 Tatjana Welzel
Tatjana Welzel Susanne M. Benseler3
Susanne M. Benseler3- 1Autoinflammation Reference Center Tuebingen (arcT) and Division of Pediatric Rheumatology, Department of Pediatrics, University Hospital Tuebingen, Tuebingen, Germany
- 2Pediatric Pharmacology and Pharmacometrics, University Children‘s Hospital Basel (UKBB), University Basel, Basel, Switzerland
- 3Rheumatology, Department of Pediatrics, Alberta Children's Hospital (ACH), ACH Research Institute, University of Calgary, Calgary, AB, Canada
Monogenic Interleukin 1 (IL-1) mediated autoinflammatory diseases (AID) are rare, often severe illnesses of the innate immune system associated with constitutively increased secretion of pro-inflammatory cytokines. Clinical characteristics include recurrent fevers, inflammation of joints, skin, and serous membranes. CNS and eye inflammation can be seen. Characteristically, clinical symptoms are coupled with elevated inflammatory markers, such as C-reactive protein (CRP) and serum amyloid A (SAA). Typically, AID affect infants and children, but late-onset and atypical phenotypes are described. An in-depth understanding of autoinflammatory pathways and progress in molecular genetics has expanded the spectrum of AID. Increasing numbers of genetic variants with undetermined pathogenicity, somatic mosaicisms and phenotype variability make the diagnosis of AID challenging. AID should be diagnosed as early as possible to prevent organ damage. The diagnostic approach includes patient/family history, ethnicity, physical examination, specific functional testing and inflammatory markers (SAA, CRP) during, and in between flares. Genetic testing should be performed, when an AID is suspected. The selection of genetic tests is guided by clinical findings. Targeted and rapid treatment is crucial to reduce morbidity, mortality and psychosocial burden after an AID diagnosis. Management includes effective treat-to-target therapy and standardized, partnered monitoring of disease activity (e.g., AIDAI), organ damage (e.g., ADDI), patient/physician global assessment and health related quality of life. Optimal AID care in childhood mandates an interdisciplinary team approach. This review will summarize the current evidence of diagnosing and managing children with common monogenic IL-1 mediated AID.
Introduction
Monogenic IL-1 mediated autoinflammatory diseases (AID) are rare, often severe disorders caused by variants in innate immunity genes resulting in a constitutive overproduction of pro-inflammatory cytokines (1, 2). Clinical characteristics can include recurrent fevers, inflammation of joints, eyes, skin, and serous membranes (3). Severe phenotypes can include inflammation of CNS, bones, inner ears with hearing loss, and kidneys (4, 5). Patients with AID frequently report fatigue, irritability, headache, abdominal pain, and musculoskeletal complaints (6–9). Characteristically, clinical symptoms are coupled with increased inflammatory markers, such as C-reactive protein (CRP) and serum amyloid A (SAA) (3). AA amyloidosis is a serious complication with a prevalence of up to 50% in untreated familiar Mediterranean fever (FMF) (10).
The genetic origin of IL-1 mediated AID was first determined for FMF in 1997 (11, 12). In 1999, mutations in the TNFRSF1A gene were shown to be associated with the Tumor necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS) previously called Familial Hibernian fever (13–15). In the following disease-causing genes were identified for several AID including the Cryopyrin-Associated Periodic Syndromes (CAPS) and the Hyperimmunoglobulinemia D Syndrome (HIDS)/Mevalonate Kinase Deficiency (MKD). The AID spectrum is continuously expanding due to advancements in genomic technologies such as next generation-sequencing (NGS) (16, 17). Translational research has further advanced the pathophysiological understanding of AID.
The management of AID patients include early diagnosis, effective therapy, treat-to-target (T2T) strategies and standardized monitoring of disease activity and damage. Therefore, a multidisciplinary team approach and attention to disease-related psychosocial burden are important. This review will summarize the available evidence focusing on common monogenic IL-1 mediated AID including CAPS, HIDS/MKD, FMF, and TRAPS.
Introducing the Inflammasome
Inflammasomes are intracellular complexes controlling inflammation and immune cell activation triggered by a variety of exogenous and endogenous triggers (2, 18) (Figure 1). The nucleotide-binding domain-like receptor (NLR) family forms a group of proteins involved in the formation of inflammasome sensors (22). These contain a pyrin domain or a caspase activation and recruitment domain (22). One of the most prominent members of NLR families in monogenic AID is NLRP3 (22). Pyrin is another important inflammasome-forming protein (23). Inflammasome assembly (Figure 1) leads to the activation of caspase-1, which is able to process the inactive pre-cursor form of IL-1β to its mature bioactive form and induce its release (22, 24). IL-1β is one of the most prominent products of inflammasome activation and a key regulator of systemic inflammation (22). Genetic variants can alter proteins involved in the inflammasome pathways (25).
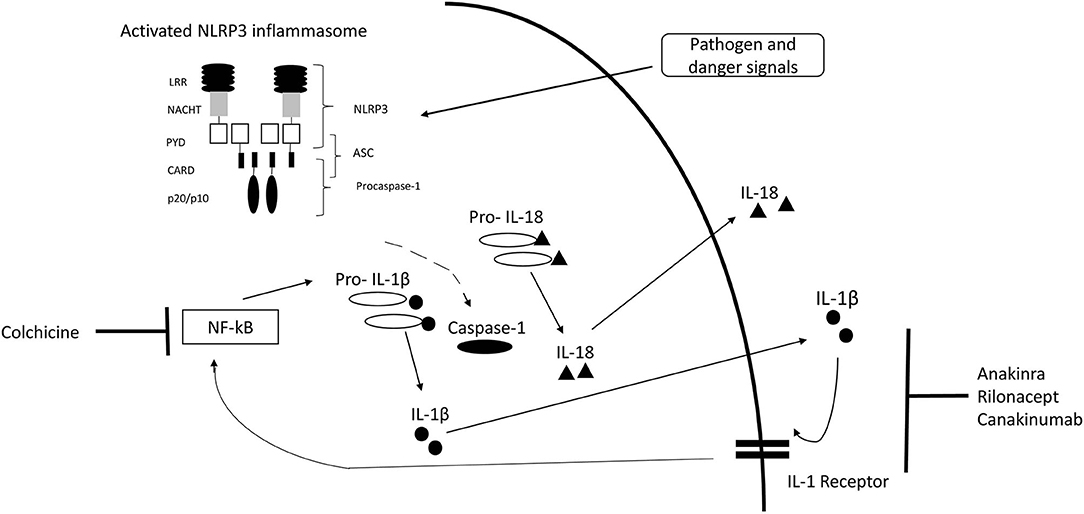
Figure 1. IL-1 inflammasome structure, cytokine release, and treatment target. Inflammasome formation is induced by a variety of triggers. Activated NLRP3 subsequently nucleates ASC forming filaments via PYD-PYD interactions and drives procaspase-1 filament formation through CARD-CARD interactions. Caspase-1 mediates cleavage of pro-IL-1β and pro-IL-18 to IL-1β and IL-18. [Modified from McCoy et al. (19), Lachmann et al. (20), Broderick (21)]. CARD, caspase activation and recruitment domain; LRR, leucine-rich-repeats; PYD, Pyrin domain; ASC, Apoptosis related speck-like protein containing CARD.
Pathogenesis in Brief of Common IL-1 Mediated Aid
CAPS pathogenic gain-of-function variants in the NLRP3 gene result in activation of the NLRP3 inflammasome with increased IL-1β secretion (26, 27). FMF is caused by autosomal recessive variants in the MEFV gene encoding for pyrin, a protein involved in the pyrin inflammasome (11, 12). HIDS/MKD results from loss-of-function mutations in the MVK gene, encoding for an enzyme of the isoprenoid biosynthesis (28, 29). The impaired isoprenoid biosynthesis leads to accumulation of mevalonate, shortage of end-products and reduced isoprenylated proteins (30, 31). Particularly, the shortage of geranylgeranyl-pyrophosphate affects small GTPases, resulting in IL-1β hypersecretion, activation of the pyrin inflammasome and the nuclear factor κB (NF-κB) pathway (32–34). TRAPS results from variants in the TNFRSF1A gene (14). The intracellular retention of the mutated receptor causes several pathological responses including autophagy, increased endoplasmatic reticulum stress, excessive mitochondrial reactive oxygen species and enhanced NF-κB activation with production of pro-inflammatory cytokines including IL-1 (35–37). Four distinctly different pathogenic variants in heterogeneous inflammatory pathways result in increased IL-1β release. It remains unclear, why the common feature of increased IL-1 β is associated with a heterogeneous clinical phenotype across diseases and even within each IL-1 mediated AID.
Diagnosis
In 2012, Toplak et al. reported a medium diagnostic delay for AID of more than 7 years (range 0.3–76). There was a rapid increase in recognition after the first AID gene discovery in 1997 (38). Diagnosis of AID should be made as early as possible to prevent organ damage (39). In patients with suspected AID, a stepwise diagnostic approach should be performed, including patient and family history, ethnicity, physical examination, and inflammatory markers during febrile attacks and symptom free-intervals and genetic testing (40–42) (Figure 2). Other differential diagnosis such as immunodeficiencies, infections, autoimmune diseases and malignancies need to be excluded. Red flags are a family history of early hearing loss or renal transplants, Mediterranean background, fever periodicity and specific flare triggers, such as cold exposure.
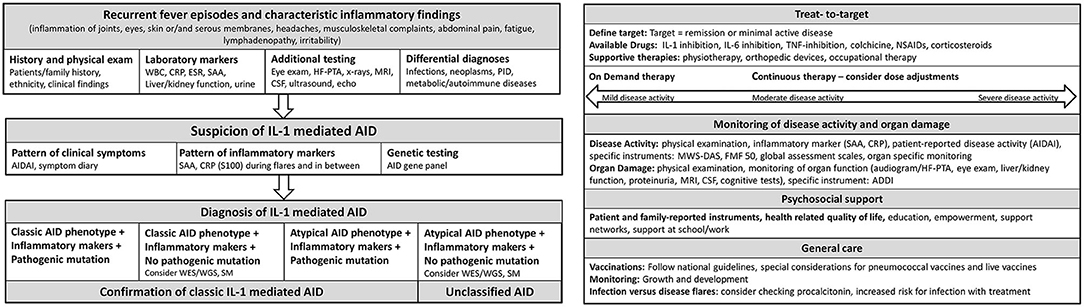
Figure 2. Flow sheet diagnostic steps and management of AID. AID, autoinflammatory disease; WBC, whole blood count; CRP, c reactive protein; ESR, erythrocyte sedimentation rate; SAA, Serum Amyloid A; HF-PTA, high frequency pure tone audiogram; MRI, magnet resonance imaging; CSF, cerebrospinal fluid; PID, primary immune deficiency; IL, Interleukin; AIDAI, Autoinflammatory Disease Activity Index; S100, S 100 proteins; WES, whole exome sequencing; WGS, whole genome sequencing; SM, somatic mosaicism; TNF, Tumor necrosis factor; NSAIDs, Nonsteroidal anti-inflammatory drugs; MWS-DAS, Muckle Wells Syndrome Disease activity score; FMF 50, Familial Mediterranean Fever 50 Score; ADDI, Autoinflammatory Disease Damage Index.
Clinical Aid Symptoms
Recurrent fever is a leading symptom varying in duration for different AID associated with specific clinical symptoms. The Autoinflammatory Disease Activity Index (AIDAI), a standardized symptom diary (43), captures AID characteristic symptoms and helps identify phenotypic patterns.
The spectrum of CAPS includes three phenotypes and their overlaps: familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and the neonatal-onset multisystem inflammatory disease (NOMID)/chronic infantile neurologic cutaneous and articular syndrome (CINCA) (44, 45). The FCAS phenotype is characterized by cold-induced neutrophilic dermatitis, fever, and chills (4). Patients with the moderate MWS may suffer from sensorineural hearing loss, urticaria-like rash and arthritis, while the severe NOMID phenotype is associated with aseptic meningitis, skeletal deformities and papilledema (4). MWS onset is often in early childhood, while NOMID is frequently recognized in the neonatal period (4).
Characteristic symptoms of FMF are recurrent fevers ranging 1–3 days, serositis, abdominal pain and/or pericarditis, arthritis, myalgia, and erysipelas-like erythema (46).
HIDS/MKD related features include malaise, fatigue, weight loss, lymphadenopathy, mucocutaneous involvement, musculoskeletal complaints, and gastrointestinal symptoms (47). Neurological involvement and life-threatening macrophage activation syndrome are observed.
TRAPS patients typically present with musculoskeletal complaints, abdominal pain, maculopapular and migratory rash, and periorbital edema (6). Serositis, pericarditis, arthritis and myalgia may be more prominent in adulthood, while abdominal pain is found more typically in childhood (6, 48). Furthermore, Gaggiano et al. highlight that high penetrance variants are associated with abdominal pain, amyloidosis and subclinical inflammation, whereas oral aphthosis is frequently observed in low-penetrance variants (48). TRAPS may start in adulthood, family history may then be non-contributory.
Laboratory Investigations
CRP, SAA and complete blood count are considered first line laboratory parameters and should be tested during febrile attacks and in symptom-free intervals (42). Liver and muscle enzymes, renal function tests, and urine analysis including 24 h evaluation for proteinuria should be performed serially (Figure 2). Immunoglobulin D (IgD) and mevalonic acid levels may be useful in suspected HIDS/MKD (47).
Functional Testing
AID can affect multiple organ systems and may result in organ damage. Functional testing is required at diagnosis and during monitoring (Figure 2). In suspected AID, a thorough physical examination including growth, development, musculoskeletal, neurological, and ophthalmologic examination should be performed (39). In suspected CAPS, high frequency audiograms (HF-PTA) including 0.5–10 kHz, formal cognitive testing, magnetic resonance imaging (MRI) brain, spinal tap with opening pressure, cerebrospinal fluid (CSF) cell count and protein, and a lesional skin biopsy should be considered (39). In patients with severe musculoskeletal involvement, x-rays and bone MRI are recommended (39). In HIDS/MKD, additional cognitive testing and muscle and liver enzyme monitoring are recommended (39). For FMF patients, particularly during colchicine treatment, renal and liver function tests should be monitored serially (49).
Genetic Testing
Molecular testing should be performed, when the clinical phenotype, laboratory and functional tests are suggestive of AID (40). Sanger sequencing had been primarily utilized to identify AID-causing variants. In recent years, NGS-AID panels have become the gold standard (50). Complete coding sequences of AID genes are enriched in these panels. The Genetic Testing Registry database (https://www.ncbi.nlm.nih.gov/gtr/) provides an overview of available panels. The reported diagnostic yield of comprehensive gene testing panels seems to range between 21 and 32% (51, 52). Whole exome sequencing (WES) should be considered in patients with negative panel testing to improve the diagnostic yield preferably using a family-based trio approach (53). In addition, WES enables the discovery of novel variants in known AID genes and in those not yet associated with diseases. Whole genome sequencing (WGS) can identify deep intronic variants and mutations in non-coding regulatory regions and therefore increases the diagnostic yield (53). Additionally, it allows a much more reliable identification of copy number variations compared with WES (53).
Somatic mosaicism results from de novo post-zygotic mutations. AID-panel testing in these patients may be negative. AID onset and phenotype may be atypical. Saito et al. first identified somatic mutations causing NOMID (54). Subsequently, Tanaka et al. reported somatic mutations in 70% of previously mutation-negative NOMID patients (55).
The identified pathogenic variants may allow prediction of disease severity; for example FMF patients with homozygous M694V were found to be at high risk for a severe phenotype including early disease-onset (56). Similarly, in HIDS/MKD combined heterozygosity for p.V377I/p.I268T was shown to be a risk factor for AA amyloidosis (47). Genetic variants are typically described as “pathogenic,” “likely pathogenic,” “uncertain significance,” “likely benign,” and “benign” (57). Some platforms can assist clinicians and geneticists in determining the pathogenicity of variants including the MOLGENIS platform (58). The Infevers database is an exhaustive registry of sequence variants identified in AID related genes (59).
Diagnostic and Classification Criteria
Diagnostic criteria are used to guide the care of individual patients, establish the correct diagnosis, and start targeted treatment (60). The diagnostic criteria for CAPS mandate raised inflammatory markers (CRP/SAA) plus at least two of the following symptoms: neutrophilic dermatitis, cold-triggered episodes, sensorineural hearing loss, musculoskeletal symptoms, chronic aseptic meningitis, and skeletal abnormalities (61). These criteria enable physicians to make a CAPS diagnosis without mandating evidence of a disease causing variant (e.g., in case of low-penetrance variants). For FMF, the first Tel Hashomer criteria were proposed for adult patients in 1967 (62). In 1997, Livneh et al. proposed a set of diagnostic FMF criteria based on the presence of one major or two minor criteria, or one minor plus five supportive criteria (63). The major criteria are typical attacks (lasting 12–72 h, >3 attacks, fever > 38°C) with any one of peritonitis, pleuritis/pericarditis, monoarthritis or unilateral “orchitis”, erysipelas-like eruption in the calf, and/or symmetric myalgia with extreme tenderness in the lower extremities. The minor criteria were defined as incomplete attacks, exertional leg pain and favorable response to colchicine. In 2009, Yalçinkaya, Ozen et al. proposed the pediatric FMF criteria requiring ≥2 of the following characteristics: fever, abdominal pain, one-sided chest pain and arthritis, each lasting 6–72 h, ≥3 attacks, and family history of FMF (64), allowing to make a clinical FMF diagnosis in case of inconclusive/negative genetics and are useful in selection which patients should be genetically tested. These criteria were developed in a Turkish population. Subsequently, Demirkaya et al. compared the performance of these existing criteria in the pediatric AID cohort of the Eurofever registry (65). The Yalchinkaya-Ozen criteria yielded a higher sensitivity (87.4%) than 1967 and 1997 criteria. The authors suggest that they can be used for FMF diagnosis in pediatric patients from European and eastern Mediterranean region (65). Classification criteria are primarily used to define patients cohorts for research (60). In 2019, Gattorno et al. published validated evidence-based classification criteria for hereditary AID including CAPS, HIDS/MKD, FMF, and TRAPS with pathogenic/likely pathogenic variants, low-penetrance variants and without genetic testing/findings (66).
Diagnostic Uncertainty
While advanced genetic testing may establish a diagnosis in some patients, testing may still be negative, inconclusive or even misleading (1, 52, 67, 68). Therefore, the correlation of clinical phenotype and genetic result is critical (67). Low-penetrance variants in AID genes can be present in the general population. As some of these low-penetrance variant carriers nevertheless express AID symptoms unlike the known classical phenotype in confirmed pathogenic variants (69), it might be possible that these are mediated by different pathways parallel to the caspase-1 activation (70). Moreover, low-penetrance variants may confer an increased susceptibility to inflammation (71).
Effective Aid Therapy: Treat-to-Target (T2T)
Therapy is comprehensive including medication, psychosocial support, physiotherapy and supportive care such as hearing aids (Figure 2). Traditional symptomatic therapy consisted of non-steroidal anti-inflammatory drugs (NSAIDS) and glucocorticoids, which often shortened disease flares but can increase their frequency (39, 49). Today, IL-1 inhibitors play a pivotal role and evidence-based AID treatment plans are available (39, 49).
Targeting of inflammatory pathways enables T2T strategies (19, 72) (Figure 1). Key component of T2T is the definition of a target such as remission or minimal disease activity. Standardized serial assessments are required to determine, if the target is achieved (72) (Figure 2). Different levels of disease activity require different treatment approaches (39, 72, 73). Frequently, dose adjustments are required, particularly in children and in severe disease subtypes (74, 75).
Colchicine
Colchicine is the established first-line FMF therapy with favorable response and risk reduction of AA amyloidosis (49, 76). Colchicine should be started as early as possible (49). Colchicine is metabolized by CYP3A4 enzymes and the P-glycoprotein (P-gp) efflux transporter, therefore, concomitant treatment with CYP3A4 and/or P-pg inhibitors should be avoided (77, 78).
IL-1 Targeting Drugs
IL-1 inhibition has been shown to be safe and effective in controlling inflammation in CAPS, TRAPS, and MKD/HIDS (75, 79–82). It is a valid therapeutic strategy in FMF patients with colchicine-intolerance/resistance (83–86). Currently, three IL-1 inhibitors are approved by the US Food and Drug Administration for AID.
Long-term efficacy and safety of the short-acting recombinant IL-1 receptor antagonist anakinra has been confirmed in several studies (79, 86–90). Anakinra is administered daily subcutaneously and blocks the binding of IL-1α and IL-1β to the IL-1 receptor. In a study of 43 CAPS patients treated with anakinra up to 5 years, serious adverse events reported most frequently were pneumonia and gastroenteritis (79). There is evidence that pediatric patients with undifferentiated AID may also benefit from anakinra (91).
The recombinant soluble IL-1 receptor rilonacept binds to IL-1α and IL-1β. Weekly subcutaneous administration has shown a good safety and efficacy profile (92).
Canakinumab is a fully humanized anti-IL-1β monoclonal antibody selectively binding soluble IL-1β. It has to be administered every 4–8 weeks subcutaneously. Several studies confirmed long-term efficacy and safety (93–98). Brogan et al., reported complete response to canakinumab in 17 CAPS patients <5 years of age (71% MWS, 24% NOMID, 6% FCAS) (96). Open label observations suggest that children require higher doses up to 8 mg/kg/4 weekly to achieve remission, particularly in severe CAPS phenotype (94). Recently, efficacy and safety of canakinumab was demonstrated for FMF, TRAPS and HIDS/MKD (75, 99). In patients with HIDS/MKD dose adjustment is frequently needed (75). The rate of serious infections was 7.4/100 patients-years in 181 patients with TRAPS, HIDS and CAPS (75).
Alternatives
In a prospective open-label dose escalation study, etanercept reduce symptoms and inflammatory markers in a dose-dependent manner in TRAPS (100). FMF patients with chronic arthritis and sacroiliitis can benefit from TNF-inhibition (101). IL-6 inhibition may be promising in TRAPS patients (102) and HIDS/MKD, particularly when refractory to anakinra/etanercept (103–105). Hematopoietic stem cell transplantation has been performed in refractory HIDS/MKD patients (39).
Psychosocial Needs in Aid
The care of patients with AID should include psycho-social support, as AID affect all areas of life (106) (Figure 2). AID are associated with depression, lower health related quality of life, anxiety and social isolation (107–110). Patients/parents have to deal with work/school-related challenges because of frequent sick-leaves (107). Long-term management should take psychological factors such as illness beliefs, coping strategies and the distribution of dependency into account (108). Patient support networks can provide important support (111).
Monitoring of Aid Activity and Damage
Regular monitoring of disease activity is crucial (39). This includes physical examination, measurement of height and weight, neurological and musculoskeletal examination, and determination of SAA and CRP levels to detect ongoing inflammation (Figure 2). Repeatedly increased SAA levels between AID flares may indicate a significant risk for AA amyloidosis. Monitoring of SAA and S100 proteins may detect subclinical diseases activity, particularly in FMF (112, 113).
The validated patient-reported AIDAI is a simple tool for assessing disease activity (43). The AIDAI contains 13 items addressing fever (>38°C), overall symptoms, specific AID symptoms, and use of NSAIDs (43). The clinical symptoms are dichotomous and scored as 0 (absent) or 1 (present) (43). The maximum score per day is 12 with a cumulative monthly score ranging from 0 to 372 (43). The clinical meaningful threshold indicating active AID is a score of at least 9 (43). The Autoinflammatory Disease Damage Index (ADDI) is a reliable instrument to assess disease-related organ damage in FMF, CAPS, TRAPS and HIDS/MKD (114). ADDI consists of 18 items grouped in eight categories of reproductive, renal/amyloidosis, developmental, serosal, neurological, auditory, ocular, and musculoskeletal damage (114, 115). Damage is defined as persistent or irreversible change in structure or function present for at least 6 months (114, 115). The ADDI can be used to monitor structural damage in individual patients, and allows outcome analysis and comparison of damage accrual in clinical trials (114).
Infections and Vaccines
Patients can experience febrile inflammatory episodes not primarily related to their AID. Particularly in atypical AID-flares, infections have to be excluded (39). While CRP does not discriminate between infection and flare, procalcitonin (PCT) may be a promising marker (116). Some infectious diseases are preventable by vaccination. However, both vaccination and infection may trigger flares, particularly in HIDS/MKD (117). Patients with CAPS may develop severe local and systemic inflammatory reactions after pneumococcal vaccination (118, 119). The 13-valent pneumococcal conjugate vaccine appears to be more favorable compared to the polysaccharide vaccine (119). In general, vaccination recommendations for patients with immunosuppressive therapy and inflammatory rheumatic diseases can be used for AID, where inactive vaccines are considered as safe and are recommended to national vaccination guidelines (120–123) (Figure 2). In accordance with the recommendations of the European League Against Rheumatism, annually influenza vaccinations are recommended for AID patients and immunosuppressive therapy (120, 124). Live -vaccines should be avoided/has to be considered individual for the patient (121, 122, 124).
Summary
AID are rare diseases associated with the risk of severe morbidity, mortality and reduced health-related quality of life. The increasing number of somatic mosaicisms and low-penetrance variants make the diagnosis of these potential live-threatening diseases challenging. A standardized diagnostic approach for suspected AID should include the clinical phenotype, inflammatory markers, functional, and genetic testing (Figure 2). AID panels should be performed and may need to be supplemented with WES/WGS. The management of AID mandates a multidisciplinary team and psychosocial support. Medication should be tailored individually using T2T strategies. In IL-1 mediated AID, colchicine, and IL-1 inhibition are effective. Alternative therapies including IL-6 inhibition and TNF-blockade can be beneficial. Regular target evaluation and standardized monitoring of disease activity and organ damage is important. Vaccines should be administered according to national vaccination guidelines, respecting general vaccination recommendations for patients with rheumatic diseases.
Author Contributions
TW, SB, and JK-D conceived the concept of the manuscript, wrote the manuscript, drafted the work, and reviewed the article critically. All authors have provided approval for publication.
Conflict of Interest
SB participated on Add-boards from Sobi and Novartis. JK-D received speaker honoraria and grant support from Sobi and Novartis.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sarrabay G, Touitou I. Autoinflammation. Management of hereditary recurrent fevers–SHARE experience. Nat Rev Rheumatol. (2015) 11:567–9. doi: 10.1038/nrrheum.2015.114
2. Broderick L. Hereditary autoinflammatory disorders: recognition and treatment. Immunol Allergy Clin North Am. (2019) 39:13–29. doi: 10.1016/j.iac.2018.08.004
3. Lachmann HJ. Periodic fever syndromes. Best Pract Res Clin Rheumatol. (2017) 31:596–609. doi: 10.1016/j.berh.2017.12.001
4. Hoffmann HM, Kuemmerle-Deschner JB, Goldbach-Mansky R. Cryopyrin-Associated Periodic Syndromes (CAPS). In: Hashkes PJ, Laxer RM, Simon A, editors. Textbook of Autoinflammation. Cham: Springer Nature Switzerland AG (2019). p. 347–65. doi: 10.1007/978-3-319-98605-0_19
5. Kuemmerle-Deschner JB, Koitschev A, Ummenhofer K, Hansmann S, Plontke SK, Koitschev C, et al. Hearing loss in Muckle-Wells syndrome. Arthritis Rheum. (2013) 65:824–31. doi: 10.1002/art.37810
6. Lachmann HJ, Papa R, Gerhold K, Obici L, Touitou I, Cantarini L, et al. The phenotype of TNF receptor-associated autoinflammatory syndrome (TRAPS) at presentation: a series of 158 cases from the Eurofever/EUROTRAPS international registry. Ann Rheum Dis. (2014) 73:2160–7. doi: 10.1136/annrheumdis-2013-204184
7. Hoffmann GF, Charpentier C, Mayatepek E, Mancini J, Leichsenring M, Gibson KM, et al. Clinical and biochemical phenotype in 11 patients with mevalonic aciduria. Pediatrics. (1993) 91:915–21.
8. Levy R, Gerard L, Kuemmerle-Deschner J, Lachmann HJ, Kone-Paut I, Cantarini L, et al. Phenotypic and genotypic characteristics of cryopyrin-associated periodic syndrome: a series of 136 patients from the Eurofever Registry. Ann Rheum Dis. (2015) 74:2043–9. doi: 10.1136/annrheumdis-2013-204991
9. Bader-Meunier B, Florkin B, Sibilia J, Acquaviva C, Hachulla E, Grateau G, et al. Mevalonate kinase deficiency: a survey of 50 patients. Pediatrics. (2011) 128:e152–9. doi: 10.1542/peds.2010-3639
10. Obici L, Merlini G. Amyloidosis in autoinflammatory syndromes. Autoimmun Rev. (2012) 12:14–7. doi: 10.1016/j.autrev.2012.07.016
11. French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. (1997) 17:25–31. doi: 10.1038/ng0997-25
12. International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. (1997) 90:797–807. doi: 10.1016/S0092-8674(00)80539-5
13. Williamson LM, Hull D, Mehta R, Reeves WG, Robinson BH, Toghill PJ. Familial Hibernian fever. Q J Med. (1982) 51:469–80.
14. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. (1999) 97:133–44. doi: 10.1016/S0092-8674(00)80721-7
15. Mulley J, Saar K, Hewitt G, Ruschendorf F, Phillips H, Colley A, et al. Gene localization for an autosomal dominant familial periodic fever to 12p13. Am J Hum Genet. (1998) 62:884–9. doi: 10.1086/301793
16. Kastner DL. Autoinflammation: past, present, and future. In: Hashkes PJ, Laxer RM, Simon A, editors. Textbook of Autoinflammation. Cham: Springer Nature Switzerland AG (2019). p. 3–15. doi: 10.1007/978-3-319-98605-0_1
17. Georgin-Lavialle S, Rodrigues F, Hentgen V, Fayand A, Quartier P, Bader-Meunier B, et al. Clinical overview of auto-inflammatory diseases. Rev Med Interne. (2018) 39:214–32. doi: 10.1016/j.revmed.2018.01.004
18. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
19. McCoy SS, Stannard J, Kahlenberg JM. Targeting the inflammasome in rheumatic diseases. Transl Res. (2016) 167:125–37. doi: 10.1016/j.trsl.2015.06.006
20. Lachmann HJ, Quartier P, So A, Hawkins PN. The emerging role of interleukin-1β in autoinflammatory diseases. Arthritis Rheum. (2011) 63:314–24. doi: 10.1002/art.30105
21. Broderick L. Inflammasomes and autoinflammation. In: Hashkes PJ, Laxer RM, Simon A, editors. Textbook of Autoinflammation. Cham: Springer Nature Switzerland AG (2019). p. 89–109. doi: 10.1007/978-3-319-98605-0
22. de Torre-Minguela C, Mesa Del Castillo P, Pelegrin P. The NLRP3 and pyrin inflammasomes: implications in the pathophysiology of autoinflammatory diseases. Front Immunol. (2017) 8:43. doi: 10.3389/fimmu.2017.00043
23. Yu JW, Wu J, Zhang Z, Datta P, Ibrahimi I, Taniguchi S, et al. Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ. (2006) 13:236–49. doi: 10.1038/sj.cdd.4401734
24. Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. (2015) 42:991–1004. doi: 10.1016/j.immuni.2015.06.003
25. Martorana D, Bonatti F, Mozzoni P, Vaglio A, Percesepe A. Monogenic autoinflammatory diseases with mendelian inheritance: genes, mutations, and genotype/phenotype correlations. Front Immunol. (2017) 8:344. doi: 10.3389/fimmu.2017.00344
26. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. (2001) 29:301–5. doi: 10.1038/ng756
27. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. (2004) 20:319–25. doi: 10.1016/S1074-7613(04)00046-9
28. Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet. (1999) 22:178–81. doi: 10.1038/9696
29. Houten SM, Kuis W, Duran M, de Koning TJ, van Royen-Kerkhof A, Romeijn GJ, et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet. (1999) 22:175–7. doi: 10.1038/9691
30. Munoz MA, Jurczyluk J, Mehr S, Chai RC, Arts RJW, Sheu A, et al. Defective protein prenylation is a diagnostic biomarker of mevalonate kinase deficiency. J Allergy Clin Immunol. (2017) 140:873–5 e6. doi: 10.1016/j.jaci.2017.02.033
31. Jurczyluk J, Munoz MA, Skinner OP, Chai RC, Ali N, Palendira U, et al. Mevalonate kinase deficiency leads to decreased prenylation of Rab GTPases. Immunol Cell Biol. (2016) 94:994–9. doi: 10.1038/icb.2016.58
32. Mandey SH, Kuijk LM, Frenkel J, Waterham HR. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum. (2006) 54:3690–5. doi: 10.1002/art.22194
33. Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol. (2016) 17:914–21. doi: 10.1038/ni.3457
34. Akula MK, Shi M, Jiang Z, Foster CE, Miao D, Li AS, et al. Control of the innate immune response by the mevalonate pathway. Nat Immunol. (2016) 17:922–9. doi: 10.1038/ni.3487
35. Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J Exp Med. (2011) 208:519–33. doi: 10.1084/jem.20102049
36. Bachetti T, Chiesa S, Castagnola P, Bani D, Di Zanni E, Omenetti A, et al. Autophagy contributes to inflammation in patients with TNFR-associated periodic syndrome (TRAPS). Ann Rheum Dis. (2013) 72:1044–52. doi: 10.1136/annrheumdis-2012-201952
37. Savic S, McDermot MF. Tumor Necrosis Factor (TNF) Receptor-Associated Periodic Syndrome (TRAPS). In: Hashkes PJ, Laxer RM, Simon A, editors. Textbook of Autoinflammation. Cham: Springer Nature Switzerland AG (2019). p. 329–46. doi: 10.1007/978-3-319-98605-0_18
38. Toplak N, Frenkel J, Ozen S, Lachmann HJ, Woo P, Kone-Paut I, et al. An international registry on autoinflammatory diseases: the Eurofever experience. Ann Rheum Dis. (2012) 71:1177–82. doi: 10.1136/annrheumdis-2011-200549
39. ter Haar NM, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA, et al. Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis. (2015) 74:1636–44. doi: 10.1136/annrheumdis-2015-207546
40. Ceccherini I, Rusmini M, Arostegui JI. Genetic aspects of investigating and understanding autoinflammation. In: Hashkes PJ, Laxer RM, Simon A, editors. Textbook of Autoinflammation. Cham: Springer Nature Switzerland AG (2019). p. 19–48. doi: 10.1007/978-3-319-98605-0_2
41. Soon GS, Laxer RM. Approach to recurrent fever in childhood. Can Fam Physician. (2017) 63:756–62.
42. Georgin-Lavialle S, Fayand A, Rodrigues F, Bachmeyer C, Savey L, Grateau G. Autoinflammatory diseases: state of the art. Presse Med. (2019) 48:e25–48. doi: 10.1016/j.lpm.2018.12.003
43. Piram M, Kone-Paut I, Lachmann HJ, Frenkel J, Ozen S, Kuemmerle-Deschner J, et al. Validation of the auto-inflammatory diseases activity index (AIDAI) for hereditary recurrent fever syndromes. Ann Rheum Dis. (2014) 73:2168–73. doi: 10.1136/annrheumdis-2013-203666
44. Aksentijevich I, Putnam CD, Remmers EF, Mueller JL, Le J, Kolodner RD, et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. (2007) 56:1273–85. doi: 10.1002/art.22491
45. Neven B, Callebaut I, Prieur AM, Feldmann J, Bodemer C, Lepore L, et al. Molecular basis of the spectral expression of CIAS1 mutations associated with phagocytic cell-mediated autoinflammatory disorders CINCA/NOMID, MWS, and FCU. Blood. (2004) 103:2809–15. doi: 10.1182/blood-2003-07-2531
46. Ozen S. Familial mediterranean fever: revisiting an ancient disease. Eur J Pediatr. (2003) 162:449–54. doi: 10.1007/s00431-003-1223-x
47. Ter Haar NM, Jeyaratnam J, Lachmann HJ, Simon A, Brogan PA, Doglio M, et al. The phenotype and genotype of mevalonate kinase deficiency: a series of 114 cases from the Eurofever registry. Arthritis Rheumatol. (2016) 68:2795–805. doi: 10.1002/art.39763
48. Gaggiano C, Vitale A, Obici L, Merlini G, Soriano A, Viapiana O, et al. Clinical features at onset and genetic characterization of pediatric and adult patients with TNF-alpha receptor-associated periodic syndrome (TRAPS): a series of 80 cases from the AIDA network. Mediators Inflamm. (2020) 2020:8562485. doi: 10.1155/2020/8562485
49. Ozen S, Demirkaya E, Erer B, Livneh A, Ben-Chetrit E, Giancane G, et al. EULAR recommendations for the management of familial Mediterranean fever. Ann Rheum Dis. (2016) 75:644–51. doi: 10.1136/annrheumdis-2015-208690
50. Rusmini M, Federici S, Caroli F, Grossi A, Baldi M, Obici L, et al. Next-generation sequencing and its initial applications for molecular diagnosis of systemic auto-inflammatory diseases. Ann Rheum Dis. (2016) 75:1550–7. doi: 10.1136/annrheumdis-2015-207701
51. Omoyinmi E, Standing A, Keylock A, Price-Kuehne F, Melo Gomes S, Rowczenio D, et al. Clinical impact of a targeted next-generation sequencing gene panel for autoinflammation and vasculitis. PLoS ONE. (2017) 12:e0181874. doi: 10.1371/journal.pone.0181874
52. Karacan I, Balamir A, Ugurlu S, Aydin AK, Everest E, Zor S, et al. Diagnostic utility of a targeted next-generation sequencing gene panel in the clinical suspicion of systemic autoinflammatory diseases: a multi-center study. Rheumatol Int. (2019) 39:911–9. doi: 10.1007/s00296-019-04252-5
53. Schnappauf O, Aksentijevich I. Current and future advances in genetic testing in systemic autoinflammatory diseases. Rheumatology. (2019) 58(Suppl.6):vi44–55. doi: 10.1093/rheumatology/kez294
54. Saito M, Fujisawa A, Nishikomori R, Kambe N, Nakata-Hizume M, Yoshimoto M, et al. Somatic mosaicism of CIAS1 in a patient with chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. (2005) 52:3579–85. doi: 10.1002/art.21404
55. Tanaka N, Izawa K, Saito MK, Sakuma M, Oshima K, Ohara O, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum. (2011) 63:3625–32. doi: 10.1002/art.30512
56. Giancane G, Ter Haar NM, Wulffraat N, Vastert SJ, Barron K, Hentgen V, et al. Evidence-based recommendations for genetic diagnosis of familial Mediterranean fever. Ann Rheum Dis. (2015) 74:635–41. doi: 10.1136/annrheumdis-2014-206844
57. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
58. Van Gijn ME, Ceccherini I, Shinar Y, Carbo EC, Slofstra M, Arostegui JI, et al. New workflow for classification of genetic variants' pathogenicity applied to hereditary recurrent fevers by the International Study Group for Systemic Autoinflammatory Diseases (INSAID). J Med Genet. (2018) 55:530–7. doi: 10.1136/jmedgenet-2017-105216
59. Milhavet F, Cuisset L, Hoffman HM, Slim R, El-Shanti H, Aksentijevich I, et al. The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat. (2008) 29:803–8. doi: 10.1002/humu.20720
60. Hashkes PJ, Barron KS, Laxer, RM. Clinical approach to the diagnosis of autoinflammatory diseases. In: Hashkes PJ, Laxer RM, Simon A, editors. Textbook of Autoinflammation. Cham: Springer Nature Switzerland AG (2019). p. 203–23. doi: 10.1007/978-3-319-98605-0_11
61. Kuemmerle-Deschner JB, Ozen S, Tyrrell PN, Kone-Paut I, Goldbach-Mansky R, Lachmann H, et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis. (2017) 76:942–7. doi: 10.1136/annrheumdis-2016-209686
62. Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med. (1967) 43:227–53. doi: 10.1016/0002-9343(67)90167-2
63. Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, Lidar T, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. (1997) 40:1879–85. doi: 10.1002/art.1780401023
64. Yalcinkaya F, Ozen S, Ozcakar ZB, Aktay N, Cakar N, Duzova A, et al. A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology. (2009) 48:395–8. doi: 10.1093/rheumatology/ken509
65. Demirkaya E, Saglam C, Turker T, Kone-Paut I, Woo P, Doglio M, et al. Performance of different diagnostic criteria for familial mediterranean fever in children with periodic fevers: results from a multicenter international registry. J Rheumatol. (2016) 43:154–60. doi: 10.3899/jrheum.141249
66. Gattorno M, Hofer M, Federici S, Vanoni F, Bovis F, Aksentijevich I, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. (2019) 78:1025–32. doi: 10.1136/annrheumdis-2019-215048
67. Boursier G, Rittore C, Georgin-Lavialle S, Belot A, Galeotti C, Hachulla E, et al. Positive impact of expert reference center validation on performance of next-generation sequencing for genetic diagnosis of autoinflammatory diseases. J Clin Med. (2019) 8:101729. doi: 10.3390/jcm8101729
68. Papa R, Rusmini M, Volpi S, Caorsi R, Picco P, Grossi A, et al. Next generation sequencing panel in undifferentiated autoinflammatory diseases identifies patients with colchicine-responder recurrent fevers. Rheumatology. (2019) 59:344–60. doi: 10.1136/annrheumdis-2019-eular.3562
69. Kuemmerle-Deschner JB, Verma D, Endres T, Broderick L, de Jesus AA, Hofer F, et al. Clinical and molecular phenotypes of low-penetrance variants of NLRP3: diagnostic and therapeutic challenges. Arthritis Rheumatol. (2017) 69:2233–40. doi: 10.1002/art.40208
70. Rieber N, Gavrilov A, Hofer L, Singh A, Oz H, Endres T, et al. A functional inflammasome activation assay differentiates patients with pathogenic NLRP3 mutations and symptomatic patients with low penetrance variants. Clin Immunol. (2015) 157:56–64. doi: 10.1016/j.clim.2015.01.003
71. D'Osualdo A, Ferlito F, Prigione I, Obici L, Meini A, Zulian F, et al. Neutrophils from patients with TNFRSF1A mutations display resistance to tumor necrosis factor-induced apoptosis: pathogenetic and clinical implications. Arthritis Rheum. (2006) 54:998–1008. doi: 10.1002/art.21657
72. Smolen JS. Treat-to-target: rationale and strategies. Clin Exp Rheumatol. (2012) 30(4 Suppl.73):S2–6.
73. Hansmann S, Lainka E, Horneff G, Holzinger D, Rieber N, Jansson AF, et al. Consensus protocols for the diagnosis and management of the hereditary autoinflammatory syndromes CAPS, TRAPS and MKD/HIDS: a German PRO-KIND initiative. Pediatr Rheumatol Online J. (2020) 18:17. doi: 10.1186/s12969-020-0409-3
74. Kuemmerle-Deschner JB, Hofer F, Endres T, Kortus-Goetze B, Blank N, Weissbarth-Riedel E, et al. Real-life effectiveness of canakinumab in cryopyrin-associated periodic syndrome. Rheumatology. (2016) 55:689–96. doi: 10.1093/rheumatology/kev416
75. De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med. (2018) 378:1908–19. doi: 10.1056/NEJMoa1706314
76. Kallinich T, Haffner D, Niehues T, Huss K, Lainka E, Neudorf U, et al. Colchicine use in children and adolescents with familial Mediterranean fever: literature review and consensus statement. Pediatrics. (2007) 119:e474–83. doi: 10.1542/peds.2006-1434
77. Slobodnick A, Shah B, Pillinger MH, Krasnokutsky S. Colchicine: old and new. Am J Med. (2015) 128:461–70. doi: 10.1016/j.amjmed.2014.12.010
78. Terkeltaub RA, Furst DE, Digiacinto JL, Kook KA, Davis MW. Novel evidence-based colchicine dose-reduction algorithm to predict and prevent colchicine toxicity in the presence of cytochrome P450 3A4/P-glycoprotein inhibitors. Arthritis Rheum. (2011) 63:2226–37. doi: 10.1002/art.30389
79. Kullenberg T, Lofqvist M, Leinonen M, Goldbach-Mansky R, Olivecrona H. Long-term safety profile of anakinra in patients with severe cryopyrin-associated periodic syndromes. Rheumatology. (2016) 55:1499–506. doi: 10.1093/rheumatology/kew208
80. Kone-Paut I, Galeotti C. Current treatment recommendations and considerations for cryopyrin-associated periodic syndrome. Expert Rev Clin Immunol. (2015) 11:1083–92. doi: 10.1586/1744666X.2015.1077702
81. Kostjukovits S, Kalliokoski L, Antila K, Korppi M. Treatment of hyperimmunoglobulinemia D syndrome with biologics in children: review of the literature and Finnish experience. Eur J Pediatr. (2015) 174:707–14. doi: 10.1007/s00431-015-2505-9
82. Ozen S, Kuemmerle-Deschner JB, Cimaz R, Livneh A, Quartier P, Kone-Paut I, et al. International retrospective chart review of treatment patterns in severe familial mediterranean fever, tumor necrosis factor receptor-associated periodic syndrome, and mevalonate kinase deficiency/hyperimmunoglobulinemia D syndrome. Arthritis Care Res. (2017) 69:578–86. doi: 10.1002/acr.23120
83. Gulez N, Makay B, Sozeri B. Long-term effectiveness and safety of canakinumab in pediatric familial mediterranean fever patients. Mod Rheumatol. (2018) 2018:1–13. doi: 10.1080/14397595.2018.1559488
84. Varan O, Kucuk H, Babaoglu H, Guven SC, Ozturk MA, Haznedaroglu S, et al. Efficacy and safety of interleukin-1 inhibitors in familial Mediterranean fever patients complicated with amyloidosis. Mod Rheumatol. (2019) 29:363–6. doi: 10.1080/14397595.2018.1457469
85. Laskari K, Boura P, Dalekos GN, Garyfallos A, Karokis D, Pikazis D, et al. Longterm beneficial effect of canakinumab in colchicine-resistant familial mediterranean fever. J Rheumatol. (2017) 44:102–9. doi: 10.3899/jrheum.160518
86. Ben-Zvi I, Kukuy O, Giat E, Pras E, Feld O, Kivity S, et al. Anakinra for colchicine-resistant familial mediterranean fever: a randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. (2017) 69:854–62. doi: 10.1002/art.39995
87. Lepore L, Paloni G, Caorsi R, Alessio M, Rigante D, Ruperto N, et al. Follow-up and quality of life of patients with cryopyrin-associated periodic syndromes treated with Anakinra. J Pediatr. (2010) 157:310–5 e1. doi: 10.1016/j.jpeds.2010.02.040
88. Neven B, Marvillet I, Terrada C, Ferster A, Boddaert N, Couloignier V, et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in 10 patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. (2010) 62:258–67. doi: 10.1002/art.25057
89. Sibley CH, Plass N, Snow J, Wiggs EA, Brewer CC, King KA, et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease treated with anakinra: a cohort study to determine 3- and 5-year outcomes. Arthritis Rheum. (2012) 64:2375–86. doi: 10.1002/art.34409
90. Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, Wittkowski H, Bialkowski A, Tzaribachev N, et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum. (2011) 63:840–9. doi: 10.1002/art.30149
91. Garg S, Wynne K, Omoyinmi E, Eleftheriou D, Brogan P. Efficacy and safety of anakinra for undifferentiated autoinflammatory diseases in children: a retrospective case review. Rheumatol Adv Pract. (2019) 3:rkz004. doi: 10.1093/rap/rkz004
92. Hoffman HM, Throne ML, Amar NJ, Sebai M, Kivitz AJ, Kavanaugh A, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. (2008) 58:2443–52. doi: 10.1002/art.23687
93. Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. (2009) 360:2416–25. doi: 10.1056/NEJMoa0810787
94. Kuemmerle-Deschner JB, Hachulla E, Cartwright R, Hawkins PN, Tran TA, Bader-Meunier B, et al. Two-year results from an open-label, multicentre, phase III study evaluating the safety and efficacy of canakinumab in patients with cryopyrin-associated periodic syndrome across different severity phenotypes. Ann Rheum Dis. (2011) 70:2095–102. doi: 10.1136/ard.2011.152728
95. Caorsi R, Lepore L, Zulian F, Alessio M, Stabile A, Insalaco A, et al. The schedule of administration of canakinumab in cryopyrin associated periodic syndrome is driven by the phenotype severity rather than the age. Arthritis Res Ther. (2013) 15:R33. doi: 10.1186/ar4184
96. Brogan PA, Hofer M, Kuemmerle-Deschner JB, Kone-Paut I, Roesler J, Kallinich T, et al. Rapid and sustained long-term efficacy and safety of canakinumab in patients with cryopyrin-associated periodic syndrome ages 5 years and younger. Arthritis Rheumatol. (2019) 71:1955–63. doi: 10.1002/art.41004
97. Russo RA, Melo-Gomes S, Lachmann HJ, Wynne K, Rajput K, Eleftheriou D, et al. Efficacy and safety of canakinumab therapy in paediatric patients with cryopyrin-associated periodic syndrome: a single-centre, real-world experience. Rheumatology. (2014) 53:665–70. doi: 10.1093/rheumatology/ket415
98. Kuemmerle-Deschner JB, Ramos E, Blank N, Roesler J, Felix SD, Jung T, et al. Canakinumab (ACZ885, a fully human IgG1 anti-IL-1beta mAb) induces sustained remission in pediatric patients with cryopyrin-associated periodic syndrome (CAPS). Arthritis Res Ther. (2011) 13:R34. doi: 10.1186/ar3266
99. Arostegui JI, Anton J, Calvo I, Robles A, Iglesias E, Lopez-Montesinos B, et al. Open-label, phase II study to assess the efficacy and safety of canakinumab treatment in active hyperimmunoglobulinemia D with periodic fever syndrome. Arthritis Rheumatol. (2017) 69:1679–88. doi: 10.1002/art.40146
100. Bulua AC, Mogul DB, Aksentijevich I, Singh H, He DY, Muenz LR, et al. Efficacy of etanercept in the tumor necrosis factor receptor-associated periodic syndrome: a prospective, open-label, dose-escalation study. Arthritis Rheum. (2012) 64:908–13. doi: 10.1002/art.33416
101. Bilgen SA, Kilic L, Akdogan A, Kiraz S, Kalyoncu U, Karadag O, et al. Effects of anti-tumor necrosis factor agents for familial mediterranean fever patients with chronic arthritis and/or sacroiliitis who were resistant to colchicine treatment. J Clin Rheumatol. (2011) 17:358–62. doi: 10.1097/RHU.0b013e31823682f5
102. Vaitla PM, Radford PM, Tighe PJ, Powell RJ, McDermott EM, Todd I, et al. Role of interleukin-6 in a patient with tumor necrosis factor receptor-associated periodic syndrome: assessment of outcomes following treatment with the anti-interleukin-6 receptor monoclonal antibody tocilizumab. Arthritis Rheumatism. (2011) 63:1151–5. doi: 10.1002/art.30215
103. Shendi HM, Devlin LA, Edgar JD. Interleukin 6 blockade for hyperimmunoglobulin D and periodic fever syndrome. J Clin Rheumatol. (2014) 20:103–5. doi: 10.1097/01.RHU.0000442576.41537.de
104. Stoffels M, Jongekrijg J, Remijn T, Kok N, van der Meer JW, Simon A. TLR2/TLR4-dependent exaggerated cytokine production in hyperimmunoglobulinaemia D and periodic fever syndrome. Rheumatology. (2015) 54:363–8. doi: 10.1093/rheumatology/keu341
105. Musters A, Tak PP, Baeten DL, Tas SW. Anti-interleukin 6 receptor therapy for hyper-IgD syndrome. BMJ Case Rep. (2015) 2015:210513. doi: 10.1136/bcr-2015-210513
106. Erbis G, Schmidt K, Hansmann S, Sergiichuk T, Michler C, Kuemmerle-Deschner JB, et al. Living with autoinflammatory diseases: identifying unmet needs of children, adolescents and adults. Pediatr Rheumatol Online J. (2018) 16:81. doi: 10.1186/s12969-018-0300-7
107. Sparud-Lundin C, Berg S, Fasth A, Karlsson A, Wekell P. From uncertainty to gradually managing and awaiting recovery of a periodic condition- a qualitative study of parents experiences of PFAPA syndrome. BMC Pediatr. (2019) 19:99. doi: 10.1186/s12887-019-1458-y
108. Cipolletta S, Giudici L, Punzi L, Galozzi P, Sfriso P. Health-related quality of life, illness perception, coping strategies and the distribution of dependency in autoinflammatory diseases. Clin Exp Rheumatol. (2019) 37(Suppl.121):156–7.
109. Makay B, Emiroglu N, Unsal E. Depression and anxiety in children and adolescents with familial Mediterranean fever. Clin Rheumatol. (2010) 29:375–9. doi: 10.1007/s10067-009-1330-9
110. Giese A, Ornek A, Kilic L, Kurucay M, Sendur SN, Lainka E, et al. Anxiety and depression in adult patients with familial Mediterranean fever: a study comparing patients living in Germany and Turkey. Int J Rheum Dis. (2017) 20:2093–100. doi: 10.1111/1756-185X.12297
111. Hausmann JS, Lomax KG, Shapiro A, Durrant K. The patient journey to diagnosis and treatment of autoinflammatory diseases. Orphanet J Rare Dis. (2018) 13:156. doi: 10.1186/s13023-018-0902-7
112. Duzova A, Bakkaloglu A, Besbas N, Topaloglu R, Ozen S, Ozaltin F, et al. Role of A-SAA in monitoring subclinical inflammation and in colchicine dosage in familial Mediterranean fever. Clin Exp Rheumatol. (2003) 21:509–14.
113. Kessel C, Holzinger D, Foell D. Phagocyte-derived S100 proteins in autoinflammation: putative role in pathogenesis and usefulness as biomarkers. Clin Immunol. (2013) 147:229–41. doi: 10.1016/j.clim.2012.11.008
114. Ter Haar NM, Annink KV, Al-Mayouf SM, Amaryan G, Anton J, Barron KS, et al. Development of the autoinflammatory disease damage index (ADDI). Ann Rheum Dis. (2017) 76:821–30.
115. Ter Haar NM, van Delft ALJ, Annink KV, van Stel H, Al-Mayouf SM, Amaryan G, et al. In silico validation of the autoinflammatory disease damage index. Ann Rheum Dis. (2018) 77:1599–605. doi: 10.1136/annrheumdis-2018-213725
116. Limper M, de Kruif MD, Duits AJ, Brandjes DP, van Gorp EC. The diagnostic role of procalcitonin and other biomarkers in discriminating infectious from non-infectious fever. J Infect. (2010) 60:409–16. doi: 10.1016/j.jinf.2010.03.016
117. Jeyaratnam J tHN, Lachmann H, Simon A, Brogan P, Doglio M, et al. Genetic and phenotypic characteristics of 114 patients with mevalonate kinase deficiency. Pediatr Rheumatol. (2015) 13(suppl.1):25. doi: 10.1186/1546-0096-13-S1-P25
118. Walker UA, Hoffman HM, Williams R, Kuemmerle-Deschner J, Hawkins PN. Brief report: severe inflammation following vaccination against Streptococcus pneumoniae in patients with cryopyrin-associated periodic syndromes. Arthritis Rheumatol. (2016) 68:516–20. doi: 10.1002/art.39482
119. Jaeger VK, Hoffman HM, van der Poll T, Tilson H, Seibert J, Speziale A, et al. Safety of vaccinations in patients with cryopyrin-associated periodic syndromes: a prospective registry based study. Rheumatology. (2017) 56:1484–91. doi: 10.1093/rheumatology/kex185
120. Heijstek MW, Ott de Bruin LM, Bijl M, Borrow R, van der Klis F, Kone-Paut I, et al. EULAR recommendations for vaccination in paediatric patients with rheumatic diseases. Ann Rheum Dis. (2011) 70:1704–12. doi: 10.1136/ard.2011.150193
121. Minden K, Speth F, Huppertz HI, Borte M. Immunization in children and adolescents with rheumatic and musculoskeletal diseases. Z Rheumatol. (2014) 73:878–89. doi: 10.1007/s00393-014-1396-x
122. Wagner N, Assmus F, Arendt G, Baum E, Baumann U, Bogdan C, et al. Impfen bei Immundefizienz : Anwendungshinweise zu den von der Ständigen Impfkommission empfohlenen Impfungen. (IV) Impfen bei Autoimmunkrankheiten, bei anderen chronisch-entzündlichen Erkrankungen und unter immunmodulatorischer Therapie. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. (2019) 62:494–515. doi: 10.1007/s00103-019-02905-1
123. Ehl S, Bogdan C, Burchard G. Impfen bei Immundefizienz Anwendungshinweise zu den von der Ständigen Impfkommission empfohlenen Impfungen. (II) Impfen bei 1. Primären Immundefekterkrankungen und 2. HIV-Infektion. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. (2018) 61:1034–51. doi: 10.1007/s00103-018-2761-8
Keywords: treat-to-target, AID management, multidisciplinary team, disease activity, monitoring, autoinflammation
Citation: Welzel T, Benseler SM and Kuemmerle-Deschner JB (2021) Management of Monogenic IL-1 Mediated Autoinflammatory Diseases in Childhood. Front. Immunol. 12:516427. doi: 10.3389/fimmu.2021.516427
Received: 30 November 2019; Accepted: 12 February 2021;
Published: 18 March 2021.
Edited by:
Marco Gattorno, Giannina Gaslini Institute (IRCCS), ItalyReviewed by:
Tadej Avcin, University Medical Centre Ljubljana, SloveniaAntonio Vitale, University of Siena, Italy
Copyright © 2021 Welzel, Benseler and Kuemmerle-Deschner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jasmin B. Kuemmerle-Deschner, SmFzbWluLmt1ZW1tZXJsZS1kZXNjaG5lckBtZWQudW5pLXR1ZWJpbmdlbi5kZQ==