaging (MRI) abnormalities, treatments and prognosis for this rare clinico-radiographic syndrome.
Results: A total of 21 cases were identified during a literature review, with a mean patient age at onset of 26.8 years. The primary clinicopathological characteristics included seizures (100%), headache (71.4%), fever (52.3%) and other cortical symptoms associated with the encephalitis location (61.9%). The common seizure types were focal to bilateral tonic-clonic seizures (28.6%) and unknown-onset tonic-clonic seizures (38.1%). The cortical abnormalities on MRI FLAIR imaging were commonly located in the frontal (58.8%), parietal (70.6%) and temporal (64.7%) lobes. In addition, pleocytosis in the cerebrospinal fluid was reported in the majority of the patients (95.2%). All patients received a treatment regimen of corticosteroids and 9 patients received anti-epileptic drugs. Clinical improvement was achieved in all patients; however, one-third of the patients reported relapse following recovery from cortical encephalitis.
Conclusions: FLAMCES is a rare phenotype of MOG-associated disease. Thus, the wider recognition of this rare syndrome may enable timely diagnosis and the development of suitable treatment regimens.
Myelin oligodendrocyte glycoprotein (MOG) is a membrane protein expressed on the surface of oligodendrocytes and in myelin sheaths (1). The primary clinical phenotypes for MOG-IgG-positive disorders are optic neuritis and myelitis (2, 3). Recent studies have suggested that MOG-associated demyelinating disease may be an entity distinct from multiple sclerosis (MS), acute disseminated encephalomyelitis (ADEM) and neuromyelitis optica spectrum disorder (NMOSD) (1, 3).
Epileptic seizures have been recorded in ~20% of patients with MOG-associated diseases (4). Furthermore, encephalitis accompanied by seizures and cortical lesions is a rare anti-MOG phenotype, which was first reported by Ogawa in 2017 (5). All 4 patients with MOG-IgG positivity described by Ogawa were observed to have experienced seizures and exhibited unilateral hyperintense cortical lesions on magnetic resonance imaging (MRI) fluid attenuated inversion recovery (FLAIR) sequences. Recently, Budhram systematically reviewed the literature and identified 20 similar cases with clinical symptoms (seizures, headache, fever and cortical symptoms); the MRI scan identified cortical FLAIR-hyperintense lesions in anti-MOG-associated encephalitis with seizures (FLAMES) (6). It is difficult to comprehend why Budhram performed investigations in some patients without seizure symptoms, despite naming this rare phenotype FLAMES (6). The present study hypothesized that the primary features of this MOG phenotype are the clinical manifestations of cortical encephalitis and seizures, as well as the radio-imaging changes in the cerebral cortical FLAIR hyperintense lesions on MRI. We named this phenotype “FLAMCES”, which stands for FLAIR-hyperintense lesions in anti-MOG antibody-associated cerebral cortical encephalitis with seizures.
The characteristics and outcome of the seizures of this rare syndrome have not been investigated to date. In the present study, two new similar cases were reported, and a systematic review of previous cases presenting with FLAMCES was conducted. The study aimed to further characterize the clinical features and outcome of this rare clinico-radiographic syndrome associated with anti-MOG antibodies.
The literature was searched for reported cases of encephalitis with seizures and cortical lesions following MRI scans in the presence of anti-MOG antibodies. The repositories PubMed, Ovid, EBSCOhost and ScienceDirect were searched for the terms’ [encephalitis] AND [MOG]’, ‘[MOG] AND [seizure]’ and ‘[MOG] AND [cortical] AND [MRI]’. All relevant published articles from January 2017 to May 2020 were reviewed for potential study inclusion. Cases were included in the present study if they met the following criteria: i) presented with encephalitis accompanied by seizures; ii) exhibited distinctive unilateral/bilateral cortical FLAIR hyperintensity on MRI without the involvement of the adjacent juxta-cortical white matter; iii) the presence of MOG-IgG antibodies were identified by cell-based assay (CBA) in the serum; and iv) infectious encephalitis [e.g., herpes simplex virus (HSV), syphilis] and other autoimmune diseases (e.g., multiple sclerosis, NMOSD, Hashimoto’s encephalitis, rheumatological diseases, autoimmune encephalitis associated with synaptic receptors/neuronal cell surface proteins antibodies) were rationally excluded. Cases were excluded if insufficient data were provided in the literature. Discrepancies between reviewers regarding the inclusion of cases were resolved by discussion. Information of the enrolled cases and our cases are presented in the Table 1. Two new cases diagnosed in our hospital were also included in the present study, for which the patients provided written informed consent. The present study was approved by the Ethics Committee of Qilu Hospital of Shandong University.
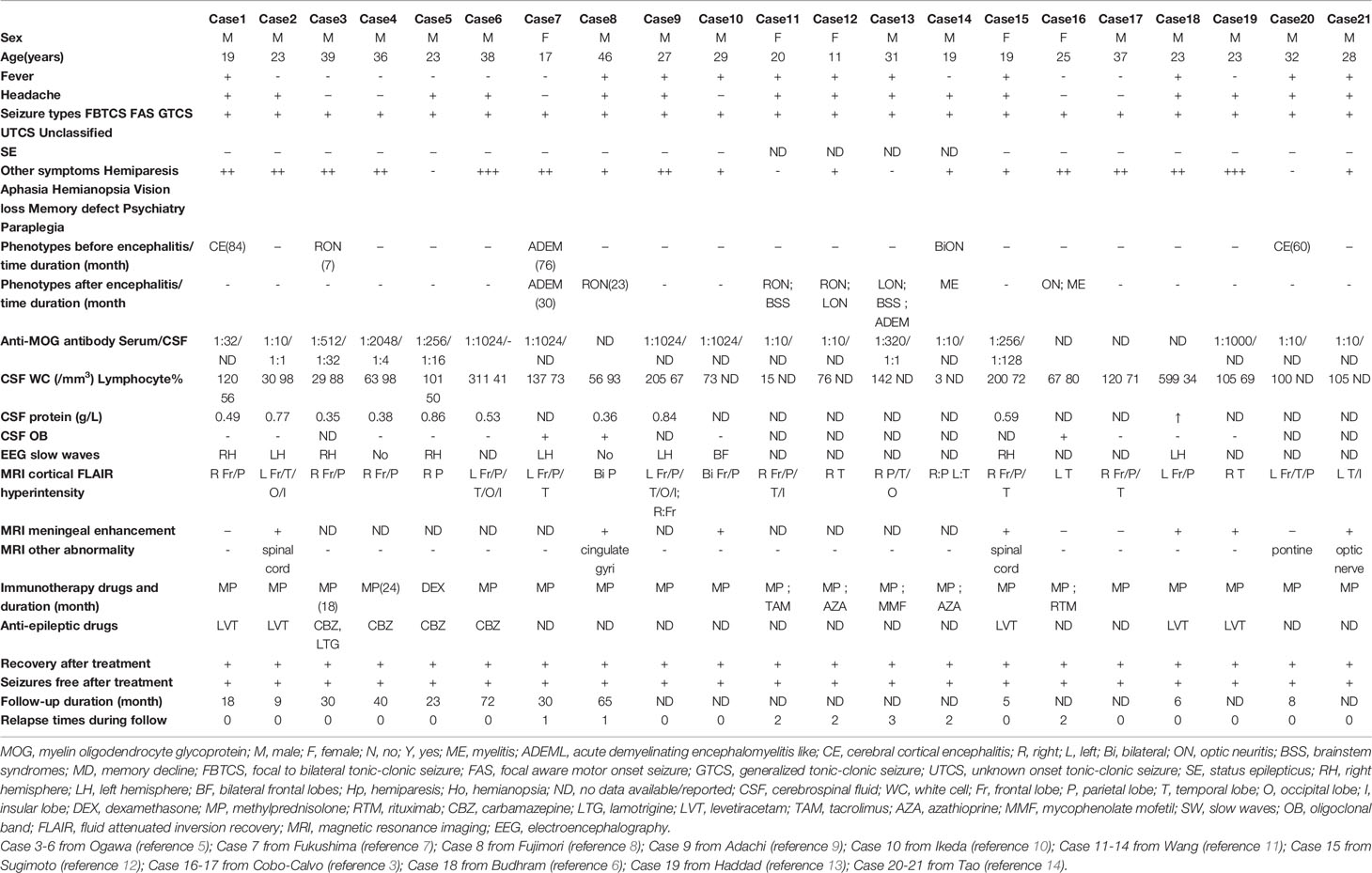
Table 1 The clinical manifestations, diagnostic examination and outcome of FLAMCES.
A 19-year-old man was admitted to the hospital due to experiencing seizures and blurred vision, with prodromal headache and fever. The patient developed a focal motor seizure and a secondary generalized tonic-clonic seizure. His vision acuity of right and left eye was 3.9 and 4.8 respectively by logarithmic visual acuity chart examination. His left upper limb power was grade 4 (MRC). The brain MRI scan revealed obvious hyperintensity on the FLAIR image not diffusion weighted imaging (DWI) in the right frontal and parietal cortex (Figures 1A, B). Examination of the cerebrospinal fluid (CSF) revealed normal pressure, but increased pleocytosis (120/μl) and protein (0.49 g/l) levels. The screening of the CSF by RT-PCR for the presence of HSV, Epstein-Barr virus, cytomegalovirus and returned negative results. The syphilis infection was excluded by TPPA and RPR testing. The presence of serum anti-MOG antibodies was positive (titer 1:32), as determined using a CBA. The results for anti-aquaporin-4 (AQP4) antibody, anti-N-methyl-D-aspartate receptor antibody, anti-leucine-rich glioma inactivated 1 (LGI1) antibody, anti-contactin-associated protein-like 2 (CASPR2) antibody, α-amino-3-hydroxy-5-methyl-isoxazolepropionic acid receptor (AMPAR) antibody, gamma-aminobutyric acid (GABA) receptor, and other autoimmune antibodies, including antinuclear, anti-ds-DNA, anti-thyroid and anti-neutrophil cytoplastic antibodies, were all negative. Subsequently, oxcarbazepine (600mg per day) and intravenous methylprednisolone (500mg per day for 5 days) were administered, followed by a dose of oral prednisone (1 mg/kg per day), with the doses gradually decreasing over time. Mycophenolate mofetil was added with a dose of 1 gram per day. The neurological symptoms of the patient resolved following treatment. In fact, a brain MRI scan after 4 weeks revealed that the abnormal cortical hyperintensity had almost disappeared (Figure 1C).
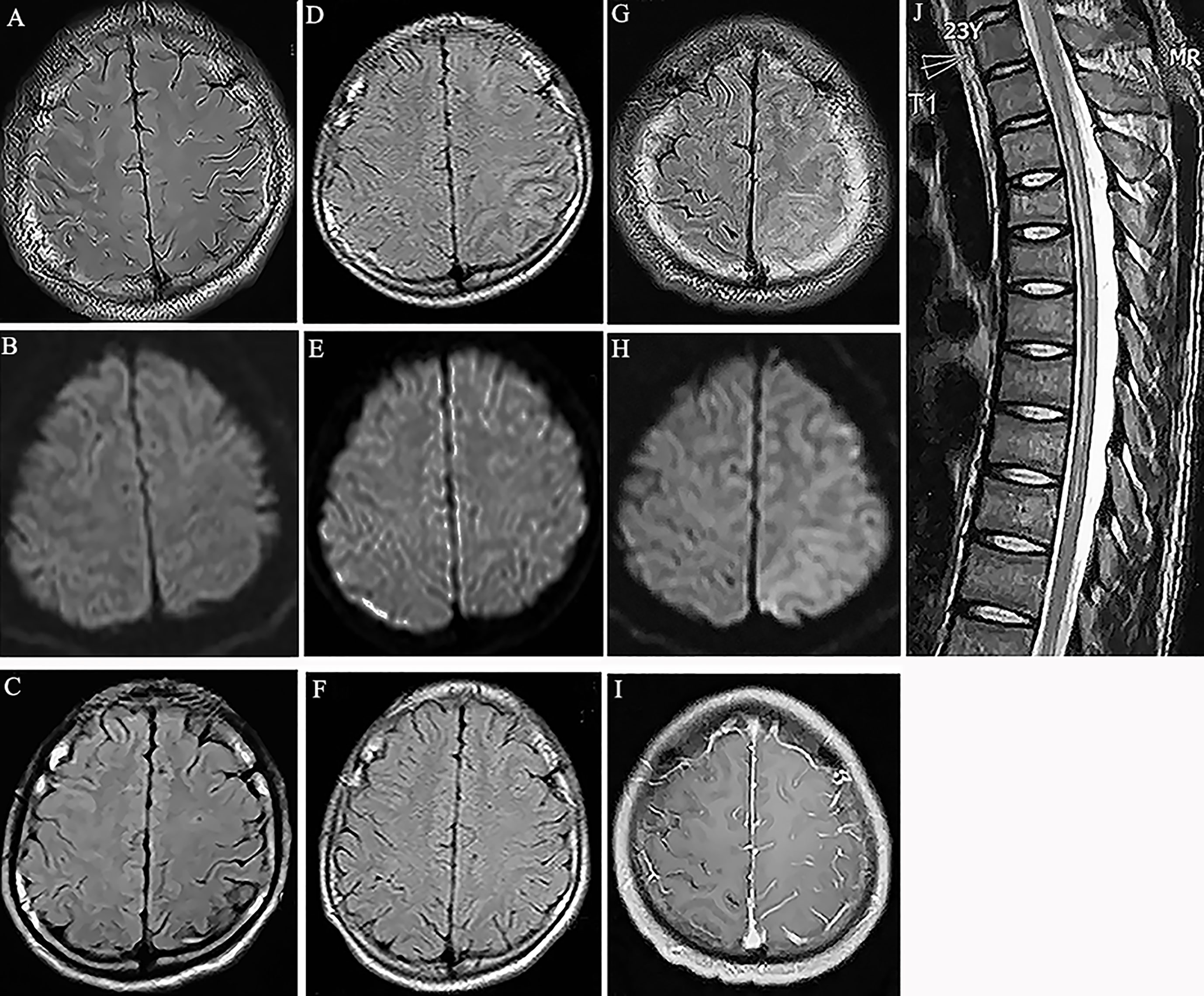
Figure 1 (A) Fluid attenuation inversion recovery (FLAIR) image showed hyperintensity in the cortical region of right frontal and parietal lobes without white matter involvement (Case 1). (B) Diffusion weighted imaging (DWI) showed no obvious signal change in the cortex (Case 1). (C) MRI scan showed the abnormality disappeared at the follow-up scan (Case 1). (D) Hyperintensity in the cortical regions of left parietal lobes in MRI FLAIR images 7 years before this episode (Case 1). (E) DWI showed no obvious signal change in the cortex (Case 1). (F) MRI scan showed the abnormal lesions disappeared at the follow-up scan (Case 1). (G) Hyperintensity in the cortical regions of left frontal and parietal lobes in MRI FLAIR images (Case 2). (H) DWI showed mild hyperintensity in the involved cortical regions (Case 2). (I) Gadolinium enhanced T1-weighted image showed meningeal linear enhancements in the sulci of left brain lobes (Case 2). (J) T2-weighted image showed hyperintensity in the spinal cord from thorax 2 to 9 segments. (Case 2).
The patient’s history revealed that he had been admitted to the hospital 7 years prior due to a seizure and headache. At the time, the brain MRI scan revealed hyperintensity in the left frontal and parietal cortex on the FLAIR images. In addition, a CSF examination revealed mildly elevated levels of white blood cells. Following treatment with glucocorticoids, the patient completely recovered and was diagnosed with steroid-responsive encephalitis of unknown etiology (Figures 1D–F).
A 23-year-old male patient presented with a headache, generalized tonic-clonic seizures and altered mental status, without a fever. In addition, no demyelination syndrome was reported. Two days later, the patient presented with weakness in both lower limbs and a brain MRI scan revealed the presence of diffuse cortical lesions in the left cerebral hemisphere (Figures 1G–I). The cognitive test score of this patient was 20 by mini-mental state examination (MMSE). His right nasolabial fold became shallow. His right and left upper limb power were grade 3 and grade 5, respectively. His right and left lower limb power were grade 2 and grade 3, respectively. He showed active bilateral knee tendon reflexes with positive bilateral Babinski sign. The spinal cord MRI scan identified the presence of myelitis from the T2 to the T9 segments (Figure 1J). In addition, the CSF examination reported 30 lymphocytes/μl and elevated protein levels of 0.77 g/l. Extensive screening by RT-PCR, TPPA, RPR and immunofluorescence testing for the presence of infections, including HSV, Epstein-Barr virus, cytomegalovirus and syphilis, and for the presence of autoimmune antibodies associated with synaptic receptors/neuronal cell surface proteins in the CSF, returned negative results. The serum antinuclear, anti-SS-A, anti-SS-B, anti-ds-DNA, anti-thyroid and anti-neutrophil cytoplastic antibodies were all negative. The anti-AQP4 antibody was also negative; however, the serum anti-MOG antibody was positive (titer 1:32). The patient was treated with intravenous methylprednisolone (500mg per day for 5 days), mycophenolate mofetil (1 g per day) and levetiracetam (1.5g per day). Then oral prednisone gradual tapered from a dose of 60 mg per day. Three weeks later, the patient had recovered fully and no longer experienced seizures. The glucocorticoid dosage was gradually reduced, and no relapse was reported over the following 9 months.
An additional 19 cases from 11 different literature reports were identified and included in the review in addition to our two new reported patients (2, 5–14).
The mean age at onset of the patients was 26.8 years (range, 11-46 years) and 76.2% of the patients were male. The type of seizure experienced included focal aware motor onset (1/21), focal to bilateral tonic-clonic (6/21), unknown onset tonic-clonic (8/21) and unclassified (6/21)-seizures (due to inadequate information or inability to place in other categories). No patients developed status epilepticus. The frequency of the other symptoms reported among these cases was as follows: 15/21 (71.4%) headache, 11/21 (52.3%) fever and 13/21 (61.9%) cortical symptoms, including aphasia, hemiparesis (8/21), hemianopsia (4/21), memory defect (4/21) and psychiatric symptoms (5/21). In addition, 7/21 patients presented with other clinical phenotypes, including optic neuritis (5/21) and myelitis (2/21), whereas 11/21 patients experienced relapse (range, 1-3 episodes) before or/and after the encephalitis episode. The previous phenotype before the onset of the encephalitis phenotype included optic neuritis (2/21), ADEM-like syndrome (1/21) and encephalitis (2/21). During the follow-up, 7 patients reported relapses, including optic neuritis (5/21), myelitis (2/21), brainstem syndrome (2/20) and ADEM-like syndrome (2/21) following the manifestation of the encephalitis phenotype (Table 1).
All patients presented with cortical hyperintensity on the MRI FLAIR sequence, without involvement of the white matter. In addition, 17/21 patients presented with unilateral cortical abnormalities, including in the frontal (10/17), parietal (12/17), temporal (11/17), insular (4/17) and occipital (3/17) lobes, and 4/21 patients exhibited bilateral abnormalities within the frontal (3/4), parietal (3/4), temporal (2/4), insular (1/4) and occipital (1/4) lobes. Brain MRI enhancement data were reported in 11 cases, and 7/11 patients presented with meningeal enhancement. Lesions in the spinal cord (2/21), brainstem (1/21) and optic nerves (1/21) were also reported on MRI. Pleocytosis in the CSF was observed in 20/21 patients. Furthermore, EEG data were available for 11 cases and slow waves were reported in 9/11 patients. The number of white blood cells in the CSF ranged from 3- to 599/ml and the protein levels ranged from 0.35- to 0.84 g/l. A total of 12 patients underwent a CSF oligoclonal band screen and 3 cases were found to be positive. The titer of the MOG antibody in the serum ranged from 1:10 to 1:2,048. In addition, 6 patients were found to have MOG antibodies in the CSF, with a titer ranging from 1:1 to 1:128 (Table 1).
All patients received treatment with corticosteroids, such as methylprednisolone (20/21) and dexamethasone (1/21). Anti-epileptic drugs, including levetiracetam (5/9), carbamazepine (4/9) and lamotrigine (1/9), were also administered as a single agent or in combination in 9 patients. Immunosuppressive drugs, including azathioprine (2/5), rituximab (1/5), tacrolimus (1/5) and mycophenolate mofetil (1/5) were also administered to 5 patients presenting with relapse. Both clinical and radiographic recovery was achieved in all patients following therapy, and no unprovoked seizures were reported in any of the patients following treatment. However, one-third of the patients (7/21) reported relapse during the follow-up period (Table 1).
In the present case report and literature review, the features of a rare phenotype of FLAMCES were characterized. In addition, the study reported two novel cases of this rare phenotype of MOG-associated disease. There were some unique clinical features of our patients compared with what had reported in literature. One patient experienced two attacks with clinical features of FLAMCES. The other one had clinical features with both FLAMCES and myelitis during one attack.
MOG-antibody-associated disease phenotypes are a new entity in the spectrum of inflammatory demyelinating diseases (1, 3). The most frequent clinical presentations include optic neuritis, myelitis, acute disseminated encephalomyelitis-like syndrome and brainstem syndrome (3). Less typical syndromes include encephalitis with seizures, cranial nerve involvement and chronic lymphocytic inflammation with pontine perivascular enhancement, which is responsive to steroids (3). Patients with MOG antibody-associated disease are more likely to manifest with encephalitis-like symptoms characterized by seizures compared with those with AQP4-IgG-associated disease (15). In one cohort study, nearly one fifth of MOG-antibody-positive patients presented with encephalitis, while unique cortical lesions were observed in 72.2% of the patients (11). In these cases, the majority of the patients with encephalitis were reported to have white matter lesions on the MRI scan. A total of 5 patients also overlapped with anti-N-methyl-D-aspartate-receptor encephalitis. Thus, only 4 patients fit the criteria of the present study. Ogawa first described 4 adult MOG antibody-positive cases presenting with unique unilateral encephalitis with epileptic seizures (5). All the cases enrolled in the present study presented with seizures and, amongst these, the focal to bilateral tonic-clonic seizure type was commonly observed. Since the data describing the characteristics of the seizures were not complete for the majority of the cases, the seizure types of these patients were classified as unknown onset tonic-clonic seizure or unclassified seizure. Furthermore, status epilepticus was not reported in any of these patients, which is a common occurrence in autoimmune encephalitis associated with antibodies against neuronal cell surface proteins. Consistent with the findings of Budhram, the present study reported the presence of headaches and fever in the majority of the patients (6). Cortical symptoms associated with the encephalitis lesions, including aphasia, hemiparesis, hemianopsia, memory impairment and psychiatric symptoms, were also reported in several cases. In addition, approximately half of the patients suffered an optic neuritis episode. Optic neuritis in the setting of the encephalitic episode or following an episode is a highly probable event (2).
MRI findings in MOG encephalomyelitis usually include an ADEM-like pattern with diffuse signal changes in the cortical grey matter, juxtacortical/subcortical/deep white matter and deep grey matter, according to FLAIR images (11, 15). The distinctive brain MRI findings in patients associated with MOG antibodies are thalamic and pontine lesions, while cortical lesions are only found in 16.3% of the cases (2, 3). Zhong reported that seizures in MOG-associated disease are commonly associated with cortical and subcortical brain lesions (16). Recently, Armangue also reported that, in several pediatric patients with non-ADEM encephalitis with positive MOG antibodies, 18% involved cortical or cortical-subcortical lesions. The majority of these pediatric patients presented with cognitive impairment, epilepsy and generalized cortical atrophy during the follow-up (17). In addition, Ogawa initially described the rare MOG clinico-radiographic syndrome as exhibiting distinctive characteristics, i.e., unilateral cortical lesions without the involvement of white matter in MRI scans. Budhram reported that several patients with bilateral cortical involvement demonstrated a broader disease spectrum (6). In the present study, ~20% patients in the literature reported bilateral cortical abnormalities on MRI FLAIR images. The most common sites of cerebral cortical lesions were identified as the frontal, temporal and parietal lobes. The cortical lesions on MRI FLAIR images should be carefully differentially diagnosed from HSV encephalitis, Creutzfeldt-Jakob disease, metabolic encephalopathy (hypoglycemia, hyperammonemia or hypoxemia) or mitochondrial encephalomyopathy.
The most interictal EEG findings are non-specific slow waves consistent with the location of the cortical lesions. CSF pleocytosis was also reported in almost all cases. In addition, the white blood cell count in the CSF is usually elevated with lymphocytic predominance in patients positive for MOG antibodies (18). Pathological examination has identified lymphocytic infiltration in both the subarachnoid space and brain parenchyma without distinct demyelinating plaques and neuronal/axonal loss (8, 10). Experimental studies have also established MOG-IgG as a pathogenic antigen rather than a secondary immune reaction (19). The present study discovered that the titer of MOG antibodies in the serum widely varied among the patients, and was not associated with disease severity or outcome. The concomitant release of autoimmune antibodies detected in the same patient with an autoimmune disease is not a rare clinical phenomenon. For example, encephalitis associated with MOG antibodies may overlap with N-methyl-Daspartate-receptor (NMDAR) antibodies (20, 21); and Jarius reported that concomitant autoimmune disorders were present in 8% of MOG-IgG-positive patients (18).
The syndrome of cerebral cortical encephalitis with seizures associated with MOG antibodies was initially described as a steroid-responsive disease (2). The acute seizures are likely triggered by an episode of demyelination, and immunotherapy was discovered to be more useful compared with anti-epileptic drugs (AEDs) for controlling the seizures (2). The present study also discovered that the seizures of several patients were completely resolved by steroids without any AEDs. AEDs, such as carbamazepine, lamotrigine and levetiracetam, were reported to be used to control the seizures. However, it remains difficult to accurately evaluate the efficacy of these AEDs alone in controlling seizures compared with immunotherapy.
The majority of patients with autoimmune antibody-mediated encephalitis develop seizures provoked by immune mechanisms; however, only a few patients develop epilepsy following recovery from encephalitis (4). The occurrence of seizures was completely abolished following recovery of encephalitis in the present literature review; no unprovoked epileptic seizures were reported following recovery from encephalitis. Thus, we prefer to name this rare MOG phenotype “cerebral cortical encephalitis with seizures” instead of “cerebral cortical encephalitis with epilepsy” (5, 14). Since MOG patients with an encephalitis phenotype experienced a sustained seizure-free period following immunotherapy, the long-term use of AEDs may be unnecessary (4, 15, 16). It remained unclear whether cortical lesions in patients were entirely inflammatory or some involve cortical demyelination. Pathologically, there was inflammatory changes in the cortex and subcortex with microglial proliferation in perivascular regions and subcortical white matter, which might suggest ‘preactive’ lesions of demyelination (10).
Although the rapid response to steroids is a distinctive feature in patients with MOG antibodies-associated disease, there is also a tendency for relapse following steroid withdrawal (3). Hamid described 5 cases of patients with MOG antibodies suffering from seizures and encephalopathy, and unprovoked seizures occurred in 2 of these patients. These cases exhibited involvement of the brain cortex, alongside hemispheric white matter and basal ganglia abnormalities on the MRI scan (15). This indicated that patients with abnormalities extending beyond the cortex on MRI scans have a high rate of seizure recurrence. Armangue reported that several patients with pediatric encephalitis-like MOG with extensive cortical involvement presented with severe brain atrophy, refractory epilepsy and cognitive changes (17). In several encephalitis cases, catastrophic brain injury and death occurred due to intracranial hypertension (7, 17). Thus, the phenotype of encephalitis with seizures should not be simply considered as a benign syndrome.
In conclusion, FLAMCES is a rare clinico-radiographic syndrome of MOG antibody-associated disease, whereby tonic-clonic seizures are common. FLAMCES may combine with myelitis or optic neuritis during one attack and relapse. Following treatment with glucocorticoids, patients may completely recover from encephalitis and cortical lesions, as demonstrated by MRI imaging. However, AEDs should not be used as long-term treatment and long-term immunosuppression should be considered instead for patients with relapse.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by The Ethics Committee of Qilu Hospital of Shandong University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Y-fW, X-wL, J-mL and J-yL collected the clinical data and reviewed literature. X-hZ and S-jW analyzed the data. Y-fW, X-wL and S-jW wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work is supported by grants from Innovative Research Project of Resident Standardization Training of Qilu Hospital, Shandong University (No.ZPZX2019A04); Undergraduate Teaching Reform and Research Project of Cheeloo College of Medicine, Shandong University (No.qlyxjy-201917), The Quality Improvement Plan for Postgraduate Education in Shandong Province (No.26010182037203), and The Taishan Scholars Program of Shandong Province.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ramanathan S, Dale RC, Brilot F. Anti-MOG Antibody: The History, Clinical Phenotype, and Pathogenicity of a Serum Biomarker for Demyelination. Autoimmun Rev (2016) 15:307–24. doi: 10.1016/j.autrev.2015.12.004
2. Cobo-Calvo A, Ruiz A, Maillart E, Audoin B, Zephir H, Bourre B, et al. Clinical Spectrum and Prognostic Value of CNS MOG Autoimmunity in Adults: The MOGADOR Study. Neurology (2018) 90:e1858–69. doi: 10.1212/WNL.0000000000005560
3. Cobo-Calvo A, Vukusic S, Marignier R. Clinical Spectrum of Central Nervous System Myelin Oligodendrocyte Glycoprotein Autoimmunity in Adults. Curr Opin Neurol (2019) 32:459–66. doi: 10.1097/WCO.0000000000000681
4. Yao Y, Xu Y, Ren H, Zhou X, Jin L, Huang Y, et al. Acute Epileptic Seizures in Myelin Oligodendrocyte Glycoprotein Encephalomyelitis and Neuromyelitis Optica Spectrum Disorder: A Comparative Cohort Study. Mult Scler Relat Disord (2019) 27:281–8. doi: 10.1016/j.msard.2018.11.007
5. Ogawa R, Nakashima I, Takahashi T, Kaneko K, Akaishi T, Takai Y, et al. MOG Antibody-Positive, Benign, Unilateral, Cerebral Cortical Encephalitis With Epilepsy. Neurol Neuroimmunol Neuroinflamm (2017) 4:e322. doi: 10.1212/NXI.0000000000000322
6. Budhram A, Mirian A, Le C, Hosseini-Moghaddam SM, Sharma M, Nicolle MW. Unilateral Cortical FLAIR-Hyperintense Lesions in Anti-MOG-Associated Encephalitis With Seizures (FLAMES): Characterization of a Distinct Clinico-Radiographic Syndrome. J Neuro (2019) 266:2481–7. doi: 10.1007/s00415-019-09440-8
7. Fukushima N, Suzuki M, Ogawa R, Hayashi K, Takanashi J, Ohashi T. A Case of Anti-MOG Antibody-Positive Multiphasic Disseminated Encephalomyelitis Co-Occurring With Unilateral Cerebral Cortical Encephalitis. Rinsho Shinkeigaku (2017) 57:723–8. doi: 10.5692/clinicalneurol.cn-001078
8. Fujimori J, Takai Y, Nakashima I, Sato DK, Takahashi T, Kaneko K, et al. Bilateral Frontal Cortex Encephalitis and Paraparesis in a Patient With Anti-MOG Antibodies. J Neurol Neurosurg Psychiatry (2017) 88:534–6. doi: 10.1136/jnnp-2016-315094
9. Adachi H, Ide Y, Takahashi T, Yoneda Y, Kageyama Y. Cerebral Cortical Encephalitis With Anti-Myelin Oligodendrocyte Glycoprotein (MOG) Antibody. Rinsho Shinkeigaku (2018) 58:767–70. doi: 10.5692/clinicalneurol.cn-001224
10. Ikeda T, Yamada K, Ogawa R, Takai Y, Kaneko K, Misu T, et al. The Pathological Features of MOG Antibody-Positive Cerebral Cortical Encephalitis as a New Spectrum Associated With MOG Antibodies: A Case Report. J Neurol Sci (2018) 392:113–5. doi: 10.1016/j.jns.2018.06.028
11. Wang L, Zhang B, Zhou L, Zhang Y, Li H, Li Y, et al. Encephalitis Is an Important Clinical Component of Myelin Oligodendrocyte Glycoprotein Antibody Associated Demyelination: A Single-Center Cohort Study in Shanghai China. Eur J Neurol (2019) 26:168–74. doi: 10.1111/ene.13790
12. Sugimoto T, Ishibashi H, Hayashi M, Tachiyama K, Fujii H, Kaneko K, et al. A Case of Anti-MOG Antibody-Positive Unilaterally Dominant Meningoencephalitis Followed by Longitudinally Extensive Transverse Myelitis. Mult Scler Relat Disord (2018) 25:128–30. doi: 10.1016/j.msard.2018.07.028
13. Haddad N, Roussel B, Pelcovits A, Rizvi S. Optic Neuritis, Encephalitis and Leptomeningeal Enhancement in a Patient With Anti-MOG Antibodies: A Case Study. Mult Scler Relat Disord (2019) 34:14–6. doi: 10.1016/j.msard.2019.06.010
14. Tao R, Qin C, Chen M, Yu HH, Wu LJ, Bu BT, et al. Unilateral Cerebral Cortical Encephalitis With Epilepsy: A Possible Special Phenotype of MOG Antibody-Associated Disorders. Int J Neurosci (2020) 2:1–5. doi: 10.1080/00207454.2020.1720676
15. Hamid SHM, Whittam D, Saviour M, Alorainy A, Mutch K, Linaker S, et al. Seizures and Encephalitis in Myelin Oligodendrocyte Glycoprotein Igg Disease vs Aquaporin 4 Igg Disease. JAMA Neurol (2018) 75:65–71. doi: 10.1001/jamaneurol.2017.3196
16. Zhong X, Zhou Y, Chang Y, Wang J, Shu Y, Sun X, et al. Seizure and Myelin Oligodendrocyte Glycoprotein Antibody-Associated Encephalomyelitis in a Retrospective Cohort of Chinese Patients. Front Neurol (2019) 10:415. doi: 10.3389/fneur.2019.00415
17. Armangue T, Olivé-Cirera G, Martínez-Hernandez E, Sepulveda M, Ruiz-Garcia R, Muñoz-Batista M, et al. Associations of Paediatric Demyelinating and Encephalitic Syndromes With Myelin Oligodendrocyte Glycoprotein Antibodies: A Multicentre Observational Study. Lancet Neuro (2020) 19(3):234–46. doi: 10.1016/S1474-4422(19)30488-0
18. Jarius S, Kleiter I, Ruprecht K, Asgari N, Pitarokoili K, Borisow N, et al. MOG-Igg in NMO and Related Disorders: A Multicenter Study of 50 Patients: Part 3, Brainstem Involvement-Frequency, Presentation and Outcome. J Neuroinflamm (2016) 13:281. doi: 10.1186/s12974-016-0719-z
19. Krishnamoorthy G, Lassmann H, Wekerle H, Holz A. Spontaneous Opticospinal Encephalomyelitis in a Double-Transgenic Mouse Model of Autoimmune T Cell/B Cell Cooperation. J Clin Invest (2006) 116:2385–92. doi: 10.1172/JCI28330
20. Titulaer MJ, Höftberger R, Iizuka T, Leypoldt F, McCracken L, Cellucci T, et al. Overlapping Demyelinating Syndromes and Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Ann Neurol (2014) 75:411–28. doi: 10.1002/ana.24117
Keywords: myelin oligodendrocyte glycoprotein, encephalitis, seizure, cortical, fluid attenuated inversion recovery
Citation: Wang Y-f, Liu X-w, Lin J-m, Liang J-y, Zhao X-h and Wang S-j (2021) The Clinical Features of FLAIR-Hyperintense Lesions in Anti-MOG Antibody Associated Cerebral Cortical Encephalitis with Seizures: Case Reports and Literature Review. Front. Immunol. 12:582768. doi: 10.3389/fimmu.2021.582768
Received: 13 July 2020; Accepted: 28 May 2021;
Published: 11 June 2021.
Edited by:
Hongzhi Guan, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Honghao Wang, Southern Medical University, ChinaCopyright © 2021 Wang, Liu, Lin, Liang, Zhao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheng-jun Wang, anVud2FuZzk5OTlAc2luYS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.