 Shanie Saghafian-Hedengren
Shanie Saghafian-Hedengren Eva Sverremark-Ekström
Eva Sverremark-Ekström Anna Nilsson
Anna Nilsson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 March 2021
Sec. T Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.582539
This article is part of the Research Topic Immunity in Infancy and Impact on Immediate and Long-Term Health View all 16 articles
The immune system plays a major role in recognizing and eliminating malignant cells, and this has been exploited in the development of immunotherapies aimed at either activating or reactivating the anti-tumor activity of a patient's immune system. A wide range of therapeutic approaches involving T lymphocytes, such as programmed cell death protein ligand-1 (PDL-1) inhibitors, cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) blockers, and CD19-targeted T-cell therapy through chimeric antigen receptor (CAR)-T cells or CD19/CD3 bi-specific T-cell engagers, have been introduced to the field of oncology, leading to significant improvements in overall survival of adult cancer patients. During the past few years, the availability and approval of T-cell based immunotherapies have become a reality also for the treatment of childhood cancers. However, the distribution, ratio of regulatory to effector cells and the quality of T-cell responses early in life are distinct from those during adolescence and adulthood, raising the possibility that these differences impact the efficacy of immunotherapy. Herein we provide a brief overview of the properties of conventional T cell subsets during early life. Focusing on the most common cancer type during childhood, acute lymphoblastic leukemia (ALL), we describe how current conventional therapies used against ALL influence the T-cell compartment of small children. We describe early life T-cell responses in relation to immunotherapies engaging T-cell anticancer reactivity and present our opinion that it is not only immaturity of the adaptive immune system, but also the impact of an immunosuppressive environment that may prove disadvantageous in the setting of immunotherapies targeting pediatric cancer cells.
Initially in the field of cancer immunology, Burnet proposed the immunosurveillance theory, suggesting that lymphocytes continuously scan and eliminate transformed cells to maintain the cells of the host in homeostasis (1). Evidence in favor of this theory came from observations in mice, where immunodeficient strains displayed high rates of spontaneous tumor development as well as higher susceptibility to induced tumors. An increased incidence of tumors has also been noted in immunosuppressed patients (2). This theory was however challenged, since many patients develop cancer in the absence of overt immunosuppression or immunodeficiency. As tumor cells are able to escape immunosurveillance, the theory was refined and the concept of “cancer immunoediting” emerged. This describes three key immunological features to combat immunogenic malignant cells: the tumor elimination phase by natural killer and T cells (incorporates immunosurveillance); the equilibrium phase between immune and malignant cells; and the escape phase resulting in clinically overt cancer as the host's immune system fails to eradicate cancer cells (3). Furthermore, in 2011 Hanahan and Weinberg (4) proposed that one additional hallmark of cancer was the ability to avoid immune destruction through the secretion of immunosuppressive factors from cancer cells and/or the recruitment of inflammatory or regulatory T cells (Tregs) to the tumor site.
During the last decades a wide range of immunotherapeutic approaches have been introduced for cancer treatment, such as programmed cell death protein ligand-1 (PDL-1) inhibitors, cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) blockers, CD22-targeting therapy with Inotuzumab ozogamicin and targeted T-cell therapy through chimeric antigen receptor (CAR)-T cells or bi-specific T-cell engagers leading to significant improvements in overall survival for a number of cancer patients. Similarly, in pediatric oncology, the last decade has seen some promising results from immunotherapy in children with relapsing disease. In this brief review, we will focus on acute lymphoblastic leukemia (ALL), the most common childhood cancer. We will describe the attributes of conventional T cells that are present during the early life of humans, and place these in the context of CD19-targeted T-cell immunotherapies against ALL.
The human new born immune system faces several challenges immediately after birth. During the very first years of life, the immune system encounters a myriad of different antigens and has to decide whether to react (against pathogens or transformed cells) or develop tolerance (against innocuous antigens or self-antigens). This balancing act has to continue throughout our entire lifespan in an environment with constantly changing antigens. In contrast to mice, human T cell development starts early during gestation (5). During the second trimester a developmental shift in lymphopoiesis occurs and the T-cell receptor (TCR) repertoire becomes diversified (5). New born children have a complete set of single CD4+ (helper) and CD8+ (cytotoxic) T cell subsets at birth, even when born pre-maturely (5–7). As a result of a marked expansion of recent thymic emigrants and naïve cells, absolute T cell numbers increase rapidly after birth to peak during the first month of life, followed by a gradual decrease during the first years of life to reach adult levels (5, 6). During this formative stage, T cells play a key role in combatting pathogens and establishing a memory T cell pool. Both the absolute numbers and frequency of Tregs are abundant in infants, where they are critical for developing tolerance. These cells decline from 5 to 8 years of age, reaching similar numbers to those of adults over time (7, 8). Focusing on findings from studies in humans, in the following section we will touch upon key attributes that distinguish conventional T cell populations in young children from those in adults.
Immune system competence is connected to the diversity of the host's T cell pool (9), which subsequently increases an individual's probability to recognize invading pathogens. This T cell diversity relies on the presence of a naïve T cell pool which is formed through thymopoiesis. Overall, it appears that naïve T cells in young children are not necessarily less responsive, but rather programmed to respond differently to activation signals compared to adults. Recent evidence suggests that neonatal T cells are biased toward non-specific defense mechanisms, which are less dependent on TCR recognition, while having an elevated capacity to react to inflammatory and danger signals, which are in part facilitated through the expression of innate receptors [reviewed in (10)]. Thymic output can be assessed on the basis of cell markers for recent thymic emigrants, such as surface CD31, or the presence of signal joint T-cell receptor excision circles (sjTRECs), both of which are higher in children (11, 12). By means of proliferation ability, naïve T cells in humans can be maintained through homeostatic peripheral expansion by IL-7 and IL-15 cytokines, which act pre-ferentially on CD4+ and CD8+, respectively (13, 14). IL-7-induced cell division does not, however, lead to helper T cell differentiation, as is the case with CD8+ T cells following IL-15 exposure and activation (15, 16). A high constitutive telomerase activity has been shown to protect neonatal T cells from proliferative stress and exhaustion (13), a phenomenon more commonly noted in adult T cells. Another interesting observation is that the increased apoptotic potential in human neonatal T cells (14, 15) can be reverted by cytokines acting via the IL-2 receptor γ-chain (17–19).
The effector T cell functions in early life are characterized by decreased IFN-γ but higher type 2 cytokine and CXCL8 production (20, 21), which in part have been explained by the epigenetic conformation of IL-13 (22) and IFN-γ loci (23) in neonatal CD4+ T cells. The transcriptome and epigenetic profiles, along with functional readouts from cord blood and adult naïve CD8+ T cells, corroborated that neonatal CD8+ T cells are more innate-like by displaying, among others, less cytotoxic functions, while being more prone toward producing antimicrobial peptides and reactive oxygen species (ROS) (24). It is not clear if costimulatory capacity through CD40 ligand on T cells is defective (25–27), or if a differential activity of the transcription factor NFAT (28), which affects T-cell costimulatory capacity and IFN-γ production, is present during early childhood years.
The involution of the human thymus begins already during the first year of life (29–31) and proceeds at a steady rate of 3% thymic volume loss until ~50-years of age, after which the involution rate decreases somewhat to a new stable level in the elderly (32, 33). If the thymus already starts to involute after birth (29) then the question is how a naïve T cell pool, that can react to new antigen challenges, is maintained from birth onwards in humans? Farber and colleagues have made significant contributions to advancing our understanding in this field. In one of their recent studies, that characterized naïve T cells in various secondary lymphoid tissues in humans (34), it was demonstrated that a dramatic decline in thymic output occurs rather from middle age onwards, whereby a four-fold decrease of double positive thymocytes is noted. Furthermore, it was shown that each lymphoid organ contains a unique set of naïve T cell clones which can expand, especially in those past 40 years of age, suggesting that naïve T cells can be sustained through in situ homeostasis and retention in lymphoid tissues.
The distribution and activity of T cells across tissues appear to be influenced by age. Upon comparing immunophenotypes of CD4+ and CD8+ T cells in tissues from pediatric and young adult organ donors (12), a higher frequency of naïve T cells was found across all pediatric tissues, including the lung and intestine, compared to adult tissue. Memory T cells in children, on the other hand, were found at local mucosal sites, such as lungs and intestine, but less frequent in blood and secondary lymphoid organs, while in adults both lymphoid and mucosal tissues contained high proportions of CD4+ and CD8+ memory T cells. In general, lymphoid pediatric T cells produce fewer effector cytokines (IL-1, IL-4, and IFN-γ) than adult tissue T cells, with the exception of pediatric memory T-cells derived from the intestinal mucosa (which secrete proinflammatory cytokines at similar levels to those in adults). Tissue resident memory T cells are key to providing local protective immunity. When examining tissue memory T cells for markers associated with tissue retention, pediatric mucosal samples contain a lower frequency of T cells expressing both the activation marker CD69 and the integrin CD103 compared to adults, which suggests that memory T cells in early life have not yet fully acquired a tissue resident phenotype (12). In response to respiratory tract infection, memory T-cell formation appears more limited during infancy, as evidenced by the accumulation of more terminally differentiated (TEMRA) CD8+ T cells in the lungs of younger patients, whereas the less-differentiated tissue-resident memory CD8+ T cells were more often seen in older children (35). Corresponding data in pediatric populations on the characteristics of tissue memory helper T cells following infection are lacking.
The microbiome is an important factor for T-cell compartment maturation, including Tregs (36, 37). There are various ways for commensals to shape the immune system, both directly with the adjacent immune system in the gut as well as on distance via metabolites (38), microbial compounds (39), and potentially also extracellular microvesicles (40). Tregs are a subset of CD4+ T cells identified through the master transcription factor, forkhead box protein P3 (FoxP3), with essential roles in maintaining self-tolerance and dampening immune responses (41). Peripheral Tregs represent around 5–10% of CD4+ T cells in cord blood (42, 43). They comprise 30–40% of the CD4+ T cells in pediatric tissues and 1–10% in adult tissues (12). Tregs display an entirely different TCR repertoire to other T cell populations and also exhibit a higher cell turnover compared to naïve T cells (44). Functionally, the depletion of Tregs in pediatric lymphoid and mucosal tissue results in potentiated T cell proliferation and cytokine release (12). By contrast, depletion of Treg cells in adult tissue does not alter T cell activation. Based on differential CD45RA expression, two distinct Treg subsets termed activated and resting have been identified among the neonatal FoxP3+ CD4+ T cells. These subsets are functionally distinct, such that activated Tregs are terminally differentiated and rapidly die after exerting suppression while resting Treg cells proliferate, become CTLA-4+ FoxP3hi and replenish the activated Treg pool (45). Both activated and resting Tregs increase over the first 6 months post-natally, but subsequently it is mainly the proportion of activated Tregs that increase (46).
To summarize, the significant observations of increased Treg/effector memory T cell ratios across various tissues in infants compared to adults, together with the elevated T cell response following Treg depletion in children, indicate that-rather than being intrinsically defective-infant T cells may simply be more inhibited than their adult counterparts in allowing for immune maturation and tolerance. We speculate that this may have important implications for T-cell based immunotherapies designated for use in younger children.
The global incidence of childhood cancer is estimated at ~152.8 per million person-years in 0 to 19-year-olds, with a slightly higher incidence in the age groups 0 to 4-year-old and 15 to 19-year-old (47). Childhood cancers span diverse clinical and biological tumor entities, including leukemia and lymphoma, bone and soft tissue sarcoma, central nervous system tumors, retinoblastoma, neuroblastoma, liver and kidney tumors, germ cell tumors, and additional rare cancers. Childhood leukemia represents ~30% of all childhood cancer cases, with ALL accounting for 80% and acute myeloid leukemia (AML) for 15–20% of cases (48). There are only rare cases of chronic myeloid leukemia (CML) in children. Dramatic improvements in the overall ALL survival rates have been accomplished over the last decades, with outcomes approaching 90% (49). However, progress has been slower for certain patient subgroups, such as infant-ALL, Down's syndrome and children with AML. For the purpose of this review, we will from now on focus our discussion on ALL.
Childhood ALL includes a number of subtypes defined by cell lineage (B- or T-cell), differentiation status and genetic alterations (50). These biological characteristics differ by age distribution and clinical outcome and are, therefore, used for tailoring therapy to the predicted risk of relapse. The majority of children with ALL are classified as non-high risk (HR) patients and stratified to standard (S) or intermediate (I) risk protocols at diagnosis. HR characteristics at diagnosis include age >10 years, disease onset with a WBC >50,000/μl, and the presence of known poor genetic alterations (51). Treatment of childhood ALL consists of various chemotherapy components (induction, consolidation, intensification, and maintenance with CNS prophylaxis), with treatment intensity increasing slightly between children in the standard (SR) and those in the intermediate (IR) risk groups, applied according to the respective treatment protocols. In children presenting with HR-features, treatment entails a more aggressive multimodal chemotherapy and may include allogeneic stem cell transplantation (HSCT) to achieve cure. The overall treatment period varies between 2 and 2.5 years for the majority of international protocols. Despite the risk-adapted tailoring of therapy, the most important prognostic factor is how quickly and to what extent the tumor burden decreases once therapy is initiated. Response to treatment is measured by flow cytometry (and/or PCR) after ~4 weeks of induction therapy and, at the same time, the presence of minimal residual disease (MRD, i.e., leukemic cells) in the bone marrow compartment is evaluated. Absence or persistence of MRD at defined treatment points will stratify the patient to the final risk group and treatment intensity. However, in 10–15% of those children who achieve remission, the disease will return and relapse. The precise mechanisms behind treatment failure are not fully elucidated. To date, it is believed that ALL relapses emerge from subclonal populations that share some of the mutations identified in the dominant population at diagnosis, but also some additional new mutations associated with chemoresistance (52–54). Whether childhood ALL relapses are in any way related to the emergence of immune regulatory cells suppressing anti-cancer immunity (55) remains to be clarified.
Overall, little is known about the normal T-lymphocyte population in the pediatric ALL setting. Several studies have reported increased T- cell proportions (both single CD4+ and CD8+) including Tregs at the diagnosis of childhood B-cell ALL (56, 57). Studies have shown that children at the time of their ALL diagnosis have both higher proportions and absolute cell numbers of bulk CD4+ and CD8+ T cells in peripheral blood as well as increased proportions of Tregs compared to age-matched controls (58, 59). The proportion of Tregs remained higher throughout the first 4–5 weeks of induction therapy. In addition, a study investigated the phenotype of (non-malignant) T cells present in the bone marrow at the time of diagnosis in 39 children with ALL. Data showed that a higher CD4:CD8 ratio correlated with a better treatment response at day 15, and that this association was caused by non-Tregs (60). However, the significance of Tregs in the anti-cancer response or the pathogenesis of childhood ALL is far from clear.
The use of more intense chemotherapy protocols has led to a dramatic improvement in prognosis, unfortunately accompanied by both acute- and long-term toxicity that cause significant treatment-related morbidity. Several long-term defects in humoral immunity in children treated for ALL have been noted [reviewed in (61)], but how chemotherapy affects conventional T cell subsets and their functions is less well-studied (summarized in Table 1). The general understanding is that T cells remain more resistant toward chemotherapy than B cells (61, 67). Studies have shown that the T cell recovery at bulk level is delayed post-chemotherapy and that this delay is less pronounced in toddlers and young children (62, 70). It has also been suggested that helper T-cells have a longer recovery time than cytotoxic T-cells (65, 66, 71), an effect that has shown to be more prominent in more intense chemotherapy settings (63, 68). There is also evidence of a lower proportion of T cells in peripheral blood during chemotherapy, with a pronounced effect on naïve compared to memory T-cells, the latter remaining overrepresented during a 3-year period of chemotherapy (64, 69). One may speculate that thymic output of naïve T cells is severely affected by conventional chemotherapy including steroids, but data in favor of this is as yet incomplete in the literature as opposed to that for post-haematopoietic stem cell transplantation (HSCT) [reviewed in (72)].
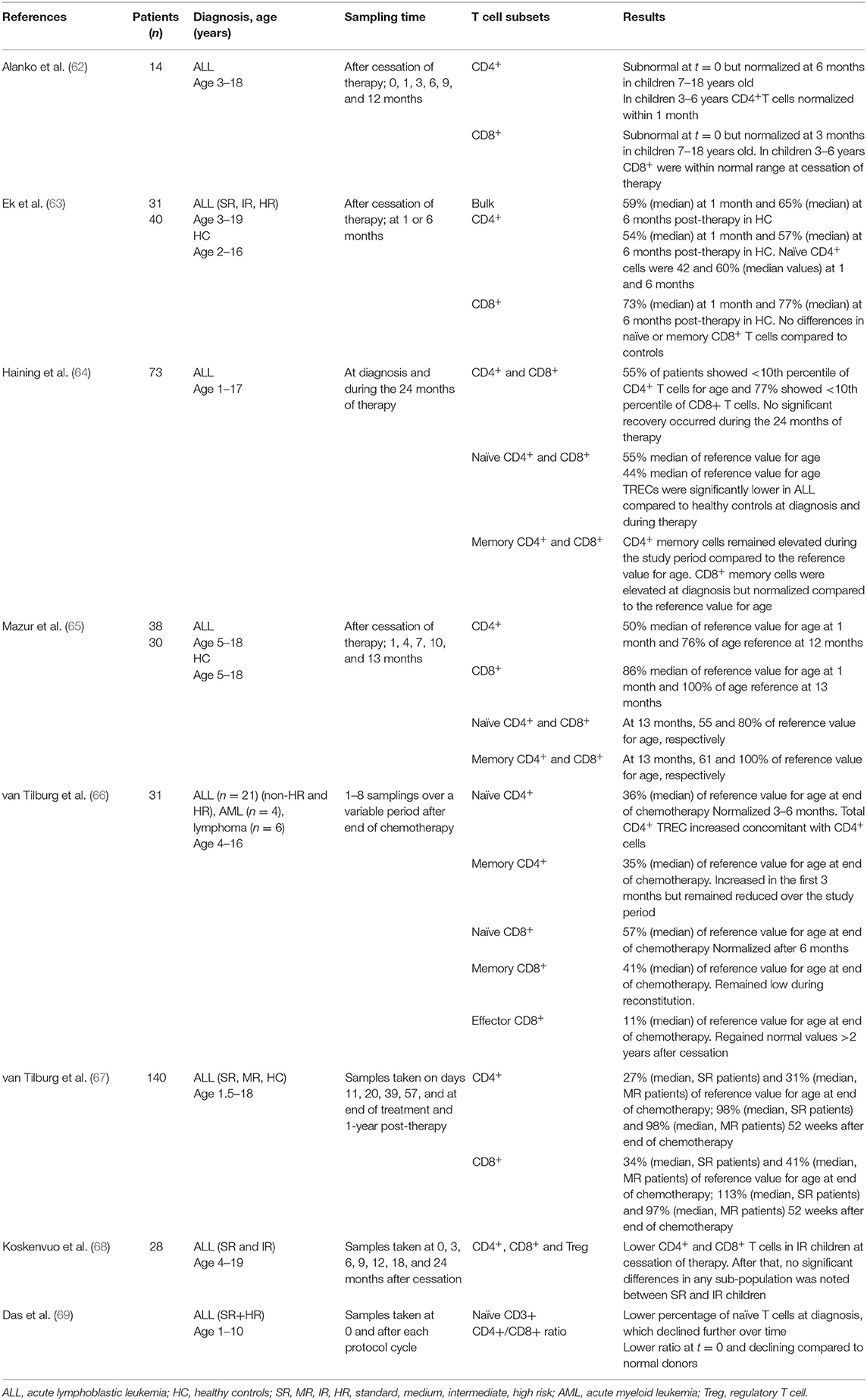
Table 1. Peripheral T cell populations in children during and after ALL therapy.
HSCT has been a curative option for a number of years for pediatric patients with relapsed ALL. Children considered for CD19-targeted immunotherapy are to a large extent in post-HSCT and possibly in their immune-reconstitution phase. There are several parameters that will impact on the kinetics of T-cell recovery after HSCT, such as the conditioning regimens, donor type and age, graft manipulation, type of graft-vs-host disease (GvHD), as well as treatment and prophylaxis [reviewed in (73)]. In children followed for T cell recovery after HSCT for hematological malignancies, studies suggest that recent thymic emigrants and TRECs recover within 6–12 months post-HSCT (11, 74, 75). Also, interventions aimed at treating GvHD itself appear to impact on thymic recovery and the naïve T cell pool, where an association between systemic steroid treatment and delayed CD4+ T-cell recovery has been noted (74). However, the current picture of post-HSCT dynamics is that Tregs are normalized within weeks followed by the recovery of memory CD4+ and CD8+ T cell within months. In contrast, the recovery of naïve T cell pool can take years, likely as this population relies on thymic output and not on peripheral homeostasis and expansion (76). Finally, while chemotherapy and HSCT have been shown to clearly impact the proportions of circulating T cells, there are almost no studies that systematically assess the functional responses of T cells (in particular the memory population) following ALL treatment. Although the proliferative potential of T cells to varicella zoster virus has been concluded as intact (64), these findings have not been corroborated by other cellular functions, such as cytotoxicity.
The introduction of new therapies for pediatric patients has been slow compared to adults, but in recent years an acceleration in both the availability and approval of novel agents for the treatment of childhood B-ALL has occurred. At least two of the most promising approaches–the CD19/CD3 bi-specific T-cell engager, blinatumomab, and the CD19-chimeric antigen receptor T (CAR-T) cell therapy–engage the patient's own T cells to bind and lyse the CD19+ B-ALL cells. During therapy, T cells are activated to unleash an aggressive immune response toward the leukemia, but also an inflammatory response that may result in severe cytokine-release syndrome and neurological toxicity. Both blinatumomab and CAR-T cell therapy have shown promising results in the treatment of relapsed/refractory pediatric B-ALL (77). von Stackelberg et al. (78) were among the first to show the efficacy of blinatumomab in heavily pre-treated pediatric ALL patients. Last year, a randomized phase 3 trial of blinatumomab vs. standard post-induction chemotherapy in high risk (early) first relapse of pediatric B-ALL showed a clear treatment benefit in the group of children who received blinatumomab: less toxicity, higher remission rates at day 29, and more children becoming eligible for HSCT (79). This study will probably shift the clinical therapeutic decisions in high risk ALL relapses in favor of immunotherapy over standard chemotherapy.
The first reported patient treated with second-generation anti-CD19 CAR T cells was an adult with refractory CLL who, after one infusion of CAR T cells, achieved remission that was sustained for 10 months (80). This was soon followed by several pediatric phase 1 trials with different CAR T cell products [reviewed in (77)]. In 2017, an important study was published in which the CAR T cell product was refined by introducing a consistent 1:1 CD4 to CD8 ratio, uniform CAR expression and limited T cell differentiation in the culture system (81). In total, 43 children and young adults with relapsed ALL were then infused and the intent-to-treat analysis showed a promising 89% response rate. In the pivotal global registration trial for tisagenlecleucel (82) the final analysis showed an outcome for 75 infused pediatric and young adult patients (total study cohort 107 screened and 92 enrolled patients). Intent-to-treat analysis showed a 66% response rate in this heavily pre-treated group of patients. In August 2017, the FDA approved tisagenlecleucel as the first commercial product for relapsed/refractory B cell malignancies in patients up to 25 years of age (83). However, even though both CAR-T cell therapy and bi-specific T-cell engagers show promising results, a significant proportion of patients do not respond or suffer severe toxicity. Even in responders, ALL immunotherapy, unless based on long-term persistent CAR T cell products, is mostly seen as a bridge to HSCT and potential cure. Whether CAR T cell therapy can replace HSCT in subsets of pediatric ALL patients is one of the most relevant questions in ALL HR therapy and will need to be addressed in prospective trials.
While immunotherapy is also a realistic option for children, one question that remains to be answered is how to improve the selection of patients for immunotherapy. Currently, T-cell expansion once immunotherapy is initiated can, to some extent, predict treatment response (84). Also, persistent B cell aplasia is a marker for long term response after anti-CD19 CAR-T cell and CD19/CD3 bi-specific T-cell engager therapy as long as CD19 expression is preserved on leukemic cells. But can we improve the selection of patients for immunotherapy upfront? In one recent study lymphocyte subsets in peripheral blood of adult patients receiving blinatumomab was investigated and responders were compared to non-responders (85). The most significant marker discriminating responders from non-responders was the percentage of Tregs, while there was no correlation in the absolute number of lymphocytes, CD4+ or CD8+ T cells, for either the naïve or effector populations. As Tregs are more abundant during childhood, similar studies in pediatric ALL patients are warranted. Recent data suggest that the T cell responses to blinatumomab may also be modulated by the expression of inhibitory molecules on the B-ALL leukemic cells (86). First, a number of co-stimulatory and inhibitory molecules regulating T cell responses were assessed by flow cytometry on patient-derived bone marrow blasts. PDL-1 was the most prominent inhibitory marker on these primary blasts while CD80 was the most highly expressed stimulatory marker. Interestingly, PDL-1 expression was significantly higher on patient leukemic cells in blinatumomab non-responders compared to responders and healthy controls. In the same report, a case regarding a 12-year-old girl with refractory ALL was presented. The patient had previous non-response to monotherapy with blinatumomab, but when combined with a PD-1 blocking antibody a partial anti-leukemic T cell effect was noted. This is in keeping with findings in adults, where blinatumomab was shown to activate Tregs to suppress effector responses by cytotoxic T cells, with the conclusion that therapeutic removal of Tregs could provide a means for converting blinatumomab non-responders to responders (85). Altogether, these studies show the potential of a detailed phenotypic characterization of T cell populations in patients that proceed to immunotherapy, which could possibly predict response and enable further tailoring of immunotherapy.
The generation, function, and regulation of immune responses by T cells during early life in humans are only partially understood. The fetal environment requires that the immune system tolerates maternal alloantigens. Following birth, a sudden shift in exposure to environmental antigens, including gut commensal bacteria, necessitates adaptation of immune responses to suit early life. Even though neonates and infants have substantial populations of T cells from birth (87), they are predominantly naïve, expressing distinct patterns of homing receptors compared to adults, and as a rule more prone to producing regulatory (14, 88) and innate-like (24) responses following activation (Figure 1). As a consequence of maintaining self-tolerance (including toward gut commensals) and to avoid immunopathology, the new born will be relatively susceptible to infections. This would be in keeping with the “disease tolerance” posit (89), which suggests that an increased susceptibility to infection in neonates is not a result of immaturity but rather one of an immunosuppressive environment, which is in part an active defense strategy likely to be disadvantageous for the young host with malignant cells.
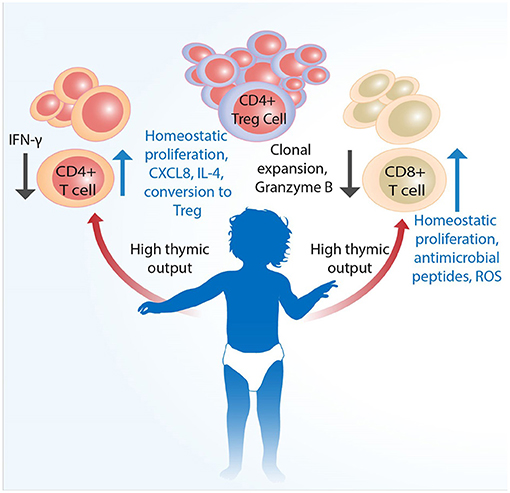
Figure 1. Infant T cells are qualitatively distinct from adult cells. Conventional T cells develop in the thymus, the size as well as output of which is largest at birth. Mature single CD4+ and CD8+ cells are proportionally less than Tregs across various tissues. Although infants have substantial populations of fully developed single CD4+ and CD8+ cells T cells from birth, these are predominantly naïve and more prone to type 2 cytokine and innate-like responses following activation. Naïve CD4+ T cells have higher propensity to develop into FoxP3+ Tregs to actively promote self-tolerance. An increased susceptibility to infection in neonates is likely not a result of only immaturity of adaptive immunity, but also that of qualitatively different functional capacity and higher level of immunosuppression, which together may negatively impact the eradication of malignant cells.
Today, it is not yet clear why certain pediatric patients respond to specific types of immunotherapies, while others do not. As we have highlighted, there are major differences in T cell populations in blood and tissue, particularly the markedly higher Treg to effector T cell ratios across the tissues of younger children compared to adults (12) that may influence response to therapy. These findings have been corroborated with recent murine studies on solid tumors showing that Tregs utilize IL-10 and IL-35 to exhaust CD8+ T cells (90, 91). In keeping with that CXCL8 and ROS impact cancer progression and T cell survival, respectively (92, 93), and that T cell expansion capability is a predictor of successful clinical expansion (94), selection of T cell subsets ahead of CAR T cell engineering could be a strategy to move the field forward (Figure 2). We envisage that the CD25+ T cell subset, containing the CD4+ effector Tregs, could be depleted prior to CAR T cell engineering without any significant risk for harmful autoimmunity as previously shown in humans (95, 96) and that the addition of IL-7 and IL-15 thereafter could optimize the ex vivo expansion of pediatric CAR T cells (69). Furthermore, there are interesting ongoing studies combining immune check-point inhibitors targeting PD-1 or CTLA-4 pathways, with CD19-CAR T cells or bi-specific T cells engagers [reviewed in (97)], which should also be considered in pediatric clinical trials. Lastly, treatment efficacy can be further enhanced through additional genetic modifications of next-generation, CAR T cells. These armored CAR T cells are genetically equipped to express cytokines, such as IL-7, IL-12, and IL-15, surface ligands, such as CD40L, or to secrete single-chain variable antibody fragments that block the PD-1/PD-1L pathway (97–99), which improve cell persistence and modulate the activity of other endogenous cells to favor T-cell mediated killing rather than suppression. All these approaches are likely to be applicable for optimization of CAR T cell efficacy and persistence in the pediatric ALL setting (Figure 2).
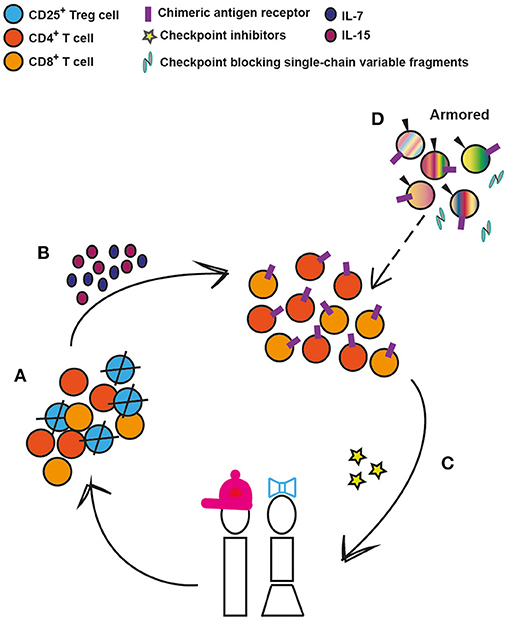
Figure 2. Conceivable strategies to enhance CAR T-cell efficacy and persistence in pediatric ALL. (A) Depletion of regulatory T cells (Tregs) prior to chimeric antigen receptor engineering. (B) Provision of proliferation and survival factors for T cells. (C) CAR T cells can be co-administered with immunomodulatory agents such as check-point inhibitors that target PD-1 or CTLA-4 pathways. (D) Armored CAR T cells are genetically modified to, along with the CAR, secrete cytokines, such as Il-7, IL-12, and IL-15, or to express immunomodulatory ligands such as CD40L or to secrete PD-1 blocking antibodies. These 4th generation cells can in the future replace the earlier-generation CAR T cells (depicted by dashed arrow).
In light of above it is necessary for future trials, to plan for follow-up of children who have received immunotherapy and become long term survivors. Firstly, it is unknown how the naïve and memory T cell pool is affected by conventional chemotherapy at various tissue sites over time. There are findings from non-human primate studies that show differential damage to the memory B-cell compartment by anti-cancer drugs according to the type of secondary lymphoid tissue (100) but equivalent data on the adaptive immune cells of humans remain to be elucidated. Lastly, there is, as yet little data on T cell recall responses in childhood cancer survivors. From our experiences in the clinic, we know that these children suffer from re-activation of herpesviruses and invasive fungal infections during ongoing therapy. Longitudinal data in this regard (especially following T cell-based immunotherapies) to assess the consequence of intense T cell activation on immunosenescence, is warranted in survivors of childhood cancer.
AN and SS-H: conceptualization, literature evaluation, figure preparation, funding acquisition, and writing up the paper. ES-E: literature evaluation, funding acquisition, and original draft writing. All authors made a substantial intellectual contribution and approved the manuscript for publication.
This work was supported by the Swedish Childhood Cancer Foundation (grant 2018-0084, AN), the Swedish Research Council (grant 2017-02001, AN), the Swedish Cancer Society (grants 2017/460, 20-1117, ES-E), the Cancer- and Allergy Foundation (ES-E), and Åke Wiberg Foundation (grant M19-0342, SS-H).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Burnet M. Cancer; a biological approach. I. The processes of control. Br Med J. (1957) 1:779–86. doi: 10.1136/bmj.1.5022.779
2. Ribatti D. The concept of immune surveillance against tumors. The first theories. Oncotarget. (2017) 8:7175–80. doi: 10.18632/oncotarget.12739
3. O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
5. Kumar BV, Connors TJ, Farber DL. Human T Cell Development, Localization, and Function throughout Life. Immunity. (2018) 48:202–13. doi: 10.1016/j.immuni.2018.01.007
6. Schatorje EJ, Gemen EF, Driessen GJ, Leuvenink J, van Hout RW, de Vries E. Paediatric reference values for the peripheral T cell compartment. Scand J Immunol. (2012) 75:436–44. doi: 10.1111/j.1365-3083.2012.02671.x
7. Huenecke S, Fryns E, Wittekindt B, Buxmann H, Konigs C, Quaiser A, et al. Percentiles of lymphocyte subsets in preterm infants according to gestational age compared to children and adolescents. Scand J Immunol. (2016) 84:291–8. doi: 10.1111/sji.12474
8. Correa-Rocha R, Perez A, Lorente R, Ferrando-Martinez S, Leal M, Gurbindo D, et al. Preterm neonates show marked leukopenia and lymphopenia that are associated with increased regulatory T-cell values and diminished IL-7. Pediatr Res. (2012) 71:590–7. doi: 10.1038/pr.2012.6
9. Nikolich-Zugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol. (2004) 4:123–32. doi: 10.1038/nri1292
10. Rudd BD. Neonatal T cells: a reinterpretation. Annu Rev Immunol. (2020) 38:229–47. doi: 10.1146/annurev-immunol-091319-083608
11. Junge S, Kloeckener-Gruissem B, Zufferey R, Keisker A, Salgo B, Fauchere JC, et al. Correlation between recent thymic emigrants and CD31+ (PECAM-1) CD4+ T cells in normal individuals during aging and in lymphopenic children. Eur J Immunol. (2007) 37:3270–80. doi: 10.1002/eji.200636976
12. Thome JJ, Bickham KL, Ohmura Y, Kubota M, Matsuoka N, Gordon C, et al. Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat Med. (2016) 22:72–7. doi: 10.1038/nm.4008
13. Schonland SO, Zimmer JK, Lopez-Benitez CM, Widmann T, Ramin KD, Goronzy JJ, et al. Homeostatic control of T-cell generation in neonates. Blood. (2003) 102:1428–34. doi: 10.1182/blood-2002-11-3591
14. Fukui T, Katamura K, Abe N, Kiyomasu T, Iio J, Ueno H, et al. IL-7 induces proliferation, variable cytokine-producing ability and IL-2 responsiveness in naive CD4+ T-cells from human cord blood. Immunol Lett. (1997) 59:21–8. doi: 10.1016/S0165-2478(97)00093-X
15. Hassan J, Reen DJ. IL-7 promotes the survival and maturation but not differentiation of human post-thymic CD4+ T cells. Eur J Immunol. (1998) 28:3057–65. doi: 10.1002/(SICI)1521-4141(199810)28:10<3057::AID-IMMU3057>3.0.CO;2-Z
16. Cookson S, Reen D. IL-15 drives neonatal T cells to acquire CD56 and become activated effector cells. Blood. (2003) 102:2195–7. doi: 10.1182/blood-2003-01-0232
17. Hassan J, Reen DJ. Human recent thymic emigrants–identification, expansion, and survival characteristics. J Immunol. (2001) 167:1970–6. doi: 10.4049/jimmunol.167.4.1970
18. Soares MV, Borthwick NJ, Maini MK, Janossy G, Salmon M, Akbar AN. IL-7-dependent extrathymic expansion of CD45RA+ T cells enables preservation of a naive repertoire. J Immunol. (1998) 161:5909–17.
19. Hazenberg MD, Otto SA, van Rossum AM, Scherpbier HJ, de Groot R, Kuijpers TW, et al. Establishment of the CD4+ T-cell pool in healthy children and untreated children infected with HIV-1. Blood. (2004) 104:3513–9. doi: 10.1182/blood-2004-03-0805
20. Wilson CB, Westall J, Johnston L, Lewis DB, Dower SK, Alpert AR. Decreased production of interferon-gamma by human neonatal cells. Intrinsic and regulatory deficiencies. J Clin Invest. (1986) 77:860–7. doi: 10.1172/JCI112383
21. Gibbons D, Fleming P, Virasami A, Michel ML, Sebire NJ, Costeloe K, et al. Interleukin-8 (CXCL8) production is a signatory T cell effector function of human newborn infants. Nat Med. (2014) 20:1206–10. doi: 10.1038/nm.3670
22. Webster RB, Rodriguez Y, Klimecki WT, Vercelli D. The human IL-13 locus in neonatal CD4+ T cells is refractory to the acquisition of a repressive chromatin architecture. J Biol Chem. (2007) 282:700–9. doi: 10.1074/jbc.M609501200
23. White GP, Watt PM, Holt BJ, Holt PG. Differential patterns of methylation of the IFN-gamma promoter at CpG and non-CpG sites underlie differences in IFN-gamma gene expression between human neonatal and adult CD45RO- T cells. J Immunol. (2002) 168:2820–7. doi: 10.4049/jimmunol.168.6.2820
24. Galindo-Albarran AO, Lopez-Portales OH, Gutierrez-Reyna DY, Rodriguez-Jorge O, Sanchez-Villanueva JA, Ramirez-Pliego O, et al. CD8(+) T cells from human neonates are biased toward an innate immune response. Cell Rep. (2016) 17:2151–60. doi: 10.1016/j.celrep.2016.10.056
25. Fuleihan R, Ahern D, Geha RS. Decreased expression of the ligand for CD40 in newborn lymphocytes. Eur J Immunol. (1994) 24:1925–8. doi: 10.1002/eji.1830240832
26. Brugnoni D, Airo P, Graf D, Marconi M, Lebowitz M, Plebani A, et al. Ineffective expression of CD40 ligand on cord blood T cells may contribute to poor immunoglobulin production in the newborn. Eur J Immunol. (1994) 24:1919–24. doi: 10.1002/eji.1830240831
27. Splawski JB, Nishioka J, Nishioka Y, Lipsky PE. CD40 ligand is expressed and functional on activated neonatal T cells. J Immunol. (1996) 156:119–27.
28. O'Neill RM, Reen DJ. Equivalent functional nuclear factor of activated T cell 1 mRNA and protein expression in cord blood and adult T cells. Transplantation. (2003) 76:1526–8. doi: 10.1097/01.TP.0000084308.19565.2B
29. Steinmann GG, Klaus B, Muller-Hermelink HK. The involution of the ageing human thymic epithelium is independent of puberty. A morphometric study. Scand J Immunol. (1985) 22:563–75. doi: 10.1111/j.1365-3083.1985.tb01916.x
30. Bertho JM, Demarquay C, Moulian N, Van Der Meeren A, Berrih-Aknin S, Gourmelon P. Phenotypic and immunohistological analyses of the human adult thymus: evidence for an active thymus during adult life. Cell Immunol. (1997) 179:30–40. doi: 10.1006/cimm.1997.1148
31. Jamieson BD, Douek DC, Killian S, Hultin LE, Scripture-Adams DD, Giorgi JV, et al. Generation of functional thymocytes in the human adult. Immunity. (1999) 10:569–75. doi: 10.1016/S1074-7613(00)80056-4
32. George AJ, Ritter MA. Thymic involution with ageing: obsolescence or good housekeeping? Immunol Today. (1996) 17:267–72. doi: 10.1016/0167-5699(96)80543-3
33. Steinmann GG. Changes in the human thymus during aging. Curr Top Pathol. (1986) 75:43–88. doi: 10.1007/978-3-642-82480-7_2
34. Thome JJ, Grinshpun B, Kumar BV, Kubota M, Ohmura Y, Lerner H, et al. Longterm maintenance of human naive T cells through in situ homeostasis in lymphoid tissue sites. Sci Immunol. (2016) 1:eaah6506. doi: 10.1126/sciimmunol.aah6506
35. Connors TJ, Baird JS, Yopes MC, Zens KD, Pethe K, Ravindranath TM, et al. Developmental regulation of effector and resident memory T cell generation during pediatric viral respiratory tract infection. J Immunol. (2018) 201:432–9. doi: 10.4049/jimmunol.1800396
36. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature. (2011) 478:250–4. doi: 10.1038/nature10434
37. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. (2013) 504:451–5. doi: 10.1038/nature12726
38. Sun M, Wu W, Chen L, Yang W, Huang X, Ma C, et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat Commun. (2018) 9:3555. doi: 10.1038/s41467-018-05901-2
39. Telesford KM, Yan W, Ochoa-Reparaz J, Pant A, Kircher C, Christy MA, et al. A commensal symbiotic factor derived from Bacteroides fragilis promotes human CD39(+)Foxp3(+) T cells and Treg function. Gut Microbes. (2015) 6:234–42. doi: 10.1080/19490976.2015.1056973
40. Mata Forsberg M, Bjorkander S, Pang Y, Lundqvist L, Ndi M, Ott M, et al. Extracellular membrane vesicles from lactobacilli dampen IFN-gamma responses in a monocyte-dependent manner. Sci Rep. (2019) 9:17109. doi: 10.1038/s41598-019-53576-6
41. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
42. Takahata Y, Nomura A, Takada H, Ohga S, Furuno K, Hikino S, et al. CD25+CD4+ T cells in human cord blood: an immunoregulatory subset with naive phenotype and specific expression of forkhead box p3 (Foxp3) gene. Exp Hematol. (2004) 32:622–9. doi: 10.1016/j.exphem.2004.03.012
43. Bjorkander S, Johansson MA, Hell L, Lasaviciute G, Nilsson C, Holmlund U, et al. FOXP3(+) CD4 T-cell maturity and responses to microbial stimulation alter with age and associate with early-life gut colonization. J Allergy Clin Immunol. (2016) 138:905–8.e4. doi: 10.1016/j.jaci.2016.04.027
44. Silva SL, Albuquerque AS, Serra-Caetano A, Foxall RB, Pires AR, Matoso P, et al. Human naive regulatory T-cells feature high steady-state turnover and are maintained by IL-7. Oncotarget. (2016) 7:12163–75. doi: 10.18632/oncotarget.7512
45. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. (2009) 30:899–911. doi: 10.1016/j.immuni.2009.03.019
46. Collier FM, Tang ML, Martino D, Saffery R, Carlin J, Jachno K, et al. The ontogeny of naive and regulatory CD4(+) T-cell subsets during the first postnatal year: a cohort study. Clin Transl Immunology. (2015) 4:e34. doi: 10.1038/cti.2015.2
47. Steliarova-Foucher E, Colombet M, Ries LAG, Moreno F, Dolya A, Bray F, et al. International incidence of childhood cancer, 2001-10: a population-based registry study. Lancet Oncol. (2017) 18:719–31. doi: 10.1016/S1470-2045(17)30186-9
48. Sexauer AN, Tasian SK. Targeting FLT3 signaling in childhood acute myeloid leukemia. Front Pediatr. (2017) 5:248. doi: 10.3389/fped.2017.00248
49. Pui CH, Yang JJ, Bhakta N, Rodriguez-Galindo C. Global efforts toward the cure of childhood acute lymphoblastic leukaemia. Lancet Child Adolesc Health. (2018) 2:440–54. doi: 10.1016/S2352-4642(18)30066-X
50. Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, et al. Childhood acute lymphoblastic leukemia: progress through collaboration. J Clin Oncol. (2015) 33:2938–48. doi: 10.1200/JCO.2014.59.1636
51. Moorman AV. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica. (2016) 101:407–16. doi: 10.3324/haematol.2015.141101
52. Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. (2008) 322:1377–80. doi: 10.1126/science.1164266
53. Oshima K, Khiabanian H, da Silva-Almeida AC, Tzoneva G, Abate F, Ambesi-Impiombato A, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Natl Acad Sci USA. (2016) 113:11306–11. doi: 10.1073/pnas.1608420113
54. Sun C, Chang L, Zhu X. Pathogenesis of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia and mechanisms underlying its relapse. Oncotarget. (2017) 8:35445–59. doi: 10.18632/oncotarget.16367
55. Colombo MP, Piconese S. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer. (2007) 7:880–7. doi: 10.1038/nrc2250
56. Ateyah ME, Hashem ME, Abdelsalam M. Epstein-Barr virus and regulatory T cells in Egyptian paediatric patients with acute B lymphoblastic leukaemia. J Clin Pathol. (2017) 70:120–5. doi: 10.1136/jclinpath-2016-203803
57. Wu CP, Qing X, Wu CY, Zhu H, Zhou HY. Immunophenotype and increased presence of CD4(+)CD25(+) regulatory T cells in patients with acute lymphoblastic leukemia. Oncol Lett. (2012) 3:421–4. doi: 10.3892/ol.2011.499
58. Salem ML, El-Shanshory MR, Abdou SH, Attia MS, Sobhy SM, Zidan MF, et al. Chemotherapy alters the increased numbers of myeloid-derived suppressor and regulatory T cells in children with acute lymphoblastic leukemia. Immunopharmacol Immunotoxicol. (2018) 40:158–67. doi: 10.1080/08923973.2018.1424897
59. Ismail AM, Abdou SM, Aty HA, Kamhawy AH, Elhinedy M, Elwageh M, et al. Autologous transplantation of CD34(+) bone marrow derived mononuclear cells in management of non-reconstructable critical lower limb ischemia. Cytotechnology. (2016) 68:771–81. doi: 10.1007/s10616-014-9828-7
60. Lustfeld I, Altvater B, Ahlmann M, Ligges S, Brinkrolf P, Rosemann A, et al. High proportions of CD4(+) T cells among residual bone marrow T cells in childhood acute lymphoblastic leukemia are associated with favorable early responses. Acta Haematol. (2014) 131:28–36. doi: 10.1159/000351429
61. Saghafian-Hedengren S, Soderstrom I, Sverremark-Ekstrom E, Nilsson A. Insights into defective serological memory after acute lymphoblastic leukaemia treatment: the role of the plasma cell survival niche, memory B-cells and gut microbiota in vaccine responses. Blood Rev. (2018) 32:71–80. doi: 10.1016/j.blre.2017.08.009
62. Alanko S, Salmi TT, Pelliniemi TT. Recovery of blood T-cell subsets after chemotherapy for childhood acute lymphoblastic leukemia. Pediatr Hematol Oncol. (1994) 11:281–92. doi: 10.3109/08880019409141671
63. Ek T, Mellander L, Andersson B, Abrahamsson J. Immune reconstitution after childhood acute lymphoblastic leukemia is most severely affected in the high risk group. Pediatr Blood Cancer. (2005) 44:461–8. doi: 10.1002/pbc.20255
64. Haining WN, Neuberg DS, Keczkemethy HL, Evans JW, Rivoli S, Gelman R, et al. Antigen-specific T-cell memory is preserved in children treated for acute lymphoblastic leukemia. Blood. (2005) 106:1749–54. doi: 10.1182/blood-2005-03-1082
65. Mazur B, Szczepanski T, Karpe J, Sonta-Jakimczyk D, Bubala H, Torbus M. Decreased numbers of CD4+ T lymphocytes in peripheral blood after treatment of childhood acute lymphoblastic leukemia. Leuk Res. (2006) 30:33–6. doi: 10.1016/j.leukres.2005.05.024
66. van Tilburg CM, van Gent R, Bierings MB, Otto SA, Sanders EA, Nibbelke EE, et al. Immune reconstitution in children following chemotherapy for haematological malignancies: a long-term follow-up. Br J Haematol. (2011) 152:201–10. doi: 10.1111/j.1365-2141.2010.08478.x
67. van Tilburg CM, van der Velden VH, Sanders EA, Wolfs TF, Gaiser JF, de Haas V, et al. Reduced versus intensive chemotherapy for childhood acute lymphoblastic leukemia: impact on lymphocyte compartment composition. Leuk Res. (2011) 35:484–91. doi: 10.1016/j.leukres.2010.10.005
68. Koskenvuo M, Ekman I, Saha E, Salokannel E, Matomaki J, Ilonen J, et al. Immunological reconstitution in children after completing conventional chemotherapy of acute lymphoblastic leukemia is marked by impaired B-cell compartment. Pediatr Blood Cancer. (2016) 63:1653–6. doi: 10.1002/pbc.26047
69. Das RK, Vernau L, Grupp SA, Barrett DM. Naive T-cell deficits at diagnosis and after chemotherapy impair cell therapy potential in pediatric cancers. Cancer Discov. (2019) 9:492–9. doi: 10.1158/2159-8290.CD-18-1314
70. Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, et al. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. N Engl J Med. (1995) 332:143–9. doi: 10.1056/NEJM199501193320303
71. Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, et al. Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood. (1997) 89:3700–7. doi: 10.1182/blood.V89.10.3700
72. Gaballa A, Clave E, Uhlin M, Toubert A, Arruda LCM. Evaluating thymic function after human hematopoietic stem cell transplantation in the personalized medicine era. Front Immunol. (2020) 11:1341. doi: 10.3389/fimmu.2020.01341
73. Boelens JJ, Hosszu KK, Nierkens S. Immune monitoring after allogeneic hematopoietic cell transplantation: toward practical guidelines and standardization. Front Pediatr. (2020) 8:454. doi: 10.3389/fped.2020.00454
74. Flinn AM, Roberts CF, Slatter MA, Skinner R, Robson H, Lawrence J, et al. Thymopoiesis following HSCT; a retrospective review comparing interventions for aGVHD in a pediatric cohort. Clin Immunol. (2018) 193:33–7. doi: 10.1016/j.clim.2018.01.006
75. Clave E, Busson M, Douay C, Peffault de Latour R, Berrou J, Rabian C, et al. Acute graft-versus-host disease transiently impairs thymic output in young patients after allogeneic hematopoietic stem cell transplantation. Blood. (2009) 113:6477–84. doi: 10.1182/blood-2008-09-176594
76. Dekker L, de Koning C, Lindemans C, Nierkens S. Reconstitution of T Cell subsets following allogeneic hematopoietic cell transplantation. Cancers (Basel). (2020) 12:1974. doi: 10.3390/cancers12071974
77. Winters A, Gore L. Moving immunotherapy into the front line in ALL. Hematology Am Soc Hematol Educ Program. (2019) 2019:209–17. doi: 10.1182/hematology.2019000017
78. von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett TM, Rizzari C, et al. Phase I/Phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. (2016) 34:4381–9. doi: 10.1200/JCO.2016.67.3301
79. Brown PA, Ji L, Xu X, Devidas M, Hogan L, Borowitz MJ, et al. A randomized phase 3 trial of blinatumomab vs. chemotherapy as post-reinduction therapy in high and intermediate risk (HR/IR) first relapse of B-Acute lymphoblastic leukemia (B-ALL) in children and adolescents/young adults (AYAs) demonstrates superior efficacy and tolerability of blinatumomab: a report from children's oncology group study AALL1331. Blood. (2019) 134 (Suppl.2): LBA-1. doi: 10.1182/blood-2019-132435
80. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
81. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
82. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
83. Lesch S, Benmebarek MR, Cadilha BL, Stoiber S, Subklewe M, Endres S, et al. Determinants of response and resistance to CAR T cell therapy. Semin Cancer Biol. (2019) 65:80–90. doi: 10.1016/j.semcancer.2019.11.004
84. Nagele V, Kratzer A, Zugmaier G, Holland C, Hijazi Y, Topp MS, et al. Changes in clinical laboratory parameters and pharmacodynamic markers in response to blinatumomab treatment of patients with relapsed/refractory ALL. Exp Hematol Oncol. (2017) 6:14. doi: 10.1186/s40164-017-0074-5
85. Duell J, Dittrich M, Bedke T, Mueller T, Eisele F, Rosenwald A, et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia. (2017) 31:2181–90. doi: 10.1038/leu.2017.41
86. Feucht J, Kayser S, Gorodezki D, Hamieh M, Doring M, Blaeschke F, et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget. (2016) 7:76902–19. doi: 10.18632/oncotarget.12357
87. Zens KD, Connors T, Farber DL. Tissue compartmentalization of T cell responses during early life. Semin Immunopathol. (2017) 39:593–604. doi: 10.1007/s00281-017-0648-7
88. Mold JE, Michaelsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science. (2008) 322:1562–5. doi: 10.1126/science.1164511
89. Harbeson D, Ben-Othman R, Amenyogbe N, Kollmann TR. Outgrowing the immaturity myth: the cost of defending from neonatal infectious disease. Front Immunol. (2018) 9:1077. doi: 10.3389/fimmu.2018.01077
90. Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, et al. Interleukin-35 limits anti-tumor immunity. Immunity. (2016) 44:316–29. doi: 10.1016/j.immuni.2016.01.013
91. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive plasticity of IL-10(+) and IL-35(+) Treg cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. (2019) 20:724–35. doi: 10.1038/s41590-019-0346-9
92. Zhao Z, Wang S, Lin Y, Miao Y, Zeng Y, Nie Y, et al. Epithelial-mesenchymal transition in cancer: role of the IL-8/IL-8R axis. Oncol Lett. (2017) 13:4577–84. doi: 10.3892/ol.2017.6034
93. Hildeman DA. Regulation of T-cell apoptosis by reactive oxygen species. Free Radic Biol Med. (2004) 36:1496–504. doi: 10.1016/j.freeradbiomed.2004.03.023
94. Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med. (2016) 8:320ra3. doi: 10.1126/scitranslmed.aad5222
95. Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci U S A. (2013) 110:17945–50. doi: 10.1073/pnas.1316796110
96. Tanaka A, Nishikawa H, Noguchi S, Sugiyama D, Morikawa H, Takeuchi Y, et al. Tyrosine kinase inhibitor imatinib augments tumor immunity by depleting effector regulatory T cells. J Exp Med. (2020) 217:e20191009. doi: 10.1084/jem.20191009
97. Cerrano M, Ruella M, Perales MA, Vitale C, Faraci DG, Giaccone L, et al. The advent of CAR T-Cell therapy for lymphoproliferative neoplasms: integrating research into clinical practice. Front Immunol. (2020) 11:888. doi: 10.3389/fimmu.2020.00888
98. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. (2018) 36:847–56. doi: 10.1038/nbt.4195
99. Srivastava S, Riddell SR. Chimeric antigen receptor T cell therapy: challenges to bench-to-bedside efficacy. J Immunol. (2018) 200:459–68. doi: 10.4049/jimmunol.1701155
Keywords: T cells, childhood, leukemia, chemotherapy, immunotherapy
Citation: Saghafian-Hedengren S, Sverremark-Ekström E and Nilsson A (2021) T Cell Subsets During Early Life and Their Implication in the Treatment of Childhood Acute Lymphoblastic Leukemia. Front. Immunol. 12:582539. doi: 10.3389/fimmu.2021.582539
Received: 12 July 2020; Accepted: 01 February 2021;
Published: 04 March 2021.
Edited by:
Monika C. Brunner-Weinzierl, University Hospital Magdeburg, GermanyReviewed by:
Sarah K. Tasian, University of Pennsylvania, United StatesCopyright © 2021 Saghafian-Hedengren, Sverremark-Ekström and Nilsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shanie Saghafian-Hedengren, c2hhbmllLmhlZGVuZ3JlbkBraS5zZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.