
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 26 February 2021
Sec. Comparative Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.568729
This article is part of the Research TopicEvolution and Comparative Immunology of Immune Systems in Marine OrganismsView all 14 articles
 Thomas J. Colgan1*†‡
Thomas J. Colgan1*†‡ Peter A. Moran1,2†‡
Peter A. Moran1,2†‡ Louise C. Archer1,2
Louise C. Archer1,2 Robert Wynne1,2
Robert Wynne1,2 Stephen A. Hutton1,2Philip McGinnity1,3
Stephen A. Hutton1,2Philip McGinnity1,3 Thomas E. Reed1,2†
Thomas E. Reed1,2†Vertebrates have evolved a complex immune system required for the identification of and coordinated response to harmful pathogens. Migratory species spend periods of their life-cycle in more than one environment, and their immune system consequently faces a greater diversity of pathogens residing in different environments. In facultatively anadromous salmonids, individuals may spend parts of their life-cycle in freshwater and marine environments. For species such as the brown trout Salmo trutta, sexes differ in their life-histories with females more likely to migrate to sea while males are more likely to stay and complete their life-cycle in their natal river. Salmonids have also undergone a lineage-specific whole genome duplication event, which may provide novel immune innovations but our current understanding of the differences in salmonid immune expression between the sexes is limited. We characterized the brown trout immune gene repertoire, identifying a number of canonical immune genes in non-salmonid teleosts to be duplicated in S. trutta, with genes involved in innate and adaptive immunity. Through genome-wide transcriptional profiling (“RNA-seq”) of male and female livers to investigate sex differences in gene expression amplitude and alternative splicing, we identified immune genes as being generally male-biased in expression. Our study provides important insights into the evolutionary consequences of whole genome duplication events on the salmonid immune gene repertoire and how the sexes differ in constitutive immune expression.
Species that migrate face a range of challenges. First, the physical act of migration can be metabolically and energetically demanding, resulting in trade-offs with other metabolically intensive physiological processes, such as immunity, when resources are limiting (1–3). Second, migratory species move through different environments and hence may be exposed to different pathogens and parasites (4, 5). For aquatic species that exhibit diadromy—the ability to move between marine and freshwater environments—an efficient immune system is required to cope with the challenges imposed by living in, and moving between, different osmotic environments with different pathogen and parasite communities.
In vertebrates, a sophisticated immune system has evolved that performs two vital functions: 1) the recognition and distinction of invasive pathogenic organisms from normal cells (“self”), and 2) coordinating an appropriate response through triggering pathways responsible for the synthesis of effector molecules that directly or indirectly reduce or remove the pathogenic threat (6, 7). Aside from detection of non-self-pathogenic organisms, the immune system also functions in the removal of abnormal cells and thus provides an important role in reducing the development and onset of disease.
Given the importance of the immune system in preventing infection and establishment of disease, there is strong selection pressure acting on immune genes. In response to these pressures, immune genes are generally fast evolving (8, 9). Additional innovations in immune potential can also arise through tandem duplication, retrotransposition, larger scale duplication of chromosomal regions or entire chromosomes, as well as whole genome duplication events (WGD). For example, two rounds of WGD events are suggested to have contributed to the genesis of the adaptive immune system in vertebrates (10). Indeed, WGD events produce duplicated copies of all genes, which selection can act on resulting in retention or removal of one or both copies. In terms of removal, as with general duplication events, gene loss can occur through reduced purifying selection resulting in functional divergence between the copies. Accumulation of deleterious mutations may eventually lead to one copy becoming non-functional (11). Gene loss may also be adaptive. For example, loss of gene function in a duplicated copy may be adaptive in response to environmental challenges (12, 13), including pathogens (14, 15). Alternatively, after duplication of a gene, functional divergence can occur whereby one copy evolves a slightly different or entirely novel function relative to the other copy. Functional divergence can result in subfunctionalization, whereby individual copies specialize on different components of the same function originally performed by the ancestral gene pre-duplication (i.e., “division of labor”), or neofunctionalization, whereby one copy may evolve a novel function (16).
As stated previously, the continuous expression and activation of immunity can be metabolically costly, resulting in trade-offs with fecundity and longevity (17). In particular, the sexes can differ in their levels of immune function with greater immunocompetence generally evident in females in comparison to males (18–23). Lower immunocompetence in males has been attributed in proximate terms to differences in circulating levels of hormones and their effects, such as androgen, and in ultimate terms to differences in life-history strategies, with males investing more in reproduction and associated secondary sexual characters while females invest in immune function and longevity (24). Moreover, in facultatively migratory species, differences are often evident between the sexes in the rate or timing of migration (25–28), which in turn has implications for exposure to pathogens and parasites and investment in immune defense. Differences in the level of immunity between the sexes can be detected at the transcriptional level (22, 29–31), whereby genes associated with the immune system may differ in their expression between the sexes. Approaches such as genome-wide transcriptomics (“RNA-seq”) are important tools for high-resolution detection and profiling of genes that differ in expression amplitude, as well as splicing, between the sexes. Such approaches have been applied to improve our understanding of genes underlying sexually dimorphic traits in a variety of taxa (32–35), including immunity (36).
An interesting study system for understanding the evolution and expression of the immune system and how it differs between the sexes are the salmonids, a group of culturally, economically and ecologically important teleost fish (37). The salmonids consist of approximately 70 species across 11 genera that have evolved flexible life-histories with a diversity of ecological adaptations that allow for migration to, and survival in, a range of freshwater and marine aquatic environments (38). There is a gradient from entirely non-anadromous species (which complete their entire life cycles in fresh water) through facultatively anadromous species to species that are almost entirely anadromous (39–44). Within facultatively anadromous species, rates of anadromy and other migratory tactics can vary between populations, and even among individuals within populations, particularly between the sexes with females more likely to undergo migration than males (42, 43, 45, 46).
Survival for prolonged periods in different aquatic environments, which contain different pathogenic threats, requires an immune system that can detect and respond to a diverse array of immune challenges. These factors likely placed strong selection pressures that shaped immune system evolution but additional immune novelty in salmonids may be the result of the salmonid-specific WGD event. The common ancestor of salmonids underwent an autotetraploidization event approximately 80–100 mya (47–49), which is believed to have contributed to genomic and phenotypic innovation as well as speciation within the salmonids (50). While the timing of the WGD event and first appearance of the anadromy during salmonid evolution are highly temporally detached, speciation rates were shown to be elevated within anadromous salmonids compared to non-anadromous salmonids with ecological factors, such as climate cooling, rather than the WGD suggested as the primary drivers of anadromy-linked diversification (47). The sequencing of the Atlantic salmon (Salmo salar) genome revealed that approximately 25% of the genome is undergoing delayed rediploidization, which is associated with major chromosomal rearrangements (48). Delayed rediploidization has been ongoing in parallel with speciation events, which has led to the proposal of ‘lineage-specific ohnolog resolution’ (LORe) as a mechanism to understand the impact of delayed rediploidization on the functional divergence of ohnologs across lineages that share a common ancestral WGD event (50). Under LORe, species divergence occurs before the rediploidization process is complete resulting in functional divergence of ohnologs independently within each lineage. The alternative model predicts rediploidization is completed prior to species diversification resulting in functional divergence of ohnologs within a shared ancestor (“Ancestral Ohnologue Resolution” or “AORe” model).
Due to shared selection pressures acting on ohnologs within a common ancestor, ohnologs that diverged in an ancestor are predicted to possess conserved functions across modern lineages (50). Recent genomic studies on salmonids have found evidence of relatively high rates of retention of duplicated genes arising from this most recent WGD (>50% of genes being found in functional ohnolog pairs), as well as evidence of neofunctionalization, whereby copies may diverge and are suggested to perform novel functions (48, 51, 52). Indeed, the evolutionary consequences of such events have served as impetus to examine functional divergence among ohnologous genes with putative immunological roles in salmonids (53–56). Despite these advances in our fundamental understanding of salmonid immunology, we understand less for species, such as the facultatively anadromous, brown trout (Salmo trutta). Recent declines in sea migration of S. trutta populations in Ireland and Scotland have raised concerns over the impact of disease and parasites, such as sea lice, on brown trout health and population performance (57–59). Given the enormous selection pressures exerted by parasites on their hosts, host defenses, including components of the host immune system, would be required to adapt to tolerate or resist so as to increase host survival and fitness (60). Therefore, increasing our understanding of brown trout immunity is warranted.
Here we had three main aims: Firstly, to characterize predicted immune genes found in the brown trout genome. For this, we used comparative genomic approaches to identify these genes in S. trutta based on homology with immune genes annotated in model organisms, such as zebrafish (Danio rerio), mouse (Mus musculus) and human (Homo sapiens). Given the overall enlarged gene repertoire in salmonids due to the salmonid-specific WGD event, as well as the strong selection pressures placed on immune genes by pathogens from both marine and freshwater environments, we would expect retention of most canonical immune genes, as well as the potential expansion of beneficial immune gene families. Secondly, we aimed to investigate evolutionary patterns of functional conservation and divergence, including gene loss in S. trutta immune ohnologs. Our final aim was to identify immune genes with sex-biased expression profiles. For this approach, we performed transcriptomic analyses on the liver, an important immunocompetent organ (61, 62), and quantified differences between males and females in gene expression amplitude, as well as alternative splicing, to identify molecular processes underlying sex differences in immune transcription and regulation.
To identify genes in brown trout with putative immune function, we obtained gene lists for annotated immune genes in the zebrafish, Danio rerio [obtained from Zebrafish Information Network (ZFIN) database; (63)], as well as for the mouse, Mus musculus (obtained from Mouse Genome Informatics (MFI) database (64) and human, Homo sapiens [obtained from ImmPort (65)]. Using biomaRt [v. 2.45.8; (66)], we parsed the Ensembl BioMart database to identify “high confidence” orthologs found in the brown trout genome based on homology (phylogenetic protein trees), as well as conserved synteny (gene order conservation score and whole genome alignment scores). The threshold for classification of a brown trout gene as a ‘high confidence’ ortholog included: 1) a minimum gene conservation score of 50, which indicates the percentage of how many of the four closest neighbors of a gene match been orthologous pairs (i.e., at least two (50%) of neighboring genes match); 2) a minimum whole genome alignment score of 50; and 3) a minimum protein percentage identity of 50%. This approach identified 2,275 brown trout genes with homology to immune genes in three model organisms (Supplemental Information Table 1).
As a preliminary measure to understand immune gene repertoires across salmonids, we investigated the presence of homologues of putative brown trout immune genes in other salmonid species. To identify homology relationships, we first followed the approach outlined by Gillard et al. (51), and obtained protein sequences for the predicted proteomes from Ensembl [release 101; (67)] for twelve teleost fish, including the zebrafish (Danio rerio; GRCz11: GCA_000002035.4), three-spined stickleback (Gasterosteus aculeatus; BROAD S1), Japanese medaka (Oryzias latipes; ASM223467v1: GCA_002234715.1), Northern pike (Esox Lucius; Eluc_v4: GCA_004634155.1), Atlantic herring (Clupea harengus; Ch_v2.02: GCA_900700415.1), Atlantic cod (Gadus morhua; gadMor1), and guppy (Poecilia reticulata; GCA_000633615.2), and the salmonids, rainbow trout (Oncorhynchus mykiss; Omyk_1.0: GCA_002163495.1), Coho salmon (Oncorhynchus kisutch; Okis_V2: GCA_002021735.2), Chinook salmon (Oncorhynchus tshawytscha; Otsh_v1.0: GCA_002872995.1), Atlantic salmon (Salmo salar; ICSASG_v2: GCA_000233375.4), and brown trout (Salmo trutta; fSalTru1.1: GCA_901001165.1). From Ensembl, we also obtained two mammalian outgroups, human, Homo sapiens (GRCh38.p13: GCA_000001405.28), and mouse, Mus musculus (GRCm38.p6: GCA_000001635.8). For protein FASTA files downloaded from Ensembl, we extracted the longest protein isoform per gene per species using the OrthoFinder script “primary_transcripts.py” [v.2.3.11; (68, 69)]. We used OrthoFinder to assign groups of orthologs based on protein sequence similarity. Multiple sequence alignment was performed for protein sequences within each orthogroup using MAFFT. Maximum-likelihood trees were estimated using FastTree as implemented within OrthoFinder. OrthoFinder constructed a total of 47,752 orthogroups of which 22,101 were species-specific groups (Supplemental Information Table 2). The percentage of genes per species represented in the orthogroups was high (92%–97.5%). We parsed these orthogroups using the putative brown trout immune genes identified through the Ensembl-based analysis (n = 2,275 genes) and identified putative immune homologs present in 1,227 orthogroups (Supplemental Information Table 3). As a secondary measure to understand immune gene expansions and losses within the salmonids, we ran OrthoFinder using only the five salmonid proteomes, as well as Northern pike, the closest related species that did not undergo a fourth WGD event (70). While other salmonid genomes are publicly available, we restricted our analysis to Ensembl-generated datasets to account for gene models being predicted using a similar annotation pipeline (Supplemental Information Table 4). The presence of single copy orthologs in both Northern pike and all sequenced salmonids suggests that gene loss occurred in a common ancestor of modern salmonids soon after the WGD event or multiple independent losses have occurred across the salmonids.
To examine the functional expression of putative immune genes of S. trutta, we obtained available RNA-seq libraries for eight tissues from the NCBI (National Center for Biotechnology Information) Short Read Archive (SRA) database (BioProject: PRJEB33055). For each sample, we quantified transcript abundance using the quasialigner, Salmon [v.0.12.0; (71)]. Using these transcript abundances, we calculated gene-level counts using tximport [v.1.14.2; (72); Supplemental Information Table 5] and corrected for library-sizes using DESeq2 [v.1.26.0; (73)]. For each tissue, we quantified the total number of immune genes expressed per tissue, as well as compared relative abundance of immune gene expression for each tissue against non-immune genes.
To identify putative immune ohnologs, we first extracted within-species paralogs for brown trout using the Ensembl BioMart database (filter: “with_strutta_paralog”). Ensembl employs a pipeline that through protein trees can time and predict the last common ancestor for paralogs. We then subsetted putative immune genes identifying putative species-specific paralogs (n = 456), genus-specific (n = 344), as well as paralogs predicted to have arisen in the Salmoninae ancestor, which may represent paralogs generated as a result of the Salmonid-specific WGD. For each of these “Salmoninae” paralogs present in the brown trout genome, we subsetted and retained paralogs that shared at least 85% protein sequence similarity. We then obtained the Northern pike ortholog of each putative paralog. We extracted paralog pairs where only a single non-duplicated pike ortholog was evident, which matched only two paralogs in brown trout. For this, we kept only Northern pike orthologs that shared at least 85% sequence similarity to each of the brown trout paralogs. We also investigated the physical genomic coordinates of each ohnolog identifying that collinearity among putative immune ohnolog pairs is a global feature of the data, consistent with these genes being ohnologs. We therefore consider these brown trout paralogs as putative ohnologs. We found no significant difference (paired two sample t-test; p > 0.05) in predicted protein length between each ohnolog pair or their respective non-duplicated ortholog in Northern pike. This approach resulted in the identification of 434 ohnolog pairs (n = 868 genes) with potential immune function. We also extracted Atlantic salmon homologs of putative immune ohnolog pairs in brown trout and compared overlap with the S. salar ohnolog pairs described by Bertolotti et al. (74). Of the 434 brown trout immune ohnolog pairs, 408 pairs shared homologs in Atlantic salmon. Of this number, 88% (n = 362) were also identified as ohnolog pairs within the analysis of Bertolotti et al. providing independent evidence for the classification of such immune genes as ohnologs.
For each ohnolog pair, we estimated the evolutionary distance between each pair and the non-duplicated ortholog in Northern pike using distmat from EMBOSS [v.6.6.0; (75)] (in terms of amino acid substitutions per 100 amino acids; Supplemental Information Table 6). This allowed for the determination of copies that were more conserved or diverged in terms of protein sequence while accounting for variation in predicted amino acid length. To explore variation in functional domain architecture, we obtained InterPro functional domains assigned to predicted proteins for each pair from Ensembl BioMart (67). We then counted and compared the number of assigned domains for each ohnolog pair to identify any differences, which may be consistent with the genes performing different functions. As variation in protein length may explain variation in protein domains, we also compared predicted protein lengths between ohnologs identifying no significant difference in length (paired two sample t-test; p = 0.6).
Using the gene-level counts generated by eight tissues, we generated co-expression clusters for putative immune ohnologs using Clust [v.1.10.8; (76)]. Here differences in expression profiles between ohnologs may reflect functional divergence, which has been determined in other salmonid species (48, 49, 51, 77). To determine divergence, we extracted ohnolog pairs that were assigned to different co-expression clusters and used corrected counts per sample generated by Clust for visualization purposes. Gene assignments to clusters are provided in Supplemental Information Table 7.
To understand differences in selection acting on each pair, we calculated dN/dS ratios between each brown trout ohnolog and their respective non-duplicated Northern pike ortholog (Supplemental Information Table 6). We first aligned predicted protein sequences using clustal Omega [v.1.2.4; (78)] and converted the aligned sequences to nucleic acids using pal2nal [v.14; (79)] to obtain codon-based nucleic acid alignments. We then ran codeML as part of PAML [v.4.9; (80)] to estimate dN/dS ratios.
To provide additional functional information on brown trout immune genes, and to examine potential sex differences in immune expression, we sampled livers from mature male and female S. trutta for transcriptomic analyses (Supplemental Information Table 8). For the present study, the fish used originated from a larger experimental aquaculture project that explored the expression of alternative life history tactics in brown trout (81, 82). This wider set-up involved 18 different tanks all connected within a recirculating aquaculture system (RAS), but in the current analysis, all our sampled fish came from two independent tanks (with each tank comprising fish from a different genetic background). Full details on the origins of the fish, fish husbandry procedures and other general information are given in Archer et al. (81, 82).
All fish were dissected between May and June 2018 (the endpoint of the larger experiment) when the fish were between two and three calendar years old. We collected livers from each individual fish and transferred the tissues to fresh 2 ml Eppendorf tubes containing RNALater solution. Samples were kept for 24 h at room temperature, and then stored at -80°C for later analysis. Samples during dissection were visually checked for mature testes or ovaries. For confirmation of genetic sex, we also obtained caudal fin clips from each fish during dissection and stored in 100% ethanol before subsequent genotyping.
Total RNA was extracted from a total of 37 tissues using TRIzol. Specifically, we extracted RNA from fifteen livers from males and from 22 livers from females. For RNA extractions, we removed each sample from -80°C long-term storage and incubated them on ice to thaw. Using a pipette, the RNALater solution was removed. We added 1ml of autoclaved phosphate-buffered saline to each tissue to briefly wash them before using sterilized forceps to transfer each washed tissue to an individual 2ml screw-cap homogenization tube. To each sample, 200ul of TRIzol was added and the sample was transferred to -80°C storage. Tissue disruption was performed using a 2 mm steel bead and a Tissuslyer II (Qiagen, UK). To each sample, a 2 mm steel bead was added and samples were homogenized at 30 Hz for 30 s. Post-homogenization, samples were visually inspected to ensure thorough disruption. Total RNA was extracted using chloroform followed by isopropanol precipitation. Precipitated RNA was washed using three washes of ethanol before elution in the elution buffer (Sigma, UK). Total RNA was purified using the Sigma GenElute Mammalian Total RNA kit. Quality assessment was initially performed using a NanoDrop ND-1000 (ThermoFisher, UK) while an accurate assessment of quantity was estimated using the Qubit fluorometer, followed by a TapeStation 2200 (Agilent, UK).
mRNA-enriched library preparation was performed for each individual sample using the NEBNext® Ultra™ RNA Library Preparation kit and sequencing performed on an Illumina NovaSeq6000. Library preparation and sequencing was performed by NovoGene, Hong Kong. Sequencing resulted in a median of 26.1 million paired-end (PE) reads (2*150 bp) per individual (min. 20.1 million PE reads; max. 34.2 million PE reads). A combined total of 980 million PE reads were generated. Summary of sample information is provided in Supplemental Information Table 8.
We quality assessed raw FASTQ sequences using FastQC [v.0.11.8; (83)] to identify adaptor contamination and sequences of low quality. Raw reads were aligned against the reference genome assembly (GCA_901001164.1) for Salmo trutta using STAR [v.2.7.0a; (84)]. Alignment statistics were calculated using samtools flagstat [v.1.9; (85)]. Summary statistics of alignments were compiled using Qualimap [v.2.2.1; (86)] and the output visualized using MultiQC [v.1.7; (87)].
We quantified transcript abundance using two complementary approaches. Similar to the approach outlined above, using Salmon, we quasialigned raw reads against cDNA sequences for coding and non-coding genes available for S. trutta from Ensembl. We calculated gene-level counts using tximport and loaded these values into a DESeq2 object using DESeq2. Raw gene-level counts are provided in Supplemental Information Table 9. We performed a Wald test implemented by DESeq2 to identify significantly differentially expressed genes between males and females (Benjamini-Hochberg (BH) adjusted p < 0.05; Supplemental Information Table 10). To explore similarities in expression profiles across all samples, a principal component analysis was performed with DESeq2 for all samples using gene-level counts for 33,228 genes expressed in the liver following a variance stabilization transformation implemented by DESeq2. As a complementary measure, for each individual, we examined gene expression using a traditional aligner-based approach. We aligned reads against the brown trout reference genome assembly using STAR and extracted gene-level counts from the resultant BAM files using HTSeq [v. 0.11.2; (88); Supplemental Information Table 11]. We found an extremely high correlation (Pearson’s correlation: r=0.9, p < 2.2e-16) between the mean gene-level counts for both this approach, as well as the Salmon approach outlined above. This is also true for comparisons for individual samples across both approaches (lowest r=0.83, highest r=0.97). As an additional measure, we analyzed and compared the number of differentially expressed genes identified with DESeq2 when using the output of either transcript quantification approach (Supplemental Information Table 12). We found that >85% of genes identified as significantly differentially expressed between the sexes by Salmon were also identified as significant by the STAR-HTSeq approach. As quasi- and pseudoaligners, such as Salmon and kallisto, have higher accuracy and consistency in transcript quantification compared to traditional aligners (89), we report only the findings of our Salmon analysis.
While the sexes may express genes at different amplitudes, they may also express different isoforms of the same gene. We used the intron-splice analyzer leafcutter [v.0.2.8; (90)] to investigate differences between the sexes. Briefly, for each sample, we aligned raw reads against the brown trout genome assembly (GCA_901001165.1) using STAR (–outSAMstrandField intronMotif, –twopassMode Basic). We extracted splice junctions using bam2junc.sh and generated intron clusters using leafcutter.py (parameters: –minclureads 50, –maxintronlen 500000, –minreads 5). We then performed differential intron analysis using leafcutter_ds.R whereby for a cluster to be included it must be identified within at least five individuals within each sex (parameters: –min_samples_per_intron 5, –min_samples_per_group 5) to allow for investigation of genes with signatures of differential intron usage between the sexes. Leafcutter implements a log likelihood ratio test comparing a null model that assumes there is no difference between groups and an alternative model, which does. We adjusted resulting p values for multiple testing (False Discovery Rate < 0.05; Supplemental Information Table 13).
Gene Ontology term enrichment analysis and visualization of outputs were performed using modified scripts generated by Colgan et al. (91). As the zebrafish is a model organism with extensive functional annotation of genes, we assigned GO terms for genes in the Danio rerio genome to putative S. trutta orthologs using biomaRt. We performed GO term enrichment analysis using topGO [v.2.34; (92)] with the ‘weight01’ algorithm and a node size of 20. The background universe consisted of all genes (n = 29,527) annotated with a Gene Ontology term.
For functional annotation of putative S. trutta immune genes, we performed a Fisher’s Exact test to identify enrichment of GO terms for species-specific duplications, genus-specific and putative immune ohnologs (Figure 1). For sex-biased immune genes, we performed Gene Ontology term enrichment analysis using a Kolmogorov-Smirnov (K-S) test. For all tests, we corrected for multiple testing using the Benjamini-Hochberg procedure and only reported terms as significant with an adjusted p < 0.05.
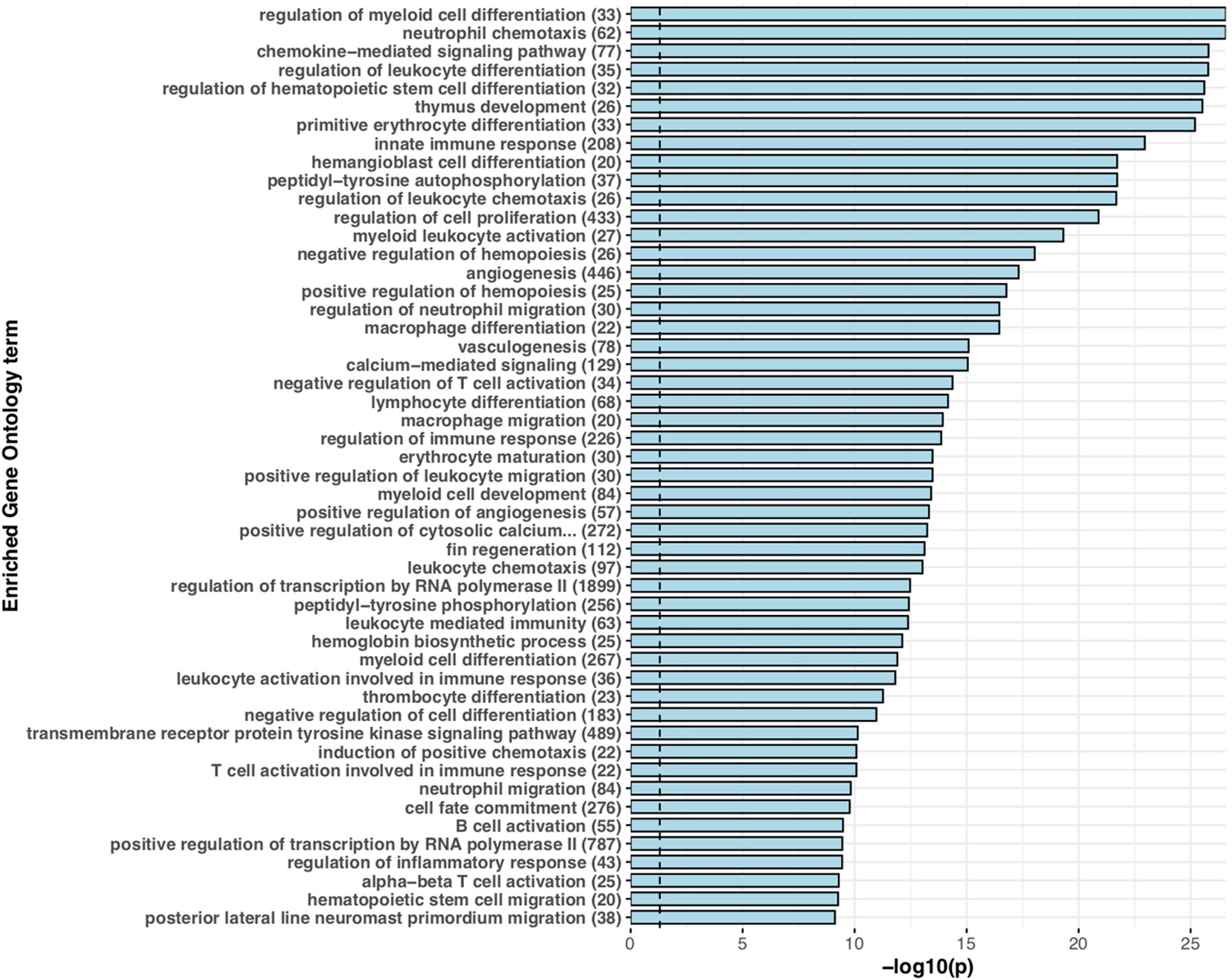
Figure 1 Expanded immune gene repertoire in the brown trout genome. Barchart displaying the top 50 significantly enriched ‘biological process’ associated Gene Ontology terms for putative immune genes with more than one copy in the brown trout genome in comparison to non-duplicated single copies in the Northern pike genome. For each significantly enriched term (Benjamini-Hochberg adjusted p < 0.05), the -log10 transformed p value is provided. For each term, the number of genes annotated with that specific term in the predicted S. trutta proteome is provided. The blue dotted vertical line represents threshold of significance (-log10 transformed p = 0.05).
For comparison between metrics associated with ohnologs, including variation in raw gene expression, predicted amino acid length, dN/dS ratios and evolutionary distance to non-duplicated ortholog outgroup, we used base statistical functions in R (v. 3.5.1). For pairwise comparison of means, we used Welch Two sample t-tests or Wilcoxon rank sum test. We tested for correlations using Pearson’s product moment correlation coefficient.
Through comparison with annotated immune genes in zebrafish, mouse and human, we identified 2,275 putative homologs encoded by the brown trout genome (Supplemental Information Table 1). As expected, due to the salmonid-specific WGD event, the number of genes with putative roles in the immune system were elevated in salmonids in comparison to non-salmonid fishes (Figure 2) with the analysis by OrthoFinder indicating an additional 1,132 homologous sequences based on sequence similarity alone. However, as the immune orthologs identified by Ensembl are assigned based on additional synteny-based information, we consider the 2,275 of higher confidence and therefore, we based the rest of our analyses on these genes.
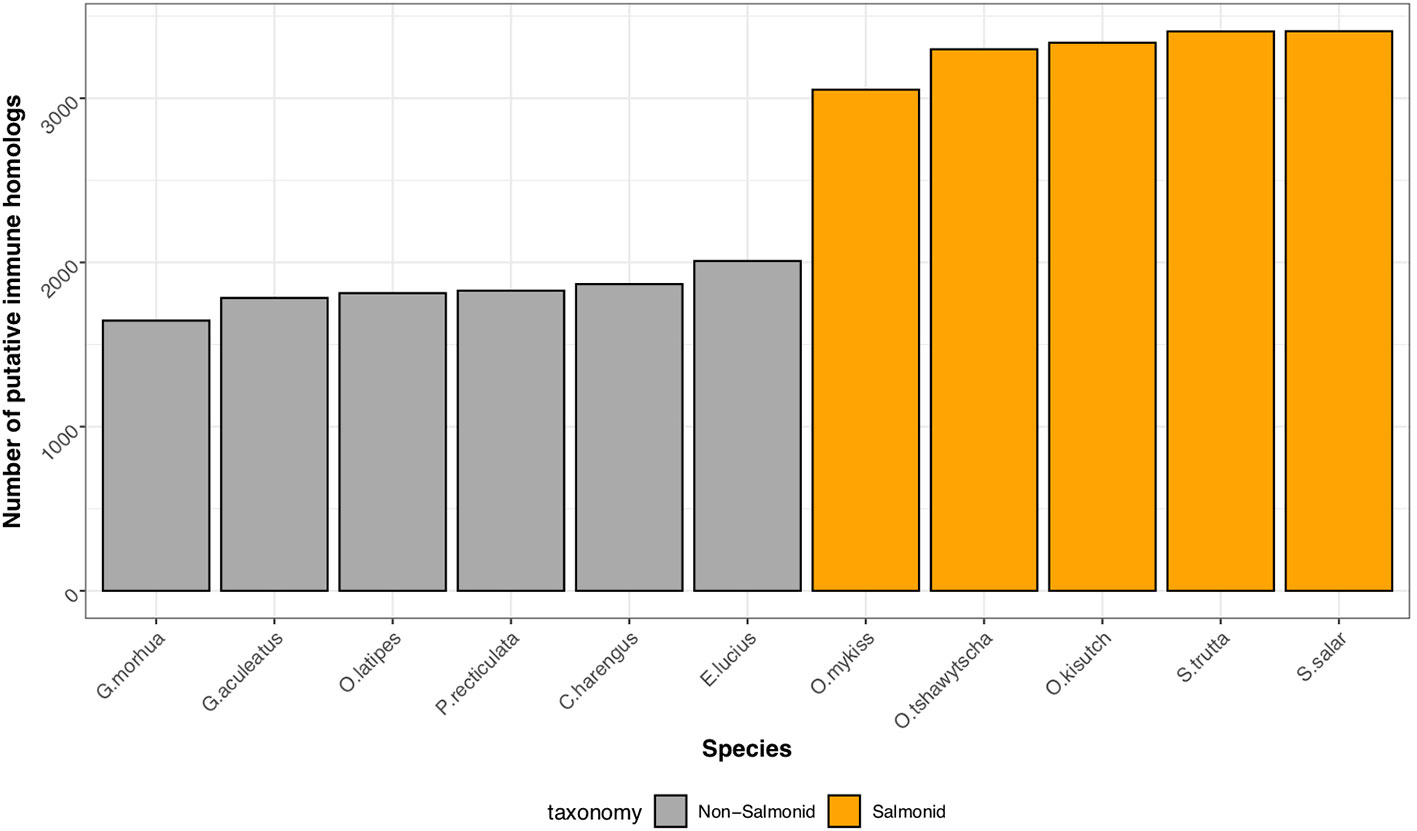
Figure 2 Conservation of immune gene repertoire in salmonid genomes. Histogram displaying the number of putative immune genes found within representative genomes of salmonid (Atlantic salmon, S. salar; Brown trout, S. trutta, Rainbow trout, O. mykiss, Coho salmon, O. kisutch, Chinook salmon, O. tshawytscha), and non-salmonid teleost fish (Northern pike, E. lucius; Atlantic herring, C. harengus; guppy, P. reticulata; Japanese medaka, O. latipes; three-spined stickleback, G. aculeatus; Atlantic cod, G. morhua).
To identify if putative S. trutta immune genes are transcribed and therefore can be considered functional, we first investigated gene expression of all potential immune genes across eight available tissues (PRJEB33055). Collectively, we identified evidence of expression for 2,233 genes out of 2,275 (98.1% of immune genes) with 1,587 genes (69.8% of putative immune genes) expressed across all tissue types (Figure 3; Supplemental Information Table 5). We identified significantly higher expression (Wilcoxon test: p < 0.05) of immune genes in comparison to non-immune genes for all tissues examined, including immunocompetent organs, such as the spleen, kidney and liver, as well as the gills, which are an entry point for infection.
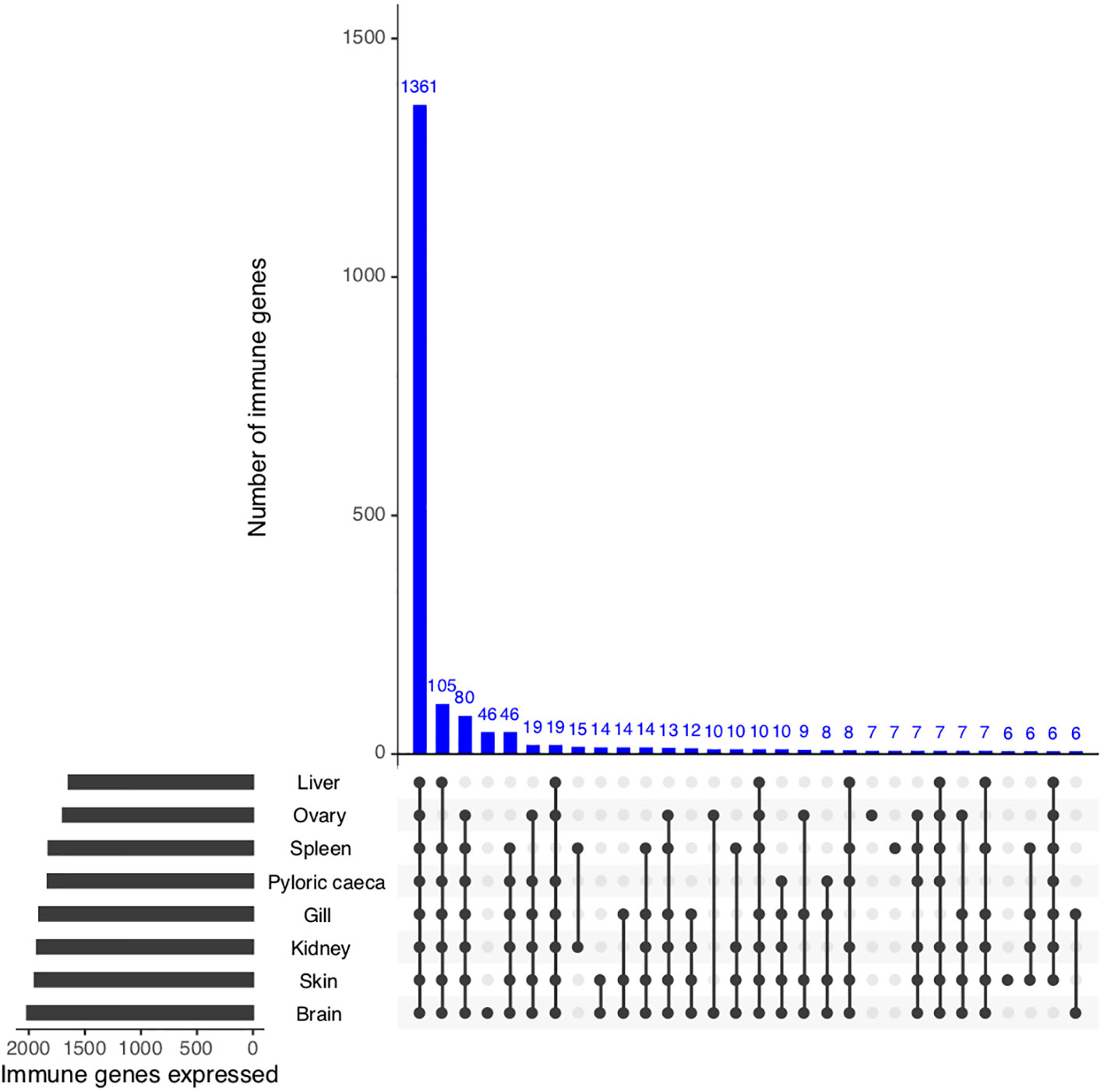
Figure 3 Functional validation of S. trutta immune expression across tissues. UpSet plot of immune gene expression across eight tissue types in brown trout. For each tissue, the plot contains a bar chart displaying the total number of immune genes expressed per tissue, as well as a histogram displaying the number of genes identified as expressed across tissues.
To understand the evolutionary consequences for the immune system post salmonid-specific WGD, we compared the predicted brown trout gene complement to that of the Northern pike, E. lucius. Using the 2,275 putative immune genes in the brown trout genome, we obtained 2,000 ‘high-confidence’ orthologs in Northern pike. Of this number, 1,444 were present as 2:1 orthologs of Northern pike genes with 868 genes (434 pairs; Supplemental Information Table 6) annotated by Ensembl as paralogs with the time of duplication estimated within the Salmoninae, suggesting that these genes may be produced by the salmonid-specific WGD and represent ohnologs. Of the other 2:1 duplicate pairs, 30 (n = 60 genes) and 27 (n = 54 genes) were estimated to have duplicated within the genus Salmo and within S. trutta, respectively. The remainder may represent older duplication events, form part of expanded gene families in brown trout or represent gene losses in Northern pike. In terms of potential immune gene loss, we identified 253 immune genes with 1:1 copies in both S. trutta and Northern pike. Of these 253 genes, OrthoFinder also identified 127 as single copy across salmonids.
To investigate patterns of functional conservation and divergence among immune ohnolog pairs, we first examined structural variation between ohnolog pairs in terms of the number of functional domains. Of the 434 immune ohnolog pairs, all proteins were annotated with at least one functional domain with 42 pairs differing in the number of functional domains with the more diverged copy generally having fewer domains suggestive that predicted proteins for at least ~11% of immune ohnolog have the potential to perform different functions. Overall, however, there was no significant difference in the number of predicted functional domains that each copy had (Wilcoxon test: p > 0.05; Figure 4). Similarly, there were no significant differences (Wilcoxon test: p > 0.05) in the mean gene, CDS or predicted protein length of ohnolog pairs. We also investigated variation between the pairs in terms of divergence from the single copy ortholog in Northern pike. Here, the more diverged copies based on evolutionary distance had significantly higher dN/dS ratios (paired two-sample t-test: p < 0.05; Figure 4) in comparison to more conserved copies but overall indicated that both copies for those pairs analyzed were under strong purifying selection (dN/dS < 0.25).
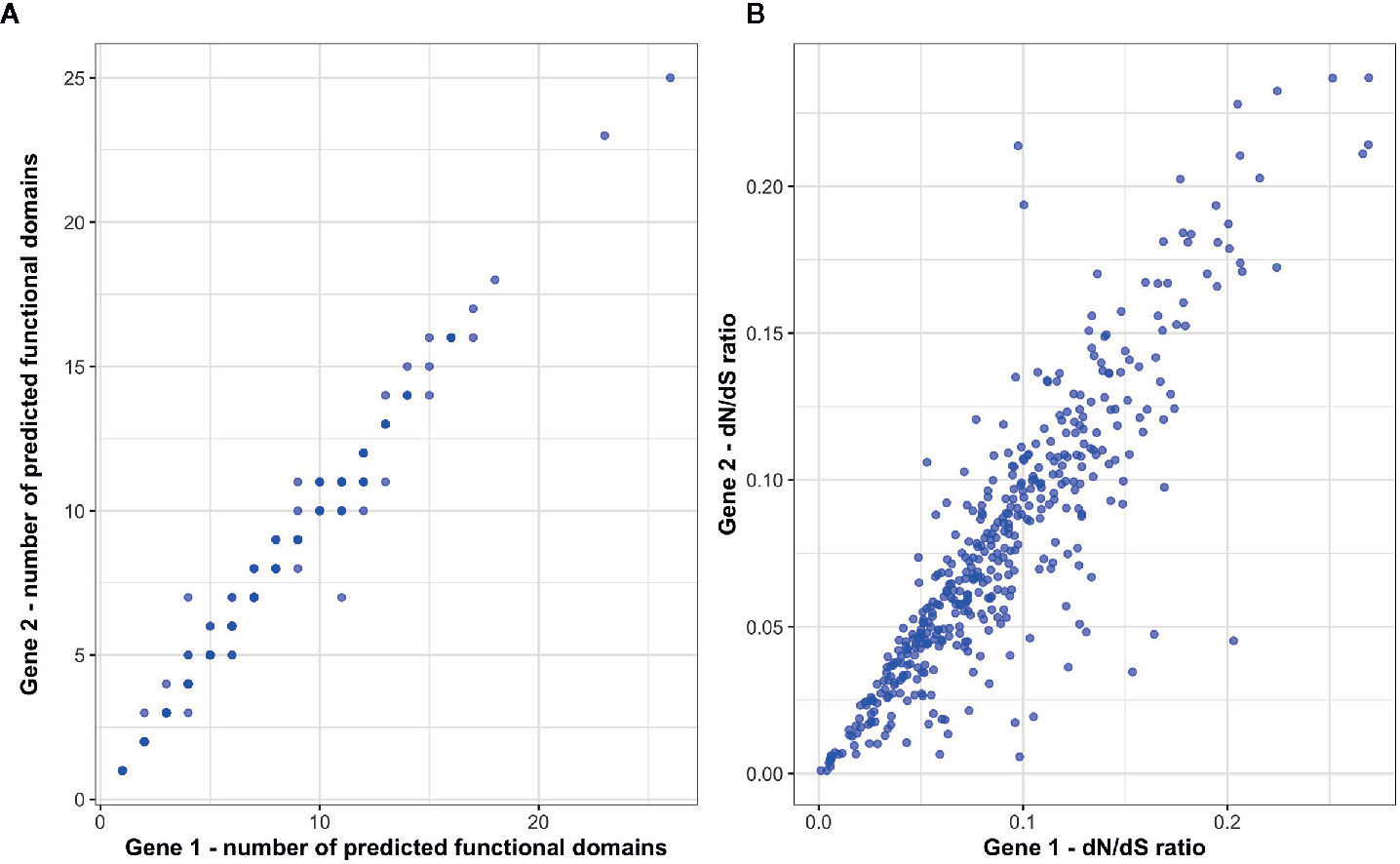
Figure 4 Assessment of functional divergence between S. trutta immune ohnolog pairs. (A) The number of predicted functional domains within the predicted protein coded for by each immune ohnolog. (B) For each ohnolog, dN/dS ratios were calculated for each and their respective non-duplicated ortholog in Northern pike.
To understand differences in gene expression profiles between immune ohnolog pairs, we performed a hierarchical clustering analysis to explore patterns of functional divergence. As a provisional measure, we first constructed co-expression networks using all putative immune genes identifying eight clusters consisting of 823 genes (smallest cluster = 20 genes; largest cluster = 257 genes). For immune ohnologs (n = 434 pairs), our analysis clustered 302 immune ohnologs into seven respective co-expression clusters (Figure 5). Of this number, 152 assigned genes belonged to ohnolog pairs (i.e., 76 pairs), where both ohnologs could be assigned to a co-expression network. The majority of these pairs (n = 55 pairs) were assigned to the same co-expression network indicating that both copies have conserved expression profiles across tissues suggestive that functions may also be conserved. Twenty one immune ohnolog pairs showed divergent co-expression profiles (Figure 5). For the remainder of the genes (n = 150) assigned to a cluster, they represent only one ohnolog from a pair where the other copy was unassigned due to lack of variation in expression or lack of expression across tissues.
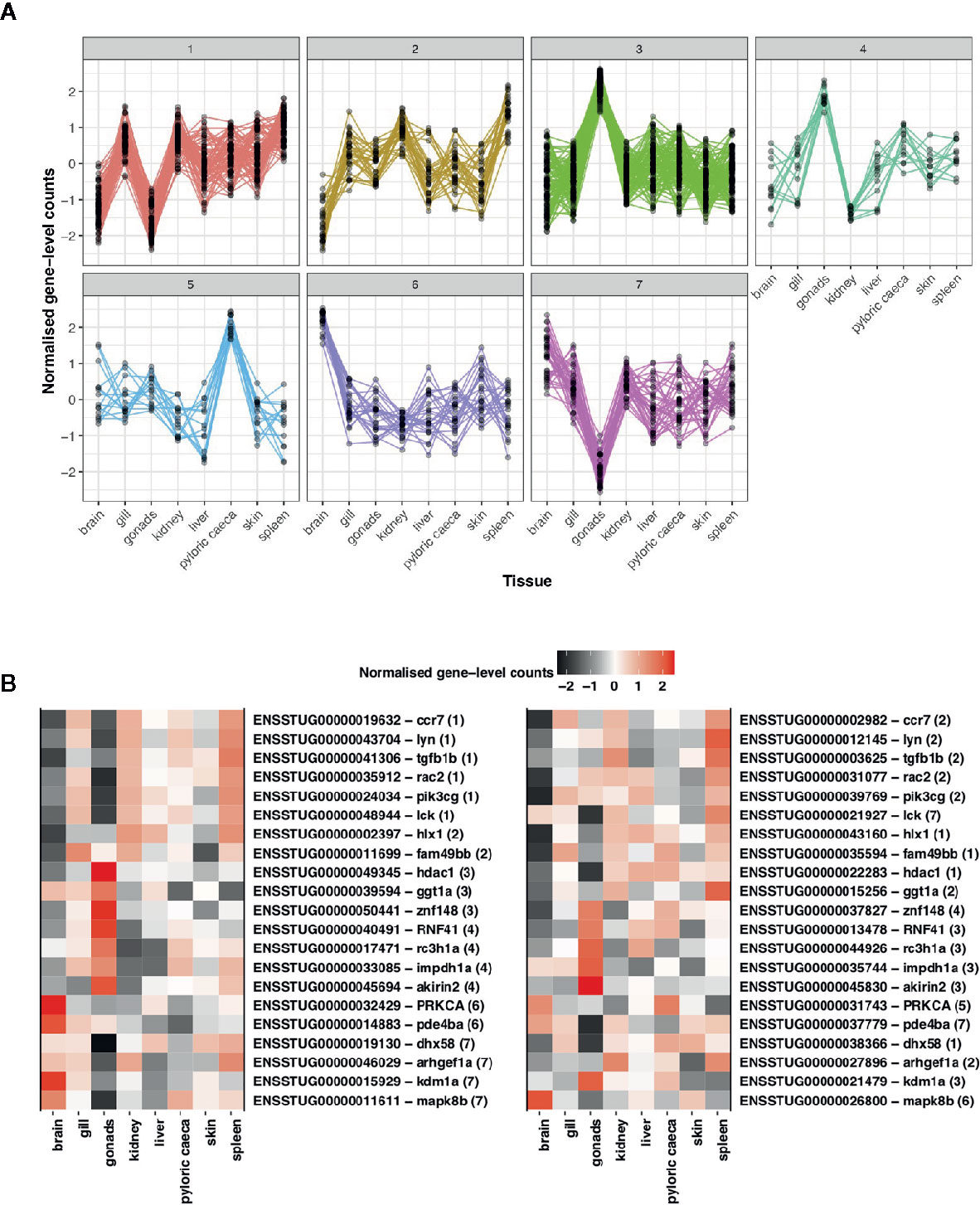
Figure 5 Assessment of divergence in S. trutta immune ohnolog expression profiles. (A) Clustering based on gene-level counts assigned 302 immune genes, which consist of one member of an ohnolog pair, to seven co-expression networks assigned based on the expression profile of each gene across eight tissue types. Each co-expression cluster is represented by a single line graph whereby the y-axis consists of gene-level counts normalized by Clust and the x-axis consists of eight tissues obtained from a single double haploid female. (B) Heatmap of expression profiles for ohnolog pairs where both ohnologs were assigned to different co-expression networks based on expression profiles across eight tissue types. Ensembl gene ID and description (separated by hyphen) are provided on the y-axis with number in parentheses indicating the cluster the gene was assigned to. The x-axis represents the eight tissues used to construct the co-expression networks.
Single copy genes present in salmonid genomes may be the result of adaptive loss or loss of a duplicated copy through neutral processes. First, using the reduced comparative dataset, we identified 4,297 single copy orthologs (SCOs) shared across the genomes of these six species. Of this number, 223 genes were annotated as putative immune genes but there was no evidence of significant enrichment of immune genes among all single copy orthologs We identified these genes as significantly enriched for the Gene Ontology term ‘toll-like receptor signaling pathway’ (GO:0002224). In total, these genes were identified as being significantly enriched (BH adjusted p < 0.05) for 26 terms with the most significant terms per GO category being ‘erythrocyte differentiation’, ‘immune response’, ‘complement activation’ and ‘cell chemotaxis’.
To investigate overall differences in gene expression between the sexes, we first performed a principal component analysis. Principal component 1 (PC1) accounted for 23% of the variance in the dataset with PC1 largely separating males and females providing evidence of sex-biased expression profiles evident in the brown trout liver (Figure 6). We also find additional variation within PC2, which is likely attributed to differences in genetic background between tanks. Second, through differential expression analysis, we identified 3,689 genes as significantly differentially expressed (BH adjusted p‐value < 0.05) between the sexes (Supplemental Information Table 10). Of this number, the majority (n = 1,969) had a female-biased expression profile, which was a significant trend (binomial test, p < 10-4). Differentially expressed genes between the sexes were significantly enriched for 12 Gene Ontology terms with the most significant per GO category being “nucleobase catabolic process” (GO:0046113), “phosphatidylethanolamine binding” (GO:0008429) and “nucleolus” (GO:0005730) (Supplemental Information Tables 14-16).
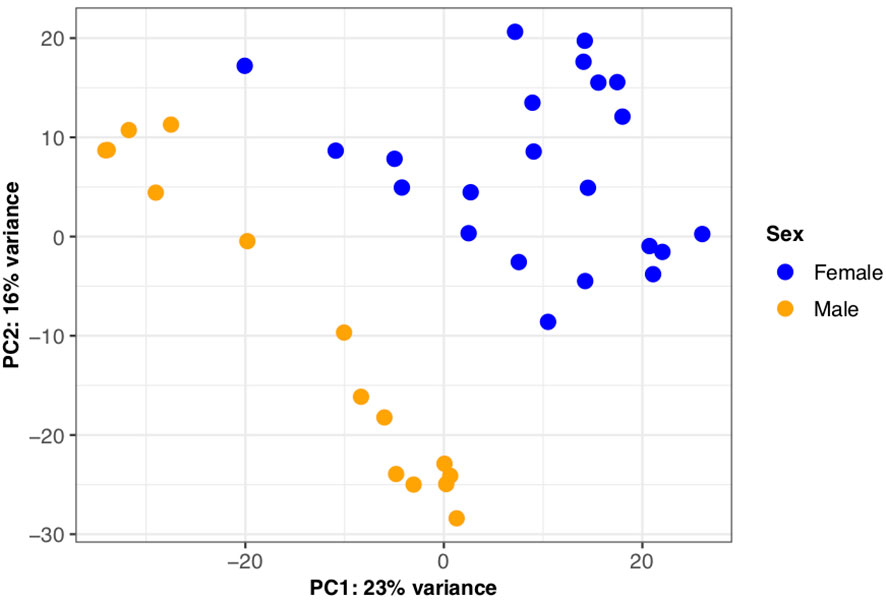
Figure 6 Sexually dimorphic gene expression in S. trutta liver. Principal component analysis for gene-level counts revealed sexually dimorphic gene expression in S. trutta liver. Principal component (PC1) explained 23% of variance with males and females clearly separating. Each dot on the scatterplot represent a single individual sample and are color-coded by sex (female = blue; male = orange).
In relation to immune expression, we detected 83% of putative immune genes (n = 1,936 of 2,275) expressed in the liver. Of this number, we identified 269 genes as significantly differentially expressed (BH p< 0.05, Figure 7, Supplementary Information Table 10) between the sexes. Thirty-five of these 269 genes were annotated as single copy immune orthologs in the brown trout genome. In contrast to the entire transcriptome, which generally demonstrated female-biased expression, we detected more immune genes with higher gene expression in males (n = 158), which was more than expected by chance (binomial test, p < 0.03). Male-biased immune genes were significantly enriched (Fisher’s exact test; BH-adjusted p < 0.05) for 18 biological process-associated Gene Ontology terms, including ‘erythrocyte development’ (GO:0048821), ‘hemopoiesis’ (GO:0030097), and ‘response to cytokine’ (GO:0034097). For female-biased immune genes, we identified seven significantly enriched Gene Ontology terms associated with hemopoiesis and neutrophil differentiation.
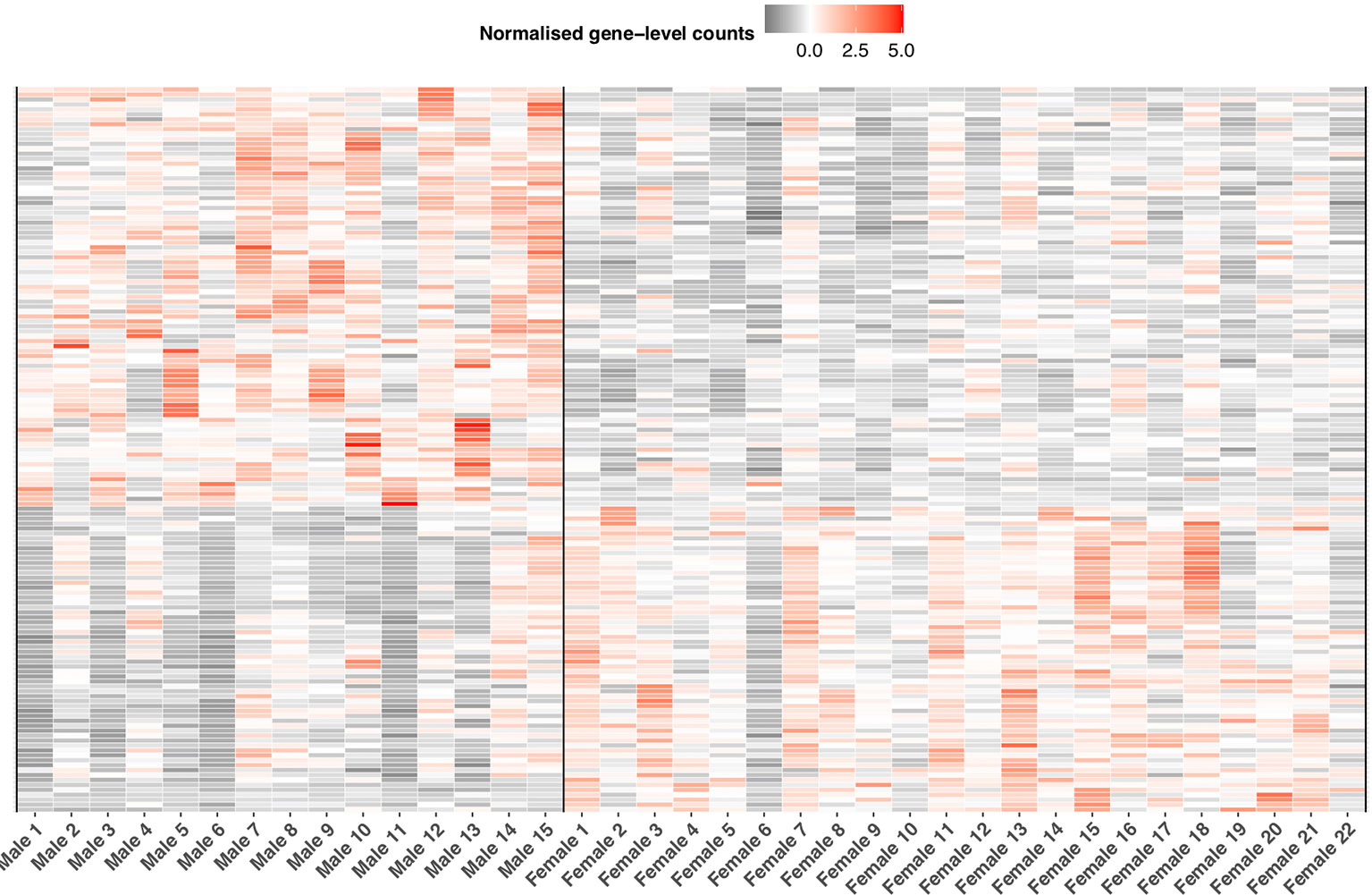
Figure 7 Sexually dimorphic immune expression in S. trutta liver. Heatmap displaying significantly differentially expressed immune genes between the sexes. Relative variation in immune gene expression is represented by a color gradient (gray = low, white = medium, red = high). Each column represents an individual trout with a vertical black line separating male from female samples. Each row represents expression level for an individual immune gene.
We identified 218 intron clusters corresponding to 176 genes with evidence of significant alternative splicing (BH adjusted p‐value < 0.05; Supplemental Information Table 13) between males and females. Of this number, 55 were also significantly differentially expressed between the two sexes. We detected 15 alternatively spliced genes with roles in the salmonid immune system. Four of these genes were also differentially expressed between the sexes.
We investigated if ohnologs demonstrated sex-biased gene expression and found 98 unique genes with sex-biased gene expression of which 12 ohnolog pairs exhibited significant differential expression for both genes (BH adjusted p < 0.05) between the sexes. For the majority (n = 10) of these ohnolog pairs, the ohnolog pairs have conserved differential expression whereby expression in both copies was elevated in the same sex compared to the other (n = 6 male-biased; n = 4 female-biased). Only for two ohnolog pairs did we identify differences in expression profiles between the ohnologs whereby the more conserved copies were all increased in males compared to females, but the less conserved copy had the opposite expression profile i.e., significantly reduced in males compared to females.
Migratory species, such as anadromous salmonids, require efficacious and adaptable immune systems to survive in a diversity of environments. Here we provide an important insight into the immune gene repertoire, as well as conservation and differences in gene expression across tissue types, for the facultatively anadromous brown trout S. trutta. Our findings indicate duplications and expansions of genes involved in immunological functions, such as chemotaxis and immune cell differentiation. Second, we assessed immune ohnologs for evidence of functional divergence in terms of domain architecture and gene expression profiles, identifying the majority to have conserved functional expression across tissue types in brown trout, while, surprisingly, only a few immune ohnolog pairs differed in terms of gene expression or the number of functional domains. We also identify evidence of immune gene loss in the salmonids. Lastly, we quantified sex-biased differences in immune gene expression in the brown trout liver, identifying the majority of differentially expressed immune genes to have male-biased expression. Our findings provide a novel insight into the immune complement of an ecologically and commercially important salmonid.
For teleost species that have undergone more recent whole genome duplication events, such as the salmonids, retention of duplicated genes may increase immune potential. In the brown trout genome, we identified high confidence orthologs for known canonical immune genes demonstrating that brown trout have essential genetic components of both innate and adaptive immune systems, as well as expansions (Figures 1 and 2). This number increases if we include protein homologs identified by OrthoFinder but additional synteny-based analyses would be required to confirm these genes as putative immune homologs. As our comparative analysis was mainly restricted to model species with well annotated immune repertoires, novel immune genes or genes highly diverged within the salmonids or restricted to Salmo trutta will not be reported. Gene expression analysis using eight available tissues indicates the vast majority of predicted immune genes as functional with expression evident across tissues (Figure 3). Retained duplicate copies were enriched for Gene Ontology terms associated with both innate and adaptive immunity. Among the most significantly enriched terms was neutrophil chemotaxis (Figure 1). Neutrophils are innate immune cells that are among the first responders to pathogen infection and inflammation and are a key aspect of the salmonid immune system. The response of these cells to chemical stimuli, known as chemotaxis, is important for the rapid response and subsequent migration of neutrophils to the site of signal origin (93). Neutrophils possess antimicrobial and phagocytic activity with the latter differing across teleost species (94). The expansion in brown trout of genes involved in chemotaxis may allow for the increased recognition of more diverse chemical stimulants, which may be beneficial for their migratory life-history that involves use of freshwater, brackish and marine environments.
Due to the complexity of the life-histories expressed by brown trout, immune repertoires are required for regulation and maintenance of immune expression during migration, an energetically stressful period, as well as to survive in both freshwater and marine environments, which can contain unique pathogenic threats (95). Variation in the strength of selection acting on duplicated genes can result in amino acid and/or regulatory divergence leading to the evolution of new functions or indeed, the process of pseudogenization and gene loss. Here, we found no significant difference in the overall number of functional domains between immune ohnologs in brown trout (Figure 4), as well as differences in expression profiles across tissues (Figure 5). However, for certain pairs, we did find evidence of domain architecture variation, as well as differences in expression profiles across tissues. We find variation in terms of functional domains for 42 pairs, and while future experimental validation is required, such pairs represent interesting candidates for investigating functional divergence in immune ohnologs. Similarly, 21 ohnolog pairs demonstrate divergence in gene expression profiles across eight tissues examined (Figure 5). As our analysis involved eight tissues rather than 15, which were previously used for Atlantic salmon (48) and rainbow trout (49), our power to detect divergent expression profiles may be reduced. Similarly, previous studies on salmonids explored conservation of ancestral function ohnologs through the construction of genome-wide co-expression networks, where one salmonid copy clustered by expression profile with that of a non-duplicated ortholog in the closely related, Northern pike, suggestive that one copy may retain and perform a conserved function across taxa. We did not explore such patterns in our analysis for brown trout due to the limited tissues available for both brown trout, of which only five were also available for Northern pike.
Of the ohnolog pairs with evidence of divergent gene expression profiles, such genes were annotated with a range of immunological function, including antiviral defense (DHX58, zinc finger protein 148), apoptosis (RAC2), inflammation regulation (MAPK8, PDE4B, Roquin-1), anti-microbial response (Akirin-2, ARHGEF2), T-cell activation (LCK, HLX) as well as tumor suppression (PRKCA, FAM49B, IMPDH1). Specific ohnolog pairs of interest included pairs where both genes were annotated as chemokine receptor type 7. In mammals, CC-chemokine receptor 7 (CCR7) is part of the G protein-coupled receptor family and can function in the activation of naive B and T lymphocytes with additional research suggesting the receptor may function in antiviral defense (96). In teleosts, the functions of chemokine receptors are less well understood but a CCR7 homolog has been previously characterized in the rainbow trout, O. mykiss, where based on sequence similarity, the predicted protein is suggested to perform a similar function to that of mammalian homologs (97, 98). The second ohnolog pair of interest were annotated as tyrosine-protein kinase Lyn. Lyn belongs to a Src- family of tyrosine kinases found in immune cells that can negatively regulate important signaling pathways (99). The gene is also a key mediator of pathways involved in B cell activation (99, 100) a function suggested as conserved in teleosts (101).
While extensive retention and functional divergence of ohnologs have been characterized in other salmonids, gene loss can also occur. Two primary molecular processes can lead to gene loss: as a consequence of an abrupt mutational event, such as an error in crossing over during meiosis, or through the slow accumulation of mutations during pseudogenization after an initial loss-of-function mutation (12). Comparative genomic studies have revealed biased patterns in gene loss in terms of functional bias with genes involved in certain cellular processes, such as DNA repair and transcription, more likely to be represented among genes where a copy has been lost (102, 103). In relation to WGD events, there is also evidence of genomic positional biases in terms of gene loss (104). Clusters of single copies may be due to reliance on similar transcriptional regulation machinery or architecture. Here through comparative analyses, we find gene loss conserved across salmonids. The conservation of synteny across salmonid genomes would suggest that loss of putative immune genes is non-random, as has been shown for other species (12). Future work will benefit from understanding the molecular, cellular and evolutionary consequences of gene loss in these species.
Sexes share largely the same genome but express it differently giving rise to different morphological and behavioral phenotypes. Here we investigated differences in gene expression in the liver, an organ previously used to understand salmonid metabolism gene expression (51), response to environmental stressors (105), as well as genes underlying sexual dimorphism (106). While the liver may have traditional roles in metabolism and antioxidant activity, a growing body of literature on mammalian and teleost immunology has provided important insights into the role of the liver in the innate immune response, immune tolerance and hematopoiesis (74, 107–112), as well as creating hostile molecular environments for parasites to migrate through (113, 114). Here we identified differential immune gene expression both in terms of expression amplitude (Figures 6, 7) and splicing between the sexes. Immune genes exhibited a general male-bias in expression, which was in contrast to overall gene expression in the liver being female-biased. Immunological studies on sex differences in immune function in brown trout have suggested reduced immune function in mature males (115, 116), and therefore, females would have been expected to have higher immune gene expression compared to males. While our fish were laboratory reared and not directly immune challenged, they were maintained in normal lab environments (i.e., clean but not sterile) and therefore, we would expect a background level of immune gene expression. Sex-biased differences in expression could be due to anticipation of immune challenge. As male brown trout are less likely to undergo sea migration, remaining resident and completing their life-cycle in freshwater environments, pathogens present in freshwater environments are more likely to encounter males than females and therefore, parasite-mediated selection in brown trout may result in variation in environment-dependent immune expression between sexes. Indeed, males do suffer more severe infestations by freshwater ectoparasites in comparison to females (117), yet our understanding of immune potential and function is lacking.
An interesting finding among the genes with sex-biased differences was the presence of putative single copy orthologs. As sexes largely share the same genome but have different fitness optima and may express some genes differently, this can result in sexually antagonistic loci, which increase fitness when expressed in one sex but are detrimental in the other. Sex-biased gene expression has been suggested as a mechanism to resolve such conflict (118, 119). Aside from transcriptional regulation, modifications in genomic architecture, such as sex-dependent dominance (120), maintenance of sexually antagonistic loci on sex chromosomes (121) or duplication events (122, 123) may also resolve conflict. It is interesting therefore that since the salmonid WGD, duplicate copies of immune genes have been lost either through adaptive or neutral processes that may now be sexually antagonistic. The application of population genetic approaches, in combination with sex-biased gene expression, have been used to reveal genomic signatures of loci associated with sexual conflict (118, 124), which could be applied to future studies in brown trout and other salmonids to provide important insights into the evolutionary processes shaping sex differences.
Salmonid genomics is a rapidly advancing field and is providing comprehensive insights into genes underlying phenotypically plastic traits, such as sea age at maturity, as well as genomic structures resolving sexually antagonistic loci (28, 120). Here we explore the consequences of a salmonid-specific WGD on immune gene repertoire in brown trout, finding that brown trout has an enlarged immune gene complement relative to non-salmonids, with many immune ohnologs retained that have conserved immune expression profiles between the pairs. We find preliminary signatures of some ohnologs coding for proteins that may have potential divergent functions between the pairs, but functional validation is required to determine the exact role these genes may play in the brown trout immune system. Lastly, we add to a growing body of research that explores key physiological differences among the sexes through the identification of differences in immune expression.
Our findings provide important insights into immune gene evolution and expression in a culturally, economically and ecologically important species. Like many species, brown trout face an uncertain future due to changing climates with increasing temperatures potentially leading to reduced sea migration rates (82) as well as potentially impacting immune function (125) while pathogens, such as sea lice, associated with increasing aquaculture, are also suggested to contribute to migratory declines (126). Improved understanding of immune potential, expression and function may benefit management strategies and conservation schemes for wild populations to assist in the maintenance of at-risk facultatively anadromous populations.
Raw sequence data files are deposited in the NCBI short read archive (BioProject ID: PRJNA667168). Scripts underpinning the data analysis are archived on GitHub (https://github.com/Joscolgan/salmonid_immune_study). Raw read counts used in the present analyses are provided as tables in Supplementary Information.
The animal study was reviewed and approved by This study was carried out in accordance with the recommendations of the Health Products Regulatory Authority (HPRA) Ireland, under HPRA project license AE19130/P034, and HPRA individual licenses AE19130/I087, AE19130/I200, AE19130/I201, and AE19130/I202.
TC, PAM, PMcG, and TR designed the experiment. LA, RW, and SH harvested the liver samples. TC performed the RNA extractions and quality checks. TC performed the majority of analyses. All authors contributed to the article and approved the submitted version.
This research was supported by an ERC Starting Grant (639192-ALH) and an SFI ERC Support Award awarded to TR. PMcG was funded by an SFI-DEL grant (2015 15/IA/3028) and the Marine Research Programme 2014–2020 RESPI/FS/16/01 (Marine Institute).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Brian Clarke, Dr. Deirdre Cotter, members of the FishEyE team at UCC, and the staff of Inland Fisheries Ireland (in particular Dr. Paddy Gargan) and the Marine Institute (in particular Dr. Russell Poole) for obtaining brood stock and for assistance in fish rearing. We also thank Luke Harman in particular for his invaluable role in designing and building a recirculating aquaculture system, and Ronan O’Sullivan and Dr. Adam Kane for assistance in fish maintenance. We thank Dr. Jamie Coughlan for genotyping fish, Dr. Eileen Dillane with assistance in logistics and Profs Tom Cross and Paulo Prodöhl for discussion on brown trout genomics. We also thank the editor and two reviewers for the beneficial and insightful comments that have greatly improved this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.568729/full#supplementary-material
1. Altizer S, Bartel R, Han BA. Animal migration and infectious disease risk. Science (2011) 331:296–302. doi: 10.1126/science.1194694
2. Eikenaar C, Hegemann A, Packmor F, Kleudgen I, Isaksson C. Not just fuel: energy stores are correlated with immune function and oxidative damage in a long-distance migrant. Curr Zool (2020) 66:21–8. doi: 10.1093/cz/zoz009
3. Eikenaar C, Hegemann A. Migratory common blackbirds have lower innate immune function during autumn migration than resident conspecifics. Biol Lett (2016) 12:20160078. doi: 10.1098/rsbl.2016.0078
4. Waldenström J, Bensch S, Kiboi S, Hasselquist D, Ottosson U. Cross-species infection of blood parasites between resident and migratory songbirds in Africa. Mol Ecol (2002) 11:1545–54. doi: 10.1046/j.1365-294X.2002.01523.x
5. MacKenzie K. Parasites as biological tags in population studies of marine organisms: an update. Parasitology (2002) 124:153–63. doi: 10.1017/S0031182002001518
6. Renshaw SA, Trede NS. A model 450 million years in the making: zebrafish and vertebrateimmunity. Dis Model Mech (2012) 5:38–47. doi: 10.1242/dmm.007138
7. Pancer Z, Cooper MD. The evolution of adaptive immunity. Annu RevImmunol (2006) 24:497–518. doi: 10.1146/annurev.immunol.24.021605.090542
8. Shultz AJ, Sackton TB. Immune genes are hotspots of shared positive selection across birds and mammals. Elife (2019) 8:e41815. doi: 10.7554/eLife.41815
9. Kosiol C, Vinar T, da Fonseca RR, Hubisz MJ, Bustamante CD, Nielsen R, et al. Patterns of positive selection in six mammaliangenomes. PLoS Genet (2008) 4:e1000144. doi: 10.1371/journal.pgen.1000144
10. Flajnik MF, Kasahara M. Origin and evolution of the adaptive immune system: genetic eventsand selective pressures. Nat Rev Genet (2010) 11:47–59. doi: 10.1038/nrg2703
11. Balakirev ES, Ayala FJ. Pseudogenes: Are they “junk” or functional DNA? Annu Rev Genet (2003) 37:123–51. doi: 10.1146/annurev.genet.37.040103.103949
12. Albalat R, Cañestro C. Evolution by gene loss. Nat Rev Genet (2016) 17:379–91. doi: 10.1038/nrg.2016.39
13. Sharma V, Hecker N, Roscito JG, Foerster L, Langer BE, Hiller M. A genomics approach reveals insights into the importance of genelosses for mammalian adaptations. Nat Commun (2018) 9:1215. doi: 10.1038/s41467-018-03667-1
14. Solbakken MH, Rise ML, Jakobsen KS, Jentoft S. Successive losses of central immune genes characterize the Gadiformes’ alternate immunity. Genome Biol Evol (2016) 8:3508–15. doi: 10.1093/gbe/evw250
15. Wang X, Grus WE, Zhang J. Gene losses during human origins. PLoS Biol (2006) 4:e52. doi: 10.1371/journal.pbio.0040052
16. Glasauer SMK, Neuhauss SCF. Whole-genome duplication in teleost fishes and its evolutionaryconsequences. Mol Genet Genomics (2014) 289:1045–60. doi: 10.1007/s00438-014-0889-2
17. Sadd BM, Schmid-Hempel P. Principles of ecological immunology. EvolAppl (2008) 2:113–21. doi: 10.1111/j.1752-4571.2008.00057.x
18. Nunn CL, Lindenfors P, Pursall ER, Rolff J. On sexual dimorphism in immune function. Philos Trans R Soc Lond B Biol Sci (2009) 364:61–9. doi: 10.1098/rstb.2008.0148
19. Zuk M, Stoehr AM. Immune defense and host life history. Am Nat (2002) 160 Suppl4:S9–S22. doi: 10.1086/342131
20. Møller AP, Sorci G, Erritzøe J. Sexual dimorphism in immune defense. Am Nat (1998) 152:605–19. doi: 10.1086/286193
21. Rolff J, Armitage SAO, Coltman DW. Genetic constraints and sexual dimorphism in immune defense. Evolution (2005) 59:1844–50. doi: 10.1111/j.0014-3820.2005.tb01831.x
22. Gal-Oz ST, Maier B, Yoshida H, Seddu K, Elbaz N, Czysz C, et al. ImmGen report: sexual dimorphism in the immune system transcriptome. Nat Commun (2019) 10:4295. doi: 10.1038/s41467-019-12348-6
23. Lawniczak MKN, Barnes AI, Linklater JR, Boone JM, Wigby S, Chapman T. Mating and immunity in invertebrates. Trends Ecol Evol (2007) 22:48–55. doi: 10.1016/j.tree.2006.09.012
24. Vincent CM, Gwynne DT. Sex-biased immunity is driven by relative differences in reproductive investment. Proc R Soc B Biol Sci (2014) 281:20140333. doi: 10.1098/rspb.2014.0333
25. Chapman BB, Brönmark C, Nilsson J-Å, Hansson L-A. The ecology and evolution of partial migration. Oikos (2011) 120:1764–75. doi: 10.1111/j.1600-0706.2011.20131.x
26. Morbey YE, Ydenberg RC. Protandrous arrival timing to breeding areas: areview. Ecol Lett (2001) 4:663–73. doi: 10.1046/j.1461-0248.2001.00265.x
27. Quinn TP, McGinnity P, Reed TE. The paradox of “premature migration” by adult anadromoussalmonid fishes: patterns and hypotheses. Can J Fish Aquat Sci (2016) 73:1015–30. doi: 10.1139/cjfas-2015-0345
28. Pearse DE, Barson NJ, Nome T, Gao G, Campbell MA, Abadía-Cardoso A, et al. Sex-dependent dominance maintains migration supergene in rainbowtrout. Nat Ecol Evol (2019) 3:1731–42. doi: 10.1038/s41559-019-1044-6
29. Fish EN. The X-files in immunity: sex-based differences predispose immuneresponses. Nat Rev Immunol (2008) 8:737–44. doi: 10.1038/nri2394
30. Rinn JL, Snyder M. Sexual dimorphism in mammalian gene expression.Trends Genet (2005) 21:298–305. doi: 10.1016/j.tig.2005.03.005
31. Krasnov A, Wesmajervi Breiland MS, Hatlen B, Afanasyev S, Skugor S. Sexual maturation and administration of 17β-estradiol and testosterone induce complex gene expression changes in skin and increase resistance of Atlantic salmon to ectoparasite salmon louse. Gen Comp Endocrinol (2015) 212:34–43. doi: 10.1016/j.ygcen.2015.01.002
32. Zheng W, Xu H, Lam SH, Luo H, Karuturi RKM, Gong Z. Transcriptomic analyses of sexual dimorphism of the zebrafish liver and the effect of sex hormones. PLoS One (2013) 8:e53562. doi: 10.1371/journal.pone.0053562
33. Melé M, Ferreira PG, Reverter F, DeLuca DS. The human transcriptome across tissues and individuals. Science (2015) 348:660–5. doi: 10.1126/science.aaa0355
34. Yang X, Schadt EE, Wang S, Wang H, Arnold AP, Ingram-Drake L, et al. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res (2006) 16:995–1004. doi: 10.1101/gr.5217506
35. Kopp A, Duncan I, Godt D, Carroll SB. Genetic control and evolution of sexually dimorphic characters in Drosophila. Nature (2000) 408:553–9. doi: 10.1038/35046017
36. Markle JG, Fish EN. SeXX matters in immunity. Trends Immunol (2014) 35:97–104. doi: 10.1016/j.it.2013.10.006
37. Waples RS, Naish KA, Primmer CR. Conservation and management of salmon in the age of genomics. Annu Rev Anim Biosci (2020) 8:117–43. doi: 10.1146/annurev-animal-021419-083617
38. Nelson JS, Grande TC, Wilson MVH. Fishes of the world. Hoboken, New Jersey, USA: John Wiley & Sons (2016).
39. Quinn TP, Myers KW. Anadromy and the marine migrations of Pacific salmon and trout: Rounsefell revisited. Rev Fish Biol Fish (2004) 14:421–42. doi: 10.1007/s11160-005-0802-5
40. Curry RA, Bernatchez L, Whoriskey F, Audet C. The origins and persistence of anadromy in brook charr. Rev Fish Biol Fish (2010) 20:557–70. doi: 10.1007/s11160-010-9160-z
41. Dodson JJ, Aubin-Horth N, Thériault V, Páez DJ. The evolutionary ecology of alternative migratory tactics in salmonid fishes. Biol Rev Camb Philos Soc (2013) 88:602–25. doi: 10.1111/brv.12019
42. Ferguson A, Reed TE, Cross TF, McGinnity P, Prodöhl PA. Anadromy, potamodromy and residency in brown trout Salmotrutta: the role of genes and the environment. J Fish Biol (2019) 95:692–718. doi: 10.1111/jfb.14005
43. Kendall NW, McMillan JR, Sloat MR, Buehrens TW, Quinn TP, Pess GR, et al. Anadromy and residency in steelhead and rainbow trout(Oncorhynchus mykiss): a review of the processes and patterns. Can J Fish Aquat Sci (2015) 72:319–42. doi: 10.1139/cjfas-2014-0192
44. Hutchings JA, Jones MEB. Life history variation and growth rate thresholds for maturity in Atlantic salmon, Salmo salar. Can J Fish Aquat Sci (1998) 55:22–47. doi: 10.1139/d98-004
45. Ohms HA, Sloat MR, Reeves GH, Jordan CE, Dunham JB. Influence of sex, migration distance, and latitude on life historyexpression in steelhead and rainbow trout (Oncorhynchus mykiss). Can J Fish Aquat Sci (2014) 71:70–80. doi: 10.1139/cjfas-2013-0274
46. Morita K, Tamate T, Kuroki M, Nagasawa T. Temperature-dependent variation in alternative migratory tactics andits implications for fitness and population dynamics in a salmonid fish. J Anim Ecol (2014) 83:1268–78. doi: 10.1111/1365-2656.12240
47. Macqueen DJ, Johnston IA. A well-constrained estimate for the timing of the salmonid whole genome duplication reveals major decoupling from species diversification. Proc Biol Sci (2014) 281:20132881. doi: 10.1098/rspb.2013.2881
48. Lien S, Koop BF, Sandve SR, Miller JR, Kent MP, Nome T, et al. The Atlantic salmon genome provides insights into rediploidization. Nature (2016) 533:200–5. doi: 10.1038/nature17164
49. Berthelot C, Brunet F, Chalopin D, Juanchich A, Bernard M, Noël B, et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun (2014) 5:3657. doi: 10.1038/ncomms4657
50. Robertson FM, Gundappa MK, Grammes F, Hvidsten TR, Redmond AK, Lien S, et al. Lineage-specific rediploidization is a mechanism to explain time-lags between genome duplication and evolutionary diversification. Genome Biol (2017) 18:111. doi: 10.1186/s13059-017-1241-z
51. Gillard G, Harvey TN, Gjuvsland A, Jin Y, Thomassen M, Lien S, et al. Life-stage-associated remodelling of lipid metabolism regulation in Atlantic salmon. Mol Ecol (2018) 27:1200–13. doi: 10.1111/mec.14533
52. Macqueen DJ, Primmer CR, Houston RD, Nowak BF, Bernatchez L, Bergseth S, et al. Functional Annotation of All Salmonid Genomes (FAASG): an international initiative supporting future salmonid research, conservation and aquaculture. BMC Genomics (2017) 18:484. doi: 10.1101/095737
53. Wang T, Hu Y, Wangkahart E, Liu F, Wang A, Zahran E, et al. Interleukin (IL)-2 Is a key regulator of T Helper 1 and T Helper 2 cytokine expression in fish: Functional characterization of two divergent IL2 paralogs in salmonids. Front Immunol (2018) 9:1683. doi: 10.3389/fimmu.2018.01683
54. Wang J, Liu M, Wu Y, Yoon S, Alnabulsi A, Liu F, et al. Immune-modulation of two BATF3 paralogues in rainbow trout Oncorhynchus mykiss. Mol Immunol (2018) 99:104–14. doi: 10.1016/j.molimm.2018.04.016
55. Lee PT, Zou J, Holland JW, Martin SAM, Kanellos T, Secombes CJ. Identification and characterization of TLR7, TLR8a2, TLR8b1 and TLR8b2 genes in Atlantic salmon (Salmo salar). Dev Comp Immunol (2013) 41:295–305. doi: 10.1016/j.dci.2013.05.013
56. Xu Q, Li R, Monte MM, Jiang Y, Nie P, Holland JW, et al. Sequence and expression analysis of rainbow trout CXCR2, CXCR3a and CXCR3b aids interpretation of lineage-specific conversion, loss and expansion of these receptors during vertebrate evolution. Dev Comp Immunol (2014) 45:201–13. doi: 10.1016/j.dci.2014.03.002
57. Penston MJ, McKibben MA, Hay DW, Gillibrand PA. Observations on open-water densities of sea lice larvae in LochShieldaig, Western Scotland. Aquac Res (2004) 35:793–805. doi: 10.1111/j.1365-2109.2004.01102.x
58. McVicar AH, Sharp LA, Walker AF, Pike AW. Diseases of wild sea trout in Scotland in relation to fishpopulation decline. Fish Res (1993)17:175–85. doi: 10.1016/0165-7836(93)90017-2
59. Poole WR, Whelan KF, Dillane MG, Cooke DJ, Matthews M. The performance of sea trout, Salmo trutta L.,stocks from the Burrishoole system western Ireland, 1970–1994. FishManag Ecol (1996) 3:73–92. doi: 10.1111/j.1365-2400.1996.tb00131.x
60. Schmid-Hempel P. Variation in immune defence as a question of evolutionaryecology. Proc Biol Sci (2003) 270:357–66. doi: 10.1098/rspb.2002.2265
61. Heymann F, Tacke F. Immunology in the liver—from homeostasis todisease. Nat Rev Gastroenterol Hepatol (2016) 13:88. doi: 10.1038/nrgastro.2015.200
62. Magnadottir B, Lange S, Gudmundsdottir S, Bøgwald J, Dalmo RA. Ontogeny of humoral immune parameters in fish. Fish Shellfish Immunol (2005) 19:429–39. doi: 10.1016/j.fsi.2005.03.010
63. Ruzicka L, Howe DG, Ramachandran S, Toro S, Van Slyke CE, Bradford YM, et al. The Zebrafish Information Network: new support for non-coding genes, richer Gene Ontology annotations and the Alliance of Genome Resources. Nucleic Acids Res (2019) 47:D867–73. doi: 10.1093/nar/gky1090
64. Bult CJ, Blake JA, Smith CL, Kadin JA, Richardson JE. Mouse Genome Database Group. Mouse Genome Database (MGD) 2019. Nucleic Acids Res (2019) 47:D801–6. doi: 10.1093/nar/gky1056
65. Bhattacharya S, Dunn P, Thomas CG, Smith B, Schaefer H, Chen J, et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci Data (2018) 5:180015.a. doi: 10.1038/sdata.2018.15
66. Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc (2009) 4:1184–91. doi: 10.1038/nprot.2009.97
67. Kinsella RJ, Kähäri A, Haider S, Zamora J, Proctor G, Spudich G, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (2011) 2011:bar030. doi: 10.1093/database/bar030
68. Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol (2015) 16:157. doi: 10.1186/s13059-015-0721-2
69. Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol (2019) 20:238. doi: 10.1186/s13059-019-1832-y
70. Rondeau EB, Minkley DR, Leong JS, Messmer AM, Jantzen JR, von Schalburg KR, et al. The genome and linkage map of the Northern pike (Esox lucius): Conserved synteny revealed between the salmonid sister group and the Neoteleostei. PLoS One (2014) 9:e102089. doi: 10.1371/journal.pone.0102089
71. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods (2017) 14:417–9. doi: 10.1038/nmeth.4197
72. Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res (2015) 4:1521. doi: 10.12688/f1000research.7563.1
73. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol (2014) 15:550. doi: 10.1186/s13059-014-0550-8
74. Bertolotti AC, Layer RM, Gundappa MK, Gallagher MD, Pehlivanoglu E, Nome T, et al. The structural variation landscape in 492 Atlantic salmon genomes. Nat Commun (2020) 11:1–16. doi: 10.1038/s41467-020-18972-x
75. Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet (2000) 16:276–7. doi: 10.1016/S0168-9525(00)02024-2
76. Abu-Jamous B, Kelly S. Clust: automatic extraction of optimal co-expressed gene clusters from gene expression data. Genome Biol (2018) 19:172. doi: 10.1186/s13059-018-1536-8
77. Campbell MA, Hale MC, McKinney GJ, Nichols KM, Pearse DE. Long-term conservation of ohnologs through partial tetrasomy following whole-genome duplication in Salmonidae. G3 (2019) 9(6):2017–28. doi: 10.1534/g3.119.400070
78. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol (2011) 7:539. doi: 10.1038/msb.2011.75
79. Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res (2006) 34:W609–12. doi: 10.1093/nar/gkl315
80. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol (2007) 24:1586–91. doi: 10.1093/molbev/msm088
81. Archer LC, Hutton SA, Harman L, O’Grady MN, Kerry JP, Russell Poole W, et al. The interplay between extrinsic and intrinsic factors in determining migration decisions in brown trout (Salmo trutta): An experimental study. Front Ecol Evol (2019) 7:222. doi: 10.3389/fevo.2019.00222
82. Archer LC, Hutton SA, Harman L, McCormick SD, O’Grady MN, Kerry JP, et al. Food and temperature stressors have opposing effects in determining flexible migration decisions in brown trout (Salmo trutta). Glob Chang Biol (2020) 26:2878–96. doi: 10.1111/gcb.14990
83. Andrews S. FastQC: a quality control tool for high throughput sequence data. In: Reference Source (2010). Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
84. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (2013) 29:15–21. doi: 10.1093/bioinformatics/bts635
85. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics (2009) 25:2078–9. doi: 10.1093/bioinformatics/btp352
86. García-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Götz S, Tarazona S, et al. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics (2012) 28:2678–9. doi: 10.1093/bioinformatics/bts503
87. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics (2016) 32:3047–8. doi: 10.1093/bioinformatics/btw354
88. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics (2015) 31:166–9. doi: 10.1093/bioinformatics/btu638
89. Sahraeian SME, Mohiyuddin M, Sebra R, Tilgner H, Afshar PT, Au KF, et al. Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis. Nat Commun (2017) 8:59. doi: 10.1038/s41467-017-00050-4
90. Li YI, Knowles DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK, et al. Annotation-free quantification of RNA splicing using LeafCutter. Nat Genet (2018) 50:151–8. doi: 10.1038/s41588-017-0004-9
91. Colgan TJ, Fletcher IK, Arce AN, Gill RJ, Rodrigues AR, Stolle E, et al. Caste- and pesticide-specific effects of neonicotinoid pesticide exposure on gene expression in bumblebees. Mol Ecol (2019) 28(8):1964–74. doi: 10.1111/mec.15047
92. Alexa A, Rahnenfuhrer J. “topGO: Enrichment analysis for Gene Ontology. R package version 2.28. 0”. In: Cranio (2016).
93. Nuzzi PA, Lokuta MA, Huttenlocher A. Analysis of neutrophil chemotaxis. Methods Mol Biol (2007) 370:23–36. doi: 10.1385/1-59745-353-6:23
94. Øverland HS, Pettersen EF, Rønneseth A, Wergeland HI. Phagocytosis by B-cells and neutrophils in Atlantic salmon (Salmo salar L.) and Atlantic cod (Gadus morhua L.). Fish Shellfish Immunol (2010) 28:193–204. doi: 10.1016/j.fsi.2009.10.021
95. Molloy S, Holland C, Poole R. Helminth parasites of brown and sea trout Salmo trutta L. from the west coast of Ireland. Biol Environ Proc R Ir Acad (1993) 93B:137–42.
96. Yoshida R, Imai T, Hieshima K, Kusuda J, Baba M, Kitaura M, et al. Molecular cloning of a novel human CC chemokine EBI1-ligand chemokine that is a specific functional ligand for EBI1, CCR7. J Biol Chem (1997) 272:13803–9. doi: 10.1074/jbc.272.21.13803
97. Bird S, Tafalla C. Teleost chemokines and their receptors. Biology (2015) 4:756–84. doi: 10.3390/biology4040756
98. Daniels GD, Zou J, Charlemagne J, Partula S, Cunningham C, Secombes CJ. Cloning of two chemokine receptor homologs (CXC-R4 andCC-R7) in rainbow trout Oncorhynchus mykiss. J Leukoc Biol (1999) 65:684–90. doi: 10.1002/jlb.65.5.684
99. Xu Y, Harder KW, Huntington ND, Hibbs ML, Tarlinton DM. Lyn tyrosine kinase: accentuating the positive and the negative. Immunity (2005) 22:9–18. doi: 10.1016/S1074-7613(04)00381-4
100. Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, et al. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell (1995) 83:301–11. doi: 10.1016/0092-8674(95)90171-X
101. Wu L, Kong L, Yang Y, Bian X, Wu S, Li B, et al. Effects of cell differentiation on the phagocytic activities of IgM+ B cells in a teleost fish. Front Immunol (2019) 10:2225. doi: 10.3389/fimmu.2019.02225
102. De Smet R, Adams KL, Vandepoele K, Van Montagu MCE, Maere S, Van de Peer Y. Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proc Natl Acad Sci U S A (2013) 110:2898–903. doi: 10.1073/pnas.1300127110
103. Librado P, Vieira FG, Sánchez-Gracia A, Kolokotronis S-O, Rozas J. Mycobacterial phylogenomics: An enhanced method for gene turnover analysis reveals uneven levels of gene gain and loss among species and gene families. Genome Biol Evol (2014) 6:1454–65. doi: 10.1093/gbe/evu117
104. Liu S, Liu Y, Yang X, Tong C, Edwards D, Parkin IAP, et al. The Brassica oleracea genome reveals theasymmetrical evolution of polyploid genomes. Nat Commun (2014) 5:3930. doi: 10.1038/ncomms4930
105. Webster TMU, Uren Webster TM, Santos EM. Global transcriptomic profiling demonstrates induction of oxidative stress and of compensatory cellular stress responses in brown trout exposed to glyphosate and Roundup. BMC Genomics (2015) 16:32. doi: 10.1186/s12864-015-1254-5
106. Sutherland BJG, Prokkola JM, Audet C, Bernatchez L. Sex-specific co-expression networks and sex-biased gene expression in the salmonid Brook Charr Salvelinus fontinalis. G3 (2019) 9:955–68. doi: 10.1534/g3.118.200910
107. Sheth K, Bankey P. The liver as an immune organ. Curr Opin Crit Care (2001) 7:99–104. doi: 10.1097/00075198-200104000-00008
108. Gao B, Jeong W-I, Tian Z. Liver: An organ with predominant innate immunity. Hepatology (2008) 47:729–36. doi: 10.1002/hep.22034
109. Deslyper G, Colgan TJ, Cooper AJR, Holland CV, Carolan JC. A proteomic investigation of hepatic resistance to Ascaris in a murine model. PLoS Negl Trop Dis (2016) 10:e0004837. doi: 10.1371/journal.pntd.0004837
110. Deslyper G, Holland CV, Colgan TJ, Carolan JC. The liver proteome in a mouse model for Ascarissuum resistance and susceptibility: evidence for an altered innate immuneresponse. Parasit Vectors (2019) 12:402. doi: 10.1186/s13071-019-3655-9
111. Kim J-H, Macqueen DJ, Winton JR, Hansen JD, Park H, Devlin RH. Effect of growth rate on transcriptomic responses to immunestimulation in wild-type, domesticated, and GH-transgenic coho salmon. BMC Genomics (2019) 20:1024. doi: 10.1186/s12864-019-6408-4
112. Skugor S, Jørgensen SM, Gjerde B, Krasnov A. Hepatic gene expression profiling reveals protective responses inAtlantic salmon vaccinated against furunculosis. BMC Genomics (2009) 10:503. doi: 10.1186/1471-2164-10-503
113. Martin SAM, Blaney SC, Houlihan DF, Secombes CJ. Transcriptome response following administration of a live bacterial vaccine in Atlantic salmon (Salmo salar). Mol Immunol (2006) 43:1900–11. doi: 10.1016/j.molimm.2005.10.007
114. Causey DR, Pohl MAN, Stead DA, Martin SAM, Secombes CJ, Macqueen DJ. High-throughput proteomic profiling of the fish liver following bacterial infection. BMC Genomics (2018) 19:719. doi: 10.1186/s12864-018-5092-0
115. Pickering AD. Cortisol-induced lymphocytopenia in brown trout, Salmo trutta L. Gen Comp Endocrinol (1984) 53:252–9. doi: 10.1016/0016-6480(84)90250-8
116. Pickering AD, Duston J. Administration of cortisol to brown trout, Salmo trutta L., and its effects on the susceptibility to Saprolegnia infection and furunculosis. J Fish Biol (1983) 23:163–75. doi: 10.1111/j.1095-8649.1983.tb02891.x
117. Pickering AD, Christie P. Sexual differences in the incidence and severity of ectoparasitic infestation of the brown trout, Salmo trutta L. J Fish Biol (1980) 16:669–83. doi: 10.1111/j.1095-8649.1980.tb03746.x
118. Chen CY, Lopes-Ramos CM, Kuijjer ML, Paulson JN. Sex differences in gene expression and regulatory networks across 29 human tissues. Cell Reports (2020) 31:107795. doi: 10.1016/j.celrep.2020.107795
119. Wright AE, Fumagalli M, Cooney CR, Bloch NI, Vieira FG, Buechel SD, et al. Male-biased gene expression resolves sexual conflict through the evolution of sex-specific genetic architecture. Evol Lett (2018) 2:52–61. doi: 10.1002/evl3.39
120. Barson NJ, Aykanat T, Hindar K, Baranski M, Bolstad GH, Fiske P, et al. Sex-dependent dominance at a single locus maintains variation in ageat maturity in salmon. Nature (2015) 528:405–8. doi: 10.1038/nature16062
121. Mank JE. Population genetics of sexual conflict in the genomicera. Nat Rev Genet (2017) 18:721–30. doi: 10.1038/nrg.2017.83
122. Gallach M, Betrán E. Intralocus sexual conflict resolved through geneduplication. Trends Ecol Evol (2011) 26:222–8. doi: 10.1016/j.tree.2011.02.004
123. VanKuren NW, Long M. Gene duplicates resolving sexual conflict rapidly evolved essentialgametogenesis functions. Nat Ecol Evol (2018) 2:705–12. doi: 10.1038/s41559-018-0471-0
124. Cheng C, Kirkpatrick M. Sex-specific selection and sex-biased gene expression in humans andflies. PLoS Genet (2016) 12:e1006170. doi: 10.1371/journal.pgen.1006170
125. Coughlan J, McGinnity P, O’Farrell B, Dillane E, Diserud O, de Eyto E, et al. Temporal variation in an immune response gene (MHC I) in anadromousSalmo trutta in an Irish river before and during aquacultureactivities. ICES J Mar Sci (2006) 63:1248–55. doi: 10.1016/j.icesjms.2006.03.025
Keywords: facultatively anadromous, immunity, gene expression, gene duplications, sexual dimorphism, salmonids
Citation: Colgan TJ, Moran PA, Archer LC, Wynne R, Hutton SA, McGinnity P and Reed TE (2021) Evolution and Expression of the Immune System of a Facultatively Anadromous Salmonid. Front. Immunol. 12:568729. doi: 10.3389/fimmu.2021.568729
Received: 02 June 2020; Accepted: 07 January 2021;
Published: 26 February 2021.
Edited by:
Sissel Jentoft, University of Oslo, NorwayReviewed by:
Daniel J. Macqueen, University of Edinburgh, United KingdomCopyright © 2021 Colgan, Moran, Archer, Wynne, Hutton, McGinnity and Reed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas J. Colgan, am9lLmNvbGdhbkB1Y2MuaWU=
†ORCID: Thomas J. Colgan orcid.org/0000-0002-0547-8228
Peter A. Moran orcid.org/0000-0002-2206-4721
Thomas E. Reed orcid.org/0000-0002-2993-0477
‡Present address: Thomas J. Colgan, Institute of Organismic and Molecular Evolution, Johannes Gutenberg University Mainz, Mainz, Germany Peter A. Moran, Department of Ecological Science—Animal Ecology, Vrije Universiteit Amsterdam, Amsterdam, Netherlands
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.