 Giulia Fumagalli
Giulia Fumagalli Laura Monza
Laura Monza Guido Cavaletti
Guido Cavaletti Roberta Rigolio
Roberta Rigolio Cristina Meregalli
Cristina Meregalli- 1Experimental Neurology Unit, School of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy
- 2NeuroMI (Milan Center for Neuroscience), University of Milano-Bicocca, Monza, Italy
Peripheral neuropathies are characterized by nerves damage and axonal loss, and they could be classified in hereditary or acquired forms. Acquired peripheral neuropathies are associated with several causes, including toxic agent exposure, among which the antineoplastic compounds are responsible for the so called Chemotherapy-Induced Peripheral Neuropathy (CIPN). Several clinical features are related to the use of anticancer drugs which exert their action by affecting different mechanisms and structures of the peripheral nervous system: the axons (axonopathy) or the dorsal root ganglia (DRG) neurons cell body (neuronopathy/ganglionopathy). In addition, antineoplastic treatments may affect the blood brain barrier integrity, leading to cognitive impairment that may be severe and long-lasting. CIPN may affect patient quality of life leading to modification or discontinuation of the anticancer therapy. Although the mechanisms of the damage are not completely understood, several hypotheses have been proposed, among which neuroinflammation is now emerging to be relevant in CIPN pathophysiology. In this review, we consider different aspects of neuro-immune interactions in several CIPN preclinical studies which suggest a critical connection between chemotherapeutic agents and neurotoxicity. The features of the neuroinflammatory processes may be different depending on the type of drug (platinum derivatives, taxanes, vinca alkaloids and proteasome inhibitors). In particular, recent studies have demonstrated an involvement of the immune response (both innate and adaptive) and the stimulation and secretion of mediators (cytokines and chemokines) that may be responsible for the painful symptoms, whereas glial cells such as satellite and Schwann cells might contribute to the maintenance of the neuroinflammatory process in DRG and axons respectively. Moreover, neuroinflammatory components have also been shown in the spinal cord with microglia and astrocytes playing an important role in CIPN development. Taking together, better understanding of these aspects would permit the development of possible strategies in order to improve the management of CIPN.
Introduction
Chemotherapy-induced peripheral neurotoxicity (CIPN) may occur in patients undergoing antineoplastic therapy, frequently being the most severe side-effect. CIPN is characterized by severe and long lasting symptoms that might affect daily activities and impact on patient quality of life. This clinical situation leads to drug schedule modification, or even withdrawal, thus potentially affecting patient’s survival and clinical outcome (1–4). The chemotherapy drugs act on different structures of the peripheral nervous system (PNS), due to both the reduced blood-nervous tissue barrier efficacy and the presence of fenestrated capillaries in dorsal root ganglia (DRG), targeting the axons, inducing a length-dependent axonopathy, or DRG neurons, leading to a neuronopathy (5, 6). Moreover, a large body of knowledge suggests the direct neurotoxic effect of antineoplastic agents to the central nervous system (CNS) (7).
CIPN is mainly a sensory and length-dependent neuropathy which progresses from the distal to the proximal regions (6): patients manifest paresthesia and dysesthesia, which may evolve into numbness, sensory loss, tingling, pins and needles sensation, with a stocking-and-glove distribution. Hyperalgesia or allodynia in limb extremities (neuropathic pain) may also occur (8). Rarely, a motor or an autonomic impairment is present (1, 4, 9). In most patients, CIPN becomes a chronic condition and the symptoms may persist or even progress for months after the end of the therapy, a phenomenon known as “coasting” (10).
Numerous factors are related to CIPN establishment: the type of employed chemotherapeutic agent, the administered dose, the period and schedule of treatment can contribute to the symptoms incidence and severity (11).
Although the antineoplastic mechanisms of action of neurotoxic drugs are well established, the reasons for axonal and ganglion damage remain unclear. Several neurotoxic mechanisms have been proposed including mitochondrial damage, impairment of axonal transport, oxidative stress, and involvement of drug transporters (3, 4, 12).
Recent findings suggest that neuroinflammation may have a role in CIPN. In fact, besides its action on dividing immune cells, chemotherapy treatment can lead to modulation of several immune system elements, from the cytokine expression to immune cell intracellular pathways, thus leading to neuroinflammation development and sensory nervous system sensitization (13–16).
In this context, an important role of glial cells has also been reported: satellite glial cells (SGCs) in DRG and Schwann cells in the axons are able to change their phenotype and secrete mediators, which cause neuronal excitability leading to pain hypersensitivity. The release of pro-inflammatory cytokines and chemokines may also recruit the monocytes population participating in the inflammatory response. Glial cells express the cytokine receptors, but they can also contribute to neuroinflammation with the same cytokine release, in a sort of self-sustaining system.
Moreover, the involvement of microglia and astrocytes in the neuropathic pain establishment has been demonstrated also in the central nervous system.
The features of the neuroinflammatory process are different and depend on the type of the anticancer drug (platinum derivatives, taxanes, vinca alkaloids, and proteasome inhibitors) (11, 14, 17).
Therefore, the aim of this work is to review the aspects of neuroinflammation in different classes of antineoplastic drugs. To follow this purpose, a review in the PubMed database has been carried out.
For each class of antineoplastic agents the searching entry was: ((((chemotherapy-induced-neuropathy) OR polyneuropathy) OR neurotoxicity) OR neuropathy) AND ((((((((((((((((((neuroinflammation) OR immune-cell-activation) OR immune-mediated-process) OR immune-system) OR immunomodulation) OR innate-immune-response) OR adaptive-immune-response) OR central-glial-activation) OR cytokine) OR chemokine) OR proinflammatory-mediators) OR neuroimmune-interaction) OR inflammasomes) OR glial-cell) OR macrophages) OR immune-cells) OR lymphocytes) OR neutrophils). The selected filters were used: English language and papers dated between January 2010 and May 2020. The resulting abstracts were carefully reviewed and relevant full-text manuscripts were selected. Moreover, the reference list of selected articles were searched for further relevant papers.
Platinum Derivatives
Cisplatin (CDDP), carboplatin (CBP) and oxaliplatin (OHP) are platinum-based antineoplastic agents used as main or adjuvant treatment for solid tumors such as germ cells, lung, colorectal, gastric, breast and head and neck cancers. They are alkylating agents thus exerting their activity forming DNA-platinum adducts (12).
Even if the incidence and severity of neurotoxicity may be different among platinum compounds, it is one of their most common dose-limiting side effects (18). In fact, prolonged CDDP exposure results in the onset of a pure sensory neuropathy with a stocking-and-glove distribution characterized by the dysfunction of fine sensory-motor coordination, numbness and paresthesia. OHP infusion may result in both chronic and acute neurotoxicity. The acute neurotoxicity occurs in about 90% patients in the next hours after OHP administration and it is characterized by dysesthesias and paresthesias exacerbated by cold exposure. The OHP chronic neurotoxicity shares the same symptoms of CDDP-associated CIPN (18, 19).
Despite during the last years several neurotoxicity mechanisms have been investigated, we will point out only the evidence for different immune system elements involvement in CIPN such as leukocytes recruitment, cytokine production and signal transduction pathways.
At the early stage of OHP treatment, pain hypersensitivity has been described together with systemic immunological response characterized by an increase in circulating CD4+ and CD8+ lymphocytes, an increase in IL-4+ splenocytes and a decrease in regulatory T-cell (T-reg) in the inguinal lymph nodes. Except for the changes in the lymph node T-cell count, all the reported altered features returned to control values at the peak stage of pain sensitization. However, systemic depletion of T-reg cells did not exacerbate mechanical allodynia in OHP-treated mice suggesting they may not contribute to the development of neuropathic pain in OHP-induced CIPN (OIPN). Moreover, at peak stage no alteration in the cytokine serum levels was reported (20). In contrast, other authors demonstrated an increased serum or plasma levels of pro-inflammatory cytokines such as TNF-α, IL-1β and IL-6 following OHP and CDDP treatment (21–28). While Li and colleagues reported that in OIPN, T-reg reduction contributed to the onset of CIPN through the increase in the pro-inflammatory response (29), Wan and coworkers observed an increase of T-reg and a decrease of Th17 levels in OHP-treated animals (24).
Besides circulating and lymph node T-cell ratio changes during CIPN course, Laumet and colleagues demonstrated that the resolution of CDDP-induced neuropathic pain and intraepidermal nerve fibers (IENF) density reduction depended on an active endogenous process that is mediated by CD8+ T-cells. However, the neuroimmune mechanisms responsible for CD8+ T-cells mediated CIPN symptoms resolution still need to be elucidated (30). T-cells were also implicated in the prevention of CDDP-induced pain-like behavior and mitochondrial dysfunction in DRG neurons, which was exerted by histone deacetylase 6 inhibitors (31).
Besides revealing direct effects on the immune cells and cytokines production, OHP and CDDP may affect inflammatory compartment modulating gene expression. In particular, it has been reported that CDDP may affect the expression of genes implicated in neuroinflammatory processes such as TNF-α and cytokine-cytokine interactions pathways in cultured rat sensory neuron-like cells (32). In agreement with these findings, the transcriptome analysis of lumbar DRG of CDDP-treated mice revealed changes in the expression of genes involved in both neuronal damage and inflammatory processes (33). Unexpectedly, OHP treatment did not induce any alteration in the transcription of inflammation related genes suggesting that in OIPN neuronal damage might precede the inflammatory process establishment, which in turn might be responsible for the development of more chronic neuropathy symptoms (33).
Studies explored the cellular signaling pathways involved in cytokine release through the activation of the Toll Like receptors (TLRs), the innate immune system key mediators (34). TLRs are transmembrane and intracytoplasmic proteins being normally implicated in the detection of pathogens. Once activated, they induce a downstream activation of several molecules such as mitogen-activated protein kinase (MAPK), phosphoinositide 3 kinase (PI3K), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and the activation transcription factor 3 (ATF3) protein. ATF3 not only regulates intracellular cascades initiated by TLRs activation, but it is also considered a marker for nerve injury. In particular, CDDP-induced tactile allodynia was associated with the upregulation in ATF3 expression in DRG neurons of CDDP-treated mice as well as in DRG neurons and sciatic nerves of OHP-treated animals (35–38). Since ATF3 plays an important role downstream TLRs activation, further studies deepened the implication of this signaling pathway in the CIPN pathogenesis. These studies on the role of TLR3 and 4 and their adapter proteins (MyD88 and TRIF) showed that CDDP-induced mechanical allodynia was reduced in trl3-/- and tlr4-/- mice compared to WT animals and was abolished in animals that completely lack TLR pathways (Myd88/Triflps2 mice). Taken together, these results suggest that MyD88 and TRIF signaling cascades triggered by the activation of TLR4 and TLR3, participated in the onset of neuropathic pain induced by CDDP (39, 40).
Moreover, TLRs-pathways seem to be also involved in OIPN. In fact, OHP determined a neuroinflammatory state in DRG neurons by the increase in matrix metalloproteinase-9 (MMP-9) levels due to TLR4/PI3K/akt signaling cascade activated by high-mobility group box 1 (HMGB-1), which was released by neurons and macrophages. Furthermore, the MMP-9-dependent inflammatory process in the DRG was also associated with the activation of microglia (increase of Iba-1 immunoreactive cells) in the spinal cord indicating central sensitization (41). In contrast, other authors suggested that HMGB-1 from non-macrophage cells played a key role in the onset of OIPN, probably through the activation of TLR4, RAGE and CXCL12/CXCR4 signaling (42). MyD88 signaling pathway results in the activation of MAPK, PI3K and NF-κB, whereas TRIF dependent signaling pathway results in the production of type I interferon and a delayed activation of NF-κB. Both pathways induced an increased expression and release of pro-inflammatory cytokines and chemokines in both the central and peripheral nervous system (17, 40).
Cytokines are small molecules involved in the immune response, which are released not only by immune cells, but also by glial and neurons. They can directly or indirectly act on primary afferent fibers, DRG and spinal dorsal horn neurons leading to pain sensitization (43). In particular, a significant increase in IL-1β, IL-6, and TNF-α was reported in DRG neurons after OHP or CDDP treatment (38, 44–47). This increased expression/release of cytokines might be also associated with changes in chemokine expression.
Chemokines are a family of chemoattractant cytokines that play an important role in the activation and infiltration of macrophages and glial cells in the onset of neuropathic pain. Several chemokine contributions in platinum compounds-dependent neurotoxicity have been shown. In fact, OHP induced an increased expression of CCL2 and its receptor (CCR2) at early time points in DRG neurons, indicating that these small proteins were involved in the onset of pain caused by antineoplastic agents (48). In addition, the NF-κB p65-mediated upregulation of CX3CL1 induced an increased neuronal excitability and contributed to the development of chronic pain after repeated OHP injections (49). Furthermore, the increased expression of CXCL12, induced by IL-1β and TNF-α -mediated activation of transcription-3 (STAT3), played a critical role in the pathogenesis of OIPN (50). On the other hand, other authors reported only a significant reduction of CCL4 in DRG neurons without any additional alteration in the profile expression of cytokines and chemokines (20).
In particular, IL-8 was identified to have a relevant role in OIPN. In fact, Brandolini and colleagues reported glial activation (increase in GFAP and Iba-1 expression) and an increase in IL-8 expression associated with the activation of different intracellular signaling pathways (p-FAK, PI3K/p-cortactin, p-STAT3, COX2 and ERK1/2). These effects were attenuated by the coadministration of DF2726A, a selective inhibitor of IL-8 receptors, indicating that the increased expression of IL-8 in DRG neurons triggered a pro-inflammatory response leading to the activation of pathways implicated in microtubule stabilization, terminal axonal arborization, synaptic plasticity and cellular damage (51). Moreover, these data confirmed previous observations which suggested COX2, PI3K/Akt2, PI3K/mTOR and pERK signaling involvement in the onset of OHP-induced pain sensitization (45, 52, 53).
Besides these findings, the increased levels of pro-inflammatory cytokines, CCR2, COX2, p-ERK and p38 MAPK were associated to the decrease in protein and mRNA levels of calcineurin (CaN) and to an increase of Nuclear Factor of Activated T cells (NFAT). These findings indicated the role of the CaN/NFAT pathway in the onset of OIPN (46).
Moreover, other and specific intracellular pathways were suggested to be implicated in platinum-induced CIPN onset, such as p38 MAPK and Sphingosine 1-phosphate (S1P) receptor pathway. The incubation of SGC culture with a p38 MAPK specific inhibitor induced a decrease in PGE2 concentration in the medium of CDDP co-treated cells. These data suggested that the CDDP-driven p38 MAPK phosphorylation led to an increase of PGE2 release by glial cells that in turn stimulated the activation of PGE receptors expressed on the ganglia neurons cell membrane. Therefore, this process could modulate the neuronal activity through the activation of second messenger cascades resulting in TRPV1 activation and thus sensitization of sensory neurons (54). This hypothesis was further supported by the results of Kuai and colleagues reporting increased levels of p-p38 together with an increased expression of TRPV1 in DRG, spinal cord, trigeminal ganglion and foot skin of CDDP-treated rats showing CIPN related symptoms (28).
On the other hand, S1Ps are lipid signaling molecules that play a crucial role in different cellular processes by interacting with one of their five receptor subtypes (S1PR1, S1PR2, S1PR3, S1PR4, and S1PR5). It was recently observed that the selective S1PR2 inhibition reduced CDDP-induced tactile allodynia and the associated activation of SGC in DRG neurons probably through the activation of stress-response proteins such as ATF3 and heme oxygenase-1 (HO-1) (55). In addition, an analogue of PGE1 reduced OHP-induced mechanical allodynia starting from early treatment stage (56).
In DRG neurons and peripheral nerves of OHP-treated animals, no signs of infiltrating inflammatory cells were reported claiming for the involvement of resident immune/glial cells inflammatory response with no roles for T-reg cells subset in OHP-induced mechanical allodynia (20, 36, 42). On the other hand, a huge macrophages infiltration was observed in DRG neurons of other OIPN models (29, 41). In particular, Li and colleagues reported macrophages infiltration and an increased release of IL-1β in lumbar DRG of OHP-treated mice, which was prevented by the co-administration with Bee Venom derived phospholipase A2 (bvPLA2). These preventive effects were reversed by the depletion of T-reg cells indicating that they were required for the anti-inflammatory response (29).
Even if DRG neurons represent the main targets of platinum neurotoxicity, SGC and Schwann cells in the PNS have been also described as relevant costars into neuroinflammation onset.
SGCs wrap the cell body of sensory neurons in the ganglia of the PNS. Following peripheral nerve injury or inflammation, SGCs are activated and release molecules that play a key role in the onset of pain conditions. In recent years, alterations in SGCs morphology and function following platinum treatment as well as their involvement in the onset of OHP-related neuropathy were evaluated. More in detail, an increased expression of GFAP-positive SGCs activated cells in DRG neurons of OHP-treated mice was reported and it was associated with an increase in gap junction-mediated coupling. Moreover, the administration of a gap junction blocker abolished the tactile allodynia and reduced the coupling percentage between SGC in OHP-treated mice. These data indicate that the increase of SGCs coupling contributed to the onset of mechanical allodynia and it is part of SGCs activation process (57). In support to these data, it has been recently reported that the incubation of SGCs primary cultures with OHP induced SGCs activation (increase of GFAP-positive cells), SGCs morphological changes, the increased expression of one of the main components of gap junctions (connexin Cx43) and pro-inflammatory cytokines release. Moreover, the incubation of DRG neurons primary cultures with the medium from OHP-treated SGCs culture enhanced neurons hyperexcitability demonstrating that these effects were due to the pro-inflammatory cytokines released by the OHP-activated SGCs (58). The activation of SGCs (increase in GFAP expression) after OHP administration was also confirmed by several other authors (36, 38, 59). In addition, it was reported that in CDDP-treated SGCs primary cultures the activation of p38 MAPK cascade enhanced the release of PGE2. Interestingly this event was attenuated by the application of drugs with glial modulatory activity, such as Ibudilast and SKF86002 (54).
With regard to peripheral nerves, the histopathological analysis of sciatic nerves of CDDP-treated rats revealed tissue damage and apoptotic alterations with axonal degeneration and myelinated fibers loss together with an increased expression of TNF-α (60). Moreover, morphological and functional alterations of Schwann cells were reported in CDDP-treated mice (61) and in vitro after OHP or CDDP incubation (37, 62).
Inflammatory processes were evaluated also at the level of skin hind paws. The reduction of IENF density associated with OHP regimen was first described in 2011 in the skin biopsies of hind paws, and it is often associated with the presence of hypersensitivity to a mechanical stimulus. Both effects were prevented by the treatment with minocycline (a drug with anti-inflammatory properties). These findings suggest that the epidermal denervation played an important role in the OHP-related neuropathic pain persistence and that it was caused by the release of pro-inflammatory cytokines induced by OHP (63). Since the IENF loss corresponded to an increase in IL-8 levels, the progressive accumulation of IL-8 in the epidermidis and the activation of the downstream pathways might be responsible for the OHP-associated epidermal denervation (51). Moreover, an increase in mast cells was reported in the dermis and subcutaneous tissue of plantar skin of OHP-treated mice (64, 65). In the authors’ opinion, the migration of mast cells in the skin may be caused by the increased expression of mast cell migration-related factors in keratinocytes, which were triggered by the Substance P (SP) released from capsaicin-sensitive sensory neurons (65). In CDDP-treated rats the decrease of IENF density and neuropathic pain symptoms were associated with the increased expression of TNF-α, IL-1β and PGE2 in paw homogenate (28).
The relationship between platinum compounds and the CNS is multi-faceted. Little information is specifically available on CDDP-induced neuroinflammation in the spinal cord whereas the most literature refers to OHP. Regarding CDDP, Park and colleagues reported no increase of Iba-1 and GFAP positive glial cells in the lumbar spinal cord of CDDP-treated animals indicating no microglial and astrocyte activation (35). In contrast to these findings, spinal microglia (increase of Iba-1 immunoreactive cells), but not astrocytes, activation associated with an increased expression of pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α, and chemokine CCL3 were also reported (66, 67). An increased expression of pro-inflammatory cytokines after CDDP treatment was confirmed also by other works (68).
With regard to OHP, no T-cell infiltration was reported in the dorsal and ventral horn of the spinal cord of OHP-treated animals (20, 69). The main interest was focused on debating about glial cells activation, inflammatory response and its cellular pathways. Therefore, it was shown that both astrocytes and microglia were activated in the early phase of OHP-related tactile and thermal hypersensitivity, whereas only astrocytes persisted in a reactive status in the late phase of OIPN (36, 37, 59, 70, 71). These data remain at least in part controversial, since some authors did not observe microglia activation at any time or demonstrated the lack of microglia activation in the late phase of OHP-induced pain sensitization (72, 73). On the other hand, other groups agreed with Di Cesare Mannelli and colleagues reporting the activation of both astrocytes and microglia (increased expression of GFAP and Iba-1 immunoreactive cells) in association with an increased release of pro-inflammatory cytokines (TNF-α and IL-1β) (38, 74). In contrast with all previous findings, Makker and colleagues did not observe neither glia (astrocytes and Iba-1 positive microglia) activation following OHP treatment, nor changes in cytokines and chemokines expression profile (20, 75). Interestingly, in the last case they showed a reduction in P2ry12 positive microglia, suggesting that homeostatic microglia reduction could result in pathological processes leading to pain hypersensitivity (20). Several studies are focused on the astrocytes activation role and the subsequent increase in pro-inflammatory cytokines (76, 77). In the spinal cord of OHP-treated rats the increase of GFAP-positive astrocytes was associated with neuropathic pain onset (72). In fact, both astrocyte activation and mechanical allodynia were reverted by the treatment with minocycline, which reverted the inflammatory response induced by activated astrocytes. Moreover, OHP-induced astrocyte activation led also to an increased expression of gap junction protein Cx43 (78). Carbenoxolone is a drug with a gap junction decoupler activity. The inhibition of astrocyte activation with carbenoxolone administration abolished the formation of astrocyte-astrocyte gap junction connections as well as the onset of allodynia in response to a mechanical stimulus. The inhibition of astrocyte-astrocyte gap junction connection with carbenoxolone pre-treatment abolished astrocytes activation as well as the onset of allodynia in response to a mechanical stimulus. In contrast, the authors did not report any protective effects when carbenoxolone was given in rats with established OIPN, suggesting that astrocyte gap junctions play a key role in the OIPN establishment, but not in its maintenance (79). Furthermore, other authors deeply elucidate the mechanism leading to pain sensitization (69, 80, 81). They observed that the astrocyte hyperactivation was accompanied by an increased release of TNF-α and IL-1β and a reduction of IL-10 and IL-4, which was not related to T-cell infiltration. Moreover, the activation of A3 adenosine receptor (A3AR) induced by the administration of a selective agonist, IB-MECA, blocked the onset of mechanical allodynia. This effect was mediated through the modulation of inflammatory processes such as inhibition of astrocyte activation and the modulation of pro-inflammatory as well as anti-inflammatory cytokines release (69). In this context, the onset of OHP-driven neuropathic pain was caused by the dysregulation of the extracellular adenosine signaling at the A3AR level. In fact, in a subsequent study, it was demonstrated that the dysregulation of this pathway depended on the increased expression of adenosine kinase (ADK) in astrocytes together with the increased expression of NLRP3 (NOD-, LRR- and Pyrin domain-containing protein 3). NLRP3 is an intracellular signaling molecule activated by danger signals to constitute the inflammasome. This resulted in the formation of the active form of IL-1β which in turn reduced the expression of anti-inflammatory molecules (81). Lastly, they excluded the role of GSK3β pathway in the onset of OIPN (81). Since GSK3β seems to be implicated in PTX-related CIPN, these results may indicate that different antineoplastic agents could activate different mechanisms leading to neuropathy (82, 83).
The increased level of pro-inflammatory cytokines in the spinal dorsal horn of OHP-treated rats was also correlated with the activation of the CaN/NFAT signaling (46). In addition to these findings, Huang and colleagues reported that among the different cytokines and chemokines investigated in the spinal cord [INF-γ, TNF-α, IL-1β, IL-6, monocyte chemoattractant protein-1 (MCP-1) and CX3CL1], NF-κB p65-mediated epigenetic upregulation of CX3CL1 played a key role in the central sensitization and the onset of acute pain like behavior following OHP administration (84).
Besides astrocytes activation (increase in GFAP-positive cells and protein levels) and the increased expression/release of pro-inflammatory cytokines, Wang and colleagues observed an increased expression of chemokines such as MCP-1 and monocyte inflammatory protein-1 (MIP-1α). This neuroinflammatory response, together with thermal and mechanical hypersensitivity, was repressed by the administration of melatonin. In the authors’ opinion, this effect could be mediated by the binding of melatonin to TLR on the surface of astrocytes. However, the involvement of spinal TLRs in the onset of OHP-associated neuropathic pain has not been elucidated yet and it should be considered for future studies (85).
Despite literature data indicate some controversial results, which may be attributed to the different animal models used, taken together all these findings suggest that OHP-induced neuroinflammation retrieved in the dorsal horn of the spinal cord (DHSC) (e.g. glial cell activation, increased release of pro-inflammatory cytokines and chemokines, reduced release of anti-inflammatory cytokines) might play a critical role in the onset of OHP-related neuropathic pain. In particular, it is undoubted that glial cells activation plays a pivotal role in the onset and maintenance of OIPN. However, literature data indicate some discrepancies since different effects have been observed at different time points on glial cells activity (increase in GFAP and/or Iba-1 positive cells density, increased expression and/or morphological changes) (86). Moreover, it is important to underline that glial cells are not only implicated in the pathological mechanism leading to CIPN, but also in the neuroprotective mechanisms. In fact, Di Cesare Mannelli and colleagues demonstrated that the modulation of astrocytes activity due to the nicotinic receptors α7 subtype (α7 nAChR) agonist effectively reduced OIPN (70). This effect was dependent on the TGF-β1 increase and glutamine synthetase release (87).
Interestingly, platinum compounds related neuroinflammatory events have been detected also in the upper CNS organs. Glial activation has been reported in some brain areas involved in pain signaling (36, 37, 70). Moreover, the increased levels of the pro-inflammatory cytokines TNF-α, IL-1, and IL-6 were reported in the cerebral cortex or whole brain homogenate of CDDP-treated rats (23, 88, 89). As suggested by the authors, this cytokines increase might have exacerbated the oxidative damage induced by CDDP through glutamate excitotoxicity or the upregulation of NF-κB expression and subsequent overproduction of cytokines. In fact, the increased transcription and translation of NF-κB gene was found to be associated with a reduction in the nuclear factor erythroid 2-related factor 2 (Nrf2) and HO-1 genes transcription and translation in the brain cortex and hippocampus of CDDP-treated animals (89, 90). A strong TNF-α and IL-1β increase associated with IL-10 decrease was also observed in the hippocampus of CDDP-treated rats with cognitive impairment (90, 91). Moreover, an increase of pro-inflammatory cytokines levels was reported in the midbrain periaqueductal gray of OHP-treated rats (92).
The Figure 1 shows the involvement of different inflammatory actors in the onset of platinum-induced CIPN according to the results of the most consistent studies.
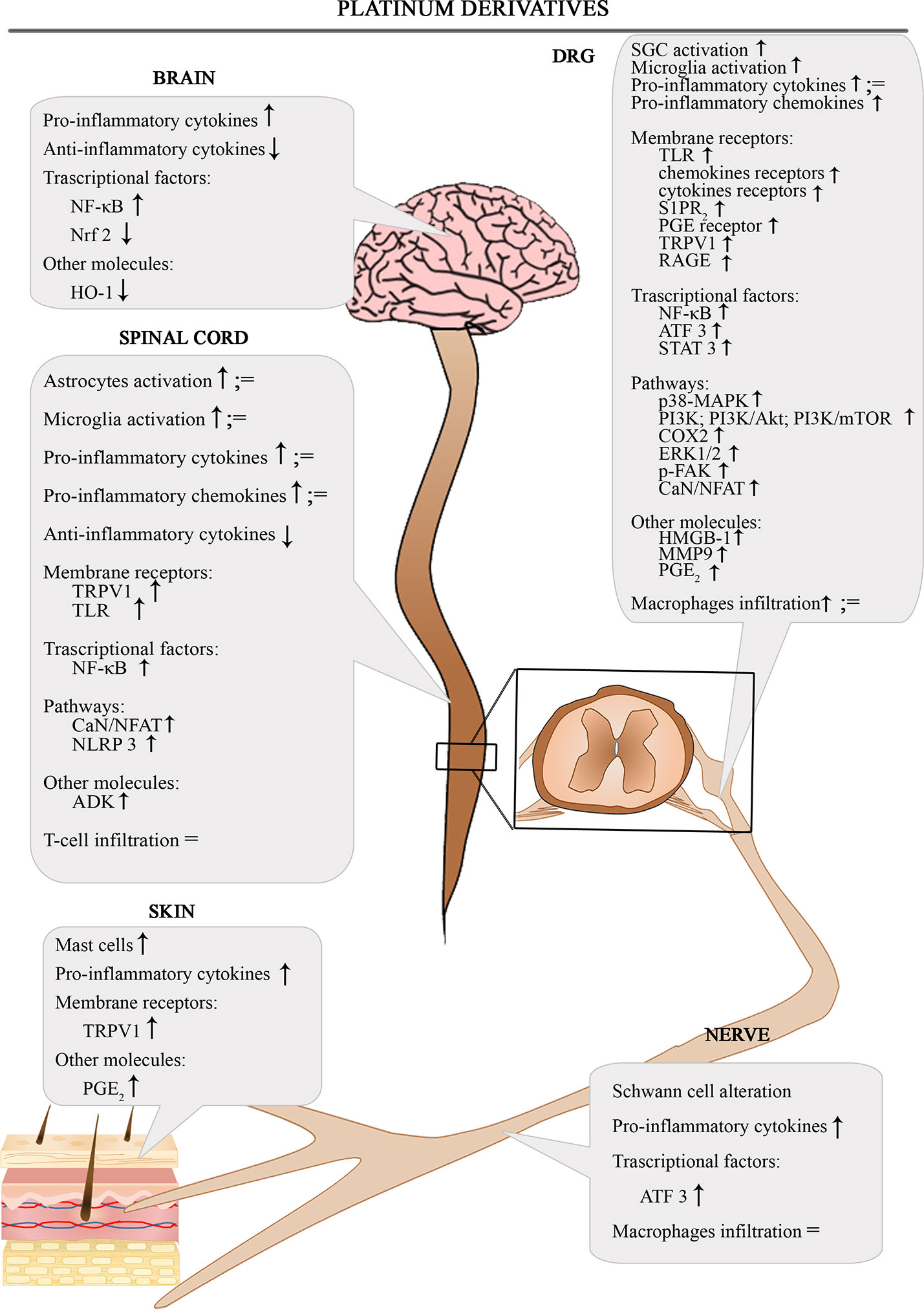
Figure 1 Schematic depiction of different inflammatory actors involved in the onset of platinum-induced CIPN.
Taxanes
Paclitaxel (PTX) and Docetaxel (DCT) are the two main members of the taxane chemotherapy drug family, a class of diterpenes affecting the microtubule dynamics. Normally, microtubules undergo a process of dynamic instability, which is characterized by depolymerization and repolymerization phenomena. Taxanes exert their toxic activity binding the heterodimer β-tubulin, stabilizing the microtubules thus leading to the arrest of the cell cycle (93, 94).
PTX is an effective drug principally used as the first line choice for the treatment of breast, ovarian and lung cancer. Despite PTX is slightly less clinically effective than DCT, it is more frequently associated with CIPN (95).
The administration of PTX results in distal axonal degeneration with nerve fiber loss which results in a sensory axonal neuropathy and it is often characterized by neuropathic pain (6).
Several preclinical studies have been conducted in order to investigate the emerging concept of neuroinflammation involvement in the onset of PTX-induced CIPN (PIPN).
Most preclinical studies deal with the role of pro- and anti- inflammatory cytokines in PTX-induced pain behaviors which are mainly assessed by mechanical or thermal thresholds tests.
At present, few evidence was published about the leukocyte contribution to taxane/PTX-induced pain. As reported previously for OIPN, PTX induced a temporary increase in CD4+ and CD8+ lymphocytes only at early treatment stages (20). The specific contribution of CD8+ T-cells in the resolution of mechanical allodynia was demonstrated elsewhere together with their mandatory role in the up-regulation of IL-10 receptors in the DRG (96).
Pro-inflammatory cytokines and chemokines IL-1α, IL-1β, IL-6, TNF-α, INF-γ, and CCL2 were significantly increased in plasma of PTX-treated rats. The relevance of some of them (TNF-α, IL-1α, IL-1β, CCL2) in sustaining hypersensitivity or pain was demonstrated by blocking their signaling using an inhibitor or a receptor antagonist (97).
Moreover, in PTX-treated mice, IL-20 serum level was increased and this phenomenon paired with IL-20 increase in serum of cancer patients undergoing PTX therapy. IL-20 plays a pivotal role acting as inflammatory mediator in activation of monocytes and astrocytes, showing a correlation with a severe sensory pain in these patients. Targeting IL-20 signaling, using anti-IL-20 monoclonal antibody, before PTX treatment, attenuated mechanical allodynia, prevented thermal hypoesthesia and also peripheral nerve damage in PTX-treated animals. Moreover, the systemic blockade of IL-20 significantly decreased systemic inflammation suppressing the pro-inflammatory cascade activation (TNF-α, IL-1β, and MCP-1) and macrophage recruitment in DRG, correlating the role of neuroimmune system to the pain behavior. The importance of IL-20 was further confirmed by the use of IL-20R1-/- mice, which were protected from PIPN and peripheral nerve degeneration (98).
Additionally, the inhibition of the IL-8 receptors (CXCR1 and CXCR2) in PTX-treated rats using a non-competitive allosteric inhibitor (reparixin) administered systemically induced a consistent antinociceptive effect (99).
Levels of pro-inflammatory elements, such as IL-6, IL-1β, TNF-α, and chemokines like CCL2, CX3CR1 were found to be modulated also in the DRG or sciatic nerves of treated animals (100–108).
Manjavachi and colleagues demonstrated an increase of the chemokine CXCL1 in both spinal cord and DRG, whereas the increase in sciatic nerve was less evident at early time points (109). In particular, Zhang and colleagues demonstrated that the blockade of MCP-1/CCR2 signaling in the DRG of PTX-treated animals attenuated mechanical hypersensitivity as well as the IENF loss (101). This observation demonstrates that mechanical hypersensitivity and IENF density reduction were dependent on the activation of MCP-1/CCR2 pathway in the DRG (101). In addition, also the treatment with minocycline prevented IENF loss as well as the development of mechanical hypersensitivity (110, 111) as previously reported for OHP. Moreover, IL-20 expression was increased in footpaw of PTX-treated animals and its inhibition prevented IENF loss caused by PTX, suggesting the role of the neuroinflammatory response in CIPN establishment also at IENF level (98).
PTX also induced the recruitment, activation and accumulation of macrophages with a pro-inflammatory M1 phenotype in DRG, leading to pro-inflammatory cytokine and chemokines release that in turn induced DRG and distal nerve ending damage (98, 112–114).
Therefore, there is a connection between neuroinflammatory elements and monocyte/macrophage infiltration: it was established that CCL2 attracts pro-inflammatory monocytes/macrophages to the DRG, causing a downstream increase of cytokines (101, 115) and M1 monocytes population triggering a pro-inflammatory cascade (98). On the other hand, despite monocyte chemoattractant CCL2 and CCL3 increased in DRG, Makker and colleagues did not observe any macrophage infiltration (20).
Distally to DRG, macrophage infiltration was also detected in sciatic nerves of PTX-treated animals (116), suggesting that macrophage activation was subsequent to the axonal degeneration (117).
Finally, it was demonstrated that PTX treatment-induced mechanical allodynia was correlated with the activation of NLRP3 inflammasome. An increased NLRP3 expression was detected in DRG and sciatic nerves of PTX-treated animals and this expression was colocalized with infiltrated macrophages (118).
Several contributions show an involvement of the CNS in the generation of the CIPN with increased levels of pro-inflammatory cytokines and chemokines in the spinal cord and the relevant role of astrocytes and microglia.
A vast number of cytokines and chemokines have been detected in the spinal cord of PTX-treated animals with main focus on IL-1β and IL-6 increase detection (20, 119, 120). In particular, the genetic interference with IL-6 signaling suggested its pivotal role in the development and maintenance of neuropathic pain (120).
Moreover, a significant increase in the expression of a robust chemoattractant molecule, CX3CL1, or of its receptor CX3CR1 (107, 121), as well as the upregulation of IL-17 expression were detected in the spinal cord of PTX-treated rats (122).
To better understand the role of cytokines and chemokines, their mRNA levels were also measured in the lumbar spinal cord of PTX-treated mice during the allodynia phase. At this later stage, an increase in the chemokine CCL2, without any concomitant change in other pro-inflammatory cytokines levels, was reported. The authors hypothesized that the alteration of those cytokines levels might have occurred at an earlier stage, contributing to the development of the neuropathy (123). An increase of TNF-α, IL-1β, IL-10, and IL-4 were instead detected in spinal cord of treated rats and associated with neuropathic pain in a study of Doyle and coworkers (124).
Among the different pathways which have been investigated in order to correlate pro-inflammatory elements and PTX-induced neuropathic pain the PKCϵ-dependent activation of TRPV1 was suggested. PKCϵ is one of the isoforms of the protein kinase C (PKC) and its activation is correlated to the augmented function of TRPV1 which enhances nociception. In particular, it was reported that mast cells release histamine which in turn induces the release of the neuropeptide SP, an inflammatory mediator that causes the sensitization of TRPV1 (125). It was demonstrated that plasma histamine levels in PTX-treated rats were higher than the control group supporting the role of histamine in PIPN. Moreover, an increased expression level of TRPV1/PKCϵ was detected in spinal cord and DRG of PTX-treated animals, hypothesizing the role of this pathway in the establishment of the animal pain behavior (126).
Besides TRPV1/PKCϵ, other studies demonstrated that the Notch signaling pathway is associated with several diseases of the nervous and immune systems. In fact, Notch signaling participates in the release of pro-inflammatory cytokines through modulation of microglial activation after nerve injury (127–130) and neuropathic pain (131). PTX treatment induced the activation of the Notch1 pathway in sciatic nerves of rats, as demonstrated by the increased expression of its receptor (106).
Moreover, IL-6 is known to activate JAK/STAT transduction pathway and MAPK cascade contributing to neuropathic pain (132). In fact, PTX treatment induced IL-6 increase in sciatic nerves, causing the increase in protein expression of its downstream molecule JAK, which in turn activated STAT3 (106, 133).
Also the activated transcription factor NF-κB was involved in inflammation, as reported in spinal cord or in sciatic nerves of PTX-treated animals. In fact, PTX caused NF-κB phosphorylation (NF-κB p65) leading to its activation and consequently to the release of pro-inflammatory cytokines that contribute to pain behavior (106, 134). More in detail, Li and colleagues suggested NF-κB p65 modulated the upregulation of CX3CL1. Interestingly, intrathecal administration of the NF-κB p65 inhibitor PDTC reduced CXCL1 expression at spinal level, as well as mechanical allodynia (121). Moreover, Akt1 (Akt family member), a downstream substrate of PI3K, once activated by CX3CR1/CX3CL1 interaction, mediates the phosphorylation of NF-κB. Akt1 levels were increased in DRG and in spinal cord after PTX treatment, playing a substantial role in painful symptoms demonstrated by the use of an intrathecal PI3K inhibitor (LY294002) which suppressed Akt1 expression and pain-related behavior (107). In addition, the inhibition of PI3K-mTOR mediated signaling led to alleviation of PTX-related neuropathic pain (102).
Moreover, the pharmacological enhancement of SIRT1 (a histone deacetylase that regulates inflammatory responses) activation reversed NF-κB p65 phosphorylation at spinal level abolishing PTX- induced pain behavior (134).
S1P has an important role in the production of pro-inflammatory mediators such as IL-1β in the inflammation process by enhancing NLRP3 inflammasome activity (135).
In a PIPN rat model, S1PR1 was found to contribute to the development and maintenance of neuropathic pain activating the neuroinflammatory process through the sphingolipid pathway in the spinal cord. In particular, using a S1PR1 antagonist (W146), the activation of NF-κB was blocked and the release of cytokines was shifted from pro-inflammatory to anti-inflammatory phenotype (136).
A3AR is expressed in inflammatory cells, glial cells and neurons and it is activated by the increased production of peroxynitrite in the spinal cord (137). The activation of A3AR prevents the enhancement of NF-κB and MAPK pathways as well as the production of pro-inflammatory cytokines suggesting the fundamental role of spinal inflammation in PIPN (138).
As previously discussed for OIPN, the use of an A3AR agonist (IB-MECA) prevented neuropathic pain in PTX-treated rats by modulating spinal glial cells neuroinflammatory process (69). In fact, adenosine can modulate many biological processes, including inflammation, by activating adenosine receptors and the activation of the A3AR inhibits inflammatory responses in different rodent models (138).
Other studies investigated the cannabinoid receptors in relationship with CIPN neuroinflammation. Cannabinoid receptors 1 (CB1) and 2 (CB2) belong to the endocannabinoid system and are mainly responsible for neuroinflammation suppressing effects. CB1 is predominantly expressed in the CNS whereas CB2 is mainly found in lymphoid organs and immune cells. CB1 has been located in several pain-related CNS regions, while CB2 mRNA increases after PNS damage (139).
Moreover, CB1 is mainly expressed in astrocytes whereas CB2 is involved in the activation of microglial cells (140, 141). In other studies, agonists for these receptors were employed successfully for PIPN treatment, modulating cytokines/chemokines expression and release. Several studies demonstrated that the use of CB2 cannabinoid receptor agonists (AM1710 and MDA7) suppressed allodynia (123, 142, 143), with a decrease of TNF-α and CCL2 mRNA levels in spinal cord of PTX-treated animals (123). In addition, glial cells activation markers and pro-inflammatory cytokines secretion were decreased in other PIPN models (143, 144) as well as a down-regulation of different pro-inflammatory coding genes was observed (145).
Moreover, the use of a synthetic cannabinoid agonist (WIN 55,212-2) to prevent PIPN development suggested the possible involvement of spinal cord glial cells in the onset of the pathology and pain modulation. Therefore, in order to study glial cell involvement in PIPN, a microglial cell activation inhibitor (minocycline) and the cannabinoid agonist were used simultaneous with the antineoplastic treatment (146). The markers of microglia cells and astrocyte activation (CD11b and GFAP, respectively) were investigated in the lumbar spinal cord of PTX-treated rats. An important astrocyte activation was detected together with the histological observations of hypertrophied cell bodies and fibrous processes, which is the typical phenotype of activated glial cells. The treatments with minocycline or cannabinoid agonist both resulted in attenuated microglia and astrocyte activation in lumbar spinal cord and in the prevention of thermal hyperalgesia and mechanical allodynia development. Furthermore, in lumbar spinal cord the IL1-β, IL-6, and TNF-α level increase was observed until the end of PTX treatment, but not later, indicating the role of these cytokines as initiators for the cascade phenomena, although at late-phase neuropathic pain and glial cell activation were maintained (123, 146).
The activation marker Iba-1 was also detected in the spinal cord of PTX-treated mice together with an increased level of IL-1β and CCL-2. In this context, the authors demonstrated the role of a selective CB2 receptor agonist for the prevention of Iba-1 up-regulation and for the reduction of IL-1β levels (147). Similarly, Wu and collaborators demonstrated an increase of Iba-1 expression in the dorsal horn of PTX-treated rats, confirming the role of CB2 receptor in the modulation of microglia dysregulation (144).
The increase of CCL2 level in spinal cord of PTX-treated mice and its contribution in hyperalgesia was also demonstrated in another study from Pevida and colleagues, in which they demonstrated a correlation with the activation of glial cells: CCL2 stimulates microglial cell activity through its receptor CCR2 (138).
A temporal correlation between microglia activation (increase of Iba-1 immunoreactive cells), the increase of chemokine CCL3 and its receptor CCR5, suggests that CCL3 release may be derived from activated microglia. Moreover, the attenuation of allodynia obtained in CCL3-neutralizing antibody-treated animals suggested that this chemokine was implicated in PTX-induced mechanical allodynia (148).
Therefore, accumulating evidence demonstrates that the activation of glial cells together with the downstream increase of pro-inflammatory cytokines and chemokines in the spinal cord is involved into the central sensitization process (20, 134, 136, 149).
Other studies demonstrated an increase of GFAP or both GFAP and Iba-1 markers in spinal cord of PTX-treated animals (124, 150–152), but controversial results have been reported. For example, other observations suggested the activation only of spinal astrocytes with no significant involvement of microglia (20, 73, 153, 154). In particular, it was suggested that the downregulation of the glial glutamate transporters GLAST and GLT-1 was responsible for the increase of GFAP-positive astrocytes (154).
The inhibition of these transporters has also been associated with neuropathic pain: in this context, the increase of GFAP expression in the spinal horn not only contributed to the mechanical and thermal allodynia in PTX-treated animals, but also to the suppressed expression of GLT-1 (82).
Finally, an increase of GFAP expression, indicating SGC activation, was demonstrated also in DRG of PTX-treated animals (57, 113).
As previously reported for OIPN, TLRs are also involved in PIPN. TLRs are differentially expressed by neurons, microglial cells and activated astrocytes (155–157).
In a PIPN rat model, TLR2 expression was downregulated and it could be involved in the antinociceptive action of the CB2 agonist (MDA7) in the attenuation of the pathology (145).
Moreover, in DRG neurons of PTX-treated rats the upregulation of TLR4 was demonstrated (158, 159). Its activation induced the increase of MCP-1 expression that consequently promoted macrophage infiltration into the DRG which was coincident to the development of the mechanical hypersensitivity (115). In addition, macrophages express an array of TLRs, which can stimulate the release of cytokines (155) such as TLR9, whose involvement in PIPN was at least in part sex dysmorphic (160). In fact, in male, but not female PTX-treated mice, the activation of TLR9 in macrophages results in the release of pro-inflammatory cytokines and chemokines that might activate A fibers driving CIPN mechanical allodynia. Moreover, differences were retrieved in male and female signaling downstream TLR9. In fact, male Tlr9 mutant (deficient) animals presented a more attenuated neuropathic pain (mechanical allodynia) compared to female mice and the pharmacological inhibition of TLR9 by intraplantar or intrathecal injection of ODN 2088 reduced the pain behavior only in male (160).
The Figure 2 shows the involvement of different inflammatory actors in the onset of PIPN according to the results of the most consistent studies.
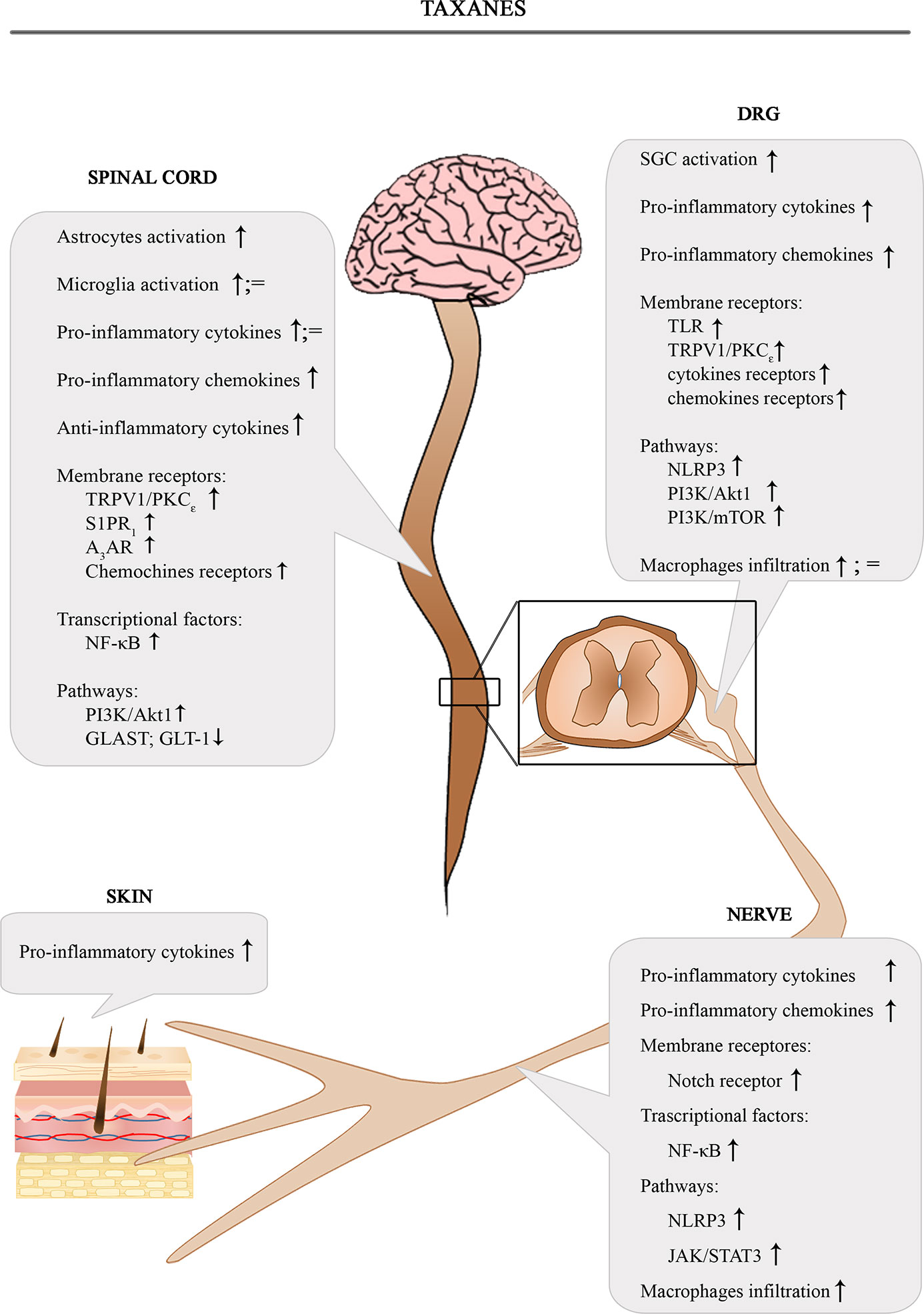
Figure 2 Schematic depiction of different inflammatory actors involved in the onset of PIPN.
Vinca Alkaloids
Vincristine (VCR) belongs to the vinca alkaloid family and is principally used in adult and pediatric hematologic cancers such as Hodgkin’s lymphoma, non-Hodgkin’s lymphoma and leukemia (161).
VCR exerts its cytotoxic effect by binding the β-subunit of tubulin heterodimers, interfering with the microtubule formation and mitotic spindle dynamics thus leading to the arrest of dividing cells in metaphase and consequently cell death (162).
VCR elicits a strong neurotoxicity, which involves both sensory and motor fibers with also autonomic impairment. The most common side effects are numbness, paresthesia, impaired balance and weakened tendon reflexes. Regarding the autonomic dysfunctions, constipation, paralytic ileus, urinary retention and orthostatic hypotension might occur (163).
The pathogenesis of VCR-induced peripheral neurotoxicity (VIPN) is not completely understood although several studies have suggested the possible contribution of the different components of the immune system.
In several studies, the alteration of the cytokine levels in blood or PNS and CNS tissue samples correlated to VCR-related neurotoxicity. In particular, the increase of TNF-α and IL-2 levels was detected in plasma of VCR-treated rats together with the increase of TNF-α, IL-1β, IL-6 levels in sciatic nerves, in the spinal cord and brain (164–171).
Information about the anti-inflammatory cytokine IL-4 was also reported: its potential protective role was highlighted in a mice model of VIPN in which a decrease of IL-4 mRNA levels in sciatic nerves of animals together with the downregulation of p-STAT6 (the downstream effector of IL-4) were detected. This downregulation was correlated with the establishment of mechanical allodynia. The correlation was confirmed using a IL-4 KO model in which an accelerated mechanical allodynia was evidenced and significantly decreased after IL-4 re-introduction. The re-introduction of IL-4 attenuated also p-STAT6 down-regulation suggesting IL-4 protective role towards VIPN via the stimulation of IL-4/STAT6 signaling pathway. Moreover, the over-expression of the pro-inflammatory cytokines IL-1β and TNF-α was reported in IL-4 KO animals compared to the wild type control mice (172).
The cytokines contribution in VIPN was also reported together with monocyte/macrophage infiltration in sciatic nerves and DRG, which was evidenced by a marked increase in F4/80 or MAC-1 (173–175). Moreover, it was reported that IL-6 co-localized with invading macrophage and the use of IL-6 neutralizing antibody suppressed VCR-induced mechanical allodynia, suggesting its role in the establishment of VIPN (173).
The role of infiltrating monocytes in VIPN was also demonstrated considering the effect of VCR on both blood-nerve/brain barriers. In fact, VCR caused endothelial cells activation and tight junction disruption leading to an increase of CCR2+ monocytes in the spinal cord. Since microglia do not express CCR2, they were probably infiltrating monocytes. Moreover, they expressed the cysteine protease Cathepsin S (CatS) (176). It is known that monocytes release CatS which in turn solubilizes the chemokine domain of CX3CL1 and activates CX3CR1 receptor in microglia, resulting in the release of pro-inflammatory cytokines (177, 178). This cascade is involved in pain condition: in fact, the inhibition of spinal CatS reversed neuropathic pain in an animal model of sciatic nerve partial ligation (179). This last observation was afterwards confirmed in a VIPN model: a centrally-penetrant CatS inhibitor reduced VCR-induced nociception confirming that CatS acted centrally in mediating VCR action (176).
The release of pro-inflammatory cytokines was investigated and associated with different activated pathways. An increased expression of Iba-1, CX3CR1 and P-p38 in the dorsal horn and high levels of TNF-α and IL-1β in the spinal cord were detected after VCR treatment. The chemokine CX3CL1 activates its microglia-specific receptor CX3CR1, leading to phosphorylation of p38MAPK kinase which then promotes the secretion and release of pro-inflammatory cytokines (130). In this contest, the important function of Notch signaling pathway was demonstrated in a VIPN rat model: using a Notch signaling inhibitor, the authors relieved pain behavior and downregulated the microglial pathway (Iba-1, CX3CR1 an P-p38 MAPK proteins) obtaining also a downregulation of the inflammatory factors TNF-α and IL-1β in the spinal cord (169).
The neuroinflammation role of the prokineticin (PK) family in a preclinical model of VIPN was also assessed (180). PK family is a chemokine family composed of two proteins: PK1 and PK2 with their receptors PK-R1 and PK-R2, respectively. PK-Rs are localized mostly in DRG and spinal cord where the highest density is found in the dorsal horn, suggesting their role in the central nociceptive signal transmission. PK-R1 is mostly expressed on astrocytes and microglia cells (181, 182). PK2 is an important linker element between inflammation and pain, in fact, it can modulate the immune system to a pro-inflammatory phenotype, releasing cytokines, and it can also sensitize the nociceptors (183). VCR induced an up-regulation of PK2 and PK-Rs in spinal cord and even DRG together with high levels of cytokines (IL-1β, TNF-α, and IL-6) and a significant increase of CD11b. The use of a PK-Rs antagonist (PC1) reduced the hypersensitivity within modulation of the neuroinflammation: it was able to downregulate the PK system, restoring a correct cytokine balance (180).
NF-κB-dependent CXCL1/CXCR2 signaling pathway was also shown to be relevant in VIPN (184). NF-κB is an important transcriptional factor, which regulates the release of pro- and anti- nociceptive factors, among which CXCL1. This chemokine acts through its receptor CXCR2 and has a role in central sensitization and pain maintenance (185, 186). VCR induced NF-κB activation and consequently the CXCL1 upregulation in the spinal cord of treated animals. The use of a NF-κB inhibitor led to an attenuation of CXCL1 immunostaining and a reduction of pain behavior, suggesting that NF-κB regulates CXCL1 upregulation (184). Moreover, p65 phosphorylation, and its increased expression, activated NF-κB pathway. The expression of p-p65 was increased in VCR-treated animals at spinal level together with the up-regulation of TNF-α and the down-regulation of IL-10, confirming NF-κB role in the neuro-immune modulation (164, 187).
In spinal cord, similarly to other drugs, microglia cells and astrocytes were involved and activated also after VCR treatment, with the release of pro-inflammatory elements. In this context, exogenous induction of HO-1 was suggested as a potential therapy approach. HO-1 is a rate-limiting enzyme of heme degradation whose induction protects against cytotoxicity and has a role in immunomodulatory and anti-inflammatory processes (188). HO-1 inducer attenuated VCR-induced pain hypersensitivity as well as it reduced GFAP and Iba-1 expression. Moreover, the HO-1 induction decreased the activation of MAPKs, which mediated the production of pro-inflammatory elements, such as TNF-α and MCP-1 (189).
The glial markers Iba-1 and GFAP colocalized with TNF-α demonstrating that TNF-α was released by these activated spinal glial cells and that it was at least in part responsible for VCR-induced mechanical allodynia, since the use of a neutralizing antibody against TNF-α reduced VIPN (190). On the contrary, neither microglia hypertrophy nor increase of Iba-1 levels were detected in the study of other authors (73).
As suggested also for the other antineoplastic agents, TLR-4 seemed to be implicated in VIPN. In fact, using a Tlr4 KO mouse model, mechanical allodynia was decreased as well as in naive minocycline-treated animals (191).
The Figure 3 shows the involvement of different inflammatory actors in the onset of VIPN according to the results of the most consistent studies.
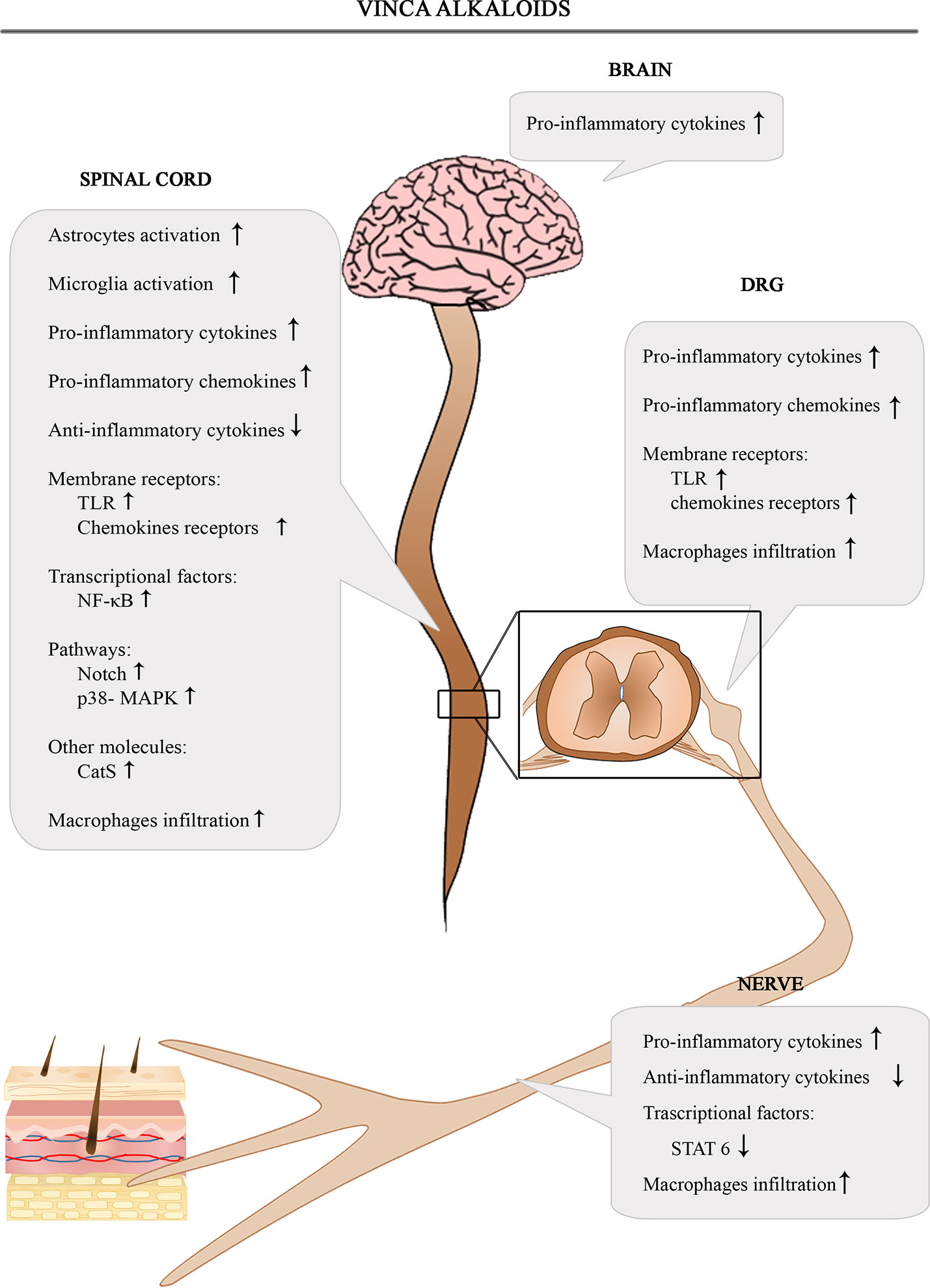
Figure 3 Schematic depiction of different inflammatory actors involved in the onset of VIPN.
Proteasome Inhibitors
Bortezomib (BTZ) is a functional proteasome inhibitor commonly used as the frontline anticancer drug in the treatment of multiple myeloma (MM). Although its clinical effectiveness has been clearly demonstrated, it frequently leads to dose limiting painful peripheral neuropathy.
Neuropathic pain associated with BTZ is often severely debilitating, including spontaneous pain as well as allodynia and hyperalgesia in the distal extremities of limbs. Unfortunately, this devastating complication often requires BTZ dose modification or discontinuation (192, 193), compromising the clinical outcome of MM patients.
The pathophysiological mechanisms by which BTZ leads to BTZ-induced peripheral neurotoxicity (BIPN) remains largely unclear and the molecular mediators of the neuropathic pain-syndrome have not been fully elucidated.
A large body of data indicates that BTZ is involved in the development of peripheral damage related to immune-mediated processes in addition to its toxic effects (194, 195). These data focus on the pivotal role of inflammation and immune response in the development and maintenance of BIPN and some of them reported also that BIPN responded to high dose steroids or intravenous immunoglobulins (IVIg) (196, 197). All together these findings provide a rationale to identify neuroinflammation as a critical key player involved in BIPN, which includes a primary sensitization process at the level of sensory neurons in DRG up to the involvement of the spinal cord (198).
In order to study neuropathic pain-syndrome, several CIPN animal models mimicking clinical features of BIPN have been reported. As previously done for the other chemotherapeutic drugs, the inflammatory immune and immune-like glial cells as well as the actions of the pro- and anti-inflammatory mediators (i.e. nerve growth factor, cytokines/chemokines or microRNA) will be considered. In this section, we will also report some attempts that were performed in order to reduce the generation of BIPN by targeting immune and glial cell responses as well as released cytokines and chemokines.
Considering first the effect of BTZ on circulating white blood cells, no changes in cell subsets and functions have been associated with BIPN, whereas the main BTZ action has been referred to PNS. Moreover, regarding cytokine circulating molecules, BTZ injection caused increased pro-inflammatory cytokines TNF-α IL-1β, IL-6 levels in plasma (199). As previously observed in the PIPN model (115), one of the initial immune responses is mediated by resident macrophages and by a large influx of infiltrated macrophages, promoting neuropathy progression (156). Of interest, M1 pro-inflammatory phenotype macrophages in the peripheral nerves have been found to play an important role in the pathogenesis of BIPN (197). These data suggest a crucial role of macrophage into neuropathic pain maintenance, which was further supported by the depletion of macrophage infiltration due to the administration of a high dose of human IVIg able to block BTZ-induced allodynia and hyperalgesia (197).
Peripheral glial cells support an active role in immune and sensory transmission by maintaining metabolic and ionic homeostasis into the peripheral nervous system (200). They were also demonstrated to play a relevant role even in BIPN by undergoing notable phenotypic changes associated with pain hypersensitivity (201). Indeed, the glial cells activation was characterized by hypertrophy and the presence of large cytoplasmic vacuoles due to mitochondrial damage and endoplasmic reticulum enlargement in both SGCs and Schwann cells in rats undergoing BTZ treatment (201).
More interestingly, the production of pro-inflammatory mediators was positively correlated to the nociceptor sensitization and neuropathic pain-syndrome derived from changes in DRG, enhancing neuronal excitability and generating pain hypersensitivity. It is noteworthy that highest and early increase of IL-6 and TNF-α in DRG of treated mice was followed with a later increase of TGF-β1 and IL-1β, which are accompanied by elevated TNF-α receptor1 (TNFR1) induction in BIPN (202). The treatment with TNF-α neutralizing antibody, significantly prevented the BTZ-induced electrophysiological alterations and the loss of myelinated and unmyelinated fibers. Furthermore, it elicited an improvement in pain behavior which was correlated to decreased expression of TNFR1, IL-6 and IL-6- corresponding signal transducing receptor (IL-6Rα) in DRG (202).
Likewise, the dose-related effects of anti-TNF-α therapy on neurotoxicity were also demonstrated in the BIPN rat model, in which the co-administration of an antibody against TNF-α was able to revert the neuropathic symptoms, although the development of the neuropathy was not prevented (203). Accordingly, a progressive increased serum level of TNF-α in patients suffering from peripheral neuropathy after several cycles of BTZ-therapy was reported. In the same study, they also demonstrated a potential neuroprotective effect of the co-administration of anti-TNF-α treatment in the BIPN rat model, showing an improvement of both electrophysiological parameters and mechanical allodynia (204). Moreover, the authors hypothesized that increased TNF-α levels caused the upregulation of heparanase (HSPE) expression, an endoglycosidase involved in the production of inflammatory cytokines, which was secondary to the development of the neuropathy (204).
In support of the crucial role of TNF-α modulation in peripheral sensitization processes involved in BTZ-induced allodynia, Zhang and colleagues demonstrated that the increase of TNF-α expression in rat DRG paired with that of the phosphorylated JNK1/2. Indeed, the suppression of TNF-α signaling induced by the TNF-synthesis inhibitor, thalidomide, as well as by TNFR1 and TNFR2 depletion in KO male mice, blocked JNK1/2 activation in DRG which was accompanied by a reduction in mechanical allodynia (205). Overall, the upregulation of TNF-α appeared to play a relevant role in orchestrating activation of JNK signaling by TNFR1 and TNFR2, mediating mechanical allodynia occurrence (205).
An ongoing interaction between transient receptor potential ankyrin 1 (TRPA1), TNF-α and its receptor TNFR1 were recently demonstrated in relationship with neuropathic pain onset. Increased TRPA1 expression in rat DRG was associated with development of mechanical pain and cold sensitivity following BTZ treatment (206). Interestingly, suppressing the expression of TRPA1 by TRPA1 antagonist HC030031 reduced allodynia and thermal hyperalgesia in BTZ-treated rats. Moreover, blocking TNF-α pathway by the action of pentoxifylline resulted in attenuated p38-MAPK and JNK signal in DRG, as well as a reduction of TRPA1 expression which was correlated with a block of neuropathic pain onset (206).
Similarly, inhibition of TNFR1-TRPA1 pathway in the dorsal horn has recently been reported, including the critical involvement of the microRNA (miR-155) in painful BIPN (207). In particular, inhibiting miR-155 signal in BTZ-treated rats displayed significantly reduced mechanical and cold sensitivity associated with the decreased TNFR1 and TRPA1 expression as well as the reduction of signals p38-MAPK and JNK in DHSC. Interestingly, administration of miR-155 mimic interfered with TNFR1-TRPA1 signal and contributed to enhanced cold hypersensitivity and allodynia (207). Therefore, interfering with this complex pathway may provide an effective treatment of neuropathic pain in BTZ-treated patients.
Moreover, in a recent study by Liu and collaborators, increased TRPA1 and IL-6 receptor levels were observed in the DRG after BTZ administration. Their contribution to BTZ-induced mechanical and cold hypersensitivity was supported inhibiting TRPA1 function, as well as blocking IL-6-mediated signal transduction. This resulted in a downregulation of intracellular signal mediated by p38-MAPK and JNK in the sensory neurons, which are correlated with a decreased TRPA1 protein expression and a consequently absence of mechanical pain and cold sensitivity. Together these results suggested that IL-6 is a signal activating TRPA1 that could be a relevant key process engaged in painful BIPN, and that interfering with this pathway might be a useful tool to reduce neuropathic pain in multiple myeloma patients (208).
To better investigate their pivotal role and responsibility in the development and maintenance of painful BIPN the chemokine family and neuroinflammation pathways were thoroughly investigated. The upregulation of chemokine CCL2 expression in DRG neurons, but not in SGCs was observed in BTZ injected rats, and it was associated with a huge infiltration of macrophage, as well as an enhanced expression of the transcription factor c-Jun. This effect might be associated with co-localization of c-Jun and ATF3 transcription factors. Furthermore, blocking c-Jun signaling prevented mechanical allodynia, as well as CCL2 upregulation. More interestingly, the pre-treatment with ATF3 siRNA suppressed the c-Jun binding to the ccl2 promoter (209).
More recently, the increasing evidence has emphasized that PKs may contribute to pain hypersensibility and neuroinflammation in a BTZ-treated mice model, as previously described in VIPN model (180). Of note, overexpression of PK system (PK2 and PK-R) in all the tissues involved in pain transmission has been proposed to contribute to the thermal hyperalgesia, mechanical and cold allodynia by inducing structural damage to DRG neurons and SGCs. Furthermore, increased macrophage activation markers and TLR4 mRNA were detected in both DRG and sciatic nerves. The increased release of pro-inflammatory cytokines and the decrease of anti-inflammatory IL-10 expression were also reported in CNS and PNS (198). In addition, the activation of the spinal cord glial cells by upregulation of glial markers GFAP led to pain hypersensibility. Moreover, DRG structural alteration together with the development of altered behavioral parameters were totally abrogated by subcutaneous administration of a PK-R antagonist (PC1) in treated mice. An evident attenuation of macrophage recruitment and prevented central sensitization into DHSC were also reported (198). Finally, the authors speculated that the upregulation of PK2 protein could be regulated by the binding between phosphorylated STAT3 and the PK2 promoter (198).
Recently, increasing attention is being paid to the role of NLRP3 inflammasome complexes as key mediators of inflammatory mechanisms involved in neuropathic pain. Accumulating evidence indicates that NLRP3 is involved in several CNS diseases, as well as in PIPN and OIPN (81, 118). In a rat model of BIPN, Liu and coworkers demonstrated an increased NLRP3 inflammasome mRNA and protein expression in DRG which could lead to painful neuropathy. This event was correlated with the upregulation of phosphorylated STAT3 signaling via increasing histone acetylation, as well as with the enhanced binding of STAT3 to Nlrp3 promoter in DRG. To further support the importance of NLRP3 in BIPN, the intrathecal injection of NLRP3 siRNA attenuated mechanical allodynia caused by BTZ. Meanwhile, specific inhibition of STAT3 activity resulted in a suppressed upregulation of NLRP3 in DRG, thus ameliorating mechanical allodynia induced by BTZ (210).
Since there is extensive evidence supporting the role of neuroinflammation in the CNS, several immune-like glial cells changes in the spinal cord have been implicated in CIPN, identifying spinal glial cells as key players that drive the establishment and maintenance of neuropathic pain (211). For instance, in animal models of PIPN, OIPN and BIPN, astrocytes became activated and proliferated (72, 154). Furthermore, since astrocytes represent the largest CNS cell population, their morphological activation in CIPN models has been debated (72). Of note, in OIPN and BIPN models, the spinal astrocytes activation occurred at multiple time points after chemotherapy treatment in parallel with the induction of mechanical sensitivity. Moreover, an immunohistochemical study which compared the activation of astrocytes and microglia in the DHSC demonstrated that only the increase in GFAP positive astrocytes was correlated to the induction of allodynia in BTZ-treated animals. In particular, changes in spinal glial morphology (arborization and hypertrophy of astrocytes) were reported, while no activation of microglia was observed. The application of minocycline, similarly to the previously reported CIPN models (63, 79, 111), totally prevented the painful symptoms and it counteracted the astrocyte activation which may result from a common underlying mechanism (72).
A more recent research from Robinson and colleagues described significant alterations in astrocytic connexins and glutamate transporters mediated neuropathic pain in the BIPN model. As previously mentioned in various forms of CIPN (79, 154), increased GFAP-positive astrocytes is believed to be one of the critical proteins correlated with the upregulation of Cx43 and the glutamate transporter dysfunction (212). These activated astrocytes (characterized also by hypertrophy and increased arborization) displayed an initial upsurge of intracellular calcium, coinciding with the presence of synaptic glutamate due to the downregulation of glutamate transporters (GLAST). In addition, after the follow up period, an increase of phosphorylated Cx43 correlated with mechanical hypersensitivity. The administration of minocycline inhibited GFAP, GLAST, and Cx43 increase and prevented the onset of mechanical allodynia (212).
Interestingly, Guo and collaborators focused on the activation of microglia and SGCs (increase of Iba-1 and GFAP immunoreactive cells) after BTZ injection, indicating the crucial association between the purinergic ligand gated ion channel 7 receptor (P2X7R) and p38 MAPK pathway as a prerequisite for BIPN (213). P2X7R is richly expressed in glial cells. The increase of P2X7R protein levels and p38 MAPK phosphorylation were reported in DRG already 2 days after chemotherapy treatment. In particular, the authors observed that P2X7R was colocalized with GFAP in DRG and with Iba-1 in DHSC, while p38 MAPK was mainly expressed in microglia cells. P2X7R downstream pathway inhibition could be able to revert neuropathic pain and the inhibition of p38 MAPK phosphorylation led to a downregulation of P2X7R expression level in BIPN-affected animals. These results indicated that blocking P2X7R-p38 signal with pharmacology therapy was beneficial to alleviate neuropathic pain resulting from BTZ treatment (213).
Moreover, the production of various pro-inflammatory cytokines as well as chemokines by spinal cord astrocytes, caused the increase of the activity of spinal cord nociceptive neurons after BTZ treatment in both mice and rat models (192, 214). Recently, the upregulation of protein expression of TNF-α as well as its mRNA level were found in rat spinal cord neurons together with increased IL-1β expression and JNK activation in astrocytes in BTZ-induced allodynia model (215). Within the spinal cord, the predominant increased JNK phosphorylation was not correlated with a similar activation of the ERK and p38-MAPK pathways. Similarly to the work of Zhang and colleagues (205), intrathecal injection of thalidomide or the IL-1 receptor antagonist (IL-1ra) in BIPN model ameliorated mechanical allodynia by downregulation the phosphorylation of JNK signal (215).
Finally, compelling researches have indicated that altered sphingolipid metabolism may be correlated with CIPN pathology and neuroinflammation in both human and animal models (199). In particular, a clinical study showed a correlation between PIPN and plasma levels of neurotoxic sphingolipids (216), and up-regulated sphingolipid metabolism were reported in PNS in a docetaxel neurotoxicity mice model (217). In addition, there was a striking upsurge in sphingosine-1-phosphate (S1P) signaling, its receptor 1 (S1PR1) and dihydro-S1P in the DHSC following BTZ treatment. The development of mechano-hypersensitivity was associated with an increased TNF-α and IL-1β and an enhancement of presynaptic glutamate release in the DHSC in mice model (218). Similarly to PTX studies (69), neuropathic pain behavior induced by BTZ was attenuated by S1PR1 antagonists (FTY720 or NIBR14 treatments) or siRNA to knockdown the expression of S1PR1. In addition, the administration of BTZ in mice with astrocyte-specific deletion of S1PR1 did not engage neuropathic pain associated with BIPN. These data suggest that astrocyte activation by S1PR1-dependent neuroinflammatory signaling is a key cellular site for S1PR1 activity. Therefore, consistent with this concept, emerging evidence proposed that S1PR1 signaling pathway in astrocytes and subsequent mechanical allodynia were regulated by the activation of NLRP3 inflammasome and IL-1 signaling in DHSC (219). Otherwise, Alè and colleagues regarding the paper of Stockstill and collaborators (218) evidenced several points to consider about the model involved in this article. In particular, the authors suggested a better understanding and attention toward the cumulative dose and the animal model employed since this acute model is not representative of the chronical painful BIPN. Moreover, blinder conditions should be employed, as well as a full investigation of the complexity of PNS (220).
The Figure 4 shows the involvement of different inflammatory actors in the onset of BIPN according to the results of the most consistent studies.
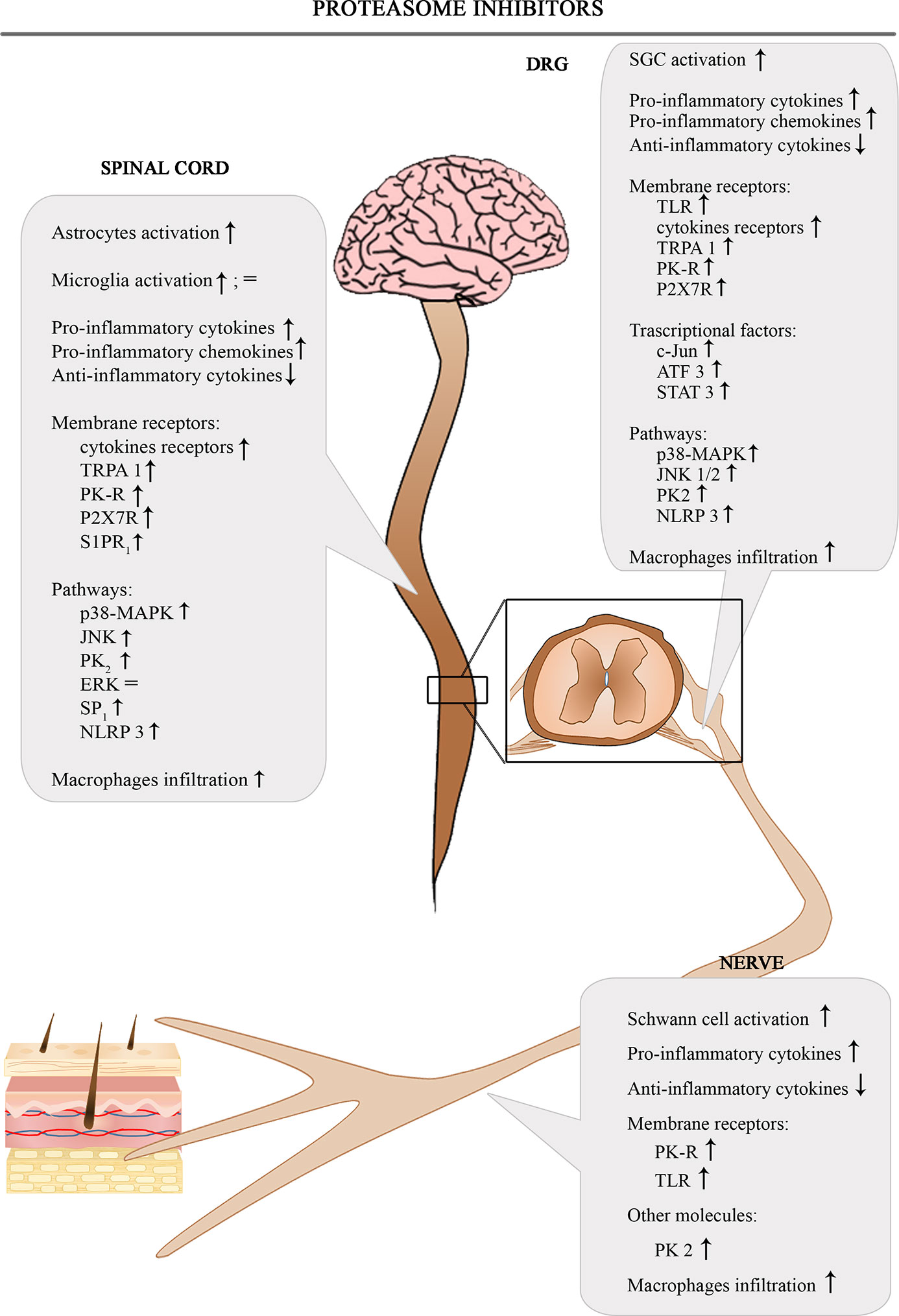
Figure 4 Schematic depiction of different inflammatory actors involved in the onset of BIPN.
Discussion
Chemotherapy represents the only available approach for fighting many different cancer types. However, this regimen is often associated with CIPN onset that frequently results in premature interruption of the treatment. Despite the recent development of some symptoms-related therapies for CIPN, no preventive or curative interventions are available. The mechanisms underlying CIPN are complex and not fully elucidated.
Here, we focused our attention on reviewing studies which pointed out the role of neuroinflammation in the onset and persistence of CIPN in rodent models. Since the generation of pain hypersensitivity may represent a common feature in the patient undergoing chemotherapy, a crosstalk between neuro-immune balance and pro-inflammatory mediators, as well as glial activation, was considered.
Despite some discrepancy in activated neuroinflammatory pathways within the same chemotherapy agent, presumably due to the different animal models used, CIPN exhibits a peculiar immune response and phenotypic cells changes in both CNS and PNS. The reliability of preclinical models is a very debated topic in CIPN, since they could negatively impact on the translatability of the results from bench to the clinical setting. The different reported effects may depend on several aspects: the species, the strain, the age, the sex, the dosage, and the treatment schedule. In particular, high doses of chemotherapy might be useful to obtain evident histopathological alterations and increased inflammatory response, but they could be far from a clinically relevant dose (221). Indeed, several CIPN-inducer agents seem to affect the same immunological targets despite the underlying pathways may be different. The identification of the peculiar neuroinflammatory pathway for each chemotherapeutic drug is required in order to be a further weapon to fight the single drug induced CIPN. Moreover, an early detection of inflammation targets in patients' blood may serve as a promising biomarker for predicting CIPN onset and course.
As we described, the inhibition of neuroinflammation pathways by immune modulation therapy in CIPN may potentially result in clinical benefits in terms of preventing and improving the painful peripheral neuropathy related to chemotherapeutic agents.
Author Contributions
CM and GF designed the review. GF and LM defined the search strategies and reviewed the literature. GF drafted the Introduction, the Taxanes, and the Vinca Alkaloids sections. LM drafted the Platinum Derivatives and Discussion sections. CM drafted the Proteasome Inhibitors and Discussion sections. GC coordinated and RR edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work has been supported by Fondazione Cariplo, grants n° 2019-1482, PRIN n° 2017ZFJCS3, and IMMUN-HUB n° 1165235.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Annalisa Canta for her assistance in illustrations.
Glossary

References
1. Zajączkowska R, Kocot-Kępska M, Leppert W, Wrzosek A, Mika J, Wordliczek J. Mechanisms of Chemotherapy-Induced Peripheral Neuropathy. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20061451
2. Miltenburg NC, Boogerd W. Chemotherapy-induced neuropathy: A comprehensive survey. Cancer Treat Rev (2014) 40:872–82. doi: 10.1016/j.ctrv.2014.04.004
3. Canta A, Pozzi E, Carozzi VA. Mitochondrial Dysfunction in Chemotherapy-Induced Peripheral Neuropathy (CIPN). Toxics (2015) 3:198–223. doi: 10.3390/toxics3020198
4. Cavaletti G, Marmiroli P. Chemotherapy-induced peripheral neurotoxicity. Curr Opin Neurol (2015) 28:500–7. doi: 10.1097/WCO.0000000000000234
5. Allodi I, Udina E, Navarro X. Specificity of peripheral nerve regeneration: interactions at the axon level. Prog Neurobiol (2012) 98:16–37. doi: 10.1016/j.pneurobio.2012.05.005
6. Carozzi VA, Canta A, Chiorazzi A. Chemotherapy-induced peripheral neuropathy: What do we know about mechanisms? Neurosci Lett (2015) 596:90–107. doi: 10.1016/j.neulet.2014.10.014
7. Branca JJV, Maresca M, Morucci G, Becatti M, Paternostro F, Gulisano M, et al. Oxaliplatin-induced blood brain barrier loosening: a new point of view on chemotherapy-induced neurotoxicity. Oncotarget (2018) 9:23426–38. doi: 10.18632/oncotarget.25193
8. Hopkins HL, Duggett NA, Flatters SJL. Chemotherapy-induced painful neuropathy: pain-like behaviours in rodent models and their response to commonly used analgesics. Curr Opin Support Palliat Care (2016) 10:119–28. doi: 10.1097/SPC.0000000000000204
9. Argyriou AA, Bruna J, Marmiroli P, Cavaletti G. Chemotherapy-induced peripheral neurotoxicity (CIPN): an update. Crit Rev Oncol Hematol (2012) 82:51–77. doi: 10.1016/j.critrevonc.2011.04.012
10. Stillman M, Cata JP. Management of chemotherapy-induced peripheral neuropathy. Curr Pain Headache Rep (2006) 10:279–87. doi: 10.1007/s11916-006-0033-z
11. Boyette-Davis JA, Hou S, Abdi S, Dougherty PM. An updated understanding of the mechanisms involved in chemotherapy-induced neuropathy. Pain Manage (2018) 8:363–75. doi: 10.2217/pmt-2018-0020
12. Starobova H, Vetter I. Pathophysiology of Chemotherapy-Induced Peripheral Neuropathy. Front Mol Neurosci (2017) 10, 174. doi: 10.3389/fnmol.2017.00174
13. Brandolini L, d’Angelo M, Antonosante A, Allegretti M, Cimini A. Chemokine Signaling in Chemotherapy-Induced Neuropathic Pain. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20122904
14. Lees JG, Makker PG, Tonkin RS, Abdulla M, Park SB, Goldstein D, et al. Immune-mediated processes implicated in chemotherapy-induced peripheral neuropathy. Eur J Cancer (2017) 73:22–9. doi: 10.1016/j.ejca.2016.12.006
15. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol (2008) 8:59–73. doi: 10.1038/nri2216
16. Javeed A, Ashraf M, Riaz A, Ghafoor A, Afzal S, Mukhtar MM. Paclitaxel and immune system. Eur J Pharm Sci (2009) 38:283–90. doi: 10.1016/j.ejps.2009.08.009
17. Flatters SJL, Dougherty PM, Colvin LA. Clinical and preclinical perspectives on Chemotherapy-Induced Peripheral Neuropathy (CIPN): a narrative review. Br J Anaesth (2017) 119:737–49. doi: 10.1093/bja/aex229
18. Calls A, Carozzi V, Navarro X, Monza L, Bruna J. Pathogenesis of platinum-induced peripheral neurotoxicity: Insights from preclinical studies. Exp Neurol (2020) 325:113141. doi: 10.1016/j.expneurol.2019.113141
19. Cavaletti G, Marmiroli P. Management of Oxaliplatin-Induced Peripheral Sensory Neuropathy. Cancers (Basel) (2020) 12. doi: 10.3390/cancers12061370
20. Makker PG, Duffy SS, Lees JG, Perera CJ, Tonkin RS, Butovsky O, et al. Characterisation of Immune and Neuroinflammatory Changes Associated with Chemotherapy-Induced Peripheral Neuropathy. PloS One (2017) 12:e0170814. doi: 10.1371/journal.pone.0170814
21. Akman T, Akman L, Erbas O, Terek MC, Taskiran D, Ozsaran A. The preventive effect of oxytocin to Cisplatin-induced neurotoxicity: an experimental rat model. BioMed Res Int (2015) 2015:167235. doi: 10.1155/2015/167235
22. Abdelsameea AA, Kabil SL. Mitigation of cisplatin-induced peripheral neuropathy by canagliflozin in rats. Naunyn Schmiedebergs Arch Pharmacol (2018) 391:945–52. doi: 10.1007/s00210-018-1521-5
23. Abdel-Wahab WM, Moussa F II. Neuroprotective effect of. Drug Des Devel Ther (2019) 13:1155–62. doi: 10.2147/DDDT.S191240
24. Wan CF, Zheng LL, Liu Y, Yu X. Houttuynia cordata Thunb reverses oxaliplatin-induced neuropathic pain in rat by regulating Th17/Treg balance. Am J Transl Res (2016) 8:1609–14.
25. Cerles O, Benoit E, Chéreau C, Chouzenoux S, Morin F, Guillaumot MA, et al. Niclosamide Inhibits Oxaliplatin Neurotoxicity while Improving Colorectal Cancer Therapeutic Response. Mol Cancer Ther (2017) 16:300–11. doi: 10.1158/1535-7163.MCT-16-0326
26. Cerles O, Gonçalves TC, Chouzenoux S, Benoit E, Schmitt A, Bennett Saidu NE, et al. Preventive action of benztropine on platinum-induced peripheral neuropathies and tumor growth. Acta Neuropathol Commun (2019) 7:9. doi: 10.1186/s40478-019-0657-y
27. Onk D, Mammadov R, Suleyman B, Cimen FK, Cankaya M, Gul V, et al. The effect of thiamine and its metabolites on peripheral neuropathic pain induced by cisplatin in rats. Exp Anim (2018) 67:259–69. doi: 10.1538/expanim.17-0090
28. Kuai CP, Ju LJ, Hu PP, Huang F. Alkaloids Attenuate Cisplatin-Induced Neuropathic Pain by Reducing Loss of IENF and Blocking TRPV1 Activation. Am J Chin Med (2020) 48:407–28. doi: 10.1142/S0192415X20500214
29. Li D, Kim W, Shin D, Jung Y, Bae H, Kim SK. Preventive Effects of Bee Venom Derived Phospholipase A₂ on Oxaliplatin-Induced Neuropathic Pain in Mice. Toxins (Basel) (2016) 8. doi: 10.3390/toxins8010027
30. Laumet G, Edralin JD, Dantzer R, Heijnen CJ, Kavelaars A. Cisplatin educates CD8+ T cells to prevent and resolve chemotherapy-induced peripheral neuropathy in mice. Pain (2019) 160:1459–68. doi: 10.1097/j.pain.0000000000001512
31. Ma J, Trinh RT, Mahant ID, Peng B, Matthias P, Heijnen CJ, et al. Cell-specific role of histone deacetylase 6 in chemotherapy-induced mechanical allodynia and loss of intraepidermal nerve fibers. Pain (2019) 160:2877–90. doi: 10.1097/j.pain.0000000000001667
32. Zhao X, Du W, Zhang M, Atiq ZO, Xia F. Sirt2-associated transcriptome modifications in cisplatin-induced neuronal injury. BMC Genomics (2020) 21:192. doi: 10.1186/s12864-020-6584-2
33. Starobova H, Mueller A, Deuis JR, Carter DA, Vetter I. Inflammatory and Neuropathic Gene Expression Signatures of Chemotherapy-Induced Neuropathy Induced by Vincristine, Cisplatin, and Oxaliplatin in C57BL/6J Mice. J Pain (2020) 21:182–94. doi: 10.1016/j.jpain.2019.06.008
34. Byrd-Leifer CA, Block EF, Takeda K, Akira S, Ding A. The role of MyD88 and TLR4 in the LPS-mimetic activity of Taxol. Eur J Immunol (2001) 31:2448–57. doi: 10.1002/1521-4141(200108)31:8<2448::aid-immu2448>3.0.co;2-n
35. Park HJ, Stokes JA, Pirie E, Skahen J, Shtaerman Y, Yaksh TL. Persistent hyperalgesia in the cisplatin-treated mouse as defined by threshold measures, the conditioned place preference paradigm, and changes in dorsal root ganglia activated transcription factor 3: the effects of gabapentin, ketorolac, and etanercept. Anesth Analg (2013) 116:224–31. doi: 10.1213/ANE.0b013e31826e1007
36. Di Cesare Mannelli L, Pacini A, Bonaccini L, Zanardelli M, Mello T, Ghelardini C. Morphologic features and glial activation in rat oxaliplatin-dependent neuropathic pain. J Pain (2013) 14:1585–600. doi: 10.1016/j.jpain.2013.08.002
37. Di Cesare Mannelli L, Pacini A, Corti F, Boccella S, Luongo L, Esposito E, et al. Antineuropathic profile of N-palmitoylethanolamine in a rat model of oxaliplatin-induced neurotoxicity. PloS One (2015) 10:e0128080. doi: 10.1371/journal.pone.0128080
38. Miguel CA, Raggio MC, Villar MJ, Gonzalez SL, Coronel MF. Anti-allodynic and anti-inflammatory effects of 17α-hydroxyprogesterone caproate in oxaliplatin-induced peripheral neuropathy. J Peripher Nerv Syst (2019) 24:100–10. doi: 10.1111/jns.12307
39. Park HJ, Stokes JA, Corr M, Yaksh TL. Toll-like receptor signaling regulates cisplatin-induced mechanical allodynia in mice. Cancer Chemother Pharmacol (2014) 73:25–34. doi: 10.1007/s00280-013-2304-9
40. Woller SA, Corr M, Yaksh TL. Differences in cisplatin-induced mechanical allodynia in male and female mice. Eur J Pain (2015) 19:1476–85. doi: 10.1002/ejp.679
41. Gu H, Wang C, Li J, Yang Y, Sun W, Jiang C, et al. High mobility group box-1-toll-like receptor 4-phosphatidylinositol 3-kinase/protein kinase B-mediated generation of matrix metalloproteinase-9 in the dorsal root ganglion promotes chemotherapy-induced peripheral neuropathy. Int J Cancer (2020) 146:2810–21. doi: 10.1002/ijc.32652
42. Tsubota M, Fukuda R, Hayashi Y, Miyazaki T, Ueda S, Yamashita R, et al. Role of non-macrophage cell-derived HMGB1 in oxaliplatin-induced peripheral neuropathy and its prevention by the thrombin/thrombomodulin system in rodents: negative impact of anticoagulants. J Neuroinflammation (2019) 16:199. doi: 10.1186/s12974-019-1581-6
43. Boyette-Davis JA, Walters ET, Dougherty PM. Mechanisms involved in the development of chemotherapy-induced neuropathy. Pain Manage (2015) 5:285–96. doi: 10.2217/pmt.15.19
44. Basu S, Sodhi A. Increased release of interleukin-1 and tumour necrosis factor by interleukin-2-induced lymphokine-activated killer cells in the presence of cisplatin and FK-565. Immunol Cell Biol (1992) 70:15–24. doi: 10.1038/icb.1992.3
45. Duan Z, Su Z, Wang H, Pang X. Involvement of pro-inflammation signal pathway in inhibitory effects of rapamycin on oxaliplatin-induced neuropathic pain. Mol Pain (2018) 14:1744806918769426. doi: 10.1177/1744806918769426
46. Huang W, Huang J, Jiang Y, Huang X, Xing W, He Y, et al. Oxaliplatin Regulates Chemotherapy Induced Peripheral Neuropathic Pain in the Dorsal Horn and Dorsal Root Ganglion via the Calcineurin/NFAT Pathway. Anticancer Agents Med Chem (2018) 18:1197–207. doi: 10.2174/1871520618666180525091158
47. Krøigård T, Metaxas A, Wirenfeldt M, Finsen B. Protective effect of ibuprofen in a rat model of chronic oxaliplatin-induced peripheral neuropathy. Exp Brain Res (2019) 237:2645–51. doi: 10.1007/s00221-019-05615-x
48. Illias AM, Gist AC, Zhang H, Kosturakis AK, Dougherty PM. Chemokine CCL2 and its receptor CCR2 in the dorsal root ganglion contribute to oxaliplatin-induced mechanical hypersensitivity. Pain (2018) 159:1308–16. doi: 10.1097/j.pain.0000000000001212
49. Wang J, Zhang XS, Tao R, Zhang J, Liu L, Jiang YH, et al. Upregulation of CX3CL1 mediated by NF-κB activation in dorsal root ganglion contributes to peripheral sensitization and chronic pain induced by oxaliplatin administration. Mol Pain (2017) 13:1744806917726256. doi: 10.1177/1744806917726256
50. Li YY, Li H, Liu ZL, Li Q, Qiu HW, Zeng LJ, et al. Activation of STAT3-mediated CXCL12 up-regulation in the dorsal root ganglion contributes to oxaliplatin-induced chronic pain. Mol Pain (2017) 13:1744806917747425. doi: 10.1177/1744806917747425
51. Brandolini L, Castelli V, Aramini A, Giorgio C, Bianchini G, Russo R, et al. DF2726A, a new IL-8 signalling inhibitor, is able to counteract chemotherapy-induced neuropathic pain. Sci Rep (2019) 9:11729. doi: 10.1038/s41598-019-48231-z
52. Jiang SP, Zhang ZD, Kang LM, Wang QH, Zhang L, Chen HP. Celecoxib reverts oxaliplatin-induced neuropathic pain through inhibiting PI3K/Akt2 pathway in the mouse dorsal root ganglion. Exp Neurol (2016) 275:11–6. doi: 10.1016/j.expneurol.2015.11.001
53. Maruta T, Nemoto T, Hidaka K, Koshida T, Shirasaka T, Yanagita T, et al. Upregulation of ERK phosphorylation in rat dorsal root ganglion neurons contributes to oxaliplatin-induced chronic neuropathic pain. PloS One (2019) 14:e0225586. doi: 10.1371/journal.pone.0225586
54. Poulsen JN, Larsen F, Duroux M, Gazerani P. Primary culture of trigeminal satellite glial cells: a cell-based platform to study morphology and function of peripheral glia. Int J Physiol Pathophysiol Pharmacol (2014) 6:1–12.
55. Wang W, Xiang P, Chew WS, Torta F, Bandla A, Lopez V, et al. Activation of sphingosine 1-phosphate receptor 2 attenuates chemotherapy-induced neuropathy. J Biol Chem (2020) 295:1143–52. doi: 10.1074/jbc.RA119.011699
56. Gauchan P, Andoh T, Kato A, Sasaki A, Kuraishi Y. Effects of the prostaglandin E1 analog limaprost on mechanical allodynia caused by chemotherapeutic agents in mice. J Pharmacol Sci (2009) 109:469–72. doi: 10.1254/jphs.08325sc
57. Warwick RA, Hanani M. The contribution of satellite glial cells to chemotherapy-induced neuropathic pain. Eur J Pain (2013) 17:571–80. doi: 10.1002/j.1532-2149.2012.00219.x
58. Schmitt L II, Leo M, Kutritz A, Kleinschnitz C, Hagenacker T. Activation and functional modulation of satellite glial cells by oxaliplatin lead to hyperexcitability of sensory neurons in vitro. Mol Cell Neurosci (2020) 105:103499. doi: 10.1016/j.mcn.2020.103499
59. Di Cesare Mannelli L, Pacini A, Micheli L, Tani A, Zanardelli M, Ghelardini C. Glial role in oxaliplatin-induced neuropathic pain. Exp Neurol (2014) 261:22–33. doi: 10.1016/j.expneurol.2014.06.016
60. Sharawy N, Rashed L, Youakim MF. Evaluation of multi-neuroprotective effects of erythropoietin using cisplatin induced peripheral neurotoxicity model. Exp Toxicol Pathol (2015) 67:315–22. doi: 10.1016/j.etp.2015.02.003
61. Oztürk G, Erdoğan E, Anlar O, Kösem M, Taşpinar M. Effect of leukemia inhibitory factor in experimental cisplatin neuropathy in mice. Cytokine (2005) 29:31–41. doi: 10.1016/j.cyto.2004.09.006
62. Imai S, Koyanagi M, Azimi Z, Nakazato Y, Matsumoto M, Ogihara T, et al. Taxanes and platinum derivatives impair Schwann cells via distinct mechanisms. Sci Rep (2017) 7:5947. doi: 10.1038/s41598-017-05784-1
63. Boyette-Davis J, Dougherty PM. Protection against oxaliplatin-induced mechanical hyperalgesia and intraepidermal nerve fiber loss by minocycline. Exp Neurol (2011) 229:353–7. doi: 10.1016/j.expneurol.2011.02.019
64. Sakamoto A, Andoh T, Kuraishi Y. Involvement of mast cells and proteinase-activated receptor 2 in oxaliplatin-induced mechanical allodynia in mice. Pharmacol Res (2016) 105:84–92. doi: 10.1016/j.phrs.2016.01.008
65. Andoh T, Sakamoto A, Kuraishi Y. 5-HT1A receptor agonists, xaliproden and tandospirone, inhibit the increase in the number of cutaneous mast cells involved in the exacerbation of mechanical allodynia in oxaliplatin-treated mice. J Pharmacol Sci (2016) 131:284–7. doi: 10.1016/j.jphs.2016.07.008
66. Hu LY, Zhou Y, Cui WQ, Hu XM, Du LX, Mi WL, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) dependent microglial activation promotes cisplatin-induced peripheral neuropathy in mice. Brain Behav Immun (2018) 68:132–45. doi: 10.1016/j.bbi.2017.10.011
67. Cataldo G, Erb SJ, Lunzer MM, Luong N, Akgün E, Portoghese PS, et al. The bivalent ligand MCC22 potently attenuates hyperalgesia in a mouse model of cisplatin-evoked neuropathic pain without tolerance or reward. Neuropharmacology (2019) 158:107598. doi: 10.1016/j.neuropharm.2019.04.004
68. Guindon J, Deng L, Fan B, Wager-Miller J, Hohmann AG. Optimization of a cisplatin model of chemotherapy-induced peripheral neuropathy in mice: use of vitamin C and sodium bicarbonate pretreatments to reduce nephrotoxicity and improve animal health status. Mol Pain (2014) 10:56. doi: 10.1186/1744-8069-10-56
69. Janes K, Wahlman C, Little JW, Doyle T, Tosh DK, Jacobson KA, et al. Spinal neuroimmune activation is independent of T-cell infiltration and attenuated by A3 adenosine receptor agonists in a model of oxaliplatin-induced peripheral neuropathy. Brain Behav Immun (2015) 44:91–9. doi: 10.1016/j.bbi.2014.08.010
70. Di Cesare Mannelli L, Pacini A, Matera C, Zanardelli M, Mello T, De Amici M, et al. Involvement of α7 nAChR subtype in rat oxaliplatin-induced neuropathy: effects of selective activation. Neuropharmacology (2014) 79:37–48. doi: 10.1016/j.neuropharm.2013.10.034
71. Micheli L, Mattoli L, Maidecchi A, Pacini A, Ghelardini C, Di Cesare Mannelli L. Effect of Vitis vinifera hydroalcoholic extract against oxaliplatin neurotoxicity: in vitro and in vivo evidence. Sci Rep (2018) 8:14364. doi: 10.1038/s41598-018-32691-w
72. Robinson CR, Zhang H, Dougherty PM. Astrocytes, but not microglia, are activated in oxaliplatin and bortezomib-induced peripheral neuropathy in the rat. Neuroscience (2014) 274:308–17. doi: 10.1016/j.neuroscience.2014.05.051
73. Zheng FY, Xiao WH, Bennett GJ. The response of spinal microglia to chemotherapy-evoked painful peripheral neuropathies is distinct from that evoked by traumatic nerve injuries. Neuroscience (2011) 176:447–54. doi: 10.1016/j.neuroscience.2010.12.052
74. Jung Y, Lee JH, Kim W, Yoon SH, Kim SK. Anti-allodynic effect of Buja in a rat model of oxaliplatin-induced peripheral neuropathy via spinal astrocytes and pro-inflammatory cytokines suppression. BMC Complement Altern Med (2017) 17:48. doi: 10.1186/s12906-017-1556-z
75. Yeo JH, Yoon SY, Kim SJ, Oh SB, Lee JH, Beitz AJ, et al. Clonidine, an alpha-2 adrenoceptor agonist relieves mechanical allodynia in oxaliplatin-induced neuropathic mice; potentiation by spinal p38 MAPK inhibition without motor dysfunction and hypotension. Int J Cancer (2016) 138:2466–76. doi: 10.1002/ijc.29980
76. Deng B, Jia L, Pan L, Song A, Wang Y, Tan H, et al. Wen-Luo-Tong Prevents Glial Activation and Nociceptive Sensitization in a Rat Model of Oxaliplatin-Induced Neuropathic Pain. Evid Based Complement Alternat Med (2016) 2016:3629489. doi: 10.1155/2016/3629489
77. Areti A, Komirishetty P, Kalvala AK, Nellaiappan K, Kumar A. Rosmarinic Acid Mitigates Mitochondrial Dysfunction and Spinal Glial Activation in Oxaliplatin-induced Peripheral Neuropathy. Mol Neurobiol (2018) 55:7463–75. doi: 10.1007/s12035-018-0920-4
78. Tonkin RS, Bowles C, Perera CJ, Keating BA, Makker PGS, Duffy SS, et al. Attenuation of mechanical pain hypersensitivity by treatment with Peptide5, a connexin-43 mimetic peptide, involves inhibition of NLRP3 inflammasome in nerve-injured mice. Exp Neurol (2018) 300:1–12. doi: 10.1016/j.expneurol.2017.10.016
79. Yoon SY, Robinson CR, Zhang H, Dougherty PM. Spinal astrocyte gap junctions contribute to oxaliplatin-induced mechanical hypersensitivity. J Pain (2013) 14:205–14. doi: 10.1016/j.jpain.2012.11.002
80. Pacini A, Micheli L, Maresca M, Branca JJ, McIntosh JM, Ghelardini C, et al. The α9α10 nicotinic receptor antagonist α-conotoxin RgIA prevents neuropathic pain induced by oxaliplatin treatment. Exp Neurol (2016) 282:37–48. doi: 10.1016/j.expneurol.2016.04.022
81. Wahlman C, Doyle TM, Little JW, Luongo L, Janes K, Chen Z, et al. Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels through astrocyte-dependent mechanisms. Pain (2018) 159:1025–34. doi: 10.1097/j.pain.0000000000001177
82. Gao M, Yan X, Weng HR. Inhibition of glycogen synthase kinase 3β activity with lithium prevents and attenuates paclitaxel-induced neuropathic pain. Neuroscience (2013) 254:301–11. doi: 10.1016/j.neuroscience.2013.09.033
83. Mo M, Erdelyi I, Szigeti-Buck K, Benbow JH, Ehrlich BE. Prevention of paclitaxel-induced peripheral neuropathy by lithium pretreatment. FASEB J (2012) 26:4696–709. doi: 10.1096/fj.12-214643
84. Huang ZZ, Li D, Ou-Yang HD, Liu CC, Liu XG, Ma C, et al. Cerebrospinal Fluid Oxaliplatin Contributes to the Acute Pain Induced by Systemic Administration of Oxaliplatin. Anesthesiology (2016) 124:1109–21. doi: 10.1097/ALN.0000000000001084
85. Wang YS, Li YY, Cui W, Li LB, Zhang ZC, Tian BP, et al. Melatonin Attenuates Pain Hypersensitivity and Decreases Astrocyte-Mediated Spinal Neuroinflammation in a Rat Model of Oxaliplatin-Induced Pain. Inflammation (2017) 40:2052–61. doi: 10.1007/s10753-017-0645-y
86. Lee JH, Kim W. The Role of Satellite Glial Cells, Astrocytes, and Microglia in Oxaliplatin-Induced Neuropathic Pain. Biomedicines (2020) 8. doi: 10.3390/biomedicines8090324
87. Di Cesare Mannelli L, Tenci B, Zanardelli M, Failli P, Ghelardini C. α7 Nicotinic Receptor Promotes the Neuroprotective Functions of Astrocytes against Oxaliplatin Neurotoxicity. Neural Plast (2015) 2015:396908. doi: 10.1155/2015/396908
88. Khadrawy YA, El-Gizawy MM, Sorour SM, Sawie HG, Hosny EN. Effect of curcumin nanoparticles on the cisplatin-induced neurotoxicity in rat. Drug Chem Toxicol (2019) 42:194–202. doi: 10.1080/01480545.2018.1504058
89. Arafa MH, Atteia HH. Protective Role of Epigallocatechin Gallate in a Rat Model of Cisplatin-Induced Cerebral Inflammation and Oxidative Damage: Impact of Modulating NF-κB and Nrf2. Neurotox Res (2020) 37:380–96. doi: 10.1007/s12640-019-00095-x
90. Jangra A, Kwatra M, Singh T, Pant R, Kushwah P, Ahmed S, et al. Edaravone alleviates cisplatin-induced neurobehavioral deficits via modulation of oxidative stress and inflammatory mediators in the rat hippocampus. Eur J Pharmacol (2016) 791:51–61. doi: 10.1016/j.ejphar.2016.08.003
91. Chen C, Zhang H, Xu H, Zheng Y, Wu T, Lian Y. Ginsenoside Rb1 ameliorates cisplatin-induced learning and memory impairments. J Ginseng Res (2019) 43:499–507. doi: 10.1016/j.jgr.2017.07.009
92. Xu D, Zhao H, Gao H, Liu D, Li J. Participation of pro-inflammatory cytokines in neuropathic pain evoked by chemotherapeutic oxaliplatin via central GABAergic pathway. Mol Pain (2018) 14:1744806918783535. doi: 10.1177/1744806918783535
93. Gornstein EL, Schwarz TL. Neurotoxic mechanisms of paclitaxel are local to the distal axon and independent of transport defects. Exp Neurol (2017) 288:153–66. doi: 10.1016/j.expneurol.2016.11.015
94. Miaskowski C, Topp K, Conley YP, Paul SM, Melisko M, Schumacher M, et al. Perturbations in neuroinflammatory pathways are associated with paclitaxel-induced peripheral neuropathy in breast cancer survivors. J Neuroimmunol (2019) 335:577019. doi: 10.1016/j.jneuroim.2019.577019
95. Chon HJ, Rha SY, Im CK, Kim C, Hong MH, Kim HR, et al. Docetaxel versus paclitaxel combined with 5-FU and leucovorin in advanced gastric cancer: combined analysis of two phase II trials. Cancer Res Treat (2009) 41:196–204. doi: 10.4143/crt.2009.41.4.196
96. Krukowski K, Eijkelkamp N, Laumet G, Hack CE, Li Y, Dougherty PM, et al. CD8+ T Cells and Endogenous IL-10 Are Required for Resolution of Chemotherapy-Induced Neuropathic Pain. J Neurosci (2016) 36:11074–83. doi: 10.1523/JNEUROSCI.3708-15.2016
97. Al-Mazidi S, Alotaibi M, Nedjadi T, Chaudhary A, Alzoghaibi M, Djouhri L. Blocking of cytokines signalling attenuates evoked and spontaneous neuropathic pain behaviours in the paclitaxel rat model of chemotherapy-induced neuropathy. Eur J Pain (2018) 22:810–21. doi: 10.1002/ejp.1169
98. Chen LH, Yeh YM, Chen YF, Hsu YH, Wang HH, Lin PC, et al. Targeting interleukin-20 alleviates paclitaxel-induced peripheral neuropathy. Pain (2020) 161:1237–54. doi: 10.1097/j.pain.0000000000001831
99. Brandolini L, Benedetti E, Ruffini PA, Russo R, Cristiano L, Antonosante A, et al. CXCR1/2 pathways in paclitaxel-induced neuropathic pain. Oncotarget (2017) 8:23188–201. doi: 10.18632/oncotarget.15533
100. Wang YC, Li N, Zhao Y, Zhang LJ. Effects of female sex hormones on chemotherapeutic paclitaxel-induced neuropathic pain and involvement of inflammatory signal. J Biol Regul Homeost Agents (2018) 32:1157–63.
101. Zhang H, Boyette-Davis JA, Kosturakis AK, Li Y, Yoon SY, Walters ET, et al. Induction of monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel-induced peripheral neuropathy. J Pain (2013) 14:1031–44. doi: 10.1016/j.jpain.2013.03.012
102. Zhang X, Jiang N, Li J, Zhang D, Lv X. Rapamycin alleviates proinflammatory cytokines and nociceptive behavior induced by chemotherapeutic paclitaxel. Neurol Res (2019) 41:52–9. doi: 10.1080/01616412.2018.1531199
103. Kim E, Hwang SH, Kim HK, Abdi S. Losartan, an Angiotensin II Type 1 Receptor Antagonist, Alleviates Mechanical Hyperalgesia in a Rat Model of Chemotherapy-Induced Neuropathic Pain by Inhibiting Inflammatory Cytokines in the Dorsal Root Ganglia. Mol Neurobiol (2019) 56:7408–19. doi: 10.1007/s12035-019-1616-0
104. Tonello R, Lee SH, Berta T. Monoclonal Antibody Targeting the Matrix Metalloproteinase 9 Prevents and Reverses Paclitaxel-Induced Peripheral Neuropathy in Mice. J Pain (2019) 20:515–27. doi: 10.1016/j.jpain.2018.11.003
105. Wu P, Chen Y. Evodiamine ameliorates paclitaxel-induced neuropathic pain by inhibiting inflammation and maintaining mitochondrial anti-oxidant functions. Hum Cell (2019) 32:251–9. doi: 10.1007/s13577-019-00238-4
106. Al-Massri KF, Ahmed LA, El-Abhar HS. Pregabalin and lacosamide ameliorate paclitaxel-induced peripheral neuropathy via inhibition of JAK/STAT signaling pathway and Notch-1 receptor. Neurochem Int (2018) 120:164–71. doi: 10.1016/j.neuint.2018.08.007
107. Li D, Chen H, Luo XH, Sun Y, Xia W, Xiong YC. CX3CR1-Mediated Akt1 Activation Contributes to the Paclitaxel-Induced Painful Peripheral Neuropathy in Rats. Neurochem Res (2016) 41:1305–14. doi: 10.1007/s11064-016-1827-y
108. Miao H, Xu J, Xu D, Ma X, Zhao X, Liu L. Nociceptive behavior induced by chemotherapeutic paclitaxel and beneficial role of antioxidative pathways. Physiol Res (2019) 68:491–500. doi: 10.33549/physiolres.933939
109. Manjavachi MN, Passos GF, Trevisan G, Araújo SB, Pontes JP, Fernandes ES, et al. Spinal blockage of CXCL1 and its receptor CXCR2 inhibits paclitaxel-induced peripheral neuropathy in mice. Neuropharmacology (2019) 151:136–43. doi: 10.1016/j.neuropharm.2019.04.014
110. Boyette-Davis J, Xin W, Zhang H, Dougherty PM. Intraepidermal nerve fiber loss corresponds to the development of taxol-induced hyperalgesia and can be prevented by treatment with minocycline. Pain (2011) 152:308–13. doi: 10.1016/j.pain.2010.10.030
111. Liu CC, Lu N, Cui Y, Yang T, Zhao ZQ, Xin WJ, et al. Prevention of paclitaxel-induced allodynia by minocycline: Effect on loss of peripheral nerve fibers and infiltration of macrophages in rats. Mol Pain (2010) 6:76. doi: 10.1186/1744-8069-6-76
112. Chen YF, Chen LH, Yeh YM, Wu PY, Chang LY, Chang JY, et al. Minoxidil is a potential neuroprotective drug for paclitaxel-induced peripheral neuropathy. Sci Rep (2017) 7:45366. doi: 10.1038/srep45366
113. Kalynovska N, Diallo M, Sotakova-Kasparova D, Palecek J. Losartan attenuates neuroinflammation and neuropathic pain in paclitaxel-induced peripheral neuropathy. J Cell Mol Med (2020) 24:7949–58. doi: 10.1111/jcmm.15427
114. Peters CM, Jimenez-Andrade JM, Jonas BM, Sevcik MA, Koewler NJ, Ghilardi JR, et al. Intravenous paclitaxel administration in the rat induces a peripheral sensory neuropathy characterized by macrophage infiltration and injury to sensory neurons and their supporting cells. Exp Neurol (2007) 203:42–54. doi: 10.1016/j.expneurol.2006.07.022
115. Zhang H, Li Y, de Carvalho-Barbosa M, Kavelaars A, Heijnen CJ, Albrecht PJ, et al. Dorsal Root Ganglion Infiltration by Macrophages Contributes to Paclitaxel Chemotherapy-Induced Peripheral Neuropathy. J Pain (2016) 17:775–86. doi: 10.1016/j.jpain.2016.02.011
116. Sekiguchi F, Domoto R, Nakashima K, Yamasoba D, Yamanishi H, Tsubota M, et al. Paclitaxel-induced HMGB1 release from macrophages and its implication for peripheral neuropathy in mice: Evidence for a neuroimmune crosstalk. Neuropharmacology (2018) 141:201–13. doi: 10.1016/j.neuropharm.2018.08.040
117. Tasnim A, Rammelkamp Z, Slusher AB, Wozniak K, Slusher BS, Farah MH. Paclitaxel causes degeneration of both central and peripheral axon branches of dorsal root ganglia in mice. BMC Neurosci (2016) 17:47. doi: 10.1186/s12868-016-0285-4
118. Jia M, Wu C, Gao F, Xiang H, Sun N, Peng P, et al. Activation of NLRP3 inflammasome in peripheral nerve contributes to paclitaxel-induced neuropathic pain. Mol Pain (2017) 13:1744806917719804. doi: 10.1177/1744806917719804
119. Miranda HF, Sierralta F, Aranda N, Poblete P, Castillo RL, Noriega V, et al. Antinociception induced by rosuvastatin in murine neuropathic pain. Pharmacol Rep (2018) 70:503–8. doi: 10.1016/j.pharep.2017.11.012
120. Huehnchen P, Muenzfeld H, Boehmerle W, Endres M. Blockade of IL-6 signaling prevents paclitaxel-induced neuropathy in C57Bl/6 mice. Cell Death Dis (2020) 11:45. doi: 10.1038/s41419-020-2239-0
121. Li D, Huang ZZ, Ling YZ, Wei JY, Cui Y, Zhang XZ, et al. Up-regulation of CX3CL1 via Nuclear Factor-κB-dependent Histone Acetylation Is Involved in Paclitaxel-induced Peripheral Neuropathy. Anesthesiology (2015) 122:1142–51. doi: 10.1097/ALN.0000000000000560
122. Luo H, Liu HZ, Zhang WW, Matsuda M, Lv N, Chen G, et al. Interleukin-17 Regulates Neuron-Glial Communications, Synaptic Transmission, and Neuropathic Pain after Chemotherapy. Cell Rep (2019) 29:2384–97.e5. doi: 10.1016/j.celrep.2019.10.085
123. Deng L, Guindon J, Cornett BL, Makriyannis A, Mackie K, Hohmann AG. Chronic cannabinoid receptor 2 activation reverses paclitaxel neuropathy without tolerance or cannabinoid receptor 1-dependent withdrawal. Biol Psychiatry (2015) 77:475–87. doi: 10.1016/j.biopsych.2014.04.009
124. Doyle T, Chen Z, Muscoli C, Bryant L, Esposito E, Cuzzocrea S, et al. Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. J Neurosci (2012) 32:6149–60. doi: 10.1523/JNEUROSCI.6343-11.2012
125. Rosa AC, Fantozzi R. The role of histamine in neurogenic inflammation. Br J Pharmacol (2013) 170:38–45. doi: 10.1111/bph.12266
126. Gao W, Zan Y, Wang ZJ, Hu XY, Huang F. Quercetin ameliorates paclitaxel-induced neuropathic pain by stabilizing mast cells, and subsequently blocking PKCϵ-dependent activation of TRPV1. Acta Pharmacol Sin (2016) 37:1166–77. doi: 10.1038/aps.2016.58
127. Yao L, Kan EM, Kaur C, Dheen ST, Hao A, Lu J, et al. Notch-1 signaling regulates microglia activation via NF-κB pathway after hypoxic exposure in vivo and in vitro. PloS One (2013) 8:e78439. doi: 10.1371/journal.pone.0078439
128. Wei Z, Chigurupati S, Arumugam TV, Jo DG, Li H, Chan SL. Notch activation enhances the microglia-mediated inflammatory response associated with focal cerebral ischemia. Stroke (2011) 42:2589–94. doi: 10.1161/STROKEAHA.111.614834
129. Cao Q, Li P, Lu J, Dheen ST, Kaur C, Ling EA. Nuclear factor-κB/p65 responds to changes in the Notch signaling pathway in murine BV-2 cells and in amoeboid microglia in postnatal rats treated with the γ-secretase complex blocker DAPT. J Neurosci Res (2010) 88:2701–14. doi: 10.1002/jnr.22429
130. Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun (2007) 21:642–51. doi: 10.1016/j.bbi.2006.11.003
131. Xie K, Qiao F, Sun Y, Wang G, Hou L. Notch signaling activation is critical to the development of neuropathic pain. BMC Anesthesiol (2015) 15:41. doi: 10.1186/s12871-015-0021-0
132. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J (2003) 374:1–20. doi: 10.1042/BJ20030407
133. Al-Massri KF, Ahmed LA, El-Abhar HS. Mesenchymal stem cells therapy enhances the efficacy of pregabalin and prevents its motor impairment in paclitaxel-induced neuropathy in rats: Role of Notch1 receptor and JAK/STAT signaling pathway. Behav Brain Res (2019) 360:303–11. doi: 10.1016/j.bbr.2018.12.013
134. Gui Y, Zhang J, Chen L, Duan S, Tang J, Xu W, et al. Icariin, a flavonoid with anti-cancer effects, alleviated paclitaxel-induced neuropathic pain in a SIRT1-dependent manner. Mol Pain (2018) 14:1744806918768970. doi: 10.1177/1744806918768970
135. Syed SN, Weigert A, Brüne B. Sphingosine Kinases are Involved in Macrophage NLRP3 Inflammasome Transcriptional Induction. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21134733
136. Janes K, Little JW, Li C, Bryant L, Chen C, Chen Z, et al. The development and maintenance of paclitaxel-induced neuropathic pain require activation of the sphingosine 1-phosphate receptor subtype 1. J Biol Chem (2014) 289:21082–97. doi: 10.1074/jbc.M114.569574
137. Ren TH, Lv MM, An XM, Leung WK, Seto WK. Activation of adenosine A3 receptor inhibits inflammatory cytokine production in colonic mucosa of patients with ulcerative colitis by down-regulating the nuclear factor-kappa B signaling. J Dig Dis (2020) 21:38–45. doi: 10.1111/1751-2980.12831
138. Pevida M, Lastra A, Hidalgo A, Baamonde A, Menéndez L. Spinal CCL2 and microglial activation are involved in paclitaxel-evoked cold hyperalgesia. Brain Res Bull (2013) 95:21–7. doi: 10.1016/j.brainresbull.2013.03.005
139. Jean-Gilles L, Gran B, Constantinescu CS. Interaction between cytokines, cannabinoids and the nervous system. Immunobiology (2010) 215:606–10. doi: 10.1016/j.imbio.2009.12.006
140. Salio C, Doly S, Fischer J, Franzoni MF, Conrath M. Neuronal and astrocytic localization of the cannabinoid receptor-1 in the dorsal horn of the rat spinal cord. Neurosci Lett (2002) 329:13–6. doi: 10.1016/s0304-3940(02)00549-9
141. Romero-Sandoval A, Nutile-McMenemy N, DeLeo JA. Spinal microglial and perivascular cell cannabinoid receptor type 2 activation reduces behavioral hypersensitivity without tolerance after peripheral nerve injury. Anesthesiology (2008) 108:722–34. doi: 10.1097/ALN.0b013e318167af74
142. Deng L, Guindon J, Vemuri VK, Thakur GA, White FA, Makriyannis A, et al. The maintenance of cisplatin- and paclitaxel-induced mechanical and cold allodynia is suppressed by cannabinoid CB₂ receptor activation and independent of CXCR4 signaling in models of chemotherapy-induced peripheral neuropathy. Mol Pain (2012) 8:71. doi: 10.1186/1744-8069-8-71
143. Naguib M, Xu JJ, Diaz P, Brown DL, Cogdell D, Bie B, et al. Prevention of paclitaxel-induced neuropathy through activation of the central cannabinoid type 2 receptor system. Anesth Analg (2012) 114:1104–20. doi: 10.1213/ANE.0b013e31824b0191
144. Wu J, Hocevar M, Bie B, Foss JF, Naguib M. Cannabinoid Type 2 Receptor System Modulates Paclitaxel-Induced Microglial Dysregulation and Central Sensitization in Rats. J Pain (2019) 20:501–14. doi: 10.1016/j.jpain.2018.10.007
145. Xu JJ, Diaz P, Bie B, Astruc-Diaz F, Wu J, Yang H, et al. Spinal gene expression profiling and pathways analysis of a CB2 agonist (MDA7)-targeted prevention of paclitaxel-induced neuropathy. Neuroscience (2014) 260:185–94. doi: 10.1016/j.neuroscience.2013.12.028
146. Burgos E, Gómez-Nicola D, Pascual D, Martín M II, Nieto-Sampedro M, Goicoechea C. Cannabinoid agonist WIN 55,212-2 prevents the development of paclitaxel-induced peripheral neuropathy in rats. Possible involvement of spinal glial cells. Eur J Pharmacol (2012) 682:62–72. doi: 10.1016/j.ejphar.2012.02.008
147. Segat GC, Manjavachi MN, Matias DO, Passos GF, Freitas CS, Costa R, et al. Antiallodynic effect of β-caryophyllene on paclitaxel-induced peripheral neuropathy in mice. Neuropharmacology (2017) 125:207–19. doi: 10.1016/j.neuropharm.2017.07.015
148. Ochi-ishi R, Nagata K, Inoue T, Tozaki-Saitoh H, Tsuda M, Inoue K. Involvement of the chemokine CCL3 and the purinoceptor P2X7 in the spinal cord in paclitaxel-induced mechanical allodynia. Mol Pain (2014) 10:53. doi: 10.1186/1744-8069-10-53
149. Janes K, Esposito E, Doyle T, Cuzzocrea S, Tosh DK, Jacobson KA, et al. A3 adenosine receptor agonist prevents the development of paclitaxel-induced neuropathic pain by modulating spinal glial-restricted redox-dependent signaling pathways. Pain (2014) 155:2560–7. doi: 10.1016/j.pain.2014.09.016
150. Singhmar P, Huo X, Li Y, Dougherty PM, Mei F, Cheng X, et al. Orally active Epac inhibitor reverses mechanical allodynia and loss of intraepidermal nerve fibers in a mouse model of chemotherapy-induced peripheral neuropathy. Pain (2018) 159:884–93. doi: 10.1097/j.pain.0000000000001160
151. Xu Y, Cheng G, Zhu Y, Zhang X, Pu S, Wu J, et al. Anti-nociceptive roles of the glia-specific metabolic inhibitor fluorocitrate in paclitaxel-evoked neuropathic pain. Acta Biochim Biophys Sin (Shanghai) (2016) 48:902–8. doi: 10.1093/abbs/gmw083
152. Ruiz-Medina J, Baulies A, Bura SA, Valverde O. Paclitaxel-induced neuropathic pain is age dependent and devolves on glial response. Eur J Pain (2013) 17:75–85. doi: 10.1002/j.1532-2149.2012.00172.x
153. Bianco MR, Cirillo G, Petrosino V, Marcello L, Soleti A, Merizzi G, et al. Neuropathic pain and reactive gliosis are reversed by dialdehydic compound in neuropathic pain rat models. Neurosci Lett (2012) 530:85–90. doi: 10.1016/j.neulet.2012.08.088
154. Zhang H, Yoon SY, Dougherty PM. Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of Paclitaxel-induced painful neuropathy. J Pain (2012) 13:293–303. doi: 10.1016/j.jpain.2011.12.002
155. Nicotra L, Loram LC, Watkins LR, Hutchinson MR. Toll-like receptors in chronic pain. Exp Neurol (2012) 234:316–29. doi: 10.1016/j.expneurol.2011.09.038
156. Montague K, Malcangio M. The Therapeutic Potential of Monocyte/Macrophage Manipulation in the Treatment of Chemotherapy-Induced Painful Neuropathy. Front Mol Neurosci (2017) 10:397:397. doi: 10.3389/fnmol.2017.00397
157. Ochoa-Cortes F, Ramos-Lomas T, Miranda-Morales M, Spreadbury I, Ibeakanma C, Barajas-Lopez C, et al. Bacterial cell products signal to mouse colonic nociceptive dorsal root ganglia neurons. Am J Physiol Gastrointest Liver Physiol (2010) 299:G723–32. doi: 10.1152/ajpgi.00494.2009
158. Li Y, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-like receptor 4 signaling contributes to Paclitaxel-induced peripheral neuropathy. J Pain (2014) 15:712–25. doi: 10.1016/j.jpain.2014.04.001
159. Li Y, Yin C, Li X, Liu B, Wang J, Zheng X, et al. Electroacupuncture Alleviates Paclitaxel-Induced Peripheral Neuropathic Pain in Rats via Suppressing TLR4 Signaling and TRPV1 Upregulation in Sensory Neurons. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20235917
160. Luo X, Huh Y, Bang S, He Q, Zhang L, Matsuda M, et al. Macrophage Toll-like Receptor 9 Contributes to Chemotherapy-Induced Neuropathic Pain in Male Mice. J Neurosci (2019) 39:6848–64. doi: 10.1523/JNEUROSCI.3257-18.2019
161. Khalilzadeh M, Panahi G, Rashidian A, Hadian MR, Abdollahi A, Afshari K, et al. The protective effects of sumatriptan on vincristine - induced peripheral neuropathy in a rat model. Neurotoxicology (2018) 67:279–86. doi: 10.1016/j.neuro.2018.06.012
162. Argyriou AA, Bruna J, Genazzani AA, Cavaletti G. Chemotherapy-induced peripheral neurotoxicity: management informed by pharmacogenetics. Nat Rev Neurol (2017) 13:492–504. doi: 10.1038/nrneurol.2017.88
163. Madsen ML, Due H, Ejskjær N, Jensen P, Madsen J, Dybkær K. Aspects of vincristine-induced neuropathy in hematologic malignancies: a systematic review. Cancer Chemother Pharmacol (2019) 84:471–85. doi: 10.1007/s00280-019-03884-5
164. Singh G, Singh A, Singh P, Bhatti R. Bergapten Ameliorates Vincristine-Induced Peripheral Neuropathy by Inhibition of Inflammatory Cytokines and NFκB Signaling. ACS Chem Neurosci (2019) 10:3008–17. doi: 10.1021/acschemneuro.9b00206
165. Barzegar-Fallah A, Alimoradi H, Mehrzadi S, Barzegar-Fallah N, Zendedel A, Abbasi A, et al. The neuroprotective effect of tropisetron on vincristine-induced neurotoxicity. Neurotoxicology (2014) 41:1–8. doi: 10.1016/j.neuro.2013.12.002
166. Muthuraman A, Singh N, Jaggi AS. Protective effect of Acorus calamus L. @ in rat model of vincristine induced painful neuropathy: an evidence of anti-inflammatory and anti-oxidative activity. Food Chem Toxicol (2011) 49:2557–63. doi: 10.1016/j.fct.2011.06.069
167. Gautam M, Ramanathan M. Saponins of Tribulus terrestris attenuated neuropathic pain induced with vincristine through central and peripheral mechanism. Inflammopharmacology (2019) 27:761–72. doi: 10.1007/s10787-018-0502-0
168. Gong SS, Li YX, Zhang MT, Du J, Ma PS, Yao WX, et al. Neuroprotective Effect of Matrine in Mouse Model of Vincristine-Induced Neuropathic Pain. Neurochem Res (2016) 41:3147–59. doi: 10.1007/s11064-016-2040-8
169. Qin B, Li Y, Liu X, Gong D, Zheng W. Notch activation enhances microglial CX3CR1/P38 MAPK pathway in rats model of vincristine-induced peripheral neuropathy. Neurosci Lett (2020) 715:134624. doi: 10.1016/j.neulet.2019.134624
170. Gautam M, Ramanathan M. Ameliorative potential of flavonoids of. Nutr Neurosci (2019), 1–11. doi: 10.1080/1028415X.2019.1627768
171. Greeshma N, Prasanth KG, Balaji B. Tetrahydrocurcumin exerts protective effect on vincristine induced neuropathy: Behavioral, biochemical, neurophysiological and histological evidence. Chem Biol Interact (2015) 238:118–28. doi: 10.1016/j.cbi.2015.06.025
172. Shi Q, Cai X, Shi G, Lv X, Yu J, Wang F. Interleukin-4 protects from chemotherapy-induced peripheral neuropathy in mice modal via the stimulation of IL-4/STAT6 signaling. Acta Cir Bras (2018) 33:491–8. doi: 10.1590/s0102-865020180060000003
173. Kiguchi N, Maeda T, Kobayashi Y, Kondo T, Ozaki M, Kishioka S. The critical role of invading peripheral macrophage-derived interleukin-6 in vincristine-induced mechanical allodynia in mice. Eur J Pharmacol (2008) 592:87–92. doi: 10.1016/j.ejphar.2008.07.008
174. Hansen N, Uçeyler N, Palm F, Zelenka M, Biko L, Lesch KP, et al. Serotonin transporter deficiency protects mice from mechanical allodynia and heat hyperalgesia in vincristine neuropathy. Neurosci Lett (2011) 495:93–7. doi: 10.1016/j.neulet.2011.03.035
175. Old EA, Nadkarni S, Grist J, Gentry C, Bevan S, Kim KW, et al. Monocytes expressing CX3CR1 orchestrate the development of vincristine-induced pain. J Clin Invest (2014) 124:2023–36. doi: 10.1172/JCI71389
176. Montague-Cardoso K, Pitcher T, Chisolm K, Salera G, Lindstrom E, Hewitt E, et al. Changes in vascular permeability in the spinal cord contribute to chemotherapy-induced neuropathic pain. Brain Behav Immun (2020) 83:248–59. doi: 10.1016/j.bbi.2019.10.018
177. Clark AK, Yip PK, Malcangio M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J Neurosci (2009) 29:6945–54. doi: 10.1523/JNEUROSCI.0828-09.2009
178. Clark AK, Gruber-Schoffnegger D, Drdla-Schutting R, Gerhold KJ, Malcangio M, Sandkühler J. Selective activation of microglia facilitates synaptic strength. J Neurosci (2015) 35:4552–70. doi: 10.1523/JNEUROSCI.2061-14.2015
179. Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A (2007) 104:10655–60. doi: 10.1073/pnas.0610811104
180. Moschetti G, Amodeo G, Paladini MS, Molteni R, Balboni G, Panerai A, et al. Prokineticin 2 promotes and sustains neuroinflammation in vincristine treated mice. Focus on pain and emotional like behavior. Brain Behav Immun (2019) 82:422–31. doi: 10.1016/j.bbi.2019.09.012
181. Negri L, Lattanzi R. Bv8/PK2 and prokineticin receptors: a druggable pronociceptive system. Curr Opin Pharmacol (2012) 12:62–6. doi: 10.1016/j.coph.2011.10.023
182. Negri L, Lattanzi R, Giannini E, Melchiorri P. Bv8/Prokineticin proteins and their receptors. Life Sci (2007) 81:1103–16. doi: 10.1016/j.lfs.2007.08.011
183. Franchi S, Sacerdote P, Panerai A. The prokineticin system: an interface between neural inflammation and pain. Neurol Sci (2017) 38:27–30. doi: 10.1007/s10072-017-2875-z
184. Zhou L, Hu Y, Li C, Yan Y, Ao L, Yu B, et al. Levo-corydalmine alleviates vincristine-induced neuropathic pain in mice by inhibiting an NF-kappa B-dependent CXCL1/CXCR2 signaling pathway. Neuropharmacology (2018) 135:34–47. doi: 10.1016/j.neuropharm.2018.03.004
185. Silva RL, Lopes AH, Guimarães RM, Cunha TM. CXCL1/CXCR2 signaling in pathological pain: Role in peripheral and central sensitization. Neurobiol Dis (2017) 105:109–16. doi: 10.1016/j.nbd.2017.06.001
186. Popiolek-Barczyk K, Makuch W, Rojewska E, Pilat D, Mika J. Inhibition of intracellular signaling pathways NF-κB and MEK1/2 attenuates neuropathic pain development and enhances morphine analgesia. Pharmacol Rep (2014) 66:845–51. doi: 10.1016/j.pharep.2014.05.001
187. Xu J, Wang W, Zhong XX, Feng Y, Wei X, Liu XG. EXPRESS: Methylcobalamin ameliorates neuropathic pain induced by vincristine in rats: Effect on loss of peripheral nerve fibers and imbalance of cytokines in the spinal dorsal horn. Mol Pain (2016) 12:1–14. doi: 10.1177/1744806916657089
188. Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol (2010) 80:1895–903. doi: 10.1016/j.bcp.2010.07.014
189. Shen Y, Zhang ZJ, Zhu MD, Jiang BC, Yang T, Gao YJ. Exogenous induction of HO-1 alleviates vincristine-induced neuropathic pain by reducing spinal glial activation in mice. Neurobiol Dis (2015) 79:100–10. doi: 10.1016/j.nbd.2015.04.012
190. Kiguchi N, Maeda T, Kobayashi Y, Kishioka S. Up-regulation of tumor necrosis factor-alpha in spinal cord contributes to vincristine-induced mechanical allodynia in mice. Neurosci Lett (2008) 445:140–3. doi: 10.1016/j.neulet.2008.09.009
191. Starobova H, Mueller A, Allavena R, Lohman RJ, Sweet MJ, Vetter I. Minocycline Prevents the Development of Mechanical Allodynia in Mouse Models of Vincristine-Induced Peripheral Neuropathy. Front Neurosci (2019) 13:653. doi: 10.3389/fnins.2019.00653
192. Robinson CR, Zhang H, Dougherty PM. Altered discharges of spinal neurons parallel the behavioral phenotype shown by rats with bortezomib related chemotherapy induced peripheral neuropathy. Brain Res (2014) 1574:6–13. doi: 10.1016/j.brainres.2014.06.013
193. Broyl A, Jongen JL, Sonneveld P. General aspects and mechanisms of peripheral neuropathy associated with bortezomib in patients with newly diagnosed multiple myeloma. Semin Hematol (2012) 49:249–57. doi: 10.1053/j.seminhematol.2012.04.001
194. Ravaglia S, Corso A, Piccolo G, Lozza A, Alfonsi E, Mangiacavalli S, et al. Immune-mediated neuropathies in myeloma patients treated with bortezomib. Clin Neurophysiol (2008) 119:2507–12. doi: 10.1016/j.clinph.2008.08.007
195. Schmitt S, Goldschmidt H, Storch-Hagenlocher B, Pham M, Fingerle-Rowson G, Ho AD, et al. Inflammatory autoimmune neuropathy, presumably induced by bortezomib, in a patient suffering from multiple myeloma. Int J Hematol (2011) 93:791–4. doi: 10.1007/s12185-011-0847-2
196. Jeter A, Kang Y. Immune modulation therapy in the management of bortezomib-induced peripheral neuropathy. Exp Hematol Oncol (2012) 1:20. doi: 10.1186/2162-3619-1-20
197. Meregalli C, Marjanovic I, Scali C, Monza L, Spinoni N, Galliani C, et al. High-dose intravenous immunoglobulins reduce nerve macrophage infiltration and the severity of bortezomib-induced peripheral neurotoxicity in rats. J Neuroinflammation (2018) 15:232. doi: 10.1186/s12974-018-1270-x
198. Moschetti G, Amodeo G, Maftei D, Lattanzi R, Procacci P, Sartori P, et al. Targeting prokineticin system counteracts hypersensitivity, neuroinflammation, and tissue damage in a mouse model of bortezomib-induced peripheral neuropathy. J Neuroinflammation (2019) 16:89. doi: 10.1186/s12974-019-1461-0
199. Singh SK, Spiegel S. Sphingosine-1-phosphate signaling: A novel target for simultaneous adjuvant treatment of triple negative breast cancer and chemotherapy-induced neuropathic pain. Adv Biol Regul (2020) 75:100670. doi: 10.1016/j.jbior.2019.100670
200. Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci (2009) 10:23–36. doi: 10.1038/nrn2533
201. Cavaletti G, Gilardini A, Canta A, Rigamonti L, Rodriguez-Menendez V, Ceresa C, et al. Bortezomib-induced peripheral neurotoxicity: a neurophysiological and pathological study in the rat. Exp Neurol (2007) 204:317–25. doi: 10.1016/j.expneurol.2006.11.010
202. Alé A, Bruna J, Morell M, van de Velde H, Monbaliu J, Navarro X, et al. Treatment with anti-TNF alpha protects against the neuropathy induced by the proteasome inhibitor bortezomib in a mouse model. Exp Neurol (2014) 253:165–73. doi: 10.1016/j.expneurol.2013.12.020
203. Chiorazzi A, Canta A, Meregalli C, Carozzi V, Sala B, Oggioni N, et al. Antibody against tumor necrosis factor-α reduces bortezomib-induced allodynia in a rat model. Anticancer Res (2013) 33:5453–9.
204. Zhao W, Wang W, Li X, Liu Y, Gao H, Jiang Y, et al. Peripheral neuropathy following bortezomib therapy in multiple myeloma patients: association with cumulative dose, heparanase, and TNF-α. Ann Hematol (2019) 98:2793–803. doi: 10.1007/s00277-019-03816-6
205. Zhang J, Su YM, Li D, Cui Y, Huang ZZ, Wei JY, et al. TNF-α-mediated JNK activation in the dorsal root ganglion neurons contributes to Bortezomib-induced peripheral neuropathy. Brain Behav Immun (2014) 38:185–91. doi: 10.1016/j.bbi.2014.01.020
206. Li C, Deng T, Shang Z, Wang D, Xiao Y. Blocking TRPA1 and TNF-α Signal Improves Bortezomib-Induced Neuropathic Pain. Cell Physiol Biochem (2018) 51:2098–110. doi: 10.1159/000495828
207. Duan Z, Zhang J, Li J, Pang X, Wang H. Inhibition of microRNA-155 Reduces Neuropathic Pain During Chemotherapeutic Bortezomib via Engagement of Neuroinflammation. Front Oncol (2020) 10:416. doi: 10.3389/fonc.2020.00416
208. Liu D, Sun M, Xu D, Ma X, Gao D, Yu H. Inhibition of TRPA1 and IL-6 signal alleviates neuropathic pain following chemotherapeutic bortezomib. Physiol Res (2019) 68:845–55. doi: 10.33549/physiolres.934015
209. Liu C, Luan S, OuYang H, Huang Z, Wu S, Ma C, et al. Upregulation of CCL2 via ATF3/c-Jun interaction mediated the Bortezomib-induced peripheral neuropathy. Brain Behav Immun (2016) 53:96–104. doi: 10.1016/j.bbi.2015.11.004
210. Liu CC, Huang ZX, Li X, Shen KF, Liu M, Ouyang HD, et al. Upregulation of NLRP3 via STAT3-dependent histone acetylation contributes to painful neuropathy induced by bortezomib. Exp Neurol (2018) 302:104–11. doi: 10.1016/j.expneurol.2018.01.011
211. Tiwari V, Guan Y, Raja SN. Modulating the delicate glial-neuronal interactions in neuropathic pain: promises and potential caveats. Neurosci Biobehav Rev (2014) 45:19–27. doi: 10.1016/j.neubiorev.2014.05.002
212. Robinson CR, Dougherty PM. Spinal astrocyte gap junction and glutamate transporter expression contributes to a rat model of bortezomib-induced peripheral neuropathy. Neuroscience (2015) 285:1–10. doi: 10.1016/j.neuroscience.2014.11.009
213. Guo Y, Xu X, Huang J, Wang Z, Li Z, Liu Z. The Actions and Mechanisms of P2X7R and p38 MAPK Activation in Mediating Bortezomib-Induced Neuropathic Pain. BioMed Res Int (2020) 2020:8143754. doi: 10.1155/2020/8143754
214. Carozzi VA, Renn CL, Bardini M, Fazio G, Chiorazzi A, Meregalli C, et al. Bortezomib-induced painful peripheral neuropathy: an electrophysiological, behavioral, morphological and mechanistic study in the mouse. PloS One (2013) 8:e72995. doi: 10.1371/journal.pone.0072995
215. Li ZY, Zhang YP, Zhang J, Zhang SB, Li D, Huang ZZ, et al. The possible involvement of JNK activation in the spinal dorsal horn in bortezomib-induced allodynia: the role of TNF-α and IL-1β. J Anesth (2016) 30:55–63. doi: 10.1007/s00540-015-2077-x
216. Kramer R, Bielawski J, Kistner-Griffin E, Othman A, Alecu I, Ernst D, et al. Neurotoxic 1-deoxysphingolipids and paclitaxel-induced peripheral neuropathy. FASEB J (2015) 29:4461–72. doi: 10.1096/fj.15-272567
217. Becker KA, Uerschels AK, Goins L, Doolen S, McQuerry KJ, Bielawski J, et al. Role of 1-Deoxysphingolipids in docetaxel neurotoxicity. J Neurochem (2020) 154:662–72. doi: 10.1111/jnc.14985
218. Stockstill K, Doyle TM, Yan X, Chen Z, Janes K, Little JW, et al. Dysregulation of sphingolipid metabolism contributes to bortezomib-induced neuropathic pain. J Exp Med (2018) 215:1301–13. doi: 10.1084/jem.20170584
219. Doyle TM, Chen Z, Durante M, Salvemini D. Activation of Sphingosine-1-Phosphate Receptor 1 in the Spinal Cord Produces Mechanohypersensitivity Through the Activation of Inflammasome and IL-1β Pathway. J Pain (2019) 20:956–64. doi: 10.1016/j.jpain.2019.02.007
220. Alé A, Argyriou AA, Bruna J. Sphingolipid metabolism products: potential new players in the pathogenesis of bortezomib-induced neuropathic pain. Ann Transl Med (2018) 6:S78. doi: 10.21037/atm.2018.10.53
Keywords: neuroinflammation, immune cell activation, neuropathic pain, chemotherapy-induced peripheral neuropathy, immune modulation
Citation: Fumagalli G, Monza L, Cavaletti G, Rigolio R and Meregalli C (2021) Neuroinflammatory Process Involved in Different Preclinical Models of Chemotherapy-Induced Peripheral Neuropathy. Front. Immunol. 11:626687. doi: 10.3389/fimmu.2020.626687
Received: 06 November 2020; Accepted: 21 December 2020;
Published: 04 February 2021.
Edited by:
Maria Maiaru, University of Reading, United KingdomReviewed by:
Lorenzo Di Cesare Mannelli, University of Florence, ItalyAlessandra Pacini, University of Florence, Italy
Copyright © 2021 Fumagalli, Monza, Cavaletti, Rigolio and Meregalli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guido Cavaletti, Z3VpZG8uY2F2YWxldHRpQHVuaW1pYi5pdA==; Roberta Rigolio, cm9iZXJ0YS5yaWdvbGlvQHVuaW1pYi5pdA==
†These authors have contributed equally to this work