 Neelakshi R. Jog
Neelakshi R. Jog Judith A. James
Judith A. James
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 03 February 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.623944
This article is part of the Research TopicPathogens, Pathobionts and AutoimmunityView all 13 articles
Systemic lupus erythematosus (SLE) is a complex systemic autoimmune disease. Infections or infectious reactivation are potential triggers for initiation of autoimmunity and for SLE flares. Epstein-Barr virus (EBV) is gamma herpes virus that has been associated with several autoimmune diseases such as SLE, multiple sclerosis, Sjogren’s syndrome, and systemic sclerosis. In this review, we will discuss the recent advances regarding how EBV may contribute to immune dysregulation, and how these mechanisms may relate to SLE disease progression.
Systemic lupus erythematosus (SLE) is a multifaceted systemic autoimmune disease (1) stemming from immune dysregulation. A characteristic feature is the presence of autoantibodies directed towards nuclear antigens (ANA), which can be detected up to a decade before disease onset. Although not completely characterized, studies suggest that cellular dysfunction, dysregulated inflammatory responses and autoantibody -mediated damage leads to progression of autoimmune disease and organ damage (2).
The underlying factors responsible for disease transition and pathogenesis likely involve an interplay between genetic and environmental factors. SLE has a twin concordance rate of 24% to 40% (3, 4) and over 100 genetic associations have been identified and confirmed (5).
Infections or pathogens have been proposed to lead to autoimmunity. Epstein Barr virus (EBV) is one such pathogen that has been repeatedly associated with SLE since the first report in 1969. EBV adopts several strategies to exploit host immune response for its persistence. Consequences to the host are increased acute inflammation and autoantibody generation, which are usually transient and self-limited, as seen in patients with infectious mononucleosis (6). However, a growing body of research suggests that these effects in certain individuals, possibly based on genetic risk factors, can cascade into a chronic inflammatory state. Due to its strong association with tumorigenesis, EBV has been studied extensively for its ability to overcome immune surveillance and approached to combat tumorigenic effects.
In this review we provide a compilation of the current understanding of how EBV may contribute to immune dysregulation, including strategies used by EBV to combat immune surveillance, and how these processes may relate to SLE pathogenesis (Figure 1).
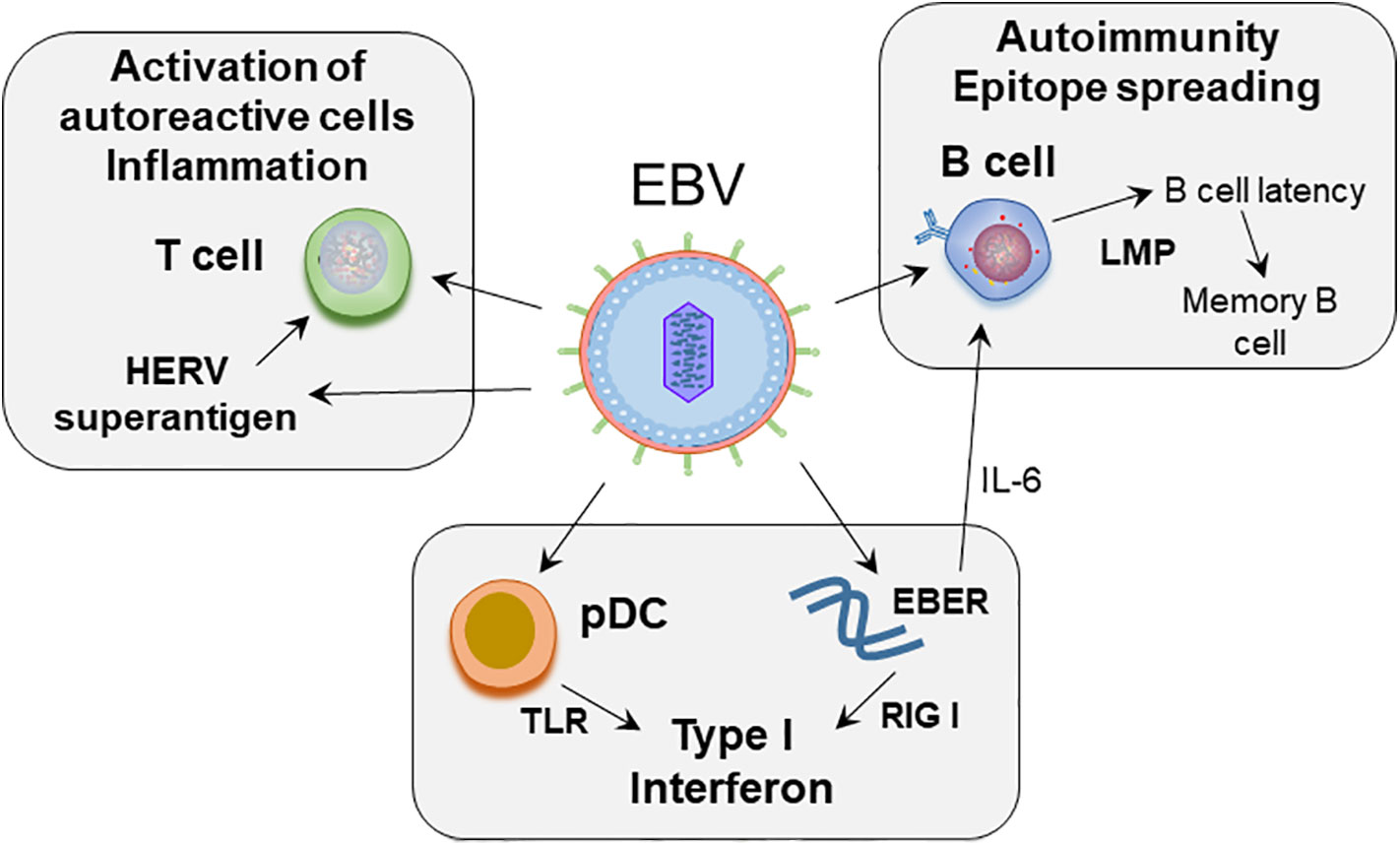
Figure 1 Proposed role of EBV in SLE pathogenesis. EBV infects naïve B cells. The infected B cells enter the memory B cell compartment through germinal center like reaction, mediated by the expression of latent membrane proteins. EBV maintains latency in the resting memory B cells. EBERs, non-coding RNA expressed by EBV, can mimic dsRNA, and activate RIG-I leading to production of type I interferons. EBERs also induce growth factor IL-6 and regulate B cell survival. EBV can act on plasmacytoid dendritic cells (pDC). Initial binding of virus is mediated by class II MHC on pDCs, following which through engagement of TLR7 and 9, EBV RNA and DNA can induce type interferon secretion by pDCs. EBV induces superantigen on HERV-K18, which can induce unregulated T cell activation.
Acute primary EBV infection, which is also a common cause of infectious mononucleosis, is characterized by fatigue, atypical lymphocytosis, splenomegaly, and lymphadenopathy. Although the host immune response eventually controls viremia, the virus maintains latency in memory B cells with occasional reactivation to infect naïve B cells. EBV genomes in latently infected B cells are thought to exist as episomes (7), although it is possible that the genomes exist as integrated DNA. EBV expresses nine latent proteins; six EBV nuclear antigens (EBNA, EBNA-1, 2, 3A, 3B, 3C, and leader protein), and three latent membrane proteins (LMP 1, 2A, and 2B). In addition to latent proteins, expression of small non-polyadenylated RNAs, EBER1 and 2, is also observed (8).
Unique forms of latency that differ in the latent protein expression have been identified (Table 1). Latency III, where all latency gene products are expressed, is the predominant latency observed in lymphoblastoid cell lines, acute infectious mononucleosis, and certain immunocompromised individuals. This form of latency can mediate naïve B cell activation. EBNA1 and LMP1/2A are expressed in the latency II program, which is observed in nasopharyngeal carcinoma and Hodgkin’s lymphoma. LMP1 and LMP2 can induce B-cell activation and growth (proliferation). Latency I, which is observed in EBV-positive Burkitt’s lymphoma tumors, expresses only EBNA-1. In this form, latent EBV genomes can multiply in dividing memory cells. The Gly-Ala repeats in EBNA-1 inhibit antigen processing, and therefore, CD8 T cells are unable to detect virally infected cells in this form. Latency 0 is observed in quiescent B cells, where no EBV proteins are expressed, however cells switch to Latency I during cell division with expression of EBNA-1, which is required for replication of the episome. Latently infected B cells occasionally reactivate EBV. This allows the virus to re-infect new B cells and epithelial cells, and acts as a source of viral transmission. Although the molecular pathways involved in viral reactivation are studied extensively, the triggers for reactivation in vivo are unclear.

Table 1 Latency forms of EBV.
The occasional reactivation of the virus can be detected serologically. A primary infection with EBV leads to an IgG response to viral capsid antigen (VCA). The VCA IgG antibodies are maintained throughout the life span of the individual. Following VCA IgG response, IgG responses toward early antigen (EA) are detected. These antibodies are detectable for 6 months to up to two years. During EBV reactivation, EA IgG levels are detectable and there is an increase in VCA IgG levels (9). Therefore, an increase in VCA IgG and presence of EA IgG indicates current or recent reactivation of the virus.
Many studies to date have demonstrated an association between SLE and EBV infection. A higher EBV seroconversion rate was observed in both pediatric and adult SLE patients compared to healthy controls (10–12). SLE patients show increased levels of IgG antibodies toward VCA and EA, both indirect markers of increased EBV reactivation. However, the IgG responses towards other herpes viruses such as cytomegalovirus (CMV) and herpes simplex virus (HSV) are similar in SLE patients and controls. These reports suggest that SLE patients may have increased reactivation of the virus. EBV viral load is elevated in SLE patients (13, 14), which may also suggest increased reactivation. A possible reason for increased reactivation is inefficient regulation of the latent phase or enhanced transition from latent to lytic phase. Interestingly, a higher percentage of patients have detectable levels of EBV gene BZLF1 (15), which is an immediate-early gene that is responsible for the switch to lytic cycle. Two other latent genes LMP1 and LMP2A were also detected in SLE patients. The type of latency maintained in SLE patients is not completely understood. LMP1/2A are expressed in latency II, and all latent genes are expressed in latency III (Table 1). The presence of two latent genes, BZLF-1 and LMP-1, which cannot be detected in seropositive healthy individuals, suggests that EBV latency may be dysregulated in some SLE patients. Based on the expression pattern of latent genes reported so far, SLE patients may have an intermediate form between latency II and III.
Based on serologic evidence and higher viral loads observed in SLE patients, the consensus is that SLE patients have increased EBV reactivation. Dysregulation of anti-viral T cell responses is a proposed mechanism for increased viral loads. SLE patients have higher interferon γ (IFNγ) secreting CD4+ T cells, but lower frequencies of EBV specific CD8+ T cell responses. EBV viral loads in peripheral blood cells positively correlated with EBV specific and IFNγ secreting CD8+ T cells (14). EBV specific CD8+ T cells in SLE patients are functionally impaired (16, 17), although CMV specific responses were unaltered (17). The upregulation of PD1 on EBV specific T cells in SLE patients may be responsible for the suppressed responses to EBV antigen, as blockade of PD1 restored IFNγ production in response to EBV antigens. Based upon these data and the observed diminished responses of SLE T cells to superantigen stimulation, the authors suggest that SLE T cells demonstrate an exhausted phenotype. However, CMV specific T cell responses were unaltered by PD1 blockade. These data suggest that the general immune surveillance mechanisms are intact in SLE patients, but there is an inherent defect in regulating EBV infection (17). Both CD4 and CD8 lytic and latent antigen specific functional T cells were lower in SLE patients. A negative correlation between SLE disease activity index (SLEDAI) and EBV specific functional T cell responses was reported (18), with decreased EBV lytic gene responsive T cells in patients with elevated disease activity. Furthermore, an inverse relationship was observed between EBV specific T cells and levels of anti-EBV antibodies (18). SLE T cells may also contribute to defective regulation of certain B cell functions (19). Absolute numbers of Th17 and Treg cells were reduced in SLE patients with EBV and/or CMV viremia compared to those without viremia or healthy controls. However, there was a direct correlation between viremia and SLEDAI, suggesting that reduction in Th17 and Treg cells may be a consequence of SLE immune dysregulation independent of viremia (20). EBV can transactivate superantigen on human endogenous retroviral (HERV)-K18, which can lead to unrestricted activation of T cells (21).
EBV induced gene 3 protein (EBI3) was identified in EBV transformed B cells (22). It serves as a beta chain for cytokines IL-27, IL-35, and IL-39, and can induce regulatory or suppressive T cells in a murine model (23). The serum IL-35 level and the percentage of CD4+EBI3+ T cells were negatively correlated with the SLE disease activity index, and both of these parameters were increased shortly after treatment of active SLE patients with methylprednisolone (24). However, levels of EBV reactivation were not determined in this study. Although EBI3 was initially reported in EBV transformed B cells and induced by LMP1, the name of the gene is misleading. EBV infection of T cells is not established unequivocally. It was later shown that EBI3 can be induced in naïve T cells by polyclonal stimulation with plate bound anti-CD3 and anti-CD28 (25). This also explains upregulation of EBI3 in experimental murine models, which lack EBV infection. Therefore, the increase in IL-35 observed in SLE patients may be independent of EBV induced gene expression. Studies evaluating the upregulation of EBI3 in SLE patients in the context of EBV infection and subsequent contribution to SLE pathogenesis are lacking.
Differences in cytokine production in response to EBV antigens have been reported. SLE patients exhibited a decreased IL-12, IFNγ, IL-17, and IL-6 response to EBNA-1, and decreased induction of IL-6, TNFβ, IL-1β, and GM-CSF upon EBV-EA-D stimulation. Serologic SLEDAI scores, based solely on anti-dsDNA, complement, thrombocyte, and leukocyte levels, correlated negatively with numerous cytokine responses against EBNA-1 and EA-D (26). These data further support impaired regulation of immune response against latent and lytic EBV antigens in SLE patients.
The numbers of infected B cells positively correlated with SLE disease activity index (15). The EBV viral load in SLE patients with active disease was found to be higher than in inactive cases (27), however, another study did not find this (17). Although this report did not find a consistent increase in EBV viral load immediately prior or at the time of a flare, the viral loads were higher in a majority of patients during elevated disease activity (17), suggesting that EBV may have a role in the pathogenesis and activity of SLE. The overall low number of EBV-infected B cells during latency and the lower numbers of B cells due to lymphopenia in SLE patients provides a technical challenge in detecting EBV DNA. Assays with higher sensitivities to detect both latent and lytic EBV genes, perhaps partnered with single cell technologies, will be helpful to understand the relation between timing of EBV reactivation and SLE flare. Detailed longitudinal analyses of a larger cohort of SLE patients will improve our understanding of viral reactivation and SLE disease activity.
Newer data have evaluated the association of EBV reactivation with SLE disease onset. Our retrospective analyses of unaffected family members of SLE patients showed that SLE relatives that subsequently transition to classified SLE (>4 ACR criteria), have increased VCA-IgG and EA-IgG at a time-point prior to the transition, when compared to relatives that do not transition to SLE (28). The responses towards CMV and HSV-1 were not different between the two groups of relatives. These data suggest that EBV reactivation observed in SLE patients is not due to immune dysregulation caused by the chronic autoimmune and inflammatory environment in patients, nor is it solely a consequence of immunosuppressive medications. However, as the study involved blood relatives of SLE patients, a genetic component may be involved in increased EBV reactivation. On similar lines, seropositivity for anti-EBV early antigen (EA), a marker of EBV reactivation, was dramatically increased in patients with SLE compared with unrelated controls (92.3% vs 40.4%; OR 17.15(95% CI 10.10, 30.66), p<0.0001) or unaffected first-degree relatives of lupus patients (49.4%; OR 12.04(7.42, 20.74), p<0.0001). The seroprevalence of VCA IgG in patients and first-degree relatives was similar suggesting same level of prior EBV exposure in these two groups (29).
Significant interactions between EBV serology and single nucleotide polymorphisms (SNPs) in genes that are associated with SLE and also involved in EBV infection were observed. The association between VCA IgG level and transitioning to SLE was modified by CD40 rs4810485 (interaction p = 0.0009). Similarly, the association between VCA IgA and transitioning to SLE was modified by IL10 rs3024493 (interaction p = 0.008) (28). In line with a genetic component contributing to increased EBV reactivation, a higher frequency of subjects with germ-line mutations in CTLA-4 had detectable EBV viral load when compared with healthy controls. None of the subjects had symptoms of EBV infection the time of analyses. However, none of the 15 subjects included in this study had a SLE diagnosis (30). Parks et al. showed a significant interaction between VCA IgA and CTLA-4 gene polymorphism (-1661AA), and increased VCA IgA seropositivity in African American SLE patients (31). CTLA-4 -1661 mutation was associated with risk of SLE in young African American patients (32).
Harley et al. recently showed that in EBV-immortalized B cells, almost half of SLE European ancestry risk alleles can be occupied by EBNA-2 protein, which is expressed in latency II and III. The authors showed that host transcriptional factors bind to SLE risk loci only in the presence of EBV, and that EBNA-2 is involved in allele dependent transcription complex formation at risk loci. These data provide another potential origin of gene/environment interaction in SLE (33).
Thus, genetic predisposition leading to immune dysregulation may contribute to EBV reactivation eventually resulting in classified SLE.
In order to evade the host immune system and to establish a persistent latent infection, EBV encodes several viral homologues of human proteins. These homologues either accentuate the effect of human proteins on immune cells, inhibit, or allow the virus to hijack the immune response to its benefit.
EBV IL-10 (vIL-10) is a late viral gene expressed during the lytic phase of virus replication encoded by the viral BCRF1 gene, which is highly homologous to the human IL-10 (hIL-10) gene (34, 35). Due to the high homology, vIL-10 shares some of the suppressive and stimulatory functions of hIL-10. vIL-10 can inhibit inflammatory cytokine (IFNγ, TNFα) production and can promote proliferation and differentiation of B cells, as well as immunoglobulin production. Functional differences between hIL-10 and vIL-10 have also been reported. vIL-10 cannot co-stimulate mouse thymocyte proliferation and mast cell proliferation and cannot up-regulate MHC class II on B cells (36–38).
We recently showed that in contrast to hIL-10, vIL-10 can induce a pro-inflammatory phenotype in monocytes. vIL-10 induced a unique gene expression profile in monocytes, and monocytes exposed to vIL-10 showed defective clearance of apoptotic cells. vIL-10 signals through the same receptor subunit as hIL-10, can act as a competitive inhibitor of hIL-10, and inhibit suppressors of immune response induced by hIL-10. vIL-10 levels were significantly higher in SLE patients plasma compared to matched controls (39). As vIL-10 is a lytic gene, these data also support increased reactivation of EBV in SLE patients.
Increased vIL-10 in SLE patients may increase pro-inflammatory responses by monocytic cells, while inhibiting hIL-10 functions. These pro-inflammatory mediators, along with a reduced clearance of apoptotic infected cells, may lead to increased antigen presentation and activation of cytotoxic T cell responses towards EBV. Indeed Stewart et al. showed that vIL-10 enhances the generation of allo-specific CTL, EBV-specific CTL, and HLA-unrestricted activated killer cells (40). Although this allows the virus to enter latency and persist, in a genetically prone individual with defective tolerance checkpoints, the defective clearance and increased antigen presentation may lead to autoimmune responses (Figure 2). Further longitudinal studies evaluating vIL-10 levels in preclinical samples, as well as in SLE patients, pre and post flare, and associations of these levels with monocyte activation status are needed to confirm the role of vIL-10 in induction of an autoimmune response.
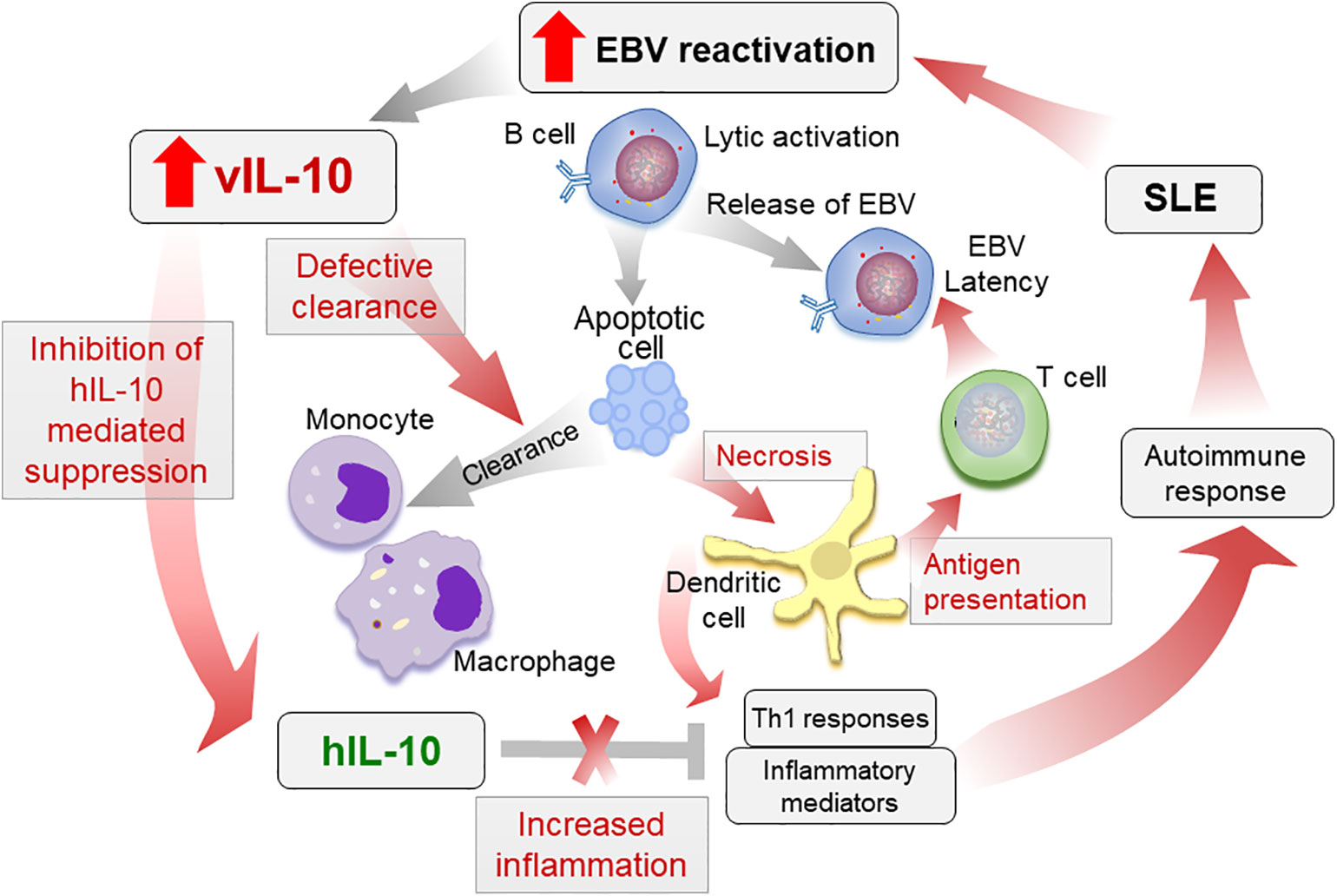
Figure 2 Proposed role of vIL-10 in SLE pathogenesis. Increased reactivation of EBV in SLE patients increases vIL-10. vIL-10 competes with hIL-10 for IL10R1 and inhibits the suppressive effects of hIL-10 on myeloid cells. vIL-10 also reduces the ability of monocytes/macrophages to clear apoptotic cells. This leads to increased secondary necrosis, increased presentation of antigens by dendritic cells (DCs), and allows virus to establish latency. Reduced clearance of apoptotic cells leading to secondary necrosis along with increased antigen presentation and inflammatory responses exacerbate autoimmune response in SLE. The processes possibly regulated by vIL-10 are shown in red arrows.
How vIL-10 induces a unique gene expression in monocytes and can inhibit hIL-10-mediated immune suppression is not clear. vIL-10 has lower affinity to IL-10R1 compared to hIL-10. However, vIL-10 is more potent than hIL-10 in inducing B cell proliferation, and therefore the lower affinity may not explain the differences in monocyte activation by vIL-10. The vIL-10: IL-10R1 interaction is very transient, while with hIL-10 is more sustained (41). A transient interaction may interfere with ligand-dependent receptor internalization and proteasomal degradation. vIL-10 may be sequestering receptors and compete with hIL-10. Although not reported in the literature yet, it is possible that the vIL-10 monomer forms a heterodimer with hIL-10 and inhibits signaling by hIL-10.
How EBV maintains latency in memory B cells is also not completely understood. It is hypothesized that EBV enters the memory B cell compartment through differentiation of the latently infected B cell blasts into resting memory B cells, also known as the germinal center (GC) model. The observations that the viral infection is strictly latent in resting memory B cells in the periphery, but active infection of naïve B cells and virus shedding can be detected in tonsillar lymphoid tissue, support this hypothesis [Reviewed in (42)].
EBV expresses three latent membrane proteins (LMP, 1, 2A, 2B) that can mimic signals necessary to rescue normal B cell differentiation in absence of T cell signals. Despite the lack of significant protein homology, LMP1 is a functional homologue of CD40, and acts as a constitutively active receptor (43). LMP1 induces B cells to express B cell-activating factor of the TNF family (BAFF) and a proliferation-inducing ligand (APRIL), which mediate B cell survival and T cell-independent antibody production, and therefore can induce class switch recombination (CSR) in absence of a GC reaction (44, 45). Thus, EBV may block B cells from entering GC, and induce extra-follicular B cell activation through the expression of LMP1. The expression of a chimeric molecule with the mouse CD40 extracellular domain and the LMP1 intracellular signaling regions in lupus-prone mouse strain accentuated the autoimmune phenotype. This suggests that LMP1 acts synergistically with host predisposing genetic factors and contributes to exacerbation of an autoimmune response (46).
LMP2A mimics the B cell receptor (BCR), and contains an immunoreceptor tyrosine based activation motif (ITAMs) which associates with downstream signaling kinases. LMP2A mimics the BCR signal and can rescue B cells lacking surface immunonoglobulin from death (47). Conditional expression of LMP2A in murine GC B cells enhanced BCR signals, facilitated plasma cell differentiation, and resulted in selection of low affinity antibody producing cells. The conditional GC expression also led to SLE-like autoimmune phenotype including anti-double stranded DNA (dsDNA) antibody production, and immune complex deposition in the kidneys (48). Expression of LMP2A transgene in anti-Sm heavy chain transgenic mice resulted in increased anti-Sm antibodies (49). In these mice transgenic for anti-Sm and LMP2A, anti-Sm B cells bypassed the pre-plasma cell tolerance checkpoint and differentiated into antibody secreting cells, suggesting that LMP2A can modify GC B-cell selection and may contribute to persistent EBV infection.
Additional genes that are expressed during EBV latency are two noncoding RNAs, EBER1 and EBER2, and 44 microRNAs (miRNAs), derived from two loci, the BART and BHRF clusters. BART transcript encodes 22 miRNA precursors (miR-BART1-22) with 40 mature miRNAs, whereas the BHRF1 transcript expresses three miRNA precursors (miR-BHRF1-1, -2, and -3) producing four mature miRNAs (50). EBV miRNA from infected cells were secreted in exosomes, which can be internalized by monocyte derived dendritic cells (51) and modulate their gene expression. In individuals with increased EBV viral load, EBV miRNA were detected in both B and non-B cells in peripheral blood. Although the levels of EBV miRNA have not been compared between SLE patients and unaffected donors, EBV miRNA may be contributing to differences in gene expression profiles observed in non-B cells in SLE patients.
EBER1 and EBER2 are present in all four latency stages (52, 53). Several reports have suggested a role for EBERs in the tumorigenic process in vivo, which are also supported by murine studies where transgenic mice expressing EBER1 developed lymphoid hyperplasia and an increase in lymphoma incidence (54). EBERs form a stem–loop structure by intramolecular base-pairing, which can give rise to dsRNA-like molecules (55, 56). EBERs can bind to dsRNA activated protein kinase PKR, inhibit its phosphorylation and can confer resistance to IFN-induced apoptosis in Burkitt’s lymphoma cells (57). EBERs can contribute to B cell transformation and growth by inducing the growth factor IL-6 (58). EBERs can regulate target regulation of several miRNAs. Expression of EBER can enhance the inhibitory effect of miR143-mediated downregulation of the inflammatory gene IL1α (59), however, the significance of these effects in the development or progression of autoimmune diseases is unclear. Expression of EBER in EBV-negative B lymphoma cell line resulted in upregulation of kinases involved in B cell pro-survival signaling, which were previously considered to be regulated solely by LMP1, suggesting a redundancy in function between EBERs and LMP1 during latency (60). EBERs are recognized by retinoic acid-inducible gene I (RIG-I) through the helicase domain and can activate signaling to induce type I interferon and interferon-stimulated genes (61).
SLE patients show increased levels of type I interferon in serum, and SLE disease activity correlates with IFNα levels and the strength of the interferon signature (62, 63). EBV increases IFNα secretion by plasmacytoid dendritic cells (pDCs) through toll-like receptors (TLR). The recognition of EBV is mediated by class II MHC molecules (64). The increased LMP1 gene expression in SLE patients correlated with SLE disease activity index (SLEDAI) and interferon induced gene expression (65). The levels of EBERs were not evaluated in this study. The contribution of EBV or EBER mediated interferon activation and the significance of this induction in progression of SLE needs further evaluation.
In SLE patients, EBV EA IgG positivity correlated with lupus antibodies (29). EBV IgG also correlated with anti-Ro and anti-La antibodies in SLE patients (66).
Molecular mimicry between SLE autoantigens and EBV antigens may lead to autoimmune response. Antibodies towards different regions of EBNA-1 protein cross-react with SLE autoantigens SmB, SmD, as well as Ro (67). Monoclonal antibodies generated from mice immunized with EBNA-1 cross-react with dsDNA (68, 69). Cross-reactivity between the anti-EBNA-1 response and anti- complement component C1q response has also been shown. Anti-C1q antibody towards A08 epitope of C1q isolated from SLE patients can bind a peptide derived from EBNA-1, EBNA348, and SLE patients that showed reactivity to EBNA348 peptide had higher levels of anti-C1q. This cross-reactivity was shown to be dependent on amino acid identity (70). Peptides derived from EBV EA and LMP1 increased ANA positivity in mice. Both these peptides increased anti-SmB and anti-SmE. While EA derived peptide, EP4, additionally increased anti-SmD and anti-Ro, LMP1 derived peptide increased anti-rRNP. Levels of EP4 antibodies were higher in SLE patients and correlated with SLEDAI. Interestingly, both these peptides had about ~60% amino-acid sequence similarities with self-peptides, but the percentage of similarities with amino-acid characteristics was 75 and 70% respectively for each peptide (71).
Immunization of experimental animals with peptides from regions of EBNA-1 lead to lupus-like autoimmune disease (72–74). In these studies, immunization with a single peptide lead to the generation of cross-reactive antibodies, but the autoimmune response also spread to several different epitopes, and was not restricted to the cross-reactive epitope. Furthermore, injection of mice with plasmids expressing either full-length EBNA-1 or EBNA-1 lacking 15 amino acids in in the Gly-Ala repeats, resulted in anti Sm, and anti-dsDNA antibodies (75). Epitope spreading has been suggested as a possible mechanism for accrual of antibody specificities, and has been shown to occur with immune response towards spliceosomal and other proteins (72, 74, 76).
Taken together, these reports suggest that molecular mimicry with EBV epitopes may allow loss of tolerance to self-antigens. Through the process of epitope spreading, these responses may target additional self-epitopes, eventually leading to pathogenic responses and to clinical SLE (Figure 3).
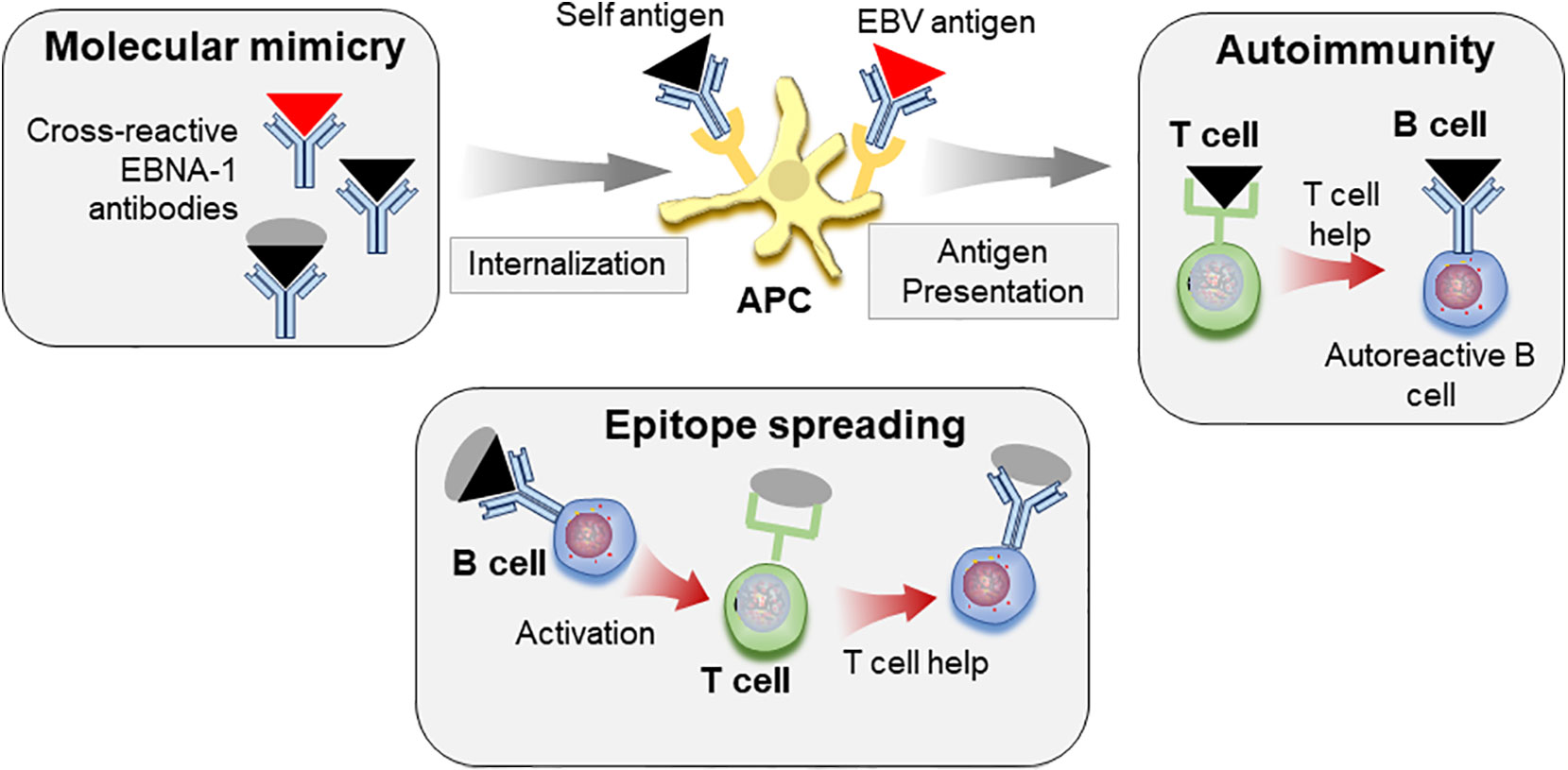
Figure 3 Molecular mimicry and epitope spreading in autoimmunity. Antibodies to viral antigens such as EBNA-1 (red triangle) cross-react with autoantigens (black triangle) due to structural similarities. Immune complexes consisting of autoantigen/antibody complexes are internalized by antigen presenting cells (APC), antigen is processed and peptides presented to T cells, which can allow for loss of tolerance. Defective T cell tolerance possibly contributed by genetic susceptibility may be responsible for this loss of tolerance. These autoreactive T cells in turn provide help to auto-reactive B cells leading to autoreactive antibody response. The self-protein bound to B cell receptor is internalized, processed, and presented to T cells. The autoimmune response can be further diversified by epitope spreading. B cells specific for viral antigen (red triangle) can recognize similar structures on self-antigen (black triangle). However, these cells can internalize and present peptides from whole protein that carries the cross-reactive epitope (black triangle+gray circle) to T cells, which then provide help for antibody response towards additional epitopes on the protein by B cells.
Although a significant effort has been made to understand EBV biology, understanding how EBV contributes to autoimmune pathogenesis, and the causal relationship between EBV infection, reactivation and autoimmunity are limited due to lack of an appropriate animal model. Peptide immunizations have been instrumental in establishing molecular mimicry between EBV antigens and autoantigens. Transgenic mouse model approaches allowed a better understanding of the ability of EBV latent proteins to modulate B cell function. However, the expression of EBV encoded oncogenes in absence of the entire EBV genome has limitations. These knowledge gaps warrant a suitable animal model that recapitulates the features of EBV infection.
Non-human primates are infected naturally with EBV-related herpesviruses, or lymphocryptoviruses (LCV), and are therefore considered as models for EBV infection [reviewed in (77)]. A primary EBV infection can be established in healthy New Zealand white rabbits, and EBV can also infect Owl monkey and marmosets (78–80). These animal models may prove to be useful for understanding role of EBV in malignancies. However, none of these are characterized as animal models for human autoimmune diseases.
A major advance in establishing a mouse model for EBV came from utilization of humanized models on an immune-deficient murine background. The reconstitution of severe combined immune-deficient (SCID) mice with human peripheral blood leukocytes results in mice with inducible human immune function (81) and development of EBV+ lymphomas by transfer of peripheral blood leukocytes from EBV positive donors (82). However, several limitations such as transient nature of the graft, low engraftment levels, and frequent graft-versus-host disease caused by human T cells attacking mouse tissues, limit the use of this model.
Reconstitution of recombination activating gene 2 (Rag2) deficient IL2 receptor gamma (IL2Rγ) deficient mice also supported EBV infection (83). The deficiency of IL2Rγ allows for T cell re-constitution, and T cells are selected on murine tissue. However, as the T cell are selected on murine and not human tissue, the response in these mice is still suboptimal. This limitation can be overcome by implanting Non-obese diabetic (NOD)/SCID mice with human fetal liver and thymic tissue to provide human T cells appropriate thymic environment, with subsequent autologous CD34+ cell implantation following sub-lethal irradiation (BLT mice) (84). BLT mice showed marked increase in memory T cells, and the T cells could respond to autologous antigen presenting cells upon EBV infection, suggesting that human T cells in BLT mice can mount human-MHC-restricted response and can be used to reproduce human T and B cell interactions. Although an attractive approach, humanized models of EBV infection have not been utilized for SLE research yet. Reconstitution of immunodeficient mice with hematopoietic stem cells from EBV positive and EBV negative SLE patients and matched controls may provide useful insights into pathways regulating increased reactivation in SLE and/or role of EBV in disease progression.
EBV infection of NOD/SCID IL2Rγ-/- (NSG) mice reconstituted with human cord blood hematopoietic cells resulted in erosive arthritis in 65% of mice (85). However, neither anti-citrullinated peptide antibodies nor rheumatoid factor were detected in the blood of affected mice. The serological response to EBV infection observed in humans was also not detected, suggesting that the arthritis observed in these mice was by mechanisms different from those in patients. However, the genetic factors associated with rheumatoid arthritis were not considered in this study.
The study does point out a possible limitation of using humanized mouse models to replicate EBV infection. During both primary infection and subsequent reactivation, lytic replication of EBV occurs in oropharyngeal epithelial cells, where infectious virus particles are produced and shed. Although EBV is hypothesized to infect and to maintain latency only in B lymphocytes (86), EBV can replicate in epithelial cells and viral gene expression patterns differ when the virus emerges from epithelial cells versus B cells, which suggests passage back and forth (87). Due to differences in routes of infection and lack of the epithelial infection, humanized mice do not recapitulate the complete life cycle of EBV infection, and therefore do not reflect the immune response to EBV infection. These models also lack final lytic replication in oropharyngeal epithelial cells, which the virus uses to amplify infectious virus production during shedding into saliva. This limitation may be overcome by human epithelial tissue grafts in humanized mice followed by infection through the natural route. However, whether the transient infection in epithelial cells that produces virus with increased tropism to B cells is necessary to establish latent EBV infection in B cells and whether this transient infection occurs during EBV reactivation are not known.
A murine virus similar to EBV is an alternate approach. The most probable is murine gamma herpes virus 68 (MHV68). Although not identical to EBV, MHV68 shares several features. MHV68 is found in class switched B cells that have undergone GC reaction and reflect memory B cells. MHV68 is a natural pathogen of free-living murid rodents. Virus neutralizing antibodies are detectable in the natural hosts (88). The infection of mice with MHV72, a gamma herpesvirus strain related to MHV68, leads to detectable anti-viral antibodies, and these correlate with viral reactivation (89).
MHV68 infection is associated with an expansion of lymphocyte populations that drives an infectious mononucleosis-like response marked by enlarged lymph nodes and splenomegaly (90, 91). Productive infection in the lungs following intranasal infection of mice with MHV68 lasts for ~10 days. During this time the virus spreads to spleen through infected B cells and establishes latency in GC B cells (92). Long term latency is detected in IgD- subset of splenic B cells (93). MHV68 has been shown to maintain latency in peritoneal macrophages, which has not been reported for EBV. However, similar to EBV, the splenic latency is solely dependent on B cells (94).
MHV68 increased anti-Sm antibodies in wild type and lupus prone mice during acute phase of infection, however, chronic infection protected mice from lupus-like disease (95). The frequency of infected cells and viral load was not determined, and single high dose of virus was used, which was administered intra-peritoneally. Lower doses of virus do not impact establishment of latency but can delay the acute-phase replication peak. Small numbers of pre-formed virus particles were detected in splenocytes of mice infected with lower doses of the virus (96). Although this small increase in the numbers of virus particles did not constitute significant reactivation in the non-autoimmune wild type C57/Bl6 strain used in that study, it may contribute to immune response in a mouse strain genetically prone to immune dysregulation. Therefore, administration of lower doses of MHV68 to lupus-prone mice by oral and/or intranasal routes, may recapitulate EBV infection in SLE patients. MHV68 does not encode a homologue for human IL-10. However, a recombinant MHV72 expressing EBV IL-10 showed exacerbated acute-phase pathogenicity (97). The effect of this recombinant virus on lupus like disease in murine models has not been evaluated. Detailed analyses of humoral response to MHV68, frequency of viral reactivation, and frequency of infected memory B cells in lupus prone mice are necessary to understand the role of MHV68 in murine lupus-like disease.
EBV can modulate immune responses in a myriad of pathways, including generation of cross-reactive antibodies, IFNα secretion, antigen independent B cell activation, gene expression modification, and anti-inflammatory response suppression. SLE patients show evidence of increased reactivation of EBV, possibly resulting from dysregulated immune responses together with genetic risk factors. Furthermore, the viral homologues such as vIL-10 modulate immune response in a manner that can exacerbate autoimmune response in genetically susceptible subjects. A longitudinal study that closely follows levels of viral latent and lytic gene expression and cellular changes, in the context of genetic risk alleles will provide an improved understanding of EBV reactivation in SLE and how this reactivation may contribute to autoimmune response.
Mouse models, either humanized or MHV infection of lupus prone mice, may be an alternate approach to decipher the role of EBV. CD34+ hematopoietic stem cells generated in vitro from induced pluripotent stem cells (iPSC), which are EBV negative, to reconstitute BLT mice described by Melkus et al. can overcome the effects of prior exposure to EBV in patient cells. The use of iPSC also allows for introducing (or reverting) specific mutations to further clarify the gene/environment interactions, and determining immune dysregulation immediately following EBV infection.
All authors contributed to the article and approved the submitted version.
This study was supported by the National Institute of Allergy, and Infectious Diseases (R03 AI139975, U19AI082714, U01AI101934), the National Institute of General Medical Sciences (U54GM104938), and the National Institute of Arthritis, Musculoskeletal and Skin Diseases (P30AR073750).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Mok CC, Lau CS. Pathogenesis of systemic lupus erythematosus. J Clin Pathol (2003) 56(7):481–90. doi: 10.1136/jcp.56.7.481
2. Justiz Vaillant AA, Goyal A, Bansal P, Varacallo M. Systemic Lupus Erythematosus. Treasure Island (FL: StatPearls (2020). doi: 10.1101/2020.09.27.20202762
3. Bogdanos DP, Smyk DS, Rigopoulou EI, Mytilinaiou MG, Heneghan MA, Selmi C, et al. Twin studies in autoimmune disease: genetics, gender and environment. J Autoimmun (2012) 38(2-3):J156–69. doi: 10.1016/j.jaut.2011.11.003
5. Hagberg N, Lundtoft C, Ronnblom L. Immunogenetics in systemic lupus erythematosus: Transitioning from genetic associations to cellular effects. Scand J Immunol (2020) 92(4):e12894. doi: 10.1111/sji.12894
6. Papesch M, Watkins R. Epstein-Barr virus infectious mononucleosis. Clin Otolaryngol Allied Sci (2001) 26(1):3–8. doi: 10.1046/j.1365-2273.2001.00431.x
7. Kieff E, Hennessy K, Fennewald S, Matsuo T, Dambaugh T, Heller M, et al. Biochemistry of latent Epstein-Barr virus infection and associated cell growth transformation. IARC Sci Publ (1985) 60):323–39.
8. Young LS, Arrand JR, Murray PG. EBV gene expression and regulation. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press (2007).
9. Gulley ML. Molecular diagnosis of Epstein-Barr virus-related diseases. J Mol Diagnostics JMD (2001) 3(1):1–10. doi: 10.1016/S1525-1578(10)60642-3
10. James JA, Kaufman KM, Farris AD, Taylor-Albert E, Lehman TJ, Harley JB. An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J Clin Invest (1997) 100(12):3019–26. doi: 10.1172/JCI119856
11. James JA, Neas BR, Moser KL, Hall T, Bruner GR, Sestak AL, et al. Systemic lupus erythematosus in adults is associated with previous Epstein-Barr virus exposure. Arthritis Rheum (2001) 44(5):1122–6. doi: 10.1002/1529-0131(200105)44:5<1122::AID-ANR193>3.0.CO;2-D
12. Li ZX, Zeng S, Wu HX, Zhou Y. The risk of systemic lupus erythematosus associated with Epstein-Barr virus infection: a systematic review and meta-analysis. Clin Exp Med (2019) 19(1):23–36. doi: 10.1007/s10238-018-0535-0
13. Moon UY, Park SJ, Oh ST, Kim WU, Park SH, Lee SH, et al. Patients with systemic lupus erythematosus have abnormally elevated Epstein-Barr virus load in blood. Arthritis Res Ther (2004) 6(4):R295–302. doi: 10.1186/ar1181
14. Kang I, Quan T, Nolasco H, Park SH, Hong MS, Crouch J, et al. Defective control of latent Epstein-Barr virus infection in systemic lupus erythematosus. J Immunol (2004) 172(2):1287–94. doi: 10.4049/jimmunol.172.2.1287
15. Gross AJ, Hochberg D, Rand WM, Thorley-Lawson DA. EBV and systemic lupus erythematosus: A new perspective. J Immunol (2005) 174(11):6599–607. doi: 10.4049/jimmunol.174.11.6599
16. Berner BR, Tary-Lehmann M, Yonkers NL, Askari AD, Lehmann PV, Anthony DD. Phenotypic and functional analysis of EBV-specific memory CD8 cells in SLE. Cell Immunol (2005) 235(1):29–38. doi: 10.1016/j.cellimm.2005.06.010
17. Larsen M, Sauce D, Deback C, Arnaud L, Mathian A, Miyara M, et al. Exhausted cytotoxic control of Epstein-Barr virus in human lupus. PLoS Pathog (2011) 7(10):e1002328. doi: 10.1371/journal.ppat.1002328
18. Draborg AH, Jacobsen S, Westergaard M, Mortensen S, Larsen JL, Houen G, et al. Reduced response to Epstein-Barr virus antigens by T-cells in systemic lupus erythematosus patients. Lupus Sci Med (2014) 1(1):e000015. doi: 10.1136/lupus-2014-000015
19. Tsokos GC, Magrath IT, Balow JE. Epstein-Barr virus induces normal B cell responses but defective suppressor T cell responses in patients with systemic lupus erythematosus. J Immunol (1983) 131(4):1797–801.
20. Su R, Li Z, Wang Y, Liu Y, Zheng X, Gao C, et al. Imbalance between Th17 and regulatory T cells in patients with systemic lupus erythematosus combined EBV/CMV viraemia. Clin Exp Rheumatol (2020) 38(5):864–73.
21. Sutkowski N, Conrad B, Thorley-Lawson DA, Huber BT. Epstein-Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity (2001) 15(4):579–89. doi: 10.1016/S1074-7613(01)00210-2
22. Devergne O, Hummel M, Koeppen H, Le Beau MM, Nathanson EC, Kieff E, et al. A novel interleukin-12 p40-related protein induced by latent Epstein-Barr virus infection in B lymphocytes. J Virol (1996) 70(2):1143–53. doi: 10.1128/JVI.70.2.1143-1153.1996
23. Shinsuke N, Hiroshi I. Overexpression of Epstein-Barr virus-induced gene 3 protein (EBI3) in MRL/lpr mice suppresses their lupus nephritis by activating regulatory T cells. Autoimmunity (2013) 46(7):446–54. doi: 10.3109/08916934.2013.809422
24. Ouyang H, Shi YB, Liu ZC, Wang Z, Feng S, Kong SM, et al. Decreased interleukin 35 and CD4+EBI3+ T cells in patients with active systemic lupus erythematosus. Am J Med Sci (2014) 348(2):156–61. doi: 10.1097/MAJ.0000000000000215
25. Mizoguchi I, Ohashi M, Hasegawa H, Chiba Y, Orii N, Inoue S, et al. EBV-induced gene 3 augments IL-23Ralpha protein expression through a chaperone calnexin. J Clin Invest (2020) 130(11):6124–40. doi: 10.1172/JCI122732
26. Draborg AH, Sandhu N, Larsen N, Lisander Larsen J, Jacobsen S, Houen G. Impaired Cytokine Responses to Epstein-Barr Virus Antigens in Systemic Lupus Erythematosus Patients. J Immunol Res (2016) 2016:6473204. doi: 10.1155/2016/6473204
27. Piroozmand A, Haddad Kashani H, Zamani B. Correlation between Epstein-Barr Virus Infection and Disease Activity of Systemic Lupus Erythematosus: a Cross-Sectional Study. Asian Pac J Cancer Prev (2017) 18(2):523–7.
28. Jog NR, Young KA, Munroe ME, Harmon MT, Guthridge JM, Kelly JA, et al. Association of Epstein-Barr virus serological reactivation with transitioning to systemic lupus erythematosus in at-risk individuals. Ann Rheum Dis (2019) 78(9):1235–41. doi: 10.1136/annrheumdis-2019-215361
29. Vista ES, Weisman MH, Ishimori ML, Chen H, Bourn RL, Bruner BF, et al. Strong viral associations with SLE among Filipinos. Lupus Sci Med (2017) 4(1):e000214. doi: 10.1136/lupus-2017-000214
30. Hoshino A, Tanita K, Kanda K, Imadome KI, Shikama Y, Yasumi T, et al. High frequencies of asymptomatic Epstein-Barr virus viremia in affected and unaffected individuals with CTLA4 mutations. Clin Immunol (2018) 195:45–8. doi: 10.1016/j.clim.2018.07.012
31. Parks CG, Cooper GS, Hudson LL, Dooley MA, Treadwell EL, St Clair EW, et al. Association of Epstein-Barr virus with systemic lupus erythematosus: effect modification by race, age, and cytotoxic T lymphocyte-associated antigen 4 genotype. Arthritis Rheum (2005) 52(4):1148–59. doi: 10.1002/art.20997
32. Parks CG, Hudson LL, Cooper GS, Dooley MA, Treadwell EL, St Clair EW, et al. CTLA-4 gene polymorphisms and systemic lupus erythematosus in a population-based study of whites and African-Americans in the southeastern United States. Lupus (2004) 13(10):784–91. doi: 10.1191/0961203304lu1085oa
33. Harley JB, Chen X, Pujato M, Miller D, Maddox A, Forney C, et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat Genet (2018) 50(5):699–707. doi: 10.1038/s41588-018-0102-3
34. Hsu DH, de Waal Malefyt R, Fiorentino DF, Dang MN, Vieira P, de Vries J, et al. Expression of interleukin-10 activity by Epstein-Barr virus protein BCRF1. Science (1990) 250(4982):830–2. doi: 10.1126/science.2173142
35. Moore KW, Vieira P, Fiorentino DF, Trounstine ML, Khan TA, Mosmann TR. Homology of cytokine synthesis inhibitory factor (IL-10) to the Epstein-Barr virus gene BCRFI. Science (1990) 248(4960):1230–4. doi: 10.1126/science.2161559
36. Go NF, Castle BE, Barrett R, Kastelein R, Dang W, Mosmann TR, et al. Interleukin 10, a novel B cell stimulatory factor: unresponsiveness of X chromosome-linked immunodeficiency B cells. J Exp Med (1990) 172(6):1625–31. doi: 10.1084/jem.172.6.1625
37. Liu Y, de Waal Malefyt R, Briere F, Parham C, Bridon JM, Banchereau J, et al. The EBV IL-10 homologue is a selective agonist with impaired binding to the IL-10 receptor. J Immunol (1997) 158(2):604–13.
38. MacNeil IA, Suda T, Moore KW, Mosmann TR, Zlotnik A. IL-10, a novel growth cofactor for mature and immature T cells. J Immunol (1990) 145(12):4167–73.
39. Jog NR, Chakravarty EF, Guthridge JM, James JA. Epstein Barr Virus Interleukin 10 Suppresses Anti-inflammatory Phenotype in Human Monocytes. Front Immunol (2018) 9:2198. doi: 10.3389/fimmu.2018.02198
40. Stewart JP, Rooney CM. The interleukin-10 homolog encoded by Epstein-Barr virus enhances the reactivation of virus-specific cytotoxic T cell and HLA-unrestricted killer cell responses. Virology (1992) 191(2):773–82. doi: 10.1016/0042-6822(92)90253-L
41. Yoon SI, Jones BC, Logsdon NJ, Harris BD, Kuruganti S, Walter MR. Epstein-Barr virus IL-10 engages IL-10R1 by a two-step mechanism leading to altered signaling properties. J Biol Chem (2012) 287(32):26586–95. doi: 10.1074/jbc.M112.376707
42. Thorley-Lawson DA, Babcock GJ. A model for persistent infection with Epstein-Barr virus: the stealth virus of human B cells. Life Sci (1999) 65(14):1433–53. doi: 10.1016/S0024-3205(99)00214-3
43. Kilger E, Kieser A, Baumann M, Hammerschmidt W. Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J (1998) 17(6):1700–9. doi: 10.1093/emboj/17.6.1700
44. Uchida J, Yasui T, Takaoka-Shichijo Y, Muraoka M, Kulwichit W, Raab-Traub N, et al. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science (1999) 286(5438):300–3. doi: 10.1126/science.286.5438.300
45. He B, Raab-Traub N, Casali P, Cerutti A. EBV-encoded latent membrane protein 1 cooperates with BAFF/BLyS and APRIL to induce T cell-independent Ig heavy chain class switching. J Immunol (2003) 171(10):5215–24. doi: 10.4049/jimmunol.171.10.5215
46. Peters AL, Stunz LL, Meyerholz DK, Mohan C, Bishop GA. Latent membrane protein 1, the EBV-encoded oncogenic mimic of CD40, accelerates autoimmunity in B6.Sle1 mice. J Immunol (2010) 185(7):4053–62. doi: 10.4049/jimmunol.0904065
47. Caldwell RG, Wilson JB, Anderson SJ, Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity (1998) 9(3):405–11. doi: 10.1016/S1074-7613(00)80623-8
48. Minamitani T, Yasui T, Ma Y, Zhou H, Okuzaki D, Tsai CY, et al. Evasion of affinity-based selection in germinal centers by Epstein-Barr virus LMP2A. Proc Natl Acad Sci U S A (2015) 112(37):11612–7. doi: 10.1073/pnas.1514484112
49. Wang H, Nicholas MW, Conway KL, Sen P, Diz R, Tisch RM, et al. EBV latent membrane protein 2A induces autoreactive B cell activation and TLR hypersensitivity. J Immunol (2006) 177(5):2793–802. doi: 10.4049/jimmunol.177.5.2793
50. Wang M, Gu B, Chen X, Wang Y, Li P, Wang K. The Function and Therapeutic Potential of Epstein-Barr Virus-Encoded MicroRNAs in Cancer. Mol Ther Nucleic Acids (2019) 17:657–68. doi: 10.1016/j.omtn.2019.07.002
51. Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MA, Hopmans ES, Lindenberg JL, et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A (2010) 107(14):6328–33. doi: 10.1073/pnas.0914843107
52. Thorley-Lawson DA, Allday MJ. The curious case of the tumour virus: 50 years of Burkitt’s lymphoma. Nat Rev Microbiol (2008) 6(12):913–24. doi: 10.1038/nrmicro2015
53. Speck SH, Ganem D. Viral latency and its regulation: lessons from the gamma-herpesviruses. Cell Host Microbe (2010) 8(1):100–15. doi: 10.1016/j.chom.2010.06.014
54. Repellin CE, Tsimbouri PM, Philbey AW, Wilson JB. Lymphoid hyperplasia and lymphoma in transgenic mice expressing the small non-coding RNA, EBER1 of Epstein-Barr virus. PLoS One (2010) 5(2):e9092. doi: 10.1371/journal.pone.0009092
55. Rosa MD, Gottlieb E, Lerner MR, Steitz JA. Striking similarities are exhibited by two small Epstein-Barr virus-encoded ribonucleic acids and the adenovirus-associated ribonucleic acids VAI and VAII. Mol Cell Biol (1981) 1(9):785–96. doi: 10.1128/MCB.1.9.785
56. Glickman JN, Howe JG, Steitz JA. Structural analyses of EBER1 and EBER2 ribonucleoprotein particles present in Epstein-Barr virus-infected cells. J Virol (1988) 62(3):902–11. doi: 10.1128/JVI.62.3.902-911.1988
57. Nanbo A, Inoue K, Adachi-Takasawa K, Takada K. Epstein-Barr virus RNA confers resistance to interferon-alpha-induced apoptosis in Burkitt’s lymphoma. EMBO J (2002) 21(5):954–65. doi: 10.1093/emboj/21.5.954
58. Wu Y, Maruo S, Yajima M, Kanda T, Takada K. Epstein-Barr virus (EBV)-encoded RNA 2 (EBER2) but not EBER1 plays a critical role in EBV-induced B-cell growth transformation. J Virol (2007) 81(20):11236–45. doi: 10.1128/JVI.00579-07
59. Alles J, Hasler D, Kazmi SMA, Tesson M, Hamilton A, Schlegel L, et al. Epstein-Barr Virus EBER Transcripts Affect miRNA-Mediated Regulation of Specific Targets and Are Processed to Small RNA Species. Noncoding RNA (2015) 1(3):170–91. doi: 10.3390/ncrna1030170
60. Pimienta G, Fok V, Haslip M, Nagy M, Takyar S, Steitz JA. Proteomics and Transcriptomics of BJAB Cells Expressing the Epstein-Barr Virus Noncoding RNAs EBER1 and EBER2. PLoS One (2015) 10(6):e0124638. doi: 10.1371/journal.pone.0124638
61. Samanta M, Iwakiri D, Kanda T, Imaizumi T, Takada K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J (2006) 25(18):4207–14. doi: 10.1038/sj.emboj.7601314
62. Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med (1979) 301(1):5–8. doi: 10.1056/NEJM197907053010102
63. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A (2003) 100(5):2610–5. doi: 10.1073/pnas.0337679100
64. Quan TE, Roman RM, Rudenga BJ, Holers VM, Craft JE. Epstein-Barr virus promotes interferon-alpha production by plasmacytoid dendritic cells. Arthritis Rheumatol (2010) 62(6):1693–701. doi: 10.1002/art.27408
65. Han L, Zhang Y, Wang Q, Xin M, Yang K, Lei K, et al. Epstein-Barr virus infection and type I interferon signature in patients with systemic lupus erythematosus. Lupus (2018) 27:947–54. doi: 10.1177/0961203317753069
66. Agmon-Levin N, Dagan A, Peri Y, Anaya JM, Selmi C, Tincani A, et al. The interaction between anti-Ro/SSA and anti-La/SSB autoantibodies and anti-infectious antibodies in a wide spectrum of auto-immune diseases: another angle of the autoimmune mosaic. Clin Exp Rheumatol (2017) 35(6):929–35.
67. James JA, Harley JB, Scofield RH. Epstein-Barr virus and systemic lupus erythematosus. Curr Opin Rheumatol (2006) 18(5):462–7. doi: 10.1097/01.bor.0000240355.37927.94
68. Yadav P, Tran H, Ebegbe R, Gottlieb P, Wei H, Lewis RH, et al. Antibodies elicited in response to EBNA-1 may cross-react with dsDNA. PLoS One (2011) 6(1):e14488. doi: 10.1371/journal.pone.0014488
69. Yadav P, Carr MT, Yu R, Mumbey-Wafula A, Spatz LA. Mapping an epitope in EBNA-1 that is recognized by monoclonal antibodies to EBNA-1 that cross-react with dsDNA. Immun Inflammation Dis (2016) 4(3):362–75. doi: 10.1002/iid3.119
70. Csorba K, Schirmbeck LA, Tuncer E, Ribi C, Roux-Lombard P, Chizzolini C, et al. Anti-C1q Antibodies as Occurring in Systemic Lupus Erythematosus Could Be Induced by an Epstein-Barr Virus-Derived Antigenic Site. Front Immunol (2019) 10:2619. doi: 10.3389/fimmu.2019.02619
71. Tu J, Wang X, Geng G, Xue X, Lin X, Zhu X, et al. The Possible Effect of B-Cell Epitopes of Epstein-Barr Virus Early Antigen, Membrane Antigen, Latent Membrane Protein-1, and -2A on Systemic Lupus Erythematosus. Front Immunol (2018) 9:187. doi: 10.3389/fimmu.2018.00187
72. James JA, Gross T, Scofield RH, Harley JB. Immunoglobulin epitope spreading and autoimmune disease after peptide immunization: Sm B/B’-derived PPPGMRPP and PPPGIRGP induce spliceosome autoimmunity. J Exp Med (1995) 181(2):453–61. doi: 10.1084/jem.181.2.453
73. Sabbatini A, Bombardieri S, Migliorini P. Autoantibodies from patients with systemic lupus erythematosus bind a shared sequence of SmD and Epstein-Barr virus-encoded nuclear antigen EBNA I. Eur J Immunol (1993) 23(5):1146–52. doi: 10.1002/eji.1830230525
74. McClain MT, Heinlen LD, Dennis GJ, Roebuck J, Harley JB, James JA. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med (2005) 11(1):85–9. doi: 10.1038/nm1167
75. Sundar K, Jacques S, Gottlieb P, Villars R, Benito ME, Taylor DK, et al. Expression of the Epstein-Barr virus nuclear antigen-1 (EBNA-1) in the mouse can elicit the production of anti-dsDNA and anti-Sm antibodies. J Autoimmun (2004) 23(2):127–40. doi: 10.1016/j.jaut.2004.06.001
76. Arbuckle MR, Reichlin M, Harley JB, James JA. Shared early autoantibody recognition events in the development of anti-Sm B/B’ in human lupus. Scand J Immunol (1999) 50(5):447–55. doi: 10.1046/j.1365-3083.1999.00640.x
77. Wang F. Nonhuman primate models for Epstein-Barr virus infection. Curr Opin Virol (2013) 3(3):233–7. doi: 10.1016/j.coviro.2013.03.003
78. Okuno K, Takashima K, Kanai K, Ohashi M, Hyuga R, Sugihara H, et al. Epstein-Barr virus can infect rabbits by the intranasal or peroral route: an animal model for natural primary EBV infection in humans. J Med Virol (2010) 82(6):977–86. doi: 10.1002/jmv.21597
79. Epstein MA, Hausen H, Ball G, Rabin H. Pilot experiments with EB virus in owl monkeys (Aotus trivirgatus). III. Serological and biochemical findings in an animal with reticuloproliferative disease. Int J Cancer (1975) 15(1):17–22. doi: 10.1002/ijc.2910150103
80. Cox C, Naylor BA, Mackett M, Arrand JR, Griffin BE, Wedderburn N. Immunization of common marmosets with Epstein-Barr virus (EBV) envelope glycoprotein gp340: effect on viral shedding following EBV challenge. J Med Virol (1998) 55(4):255–61. doi: 10.1002/(SICI)1096-9071(199808)55:4<255::AID-JMV1>3.0.CO;2-#
81. Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature (1988) 335(6187):256–9. doi: 10.1038/335256a0
82. Mosier DE, Baird SM, Kirven MB, Gulizia RJ, Wilson DB, Kubayashi R, et al. EBV-associated B-cell lymphomas following transfer of human peripheral blood lymphocytes to mice with severe combined immune deficiency. Curr Top Microbiol Immunol (1990) 166:317–23. doi: 10.1007/978-3-642-75889-8_39
83. Cocco M, Bellan C, Tussiwand R, Corti D, Traggiai E, Lazzi S, et al. CD34+ cord blood cell-transplanted Rag2-/- gamma(c)-/- mice as a model for Epstein-Barr virus infection. Am J Pathol (2008) 173(5):1369–78. doi: 10.2353/ajpath.2008.071186
84. Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med (2006) 12(11):1316–22. doi: 10.1038/nm1431
85. Kuwana Y, Takei M, Yajima M, Imadome K, Inomata H, Shiozaki M, et al. Epstein-Barr virus induces erosive arthritis in humanized mice. PLoS One (2011) 6(10):e26630. doi: 10.1371/journal.pone.0026630
86. Babcock GJ, Decker LL, Volk M, Thorley-Lawson DA. EBV persistence in memory B cells in vivo. Immunity (1998) 9(3):395–404. doi: 10.1016/S1074-7613(00)80622-6
87. Borza CM, Hutt-Fletcher LM. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat Med (2002) 8(6):594–9. doi: 10.1038/nm0602-594
88. Mistrikova J, Blaskovic D. Ecology of the murine alphaherpesvirus and its isolation from lungs of rodents in cell culture. Acta Virol (1985) 29(4):312–7.
89. Mistrikova J, Remenova A, Lesso J, Stancekova M. Replication and persistence of murine herpesvirus 72 in lymphatic system and peripheral blood mononuclear cells of Balb/C mice. Acta Virol (1994) 38(3):151–6.
90. Usherwood EJ, Ross AJ, Allen DJ, Nash AA. Murine gammaherpesvirus-induced splenomegaly: a critical role for CD4 T cells. J Gen Virol (1996) 77(Pt 4):627–30. doi: 10.1099/0022-1317-77-4-627
91. Tripp RA, Hamilton-Easton AM, Cardin RD, Nguyen P, Behm FG, Woodland DL, et al. Pathogenesis of an infectious mononucleosis-like disease induced by a murine gamma-herpesvirus: role for a viral superantigen? J Exp Med (1997) 185(9):1641–50. doi: 10.1084/jem.185.9.1641
92. Collins CM, Boss JM, Speck SH. Identification of infected B-cell populations by using a recombinant murine gammaherpesvirus 68 expressing a fluorescent protein. J Virol (2009) 83(13):6484–93. doi: 10.1128/JVI.00297-09
93. Willer DO, Speck SH. Long-term latent murine Gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J Virol (2003) 77(15):8310–21. doi: 10.1128/JVI.77.15.8310-8321.2003
94. Usherwood EJ, Stewart JP, Robertson K, Allen DJ, Nash AA. Absence of splenic latency in murine gammaherpesvirus 68-infected B cell-deficient mice. J Gen Virol (1996) 77(Pt 11):2819–25. doi: 10.1099/0022-1317-77-11-2819
95. Larson JD, Thurman JM, Rubtsov AV, Claypool D, Marrack P, van Dyk LF, et al. Murine gammaherpesvirus 68 infection protects lupus-prone mice from the development of autoimmunity. Proc Natl Acad Sci U S A (2012) 109(18):E1092–100. doi: 10.1073/pnas.1203019109
96. Tibbetts SA, Loh J, Van Berkel V, McClellan JS, Jacoby MA, Kapadia SB, et al. Establishment and maintenance of gammaherpesvirus latency are independent of infective dose and route of infection. J Virol (2003) 77(13):7696–701. doi: 10.1128/JVI.77.13.7696-7701.2003
Keywords: Epstein-Barr virus, systemic lupus erythematosus, inflammation, autoimmune disease, viral homologs of host genes, molecular mimicry
Citation: Jog NR and James JA (2021) Epstein Barr Virus and Autoimmune Responses in Systemic Lupus Erythematosus. Front. Immunol. 11:623944. doi: 10.3389/fimmu.2020.623944
Received: 30 October 2020; Accepted: 21 December 2020;
Published: 03 February 2021.
Edited by:
J. Michelle Kahlenberg, University of Michigan, United StatesReviewed by:
Andras Perl, Upstate Medical University, United StatesCopyright © 2021 Jog and James. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neelakshi R. Jog, TmVlbGFrc2hpLUpvZ0BvbXJmLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.