
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 February 2021
Sec. Microbial Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.628432
 Rebecca R. Crowther1,2,3,4
Rebecca R. Crowther1,2,3,4 Joseph E. Qualls1,2*
Joseph E. Qualls1,2*Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), is a leading cause of death worldwide. Despite decades of research, there is still much to be uncovered regarding the immune response to Mtb infection. Here, we summarize the current knowledge on anti-Mtb immunity, with a spotlight on immune cell amino acid metabolism. Specifically, we discuss L-arginine and L-tryptophan, focusing on their requirements, regulatory roles, and potential use as adjunctive therapy in TB patients. By continuing to uncover the immune cell contribution during Mtb infection and how amino acid utilization regulates their functions, it is anticipated that novel host-directed therapies may be developed and/or refined, helping to eradicate TB.
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is the leading worldwide cause of death due to a single infectious agent (1). Approximately one quarter of the world’s population is infected with Mtb, with 10 million new infections each year (1). In 2019, Mtb claimed nearly 1.3 million deaths, with over 250,000 deaths in HIV-positive patients – representing a greater than 20% reduction in TB-related deaths since 2000 (1). Progress in ending the TB epidemic is slow, however, and at this rate TB is unlikely to be eradicated this century (2). The COVID-19 pandemic has significantly curtailed this progress and is estimated to result in an additional 400,000 TB-related deaths in 2020 (1). Additionally, drug-resistant Mtb strains continue to pose a public health problem, especially in developing countries with the highest rates of infection (1, 2). While the live attenuated M. bovis Bacille Calmette-Guérin (Mb BCG) vaccine is administered to newborns in TB endemic countries, little protection is formed against pulmonary TB (3). Additionally, Mb BCG cannot be administered to immunocompromised patients due to the high risk of disseminated infection (1–3). As such, new strategies are urgently needed to end the fight against TB.
In response to infection, immune cells undergo metabolic changes. Following LPS stimulation, macrophages utilize aerobic glycolysis to generate the energy needed to fuel their effector functions (4). By contrast, while Mtb-infected inflammatory macrophages seeming rely on glycolysis, Mtb-infected alveolar macrophages utilize fatty acid β-oxidation for energy (5). Additionally, following activation T cells undergo metabolic reprogramming, switching from a naïve state where energy is derived mainly from the TCA cycle and oxidative phosphorylation to an activated state dominated by aerobic glycolysis (6). Thus, precise alterations in metabolic function depend on the pathogen and cell type involved. Amino acid metabolism is often affected following infection. Amino acids are the building blocks of proteins, but also regulate cellular responses via nutrient sensor signaling or serve as anapleurotic precursors for energy-producing pathways, such as the tricarboxylic acid (TCA) cycle. Therefore, adjustments in amino acid concentrations regulate how immune cells respond to infection.
Targeted metabolomics studies have proposed metabolite changes, particularly in amino acid abundance, may serve as biomarkers following Mtb infection. Multiple studies have observed decreased L-citrulline (L-CIT) and L-ornithine (L-ORN) – metabolites of L-arginine (L-ARG) – in the sera of active TB patients compared to healthy controls (7–9). Interestingly, following antibiotic treatment, L-CIT and L-ORN levels in TB patients increase to those of healthy controls (8). Additionally, active TB patients display decreased L-tryptophan (L-TRP) and increased levels of its metabolites, including L-kynurenine (KYN), in their sera and urine as compared to healthy controls (7–11). In fact, metabolite tracking may predict a patient’s TB status. One study found just 20 serum metabolites, 11 of which were amino acids or derivatives, were required to discriminate active TB patients from healthy controls (7). When tracking household contacts of TB patients, amino acid alterations in the serum could discriminate between patients who later developed TB and those who remained healthy (12).
Given these data, it is important to understand how amino acids contribute to immune cell function following Mtb infection. Here, we review immune responses during Mtb infection and immune cell metabolism of two key amino acids: L-ARG and L-TRP.
Upon inhalation, Mtb bacilli are phagocytosed by alveolar macrophages (Figure 1A), resident phagocytes within alveoli (13, 14). Alveolar macrophages make up the majority of mycobacteria-laden cells in the lung during early Mtb infection, with neutrophils not appearing until 10-14 days post-infection (14–16). Additionally, accumulation of group 3 innate lymphoid cells (ILC3s) parallels that of alveolar macrophages and precedes infiltration of inflammatory macrophages (17).
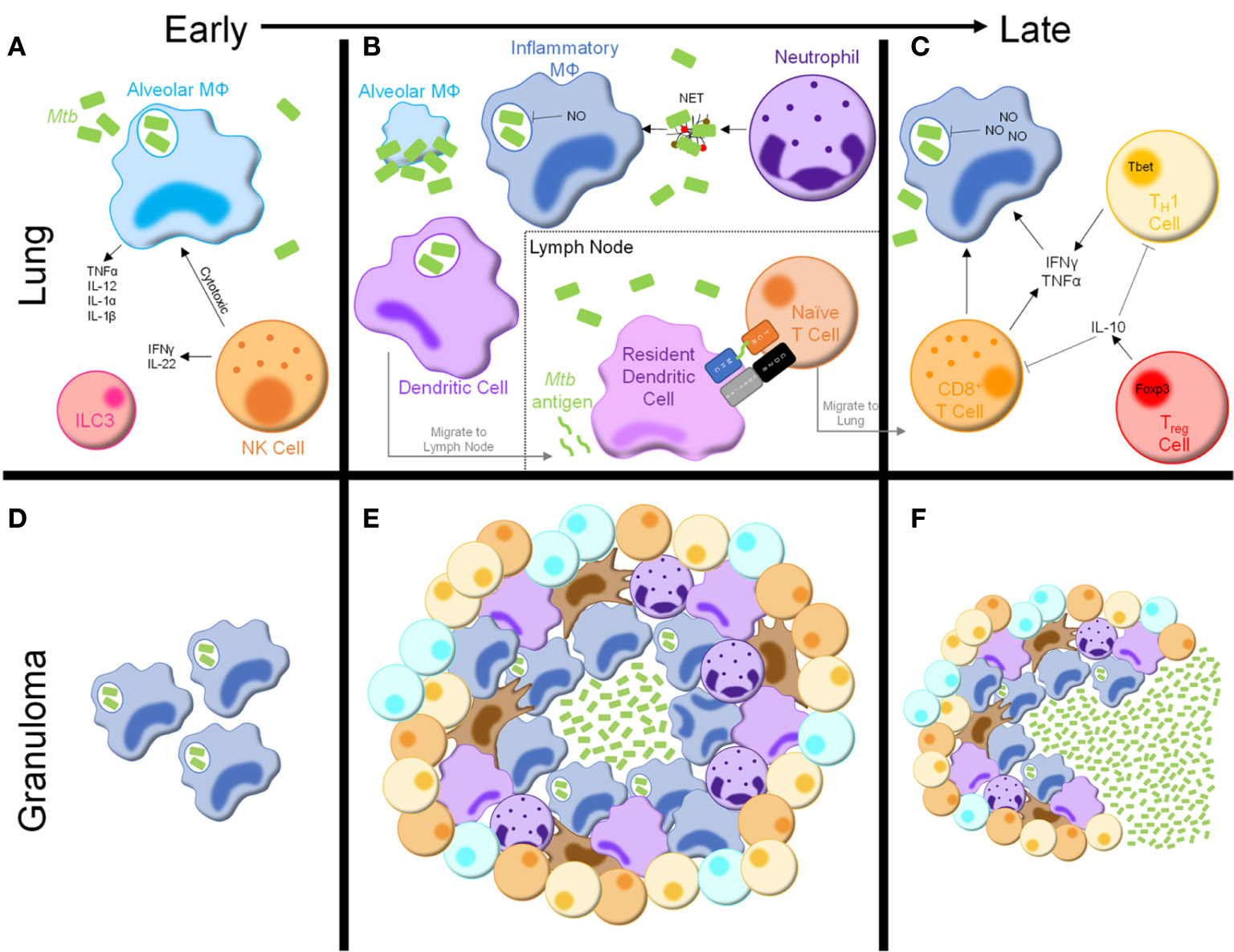
Figure 1 Immune response to Mycobacterium tuberculosis. (A) Following inhalation, Mtb bacilli are phagocytosed by alveolar macrophages in the lung, resulting in cytokine and chemokine production. NK cells are innate lymphocytes that are cytotoxic to infected alveolar macrophages and produce inflammatory cytokines. Group 3 innate lymphoid cells (ILC3s) are also present in the lung early following Mtb infection. (B) As infection progresses, alveolar macrophages are overrun with Mtb bacilli and undergo cell death. Inflammatory macrophages, neutrophils, and dendritic cells are recruited from the bloodstream. Inflammatory macrophages produce mycobactericidal nitric oxide (NO) and phagocytose Mtb-containing neutrophil extracellular traps (NETs), secreted by neutrophils. Dendritic cells phagocytose Mtb bacilli and migrate to the draining lymph node, where they secrete Mtb antigens to be presented by resident dendritic cells, initiating naïve T cell activation. (C) After migrating to the lymph node, activated T cells secrete inflammatory cytokines, including interferon (IFN)γ and tumor necrosis factor (TNF)α, which further stimulate macrophage anti-mycobacterial activity. Activated CD8+ T cells are also cytotoxic to infected macrophages. Treg cells produce cytokines, such as IL-10, to inhibit the activity of other lymphocytes in the lung. (D–F) Concurrently, granulomas begin to develop in the lung. (D) Early granulomas consist of aggregations of infected phagocytes. (E) Mature granulomas consist of a core of Mtb bacilli, infected macrophages, and multinucleated giant cells surrounded by more macrophages, neutrophils, dendritic cells, and fibroblasts. The outside of the granuloma consists of T and B cells, forming a lymphocytic cuff. (F) As infection progresses and potentially following some degree of immunosuppression the granulomas begin to break down, leading to dissemination of Mtb bacilli and reactivation of TB disease.
Alveolar macrophages express numerous pattern recognition receptors involved in Mtb recognition, stimulation of which induces an inflammatory response and recruitment of additional innate immune cells, including neutrophils, inflammatory macrophages, and dendritic cells (DCs) (16, 18, 19). Although alveolar macrophages rapidly phagocytose Mtb bacilli, the immune response is slow, contributing to impaired mycobacterial clearance. Mtb bacilli inhibit a variety of effector responses, including phagolysosome fusion and cytokine production, resulting in mycobacterial overload, cell death, and widespread distribution of infection throughout the lung (Figure 1B) (20–23).
The myeloid population responding to Mtb infection is diverse, marked by production of various anti-mycobacterial effectors including nitric oxide (NO), tumor necrosis factor (TNF) α, interleukin (IL)-12, IL-1α, and IL-1β, all of which are critical for infection control (24–26). An abundance of literature supports a dual role for macrophages as the main effector killing cells and a growth niche for mycobacteria. Inflammatory macrophages, as the main reservoir for Mtb growth, are the most well studied. Like their alveolar counterparts, inflammatory macrophages are impaired in combating Mtb infection due to inhibition of effector functions, as well as Mtb adaptations to a harsh intracellular environment (20–22).
Despite this inhibition, inflammatory macrophages harness multiple pathways to restrict Mtb growth. Combined TLR2 and IFNγR stimulation leads to transcription of Nos2, which encodes inducible NO synthase (iNOS), resulting in NO production (Figure 1B) (19). In mice, NO positively correlates with Mtb killing and mice lacking NO synthesis fail to control mycobacterial infections, succumbing even to Mb BCG (27–30). iNOS and NO production have been detected in human macrophages in vitro and the lungs of Mtb-infected patients (31–34). The contribution of NO in the pathogenesis of human Mtb infection, however, is highly debated. Some studies have found NO production does not play a central role in control of human Mtb infection, while others suggest clearance of Mtb is NO-dependent (32, 35–39). Nevertheless, the presence of iNOS and NO in the Mtb-infected lung suggests NO is involved in human immune responses to Mtb infection, albeit not entirely similar to those in mice.
While NO is a predominant mycobactericidal mechanism, macrophages also utilize nutrient limitation, reactive oxygen species, and apoptotic and autophagic pathways for Mtb elimination (19, 40). However, Mtb harnesses various inhibitory mechanisms to prevent its clearance via these mechanisms (23, 41). Therefore, it is evident macrophages play an important role in combating Mtb infection, but their efforts are thwarted by microbial adaptations.
Neutrophils are short-lived innate immune cells that rapidly migrate to the lungs upon Mtb infection (Figure 1B) (42). Neutrophils phagocytose Mtb bacilli, yet evidence of their ability to kill mycobacteria is conflicting (43–45). While studies have shown neutrophil-derived molecules are mycobactericidal, neutrophils display poor antimycobacterial activity and may contribute to destruction of the lung parenchyma (43, 44, 46–49). Upon activation, neutrophils release neutrophil extracellular traps (NETs) composed of nuclear proteins and proteases. Nucleosomes and elastase – both found within NETs – have been detected in plasma of active TB patients, and Mtb can induce NET formation in vitro (50–53). Despite evidence that Mtb-induced NETs are phagocytosed and stimulate macrophage cytokine production, NETs also provide a niche for Mtb growth (52, 53). Therefore, a clear role for neutrophil mediated Mtb defense has not yet been elucidated.
Neutrophil activity enhances anti-mycobacterial responses in peripheral immune cells. Infected neutrophils interact with and increase DC antigen presentation and T cell stimulatory capacity (54). Following administration of a M. smegmatis-derived vaccine in mice, neutrophil depletion decreased T cell responses and increased mycobacterial burden upon Mtb challenge (55). As such, neutrophils may play an important role in coordinating innate and adaptive immune responses following Mtb infection.
Natural killer (NK) cells – cytolytic innate lymphocytes recognize ligands present in the Mtb cell wall and molecules expressed by Mtb-infected phagocytes, leading to cytolytic killing of infected cells (Figure 1A) (56–58). Additionally, NK cells secrete IFNγ and IL-22 following Mtb infection, stimulating phagosome maturation in Mtb-infected macrophages (59). Recent studies have identified changes in both human and mouse NK cells following Mtb infection, resulting in decreased NK cell number and functionality (60–62). Taken together, NK cells are likely an important immune contributor during Mtb infection, but their definitive role remains to be elucidated.
DCs are essential in linking innate and adaptive immunity. In mice, DCs make up one of the largest proportions of Mtb-infected phagocytes by 4 weeks post-infection (63). Patients with DC defects are highly susceptible to disseminated mycobacterial infections, likely due to impaired adaptive immune activation (64). To initiate adaptive immunity, DCs phagocytose Mtb bacilli and migrate to draining lymph nodes to present antigenic peptides (Figure 1B) (13, 63, 65). Interestingly, this priming requires live Mtb bacilli within the lymph node (63, 65). However, Mtb-laden DCs are poor antigen presenters and instead secrete soluble, unprocessed Mtb antigens, which are presented by uninfected lymph node resident DCs (63, 66, 67). While this process circumvents inefficient antigen presentation by Mtb-infected DCs, it may also lead to decreased T cell priming (67, 68). Regardless, DCs serve a central role in priming the adaptive immune response following Mtb infection.
CD4+ T cells are essential for anti-mycobacterial immunity, as HIV/AIDS patients with low CD4+ T cell counts and animal models lacking CD4+ T cells quickly succumb to infection, even with attenuated mycobacterial strains (69–71). Naïve T cell activation is delayed following infection due to a variety of evasive mechanisms harnessed by Mtb (67, 72). In mouse models, antigen-specific CD4+ T cells are detected in the draining mediastinal lymph nodes and lungs 2 and 3 weeks post-Mtb infection, respectively (19). In non-human primates, antigen-specific CD4+ T cells are not detected in the lungs until 4 weeks post-infection (73). Adoptive transfer studies indicated CD4+ T cell activation in the lymph node does not begin until 7 days post-infection, despite an abundance of antigen-specific cells (74). These findings indicate a combination of ineffective antigen presentation and immune evasion significantly delay priming of anti-Mtb adaptive immunity.
Activated CD4+ T cells migrate to the infected lung and interact with innate cells to reinforce anti-Mtb immune responses (Figure 1C). IFNγ- and TNFα-producing CD4+ TH1 cells are required for optimal anti-mycobacterial responses, as humans and mice with defects in the IFNγ/IL-12/TH1 axis are highly susceptible to mycobacterial infection (75–82). Mice lacking TH17 immunity display increased Mtb burden following infection (83, 84). Adoptive transfer of antigen-specific TH17 cells into T cell-deficient mice prolongs survival, even when IFNγ-deficient, suggesting TH17 cells act independently of TH1 cells to control Mtb infection (85). Despite this, TH17 responses are negligible in active TB patients, indicating work is still needed to determine the impact of TH17 immunity in human Mtb infection (86). Thus, while the TH1 cells are essential, further investigation into other CD4+ T cell subtypes is required to better understand the anti-mycobacterial immune response.
IFNγ is a main driver of Mtb growth restriction and prevention of extrapulmonary dissemination through induction of iNOS, inflammatory cytokines, and antimicrobial peptides in infected macrophages (87). Despite its protective role, IFNγ has been implicated in the pathogenesis of prolonged Mtb infection as it promotes cavitation and destruction of lung tissue (88, 89). As such, TH1 activity is restricted by IL-10, an inhibitory cytokine produced chiefly by macrophages and Foxp3+ T regulatory (Treg) cells (Figure 1C) (90–92). Treg expansion has been detected in Mtb-infected macaques and humans (93–96). TH1 cells co-secreting IFNγ and IL-10 have also been detected in bronchoalveolar lavage fluid from active TB patients and mice with high infectious doses, though the contribution of these cells remains unclear (97, 98). In mice, Treg depletion enhances mycobacterial clearance at the expense of increased pulmonary immunopathology, while Treg expansion dampens TH1 responses (99–101). Taken together, TH1 immunity, while critical for Mtb clearance, is dampened by Treg cells in order to preserve lung architecture and function following Mtb infection.
Loss of CD8+ T cell responses impairs anti-mycobacterial immunity in animal models (102–104). Though an exact role remains unclear, CD8+ T cells prevent TB reactivation via cytokine production, perforin-mediated lysis of Mtb-infected phagocytes, and secretion of cytotoxic granules for direct killing of intracellular Mtb (Figure 1C) (105–107). TNFα inhibitors selectively deplete a subset of granulysin-secreting memory CD8+ T cells, which may explain why patients with autoimmune diseases prescribed anti-TNFα drugs are more susceptible to mycobacterial infections (108). Studying the role of CD8+ T cells in Mtb infection has been difficult, however, as CD8+ T cells cannot recognize Mtb-infected macrophages to the same extent as their CD4+ counterparts (109). While more work is needed, these studies show CD8+ T cells help control mycobacterial infection.
Despite being an intracellular pathogen, growing evidence indicates humoral immunity is essential for anti-Mtb immune responses. Anti-Mtb antibodies have been detected in humans exposed to Mtb and provide protection in mouse models (110). However, the antibody pool in sera of active TB patients is small and limited to membrane-associated and extracellular antigens (111). Interestingly, Fc glycosylation of anti-Mtb antibodies differs depending on the stage of infection and can discriminate between latent and active TB patients (112, 113). Antibodies secreted in latent TB promote increased NK cell responses and inflammasome activation of human monocyte derived macrophages in vitro (112). Additionally, human monocyte derived macrophages cultured with antibodies from latent TB patients displayed decreased mycobacterial burden as compared to those cultured with antibodies from active TB patients (112). These findings are complemented by reports that IgG glycosylation of anti-Mtb antibodies is required to observe protection upon transfer into mouse models (114, 115). Still, the role of humoral immunity against Mtb requires additional investigation.
Despite a coordinated immune response, patients are generally unable to clear Mtb infection. As such, the immune system walls off Mtb bacilli in granulomas, a hallmark of human TB disease (Figures 1D–F). Granulomas are characterized by a central core of Mtb-infected multinucleated giant cells and uninfected macrophages at varying stages of differentiation, surrounded by macrophages, neutrophils, DCs, and fibroblasts, along with a lymphocytic cuff composed of T and B cells (116–119). Granulomas can contain Mtb infection for decades. However, hypoxia causes the core necrosis, allowing for dissemination and reactivation of pulmonary TB infection and/or seeding in extrapulmonary sites (120, 121). Additionally, the granuloma structure precludes drug penetration and elimination of Mtb bacilli (122). Thus, while protective against active disease, granulomas present a therapeutic issue contributing to the difficulty of eradicating TB.
The immune response against Mtb is multifaceted and highly dependent on the cell’s metabolic state and ability to acquire nutrients. Minor alterations in immune cell metabolism have the potential to fundamentally change outcomes throughout Mtb infection. Changes in carbohydrate and fatty acid metabolism following Mtb infection, particularly in macrophages, have been well described and reviewed (5, 118, 123, 124). Amino acids can impact cellular nutrient signaling and be utilized by immune cells in a variety of metabolic pathways. For example, L-glutamine can signal nutrient sufficiency through mTOR or be oxidized and converted to other energy sources like citrate, which can enter the TCA cycle or be utilized for fatty acid synthesis (123). Meanwhile, L-serine, which plays a main role in 1 carbon metabolism via the folate cycle, has been shown to support macrophage functions through mTOR activation (125). Here we will focus on L-ARG and L-TRP metabolism in immune cells following Mtb infection, and how their acquisition and utilization change over the disease course.
L-ARG is an amino acid obtained from the diet, generated via de novo synthesis, or scavenged following protein turnover (126). At homeostasis, the intestinal-renal axis of L-ARG synthesis maintains the body’s L-ARG supply (127, 128). However, in early life, during chronic infection or inflammation, or following renal or intestinal injury, synthesis is not sufficient to meet the body’s needs, classifying L-ARG as a semi-essential amino acid (126). L-ARG is also part of the urea cycle, a pathway utilized to eliminate nitrogenous waste products. Patients with most urea cycle disorders exhibit impaired de novo synthesis, so L-ARG is considered essential (126).
L-ARG is the sole substrate for NO synthase enzymes (126). In myeloid cells, L-ARG is converted to NO via iNOS, forming L-citrulline (L-CIT) as a by-product (Figure 2A) (129, 130). L-CIT is a noncanonical amino acid utilized in the urea cycle and found in peptides/proteins following posttranslational modification of peptidyl-L-ARG (127, 130). In some cells, including immune cells, L-CIT serves to generate L-ARG. Through the sequential activity of argininosuccinate synthase 1 (Ass1) and argininosuccinate lyase (Asl), L-CIT is metabolized to form L-ARG (130). These reactions require L-aspartate and generate L-fumarate, which are intermediates in the TCA cycle. L-ARG can also be metabolized by arginases to form L-ornithine (L-ORN), a noncanonical amino acid formed in the urea cycle and a polyamine precursor (126, 131). There are two known arginase isoforms – cytosolic arginase 1 (Arg1) and mitochondrial arginase 2 (126).
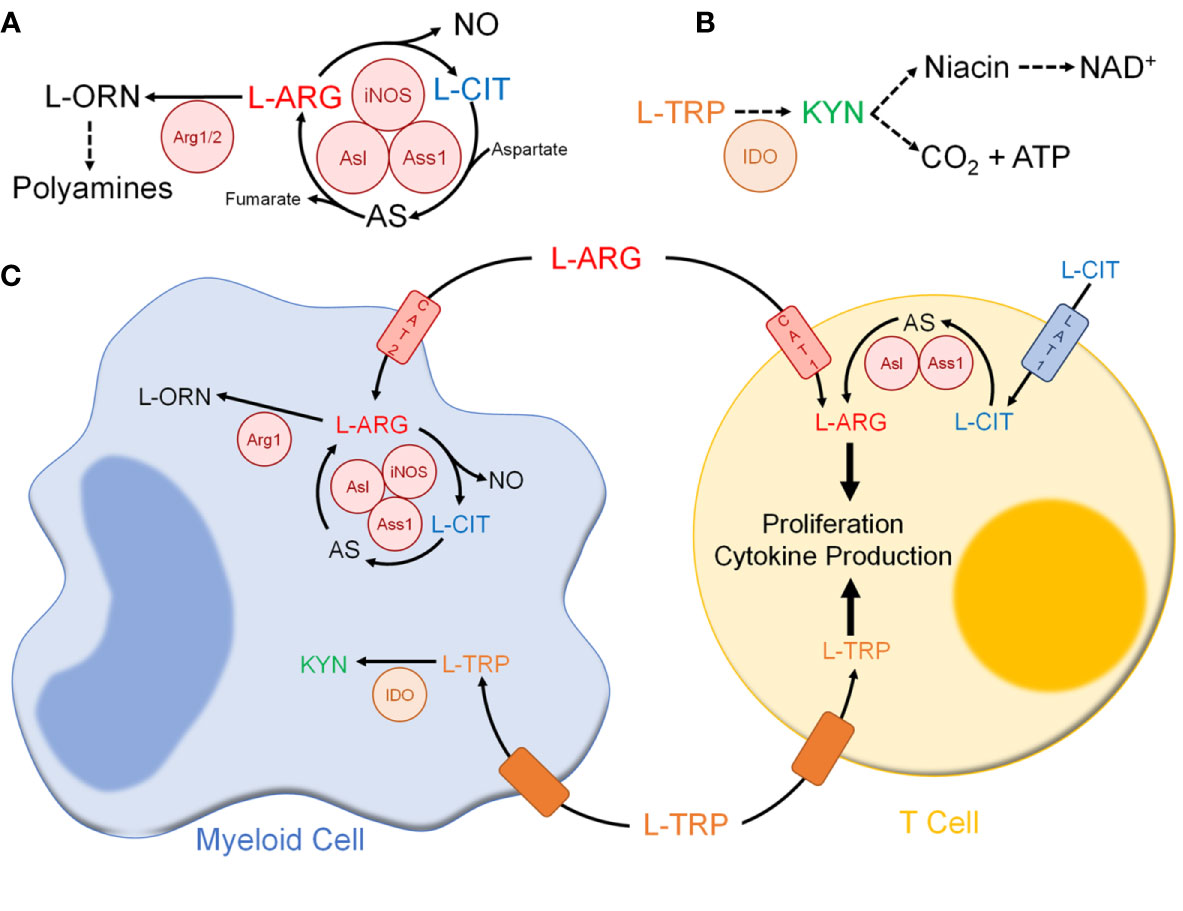
Figure 2 Immune cell amino acid metabolism following mycobacterial infection. (A) L-ARG metabolism pathway in immune cells. (B) L-TRP metabolism pathway in immune cells. (C) Amino acid metabolism between myeloid and T cells. Abbreviations: L-ARG, L-arginine; L-CIT, L-citrulline; L-ORN, L-ornithine; iNOS, inducible nitric oxide synthase; Ass1, argininosuccinate synthase 1; Asl, argininosuccinate lyase; Arg1/2, arginase 1/2; L-TRP, L-tryptophan; KYN, L-kynurenine; IDO, indolamine 2,3-dioxygenase; CAT1, cationic amino acid transport 1; CAT 2, cationic amino acid transporter 2; LAT1. L-type amino acid transporter 1.
L-ARG is also a precursor of creatine, L-proline, L-glutamate, and agmatine (126). Additionally, L-ARG regulates the protein kinases and cellular nutrient sensors general control nonderepressible 2 (GCN2) and mammalian target of rapamycin complex 1 (mTORC1) (132). The roles of GCN2 and mTORC1 in immune cells, and their regulation by L-ARG, have been reviewed elsewhere (133–136) and will not be discussed here.
Macrophages are often characterized as M1 (“classically activated”) or M2 (“alternatively activated”) based on their metabolic state and effector molecules. In fact, L-ARG utilization has often been used to classify macrophages, where iNOS or Arg1-expressing macrophages are considered “M1” or “M2”, respectively (118, 124). Following mycobacterial infection, however, macrophages co-express iNOS and Arg1 (137–139). Thus, macrophages that respond to Mtb infection do not concisely fit within the “M1” versus “M2” binary with respect to L-ARG utilization.
In mice, macrophages have been shown to utilize L-ARG to fuel NO production via iNOS (Figure 2) (124, 129). However, following Mtb infection extracellular L-ARG is not sufficient to sustain NO synthesis. As such, macrophages convert L-CIT into L-ARG, which is instrumental for continued NO production (139, 140). Amino acid tracing showed regeneration of L-ARG from L-CIT preferentially feeds into NO synthesis via iNOS, as opposed to L-ORN synthesis via Arg1 (140). This corresponds with previous studies indicating structural colocalization of NOS enzymes with Asl, one of the L-ARG synthesis enzymes, during systemic NO production (141). Evidence of colocalization of Ass1, Asl, and iNOS has not been definitively shown, though a combination of immunoprecipitation and tandem mass spectrometry suggests such a complex is formed within RAW 264.7 murine macrophages (141). Thus, macrophage L-ARG synthesis from L-CIT likely serves to fuel NO synthesis. Still, there are likely additional macrophage anti-mycobacterial mechanisms that require a sufficient L-ARG supply, and investigation outside of NO production will be beneficial since NO does not play a dominant role in human macrophages.
Immune cell L-ARG synthesis is vital for controlling mycobacterial burden. Bone marrow chimeric mice with hematopoietic cells from Ass1 hypomorphs were unable to control Mtb infection as compared to controls (139). Additionally, mice with a conditional hematopoietic or myeloid deletion of Asl or Ass1 displayed increased burden following Mb BCG or Mtb infection (142). Therefore, L-ARG and its synthesis contribute to macrophage defense following mycobacterial infection.
Arg1-expressing macrophages are present following Mtb infection in humans, non-human primates, and mice (138, 143–145). Though L-ORN is a polyamine precursor, these metabolites have little effect on Mtb growth in vitro (145). Instead, by synthesizing L-ORN, Arg1 depletes the microenvironment of L-ARG (Figure 2) (129). Arg1 expression occurs alongside upregulation of CAT-2B, the high-affinity isoform of L-ARG transporter CAT-2 (146, 147). Though iNOS has been shown to have a higher affinity for L-ARG, Arg1 has greater maximal activity, and as such imported L-ARG is preferentially shuttled for L-ORN synthesis rather than NO production (148). In mice co-infected with Schistosoma mansoni and Mtb, Arg1 impaired mycobacterial control and worsened lung pathology (149). A mouse model lacking Arg1 in hematopoietic cells displayed increased macrophage NO production and enhanced mycobacterial clearance (137). Thus, Arg1-expressing macrophages likely play an inhibitory role, limiting NO production and Mtb clearance.
Arg1-expressing macrophages are found in granulomas from Mtb-infected patients and non-human primates (138, 143). Using an intradermal Mtb infection model in Nos2-/- mice, Arg1-expressing macrophages were detected in granulomas (145). Interestingly, loss of Arg1 in hematopoietic cells of Nos2-/- mice resulted in more enlarged, necrotic granulomas and increased mycobacterial burden (145). Additionally, presence of Arg1 in granulomas correlated with decreased T cell proliferation (145). Thus, it is likely Arg1 regulates T cell-mediated lung immunopathology following Mtb infection.
Human neutrophils store and release Arg1 upon activation in vitro (150). In sepsis patients, circulating neutrophil counts positively correlate with plasma Arg1 activity (151). Following phagocytosis of fungi, Arg1 is upregulated in neutrophils (152). Airway supernatant from cystic fibrosis patients suppresses T cell responses in vitro, which was reversed following addition of Arg1 inhibitor N-ω-Hydroxy-L-norarginine (153). This was attributed to neutrophil Arg1 release, as neutrophil counts positively correlated with airway supernatant Arg1 activity (153). Thus, multiple mechanisms of neutrophil Arg1-mediated immune regulation exist, yet neutrophil Arg1 activity in Mtb infection still requires investigation.
The role of neutrophil L-ARG synthesis has not been studied in Mtb infection. Our work investigating myeloid L-ARG synthesis following mycobacterial infection utilized Lyz2-cre for conditional deletion, also targeting neutrophils (142). However, the role of neutrophil-specific L-ARG metabolism following mycobacterial infection has not been addressed and warrants further investigation.
NK cells require L-ARG for proliferation, expression of activating receptors, and IFNγ production in vitro (154). Human NK cells constitutively express endothelial NOS, inhibition of which impairs cytotoxicity (155, 156). Like T cells, NK cells are inhibited by myeloid-derived Arg1 (157–159). In hepatitis C virus infection, Arg1-producing myeloid-derived suppressor cells decreased NK cell IFNγ production through mTOR inhibition, which was reversed upon in vitro L-ARG supplementation (158). Despite this, neither NK cell L-ARG metabolism nor Arg1-mediated inhibition have been explored following Mtb infection.
Mouse bone marrow derived DCs (BMDCs) express Arg1 during development and have active GM-CSF-dependent L-ARG metabolism (160, 161). Additionally, mouse BMDCs grown without L-ARG decreased MHC II surface expression, while MHC I expression was unaffected (162). Thus, L-ARG is involved in BMDC development, though more thorough investigation is needed.
Little is known about DC L-ARG metabolism following infection. TNF/iNOS-producing DCs are one of the main inflammatory myeloid cell types present following bacterial and parasitic infections, though they have not been investigated following Mtb infection (163–165). Additionally, Giardia duodenalis-infected human monocyte-derived DCs decrease expression of the co-stimulatory molecule CD86 in the absence of L-ARG (166). However, DC L-ARG metabolism during Mtb infection has not yet been investigated.
T cells rely on L-ARG for cell cycle progression, proliferation, and effector functions (Figure 2) (167–172). T cells express the L-ARG synthesis enzymes, with Asl and Ass1 induced upon in vitro activation (173, 174). Functions requiring L-ARG can be rescued by L-CIT when L-ARG is limiting in vitro (173–175). Human CD4+ T cells utilize CAT-1 and LAT-1 to import L-ARG and L-CIT, respectively (174, 176).
T cell responses are suppressed when L-ARG is limiting. Myeloid Arg1 has been implicated in T cell suppression observed following mycobacterial infection, vaccination, and cancer (Figure 2) (153, 177–180). Following in vitro stimulation with heat-killed Mb BCG, macrophage Arg1 suppressed T cell proliferation, which was restored in cultures supplemented with L-CIT (173). These data suggest L-ARG synthesis from L-CIT may circumvent T cell suppression following L-ARG depletion. Still, mice with a T cell-specific loss of L-ARG synthesis did not display impaired mycobacterial clearance with either Mtb Erdman or Mb BCG, yet displayed a slight, although significant, decrease in CD4+ T cell accumulation in the lung and draining lymph nodes, compared to controls (173). Given the accumulation impairment, mycobacterial burden and/or T cell mediated lung immunopathology should be investigated during multiple stages of infection.
L-ARG deficiency impairs B cell maturation at the pro- to pre-B cell stage (181). Arg1-treated 697 pre-B lymphoblastic cells displayed increased cell death, which was reversed or attenuated upon addition of L-ARG or L-CIT, respectively (182). Therefore, it is likely B cells express L-ARG synthesis machinery, although this has not been definitively determined. Regardless, the impact of B cell L-ARG utilization and metabolism have not yet been explored following Mtb infection.
Given L-ARG is required for optimal immune cell function, L-ARG supplementation has been attempted in vitro and in vivo and has been considered as a potential vaccine adjuvant or therapeutic (139, 140, 183–187). L-ARG supplementation has been investigated in the treatment of active TB with mixed results. Some studies have shown oral L-ARG decreases constitutional symptoms and increases sputum conversion in active TB patients (188, 189). However, another study in which active TB patients were given a high L-ARG diet found no benefit when compared to normal diet, even though HIV-positive patients showed an increased cure rate (190). Regardless, oral L-ARG is subject to metabolism in the small intestine, resulting in excess water and electrolyte secretion and severe gastrointestinal distress, making it unlikely for oral L-ARG supplementation to be used clinically in TB treatment (191).
L-CIT presents a potential therapeutic alternative as it increases circulating L-ARG and NO following oral supplementation and has not been shown to induce gastrointestinal side effects (191–194). Additionally, use of L-CIT as an adjuvant improved antibody production following viral vaccination in mice (195). Addition of L-ARG alone or with L-CIT, however, did not increase antibody titers (195). This indicates L-CIT supplementation may be considered for boosting immune function when L-ARG is not recommended.
In summary, L-ARG plays an important role in immune cells following mycobacterial infection. In macrophages, L-ARG is needed to fuel NO synthesis and its synthesis from precursor L-CIT is required to control mycobacterial infection. Additionally, Arg1-expressing macrophages likely control T cell mediated immunopathology following Mtb infection via L-ARG depletion. T cells require L-ARG to fuel their effector functions, setting up an effective competition for nutrients with myeloid cells (Figure 2C). The requirement for L-ARG in both macrophages and T cells has been extensively studied, and likely serves as an immunoregulatory mechanism following Mtb infection. The role of L-ARG in other myeloid cells, including neutrophils and dendritic cells, as well as NK cells and B cells has not been extensively studied in the context of Mtb infection. Additionally, the use of L-ARG and/or L-CIT as potential therapeutic or preventative approaches should be further investigated in the context of Mtb infection and vaccination.
L-TRP is an essential amino acid obtained from the diet (196). However, in cases where L-TRP cannot be properly absorbed or is in low abundance, L-TRP may be in short supply (196). Proteolysis allows for recycling of L-TRP to meet cellular nutrient requirements, but the contribution of protein catabolism to L-TRP plasma concentrations is unclear (197).
When in excess, L-TRP is metabolized to form niacin (Figure 2B). In fact, L-TRP is second only to niacin itself as the body’s main source of NAD+, a reducing equivalent that is required for many metabolic reactions, such as glycolysis and the TCA cycle (198). To form NAD+, L-TRP is metabolized via the KYN pathway (196, 199). In hepatocytes, steroids and excess L-TRP upregulate tryptophan 2,3-dioxygenase (TDO), which converts L-TRP to N-formylkynurenine, a KYN precursor (196, 200). In innate immune cells, TLR and IFNγR ligation increase indoleamine 2,3-dioxygenase (IDO), which catalyzes the same reaction (196, 200). Unlike TDO, IDO allows for more promiscuous substrate binding, including 5-hydroxytryptophan, serotonin, and D-tryptophan (201). While both require oxygen, TDO requires heme-associated molecular oxygen and IDO requires superoxide anions (199, 201). Of the enzymes that metabolize L-TRP through the KYN pathway, only IDO is present and active in immune cells.
Two IDO isoforms, IDO1 and IDO2, are expressed by antigen presenting cells, epithelial cells, vascular endothelial cells, and neoplastic cells (199, 200). Discriminating between contributions of IDO1 and IDO2 is difficult, as available inhibitors are not isoform-specific (196, 199, 202). While knockout mouse lines for each isoform exist, only the Ido1-/- line is commercially available (203, 204). As such, we will refer to IDO1 and IDO2 collectively as “IDO” and provide information about specific isoforms when available.
L-TRP also forms serotonin, melatonin, and picolinate (Figure 2B) (196, 200). Like L-ARG, L-TRP regulates mTOR and GCN2; this has been reviewed elsewhere and will not be discussed here (133–136).
IDO activity, measured by L-TRP/KYN ratio, can determine TB severity. Active TB patients with low L-TRP/KYN ratios (high IDO activity), have poorer outcomes compared to those with low IDO activity (205). Additionally, active TB patients with pleurisy had increased pleural fluid IDO activity compared to patients with non-infectious pleuritis (206). Active TB patients also exhibit increased sputum IDO compared to patients with other lung diseases (207). In HIV+ patients, plasma IDO activity is increased upon Mtb co-infection and could be detected 6 months prior to TB diagnosis (208). Thus, measuring IDO activity may have prognostic value in TB diagnosis.
Upon Mtb infection, macrophages expression of IDO is IFNγ-dependent (Figure 2) (209, 210). In macaques, lung IDO expression is limited to macrophage-rich areas of granulomas (211). Some have postulated macrophage IDO-mediated L-TRP depletion exerts an anti-microbial effect; however Mtb can synthesize L-TRP, making this an unlikely anti-mycobacterial mechanism (212–214). Macaques treated with IDO inhibitor 1-methyl-DL-tryptophan (1-MT) showed decreased mycobacterial burden and lung pathology compared to controls following Mtb CDC1551 infection, while there was no difference in mycobacterial burden between Ido1-/- and wild-type mice (209, 215). Thus, a central anti-mycobacterial role for IDO has yet to be elucidated.
IDO forms metabolites that bind and activate the aryl hydrocarbon receptor (AHR), a ligand-regulated transcription factor that modulates immune activity. Following Mtb H37Rv infection, Ahr-/- mice displayed increased systemic mycobacterial burden, attributed to loss of myeloid cell AHR (216). Macrophage AHR activation induces IL-1β and IL-23 transcription, supporting IL-22 producing lymphocytes (217). Thus, the role of IDO is not limited to L-TRP depletion, as L-TRP metabolites modulate immune responses through AHR activation.
While unexplored in TB, L-TRP metabolism in neutrophils has been examined in other infection models. Neutrophil IDO expression is IFNγ-dependent (218). Following Pseudomonas aeruginosa infection, KYN blunted neutrophil-mediated bacterial killing by acting as a reactive oxygen species scavenger (219). In mice, KYN impaired neutrophil chemotaxis in an AHR-dependent manner following uropathogenic Escherichia coli infection (220). Thus, IDO activity inhibits neutrophil responses, and its contributions to neutrophil function following Mtb infection require further investigation.
L-TRP metabolism in NK cells is unexplored during Mtb infection. In NK92 MI cells, KYN induces apoptosis, an effect dependent upon the presence of reactive oxygen species (221). In a mouse B16 melanoma model, 1-MT administration decreased NK cell cytotoxicity, reducing their ability to restrict tumor size (222). Following Paracoccidioides brasiliensis infection, Ido1-/- mice displayed decreased lung NK cell expansion as compared to controls (223). Thus, the available data are limited, requiring further exploration to elucidate the role of L-TRP metabolism in general NK cell biology and following Mtb infection.
DCs express IDO in an IFNγ-dependent manner (218). BMDCs lacking IDO supported increased proliferation of mycobacterial-specific CD4+ T cells in vitro (209). Thus, DC IDO expression likely plays a tolerogenic role following Mtb infection. Interestingly, a feedback loop involving Arg1 and IDO1 has been uncovered in DCs (224). In mouse CD11c+ splenic DCs, TGF-β induces Arg1 expression, followed by Ido1 upregulation (224). Interestingly, increased Ido1 expression was lost in Arg1-deficient DCs, suggesting Arg1 is required for Ido1 upregulation (224). This was mediated by spermidine, a polyamine, via activation of Src kinase (224). Thus, formation of tolerogenic DCs requires a coordinated effort between the L-ARG and L-TRP metabolic pathways. This relationship should be further explored in other myeloid cells and in Mtb infection.
L-TRP is essential for supporting T cell functions, which are dampened by myeloid IDO activity (Figure 2) (225–227). When co-cultured with human CD3+ T cells, pleural fluid from active TB patients inhibited cell cycle progression, TH1 polarization, and cytokine production (228). The addition of 1-MT reversed the effect on cytokine production, though other T cell functions were not assessed (228). In vitro, IDO1 siRNA silencing in a macrophage/CD4+ T cell co-culture reduced Mtb burden (215). IDO1 silencing in macrophages alone had no effect on Mtb burden, indicating IDO1 inhibited anti-mycobacterial T cell functions (215). IDO1 silencing did not increase IFNγ production, though increased IFNβ was detected (215). In Mtb-infected macaques, IDO inhibition increased T cell proliferation and memory formation, leading to granuloma reorganization, with CD4+ T cell migration to the core (215). Whether CD4+ T cell influx into the granuloma is responsible for decreased Mtb burden and/or improved Mtb clearance has yet to be explored.
The inhibitory effect of IDO on T cells has been attributed to L-TRP depletion. However, metabolites produced by IDO can suppress TH1 and TH17 responses and promote Treg differentiation, a phenomenon that has been explored in autoimmune diseases (229, 230). As Treg activity inhibits anti-mycobacterial TH1 responses, the immunoregulatory role of L-TRP metabolites should be further explored.
Unlike T cells, L-TRP metabolism in B cells during Mtb infection has not been explored, though it has been studied in other infection models. In human PBMCs stimulated with Dengue virus, increasing KYN directly correlated with increased IgG production (231). However, Ido1-/- mice exhibit increased intestinal IgA, which afforded protection against Citrobacter rodentium infection (232). KYN and other L-TRP metabolites inhibit splenic B cell antibody production and promote apoptosis (232). In mice, B cell IDO1 is upregulated following T cell-independent antigen immunization, leading to impaired antibody production (233). Further, B cells expressing Epstein-Barr virus latent membrane protein 1 (LMP1) suppressed antibody secretion in LMP1-negative B cells, which was reversed upon 1-MT treatment (234). In a mouse model of Helicobacter felis-induced gastritis, Ido1-/- mice displayed decreased B cell frequency within the gastric mucosa (235). Thus, available data on the role of L-TRP metabolism in B cells are conflicting, with the majority of evidence favoring an inhibitory role on the part of IDO.
Unlike L-ARG, L-TRP supplementation has not been explored in TB. Studies indicate isoniazid (INH) – a front-line Mtb antibiotic – interferes with B vitamin metabolism, resulting in low niacin (Vitamin B3) levels and development of pellagra (236, 237). In one case study, an HIV+ patient showed signs of pellagra after long-term INH prophylaxis, which was reversed upon niacin supplementation and INH discontinuation (237). Considering the link between niacin and L-TRP, it would have been of interest to correlate niacin levels with serum L-TRP, yet this was not measured. Thus, further studies on L-TRP supplementation to prevent pellagra in TB patients taking INH, and subsequent effects on anti-mycobacterial immune responses, are needed.
L-TRP supplementation has been investigated in other infection models. In teleost fish, L-TRP supplementation increased plasma cortisol levels, which decreased circulating leukocytes following Photobacterium infection (238). However, infected fish receiving excess dietary L-TRP displayed increased survival (238). In a pig model of rotavirus infection, L-TRP supplementation expanded the Foxp3+ Treg population (239). Further studies are needed to determine the effect of L-TRP supplementation on immune responses to Mtb infection.
In summary, following infection myeloid cells express IDO, thereby depleting the microenvironment of L-TRP. Similar to L-ARG, T cells require L-TRP to fuel their effector functions, thus setting up yet another competition for nutrients following infection (Figure 2C). The role of L-TRP following Mtb infection in other immune cells requires further investigation. Additionally, the therapeutic potential of L-TRP supplementation has not yet been explored in the context of Mtb infection, but may prove beneficial in patients prescribed INH.
In this review, we summarized current knowledge on the immune response to Mtb and the contribution of immune cell L-ARG and L-TRP metabolism. While availability of other amino acids likely influences immune responses following infection, the L-ARG and L-TRP pathways are the most comprehensive and have identifiable regulatory roles. Available data suggest amino acid metabolism is central in the regulation of immune responses following infection. Infection leads to upregulation of pathways that limit nutrient availability, which in turn inhibits productive immune responses. Effects of nutrient limitation have been most extensively studied in macrophages and T cells, suggesting amino acid restriction by myeloid cells contributes to T cell suppression following Mtb infection. However, the immune system must balance amino acid requirements for T cell-mediated Mtb control with restriction to suppress T cell-mediated immunopathology. Thus, further research into competition for amino acids between myeloid and T cells should be further explored during Mtb infection.
Considering many TB-endemic areas also suffer from malnourishment, continued research into nutrient restriction and its effects on anti-Mtb immune responses is needed. Recently, it was shown that latent TB patients with low body mass index have altered immune cell profiles, including decreased memory T and B cells, as compared to latent TB patients with normal body mass index (240). This underscores the importance of nutrient availability in the immune system and parallels studies suggesting nutrient supplementation boosts immune responses following infection. Given their importance in immune cell function, the role of amino acid supplementation should be further explored as a potential therapeutic option in TB, especially in undernourished populations. Better understanding nutrient requirements of immune cells following Mtb infection may aid in development of future host-directed therapies aimed to eradicate TB.
Both authors contributed to conceptualization and writing (original draft, RC; reviewing and editing, RC and JQ). All authors contributed to the article and approved the submitted version.
This work was supported by the National Institutes of Health R01AI116668 (JQ), R21AI148612 (JQ), and the Division of Infectious Diseases at Cincinnati Children’s Hospital Medical Center. The content is solely the responsibility of the authors and does not necessarily represent the views of the funding sources.
The authors declare the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Melanie C. McKell and Stephanie M. Schmidt for additional proofreading and editing.
1. WHO. Global Tuberculosis Report 2020: World Health Organization. World Health Organization (2020).
2. Reid MJA, Arinaminpathy N, Bloom A, Bloom BR, Boehme C, Chaisson R, et al. Building a tuberculosis-free world: The Lancet Commission on tuberculosis. Lancet (2019) 393(10178):1331–84. doi: 10.1016/s0140-6736(19)30024-8
3. WHO. BCG Vaccines: WHO Position Paper. World Health Organization, Vol. 93. World Health Organization (2018). pp. 73–96.
4. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab (2015) 21(1):65–80. doi: 10.1016/j.cmet.2014.12.005
5. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med (2018) 215(4):1135–52. doi: 10.1084/jem.20172020
6. Bantug GR, Galluzzi L, Kroemer G, Hess C. The spectrum of T cell metabolism in health and disease. Nat Rev Immunol (2018) 18(1):19–34. doi: 10.1038/nri.2017.99
7. Weiner J 3rd, Parida SK, Maertzdorf J, Black GF, Repsilber D, Telaar A, et al. Biomarkers of inflammation, immunosuppression and stress with active disease are revealed by metabolomic profiling of tuberculosis patients. PLoS One (2012) 7(7):e40221. doi: 10.1371/journal.pone.0040221
8. Vrieling F, Alisjahbana B, Sahiratmadja E, van Crevel R, Harms AC, Hankemeier T, et al. Plasma metabolomics in tuberculosis patients with and without concurrent type 2 diabetes at diagnosis and during antibiotic treatment. Sci Rep (2019) 9(1):18669. doi: 10.1038/s41598-019-54983-5
9. Cho Y, Park Y, Sim B, Kim J, Lee H, Cho SN, et al. Identification of serum biomarkers for active pulmonary tuberculosis using a targeted metabolomics approach. Sci Rep (2020) 10(1):3825. doi: 10.1038/s41598-020-60669-0
10. Feng S, Du YQ, Zhang L, Feng RR, Liu SY. Analysis of serum metabolic profile by ultra-performance liquid chromatography-mass spectrometry for biomarkers discovery: application in a pilot study to discriminate patients with tuberculosis. Chin Med J (Engl) (2015) 128(2):159–68. doi: 10.4103/0366-6999.149188
11. Isa F, Collins S, Lee MH, Decome D, Dorvil N, Joseph P, et al. Mass Spectrometric Identification of Urinary Biomarkers of Pulmonary Tuberculosis. EBioMedicine (2018) 31:157–65. doi: 10.1016/j.ebiom.2018.04.014
12. Weiner J 3rd, Maertzdorf J, Sutherland JS, Duffy FJ, Thompson E, Suliman S, et al. Metabolite changes in blood predict the onset of tuberculosis. Nat Commun (2018) 9(1):5208. doi: 10.1038/s41467-018-07635-7
13. Guilliams M, Lambrecht BN, Hammad H. Division of labor between lung dendritic cells and macrophages in the defense against pulmonary infections. Mucosal Immunol (2013) 6(3):464–73. doi: 10.1038/mi.2013.14
14. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar Macrophages Provide an Early Mycobacterium tuberculosis Niche and Initiate Dissemination. Cell Host Microbe (2018) 24(3):439–46. doi: 10.1016/j.chom.2018.08.001
15. Ryder BM, Sandford SK, Manners KM, Dalton JP, Wiles S, Kirman JR. Gr1(int/high) Cells Dominate the Early Phagocyte Response to Mycobacterial Lung Infection in Mice. Front Microbiol (2019) 10:402. doi: 10.3389/fmicb.2019.00402
16. Rothchild AC, Olson GS, Nemeth J, Amon LM, Mai D, Gold ES, et al. Alveolar macrophages generate a noncanonical NRF2-driven transcriptional response to Mycobacterium tuberculosis in vivo. Sci Immunol (2019) 4(37). doi: 10.1126/sciimmunol.aaw6693
17. Ardain A, Domingo-Gonzalez R, Das S, Kazer SW, Howard NC, Singh A, et al. Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature (2019) 570(7762):528–32. doi: 10.1038/s41586-019-1276-2
18. Srivastava S, Ernst JD, Desvignes L. Beyond macrophages: the diversity of mononuclear cells in tuberculosis. Immunol Rev (2014) 262(1):179–92. doi: 10.1111/imr.12217
19. Sia JK, Rengarajan J. Immunology of Mycobacterium tuberculosis Infections. Microbiol Spectr (2019) 7(4). doi: 10.1128/microbiolspec.GPP3-0022-2018
20. Ernst JD. Mechanisms of M. tuberculosis Immune Evasion as Challenges to TB Vaccine Design. Cell Host Microbe (2018) 24(1):34–42. doi: 10.1016/j.chom.2018.06.004
21. von Both U, Berk M, Agapow PM, Wright JD, Git A, Hamilton MS, et al. Mycobacterium tuberculosis Exploits a Molecular Off Switch of the Immune System for Intracellular Survival. Sci Rep (2018) 8(1):661. doi: 10.1038/s41598-017-18528-y
22. Carranza C, Chavez-Galan L. Several Routes to the Same Destination: Inhibition of Phagosome-Lysosome Fusion by Mycobacterium tuberculosis. Am J Med Sci (2019) 357(3):184–94. doi: 10.1016/j.amjms.2018.12.003
23. Zhai W, Wu F, Zhang Y, Fu Y, Liu Z. The Immune Escape Mechanisms of Mycobacterium Tuberculosis. Int J Mol Sci (2019) 20(2). doi: 10.3390/ijms20020340
24. Kleinnijenhuis J, Oosting M, Joosten LA, Netea MG, Van Crevel R. Innate immune recognition of Mycobacterium tuberculosis. Clin Dev Immunol (2011) 2011:405310. doi: 10.1155/2011/405310
25. Mayer-Barber KD, Barber DL. Innate and Adaptive Cellular Immune Responses to Mycobacterium tuberculosis Infection. Cold Spring Harb Perspect Med (2015) 5(12). doi: 10.1101/cshperspect.a018424
26. Krakauer T. Inflammasomes, Autophagy, and Cell Death: The Trinity of Innate Host Defense against Intracellular Bacteria. Mediators Inflam (2019) 2019:2471215. doi: 10.1155/2019/2471215
27. MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci U S A (1997) 94(10):5243–8. doi: 10.1073/pnas.94.10.5243
28. Garcia I, Guler R, Vesin D, Olleros ML, Vassalli P, Chvatchko Y, et al. Lethal Mycobacterium bovis Bacillus Calmette Guérin infection in nitric oxide synthase 2-deficient mice: cell-mediated immunity requires nitric oxide synthase 2. Lab Invest (2000) 80(9):1385–97. doi: 10.1038/labinvest.3780146
29. Scanga CA, Mohan VP, Tanaka K, Alland D, Flynn JL, Chan J. The inducible nitric oxide synthase locus confers protection against aerogenic challenge of both clinical and laboratory strains of Mycobacterium tuberculosis in mice. Infect Immun (2001) 69(12):7711–7. doi: 10.1128/iai.69.12.7711-7717.2001
30. Chan J, Tanaka K, Carroll D, Flynn J, Bloom BR. Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infect Immun (1995) 63(2):736–40. doi: 10.1128/iai.63.2.736-740.1995
31. Nicholson S, Bonecini-Almeida Mda G, Lapa e Silva JR, Nathan C, Xie QW, Mumford R, et al. Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J Exp Med (1996) 183(5):2293–302. doi: 10.1084/jem.183.5.2293
32. Rich EA, Torres M, Sada E, Finegan CK, Hamilton BD, Toossi Z. Mycobacterium tuberculosis (MTB)-stimulated production of nitric oxide by human alveolar macrophages and relationship of nitric oxide production to growth inhibition of MTB. Tuber Lung Dis (1997) 78(5-6):247–55. doi: 10.1016/s0962-8479(97)90005-8
33. Kuo HP, Wang CH, Huang KS, Lin HC, Yu CT, Liu CY, et al. Nitric oxide modulates interleukin-1beta and tumor necrosis factor-alpha synthesis by alveolar macrophages in pulmonary tuberculosis. Am J Respir Crit Care Med (2000) 161(1):192–9. doi: 10.1164/ajrccm.161.1.9902113
34. Wang CH, Liu CY, Lin HC, Yu CT, Chung KF, Kuo HP. Increased exhaled nitric oxide in active pulmonary tuberculosis due to inducible NO synthase upregulation in alveolar macrophages. Eur Respir J (1998) 11(4):809–15. doi: 10.1183/09031936.98.11040809
35. Aston C, Rom WN, Talbot AT, Reibman J. Early inhibition of mycobacterial growth by human alveolar macrophages is not due to nitric oxide. Am J Respir Crit Care Med (1998) 157(6 Pt 1):1943–50. doi: 10.1164/ajrccm.157.6.9705028
36. López JW, Loader MI, Smith D, Pastorius D, Bravard M, Caviedes L, et al. Exhaled Nitric Oxide is Not a Biomarker for Pulmonary Tuberculosis. Am J Trop Med Hyg (2018) 98(6):1637–9. doi: 10.4269/ajtmh.17-0425
37. Nozaki Y, Hasegawa Y, Ichiyama S, Nakashima I, Shimokata K. Mechanism of nitric oxide-dependent killing of Mycobacterium bovis BCG in human alveolar macrophages. Infect Immun (1997) 65(9):3644–7. doi: 10.1128/iai.65.9.3644-3647.1997
38. Sharma S, Sharma M, Roy S, Kumar P, Bose M. Mycobacterium tuberculosis induces high production of nitric oxide in coordination with production of tumour necrosis factor-alpha in patients with fresh active tuberculosis but not in MDR tuberculosis. Immunol Cell Biol (2004) 82(4):377–82. doi: 10.1111/j.0818-9641.2004.01245.x
39. Ralph AP, Yeo TW, Salome CM, Waramori G, Pontororing GJ, Kenangalem E, et al. Impaired pulmonary nitric oxide bioavailability in pulmonary tuberculosis: association with disease severity and delayed mycobacterial clearance with treatment. J Infect Dis (2013) 208(4):616–26. doi: 10.1093/infdis/jit248
40. Sakowski ET, Koster S, Portal Celhay C, Park HS, Shrestha E, Hetzenecker SE, et al. Ubiquilin 1 Promotes IFN-γ-Induced Xenophagy of Mycobacterium tuberculosis. PLoS Pathog (2015) 11(7):e1005076. doi: 10.1371/journal.ppat.1005076
41. Lam A, Prabhu R, Gross CM, Riesenberg LA, Singh V, Aggarwal S. Role of apoptosis and autophagy in tuberculosis. Am J Physiol Lung Cell Mol Physiol (2017) 313(2):L218–l29. doi: 10.1152/ajplung.00162.2017
42. Lyadova IV. Neutrophils in Tuberculosis: Heterogeneity Shapes the Way? Mediators Inflamm (2017) 2017:8619307. doi: 10.1155/2017/8619307
43. Lombard R, Doz E, Carreras F, Epardaud M, Le Vern Y, Buzoni-Gatel D, et al. IL-17RA in Non-Hematopoietic Cells Controls CXCL-1 and 5 Critical to Recruit Neutrophils to the Lung of Mycobacteria-Infected Mice during the Adaptive Immune Response. PLoS One (2016) 11(2):e0149455. doi: 10.1371/journal.pone.0149455
44. Kisich KO, Higgins M, Diamond G, Heifets L. Tumor necrosis factor alpha stimulates killing of Mycobacterium tuberculosis by human neutrophils. Infect Immun (2002) 70(8):4591–9. doi: 10.1128/iai.70.8.4591-4599.2002
45. Ganbat D, Seehase S, Richter E, Vollmer E, Reiling N, Fellenberg K, et al. Mycobacteria infect different cell types in the human lung and cause species dependent cellular changes in infected cells. BMC Pulm Med (2016) 16:19. doi: 10.1186/s12890-016-0185-5
46. Sharma S, Verma I, Khuller GK. Antibacterial activity of human neutrophil peptide-1 against Mycobacterium tuberculosis H37Rv: in vitro and ex vivo study. Eur Respir J (2000) 16(1):112–7. doi: 10.1034/j.1399-3003.2000.16a20.x
47. Martineau AR, Newton SM, Wilkinson KA, Kampmann B, Hall BM, Nawroly N, et al. Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest (2007) 117(7):1988–94. doi: 10.1172/jci31097
48. Eruslanov EB, Lyadova IV, Kondratieva TK, Majorov KB, Scheglov IV, Orlova MO, et al. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun (2005) 73(3):1744–53. doi: 10.1128/iai.73.3.1744-1753.2005
49. Corleis B, Korbel D, Wilson R, Bylund J, Chee R, Schaible UE. Escape of Mycobacterium tuberculosis from oxidative killing by neutrophils. Cell Microbiol (2012) 14(7):1109–21. doi: 10.1111/j.1462-5822.2012.01783.x
50. van der Meer AJ, Zeerleder S, Blok DC, Kager LM, Lede IO, Rahman W, et al. Neutrophil extracellular traps in patients with pulmonary tuberculosis. Respir Res (2017) 18:181. doi: 10.1186/s12931-017-0663-1
51. Ramos-Kichik V, Mondragón-Flores R, Mondragón-Castelán M, Gonzalez-Pozos S, Muñiz-Hernandez S, Rojas-Espinosa O, et al. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (Edinb) (2009) 89(1):29–37. doi: 10.1016/j.tube.2008.09.009
52. Braian C, Hogea V, Stendahl O. Mycobacterium tuberculosis- induced neutrophil extracellular traps activate human macrophages. J Innate Immun (2013) 5(6):591–602. doi: 10.1159/000348676
53. Ong CW, Elkington PT, Brilha S, Ugarte-Gil C, Tome-Esteban MT, Tezera LB, et al. Neutrophil-Derived MMP-8 Drives AMPK-Dependent Matrix Destruction in Human Pulmonary Tuberculosis. PLoS Pathog (2015) 11(5):e1004917. doi: 10.1371/journal.ppat.1004917
54. Blomgran R, Ernst JD. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J Immunol (2011) 186(12):7110–9. doi: 10.4049/jimmunol.1100001
55. Trentini MM, de Oliveira FM, Kipnis A, Junqueira-Kipnis AP. The Role of Neutrophils in the Induction of Specific Th1 and Th17 during Vaccination against Tuberculosis. Front Microbiol (2016) 7:898. doi: 10.3389/fmicb.2016.00898
56. Esin S, Batoni G, Counoupas C, Stringaro A, Brancatisano FL, Colone M, et al. Direct binding of human NK cell natural cytotoxicity receptor NKp44 to the surfaces of mycobacteria and other bacteria. Infect Immun (2008) 76(4):1719–27. doi: 10.1128/iai.00870-07
57. Esin S, Counoupas C, Aulicino A, Brancatisano FL, Maisetta G, Bottai D, et al. Interaction of Mycobacterium tuberculosis cell wall components with the human natural killer cell receptors NKp44 and Toll-like receptor 2. Scand J Immunol (2013) 77(6):460–9. doi: 10.1111/sji.12052
58. Vankayalapati R, Garg A, Porgador A, Griffith DE, Klucar P, Safi H, et al. Role of NK cell-activating receptors and their ligands in the lysis of mononuclear phagocytes infected with an intracellular bacterium. J Immunol (2005) 175(7):4611–7. doi: 10.4049/jimmunol.175.7.4611
59. Dhiman R, Indramohan M, Barnes PF, Nayak RC, Paidipally P, Rao LV, et al. IL-22 produced by human NK cells inhibits growth of Mycobacterium tuberculosis by enhancing phagolysosomal fusion. J Immunol (2009) 183(10):6639–45. doi: 10.4049/jimmunol.0902587
60. Harris LD, Khayumbi J, Ongalo J, Sasser LE, Tonui J, Campbell A, et al. Distinct Human NK Cell Phenotypes and Functional Responses to Mycobacterium tuberculosis in Adults From TB Endemic and Non-endemic Regions. Front Cell Infect Microbiol (2020) 10:120. doi: 10.3389/fcimb.2020.00120
61. Moreira-Teixeira L, Tabone O, Graham CM, Singhania A, Stavropoulos E, Redford PS, et al. Mouse transcriptome reveals potential signatures of protection and pathogenesis in human tuberculosis. Nat Immunol (2020) 21(4):464–76. doi: 10.1038/s41590-020-0610-z
62. Cai Y, Dai Y, Wang Y, Yang Q, Guo J, Wei C, et al. Single-cell transcriptomics of blood reveals a natural killer cell subset depletion in tuberculosis. EBioMedicine (2020) 53:102686. doi: 10.1016/j.ebiom.2020.102686
63. Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol (2007) 179(4):2509–19. doi: 10.4049/jimmunol.179.4.2509
64. Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med (2011) 365(2):127–38. doi: 10.1056/NEJMoa1100066
65. Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, et al. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med (2008) 205(1):105–15. doi: 10.1084/jem.20071367
66. Srivastava S, Ernst JD. Cell-to-cell transfer of M. tuberculosis antigens optimizes CD4 T cell priming. Cell Host Microbe (2014) 15(6):741–52. doi: 10.1016/j.chom.2014.05.007
67. Srivastava S, Grace PS, Ernst JD. Antigen Export Reduces Antigen Presentation and Limits T Cell Control of M. tuberculosis. Cell Host Microbe (2016) 19(1):44–54. doi: 10.1016/j.chom.2015.12.003
68. Grace PS, Ernst JD. Suboptimal Antigen Presentation Contributes to Virulence of Mycobacterium tuberculosis In Vivo. J Immunol (2016) 196(1):357–64. doi: 10.4049/jimmunol.1501494
69. Leveton C, Barnass S, Champion B, Lucas S, De Souza B, Nicol M, et al. T-cell-mediated protection of mice against virulent Mycobacterium tuberculosis. Infect Immun (1989) 57(2):390–5. doi: 10.1128/IAI.57.2.390-395.1989
70. Flory CM, Hubbard RD, Collins FM. Effects of in vivo T lymphocyte subset depletion on mycobacterial infections in mice. J Leukoc Biol (1992) 51(3):225–9. doi: 10.1002/jlb.51.3.225
71. Lin PL, Rutledge T, Green AM, Bigbee M, Fuhrman C, Klein E, et al. CD4 T cell depletion exacerbates acute Mycobacterium tuberculosis while reactivation of latent infection is dependent on severity of tissue depletion in cynomolgus macaques. AIDS Res Hum Retroviruses (2012) 28(12):1693–702. doi: 10.1089/aid.2012.0028
72. Ankley L, Thomas S, Olive AJ. Fighting persistence; how chronic infections with Mycobacterium tuberculosis evade T cell-mediated clearance and new strategies to defeat them. Infect Immun (2020) 88(7):e00916-19. doi: 10.1128/iai.00916-19
73. Lin PL, Pawar S, Myers A, Pegu A, Fuhrman C, Reinhart TA, et al. Early events in Mycobacterium tuberculosis infection in cynomolgus macaques. Infect Immun (2006) 74(7):3790–803. doi: 10.1128/iai.00064-06
74. Gallegos AM, Pamer EG, Glickman MS. Delayed protection by ESAT-6-specific effector CD4+ T cells after airborne M. tuberculosis infection. J Exp Med (2008) 205(10):2359–68. doi: 10.1084/jem.20080353
75. Jouanguy E, Altare F, Lamhamedi S, Revy P, Emile JF, Newport M, et al. Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guérin infection. N Engl J Med (1996) 335(26):1956–61. doi: 10.1056/nejm199612263352604
76. Altare F, Durandy A, Lammas D, Emile JF, Lamhamedi S, Le Deist F, et al. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science (1998) 280(5368):1432–5. doi: 10.1126/science.280.5368.1432
77. Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest (1998) 101(11):2364–9. doi: 10.1172/jci2901
78. Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science (2001) 293(5528):300–3. doi: 10.1126/science.1061154
79. Moradi L, Cheraghi T, Yazdani R, Azizi G, Rasouli S, Zavareh FT, et al. Mendelian susceptibility to mycobacterial disease: Clinical and immunological findings of patients suspected for IL12Rbeta1 deficiency. Allergol Immunopathol (Madr) (2019) 47(5):491–8. doi: 10.1016/j.aller.2019.02.004
80. Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med (1993) 178(6):2243–7. doi: 10.1084/jem.178.6.2243
81. Cooper AM, Magram J, Ferrante J, Orme IM. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with mycobacterium tuberculosis. J Exp Med (1997) 186(1):39–45. doi: 10.1084/jem.186.1.39
82. Sullivan BM, Jobe O, Lazarevic V, Vasquez K, Bronson R, Glimcher LH, et al. Increased susceptibility of mice lacking T-bet to infection with Mycobacterium tuberculosis correlates with increased IL-10 and decreased IFN-gamma production. J Immunol (2005) 175(7):4593–602. doi: 10.4049/jimmunol.175.7.4593
83. Freches D, Korf H, Denis O, Havaux X, Huygen K, Romano M. Mice genetically inactivated in interleukin-17A receptor are defective in long-term control of Mycobacterium tuberculosis infection. Immunology (2013) 140(2):220–31. doi: 10.1111/imm.12130
84. Gopal R, Monin L, Slight S, Uche U, Blanchard E, Fallert Junecko BA, et al. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog (2014) 10(5):e1004099. doi: 10.1371/journal.ppat.1004099
85. Wozniak TM, Saunders BM, Ryan AA, Britton WJ. Mycobacterium bovis BCG-specific Th17 cells confer partial protection against Mycobacterium tuberculosis infection in the absence of gamma interferon. Infect Immun (2010) 78(10):4187–94. doi: 10.1128/iai.01392-09
86. Perreau M, Rozot V, Welles HC, Belluti-Enders F, Vigano S, Maillard M, et al. Lack of Mycobacterium tuberculosis-specific interleukin-17A-producing CD4+ T cells in active disease. Eur J Immunol (2013) 43(4):939–48. doi: 10.1002/eji.201243090
87. Sakai S, Kauffman KD, Schenkel JM, McBerry CC, Mayer-Barber KD, Masopust D, et al. Cutting Edge: Control of Mycobacterium tuberculosis Infection by a Subset of Lung Parenchyma–Homing CD4 T Cells. J Immunol (2014) 192(7):2965–9. doi: 10.4049/jimmunol.1400019
88. Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T Cell-Derived IFN-γ Plays a Minimal Role in Control of Pulmonary Mycobacterium tuberculosis Infection and Must Be Actively Repressed by PD-1 to Prevent Lethal Disease. PLoS Pathog (2016) 12(5):e1005667. doi: 10.1371/journal.ppat.1005667
89. Chamie G, Luetkemeyer A, Walusimbi-Nanteza M, Okwera A, Whalen CC, Mugerwa RD, et al. Significant variation in presentation of pulmonary tuberculosis across a high resolution of CD4 strata. Int J Tuberc Lung Dis (2010) 14(10):1295–302.
90. Wang X, Wu Y, Jiao J, Huang Q. Mycobacterium tuberculosis infection induces IL-10 gene expression by disturbing histone deacetylase 6 and histonedeacetylase 11 equilibrium in macrophages. Tuberculosis (Edinb) (2018) 108:118–23. doi: 10.1016/j.tube.2017.11.008
91. Bouzeyen R, Haoues M, Barbouche MR, Singh R, Essafi M. FOXO3 Transcription Factor Regulates IL-10 Expression in Mycobacteria-Infected Macrophages, Tuning Their Polarization and the Subsequent Adaptive Immune Response. Front Immunol (2019) 10:2922. doi: 10.3389/fimmu.2019.02922
92. Redford PS, Boonstra A, Read S, Pitt J, Graham C, Stavropoulos E, et al. Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. Eur J Immunol (2010) 40(8):2200–10. doi: 10.1002/eji.201040433
93. Guyot-Revol V, Innes JA, Hackforth S, Hinks T, Lalvani A. Regulatory T cells are expanded in blood and disease sites in patients with tuberculosis. Am J Respir Crit Care Med (2006) 173(7):803–10. doi: 10.1164/rccm.200508-1294OC
94. Hougardy JM, Place S, Hildebrand M, Drowart A, Debrie AS, Locht C, et al. Regulatory T cells depress immune responses to protective antigens in active tuberculosis. Am J Respir Crit Care Med (2007) 176(4):409–16. doi: 10.1164/rccm.200701-084OC
95. Green AM, Mattila JT, Bigbee CL, Bongers KS, Lin PL, Flynn JL. CD4(+) regulatory T cells in a cynomolgus macaque model of Mycobacterium tuberculosis infection. J Infect Dis (2010) 202(4):533–41. doi: 10.1086/654896
96. Geffner L, Basile JI, Yokobori N, Sabio YGC, Musella R, Castagnino J, et al. CD4(+) CD25(high) forkhead box protein 3(+) regulatory T lymphocytes suppress interferon-γ and CD107 expression in CD4(+) and CD8(+) T cells from tuberculous pleural effusions. Clin Exp Immunol (2014) 175(2):235–45. doi: 10.1111/cei.12227
97. Moreira-Teixeira L, Redford PS, Stavropoulos E, Ghilardi N, Maynard CL, Weaver CT, et al. T Cell-Derived IL-10 Impairs Host Resistance to Mycobacterium tuberculosis Infection. J Immunol (2017) 199(2):613–23. doi: 10.4049/jimmunol.1601340
98. Gerosa F, Nisii C, Righetti S, Micciolo R, Marchesini M, Cazzadori A, et al. CD4(+) T cell clones producing both interferon-gamma and interleukin-10 predominate in bronchoalveolar lavages of active pulmonary tuberculosis patients. Clin Immunol (1999) 92(3):224–34. doi: 10.1006/clim.1999.4752
99. Scott-Browne JP, Shafiani S, Tucker-Heard G, Ishida-Tsubota K, Fontenot JD, Rudensky AY, et al. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J Exp Med (2007) 204(9):2159–69. doi: 10.1084/jem.20062105
100. Ordway D, Henao-Tamayo M, Harton M, Palanisamy G, Troudt J, Shanley C, et al. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol (2007) 179(1):522–31. doi: 10.4049/jimmunol.179.1.522
101. Cardona P, Marzo-Escartín E, Tapia G, Díaz J, García V, Varela I, et al. Oral Administration of Heat-Killed Mycobacterium manresensis Delays Progression toward Active Tuberculosis in C3HeB/FeJ Mice. Front Microbiol (2015) 6:1482. doi: 10.3389/fmicb.2015.01482
102. Lalvani A, Brookes R, Wilkinson RJ, Malin AS, Pathan AA, Andersen P, et al. Human cytolytic and interferon gamma-secreting CD8+ T lymphocytes specific for Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (1998) 95(1):270–5. doi: 10.1073/pnas.95.1.270
103. Behar SM, Dascher CC, Grusby MJ, Wang CR, Brenner MB. Susceptibility of mice deficient in CD1D or TAP1 to infection with Mycobacterium tuberculosis. J Exp Med (1999) 189(12):1973–80. doi: 10.1084/jem.189.12.1973
104. Sousa AO, Mazzaccaro RJ, Russell RG, Lee FK, Turner OC, Hong S, et al. Relative contributions of distinct MHC class I-dependent cell populations in protection to tuberculosis infection in mice. Proc Natl Acad Sci U S A (2000) 97(8):4204–8. doi: 10.1073/pnas.97.8.4204
105. van Pinxteren LA, Cassidy JP, Smedegaard BH, Agger EM, Andersen P. Control of latent Mycobacterium tuberculosis infection is dependent on CD8 T cells. Eur J Immunol (2000) 30(12):3689–98. doi: 10.1002/1521-4141(200012)30:12<3689::aid-immu3689>3.0.co;2-4
106. Stenger S, Mazzaccaro RJ, Uyemura K, Cho S, Barnes PF, Rosat JP, et al. Differential effects of cytolytic T cell subsets on intracellular infection. Science (1997) 276(5319):1684–7. doi: 10.1126/science.276.5319.1684
107. Stenger S, Hanson DA, Teitelbaum R, Dewan P, Niazi KR, Froelich CJ, et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science (1998) 282(5386):121–5. doi: 10.1126/science.282.5386.121
108. Bruns H, Meinken C, Schauenberg P, Härter G, Kern P, Modlin RL, et al. Anti-TNF immunotherapy reduces CD8+ T cell-mediated antimicrobial activity against Mycobacterium tuberculosis in humans. J Clin Invest (2009) 119(5):1167–77. doi: 10.1172/jci38482
109. Yang JD, Mott D, Sutiwisesak R, Lu YJ, Raso F, Stowell B, et al. Mycobacterium tuberculosis-specific CD4+ and CD8+ T cells differ in their capacity to recognize infected macrophages. PLoS Pathog (2018) 14(5):e1007060. doi: 10.1371/journal.ppat.1007060
110. Li H, Wang XX, Wang B, Fu L, Liu G, Lu Y, et al. Latently and uninfected healthcare workers exposed to TB make protective antibodies against Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (2017) 114(19):5023–8. doi: 10.1073/pnas.1611776114
111. Kunnath-Velayudhan S, Salamon H, Wang HY, Davidow AL, Molina DM, Huynh VT, et al. Dynamic antibody responses to the Mycobacterium tuberculosis proteome. Proc Natl Acad Sci U S A (2010) 107(33):14703–8. doi: 10.1073/pnas.1009080107
112. Lu LL, Chung AW, Rosebrock TR, Ghebremichael M, Yu WH, Grace PS, et al. A Functional Role for Antibodies in Tuberculosis. Cell (2016) 167(2):433–43. doi: 10.1016/j.cell.2016.08.072
113. Lu LL, Das J, Grace PS, Fortune SM, Restrepo BI, Alter G. Antibody Fc Glycosylation Discriminates Between Latent and Active Tuberculosis. J Infect Dis (2020) 222(12):2093–102. doi: 10.1093/infdis/jiz643
114. Kumagai T, Palacios A, Casadevall A, García MJ, Toro C, Tiemeyer M, et al. Serum IgM Glycosylation Associated with Tuberculosis Infection in Mice. mSphere (2019) 4(2). doi: 10.1128/mSphere.00684-18
115. Olivares N, Marquina B, Mata-Espinoza D, Zatarain-Barron ZL, Pinzón CE, Estrada I, et al. The protective effect of immunoglobulin in murine tuberculosis is dependent on IgG glycosylation. Pathog Dis (2013) 69(3):176–83. doi: 10.1111/2049-632x.12069
116. Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol (2012) 12(5):352–66. doi: 10.1038/nri3211
117. Cadena AM, Fortune SM, Flynn JL. Heterogeneity in tuberculosis. Nat Rev Immunol (2017) 17(11):691–702. doi: 10.1038/nri.2017.69
118. Wilson JL, Mayr HK, Weichhart T. Metabolic Programming of Macrophages: Implications in the Pathogenesis of Granulomatous Disease. Front Immunol (2019) 10:2265. doi: 10.3389/fimmu.2019.02265
119. Ulrichs T, Kosmiadi GA, Trusov V, Jörg S, Pradl L, Titukhina M, et al. Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J Pathol (2004) 204(2):217–28. doi: 10.1002/path.1628
120. Tsai MC, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, et al. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell Microbiol (2006) 8(2):218–32. doi: 10.1111/j.1462-5822.2005.00612.x
121. Via LE, Lin PL, Ray SM, Carrillo J, Allen SS, Eum SY, et al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun (2008) 76(6):2333–40. doi: 10.1128/iai.01515-07
122. Prideaux B, Via LE, Zimmerman MD, Eum S, Sarathy J, O’Brien P, et al. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat Med (2015) 21(10):1223–7. doi: 10.1038/nm.3937
123. Hackett EE, Sheedy FJ. An Army Marches on Its Stomach: Metabolic Intermediates as Antimicrobial Mediators in Mycobacterium tuberculosis Infection. Front Cell Infect Microbiol (2020) 10:446. doi: 10.3389/fcimb.2020.00446
124. Qualls JE, Murray PJ. Immunometabolism within the tuberculosis granuloma: amino acids, hypoxia, and cellular respiration. Semin Immunopathology (2016) 38(2):139–52. doi: 10.1007/s00281-015-0534-0
125. Chen S, Xia Y, He F, Fu J, Xin Z, Deng B, et al. Serine Supports IL-1beta Production in Macrophages Through mTOR Signaling. Front Immunol (2020) 11:1866. doi: 10.3389/fimmu.2020.01866
126. Morris SM Jr. Arginine Metabolism Revisited. J Nutr (2016) 146(12):2579s–86s. doi: 10.3945/jn.115.226621
127. Windmueller HG, Spaeth AE. Source and fate of circulating citrulline. Am J Physiol-Endocrinol Metab (1981) 241(6):E473–80. doi: 10.1152/ajpendo.1981.241.6.E473
128. Brosnan ME, Brosnan JT. Renal arginine metabolism. J Nutr (2004) 134(10 Suppl):2791S–5S; discussion 6S-7S. doi: 10.1093/jn/134.10.2791S
129. Popovic PJ, Zeh IIIHJ, Ochoa JB. Arginine and Immunity. J Nutr (2007) 137(6):1681S–6S. doi: 10.1093/jn/137.6.1681S
130. Curis E, Nicolis I, Moinard C, Osowska S, Zerrouk N, Bénazeth S, et al. Almost all about citrulline in mammals. Amino Acids (2005) 29(3):177–205. doi: 10.1007/s00726-005-0235-4
131. Hesterberg RS, Cleveland JL, Epling-Burnette PK. Role of Polyamines in Immune Cell Functions. Med Sci (Basel) (2018) 6(1). doi: 10.3390/medsci6010022
132. Broer S, Broer A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem J (2017) 474(12):1935–63. doi: 10.1042/bcj20160822
133. Liu Y, Zhang DT, Liu XG. mTOR signaling in T cell immunity and autoimmunity. Int Rev Immunol (2015) 34(1):50–66. doi: 10.3109/08830185.2014.933957
134. Saleiro D, Platanias LC. Intersection of mTOR and STAT signaling in immunity. Trends Immunol (2015) 36(1):21–9. doi: 10.1016/j.it.2014.10.006
135. Keating R, McGargill MA. mTOR Regulation of Lymphoid Cells in Immunity to Pathogens. Front Immunol (2016) 7:180. doi: 10.3389/fimmu.2016.00180
136. Jones RG, Pearce EJ. MenTORing Immunity: mTOR Signaling in the Development and Function of Tissue-Resident Immune Cells. Immunity (2017) 46(5):730–42. doi: 10.1016/j.immuni.2017.04.028
137. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, et al. Toll-like receptor–induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol (2008) 9:1399. doi: 10.1038/ni.1671
138. Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol (2013) 191(2):773–84. doi: 10.4049/jimmunol.1300113
139. Qualls JE, Subramanian C, Rafi W, Smith AM, Balouzian L, DeFreitas AA, et al. Sustained generation of nitric oxide and control of mycobacterial infection requires argininosuccinate synthase 1. Cell Host Microbe (2012) 12(3):313–23. doi: 10.1016/j.chom.2012.07.012
140. Rapovy SM, Zhao J, Bricker RL, Schmidt SM, Setchell KD, Qualls JE. Differential Requirements for L-Citrulline and L-Arginine during Antimycobacterial Macrophage Activity. J Immunol (2015) 195(7):3293–300. doi: 10.4049/jimmunol.1500800
141. Erez A, Nagamani SCS, Shchelochkov OA, Premkumar MH, Campeau PM, Chen Y, et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat Med (2011) 17:1619. doi: 10.1038/nm.2544
142. Lange SM, McKell MC, Schmidt SM, Zhao J, Crowther RR, Green LC, et al. L-Arginine Synthesis from L-Citrulline in Myeloid Cells Drives Host Defense against Mycobacteria In Vivo. J Immunol (2019) 202(6):1747–54. doi: 10.4049/jimmunol.1801569
143. Pessanha AP, Martins RA, Mattos-Guaraldi AL, Vianna A, Moreira LO. Arginase-1 expression in granulomas of tuberculosis patients. FEMS Immunol Med Microbiol (2012) 66(2):265–8. doi: 10.1111/j.1574-695X.2012.01012.x
144. Qualls JE, Neale G, Smith AM, Koo MS, DeFreitas AA, Zhang H, et al. Arginine usage in mycobacteria-infected macrophages depends on autocrine-paracrine cytokine signaling. Sci Signal (2010) 3(135):ra62. doi: 10.1126/scisignal.2000955
145. Duque-Correa MA, Kühl AA, Rodriguez PC, Zedler U, Schommer-Leitner S, Rao M, et al. Macrophage arginase-1 controls bacterial growth and pathology in hypoxic tuberculosis granulomas. Proc Natl Acad Sci (2014) 111(38):E4024–E32. doi: 10.1073/pnas.1408839111
146. Yeramian A, Martin L, Serrat N, Arpa L, Soler C, Bertran J, et al. Arginine Transport via Cationic Amino Acid Transporter 2 Plays a Critical Regulatory Role in Classical or Alternative Activation of Macrophages. J Immunol (2006) 176(10):5918–24. doi: 10.4049/jimmunol.176.10.5918
147. Yeramian A, Martin L, Arpa L, Bertran J, Soler C, McLeod C, et al. Macrophages require distinct arginine catabolism and transport systems for proliferation and for activation. Eur J Immunol (2006) 36(6):1516–26. doi: 10.1002/eji.200535694
148. Wu G, Morris SM Jr. Arginine metabolism: nitric oxide and beyond. Biochem J (1998) 336(Pt 1):1–17. doi: 10.1042/bj3360001
149. Monin L, Griffiths KL, Lam WY, Gopal R, Kang DD, Ahmed M, et al. Helminth-induced arginase-1 exacerbates lung inflammation and disease severity in tuberculosis. J Clin Invest (2015) 125(12):4699–713. doi: 10.1172/jci77378
150. Jacobsen LC, Theilgaard-Mönch K, Christensen EI, Borregaard N. Arginase 1 is expressed in myelocytes/metamyelocytes and localized in gelatinase granules of human neutrophils. Blood (2007) 109(7):3084–7. doi: 10.1182/blood-2006-06-032599
151. Darcy CJ, Woodberry T, Davis JS, Piera KA, McNeil YR, Chen Y, et al. Increased plasma arginase activity in human sepsis: association with increased circulating neutrophils. Clin Chem Lab Med (2014) 52(4):573–81. doi: 10.1515/cclm-2013-0698
152. Rubin-Bejerano I, Fraser I, Grisafi P, Fink GR. Phagocytosis by neutrophils induces an amino acid deprivation response in Saccharomyces cerevisiae and Candida albicans. Proc Natl Acad Sci U S A (2003) 100(19):11007–12. doi: 10.1073/pnas.1834481100
153. Ingersoll SA, Laval J, Forrest OA, Preininger M, Brown MR, Arafat D, et al. Mature Cystic Fibrosis Airway Neutrophils Suppress T Cell Function: Evidence for a Role of Arginase 1 but Not Programmed Death-Ligand 1. J Immunol (2015) 194(11):5520–8. doi: 10.4049/jimmunol.1500312
154. Lamas B, Vergnaud-Gauduchon J, Goncalves-Mendes N, Perche O, Rossary A, Vasson MP, et al. Altered functions of natural killer cells in response to L-Arginine availability. Cell Immunol (2012) 280(2):182–90. doi: 10.1016/j.cellimm.2012.11.018
155. Furuke K, Burd PR, Horvath-Arcidiacono JA, Hori K, Mostowski H, Bloom ET. Human NK cells express endothelial nitric oxide synthase, and nitric oxide protects them from activation-induced cell death by regulating expression of TNF-alpha. J Immunol (1999) 163(3):1473–80.
156. Xiao L, Eneroth PH, Qureshi GA. Nitric oxide synthase pathway may mediate human natural killer cell cytotoxicity. Scand J Immunol (1995) 42(5):505–11. doi: 10.1111/j.1365-3083.1995.tb03687.x
157. Oberlies J, Watzl C, Giese T, Luckner C, Kropf P, Müller I, et al. Regulation of NK Cell Function by Human Granulocyte Arginase. J Immunol (2009) 182(9):5259–67. doi: 10.4049/jimmunol.0803523
158. Goh CC, Roggerson KM, Lee HC, Golden-Mason L, Rosen HR, Hahn YS. Hepatitis C Virus-Induced Myeloid-Derived Suppressor Cells Suppress NK Cell IFN-γ Production by Altering Cellular Metabolism via Arginase-1. J Immunol (2016) 196(5):2283–92. doi: 10.4049/jimmunol.1501881
159. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer (2017) 5(1):101. doi: 10.1186/s40425-017-0308-4
160. Yang Q, Wei J, Zhong L, Shi M, Zhou P, Zuo S, et al. Cross talk between histone deacetylase 4 and STAT6 in the transcriptional regulation of arginase 1 during mouse dendritic cell differentiation. Mol Cell Biol (2015) 35(1):63–75. doi: 10.1128/mcb.00805-14
161. Fabrik I, Link M, Hartlova A, Dankova V, Rehulka P, Stulik J. Application of SILAC labeling to primary bone marrow-derived dendritic cells reveals extensive GM-CSF-dependent arginine metabolism. J Proteome Res (2014) 13(2):752–62. doi: 10.1021/pr4007798
162. Narita Y, Kitamura H, Wakita D, Sumida K, Masuko K, Terada S, et al. The key role of IL-6-arginase cascade for inducing dendritic cell-dependent CD4(+) T cell dysfunction in tumor-bearing mice. J Immunol (2013) 190(2):812–20. doi: 10.4049/jimmunol.1103797
163. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity (2003) 19(1):59–70. doi: 10.1016/s1074-7613(03)00171-7
164. Bosschaerts T, Guilliams M, Stijlemans B, Morias Y, Engel D, Tacke F, et al. Tip-DC development during parasitic infection is regulated by IL-10 and requires CCL2/CCR2, IFN-gamma and MyD88 signaling. PLoS Pathog (2010) 6(8):e1001045. doi: 10.1371/journal.ppat.1001045
165. Galvão-Filho B, de Castro JT, Figueiredo MM, Rosmaninho CG, Antonelli L, Gazzinelli RT. The emergence of pathogenic TNF/iNOS producing dendritic cells (Tip-DCs) in a malaria model of acute respiratory distress syndrome (ARDS) is dependent on CCR4. Mucosal Immunol (2019) 12(2):312–22. doi: 10.1038/s41385-018-0093-5
166. Banik S, Renner Viveros P, Seeber F, Klotz C, Ignatius R, Aebischer T. Giardia duodenalis arginine deiminase modulates the phenotype and cytokine secretion of human dendritic cells by depletion of arginine and formation of ammonia. Infect Immun (2013) 81(7):2309–17. doi: 10.1128/iai.00004-13
167. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T Cell Receptor CD3ζ Chain Expression byL-Arginine. J Biol Chem (2002) 227(24):21123–9. doi: 10.1074/jbc.M110675200
168. Zea AH, Rodriguez PC, Culotta KS, Hernandez CP, DeSalvo J, Ochoa JB, et al. l-Arginine modulates CD3ζ expression and T cell function in activated human T lymphocytes. Cell Immunol (2004) 232(1):21–31. doi: 10.1016/j.cellimm.2005.01.004
169. Rodriguez PC, Quiceno DG. Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109(4):1568–73. doi: 10.1182/blood-2006-06-031856
170. Rodriguez PC, Hernandez CP, Morrow K, Sierra R, Zabaleta J, Wyczechowska DD, et al. L-Arginine Deprivation Regulates Cyclin D3 mRNA Stability in Human T Cells by Controlling HuR Expression. J Immunol (2010) 185(9):5198–204. doi: 10.4049/jimmunol.1001224
171. Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, et al. L-Arginine Depletion Blunts Antitumor T-cell Responses by Inducing Myeloid-Derived Suppressor Cells. Cancer Res (2015) 75(2):275–83. doi: 10.1158/0008-5472.can-14-1491
172. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell (2016) 167(3):829–42. doi: 10.1016/j.cell.2016.09.031
173. Lange SM, McKell MC, Schmidt SM, Hossfeld AP, Chaturvedi V, Kinder JM, et al. l-Citrulline Metabolism in Mice Augments CD4+ T Cell Proliferation and Cytokine Production In Vitro, and Accumulation in the Mycobacteria-Infected Lung. Front Immunol (2017) 8:1561(1561). doi: 10.3389/fimmu.2017.01561
174. Werner A, Koschke M, Leuchtner N, Luckner-Minden C, Habermeier A, Rupp J, et al. Reconstitution of T Cell Proliferation under Arginine Limitation: Activated Human T Cells Take Up Citrulline via L-Type Amino Acid Transporter 1 and Use It to Regenerate Arginine after Induction of Argininosuccinate Synthase Expression. Front Immunol (2017) 8:864(864). doi: 10.3389/fimmu.2017.00864
175. Bansal V, Rodriguez P, Wu G, Eichler DC, Zabaleta J, Taheri F, et al. Citrulline Can Preserve Proliferation and Prevent the Loss of CD3 ζ Chain Under Conditions of Low Arginine. J Parenteral Enteral Nutr (2004) 28(6):423–30. doi: 10.1177/0148607104028006423
176. Werner A, Amann E, Schnitzius V, Habermeier A, Luckner-Minden C, Leuchtner N, et al. Induced arginine transport via cationic amino acid transporter-1 is necessary for human T-cell proliferation. Eur J Immunol (2016) 46(1):92–103. doi: 10.1002/eji.201546047
177. Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, et al. L-Arginine Consumption by Macrophages Modulates the Expression of CD3ζ Chain in T Lymphocytes. J Immunol (2003) 171(3):1232–9. doi: 10.4049/jimmunol.171.3.1232
178. Bronte V, Serafini P, Mazzoni A, Segal DM. Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol (2003) 24(6):301–5. doi: 10.1016/S1471-4906(03)00132-7
179. Munder M, Schneider H, Luckner C, Giese T, Langhans C-D, Fuentes JM, et al. Suppression of T-cell functions by human granulocyte arginase. Blood (2006) 108(5):1627–34. doi: 10.1182/blood-2006-11-010389
180. Abdissa K, Nerlich A, Beineke A, Ruangkiattikul N, Pawar V, Heise U, et al. Presence of Infected Gr-1(int)CD11b(hi)CD11c(int) Monocytic Myeloid Derived Suppressor Cells Subverts T Cell Response and Is Associated With Impaired Dendritic Cell Function in Mycobacterium avium-Infected Mice. Front Immunol (2018) 9:2317. doi: 10.3389/fimmu.2018.02317
181. de Jonge WJ, Kwikkers KL, te Velde AA, van Deventer SJ, Nolte MA, Mebius RE, et al. Arginine deficiency affects early B cell maturation and lymphoid organ development in transgenic mice. J Clin Invest (2002) 110(10):1539–48. doi: 10.1172/jci16143
182. Métayer LE, Brown RD, Carlebur S, Burke GAA, Brown GC. Mechanisms of cell death induced by arginase and asparaginase in precursor B-cell lymphoblasts. Apoptosis (2019) 24(1-2):145–56. doi: 10.1007/s10495-018-1506-3
183. Gonce SJ, Peck MD, Alexander JW, Miskell PW. Arginine Supplementation and Its Effect on Established Peritonitis in Guinea Pigs. J Parenteral Enteral Nutr (1990) 14(3):237–44. doi: 10.1177/0148607190014003237
184. Norris KA, Schrimpf JE, Flynn JL, Morris SM. Enhancement of macrophage microbicidal activity: supplemental arginine and citrulline augment nitric oxide production in murine peritoneal macrophages and promote intracellular killing of Trypanosoma cruzi. Infect Immun (1995) 63(7):2793–6. doi: 10.1128/IAI.63.7.2793-2796.1995
185. Grasemann H, Grasemann C, Kurtz F, Tietze-Schillings G, Vester U, Ratjen F. Oral L-arginine supplementation in cystic fibrosis patients: a placebo-controlled study. Eur Respir J (2005) 25(1):62–8. doi: 10.1183/09031936.04.00086104
186. Munir K, Muneer MA, Masaoud E, Tiwari A, Mahmud A, Chaudhry RM, et al. Dietary arginine stimulates humoral and cell-mediated immunity in chickens vaccinated and challenged against hydropericardium syndrome virus. Poultry Sci (2009) 88(8):1629–38. doi: 10.3382/ps.2009-00152
187. Grasemann H, Tullis E, Ratjen F. A randomized controlled trial of inhaled l-Arginine in patients with cystic fibrosis. J Cystic Fibrosis (2013) 12(5):468–74. doi: 10.1016/j.jcf.2012.12.008
188. Schön T, Elias D, Moges F, Melese E, Tessema T, Stendahl O, et al. Arginine as an adjuvant to chemotherapy improves clinical outcome in active tuberculosis. Eur Respiratory J (2003) 21(3):483–8. doi: 10.1183/09031936.03.00090702
189. Farazi A, Shafaat O, Sofian M, Kahbazi M. Arginine Adjunctive Therapy in Active Tuberculosis. Tuberculosis Res Treat (2015) 2015:5. doi: 10.1155/2015/205016
190. Schön T, Idh J, Westman A, Elias D, Abate E, Diro E, et al. Effects of a food supplement rich in arginine in patients with smear positive pulmonary tuberculosis–a randomised trial. Tuberculosis (Edinb) (2011) 91(5):370–7. doi: 10.1016/j.tube.2011.06.002
191. Grimble GK. Adverse Gastrointestinal Effects of Arginine and Related Amino Acids. J Nutr (2007) 137(6):1693S–701S. doi: 10.1093/jn/137.6.1693S
192. Allerton TD, Proctor DN, Stephens JM, Dugas TR, Spielmann G, Irving BA. l-Citrulline Supplementation: Impact on Cardiometabolic Health. Nutrients (2018) 10(7). doi: 10.3390/nu10070921
193. Wijnands KAP, Vink H, Briedé JJ, van Faassen EE, Lamers WH, Buurman WA, et al. Citrulline a More Suitable Substrate than Arginine to Restore NO Production and the Microcirculation during Endotoxemia. PLoS One (2012) 7(5):e37439. doi: 10.1371/journal.pone.0037439
194. Wijnands KAP, Meesters DM, Van Barneveld KWY, Visschers RGJ, Briedé JJ, Vandendriessche B, et al. Citrulline Supplementation Improves Organ Perfusion and Arginine Availability under Conditions with Enhanced Arginase Activity. Nutrients (2015) 7(7):5217. doi: 10.3390/nu7075217
195. Kaminaka K, Matsuda J, Nozaki C. Citrulline as a novel adjuvant candidate for vaccines. BioMed Res (2019) 40(1):1–7. doi: 10.2220/biomedres.40.1
196. Comai S, Bertazzo A, Brughera M, Crotti S. Tryptophan in health and disease. Adv Clin Chem (2020) 95:165–218. doi: 10.1016/bs.acc.2019.08.005
197. Palego L, Betti L, Rossi A, Giannaccini G. Tryptophan Biochemistry: Structural, Nutritional, Metabolic, and Medical Aspects in Humans. J Amino Acids (2016) 2016:8952520. doi: 10.1155/2016/8952520
198. Moffett JR, Namboodiri MA. Tryptophan and the immune response. Immunol Cell Biol (2003) 81(4):247–65. doi: 10.1046/j.1440-1711.2003.t01-1-01177.x
199. Cervenka I, Agudelo LZ, Ruas JL. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science (2017) 357(6349). doi: 10.1126/science.aaf9794
200. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol (2013) 34(3):137–43. doi: 10.1016/j.it.2012.10.001
201. Le Floc’h N, Otten W, Merlot E. Tryptophan metabolism, from nutrition to potential therapeutic applications. Amino Acids (2011) 41(5):1195–205. doi: 10.1007/s00726-010-0752-7
202. Van de Velde LA, Gingras S, Pelletier S, Murray PJ. Issues with the Specificity of Immunological Reagents for Murine IDO1. Cell Metab (2016) 23(3):389–90. doi: 10.1016/j.cmet.2016.02.004
203. Metz R, Smith C, DuHadaway JB, Chandler P, Baban B, Merlo LM, et al. IDO2 is critical for IDO1-mediated T-cell regulation and exerts a non-redundant function in inflammation. Int Immunol (2014) 26(7):357–67. doi: 10.1093/intimm/dxt073
204. Baban B, Chandler P, McCool D, Marshall B, Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J Reprod Immunol (2004) 61(2):67–77. doi: 10.1016/j.jri.2003.11.003
205. Suzuki Y, Suda T, Asada K, Miwa S, Suzuki M, Fujie M, et al. Serum indoleamine 2,3-dioxygenase activity predicts prognosis of pulmonary tuberculosis. Clin Vaccine Immunol (2012) 19(3):436–42. doi: 10.1128/cvi.05402-11
206. Suzuki Y, Miwa S, Akamatsu T, Suzuki M, Fujie M, Nakamura Y, et al. Indoleamine 2,3-dioxygenase in the pathogenesis of tuberculous pleurisy. Int J Tuberc Lung Dis (2013) 17(11):1501–6. doi: 10.5588/ijtld.13.0082
207. Almeida AS, Lago PM, Boechat N, Huard RC, Lazzarini LC, Santos AR, et al. Tuberculosis is associated with a down-modulatory lung immune response that impairs Th1-type immunity. J Immunol (2009) 183(1):718–31. doi: 10.4049/jimmunol.0801212
208. Adu-Gyamfi CG, Snyman T, Hoffmann CJ, Martinson NA, Chaisson RE, George JA, et al. Plasma Indoleamine 2, 3-Dioxygenase, a Biomarker for Tuberculosis in Human Immunodeficiency Virus-Infected Patients. Clin Infect Dis (2017) 65(8):1356–8. doi: 10.1093/cid/cix550
209. Blumenthal A, Nagalingam G, Huch JH, Walker L, Guillemin GJ, Smythe GA, et al. M. tuberculosis induces potent activation of IDO-1, but this is not essential for the immunological control of infection. PLoS One (2012) 7(5):e37314. doi: 10.1371/journal.pone.0037314
210. Mehra S, Pahar B, Dutta NK, Conerly CN, Philippi-Falkenstein K, Alvarez X, et al. Transcriptional reprogramming in nonhuman primate (rhesus macaque) tuberculosis granulomas. PLoS One (2010) 5(8):e12266. doi: 10.1371/journal.pone.0012266
211. Mehra S, Alvarez X, Didier PJ, Doyle LA, Blanchard JL, Lackner AA, et al. Granuloma correlates of protection against tuberculosis and mechanisms of immune modulation by Mycobacterium tuberculosis. J Infect Dis (2013) 207(7):1115–27. doi: 10.1093/infdis/jis778
212. Virok DP, Raffai T, Kókai D, Paróczai D, Bogdanov A, Veres G, et al. Indoleamine 2,3-Dioxygenase Activity in Chlamydia muridarum and Chlamydia pneumoniae Infected Mouse Lung Tissues. Front Cell Infect Microbiol (2019) 9:192. doi: 10.3389/fcimb.2019.00192
213. Ganesan S, Roy CR. Host cell depletion of tryptophan by IFNγ-induced Indoleamine 2,3-dioxygenase 1 (IDO1) inhibits lysosomal replication of Coxiella burnetii. PLoS Pathog (2019) 15(8):e1007955. doi: 10.1371/journal.ppat.1007955
214. Consalvi S, Scarpecci C, Biava M, Poce G. Mycobacterial tryptophan biosynthesis: A promising target for tuberculosis drug development? Bioorg Med Chem Lett (2019) 29(23):126731. doi: 10.1016/j.bmcl.2019.126731
215. Gautam US, Foreman TW, Bucsan AN, Veatch AV, Alvarez X, Adekambi T, et al. In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune-mediated control of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (2018) 115(1):E62–e71. doi: 10.1073/pnas.1711373114
216. Moura-Alves P, Fae K, Houthuys E, Dorhoi A, Kreuchwig A, Furkert J, et al. AhR sensing of bacterial pigments regulates antibacterial defence. Nature (2014) 512(7515):387–92. doi: 10.1038/nature13684
217. Memari B, Bouttier M, Dimitrov V, Ouellette M, Behr MA, Fritz JH, et al. Engagement of the Aryl Hydrocarbon Receptor in Mycobacterium tuberculosis-Infected Macrophages Has Pleiotropic Effects on Innate Immune Signaling. J Immunol (2015) 195(9):4479–91. doi: 10.4049/jimmunol.1501141
218. Bozza S, Fallarino F, Pitzurra L, Zelante T, Montagnoli C, Bellocchio S, et al. A crucial role for tryptophan catabolism at the host/Candida albicans interface. J Immunol (2005) 174(5):2910–8. doi: 10.4049/jimmunol.174.5.2910
219. Genestet C, Le Gouellec A, Chaker H, Polack B, Guery B, Toussaint B, et al. Scavenging of reactive oxygen species by tryptophan metabolites helps Pseudomonas aeruginosa escape neutrophil killing. Free Radic Biol Med (2014) 73:400–10. doi: 10.1016/j.freeradbiomed.2014.06.003
220. Loughman JA, Yarbrough ML, Tiemann KM, Hunstad DA. Local Generation of Kynurenines Mediates Inhibition of Neutrophil Chemotaxis by Uropathogenic Escherichia coli. Infect Immun (2016) 84(4):1176–83. doi: 10.1128/iai.01202-15
221. Song H, Park H, Kim YS, Kim KD, Lee HK, Cho DH, et al. L-kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int Immunopharmacol (2011) 11(8):932–8. doi: 10.1016/j.intimp.2011.02.005
222. Kai S, Goto S, Tahara K, Sasaki A, Kawano K, Kitano S. Inhibition of indoleamine 2,3-dioxygenase suppresses NK cell activity and accelerates tumor growth. J Exp Ther Oncol (2003) 3(6):336–45. doi: 10.1111/j.1533-869x.2003.01108.x
223. de Araújo EF, Feriotti C, Galdino NAL, Preite NW, Calich VLG, Loures FV. The IDO-AhR Axis Controls Th17/Treg Immunity in a Pulmonary Model of Fungal Infection. Front Immunol (2017) 8:880. doi: 10.3389/fimmu.2017.00880
224. Mondanelli G, Bianchi R, Pallotta MT, Orabona C, Albini E, Iacono A, et al. A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity (2017) 46(2):233–44. doi: 10.1016/j.immuni.2017.01.005
225. Ufermann CM, Domröse A, Babel T, Tersteegen A, Cengiz SC, Eller SK, et al. Indoleamine 2,3-Dioxygenase Activity During Acute Toxoplasmosis and the Suppressed T Cell Proliferation in Mice. Front Cell Infect Microbiol (2019) 9:184. doi: 10.3389/fcimb.2019.00184
226. Li R, Li H, Sun Q, Liu L, Zhang C, Ren X. Indoleamine 2,3-dioxygenase regulates T cell activity through Vav1/Rac pathway. Mol Immunol (2017) 81:102–7. doi: 10.1016/j.molimm.2016.11.018
227. Sage LK, Fox JM, Mellor AL, Tompkins SM, Tripp RA. Indoleamine 2,3-dioxygenase (IDO) activity during the primary immune response to influenza infection modifies the memory T cell response to influenza challenge. Viral Immunol (2014) 27(3):112–23. doi: 10.1089/vim.2013.0105
228. Li Q, Li L, Liu Y, Fu X, Qiao D, Wang H, et al. Pleural fluid from tuberculous pleurisy inhibits the functions of T cells and the differentiation of Th1 cells via immunosuppressive factors. Cell Mol Immunol (2011) 8(2):172–80. doi: 10.1038/cmi.2010.80
229. Yan Y, Zhang GX, Gran B, Fallarino F, Yu S, Li H, et al. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol (2010) 185(10):5953–61. doi: 10.4049/jimmunol.1001628
230. Platten M, Ho PP, Youssef S, Fontoura P, Garren H, Hur EM, et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science (2005) 310(5749):850–5. doi: 10.1126/science.1117634
231. Bonezi V, Cataneo AHD, Branquinho MSF, Silva MBB, Gonzalez-Dias P, Pereira SS, et al. Flavivirus-Mediating B Cell Differentiation Into Antibody-Secreting Cells in Humans Is Associated With the Activation of the Tryptophan Metabolism. Front Immunol (2020) 11:20. doi: 10.3389/fimmu.2020.00020
232. Harrington L, Srikanth CV, Antony R, Rhee SJ, Mellor AL, Shi HN, et al. Deficiency of indoleamine 2,3-dioxygenase enhances commensal-induced antibody responses and protects against Citrobacter rodentium-induced colitis. Infect Immun (2008) 76(7):3045–53. doi: 10.1128/iai.00193-08
233. Shinde R, Shimoda M, Chaudhary K, Liu H, Mohamed E, Bradley J, et al. B Cell-Intrinsic IDO1 Regulates Humoral Immunity to T Cell-Independent Antigens. J Immunol (2015) 195(5):2374–82. doi: 10.4049/jimmunol.1402854
234. Tsai CY, Sakakibara S, Yasui T, Minamitani T, Okuzaki D, Kikutani H. Bystander inhibition of humoral immune responses by Epstein-Barr virus LMP1. Int Immunol (2018) 30(12):579–90. doi: 10.1093/intimm/dxy053
235. El-Zaatari M, Bass AJ, Bowlby R, Zhang M, Syu LJ, Yang Y, et al. Indoleamine 2,3-Dioxygenase 1, Increased in Human Gastric Pre-Neoplasia, Promotes Inflammation and Metaplasia in Mice and Is Associated With Type II Hypersensitivity/Autoimmunity. Gastroenterology (2018) 154(1):140–53.e17. doi: 10.1053/j.gastro.2017.09.002
236. Bilgili SG, Karadag AS, Calka O, Altun F. Isoniazid-induced pellagra. Cutan Ocul Toxicol (2011) 30(4):317–9. doi: 10.3109/15569527.2011.574303
237. Kipsang JK, Choge JK, Marinda PA, Khayeka-Wandabwa C. Pellagra in isoniazid preventive and antiretroviral therapy. IDCases (2019) 17:e00550. doi: 10.1016/j.idcr.2019.e00550
238. Machado M, Azeredo R, Domingues A, Fernandez-Boo S, Dias J, Conceição LEC, et al. Dietary tryptophan deficiency and its supplementation compromises inflammatory mechanisms and disease resistance in a teleost fish. Sci Rep (2019) 9(1):7689. doi: 10.1038/s41598-019-44205-3
239. Fischer DD, Kandasamy S, Paim FC, Langel SN, Alhamo MA, Shao L, et al. Protein Malnutrition Alters Tryptophan and Angiotensin-Converting Enzyme 2 Homeostasis and Adaptive Immune Responses in Human Rotavirus-Infected Gnotobiotic Pigs with Human Infant Fecal Microbiota Transplant. Clin Vaccine Immunol (2017) 24(8). doi: 10.1128/cvi.00172-17
Keywords: tuberculosis, mycobacteria, L-arginine, L-tryptophan, amino acid
Citation: Crowther RR and Qualls JE (2021) Metabolic Regulation of Immune Responses to Mycobacterium tuberculosis: A Spotlight on L-Arginine and L-Tryptophan Metabolism. Front. Immunol. 11:628432. doi: 10.3389/fimmu.2020.628432
Received: 12 November 2020; Accepted: 30 December 2020;
Published: 09 February 2021.
Edited by:
Maziar Divangahi, McGill University, CanadaReviewed by:
Frederick J. Sheedy, Trinity College Dublin, IrelandCopyright © 2021 Crowther and Qualls. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph E. Qualls, am9zZXBoLnF1YWxsc0BjY2htYy5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.