 Allison M. Owen
Allison M. Owen Jessica B. Fults
Jessica B. Fults Naeem K. Patil
Naeem K. Patil Antonio Hernandez1
Antonio Hernandez1 Julia K. Bohannon
Julia K. Bohannon- 1Department of Anesthesiology, Vanderbilt University Medical Center, Nashville, TN, United States
- 2University of Texas Southwestern Medical School, Dallas, TX, United States
- 3Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, TN, United States
Despite advances in critical care medicine, infection remains a significant problem that continues to be complicated with the challenge of antibiotic resistance. Immunocompromised patients are highly susceptible to development of severe infection which often progresses to the life-threatening condition of sepsis. Thus, immunotherapies aimed at boosting host immune defenses are highly attractive strategies to ward off infection and protect patients. Recently there has been mounting evidence that activation of the innate immune system can confer long-term functional reprogramming whereby innate leukocytes mount more robust responses upon secondary exposure to a pathogen for more efficient clearance and host protection, termed trained immunity. Toll-like receptor (TLR) agonists are a class of agents which have been shown to trigger the phenomenon of trained immunity through metabolic reprogramming and epigenetic modifications which drive profound augmentation of antimicrobial functions. Immunomodulatory TLR agonists are also highly beneficial as vaccine adjuvants. This review provides an overview on TLR signaling and our current understanding of TLR agonists which show promise as immunotherapeutic agents for combating infection. A brief discussion on our current understanding of underlying mechanisms is also provided. Although an evolving field, TLR agonists hold strong therapeutic potential as immunomodulators and merit further investigation for clinical translation.
Introduction
Nosocomial infections, or healthcare associated infections (HCAI), represent a significant cause of global morbidity and mortality, and the United States is no exception. Each year, approximately one out of 25 hospitalized patients in the United States is diagnosed with at least one infection related to hospital care (1). Infection leading to sepsis remains one of the leading causes of death in U.S. hospitals, affecting more than 1.7 million and causing 270,000 deaths annually. Sepsis is also a major contributor to re-hospitalizations and is one of the most expensive conditions treated in U.S. hospitals, costing more than $2 billion per year (2–4). The overall 30-day mortality rate for patients in the intensive care unit (ICU) is approximately 20%, and for patients with sepsis and accompanying organ dysfunction this statistic is 30%–50% (5). Despite advancements in healthcare overall, this clinical outcome has not improved over the past 25 years (6).
Critically ill patients are at a significantly increased risk of infection due to injury- or illness-induced immune dysfunction and pathogen exposure through invasive life-saving procedures in the healthcare setting (7). Further complicating the risk of HCAI is the continuing rise of antibiotic-resistant pathogens. Infections secondary to resistant pathogens are one of the most critical threats to modern medicine, and this situation is being exacerbated by dwindling effective treatment options (8). The United States has more than 2.8 million antibiotic-resistant infections annually, resulting in more than 35,000 deaths (1). Solely focusing on the development of new antibiotics is not a permanent solution as pathogens will continue to evolve and become resistant to new drugs (9). Thus, immunomodulatory therapies that boost host immune responses and protect immunocompromised patients against infections are a highly attractive strategy.
One promising approach to restore immune responses relies on the induction of innate immune memory, also termed trained immunity. Classically, the role of the innate immune system is to recognize a pathogen and mount a broad and rapid response with immunological memory being considered specific to the adaptive immune system. Recent evidence demonstrates that innate immune cells also display long-term adaptive characteristics after initial challenge with pathogens or their products, which results in enhanced capacity to eliminate infections upon subsequent challenge (10, 11). Trained immunity refers to the phenomenon of activating the innate immune system through exposure to pathogen associated molecular patterns (PAMPs), triggering long-term functional reprogramming by which innate leukocytes mount an enhanced antimicrobial response upon exposure to a secondary microbial pathogen (12). This protection is broad whereby the host is resistant to an array of pathogens for weeks to months once the altered functional state of innate immune training is initiated. Important to note, the terminology referring to different adaptive programs of innate immunity has evolved with the field. Although used somewhat interchangeably in the literature, a consensus has recently been made to clearly differentiate between the four different adaptive programs: differentiation, priming, tolerance, and training (13). Trained immunity specifically refers to the phenomenon in which the activation status of innate cells returns to baseline after primary stimulation prior to the secondary stimulation. Nevertheless, this active new field of research is rapidly evolving, an arm of which is aimed at taking advantage of trained immunity as an innovative strategy to combat infection (14).
Emerging evidence suggests that Toll-like receptor (TLR) agonists are a promising class of immunomodulatory agents that confer long-term protection against subsequent infectious challenge via enhanced innate immunity (15, 16). TLRs play a crucial role in activation of innate immune responses by recognizing PAMPs which then trigger downstream signaling pathways and ultimately stimulate the production of proinflammatory cytokines and type I interferons. Several TLR agonists are recognized widely for their vaccine adjuvant properties and several are FDA-approved for this use, but their ability to induce trained immunity is becoming more recognized. Here, we review recent progress in our understanding of mechanisms of TLR agonist-mediated trained immunity and its strong potential for clinical translation to protect patients against life-threatening infection.
Toll-Like Receptor Signaling Pathways
As the first line of defense against pathogens, the innate immune system utilizes pattern recognition receptors (PRRs) to rapidly detect microbes and deploy antimicrobial responses. TLRs are a well characterized family of PRRs comprised of 10 members in humans (TLR1-TLR10) and 12 members in mice (TLR1-9, TLR11-13) that are expressed in innate immune cells (i.e. dendritic cells, DCs; and macrophages) and non-immune cells (i.e. fibroblasts and epithelial cells) (17). These receptors are synthesized in the endoplasmic reticulum (ER), processed in the Golgi apparatus, and transported to the plasma membrane or intracellular compartment depending on the localization of the PAMP they recognize (18, 19). TLRs detect a wide array of PAMPs, including Gram negative and positive bacteria, viruses, flagellin proteins, lipids, nucleic acids, and damage-associated molecular patterns (DAMPs). This is in part accomplished by receptor localization to the cell surface or intracellular compartments (20). TLRs which recognize nucleic acids are localized to intracellular compartments for decreased risk of contact with “self” nucleic acids whereas cell surface TLRs largely recognize microbial membrane compartments and therefore do not require this protective strategy (21).
TLRs are composed of a horseshoe-like leucine-rich repeat (LRR) ectodomain which interacts with the respective PAMP or DAMP, a transmembrane helix domain, and a cytoplasmic Toll/IL-1 receptor (TIR) domain which is involved in activation of downstream signaling (22, 23). Upon ligand binding, TLRs homo- or hetero-dimerize which dictates recruitment of specific TIR containing adaptor proteins for activation of downstream signaling. TLRs signal either by recruiting the adapter molecule myeloid differentiation primary response differentiation gene 88 (MyD88) or the MyD88-independent Toll/IL-1R (TIR) domain-containing adapter producing interferon-β (TRIF) signaling. Initiation of MyD88- or TRIF-dependent signaling activates mitogen-activated protein kinases (MAPKs) and IkB kinases (IKKs) that then activate transcription factors to regulate the expression of pro-inflammatory cytokines and chemokines as well as type I interferons (Figure 1). Therefore, these pathways trigger unique antimicrobial defenses to confer protection against diverse pathogens. MyD88- and TRIF-dependent signaling cascades are important to consider in the understanding and application of TLR-mediated trained immunity.
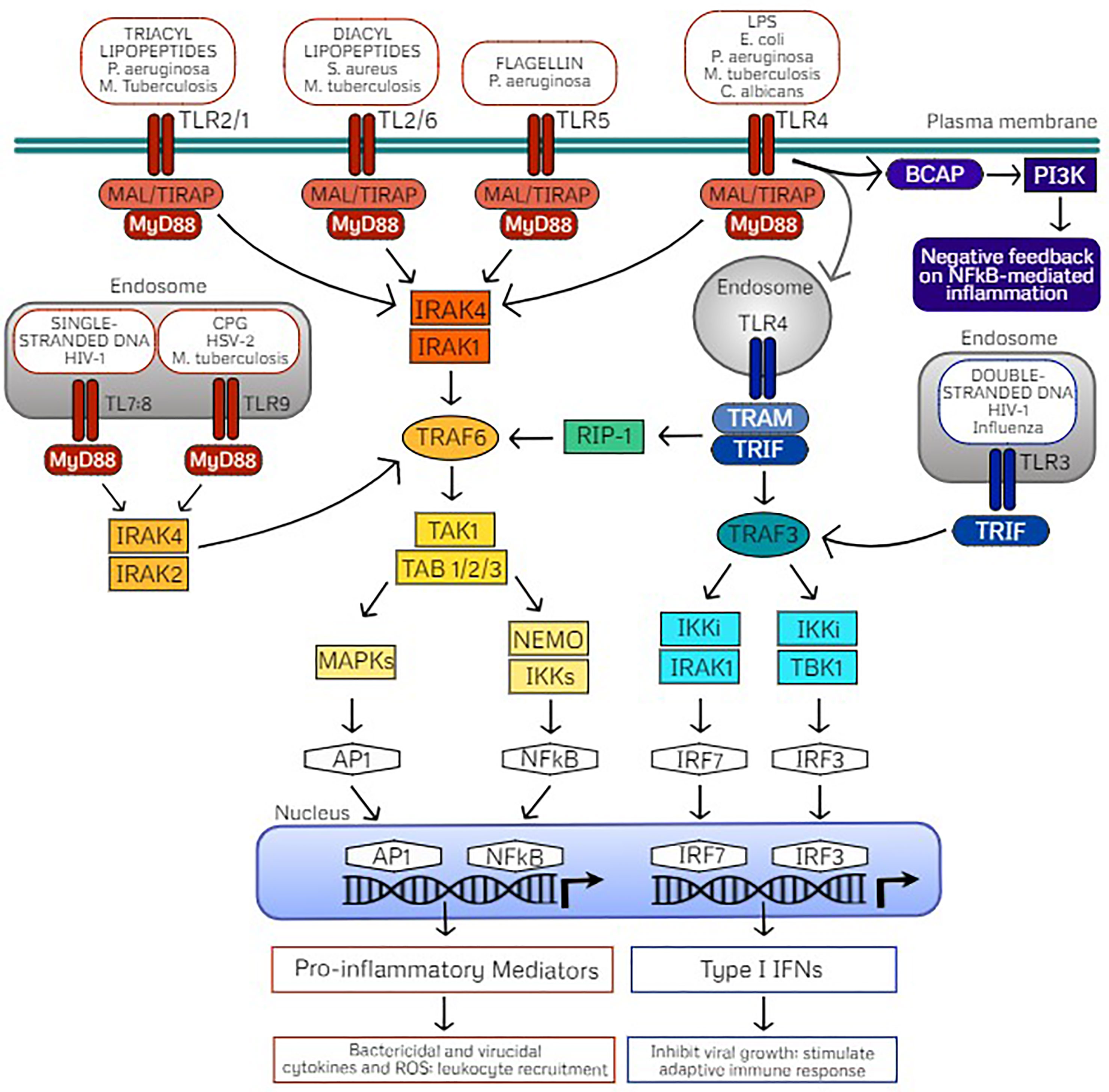
Figure 1 TLR signaling pathways mediated through MyD88- or TRIF-dependent cascades. Cell surface Toll-like receptors (TLRs) include TLR4 and TLR5 which form homodimers upon recognition of their classic ligands lipopolysaccharide (LPS) and flagellin, respectively. TLR2 is also localized on the cell surface which heterodimerizes with TLR1 or TLR6, dependent on ligand recognition with either triacyl or diacyl lipopeptides. TLR recognition of PAMPs on common pathogens are indicated. These cell surface TLRs signal via the adaptor protein myeloid differentiation primary response protein (MyD88) through MyD88-adapter-like protein (MAL) also referred to as TIR Domain Containing Adaptor Protein (TIRAP). Intracellular TLRs include TLR9, stimulated by agonist CpG oligodeoxynucleotides (CPG) and the heterodimer TLR7/TLR8, stimulated by single stranded DNA (ssDNA) which signal through direct interaction with MyD88. Activation of MyD88 signaling induces phosphorylation of IL-R-associated kinases (IRAKs) dependent on TLR localization which in turn interacts with TNF receptor-associated factor 6 (TRAF6) and downstream activation of the TAK1 (transforming growth factor β–activated kinase 1)/TAB (TAK1-binding protein) complex. The TAK1/TAB complex activates mitogen-activated protein kinases (MAPKs) or the IkB kinase (IKK) complex of which NF-κB essential modulator (NEMO) is the regulatory subunit. These signaling events activate transcription factors activator protein-1 (AP1) and nuclear factor-κB (NFκB), which translocate into the nucleus for transcription of inflammatory mediators. As a result, pro-inflammatory cytokines and reactive oxygen species drive bacterial killing and limit viral replication as well as stimulate leukocyte recruitment to clear the infectious pathogen. Alternatively, TLR3 recognizes double stranded DNA (dsDNA) and uniquely signals through the adaptor protein Toll/IL-1R (TIR) domain-containing adapter producing interferon-β (TRIF) which interacts with TRAF3. TLR4 also activates TRIF through endocytosis, which activates TRIF signaling via the sorting adaptor protein TRIF-related adaptor molecule (TRAM). Activated TRAF3 signals through IKKi/IRAK1 or IKKi/TBK1 which activate the transcription factors interferon-regulatory factor (IRF) 7 and IRF3, respectively, which translocate to the nucleus for transcription of type I interferons (IFNs). Type I interferons act to inhibit viral replication as well as stimulate adaptive immunity. TRIF-dependent TLR4 signaling can also activate TRAF6 via receptor interacting protein (RIP)-1 for late NFκB signaling. B-cell adaptor for phosphatidylinositol 3-kinase (PI3K) (BCAP) also seems to be an adaptor protein that confers negative feedback on NFκB-mediated inflammation via PI3K as a regulatory mechanism.
MyD88-Dependent Signaling Cascades
Upon ligand binding, all TLRs except TLR3 initiate downstream signaling by recruiting adaptor protein MyD88 either directly (TLR9, TLR11, TLR13, TLR7/TLR8, and TLR2/TLR10) or indirectly via the sorting adaptor TIR domain containing adaptor protein (TIRAP; also termed MyD88-adaptor-like, MAL; TLR4, TLR5, TLR2/TLR1, and TLR2/TLR6) (18, 24). TIRAP binds with different lipids depending on TLR localization which mediates assembly of kinases (IRAK4 and either IRAK2 or IRAK1) termed the “Myddosome” (17, 25, 26). Through formation of this oligomeric signaling complex, the kinase domains of IL-1 receptor-associated kinases (IRAKs) are phosphorylated. Activated IRAK1 associates with TRAF6 and the TAK1 protein kinase complex which culminates in activation of the transcription factors nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) via IKKs and MAPK, respectively. Thus, MyD88-dependent TLR signaling results in translocation of NF-κB and AP-1 to the nucleus for production of pro-inflammatory mediators, playing a critical role in triggering an inflammatory response for defense against an invading pathogen.
Beyond its role in stimulating inflammation, MyD88 is a key regulator of phagocytosis of bacteria by macrophages (27) and DCs (28) via IRAK4 and p38 MAPK (29) which lead to the expression of scavenger receptors. This pathway also influences phagocytosis via NADPH oxidase assembly and thus superoxide production for bacterial killing (30). Upon TLR4 stimulation by lipopolysaccharide (LPS) in macrophages, it has been found that MyD88 signaling also activates Src tyrosine kinase via the cytoplasmic protein EGF receptor pathway substrate number 9 [Eps8 (31)] which causes actin cytoskeleton rearrangement. Although alternate MyD88-independent pathways have been identified [actin-Cdc42/Rac pathway (32); CD14/complement receptor 3 (33)], the MyD88 pathway seems to be the main driver of phagocytosis of Borrelia burgdorferi (34). It also plays a role in phagocytosis of fungal pathogens, although this differs depending on the fungal challenge (35). The role of MyD88 signaling in innate immunity is highlighted by high incidence of life-threatening infection in patients with MyD88 and IRAK4 deficiencies (36, 37).
TRIF-Dependent Signaling Cascades
TLR3 exclusively signals through the TRIF-dependent pathway through direct interaction, whereas TLR4 uniquely signals through MyD88 at the cell surface or through TRIF (also referred to as TICAM-1) upon internalization of the receptor complex after ligand binding (20). Trafficking of TLR4 to the endosomal compartment is dependent on CD14 (38, 39). After TLR4 endocytosis, the recruitment of the adaptor protein TRAM (also termed TICAM-2) is coordinated with the release of the TIRAP-MyD88 complex (19, 40). From the endosomal compartment, TLR3 and TLR4 associate with the TRAM-TRIF complex which interact with TNF receptor-associated factor 6 (TRAF6) and lead to activation of NF-κB or AP-1 and downstream production of inflammatory cytokines. Alternatively, interaction with TRAF3 induces interferon regulatory factors 3 (IRF3) or IRF7 and downstream production of type I interferons (17, 40). Briefly, TRAF6 activates the TAK1 complex that subsequently activates NF-κB and MAPKs via the IKK family member NF-kB essential modifier (NEMO). On the other hand, TRAF3 recruits TBK1 and IKKi resulting in phosphorylation of IRF3 or IRF7 which dimerize and translocate to the nucleus to induce to transcription of IFNs. Additional intricacies of the TRIF-dependent signaling cascade are reviewed by Ullah et al. (18). Production of type I IFNs is most often associated with defense against double-stranded RNA (dsRNA) viruses; however, they are also important for response to single-stranded RNA (ssRNA) viruses, DNA viruses, and bacteria (41, 42). TRIF signaling seems to play a role in activation of the adaptive immune system via T cell stimulation. Importantly, TRIF signaling mediates caspase activation, apoptosis, and necroptosis which may play a role in removing infected cells, thus preventing pathogen dissemination via cell death (43–45).
Balance Among MyD88- and TRIF-Dependent-Signaling
Activation of MyD88-dependent and TRIF-dependent signaling cascades allows for immune functional responses specific to the pathogen sensed by TLR ligand binding, as discussed above. Thus, these pathways are subject to several regulatory strategies for the balanced production of inflammatory cytokines and type I IFNs for elimination of pathogens but also to control the magnitude of the response to prevent pathogenic inflammation and autoimmune disease (46). Such regulatory controls of TLR-mediated inflammatory responses include cooperation with coreceptors, post-translational modifications, cellular trafficking, and negative feedback, which are reviewed in detail by Leifer and Medvedev (47). Commonly, regulatory molecules (1) interfere with signaling complex formation via TIR domain-containing molecules, (2) prevent association of TRAF6 or TRAF3 with their respective signaling complexes via deubiqutinases (48–50), (3) competitively inhibit downstream signaling (51), or (4) provide mRNA stability of signaling molecules regulated by miRNAs (50), or cytokines by RNA-binding proteins (52).
Interestingly, both MyD88-dependent and TRIF-dependent pathways are required for maximal antimicrobial responses upon LPS-activation of TLR4 (53), demonstrating that they work in concert rather than being redundant. As such, TLR4 signaling is tightly controlled by localization, internalization upon ligand binding (thus acting as a temporal regulator), as well as influence of regulatory molecules. In addition to MAPK and NF-κB pathway activation upon TLR stimulation, the PI3K pathway is also activated via B-cell adaptor for PI3K (BCAP) in macrophages (54, 55) and DCs (56). BCAP/PI3K signaling serves as a negative feedback arm which limits NF-κB induced inflammation and acts as an endogenous regulatory mechanism (57). Additionally, the adaptor TRAF3 has been found to play an inhibitory role on TLR-mediated MAPK activity through preventing the release of the TAK1 signaling complex (58, 59), while peroxiredoxin-1 (PRDX1) attenuates NF-κB activation via attenuation of ubiquitin-ligase activity of TRAF6 (60). Understanding these endogenous negative feedback mechanisms will be highly useful while translating TLR-mediated trained immunity for clinical application to protect patients against infection with careful attention as to limit inflammatory responses.
Toll-Like Receptor Agonist-Mediated Trained Immunity and Protection Against Infection
Innate Immune Cell Types Which Drive Toll-Like Receptor-Induced Trained Immunity
TLR agonists have exhibited highly attractive immunomodulatory properties whereby they induce augmentation of cell recruitment, antimicrobial effector functions (i.e. phagocytosis, respiratory burst, production of proinflammatory cytokines and chemokines), bacterial clearance, attenuate inflammation, and trigger cross-protection to infection with clinically relevant pathogens (61). Importantly, trained immunity is not dependent on T and B lymphocytes as evidenced by preserved protection against several models of infection in transgenic RAG2 knockout mice comparative to survival benefit observed in wild type animals (62, 63).
To date, TLR-mediated innate immune cellular responses have largely been studied in monocytes, macrophages, and natural killer (NK) cells which show long-term functional reprogramming with increased responses to secondary stimulation by bacterial, parasitic, or viral microbes (12, 62, 64) (Figure 2). As the ‘first responders’ to infection, neutrophils play a key role in TLR-mediated resistance to infection via increased recruitment and function (65–67). Similarly, activation of macrophages by TLR signaling results in increased antimicrobial effector functions (phagocytic capacity, respiratory burst, altered production of inflammatory mediators) (63). Importantly, both neutrophils and macrophages are required for TLR4-mediated resistance to infection. DCs can be activated by TLR signaling or secondary to TLR-activation of NK cells which ‘bridge the gap’ between innate and adaptive immunity whereby activated and matured DCs migrate to the lymph nodes and subsequently activate naïve T-cells (15, 68). Thus, immunomodulation of DCs by TLR signaling may prove to be an effective vaccine adjuvant strategy (69). Continued investigation regarding innate immune cell responses to TLR-mediated trained immunity will help refine therapeutic strategies to address specific clinical scenarios.
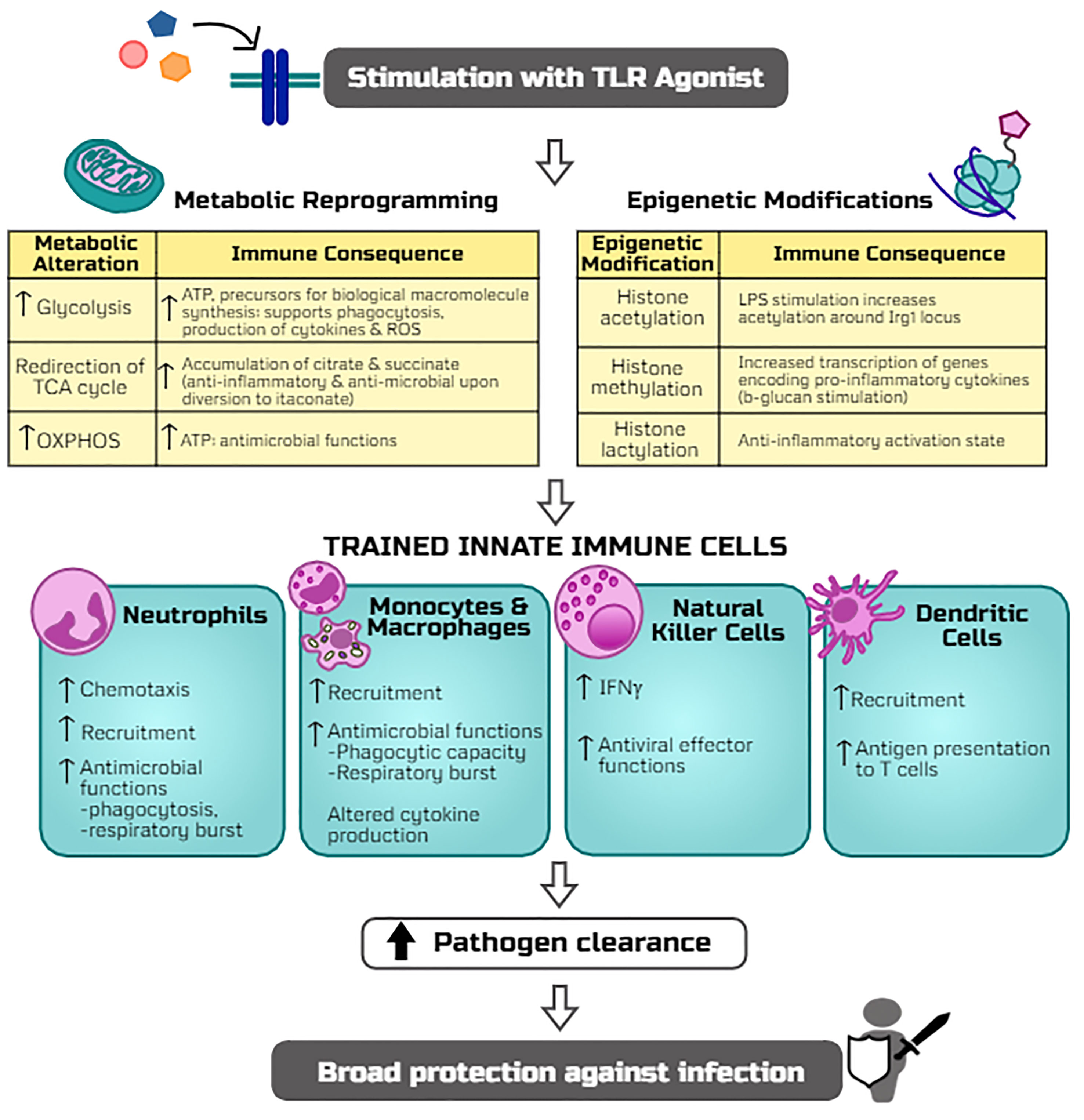
Figure 2 Potential mechanisms by which TLR agonists trigger trained immunity and host resistance to infection. The stimulation of TLR signaling induces metabolic reprogramming (including alterations in glycolysis, TCA cycle, and oxidative phosphorylation, i.e. OXPHOS) and epigenetic modifications (histone acetylation, methylation, and lactylation) which rewire innate leukocytes for more robust antimicrobial functions upon a secondary infectious challenge. Adaptations of innate cell programs thereby allow for more efficient clearance of pathogens and thus protection against a broad array of infections.
Immunomodulation via Targeting Cell Surface Toll-Like Receptors
Agonists which stimulate cell surface TLRs 2, 4, and 5 have been widely studied for their potential as immunomodulators and conferring host resistance to infection. Such studies are discussed below and are summarized in Table 1.
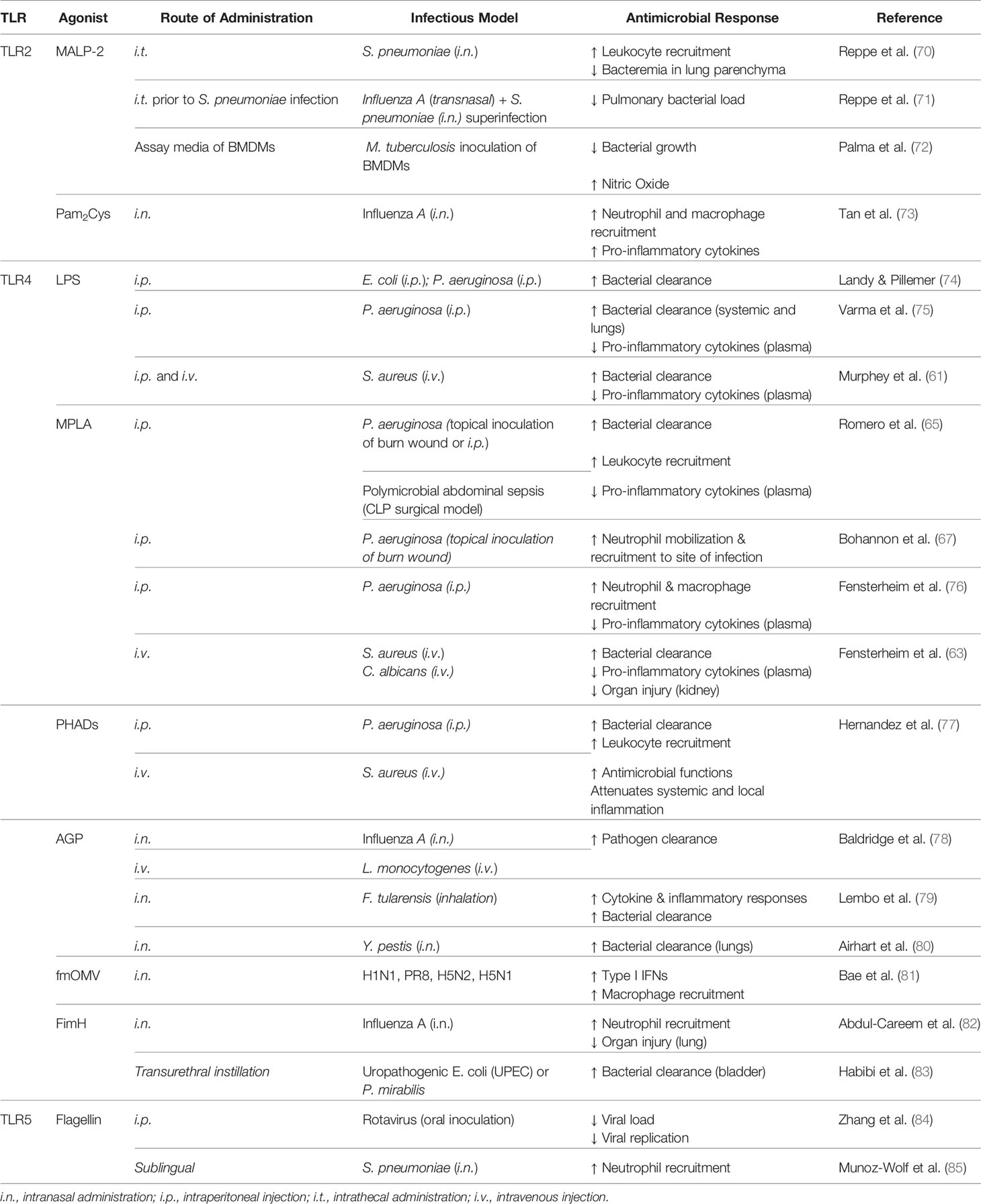
Table 1 Agonists which trigger trained immunity via cell surface TLRs.
Toll-Like Receptor 2
TLR2 recognizes lipid-containing PAMPs of Gram positive bacteria (i.e. lipopeptides, peptidoglycan), as well as viral (i.e. HSV glycoproteins) and fungal (i.e. zymosan) pathogens, and upon activation it forms heterodimers with TLR1, TLR6, or other cell surface molecules such as Dectin-1 and CD36 (86). Both TLR2/TLR1 and TLR2/TLR6 result in MyD88-dependent signaling via MAL/TIRAP; however, the TLR2/TLR1 heterodimer is activated by triacyl lipopeptides whereas the TLR2/TLR6 heterodimer is stimulated by diacyl lipopeptides (87). Several TLR2 agonists have shown promising immunomodulatory effects.
First isolated from Mycoplasma fermentans in 1997 (88), macrophage-activating lipopeptide-2 (MALP-2) has become a well-studied immunomodulator which activates TLR2/TLR6 heterodimer. When administered 24 h prior to challenge with Streptococcus pneumoniae, MALP-2 treatment reduced bacterial load and enhanced leukocyte migration in the lungs (70). Interestingly, treatment of influenza A virus-infected mice with MALP-2 prior to challenge with S. pneumoniae enhanced leukocyte recruitment and reduced bacterial load in the lungs, and was associated with increased survival and improved body condition (71). MALP-2 immunomodulation is presumably driven by its ability to rapidly stimulate neutrophil chemotactic activity followed by induction of monocyte chemoattractant protein-1 (MCP-1) activity in the lungs (89). Further, MALP-2 induces production of proinflammatory cytokines (IL-6, TNF-α) and chemokines (macrophage inflammatory protein-1α and -1β; MIP) (89, 90). Palma et al. also postulated that the microbicidal effect which they observed of MALP-2 on Mycobacterium tuberculosis was mediated by nitric oxide (NO) production (72).
As MALP-2 demonstrates attractive immunomodulatory potential, several synthetic analogs of the molecule, termed palmitoylated peptides, have been investigated in the recent decade. The peptide dipalmitoyl-S-glyceryl cysteine (Pam2Cys) targets TLR2/TLR1, whereas the peptide tripalmitoyl-S-glyceryl cysteine (Pam3Cys) stimulates TLR2/TLR6. These peptides and their derivatives hold strong potential as vaccine adjuvants, with Pam2Cys seeming to be more ideal due to increased solubility and potency compared to Pam3Cys (91, 92). A body of evidence suggests that immunostimulation with Pam2Cys provides immediate protection against acute infection with influenza virus but also allows for the development of specific immune responses for long-term protection (73). Such protection is mediated via activation of DCs, increased leukocyte recruitment, and increased production of inflammatory cytokines (73). Mifsud and colleagues demonstrated that a derivative of Pam2Cys mediates potent anti-viral activity against influenza infection but also protects against secondary infections with S. pneumoniae by reducing bacterial burden and inflammation (93). Although these compounds display promise, their synthesis is expensive; thus, recently a novel inexpensive synthesis strategy of an N-acetylated Pam2Cys analog has been developed, which seems to maintain high potency (94).
The potential application of another TLR2/TLR6 agonist, GSK3277329, was studied in the context of chemotherapy-induced neutropenia which demonstrated that repeated daily injections of the compound for 2 weeks effectively restored neutrophil loss in monkeys given chemotherapy treatment (95). This evidence further illustrates the potential application of TLR agonists in the context of protecting immunocompromised patients from potentially life-threatening infection by boosting innate immunity.
Toll-Like Receptor 4
TLR4 recognizes lipopolysaccharide (LPS) and uniquely signals through both MyD88- and TRIF-dependent pathways. Discovered in 1956 by Landy and Pillemer, mice treated with LPS were resistant to subsequent challenge with Gram negative pathogens (74). Following this work, it became evident that LPS conferred resistance to a broad array of microbes beyond Gram negative bacteria (75, 96) to include Gram positive Staphylococcus aureus (61) and fungal pathogens (97) as well as polymicrobial sepsis (98). LPS-mediated resistance to infection is associated with reduced bacterial burden (75, 99), increased leukocyte recruitment (61, 100), and attenuated inflammation (101).
Despite several studies demonstrating its potential therapeutic benefit, the application of LPS as an immunomodulator for translation to the clinical scenario was largely abandoned due to its toxicity (16, 102). More recently, derivatives of LPS as well as synthetic molecules have demonstrated potent induction of trained immunity with significantly reduced toxicity, thus holding strong therapeutic potential. Notably, monophosphoryl lipid A (MPLA) is structurally identical to LPS with the exception of the cleaved C1 phosphate group from lipid A which reduces its toxicity 100-fold (103, 104). Binding of MPLA by TLR4 induces both MyD88- and TRIF-dependent signaling, although MyD88 signaling seems to be predominant (105). Similarly to protection conferred by LPS immunomodulation, administration of MPLA prior to infectious challenge provides a survival benefit to an array of pathogens including Gram negative P. aeruginosa, Gram positive S. aureus, viral influenza, and fungal C. albicans (63, 65, 106). Importantly, MPLA also protects burn-injured mice from wound infection with the clinically relevant pathogen P. aeruginosa (67) and in large animals (sheep (107) demonstrating its capacity to induce protection in the immunocompromised host. MPLA-driven survival benefit lasts for at least 10 days following administration (76). Evidence suggests that B and T cells are not required for MPLA-mediated protection to S. aureus, neither were recruited monocytes; conversely, depletion of macrophages or neutrophils resulted in loss of MPLA-induced survival benefit (63). MPLA treatment enhances leukocyte recruitment, bacterial clearance, antimicrobial functions, and attenuates inflammation which all likely play a role in resistance to infection (65, 67). It is important to highlight that MPLA similarly enhances human neutrophil responses characterized by increased chemotaxis and bacterial killing (108). Additionally, MPLA stimulates the adaptive immune response whereby it increases antibody titers up to 20-fold (109) and thus is used as an adjuvant in malaria (AS01), human papillomavirus (HPV), and hepatitis B (AS04) vaccines (110, 111).
With their immunostimulatory properties, it is unsurprising that several synthetic TLR4 agonists have been developed with the goal of clinical translation. One promising class of synthetic TLR4 agonists are phosphorylated hexa-acyl disaccharides (PHADs) which are similar in structure to MPLA as they have only one phosphate group (16). PHADs similarly bind TLR4 and activate both MyD88- and TRIF-dependent signaling. Hernandez and colleagues recently showed that treating mice with PHADs confers protection to P. aeruginosa and S. aureus, both of which are of high clinical relevance (77). They found the survival benefit to be associated with increased bacterial clearance, an effect which was observed up to 10 days after treatment. Further, treatment with PHADs increased leukocyte recruitment and antimicrobial functions while attenuating systemic and local levels of proinflammatory cytokines (77).
Another synthetic lipid A TLR4 agonist, aminoalkyl glucosamine 4-phosphate (AGP), was first found to possess immunostimulatory properties two decades ago (112). Initially studied for their potential as a vaccine adjuvant (80), later studies demonstrated that AGPs confer protection to an otherwise lethal influenza challenge as well as to Listeria monocytogenes infection which were associated with increased bacterial clearance (78). Intranasal administration of AGP either before or after infection with the Gram-negative pathogen Francisella tularensis resulted in increased survival, and interestingly the survivors were protected against rechallenge with aerosolized Francisella novicida (113). Likewise, intranasal administration of AGPs prior to challenge with Yersinia pestis also extended time to death which was correlated with cytokine production and decreased bacterial load in the lung (80). Evidence suggests that TLR4 signaling via activation by AGPs induces NF-κB and IRF-3 signaling independent of the co-receptor CD14 (114).
Additionally, outer membrane vesicles (OMVs) with low endotoxicity by modification of lipid A of LPS (fmOMV) may increase protective benefit of intranasally administered influenza vaccine (115). Further, fmOMV confers protection against a lethal dose of pandemic viruses (H1N1, PR8, H5N2, and highly pathogenic H5N1) which is dependent on macrophages but independent of neutrophils. Treatment with fmOMV increased macrophage recruitment and production of type I IFNs without observance of adverse effects (81).
Beyond lipid A compounds, other natural and synthetic TLR4 ligands have been investigated. Fimbriae H protein (FimH) is the receptor-recognizing element of the adhesive organelle type 1 fimbriae on uropathogenic E. coli (UPEC) (116). Mice which receive FimH intranasally are resistant to influenza infection through increased recruitment of neutrophils and production of proinflammatory cytokines (TNF-α and IL-12) and chemokines (RANTES) in a macrophage-independent manner (82). Fusion of fimH with the MR/P fimbriae protein MrpH from P. mirabilis (MrpH.FimH fusion protein) conferred higher protection to UPEC and P. mirabilis, associated with reduced bacterial burden in the bladder and kidney as well as increased neutrophil recruitment (83).
Two classes of synthetic small molecule agonists have been studied with modest therapeutic potential. Neoseptin-3 is a more potent TLR4 agonist than LPS despite being structurally unique (117, 118). Neoseptin-3-activation of TLR4 signaling results in MyD88- and TRIF-dependent signaling and downstream activation of NF-κB and IFN-β but not IFN-α (118). Evidence suggests that Neoseptin-3 may hold strong vaccine adjuvant properties whereby mice immunized by ovalbumin (OVA) together with the compound had increased OVA-specific IgG production 21 days later when compared to immunized vehicle controls mice. However, these compounds failed to induce TLR4 signaling in human THP-1 monocytes, thus calling into question their therapeutic potential (118).
Toll-Like Receptor 5
TLR5 recognizes bacterial flagellin (119) and signals through MyD88, culminating in the production of inflammatory mediators. Studies have shown that bacterial flagellin is indeed an immunomodulatory agent. Mucosal administration of flagellin conferred resistance to S. pneumoniae lung infection whereby flagellin treatment increased bacterial clearance which was associated with increased neutrophil mobilization independent of B- and T-cells (120). More recently, this research group showed that sublingual administration of flagellin also effectively protects against pneumonia (85). Demonstrating cross-protection, treatment with bacterial flagellin was able to prevent infection with or cure ongoing infection of rotavirus in mice independent of adaptive immunity (84).
Bacterial flagellin also restores antibiotic-impaired innate immunity (121) and improves efficacy of antibiotics in the treatment of influenza virus or pneumonia (122). Thus, bacterial flagellin may be highly useful in prevention of infection in immunocompetent and immunocompromised individuals, as well as a strategy to combat antibiotic resistant microbes and boost efficacy of current antibiotic drugs.
Immunomodulation via Targeting Intracellular Toll-Like Receptors
Numerous TLR agonists which activate intracellular TLRs have also been widely studied for their potential application as immunomodulators that trigger trained immunity, which are summarized in Table 2.
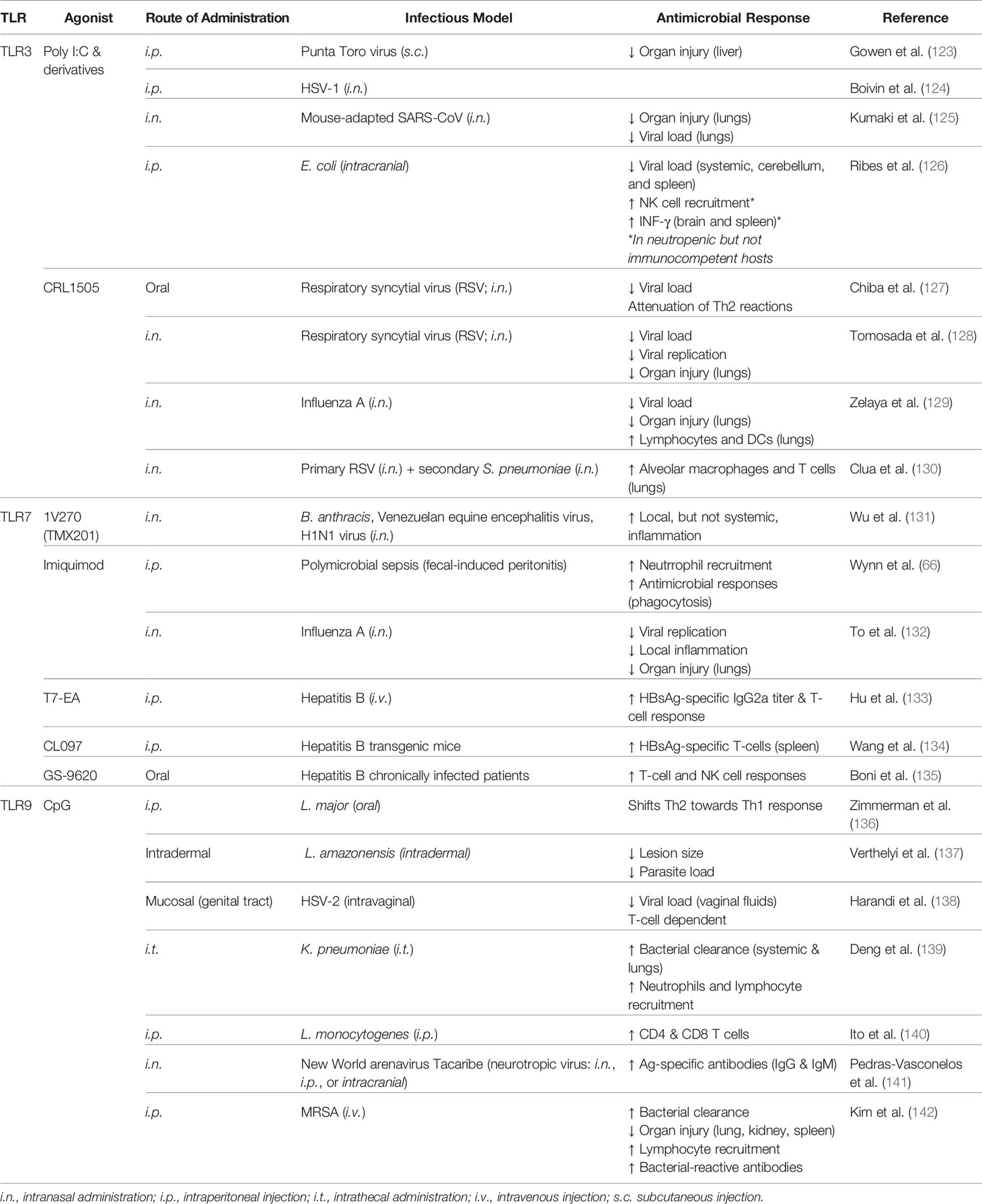
Table 2 Agonists which trigger trained immunity via intracellular TLRs.
Toll-Like Receptor 3
TLR3 is localized to the intracellular compartment and recognizes viral dsRNA, including that produced during replication of ssRNA viruses or self-RNAs released from damaged cells (86, 143). Stimulation of TLR3 results in direct interaction with TRIF for downstream activation of IRF3 and modest activation of NF-κB (144). TLR3/TRIF activation culminates in production of type I IFNs and inflammatory cytokines for killing of invading viruses and has also been found to be important in cross-priming CD8+ T cell responses in a virus-specific manner (21, 145). As TLR3 signaling is MyD88-independent, the safety and immunostimulatory properties of TLR3-specific agonists is unique among other TLR immunomodulators (146).
The TLR3-activating synthetic dsRNA molecule poly-inosinic:poly-cytidylic acid (poly I:C) was first discovered to confer protection against subsequent viral challenge in 1969 (147). More recently, it was found that intraperitoneal administration of poly I:C 3 days prior to challenge with E. coli K1 meningitis in neutropenic mice resulted in increased recruitment of NK cells, production of RANTES and IFN-γ, and decreased bacterial burden (126). Pre-treatment with poly I:C resulted in survival benefit of neutropenic but not immunocompetent mice. In another model of infection, poly I:C conferred anti-viral properties (148) which lead to neuroprotection in a mouse model of HSV-1 encephalitis (124). In a unique oyster model, poly I:C injection protected the organism against subsequent environmental infection by mitigating viral replication which persisted for at least 5 months (148). It is suggested that the length of the dsRNA dictates distinct antimicrobial functions dependent on cell type which should be considered during study design and data interpretation (149). It has been posed that the difference in TLR3-mediated protection against viral infection compared to bacterial infection may be due to the production of type I IFNs which impairs bacterial clearance (15). Indeed, intranasal administration of poly I:C prior to challenge with S. pneumoniae and methicillin-resistant S. aureus increased susceptibility to infection (150).
Since its creation 5 decades ago, analogs of poly I:C have been rapidly developed in effort to reduce toxicity (146). The substitution of a uridylic acid at a molar ratio of 12:1 in the synthesis of the poly C strand results in poly I:C12U which is more rapidly metabolized in vivo (151). Pre-treatment with poly I:C12U protects against subsequent viral challenge more effectively than poly I:C (123). Interestingly, a protective benefit has also been observed when the molecule was administered 2 days after viral myocarditis infection (152). In a clinical trial in which poly I:C12U was administered to HIV-infected patients, immune function was restored or stabilized (153); however, clinical investigation did not progress past phase II clinical trials (154). Poly I:C12U also holds promise as a vaccine adjuvant as it increases efficacy of intranasal H5N1 immunization (155) and intradermal HSV-2 immunization, which conferred resistance to subsequent otherwise lethal HSV-2 infectious challenge (156). Demonstrating its safety, this compound has been developed as a therapy for chronic fatigue syndrome (157, 158) that is approved in Argentina and has been approved for early access program in the European Union and Turkey, although it does not currently have FDA approval in the United States (Rintatolimod, tradename Ampligen).
Two other chemically stabilized analogs of poly I:C have demonstrated promising immunostimulatory properties. The first being poly IC : LC (termed Hiltonol) which has been shown to protect rhesus monkeys from several viruses including yellow fever, Rift Valley fever, and rabies (159, 160) and has conferred protection against highly viral strains of H4N1 and influenza in mice (161, 162). Importantly, it was found in 2017 that intranasal administration of poly IC : LC 24 h prior to or 8 h after an otherwise lethal challenge with SARS-CoV conferred survival benefit as well as reduced lung hemorrhage scores and lung viral titers in mice (125). Additionally, this molecule has been investigated for its potential in boosting immunity of HIV-infected patients which induced transient innate immune responses, suggesting application as a vaccine adjuvant may be appropriate (163). This line of investigation is being pursued whereby administration of poly IC : LC alongside an antigen stimulates a ‘live virus vaccine equivalent’ effect whereby antigen-presenting cells (APCs) are activated, T lymphocyte response is elicited, memory T and B cells are generated, and Teff/Treg ratios are increased (164). Keyhole limpet hemocyanin (KHL) or HPV vaccines elicited significantly elevated antibody responses and Th1 immune responses when administered with poly IC : LC in rhesus Macaques (165).
The second poly I:C stabilized analog is PIKA which has been shown to protect mice against an array of influenza viruses along with decreased viral burden in the lungs and increased recruitment of macrophages, neutrophils, and plasmacytoid DCs (166). However, application of PIKA has mostly focused on its potential as a vaccine adjuvant for H5N1 (167–169), Hepatitis B (HBsAg) (170), and rabies (171), the latter of which underwent phase II clinical trials with moderate success (172).
Beyond the poly I:C class of TLR3-stimulating molecules, oral administration of purified L. rhamnosus CRL1505 peptidoglycan confers resistance to RSV infection associated with decreased viral loads in the lungs and augmented cytokine responses (127). Protection against RSV and subsequent secondary infection to pneumococcal pneumonia was found to be dependent on TLR3 (128, 130) and macrophages (173). In an immunocompromised-malnourished model, it was found that immunostimulatory properties of CRL1505 peptidoglycan extended beyond augmentation of innate immunity. Administration of CRL1505 enhanced the Th2 response and recovery of B cells after S. pneumoniae infection (174). The investigators also found that intranasal administration of CRL1505 prior to challenge with influenza virus was associated with reduced pulmonary injury and viral loads in the lungs via regulation of pro-inflammatory cytokines and increased levels of type I IFNs (129).
Together, TLR3 agonists hold strong promise, especially in protection against viral infections and for vaccine adjuvant strategies. However, careful attention needs to be paid as to the potential propagation of bacterial infections by TLR3-induced production of type I IFNs.
Toll-Like Receptor 7
The endosomally located TLR7 recognizes ssRNA and often plays a role in responding to viral infections through MyD88-dependent signaling. The small molecule 1V270 (also designated TMX201) is a TLR7 ligand conjugated with a phospholipid that has been shown to protect mice from an otherwise lethal infection with Bacillus anthracis, Venezuelan equine encephalitis virus, and H1N1 influenza virus (131). 1V270-mediated protection was associated with increased cytokines and chemokines in bronchial alveolar lavage fluids but not in circulation.
The Imidazoquinoline compound Imiquimod is a low molecular weight compound which selectively activates TLR7. Imiquimod is an FDA approved immune response modifier for the topical treatment of genital warts caused by HPV (175). Imiquimod has also been found to be protective against influenza A infection in mice which was associated with reduced viral replication, airway inflammation, proinflammatory cytokine production, and preservation of body weight (132). Neonates were also protected against polymicrobial sepsis when infection was initiated 24 h after treatment (66). It is also important to note that imiquimod may be an effective vaccine adjuvant strategy for influenza (176).
Additionally, several TLR7 agonists have been shown to improve immunity of hepatitis B-infected hosts and are implicated as potential HBV vaccine adjuvants. When administered together with an alum adjuvant and recombinant hepatitis B surface antigen (HBsAg) protein, the novel TLR7 agonist T7-EA induced HBsAg-specific antibody and restored T-cell responses in a murine model (133). Similar improvement of HBsAg-specific T-cell function was observed by immunizing HBV-transgenic mice with a TLR7/TLR8 agonist (CL097)-conjugated HBV protein (134). In a prospective clinical study, it was found that oral administration of the TLR agonist GS-9620 increased T-cell responses to HBV peptides demonstrated by increased cytokine production; however, it failed to reduce serum HBsAg levels (135).
Toll-Like Receptor 9
TLR9 is expressed in DCs, monocytes, macrophages, and B cells and recognizes bacterial and viral DNA. Upon ligand binding, it signals through MyD88 directly; however, it is important to note that signaling depends on intracellular localization as a mechanism to fine-tune the immune response. In resting cells, TLR9 is localized to the ER (177). Recently described in detail by Marongiu et al., trafficking of TLR9 is controlled by the multimembrane protein unc-93 homolog B1 (UNCB1) which is required for the receptor to leave the ER and traffic to the Golgi (178). After delivery to the plasma membrane, the adaptor protein AP-2 is recruited to mediate internalization of the receptor in a clathrin-dependent mechanism (179). In parallel to internalization of TLR9, the ligand must also be endocytosed. TLRs are then localized to early endosomal compartments and the pathway bifurcates to either IRF7 signaling endosomes or NF-κB signaling endosomes (180), which are determined by AP-3 (181). Thus, the localization of TLR9, presence of co-receptors and co-factors, as well as trafficking of the ligand itself all influence downstream signaling and determine whether pro-inflammatory cytokines or type I IFNs are produced. Such intricate control also prevents recognition of self-DNA to prevent autoimmune dysfunction.
CpG oligodeoxynucleotides (CpG ODNs) are synthetic molecules which mimic bacterial DNA and stimulate TLR9. Pre-treatment with CpG has been found to protect mice from Leishmania major infection by shifting immune responses from Th2 towards Th1 (136). CpG conferred survival benefit to Leishmania major and F. tularensis for up to 2 weeks independent of the route of infection (182). Intrathecal administration of CpG 48 h prior to Klebsiella pneumoniae infection resulted in increased survival, associated with reduced bacterial burden in the lungs and circulation, increased recruitment of neutrophils, NK cells, γδ-T cells, and augmented inflammatory response (139). Interestingly, treatment of methicillin-resistant S. aureus (MRSA)-infected mice with CpG improved survival (142). Beyond protection against bacterial microbes, CpG conferred survival benefit to viral challenge by HSV-2 which was associated with decreased viral replication (138) via augmentation of the innate immunity (183). CpG has also conferred protection of neonate mice challenged with Listeria infection (140) as well as neurotropic Tacaribe Arenavirus which was associated with decreased viral load, increased antigen-specific antibodies, and NO production via NO synthase expression (141). CpG-mediated protection was preserved in T cell-depleted immunocompromised mice (182). HIV-infected macaques treated with CpG prior to challenge with Leishmania exhibited decreased lesion size and parasite load (137). The two latter studies clearly demonstrate the clinical potential of CpG to protect immunocompromised populations against opportunistic infections.
In 2017, the FDA approved the use of CpG 1018 as a vaccine adjuvant in a hepatitis B vaccine (Heplisav-B) which has increased efficacy of the vaccine, thus reducing the prior three-dose strategy to a two-dose strategy (184). CpG 1018 increases antibody concentrations, stimulates helper (CD4+) and cytotoxic (CD8+) T cells, boosts T and B cell memory responses, and shifts T cells towards a Th1 response. Researchers have found that the 2-dose HBV vaccine strategy with CpG as the adjuvant compared to the 3-dose strategy with aluminum hydroxide was more effective in patients aged 60–70 years old with type 2 diabetes mellitus (185), a population which typically demonstrates reduced immunogenicity compared to younger and/or non-diabetic populations. Thus, CpG may be a beneficial agent to boost immune responses in vulnerable patient populations.
Other Pathogen Associated Molecular Patterns Which Trigger Trained Immunity in a Toll-Like Receptor-Associated Mechanism
Bacillus Calmette-Guerin Vaccine
The Bacillus Calmette–Guerin (BCG) tuberculosis vaccine is the most used vaccine globally which has been demonstrated to confer T cell-independent cross-protection against fungal infection with C. ablicans or with the parasite Schistosomiasis mansoni (186, 187). After adjusting for age and other vaccines, the BCG vaccine is associated with a significantly lower mortality ratio among infants in Ginea-Bissau (188). In animal studies, BCG-mediated non-specific protection lasts for at least 3 months and is independent of T and B cells (189) and there is evidence that protection may last up to a year (190). Although the immunization effect of the BCG vaccine against M. tuberculosis requires adaptive immunity, namely T cell activation, the initial response to BCG is through the innate immunity whereby TLR2 and TLR4 and downstream MyD88-dependent signaling are activated (191). The resulting activation of the NF-κB pathway serves as the link between the innate and adaptive response. Therefore, the BCG vaccine does activate the innate immune response and may be responsible for driving broad protection.
β-Glucan
Fungal β-glucans are a promising class of molecules which trigger trained immunity, although the immunostimulatory properties differ depending on the strain from which they were isolated (192). These naturally derived molecules have been found to confer protection against a model of E. coli peritonitis (193), S. aureus (194), influenza (195), and MRSA (196) and were associated with increased leukocyte recruitment and antimicrobial functions. Treatment of burn-injured mice with glucan phosphate prior to wound infection with P. aeruginosa improved survival, attenuated cytokine production, and decreased bacterial load at the burn wound (197).
β-glucans bind their specific PRR dectin-1 which results in downstream inflammasome activation. However, β-glucan-mediated production of inflammatory cytokines and reactive oxygen species (ROS) is dependent on the cooperation between dectin-1 and TLR2 (198). Further, TNF-α production in response to zymosan or live fungi is dependent on MyD88 (199). Thus, β-glucan-mediated trained immunity is dependent on the synergism of dectin-1 and TLR2.
CL429
One research group is investigating the potential immunostimulant properties of CL429, which is a novel chimeric compound that was designed to stimulate both TLR2 and NOD2 by covalently linking the NOD2 ligand Murabutide with the TLR2 ligand Pam2C (200). Initially studied as a vaccine adjuvant, they went on to find that CL429 confers protection against pneumovirus (PVM) infection associated with attenuated inflammation (201) and against leptospiral infection for up to 3 months via increased proinflammatory cytokine and chemokine production (202).
CpG-Oligodeoxynucleotide : AG-OVA Nanoparticles
Similar in approach as the CL429 molecule, the TLR9-activating ligand CpG ODN was crosslinked with the dectin-1/TLR2 stimulating agonist β-glucan-Ovalbumin resulting in CpG-OND : AG-OVA dual-targeting nanoparticles as a vaccine adjuvant strategy (203). Investigators found that the nanoparticles enhanced APC maturation and induced robust Th1 and Th2 responses similar to that triggered by Freund’s adjuvant but without the toxicity. Although this novel compound demonstrates promise as a vaccine adjuvant, it would also be of interest to investigate whether it confers broad protection to infection which may be more profound than that mediated by CpG or β-glucan alone, both of which are strong immunomodulators.
Cross-Protection Between Infections
As the BCG vaccine seemingly mediates cross-protection against pathogens besides tuberculosis, evidence suggests that some infections also confer cross-protection which is, at least in part, due to trained immunity (10). For example, administration of an attenuated strain of C. albicans conferred host resistance to subsequent challenge with the Gram-positive bacteria S. aureus; this phenomenon was found to be independent of T cells but was dependent on macrophages (204). Interestingly, several observations have suggested that viral infections may trigger a similar cross-protection benefit. Barton et al. demonstrated that latent herpesvirus was associated with protection against bacterial L. monocytogenes and against bacterial L. monocytogenes and Y. pestis which was similarly dependent on macrophages (205). This observation was confirmed by others who elucidated that herpesvirus-induced protection against bacterial infection is transient (approximately 5 months) despite stable viral load (206). NK cells are also key mediators of cross-protection whereby they expand during the initial infection and are primed to undergo a second expansion as well as produce more cytokines upon a secondary infection (207). More recently, a Singapore study of military recruits within a 5-year period showed that men infected with influenza were protected against subsequent infection with adenovirus (79). It should be noted, however, that cross-protection between infections is generally considered to be dependent on both non-specific reprogramming of innate immunity as well as activation of memory T cells.
Toll-Like Receptor Antagonists as Potential Immunomodulatory Strategies for Treatment of Chronic Infectious Diseases
It is clear that TLR agonists hold strong therapeutic potential to mediate host resistance to subsequent infection; however, this is only one of many potential therapeutic applications of TLR immunomodulators. As TLR signaling cascades culminate in robust inflammation, TLR antagonists are under development for the treatment of chronic infectious and inflammatory diseases (208). To date, these compounds are generally designed to bind the TLR, thus preventing the binding of agonists responsible for driving inflammation (209–211). The TLR4 antagonist Eritoran (E5564) reached Phase III clinical trials for the treatment of sepsis. This synthetic lipid A analogue which prevents LPS from activating TLR4, hypothetically preventing propagation of systemic inflammatory response syndrome (SIRS) characteristic of sepsis. Preclinical and early clinical studies with the compound showed promising anti-inflammatory results in response to LPS (212–214), however the study failed to meet its target end-point in phase III (215). Similarly, another TLR4 antagonist designated Resatorvid (TAK-242) was also studied in the treatment of sepsis and reached Phase III of the clinical trials, however it failed to attenuate inflammation in septic patients (216).
Recently, the antimalarial drugs chloroquine and hydroxychloroquine have been under investigation for treatment of COVID-19 with the hypothesis that these drugs will prevent glycosylation of the angiotensin-converting-enzyme 2 (ACE2) as well as inhibit endosomal TLR activation (217, 218). Initial in vitro studies showed potent antiviral activity (219). Randomized trials have not shown improved clinical outcomes in the hydroxychloroquine-treated COVID patients (218, 220, 221). In another study, the Bruton tyrosine kinase (BTK) inhibitor acalabrutinib was administered to COVID patients for 10–14 days which seemed to improve patient outcomes as indicated by oxygenation (222).
Although clinical trials using TLR antagonists in the treatment of severe infection have been unsuccessful to date, critical information has been gained from these investigations which provide a strong foundation for future studies. Importantly, this class of compounds is also being widely studied for treatment of chronic inflammatory conditions such as rheumatoid arthritis and autoimmune disorders. One key finding of the clinical trials studying the TLR4 antagonists Eritoran and Resatorvid was that the compounds were well-tolerated (215). Continued drug discovery efforts via high throughput screening and alternative approaches such as targeting the transcriptional regulation of TLRs to suppress their expression rather than direct inhibition of the receptor may move the field forward (223). These efforts will be also be supported by continued elucidation of TLR signaling mechanisms and immune responses.
Metabolic and Epigenetic Reprogramming as the Basis for Toll-Like Receptor Agonist-Induced Trained ImmunitY
Upon inflammatory stimulation, innate leukocytes undergo metabolic reprogramming that is characterized by augmentation of glycolysis and mitochondrial oxidative phosphorylation to meet the increased energy demands for combating an infection (14, 224). Previous investigations aimed at deciphering the molecular mechanisms of trained immunity-mediated protection against infections have predominantly used the fungal ligand β-glucan (225, 226). Reviewed in detail by Netea and colleagues, metabolic reprogramming and epigenetic modifications are the key mechanisms of β-glucan-induced trained immunity (226). It has been shown that Akt/mTOR/HIF-1α signaling is critical in β-glucan induced augmentation of glycolysis in monocytes (225). As opposed to the breadth of mechanistic understanding of trained immunity induced by β-glucan, the molecular mechanisms underlying TLR ligand-induced training of leukocytes are an evolving field. Our studies show that treatment with the TLR4 ligand MPLA not only increases glycolysis but also augments mitochondrial oxidative phosphorylation and mitochondrial biogenesis in concert with increased antimicrobial functions of macrophages (63). Further, stimulation of macrophages with the classic TLR4 ligand LPS reprograms mitochondrial metabolism leading to increased accumulation of tricarboxylic citric acid (TCA) cycle metabolites that play an important role in TLR agonist-mediated trained immunity (227, 228).
The finding that a variety of TLR ligands have the ability to mediate protection against infection from a broad array of organisms which activate distinct TLRs, including Gram-positive and Gram-negative bacteria, fungi, and viruses, demonstrates that TLR activation has the ability to offer cross-protection against diverse pathogens. Both MyD88 and TRIF activation have been implicated in facilitating signaling-driven metabolic and epigenetic alterations induced by TLR ligands (229), but little is understood about the roles of these signaling pathways in initiating trained immunity. This raises the question as to whether activation of trained immunity by TLR ligands is mediated through common signaling pathways. Future studies providing insight into these common pathways would pave the way for the discovery of a multitude of potential TLR ligand-based therapeutics to improve resistance to infection. The following sections will provide a succinct overview of leukocyte metabolic reprogramming and epigenetic modifications as the basis for TLR ligand induced trained immunity (Figure 2).
Toll-Like Receptor Ligand-Induced Metabolic Reprogramming of Innate Leukocytes
Stimulation of macrophages with LPS increases glucose uptake and glycolytic capacity mediated via stabilization and upregulation of hypoxia-inducible factor 1- α (HIF-1α) (230–232). Increased glycolytic capacity serves to rapidly generate ATP and provide essential precursors for synthesis of amino acids, lipids, and nucleotides that are necessary for optimal effector activities and cell viability under stress conditions (233). Our studies have shown that deletion of HIF-1α or inhibition of mammalian target of rapamycin (mTOR; known to stabilize HIF-1α) attenuates MPLA-induced increased glycolysis, abolishing the protective effect of MPLA against infection (63, 76).
Along with augmented glycolysis, reprogramming of mitochondrial metabolism plays a key role in modulating the inflammatory response of innate leukocytes. LPS-induced activation of macrophages introduces ‘breaks’ in the TCA cycle at the levels of isocitrate dehydrogenase and succinate dehydrogenase (SDH), leading to increased accumulation of citrate and succinate (234). Studies from our laboratory also show that MPLA treatment cause an early reduction in TCA cycle flux between citrate and α-ketoglutarate leading to increased accumulation of citrate (63). Citrate is diverted towards generation of itaconate via increased immunoresponsive gene 1 (Irg1) enzyme expression (235). Itaconate is being widely investigated for its direct antimicrobial and anti-inflammatory effects. The direct antimicrobial effect of itaconate is mediated via inhibition of the microbial enzyme isocitrate lyase (236). Itaconate has been shown to inhibit the growth of numerous pathogens including M. tuberculosis, S. aureus, Legionella pneumoniae, Acinetobacter baumanii, and Salmonella enterica (235, 237, 238). Itaconate can be transported into the phagosome, where it limits microbial growth (239), implying a role for phagolysosomes as a critical site for itaconate’s antimicrobial effects. The synthetic itaconate analog, 4-octyl-itaconate (4OI) exerts anti-inflammatory effects and potently activates the NF-E2-related factor 2 (Nrf2) pathway (240) which regulates the expression of cytoprotective proteins and plays a critical role in redox homeostasis (241). A study by Swain et al. shows that endogenous itaconic acid fails to activate Nrf2 as compared to 4OI, and that synthetic itaconate analogs do not recapitulate the effects of endogenous itaconic acid (242). However, endogenous itaconic acid is anti-inflammatory and reduces IL-1β production similar to 4OI (240, 242). In contrast to these findings, treatment with β-glucan does not induce significant levels of Irg1 and itaconate in human monocytes, and pretreatment with 4OI diminishes β-glucan induced trained phenotype (243). However, it is important to note that β-glucan focused studies relied on measuring cytokine responses alone as a key for demonstrating the lack of effect of itaconate on β-glucan-induced trained immunity although cytokine responses to an inflammatory stimuli may not correlate with actual protective response (76).
Beyond the role of citrate in serving as a precursor for itaconate, our studies using MPLA-stimulated macrophages have shown that citrate transported into the cytosol replenishes mitochondrial oxaloacetate pools and fuels a sustained increase in mitochondrial TCA cycle flux (63). Upon TLR4 stimulation of macrophages, inhibition of SDH activity by itaconate and increased TCA cycle flux also results in succinate accumulation (231, 244). Increased succinate levels and inhibition of SDH activity stabilize HIF-1α and increase mitochondrial ROS generation which lead to an enhanced inflammatory response (231, 244). Mitochondrial ROS aid in microbial clearance (245); however, this phenomenon requires further investigation. Importantly, MPLA-induced increase in TCA cycle flux is associated with enhanced macrophage antimicrobial effects and protection against infections (63). Therefore, metabolic reprogramming plays a critical role in TLR ligand induced trained immunity-mediated protection against infections.
Role of Epigenetic Modifications in Toll-Like Receptor Ligand-Induced Trained Immunity
Exposure to inflammatory stimulus or pathogens also causes epigenetic reprogramming in innate leukocytes reflected by alterations in the histone acetylation and methylation status (246). The major histone modifications including acetylation (H3K27ac) and methylation (H3K4me3) in monocytes exposed to β-glucan persist even 7 days after removal of the initial stimulus and are strongly associated with metabolic reprogramming (225). A study by Saeed et al. showed that LPS and β-glucan induce diverse and opposing alterations in the epigenome with β-glucan showing a greater degree of de novo H3K27ac modifications in gene loci encoding for inflammatory responses (247). Alternatively, LPS stimulation of human monocytes acutely induces a strong acetylation of H3K27 around Irg1 gene locus within 1 h of stimulation which is associated with increased expression of Irg1 (243).
Metabolic reprogramming and epigenetic modifications are tightly interconnected. Fumarate accumulates in monocytes stimulated with β-glucan via glutamine anaplerosis. Increased fumarate levels have been shown to downregulate the activity of histone demethylase KDM5 (248). In turn, decreased KDM5 activity upregulates trimethylation of H3K4 at promoters of genes encoding pro-inflammatory cytokines (248). α-ketoglutarate stimulates the jumonji domain containing family of the lysine demethylase enzyme JMJD33. Further, a high α-ketoglutarate/succinate ratio favors anti-inflammatory phenotype in macrophages (249).
The post-translational modification of succinylation arising from the addition of succinate to the protein lysine residues supports a pro-inflammatory state in macrophages (231). Succinylation is known to occur on histone lysine residues in human cells (250). However, the role of histone succinylation in the context of trained immunity is currently unknown and remains an important question to be addressed in future studies. A study by Zhang et al. showed that lactate can also bind to histone lysine residues (lactylation) and demonstrated 28 distinct histone lactylation sites (251). This study showed that LPS-induced increase in macrophage lactate levels via increased glycolysis sets in motion a lactylation epigenetic program which directs the expression of genes involved in alternative anti-inflammatory activation state in macrophages (251). The influence of accumulated metabolites during trained immunity is just beginning to be explored and needs further characterization.
The traditional school of thought is that treatment with TLR ligands such as LPS induces a state of immune tolerance, while leukocytes exposed of β-glucan produce a heightened response to secondary stimulation (trained phenotype) classically reflected by cytokine production (252). However, as discussed, treatment with clinically applicable TLR agonists protect against a broad array of infections, implying that TLR ligands also induce robust trained immunity. One potential explanation for this discrepancy in the literature regarding whether TLR ligands induce a state of tolerance or training may be due to the reliance on cytokine production upon exposure to a secondary stimuli which has more recently been shown to not be uniformly indicative of antimicrobial immunity (76). Future studies aimed at detailed characterization of TLR agonist-induced epigenetic reprogramming and defining its link with metabolic reprogramming will be critical in elucidating the mechanisms of TLR agonist-induced trained immunity.
Toll-Like Receptor Agonist-Induced Trained Immunity: A Clinical Perspective
Potential Adverse Consequences of Immunostimulation by Toll-Like Receptor Agonists
It is important to thoroughly consider that activation of the immune system may have deleterious consequences and requires careful study to identify the clinical situations in which TLR-mediated immunomodulation are most appropriate. Sepsis and septic shock yield a proinflammatory response that results in organ injury; however, survivors demonstrate an immunosuppressive phenotype that results in secondary infections and increased mortality (253, 254). Likewise, a potentially harmful outcome of TLR agonist treatment is tolerance to subsequent exposure of endotoxin, particularly in the setting of prolonged LPS exposure or treatment, a phenomena also termed as immunoparalysis (255, 256). Aberrant activation of TLR signaling by PAMPs or DAMPs, mutations of TLR signaling molecules, or failure of self-recognition mechanisms are responsible for development of several diseases such as autoimmune, chronic inflammatory, and allergic diseases (257). In the field of oncology research, adverse effects from TLR immunotherapy have been linked to unintended expansion of adaptive leukocytes, such in B-cell lymphoma, where activation of TLR4 MyD88-dependent signaling may exacerbate the disease (258–260). Other adverse effects of treatment with TLR agonists have been described in cardiovascular medicine research where treatment with oxidized low-density lipoprotein and the BCG vaccine yield a dose-dependent response of proinflammatory cytokines. These mediators damage human coronary smooth muscle cells and increase atherosclerosis which was found to be TLR2- and TLR4-dependent (261).
Therapeutic Potential of Toll-Like Receptor Ligands Beyond Infection Resistance
Although there are some potential adverse effects of TLR immunotherapy which require consideration, there are several patient populations which may benefit from novel TLR agonist strategies. The oncology patient population presents a challenge due to immunosuppression. Most cancer-associated antigens are self-antigens and require immunostimulant adjuvants in addition to cancer-targeting strategies (262). In an H22 liver cancer murine model, administration of curdlan sulfate-matured tumor cell lysate-pulsed DCs was associated with an increase in CD80, MHC-1 and MHC-II expression, CD8+T cell infiltration, upregulated TNF-α and INF-γ transcription, and downregulation of TGF-β transcription in tumor tissues, and improved survival (263). In a separate study, the use of the TLR3 agonist Ampligen, a GMP-grade synthetic poly I:C derivative, was shown to mature human monocytes derived from DCs and sustained bioactive IL-12 production, and generate Th1 specific anti-cancer responses in peripheral blood T-cells obtained from cancer patients (262). In a study by Breckpot et al., the zinc finger protein A20 was downregulated in poly I:C treated DCs which led to sustained production of IL-6, IL-10, and IL-12p70, thus making poly I:C a candidate adjuvant for an anti-cancer immunotherapy (264). Likewise, intratumor administration of the TLR7 agonist 1V270 increased the ratio of M1 to M2 tumor-associated macrophages and was associated with improved survival (265). In addition to anti-cancer immunotherapy, TLR agonists have shown promise in reduction of ischemia reperfusion injury in cardiac myocytes (266, 267). Further, TLR therapy has also been studied in progressive diseases such as Alzheimer’s disease, where single or repeated treatment has been shown to reduce evidence of disease progression (268, 269).
Aside from innate immune cells, non-immune cells are capable of long-term memory, including hematopoietic, mesenchymal, and epithelial stem cells (270). There is an increasing body of evidence on how the microbiome influences immunity and how probiotic therapy modulates innate immunity (271). Interestingly, trained immunity may be a mechanism of the beneficial effects of probiotics. Probiotics have been found to augment innate immune function via receptor antagonism or expression, binding or expression of adaptor molecules, expression of regulatory signaling molecules, induction of micro-RNAs, and secretion of immunomodulatory proteins, lipids, and metabolites (271).
Trained immunity may even play a role in combating the ongoing COVID-19 pandemic. As one of the sequelae of COVID-19 infections include secondary respiratory infections, the BCG vaccine or β-glucan may be adjunct strategies to reduce morbidity and mortality by enhancing immunity (272, 273). Imiquimod, a TLR7 agonist, has also been proposed as a therapeutic adjunct for COVID-19 and related infections (274). In summary, there is a growing body of evidence focusing on trained immunity as a mechanism to enhance immunity against a broad array of infections which are common in critically ill patients, but also for several other patient populations as discussed above. Such application requires further investigation to elucidate the full potential of TLR agonist-based immunotherapies (Table 3).
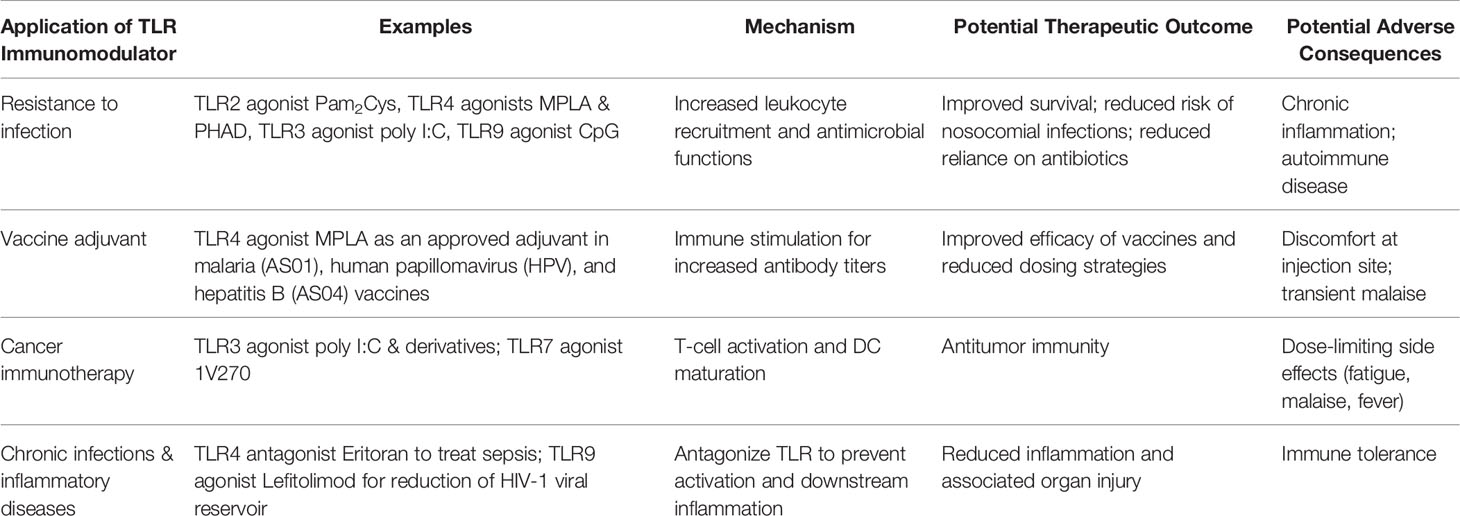
Table 3 Clinical application of TLR immunomodulators.
Clinical Trials Investigating Toll-Like Receptor Immunomodulators
Ongoing clinical trials investigating the adjuvant properties of TLR agonists constitute approximately double of those studying them as therapeutics (275), demonstrating that the immunomodulatory properties of these compounds are largely being harnessed for vaccine development. The application of trained immunity for vaccine development is highly attractive due to its potential to (1) increase nonspecific effector responses of innate immune cells, and (2) to activate DCs to enhance adaptive T cell responses to specific and nonrelated antigens (276). For example, a novel synthetic small molecule TLR7/8 agonist 3M-052 is now in a phase I clinical trial studying the safety and immunogenicity of the HIV-1 BG505 SOSIP.664 gp140 vaccine candidate (NCT04177355). Other TLR agonists currently in clinical trials as vaccine adjuvant strategies for HIV-1 include TLR3 agonist Poly ICLC (NCT02071095) and TLR9 agonists MGN1703 (NCT02443935) and CpG-7909 (NCT00562939). TLR7 compounds are under investigation as vaccine adjuvant strategies for hepatitis B as well, including Vesatolimod (GS-9620; NCT02166047) and R07020531 (NCT02956850). The TLR9 compound SD-101 is being studied as an adjuvant for chronic hepatitis C (NCT00823862).
On the other hand, the TLR9 agonist Lefitolimod in combination with neutralizing antibodies is in phase II trials studying its effectiveness in conferring reservoir reduction in HIV infection (NCT03837756) after phase I demonstrated its safety and effectiveness in improving both innate and adaptive immunity in HIV-1 infected patients (277). The TLR7 compound Imiquimod has completed phase II trials for its efficacy in treating human papillomavirus (HPV) when applied topically (NCT00941811).
In addition to the above listed clinical trials regarding investigation of TLR agonists as vaccine adjuvant strategies or drugs to fight infection, numerous TLR ligands are being investigated as immunomodulators to treat chronic inflammatory diseases, cancer, and autoimmune disorders, which can be found listed in Anwar et al. (275).
Current Challenges to the Clinical Translation of Toll-Like Receptor Immunomodulators
Although there are several potential clinical applications of TLR immunomodulators as stand-alone therapies which are supported by a growing body of strong preclinical evidence, several knowledge gaps hinder progress of clinical translation. Much remains to be elucidated regarding duration of protection mediated by TLR agonists, and whether protection could be continued by repeated treatment once protection wanes. Further, the most effective but feasible route of administration is essential to identify, with oral and intranasal administrations likely most practical. Dosing and efficacy for various patient populations is also essential to understand, such as whether aged patients or those with comorbidities respond similarly to healthy young patients. In this regard, one limiting factor remains the use of healthy young animals for the vast majority of preclinical studies which does not recapitulate the clinical situation and therefore limits the amount of information that could be gained prior to starting clinical trials.
Another limiting factor is the striking lack of reporting of clinical trial data; therefore, scientists do not have all of the tools that otherwise could refine ongoing and future studies. Finally, as TLR agonists initiate inflammatory cytokine pathways, one glaring concern remains the potential of these compounds to trigger inflammatory or autoimmune disease. Thus, it is critical to continue elucidating TLR signaling mechanisms to identify potential therapeutic targets which may circumvent this concern in addition to conducting proper dosing studies aimed at avoiding induction of inflammation. Rapid scientific progress has been made since the discovery of the phenomenon of trained immunity and its potential therapeutic application. As the field drives forward to fill in these knowledge gaps, the goal of clinical translation of TLRs as immunomodulators holds strong promise to be realized.
Concluding Remarks
Unfortunately highlighted during the ongoing SARS-CoV-2 pandemic, immunocompromised patients are highly susceptible to life-threatening infections. Beyond the current healthcare crisis, populations with insufficient immune responses fail to clear pathogens which results in opportunistic infections that are often difficult to combat due to the increasing prevalence of antibiotic resistance. With limited pharmacological tools, it is critical to develop new strategies aimed at combating infection. Here we have reviewed the potential application of TLR agonists as immunotherapies which trigger trained immunity and confer broad protection to microbes. Importantly, since immunomodulation targets the host response rather than the pathogen, development of microbial resistance is unlikely. TLR agonists have also shown promise as adjuvants for cancer-targeting immunotherapies. Moreover, our lab and others have demonstrated that such agonists may be highly useful as stand-alone therapies to protect against infection through boosting antimicrobial responses of innate leukocytes.
With their instrumental role in stimulating innate immunity and in activation of inflammatory responses, TLRs are tightly controlled by localization to the cell surface or endosomal compartment as well as complex downstream signaling pathways via MyD88- or TRIF-dependent cascades. Although much remains to be elucidated, TLR-mediated trained immunity seems driven by metabolic reprogramming and epigenetic modifications. It is critical to further elucidate the cell types, signaling pathways, and intracellular mechanisms responsible for conferring the beneficial protective effects of TLR agonists via trained immunity. Doing so will aid the translation of TLR-based immunotherapies to protect patients from potentially life-threatening infections.
Author Contributions
The manuscript was conceptualized by AO and JB. AO wrote the sections on TLR signaling pathways, trained immunity triggered by TLR agonists, and drafted the figures. JF wrote the introduction and drafted the tables. NP wrote the sections on metabolic and epigenetic modifications, and AH contributed the section on clinical perspectives. JB supervised the drafting and performed the final editing. All authors contributed to the article and approved the submitted version.
Funding
AO is supported by 5T32AI38932-02, NP is supported by 5T32GM108554-05, Shock Society Faculty Research Award, and Vanderbilt Faculty Research Scholar Award, AH by K08 GM123345, and JB by R01 GM121711.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Glossary
ACE2: Angiotensin-converting-enzyme 2
AGP: Aminoalkyl glucosamine 4-phosphate
AP-1: Activator protein-1
APC: Antigen-presenting cell
BCAP: B-cell adaptor for PI3K
BCG vaccine: Bacillus Calmette-Guerin tuberculosis vaccine
BTK: Bruton’s tyrosine kinase
CDC: Centers for Disease Control and Prevention
CpG ODN: Cytosine-phosphate-guanine dinucleotides oligodeoxynucleotides
CLP: Cecal ligation and puncture
DAMP: Damage-associated molecular pattern
DCs: Dendritic cells
dsRNA: Double-stranded RNA
DUBA: Deubiquitinating Enzyme A
EGF: Epidermal growth factor
ER: Endoplasmic reticulum
FDA: U.S. Food and Drug Administration
FimH: Fimbriae H protein
fmOMV: Low endotoxicity outer membrane vesicles
H1N1: Influenza A subtype
H5N1: Influenza A subtype
avian flu: bird flu
H5N2: Influenza A subtype
avian flu: bird flu
HBV: Hepatitis B virus
HBsAg: Hepatitis B surface antigen
HCAI: Healthcare associated infections
HIV: Human immunodeficiency virus
HPV: Human papillomavirus
HSV: Herpes simplex virus
ICU: Intensive care unit
Ig: Immunoglobulin
IKK: IkB kinase
IKKi: IKK inducible gene
IRAK: Interleukin-1 receptor-associated kinases
IRF: Interferon regulatory factor
KHL: Keyhole limpet hemocyanin
LPS: Lipopolysaccharide
LRR: Leucine-rich repeat
MAL: MyD88-adaptor-like
MALP: Macrophage-activating lipopeptide
MAPK: Mitogen-activated protein kinase
MCP: Monocyte chemoattractant protein
miRNA: Micro RNA
MPLA: Monophosphoryl lipid A
mRNA: Messenger RNA
MrpH: MR/P fimbriae protein
MRSA: Methicillin-resistant S. aureus
MyD88: Myeloid differentiation 88
NADPH: Nicotinamide adenine dinucleotide phosphate
NEMO: NF-kB essential modifier
NF-κB: Nuclear factor-κb
NK cells: Natural killer cells
NO: Nitric oxide
NOD: Nucleotide oligomerization domain
OMV: Outer membrane vesicles
OVA: Ovalbumin
Pam2Cys: Peptide dipalmitoyl-S-glyceryl cysteine
Pam3Cys: Peptide tripalmitoyl-S-glyceryl cysteine
PAMP: Pathogen-associated molecular pattern
PHAD: Phosphorylated hexa-acyl disaccharides
PI3K: Phosphatidylinositol 3-kinase
Poly I:C: Polyinosinic:polycytidylic acid
PR8: Mouse-adapted H1N1
PRDX: Peroxiredoxin
PRR: Pattern recognition receptor
PVM: Pneumovirus
RAG: Recombination-activating gene
RANTES: Regulated upon activation, normal T cell expressed and secreted
RIP: Receptor-interacting protein
RSV: Respiratory syncytial virus
SDH: Succinate dehydrogenase
ssRNA: Single-stranded RNA
TAK: Transforming growth factor β–activated kinase 1
TANK: TRAF-associated NFκB activator
TBK: TANK-binding kinase
TCA cycle: Tricarboxylic citric acid cycle
Teff: Effector T cell
Th: T helper cell
THP: Tamm-Horsfall protein
TICAM: TIR domain-containing adaptor molecule
TIR: Toll/interleukin receptor
TIRAP: TIR domain-containing adaptor protein
TLR: Toll-like receptor
TMX201: Small molecule 1V270
TNF: Tumor necrosis factor
TRAF: TNF receptor-associated factor
TRAM: TRIF-related adaptor molecule
Treg: Regulatory T cell
TRIF: TIR-domain-containing adapter-inducing interferon-B
UNCB1: Unc-93 homolog B1
UPEC: Uropathogenic E. coli
References
1. CDC. Antibiotic Resistance Threats in the United States, 2019. Atlanta, GA: U.S. Department of Health and Human Services (2019). Available at: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf.
2. Statistics NCfH. Health, United States, 2017: With special feature on mortality. Hyattsville, MD: Centers for Disease Control and Prevention - National Center for Health Statistics (2017). Available at: https://www.cdc.gov/nchs/data/hus/hus17.pdf.
3. Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med (2007) 35(5):1244–50. doi: 10.1097/01.CCM.0000261890.41311.E9
4. Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, et al. Program CDCPE. Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009-2014. JAMA (2017) 318(13):1241–9. doi: 10.1001/jama.2017.13836
5. Vincent JL, Abraham E, Annane D, Bernard G, Rivers E, Van den Berghe G. Reducing mortality in sepsis: new directions. Crit Care (2002) 6(Suppl 3):S1–18. doi: 10.1186/cc1860
6. Torio CM, Moore BJ. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2013: Statistical Brief 204. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville MD: Agency for Healthcare Research and Quality (2006).
7. Ehlenbach WJ, Curtis JR. Noninvasive ventilation for patients near the end of life: what do we know and what do we need to know? Crit Care Med (2008) 36(3):1003–4. doi: 10.1097/CCM.0B013E318165FD78
8. WHO. Antimicrobial resistance: global report on surveillance 2014. World Health Organization (2014).
9. Reardon S. Antibiotic resistance sweeping developing world. Nature (2014) 509(7499):141–2. doi: 10.1038/509141a
10. Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe (2011) 9(5):355–61. doi: 10.1016/j.chom.2011.04.006
11. Dominguez-Andres J, Netea MG. Long-term reprogramming of the innate immune system. J Leukoc Biol (2019) 105(2):329–38. doi: 10.1002/JLB.MR0318-104R
12. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: A program of innate immune memory in health and disease. Science (2016) 352(6284):aaf1098. doi: 10.1126/science.aaf1098
13. Divangahi M, Aaby P, Khader SA, Barreiro LB, Bekkering S, Chavakis T, et al. Trained immunity, tolerance, priming and differentiation: distinct immunological processes. Nat Immunol (2021) 22(1):2–6. doi: 10.1038/s41590-020-00845-6
14. McBride MA, Owen AM, Stothers CL, Hernandez A, Luan L, Burelbach KR, et al. The Metabolic Basis of Immune Dysfunction Following Sepsis and Trauma. Front Immunol (2020) 11:1043. doi: 10.3389/fimmu.2020.01043
15. Mifsud EJ, Tan AC, Jackson DC. TLR Agonists as Modulators of the Innate Immune Response and Their Potential as Agents Against Infectious Disease. Front Immunol (2014) 5:79. doi: 10.3389/fimmu.2014.00079
16. Hernandez A, Patil NK, Stothers CL, Luan L, McBride MA, Owen AM, et al. Immunobiology and application of toll-like receptor 4 agonists to augment host resistance to infection. Pharmacol Res (2019) 150:104502. doi: 10.1016/j.phrs.2019.104502
17. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
18. Ullah MO, Sweet MJ, Mansell A, Kellie S, Kobe B. TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J Leukoc Biol (2016) 100(1):27–45. doi: 10.1189/jlb.2RI1115-531R
19. Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol (2014) 14(8):546–58. doi: 10.1038/nri3713
20. Marongiu L, Gornati L, Artuso I, Zanoni I, Granucci F. Below the surface: The inner lives of TLR4 and TLR9. J Leukoc Biol (2019) 106(1):147–60. doi: 10.1002/JLB.3MIR1218-483RR
21. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34(5):637–50. doi: 10.1016/j.immuni.2011.05.006
22. Botos I, Segal DM, Davies DR. The structural biology of Toll-like receptors. Structure (2011) 19(4):447–59. doi: 10.1016/j.str.2011.02.004
23. Krishnan J, Selvarajoo K, Tsuchiya M, Lee G, Choi S. Toll-like receptor signal transduction. Exp Mol Med (2007) 39(4):421–38. doi: 10.1038/emm.2007.47
24. O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol (2013) 13(6):453–60. doi: 10.1038/nri3446
25. Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell (2006) 125(5):943–55. doi: 10.1016/j.cell.2006.03.047
26. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature (2010) 465(7300):885–90. doi: 10.1038/nature09121
27. Doyle SE, O’Connell RM, Miranda GA, Vaidya SA, Chow EK, Liu PT, et al. Toll-like receptors induce a phagocytic gene program through p38. J Exp Med (2004) 199(1):81–90. doi: 10.1084/jem.20031237
28. Mantegazza AR, Zajac AL, Twelvetrees A, Holzbaur EL, Amigorena S, Marks MS. TLR-dependent phagosome tubulation in dendritic cells promotes phagosome cross-talk to optimize MHC-II antigen presentation. Proc Natl Acad Sci U S A (2014) 111(43):15508–13. doi: 10.1073/pnas.1412998111
29. Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science (2004) 304(5673):1014–8. doi: 10.1126/science.1096158
30. Laroux FS, Romero X, Wetzler L, Engel P, Terhorst C. Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of gram-negative bacteria. J Immunol (2005) 175(9):5596–600. doi: 10.4049/jimmunol.175.9.5596
31. Chen YJ, Hsieh MY, Chang MY, Chen HC, Jan MS, Maa MC, et al. Eps8 protein facilitates phagocytosis by increasing TLR4-MyD88 protein interaction in lipopolysaccharide-stimulated macrophages. J Biol Chem (2012) 287(22):18806–19. doi: 10.1074/jbc.M112.340935
32. Kong L, Ge BX. MyD88-independent activation of a novel actin-Cdc42/Rac pathway is required for Toll-like receptor-stimulated phagocytosis. Cell Res (2008) 18(7):745–55. doi: 10.1038/cr.2008.65
33. Hawley KL, Olson CM Jr., Iglesias-Pedraz JM, Navasa N, Cervantes JL, Caimano MJ, et al. CD14 cooperates with complement receptor 3 to mediate MyD88-independent phagocytosis of Borrelia burgdorferi. Proc Natl Acad Sci U S A (2012) 109(4):1228–32. doi: 10.1073/pnas.1112078109
34. Shin OS, Isberg RR, Akira S, Uematsu S, Behera AK, Hu LT. Distinct roles for MyD88 and Toll-like receptors 2, 5, and 9 in phagocytosis of Borrelia burgdorferi and cytokine induction. Infect Immun (2008) 76(6):2341–51. doi: 10.1128/IAI.01600-07
35. Marr KA, Balajee SA, Hawn TR, Ozinsky A, Pham U, Akira S, et al. Differential role of MyD88 in macrophage-mediated responses to opportunistic fungal pathogens. Infect Immun (2003) 71(9):5280–6. doi: 10.1128/IAI.71.9.5280-5286.2003
36. Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Med (Baltimore) (2010) 89(6):403–25. doi: 10.1097/MD.0b013e3181fd8ec3
37. Ku CL, von Bernuth H, Picard C, Zhang SY, Chang HH, Yang K, et al. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med (2007) 204(10):2407–22. doi: 10.1084/jem.20070628
38. Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell (2011) 147(4):868–80. doi: 10.1016/j.cell.2011.09.051
39. Chiang CY, Veckman V, Limmer K, David M. Phospholipase Cgamma-2 and intracellular calcium are required for lipopolysaccharide-induced Toll-like receptor 4 (TLR4) endocytosis and interferon regulatory factor 3 (IRF3) activation. J Biol Chem (2012) 287(6):3704–9. doi: 10.1074/jbc.C111.328559
40. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol (2008) 9(4):361–8. doi: 10.1038/ni1569
41. Hyun J, Kanagavelu S, Fukata M. A unique host defense pathway: TRIF mediates both antiviral and antibacterial immune responses. Microbes Infect (2013) 15(1):1–10. doi: 10.1016/j.micinf.2012.10.011
42. Zanoni I, Spreafico R, Bodio C, Di Gioia M, Cigni C, Broggi A, et al. IL-15 cis presentation is required for optimal NK cell activation in lipopolysaccharide-mediated inflammatory conditions. Cell Rep (2013) 4(6):1235–49. doi: 10.1016/j.celrep.2013.08.021
43. Han KJ, Su X, Xu LG, Bin LH, Zhang J, Shu HB. Mechanisms of the TRIF-induced interferon-stimulated response element and NF-kappaB activation and apoptosis pathways. J Biol Chem (2004) 279(15):15652–61. doi: 10.1074/jbc.M311629200
44. He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A (2011) 108(50):20054–9. doi: 10.1073/pnas.1116302108
45. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem (2013) 288(43):31268–79. doi: 10.1074/jbc.M113.462341
46. O’Neill LA, Bryant CE, Doyle SL. Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol Rev (2009) 61(2):177–97. doi: 10.1124/pr.109.001073
47. Leifer CA, Medvedev AE. Molecular mechanisms of regulation of Toll-like receptor signaling. J Leukoc Biol (2016) 100(5):927–41. doi: 10.1189/jlb.2MR0316-117RR
48. Skaug B, Chen J, Du F, He J, Ma A, Chen ZJ. Direct, noncatalytic mechanism of IKK inhibition by A20. Mol Cell (2011) 44(4):559–71. doi: 10.1016/j.molcel.2011.09.015
49. Yuk JM, Shin DM, Lee HM, Kim JJ, Kim SW, Jin HS, et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat Immunol (2011) 12(8):742–51. doi: 10.1038/ni.2064
50. Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol (2012) 33(9):449–58. doi: 10.1016/j.it.2012.05.002
51. Guven-Maiorov E, Keskin O, Gursoy A, Nussinov R. A Structural View of Negative Regulation of the Toll-like Receptor-Mediated Inflammatory Pathway. Biophys J (2015) 109(6):1214–26. doi: 10.1016/j.bpj.2015.06.048
52. Maeda K, Akira S. Regulation of mRNA stability by CCCH-type zinc-finger proteins in immune cells. Int Immunol (2017) 29(4):149–55. doi: 10.1093/intimm/dxx015
53. Shen H, Tesar BM, Walker WE, Goldstein DR. Dual signaling of MyD88 and TRIF is critical for maximal TLR4-induced dendritic cell maturation. J Immunol (2008) 181(3):1849–58. doi: 10.4049/jimmunol.181.3.1849
54. Ni M, MacFarlane AWT, Toft M, Lowell CA, Campbell KS, Hamerman JA. B-cell adaptor for PI3K (BCAP) negatively regulates Toll-like receptor signaling through activation of PI3K. Proc Natl Acad Sci U S A (2012) 109(1):267–72. doi: 10.1073/pnas.1111957108
55. Troutman TD, Hu W, Fulenchek S, Yamazaki T, Kurosaki T, Bazan JF, et al. Role for B-cell adapter for PI3K (BCAP) as a signaling adapter linking Toll-like receptors (TLRs) to serine/threonine kinases PI3K/Akt. Proc Natl Acad Sci U S A (2012) 109(1):273–8. doi: 10.1073/pnas.1118579109
56. Miao Y, Jiang M, Qi L, Yang, Xiao W, Fang F. BCAP Regulates Dendritic Cell Maturation Through the Dual-Regulation of NF-kappaB and PI3K/AKT Signaling During Infection. Front Immunol (2020) 11:250. doi: 10.3389/fimmu.2020.00250
57. Pourrajab F, Yazdi MB, Zarch MB, Zarch MB, Hekmatimoghaddam S. Cross talk of the first-line defense TLRs with PI3K/Akt pathway, in preconditioning therapeutic approach. Mol Cell Ther (2015) 3:4. doi: 10.1186/s40591-015-0041-7
58. Ostuni R, Zanoni I, Granucci F. Deciphering the complexity of Toll-like receptor signaling. Cell Mol Life Sci (2010) 67(24):4109–34. doi: 10.1007/s00018-010-0464-x
59. Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol (2010) 11(1):70–5. doi: 10.1038/ni.1819
60. Min Y, Kim MJ, Lee S, Chun E, Lee KY. Inhibition of TRAF6 ubiquitin-ligase activity by PRDX1 leads to inhibition of NFKB activation and autophagy activation. Autophagy (2018) 14(8):1347–58. doi: 10.1080/15548627.2018.1474995
61. Murphey ED, Fang G, Sherwood ER. Endotoxin pretreatment improves bacterial clearance and decreases mortality in mice challenged with Staphylococcus aureus. Shock (2008) 29(4):512–8. doi: 10.1097/SHK.0b013e318150776f
62. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe (2012) 12(2):223–32. doi: 10.1016/j.chom.2012.06.006
63. Fensterheim BA, Young JD, Luan L, Kleinbard RR, Stothers CL, Patil NK, et al. The TLR4 Agonist Monophosphoryl Lipid A Drives Broad Resistance to Infection via Dynamic Reprogramming of Macrophage Metabolism. J Immunol (2018) 200(11):3777–89. doi: 10.4049/jimmunol.1800085
64. Chen F, Wu W, Millman A, Craft JF, Chen E, Patel N, et al. Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat Immunol (2014) 15(10):938–46. doi: 10.1038/ni.2984
65. Romero CD, Varma TK, Hobbs JB, Reyes A, Driver B, Sherwood ER. The Toll-like receptor 4 agonist monophosphoryl lipid a augments innate host resistance to systemic bacterial infection. Infect Immun (2011) 79(9):3576–87. doi: 10.1128/IAI.00022-11
66. Wynn JL, Scumpia PO, Winfield RD, Delano MJ, Kelly-Scumpia K, Barker T, et al. Defective innate immunity predisposes murine neonates to poor sepsis outcome but is reversed by TLR agonists. Blood (2008) 112(5):1750–8. doi: 10.1182/blood-2008-01-130500
67. Bohannon JK, Luan L, Hernandez A, Afzal A, Guo Y, Patil NK, et al. Role of G-CSF in monophosphoryl lipid A-mediated augmentation of neutrophil functions after burn injury. J leukocyte Biol (2016) 99(4):629–40. doi: 10.1189/jlb.4A0815-362R
68. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392(6673):245–52. doi: 10.1038/32588
69. Munz C, Steinman RM, Fujii S. Dendritic cell maturation by innate lymphocytes: coordinated stimulation of innate and adaptive immunity. J Exp Med (2005) 202(2):203–7. doi: 10.1084/jem.20050810
70. Reppe K, Tschernig T, Luhrmann A, van Laak V, Grote K, Zemlin MV, et al. Immunostimulation with macrophage-activating lipopeptide-2 increased survival in murine pneumonia. Am J Respir Cell Mol Biol (2009) 40(4):474–81. doi: 10.1165/rcmb.2008-0071OC
71. Reppe K, Radunzel P, Dietert K, Tschernig T, Wolff T, Hammerschmidt S, et al. Pulmonary immunostimulation with MALP-2 in influenza virus-infected mice increases survival after pneumococcal superinfection. Infect Immun (2015) 83(12):4617–29. doi: 10.1128/IAI.00948-15
72. Palma C, Iona E, Ebensen T, Guzman CA, Cassone A. The toll-like receptor 2/6 ligand MALP-2 reduces the viability of Mycobacterium tuberculosis in murine macrophages. Open Microbiol J (2009) 3:47–52. doi: 10.2174/1874285800903010047
73. Tan AC, Mifsud EJ, Zeng W, Edenborough K, McVernon J, Brown LE, et al. Intranasal administration of the TLR2 agonist Pam2Cys provides rapid protection against influenza in mice. Mol Pharm (2012) 9(9):2710–8. doi: 10.1021/mp300257x
74. Landy M, Pillemer L. Increased resistance to infection and accompanying alteration in properidin levels following administration of bacterial lipopolysaccharides. J Exp Med (1956) 104(3):383–409. doi: 10.1084/jem.104.3.383
75. Varma TK, Durham M, Murphey ED, Cui W, Huang Z, Lin CY, et al. Endotoxin priming improves clearance of Pseudomonas aeruginosa in wild-type and interleukin-10 knockout mice. Infect Immun (2005) 73(11):7340–7. doi: 10.1128/IAI.73.11.7340-7347.2005
76. Fensterheim BA, Guo Y, Sherwood ER, Bohannon JK. The Cytokine Response to Lipopolysaccharide Does Not Predict the Host Response to Infection. J Immunol (2017) 198(8):3264–73. doi: 10.4049/jimmunol.1602106
77. Hernandez A, Luan L, Stothers CL, Patil NK, Fults JB, Fensterheim BA, et al. Phosphorylated Hexa-Acyl Disaccharides Augment Host Resistance Against Common Nosocomial Pathogens. Crit Care Med (2019) 47(11):e930–8. doi: 10.1097/CCM.0000000000003967
78. Baldridge JR, Cluff CW, Evans JT, Lacy MJ, Stephens JR, Brookshire VG, et al. Immunostimulatory activity of aminoalkyl glucosaminide 4-phosphates (AGPs): induction of protective innate immune responses by RC-524 and RC-529. J Endotoxin Res (2002) 8(6):453–8. doi: 10.1177/09680519020080061501
79. Chen IM, Loh JP, Chuah CXP, Gao QHC, Sun Y, Ng SH, et al. Evidence for Cross-Protection Against Subsequent Febrile Respiratory Illness Episodes From Prior Infections by Different Viruses Among Singapore Military Recruits 2009-2014. J Infect Dis (2019) 219(12):1913–23. doi: 10.1093/infdis/jiz046
80. Airhart CL, Rohde HN, Bohach GA, Hovde CJ, Deobald CF, Lee SS, et al. Induction of innate immunity by lipid A mimetics increases survival from pneumonic plague. Microbiol (Reading) (2008) 154(Pt 7):2131–8. doi: 10.1099/mic.0.2008/017566-0
81. Bae EH, Seo SH, Kim CU, Jang MS, Song MS, Lee TY, et al. Bacterial Outer Membrane Vesicles Provide Broad-Spectrum Protection against Influenza Virus Infection via Recruitment and Activation of Macrophages. J Innate Immun (2019) 11(4):316–29. doi: 10.1159/000494098
82. Abdul-Careem MF, Firoz Mian M, Gillgrass AE, Chenoweth MJ, Barra NG, Chan T, et al. FimH, a TLR4 ligand, induces innate antiviral responses in the lung leading to protection against lethal influenza infection in mice. Antiviral Res (2011) 92(2):346–55. doi: 10.1016/j.antiviral.2011.09.004
83. Habibi M, Asadi Karam MR, Bouzari S. Transurethral instillation with fusion protein MrpH.FimH induces protective innate immune responses against uropathogenic Escherichia coli and Proteus mirabilis. APMIS (2016) 124(6):444–52. doi: 10.1111/apm.12523
84. Zhang B, Chassaing B, Shi Z, Uchiyama R, Zhang Z, Denning TL, et al. Viral infection. Prevention and cure of rotavirus infection via TLR5/NLRC4-mediated production of IL-22 and IL-18. Science (2014) 346(6211):861–5. doi: 10.1126/science.1256999
85. Munoz-Wolf N, Rial A, Fougeron D, Tabareau J, Sirard JC, Chabalgoity JA. Sublingual flagellin protects against acute pneumococcal pneumonia in a TLR5-dependent and NLRC4-independent fashion. Future Microbiol (2016) 11:1167–77. doi: 10.2217/fmb-2016-0045
86. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124(4):783–801. doi: 10.1016/j.cell.2006.02.015
87. Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, et al. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity (2009) 31(6):873–84. doi: 10.1016/j.immuni.2009.09.018
88. Muhlradt PF, Kiess M, Meyer H, Sussmuth R, Jung G. Isolation, structure elucidation, and synthesis of a macrophage stimulatory lipopeptide from Mycoplasma fermentans acting at picomolar concentration. J Exp Med (1997) 185(11):1951–8. doi: 10.1084/jem.185.11.1951
89. Luhrmann A, Deiters U, Skokowa J, Hanke M, Gessner JE, Muhlradt PF, et al. In vivo effects of a synthetic 2-kilodalton macrophage-activating lipopeptide of Mycoplasma fermentans after pulmonary application. Infect Immun (2002) 70(7):3785–92. doi: 10.1128/IAI.70.7.3785-3792.2002
90. Kaufmann A, Muhlradt PF, Gemsa D, Sprenger H. Induction of cytokines and chemokines in human monocytes by Mycoplasma fermentans-derived lipoprotein MALP-2. Infect Immun (1999) 67(12):6303–8. doi: 10.1128/IAI.67.12.6303-6308.1999
91. Zaman M, Toth I. Immunostimulation by synthetic lipopeptide-based vaccine candidates: structure-activity relationships. Front Immunol (2013) 4:318. doi: 10.3389/fimmu.2013.00318
92. Muhlradt PF, Kiess M, Meyer H, Sussmuth R, Jung G. Structure and specific activity of macrophage-stimulating lipopeptides from Mycoplasma hyorhinis. Infect Immun (1998) 66(10):4804–10. doi: 10.1128/IAI.66.10.4804-4810.1998
93. Mifsud EJ, Tan AC, Short KR, Brown LE, Chua BY, Jackson DC. Reducing the impact of influenza-associated secondary pneumococcal infections. Immunol Cell Biol (2016) 94(1):101–8. doi: 10.1038/icb.2015.71
94. Zhou Y, Banday AH, Hruby VJ, Cai M. Development of N-Acetylated Dipalmitoyl-S-Glyceryl Cysteine Analogs as Efficient TLR2/TLR6 Agonists. Molecules (2019) 24(19):5161–7. doi: 10.3390/molecules24193512
95. Laping NJ, DeMartino MP, Cottom JE, Axten JM, Emery JG, Guss JH, et al. TLR2 agonism reverses chemotherapy-induced neutropenia in Macaca fascicularis. Blood Adv (2017) 1(26):2553–62. doi: 10.1182/bloodadvances.2017010611
96. Lehner MD, Morath S, Michelsen KS, Schumann RR, Hartung T. Induction of cross-tolerance by lipopolysaccharide and highly purified lipoteichoic acid via different Toll-like receptors independent of paracrine mediators. J Immunol (2001) 166(8):5161–7. doi: 10.4049/jimmunol.166.8.5161
97. Rayhane N, Fitting C, Lortholary O, Dromer F, Cavaillon JM. Administration of endotoxin associated with lipopolysaccharide tolerance protects mice against fungal infection. Infect Immun (2000) 68(6):3748–53. doi: 10.1128/IAI.68.6.3748-3753.2000
98. Wheeler DS, Lahni PM, Denenberg AG, Poynter SE, Wong HR, Cook JA, et al. Induction of endotoxin tolerance enhances bacterial clearance and survival in murine polymicrobial sepsis. Shock (2008) 30(3):267–73. doi: 10.1097/shk.0b013e318162c190
99. Deng M, Scott MJ, Loughran P, Gibson G, Sodhi C, Watkins S, et al. Lipopolysaccharide clearance, bacterial clearance, and systemic inflammatory responses are regulated by cell type-specific functions of TLR4 during sepsis. J Immunol (2013) 190(10):5152–60. doi: 10.4049/jimmunol.1300496
100. Shahin RD, Engberg I, Hagberg L, Svanborg Eden C. Neutrophil recruitment and bacterial clearance correlated with LPS responsiveness in local gram-negative infection. J Immunol (1987) 138(10):3475–80.
101. Rusek P, Wala M, Druszczynska M, Fol M. Infectious Agents as Stimuli of Trained Innate Immunity. Int J Mol Sci (2018) 19(2):456. doi: 10.3390/ijms19020456
102. Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, et al. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J (1994) 8(2):217–25. doi: 10.1096/fasebj.8.2.8119492
103. Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature (2009) 458(7242):1191–5. doi: 10.1038/nature07830
104. Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, Miyake K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem Biophys Res Commun (2008) 368(1):94–9. doi: 10.1016/j.bbrc.2008.01.061
105. Hernandez A, Bohannon JK, Luan L, Fensterheim BA, Guo Y, Patil NK, et al. The role of MyD88- and TRIF-dependent signaling in monophosphoryl lipid A-induced expansion and recruitment of innate immunocytes. J leukocyte Biol (2016) 100(6):1311–22. doi: 10.1189/jlb.1A0216-072R
106. Cluff CW, Baldridge JR, Stover AG, Evans JT, Johnson DA, Lacy MJ, et al. Synthetic toll-like receptor 4 agonists stimulate innate resistance to infectious challenge. Infect Immun (2005) 73(5):3044–52. doi: 10.1128/IAI.73.5.3044-3052.2005
107. Fukuda S, Ihara K, Bohannon JK, Hernandez A, Patil NK, Luan L, et al. Monophosphoryl Lipid a Attenuates Multiorgan Dysfunction During Post-Burn Pseudomonas Aeruginosa Pneumonia in Sheep. Shock (2020) 53(3):307–16. doi: 10.1097/SHK.0000000000001364
108. Ruchaud-Sparagano MH, Mills R, Scott J, Simpson AJ. MPLA inhibits release of cytotoxic mediators from human neutrophils while preserving efficient bacterial killing. Immunol Cell Biol (2014) 92(9):799–809. doi: 10.1038/icb.2014.55
109. Baldridge JR, McGowan P, Evans JT, Cluff C, Mossman S, Johnson D, et al. Taking a Toll on human disease: Toll-like receptor 4 agonists as vaccine adjuvants and monotherapeutic agents. Expert Opin Biol Ther (2004) 4(7):1129–38. doi: 10.1517/14712598.4.7.1129
110. Didierlaurent AM, Morel S, Lockman L, Giannini SL, Bisteau M, Carlsen H, et al. AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J Immunol (2009) 183(10):6186–97. doi: 10.4049/jimmunol.0901474
111. Garcon N, Di Pasquale A. From discovery to licensure, the Adjuvant System story. Hum Vaccin Immunother (2017) 13(1):19–33. doi: 10.1080/21645515.2016.1225635
112. Johnson AT, Wang L, Gillett SJ, Chandraratna RA. High affinity retinoic acid receptor antagonists: analogs of AGN 193109. Bioorg Med Chem Lett (1999) 9(4):573–6. doi: 10.1016/S0960-894X(99)00047-5
113. Lembo A, Pelletier M, Iyer R, Timko M, Dudda JC, West TE, et al. Administration of a synthetic TLR4 agonist protects mice from pneumonic tularemia. J Immunol (2008) 180(11):7574–81. doi: 10.4049/jimmunol.180.11.7574
114. Legat A, Thomas S, Hermand P, Van Mechelen M, Goldman M, De Wit D. CD14-independent responses induced by a synthetic lipid A mimetic. Eur J Immunol (2010) 40(3):797–802. doi: 10.1002/eji.200939992
115. Lee TY, Kim CU, Bae EH, Seo SH, Jeong DG, Yoon SW, et al. Outer membrane vesicles harboring modified lipid A moiety augment the efficacy of an influenza vaccine exhibiting reduced endotoxicity in a mouse model. Vaccine (2017) 35(4):586–95. doi: 10.1016/j.vaccine.2016.12.025
116. Schembri MA, Kjaergaard K, Sokurenko EV, Klemm P. Molecular characterization of the Escherichia coli FimH adhesin. J Infect Dis (2001) 183(Suppl 1):S28–31. doi: 10.1086/318847
117. Wang Y, Su L, Morin MD, Jones BT, Whitby LR, Surakattula MM, et al. TLR4/MD-2 activation by a synthetic agonist with no similarity to LPS. Proc Natl Acad Sci U S A (2016) 113(7):E884–893. doi: 10.1073/pnas.1525639113
118. Morin MD, Wang Y, Jones BT, Su L, Surakattula MM, Berger M, et al. Discovery and Structure-Activity Relationships of the Neoseptins: A New Class of Toll-like Receptor-4 (TLR4) Agonists. J Med Chem (2016) 59(10):4812–30. doi: 10.1021/acs.jmedchem.6b00177
119. Hajam IA, Dar PA, Shahnawaz I, Jaume JC, Lee JH. Bacterial flagellin-a potent immunomodulatory agent. Exp Mol Med (2017) 49(9):e373. doi: 10.1038/emm.2017.172
120. Munoz N, Van Maele L, Marques JM, Rial A, Sirard JC, Chabalgoity JA. Mucosal administration of flagellin protects mice from Streptococcus pneumoniae lung infection. Infect Immun (2010) 78(10):4226–33. doi: 10.1128/IAI.00224-10
121. Kinnebrew MA, Ubeda C, Zenewicz LA, Smith N, Flavell RA, Pamer EG. Bacterial flagellin stimulates Toll-like receptor 5-dependent defense against vancomycin-resistant Enterococcus infection. J Infect Dis (2010) 201(4):534–43. doi: 10.1086/650203
122. Porte R, Fougeron D, Munoz-Wolf N, Tabareau J, Georgel AF, Wallet F, et al. A Toll-Like Receptor 5 Agonist Improves the Efficacy of Antibiotics in Treatment of Primary and Influenza Virus-Associated Pneumococcal Mouse Infections. Antimicrob Agents Chemother (2015) 59(10):6064–72. doi: 10.1128/AAC.01210-15
123. Gowen BB, Wong MH, Jung KH, Sanders AB, Mitchell WM, Alexopoulou L, et al. TLR3 is essential for the induction of protective immunity against Punta Toro Virus infection by the double-stranded RNA (dsRNA), poly(I:C12U), but not Poly(I:C): differential recognition of synthetic dsRNA molecules. J Immunol (2007) 178(8):5200–8. doi: 10.4049/jimmunol.178.8.5200
124. Boivin N, Sergerie Y, Rivest S, Boivin G. Effect of pretreatment with toll-like receptor agonists in a mouse model of herpes simplex virus type 1 encephalitis. J Infect Dis (2008) 198(5):664–72. doi: 10.1086/590671
125. Kumaki Y, Salazar AM, Wandersee MK, Barnard DL. Prophylactic and therapeutic intranasal administration with an immunomodulator, Hiltonol((R)) (Poly IC:LC), in a lethal SARS-CoV-infected BALB/c mouse model. Antiviral Res (2017) 139:1–12. doi: 10.1016/j.antiviral.2016.12.007
126. Ribes S, Arcilla C, Ott M, Schutze S, Hanisch UK, Nessler S, et al. Pre-treatment with the viral Toll-like receptor 3 agonist poly(I:C) modulates innate immunity and protects neutropenic mice infected intracerebrally with Escherichia coli. J Neuroinflamm (2020) 17(1):24. doi: 10.1186/s12974-020-1700-4
127. Chiba E, Tomosada Y, Vizoso-Pinto MG, Salva S, Takahashi T, Tsukida K, et al. Immunobiotic Lactobacillus rhamnosus improves resistance of infant mice against respiratory syncytial virus infection. Int Immunopharmacol (2013) 17(2):373–82. doi: 10.1016/j.intimp.2013.06.024
128. Tomosada Y, Chiba E, Zelaya H, Takahashi T, Tsukida K, Kitazawa H, et al. Nasally administered Lactobacillus rhamnosus strains differentially modulate respiratory antiviral immune responses and induce protection against respiratory syncytial virus infection. BMC Immunol (2013) 14:40. doi: 10.1186/1471-2172-14-40
129. Zelaya H, Tada A, Vizoso-Pinto MG, Salva S, Kanmani P, Aguero G, et al. Nasal priming with immunobiotic Lactobacillus rhamnosus modulates inflammation-coagulation interactions and reduces influenza virus-associated pulmonary damage. Inflammation Res (2015) 64(8):589–602. doi: 10.1007/s00011-015-0837-6
130. Clua P, Kanmani P, Zelaya H, Tada A, Kober A, Salva S, et al. Peptidoglycan from Immunobiotic Lactobacillus rhamnosus Improves Resistance of Infant Mice to Respiratory Syncytial Viral Infection and Secondary Pneumococcal Pneumonia. Front Immunol (2017) 8:948. doi: 10.3389/fimmu.2017.00948
131. Wu CC, Crain B, Yao S, Sabet M, Lao FS, Tawatao RI, et al. Innate immune protection against infectious diseases by pulmonary administration of a phospholipid-conjugated TLR7 ligand. J Innate Immun (2014) 6(3):315–24. doi: 10.1159/000355217
132. To EE, Erlich J, Liong F, Luong R, Liong S, Bozinovski S, et al. Intranasal and epicutaneous administration of Toll-like receptor 7 (TLR7) agonists provides protection against influenza A virus-induced morbidity in mice. Sci Rep (2019) 9(1):2366. doi: 10.1038/s41598-019-38864-5
133. Hu Y, Tang L, Zhu Z, Meng H, Chen T, Zhao S, et al. A novel TLR7 agonist as adjuvant to stimulate high quality HBsAg-specific immune responses in an HBV mouse model. J Transl Med (2020) 18(1):112. doi: 10.1186/s12967-020-02275-2
134. Wang Y, Chen K, Wu Z, Liu Y, Liu S, Zou Z, et al. Immunizations with hepatitis B viral antigens and a TLR7/8 agonist adjuvant induce antigen-specific immune responses in HBV-transgenic mice. Int J Infect Dis (2014) 29:31–6. doi: 10.1016/j.ijid.2014.07.015
135. Boni C, Vecchi A, Rossi M, Laccabue D, Giuberti T, Alfieri A, et al. TLR7 Agonist Increases Responses of Hepatitis B Virus-Specific T Cells and Natural Killer Cells in Patients With Chronic Hepatitis B Treated With Nucleos(T)Ide Analogues. Gastroenterology (2018) 154(6):1764–77.e1767. doi: 10.1053/j.gastro.2018.01.030
136. Zimmermann S, Egeter O, Hausmann S, Lipford GB, Rocken M, Wagner H, et al. CpG oligodeoxynucleotides trigger protective and curative Th1 responses in lethal murine leishmaniasis. J Immunol (1998) 160(8):3627–30.
137. Verthelyi D, Gursel M, Kenney RT, Lifson JD, Liu S, Mican J, et al. CpG oligodeoxynucleotides protect normal and SIV-infected macaques from Leishmania infection. J Immunol (2003) 170(9):4717–23. doi: 10.4049/jimmunol.170.9.4717
138. Harandi AM, Eriksson K, Holmgren J. A protective role of locally administered immunostimulatory CpG oligodeoxynucleotide in a mouse model of genital herpes infection. J Virol (2003) 77(2):953–62. doi: 10.1128/JVI.77.2.953-962.2003
139. Deng JC, Moore TA, Newstead MW, Zeng X, Krieg AM, Standiford TJ. CpG oligodeoxynucleotides stimulate protective innate immunity against pulmonary Klebsiella infection. J Immunol (2004) 173(8):5148–55. doi: 10.4049/jimmunol.173.8.5148
140. Ito S, Ishii KJ, Gursel M, Shirotra H, Ihata A, Klinman DM. CpG oligodeoxynucleotides enhance neonatal resistance to Listeria infection. J Immunol (2005) 174(2):777–82. doi: 10.4049/jimmunol.174.2.777
141. Pedras-Vasconcelos JA, Goucher D, Puig M, Tonelli LH, Wang V, Ito S, et al. CpG oligodeoxynucleotides protect newborn mice from a lethal challenge with the neurotropic Tacaribe arenavirus. J Immunol (2006) 176(8):4940–9. doi: 10.4049/jimmunol.176.8.4940
142. Kim TH, Kim D, Gautam A, Lee H, Kwak MH, Park MC, et al. CpG-DNA exerts antibacterial effects by protecting immune cells and producing bacteria-reactive antibodies. Sci Rep (2018) 8(1):16236. doi: 10.1038/s41598-018-34722-y
143. Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity (2010) 32(3):305–15. doi: 10.1016/j.immuni.2010.03.012
144. Matsumoto M, Kikkawa S, Kohase M, Miyake K, Seya T. Establishment of a monoclonal antibody against human Toll-like receptor 3 that blocks double-stranded RNA-mediated signaling. Biochem Biophys Res Commun (2002) 293(5):1364–9. doi: 10.1016/S0006-291X(02)00380-7
145. Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, Alexopoulou L, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature (2005) 433(7028):887–92. doi: 10.1038/nature03326
146. Nicodemus CF, Berek JS. TLR3 agonists as immunotherapeutic agents. Immunotherapy (2010) 2(2):137–40. doi: 10.2217/imt.10.8
147. Richmond JY, Hamilton LD. Foot-and-mouth disease virus inhibition induced in mice by synthetic double-stranded RNA (polyriboinosinic and polyribocytidylic acids). Proc Natl Acad Sci U S A (1969) 64(1):81–6. doi: 10.1073/pnas.64.1.81
148. Lafont M, Petton B, Vergnes A, Pauletto M, Segarra A, Gourbal B, et al. Long-lasting antiviral innate immune priming in the Lophotrochozoan Pacific oyster, Crassostrea gigas. Sci Rep (2017) 7(1):13143. doi: 10.1038/s41598-017-13564-0
149. Mian MF, Ahmed AN, Rad M, Babaian A, Bowdish D, Ashkar AA. Length of dsRNA (poly I:C) drives distinct innate immune responses, depending on the cell type. J Leukoc Biol (2013) 94(5):1025–36. doi: 10.1189/jlb.0312125
150. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest (2009) 119(7):1910–20. doi: 10.1172/JCI35412
151. Carter WA, Pitha PM, Marshall LW, Tazawa I, Tazawa S, Ts’o PO. Structural requirements of the rI n -rC n complex for induction of human interferon. J Mol Biol (1972) 70(3):567–87. doi: 10.1016/0022-2836(72)90560-8
152. Padalko E, Nuyens D, De Palma A, Verbeken E, Aerts JL, De Clercq E, et al. The interferon inducer ampligen [poly(I)-poly(C12U)] markedly protects mice against coxsackie B3 virus-induced myocarditis. Antimicrob Agents Chemother (2004) 48(1):267–74. doi: 10.1128/AAC.48.1.267-274.2004
153. Thompson KA, Strayer DR, Salvato PD, Thompson CE, Klimas N, Molavi A, et al. Results of a double-blind placebo-controlled study of the double-stranded RNA drug polyI:polyC12U in the treatment of HIV infection. Eur J Clin Microbiol Infect Dis (1996) 15(7):580–7. doi: 10.1007/BF01709367
154. Mismatched double-stranded RNA: polyI:polyC12U. Drugs RD (2004) 5(5):297–304. doi: 10.2165/00126839-200405050-00006
155. Ichinohe T, Kawaguchi A, Tamura S, Takahashi H, Sawa H, Ninomiya A, et al. Intranasal immunization with H5N1 vaccine plus Poly I:Poly C12U, a Toll-like receptor agonist, protects mice against homologous and heterologous virus challenge. Microbes Infect (2007) 9(11):1333–40. doi: 10.1016/j.micinf.2007.06.007
156. Bardel E, Doucet-Ladeveze R, Mathieu C, Harandi AM, Dubois B, Kaiserlian D. Intradermal immunisation using the TLR3-ligand Poly (I:C) as adjuvant induces mucosal antibody responses and protects against genital HSV-2 infection. NPJ Vaccines (2016) 1:16010. doi: 10.1038/npjvaccines.2016.10
157. Strayer DR, Carter WA, Brodsky I, Cheney P, Peterson D, Salvato P, et al. A controlled clinical trial with a specifically configured RNA drug, poly(I).poly(C12U), in chronic fatigue syndrome. Clin Infect Dis (1994) 18(Suppl 1):S88–95. doi: 10.1093/clinids/18.Supplement_1.S88
158. Suhadolnik RJ, Reichenbach NL, Hitzges P, Adelson ME, Peterson DL, Cheney P, et al. Changes in the 2-5A synthetase/RNase L antiviral pathway in a controlled clinical trial with poly(I)-poly(C12U) in chronic fatigue syndrome. In Vivo (1994) 8(4):599–604.
159. Stephen EL, Sammons ML, Pannier WL, Baron S, Spertzel RO, Levy HB. Effect of a nuclease-resistant derivative of polyriboinosinic-polyribocytidylic acid complex on yellow fever in rhesus monkeys (Macaca mulatta). J Infect Dis (1977) 136(1):122–6. doi: 10.1093/infdis/136.1.122
160. Kende M. Prophylactic and therapeutic efficacy of poly(I,C)-LC against Rift Valley fever virus infection in mice. J Biol Response Mod (1985) 4(5):503–11.
161. Wong JP, Christopher ME, Viswanathan S, Karpoff N, Dai X, Das D, et al. Activation of toll-like receptor signaling pathway for protection against influenza virus infection. Vaccine (2009) 27(25-26):3481–3. doi: 10.1016/j.vaccine.2009.01.048
162. Wong JP, Yang H, Nagata L, Kende M, Levy H, Schnell G, et al. Liposome-mediated immunotherapy against respiratory influenza virus infection using double-stranded RNA poly ICLC. Vaccine (1999) 17(13-14):1788–95. doi: 10.1016/S0264-410X(98)00439-3
163. Saxena M, Sabado RL, La Mar M, Mohri H, Salazar AM, Dong H, et al. Poly-ICLC, a TLR3 Agonist, Induces Transient Innate Immune Responses in Patients With Treated HIV-Infection: A Randomized Double-Blinded Placebo Controlled Trial. Front Immunol (2019) 10:725. doi: 10.3389/fimmu.2019.00725
164. Martins KA, Bavari S, Salazar AM. Vaccine adjuvant uses of poly-IC and derivatives. Expert Rev Vaccines (2015) 14(3):447–59. doi: 10.1586/14760584.2015.966085
165. Stahl-Hennig C, Eisenblatter M, Jasny E, Rzehak T, Tenner-Racz K, Trumpfheller C, et al. Synthetic double-stranded RNAs are adjuvants for the induction of T helper 1 and humoral immune responses to human papillomavirus in rhesus macaques. PLoS Pathog (2009) 5(4):e1000373. doi: 10.1371/journal.ppat.1000373
166. Lau YF, Tang LH, Ooi EE. A TLR3 ligand that exhibits potent inhibition of influenza virus replication and has strong adjuvant activity has the potential for dual applications in an influenza pandemic. Vaccine (2009) 27(9):1354–64. doi: 10.1016/j.vaccine.2008.12.048
167. Lau YF, Tang LH, McCall AW, Ooi EE, Subbarao K. An adjuvant for the induction of potent, protective humoral responses to an H5N1 influenza virus vaccine with antigen-sparing effect in mice. J Virol (2010) 84(17):8639–49. doi: 10.1128/JVI.00596-10
168. Desheva JA, Lu XH, Rekstin AR, Rudenko LG, Swayne DE, Cox NJ, et al. Characterization of an influenza A H5N2 reassortant as a candidate for live-attenuated and inactivated vaccines against highly pathogenic H5N1 viruses with pandemic potential. Vaccine (2006) 24(47-48):6859–66. doi: 10.1016/j.vaccine.2006.06.023
169. Steel J. New strategies for the development of H5N1 subtype influenza vaccines: progress and challenges. BioDrugs (2011) 25(5):285–98. doi: 10.1007/BF03256169
170. Shen E, Li L, Li L, Feng L, Lu L, Yao Z, et al. PIKA as an adjuvant enhances specific humoral and cellular immune responses following the vaccination of mice with HBsAg plus PIKA. Cell Mol Immunol (2007) 4(2):113–20.
171. Zhang Y, Zhang S, Li L, Hu R, Lin H, Liu H, et al. Ineffectiveness of rabies vaccination alone for post-exposure protection against rabies infection in animal models. Antiviral Res (2016) 135:56–61. doi: 10.1016/j.antiviral.2016.10.002
172. Kalimuddin S, Wijaya L, Chan YFZ, Wong AWL, Oh HML, Wang LF, et al. A phase II randomized study to determine the safety and immunogenicity of the novel PIKA rabies vaccine containing the PIKA adjuvant using an accelerated regimen. Vaccine (2017) 35(51):7127–32. doi: 10.1016/j.vaccine.2017.10.097
173. Clua P, Tomokiyo M, Raya Tonetti F, Islam MA, Garcia Castillo V, Marcial G, et al. The Role of Alveolar Macrophages in the Improved Protection against Respiratory Syncytial Virus and Pneumococcal Superinfection Induced by the Peptidoglycan of Lactobacillus rhamnosus CRL1505. Cells (2020) 9(7):1653. doi: 10.3390/cells9071653
174. Kolling Y, Salva S, Villena J, Alvarez S. Are the immunomodulatory properties of Lactobacillus rhamnosus CRL1505 peptidoglycan common for all Lactobacilli during respiratory infection in malnourished mice? PLoS One (2018) 13(3):e0194034. doi: 10.1371/journal.pone.0194034
175. Miller RL, Gerster JF, Owens ML, Slade HB, Tomai MA. Imiquimod applied topically: a novel immune response modifier and new class of drug. Int J Immunopharmacol (1999) 21(1):1–14. doi: 10.1016/S0192-0561(98)00068-X
176. Miller SM, Cybulski V, Whitacre M, Bess LS, Livesay MT, Walsh L, et al. Novel Lipidated Imidazoquinoline TLR7/8 Adjuvants Elicit Influenza-Specific Th1 Immune Responses and Protect Against Heterologous H3N2 Influenza Challenge in Mice. Front Immunol (2020) 11:406. doi: 10.3389/fimmu.2020.00406
177. Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature (2008) 452(7184):234–8. doi: 10.1038/nature06726
178. Pelka K, Bertheloot D, Reimer E, Phulphagar K, Schmidt SV, Christ A, et al. The Chaperone UNC93B1 Regulates Toll-like Receptor Stability Independently of Endosomal TLR Transport. Immunity (2018) 48(5):911–22.e917. doi: 10.1016/j.immuni.2018.04.011
179. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol (2004) 5(2):190–8. doi: 10.1038/ni1028
180. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434(7034):772–7. doi: 10.1038/nature03464
181. Sasai M, Linehan MM, Iwasaki A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science (2010) 329(5998):1530–4. doi: 10.1126/science.1187029
182. Elkins KL, Rhinehart-Jones TR, Stibitz S, Conover JS, Klinman DM. Bacterial DNA containing CpG motifs stimulates lymphocyte-dependent protection of mice against lethal infection with intracellular bacteria. J Immunol (1999) 162(4):2291–8.
183. Gill N, Deacon PM, Lichty B, Mossman KL, Ashkar AA. Induction of innate immunity against herpes simplex virus type 2 infection via local delivery of Toll-like receptor ligands correlates with beta interferon production. J Virol (2006) 80(20):9943–50. doi: 10.1128/JVI.01036-06
184. A two-dose hepatitis B vaccine for adults (Heplisav-B). JAMA (2018) 319(8):822–3. doi: 10.1001/jama.2018.1097.
185. Hyer RN, Janssen RS. Immunogenicity and safety of a 2-dose hepatitis B vaccine, HBsAg/CpG 1018, in persons with diabetes mellitus aged 60-70years. Vaccine (2019) 37(39):5854–61. doi: 10.1016/j.vaccine.2019.08.005
186. van ‘t Wout JW, Poell R, van Furth R. The role of BCG/PPD-activated macrophages in resistance against systemic candidiasis in mice. Scand J Immunol (1992) 36(5):713–9. doi: 10.1111/j.1365-3083.1992.tb03132.x
187. Tribouley J, Tribouley-Duret J, Appriou M. [Effect of Bacillus Callmette Guerin (BCG) on the receptivity of nude mice to Schistosoma mansoni]. C R Seances Soc Biol Fil. (1978) 172(5):902–4.
188. Kristensen I, Aaby P, Jensen H. Routine vaccinations and child survival: follow up study in Guinea-Bissau, West Africa. BMJ (2000) 321(7274):1435–8. doi: 10.1136/bmj.321.7274.1435
189. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A (2012) 109(43):17537–42. doi: 10.1073/pnas.1202870109
190. Kleinnijenhuis J, Quintin J, Preijers F, Benn CS, Joosten LA, Jacobs C, et al. Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J Innate Immun (2014) 6(2):152–8. doi: 10.1159/000355628
191. Tsuji S, Matsumoto M, Takeuchi O, Akira S, Azuma I, Hayashi A, et al. Maturation of human dendritic cells by cell wall skeleton of Mycobacterium bovis bacillus Calmette-Guerin: involvement of toll-like receptors. Infect Immun (2000) 68(12):6883–90. doi: 10.1128/IAI.68.12.6883-6890.2000
192. Camilli G, Tabouret G, Quintin J. The Complexity of Fungal beta-Glucan in Health and Disease: Effects on the Mononuclear Phagocyte System. Front Immunol (2018) 9:673. doi: 10.3389/fimmu.2018.00673
193. Williams DL, Browder IW, Di Luzio NR. Immunotherapeutic modification of Escherichia coli–induced experimental peritonitis and bacteremia by glucan. Surgery (1983) 93(3):448–54.
194. Di Luzio NR, Williams DL. Protective effect of glucan against systemic Staphylococcus aureus septicemia in normal and leukemic mice. Infect Immun (1978) 20(3):804–10. doi: 10.1128/IAI.20.3.804-810.1978
195. Vetvicka V, Vetvickova J. Glucan supplementation enhances the immune response against an influenza challenge in mice. Ann Transl Med (2015) 3(2):22. doi: 10.3978/j.issn.2305-5839.2015.01.08
196. Liang J, Melican D, Cafro L, Palace G, Fisette L, Armstrong R, et al. Enhanced clearance of a multiple antibiotic resistant Staphylococcus aureus in rats treated with PGG-glucan is associated with increased leukocyte counts and increased neutrophil oxidative burst activity. Int J Immunopharmacol (1998) 20(11):595–614. doi: 10.1016/S0192-0561(98)00007-1
197. Lyuksutova OI, Murphey ED, Toliver-Kinsky TE, Lin CY, Cui W, Williams DL, et al. Glucan phosphate treatment attenuates burn-induced inflammation and improves resistance to Pseudomonas aeruginosa burn wound infection. Shock (2005) 23(3):224–32. doi: 10.1097/01.shk.0000151864.79293.41
198. Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med (2003) 197(9):1107–17. doi: 10.1084/jem.20021787
199. Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. Dectin-1 mediates the biological effects of beta-glucans. J Exp Med (2003) 197(9):1119–24. doi: 10.1084/jem.20021890
200. Pavot V, Rochereau N, Resseguier J, Gutjahr A, Genin C, Tiraby G, et al. Cutting edge: New chimeric NOD2/TLR2 adjuvant drastically increases vaccine immunogenicity. J Immunol (2014) 193(12):5781–5. doi: 10.4049/jimmunol.1402184
201. Rice TA, Brenner TA, Percopo CM, Ma M, Keicher JD, Domachowske JB, et al. Signaling via pattern recognition receptors NOD2 and TLR2 contributes to immunomodulatory control of lethal pneumovirus infection. Antiviral Res (2016) 132:131–40. doi: 10.1016/j.antiviral.2016.06.002
202. Santecchia I, Vernel-Pauillac F, Rasid O, Quintin J, Gomes-Solecki M, Boneca IG, et al. Innate immune memory through TLR2 and NOD2 contributes to the control of Leptospira interrogans infection. PLoS Pathog (2019) 15(5):e1007811. doi: 10.1371/journal.ppat.1007811
203. Jin JW, Tang SQ, Rong MZ, Zhang MQ. Synergistic effect of dual targeting vaccine adjuvant with aminated beta-glucan and CpG-oligodeoxynucleotides for both humoral and cellular immune responses. Acta Biomater (2018) 78:211–23. doi: 10.1016/j.actbio.2018.08.002
204. Bistoni F, Verducci G, Perito S, Vecchiarelli A, Puccetti P, Marconi P, et al. Immunomodulation by a low-virulence, agerminative variant of Candida albicans. Further evidence for macrophage activation as one of the effector mechanisms of nonspecific anti-infectious protection. J Med Vet Mycol (1988) 26(5):285–99. doi: 10.1080/02681218880000401
205. Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature (2007) 447(7142):326–9. doi: 10.1038/nature05762
206. Yager EJ, Szaba FM, Kummer LW, Lanzer KG, Burkum CE, Smiley ST, et al. gamma-Herpesvirus-induced protection against bacterial infection is transient. Viral Immunol (2009) 22(1):67–72. doi: 10.1089/vim.2008.0086
207. Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature (2009) 457(7229):557–61. doi: 10.1038/nature07665
208. Hennessy EJ, Parker AE, O’Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov (2010) 9(4):293–307. doi: 10.1038/nrd3203
209. Makkouk A, Abdelnoor AM. The potential use of Toll-like receptor (TLR) agonists and antagonists as prophylactic and/or therapeutic agents. Immunopharmacol Immunotoxicol (2009) 31(3):331–8. doi: 10.1080/08923970902802926
210. Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med (2007) 13(5):552–9. doi: 10.1038/nm1589
211. Rezaei N. Therapeutic targeting of pattern-recognition receptors. Int Immunopharmacol (2006) 6(6):863–9. doi: 10.1016/j.intimp.2006.02.005
212. Savov JD, Brass DM, Lawson BL, McElvania-Tekippe E, Walker JK, Schwartz DA. Toll-like receptor 4 antagonist (E5564) prevents the chronic airway response to inhaled lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol (2005) 289(2):L329–337. doi: 10.1152/ajplung.00014.2005
213. Sun Y, Pearlman E. Inhibition of corneal inflammation by the TLR4 antagonist Eritoran tetrasodium (E5564). Invest Ophthalmol Vis Sci (2009) 50(3):1247–54. doi: 10.1167/iovs.08-2628
214. Lynn M, Rossignol DP, Wheeler JL, Kao RJ, Perdomo CA, Noveck R, et al. Blocking of responses to endotoxin by E5564 in healthy volunteers with experimental endotoxemia. J Infect Dis (2003) 187(4):631–9. doi: 10.1086/367990
215. Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA (2013) 309(11):1154–62. doi: 10.1001/jama.2013.2194
216. Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med (2010) 38(8):1685–94. doi: 10.1097/CCM.0b013e3181e7c5c9
217. Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol (2011) 186(8):4794–804. doi: 10.4049/jimmunol.1000702
218. Boulware DR, Pullen MF, Bangdiwala AS, Pastick KA, Lofgren SM, Okafor EC, et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N Engl J Med (2020) 383(6):517–25. doi: 10.1056/NEJMoa2016638
219. Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P, et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin Infect Dis (2020) 71(15):732–9. doi: 10.1093/cid/ciaa237
220. Skipper CP, Pastick KA, Engen NW, Bangdiwala AS, Abassi M, Lofgren SM, et al. Hydroxychloroquine in Nonhospitalized Adults With Early COVID-19 : A Randomized Trial. Ann Intern Med (2020) 173(8):623–31. doi: 10.7326/M20-4207
221. Mitja O, Corbacho-Monne M, Ubals M, Tebe C, Penafiel J, Tobias A, et al. Hydroxychloroquine for Early Treatment of Adults with Mild Covid-19: A Randomized-Controlled Trial. Clin Infect Dis (2020). doi: 10.1093/cid/ciaa1009
222. Roschewski M, Lionakis MS, Sharman JP, Roswarski J, Goy A, Monticelli MA, et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci Immunol (2020) 5(48):eabd0110. doi: 10.1126/sciimmunol.abd0110
223. Kox M, van Velzen JF, Pompe JC, Hoedemaekers CW, van der Hoeven JG, Pickkers P. GTS-21 inhibits pro-inflammatory cytokine release independent of the Toll-like receptor stimulated via a transcriptional mechanism involving JAK2 activation. Biochem Pharmacol (2009) 78(7):863–72. doi: 10.1016/j.bcp.2009.06.096
224. Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res (2015) 25(7):771–84. doi: 10.1038/cr.2015.68
225. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345(6204):1250684. doi: 10.1126/science.1250684
226. Netea MG, Dominguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol (2020) 20(6):375–88. doi: 10.1038/s41577-020-0285-6
227. Patil NK, Bohannon JK, Hernandez A, Patil TK, Sherwood ER. Regulation of leukocyte function by citric acid cycle intermediates. J Leukocyte Biol (2019) 106(1):105–17. doi: 10.1002/JLB.3MIR1118-415R
228. Murphy MP, O’Neill LAJ. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell (2018) 174(4):780–4. doi: 10.1016/j.cell.2018.07.030
229. Lauterbach MA, Hanke JE, Serefidou M, Mangan MSJ, Kolbe CC, Hess T, et al. Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity (2019) 51(6):997–1011.e1017. doi: 10.1016/j.immuni.2019.11.009
230. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab (2012) 15(6):813–26. doi: 10.1016/j.cmet.2012.04.023
231. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496(7444):238–42. doi: 10.1038/nature11986
232. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell (2003) 112(5):645–57. doi: 10.1016/S0092-8674(03)00154-5
233. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell (2017) 169(4):570–86. doi: 10.1016/j.cell.2017.04.004
234. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity (2015) 42(3):419–30. doi: 10.1016/j.immuni.2015.02.005
235. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A (2013) 110(19):7820–5. doi: 10.1073/pnas.1218599110
236. Cordes T, Michelucci A, Hiller K. Itaconic Acid: The Surprising Role of an Industrial Compound as a Mammalian Antimicrobial Metabolite. Annu Rev Nutr (2015) 35:451–73. doi: 10.1146/annurev-nutr-071714-034243
237. Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, et al. IFNs Modify the Proteome of Legionella-Containing Vacuoles and Restrict Infection Via IRG1-Derived Itaconic Acid. PLoS Pathog (2016) 12(2):e1005408. doi: 10.1371/journal.ppat.1005408
238. Sasikaran J, Ziemski M, Zadora PK, Fleig A, Berg IA. Bacterial itaconate degradation promotes pathogenicity. Nat Chem Biol (2014) 10(5):371–7. doi: 10.1038/nchembio.1482
239. Chen M, Sun H, Boot M, Shao L, Chang SJ, Wang W, et al. Itaconate is an effector of a Rab GTPase cell-autonomous host defense pathway against Salmonella. Science (2020) 369(6502):450–5. doi: 10.1126/science.aaz1333
240. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature (2018) 556(7699):113–7. doi: 10.1038/nature25986
241. Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med (2015) 88(Pt B):179–88. doi: 10.1016/j.freeradbiomed.2015.04.036
242. Swain A, Bambouskova M, Kim H, Andhey PS, Duncan D, Auclair K, et al. Comparative evaluation of itaconate and its derivatives reveals divergent inflammasome and type I interferon regulation in macrophages. Nat Metab (2020) 2(7):594–602. doi: 10.1038/s42255-020-0210-0
243. Dominguez-Andres J, Novakovic B, Li Y, Scicluna BP, Gresnigt MS, Arts RJW, et al. The Itaconate Pathway Is a Central Regulatory Node Linking Innate Immune Tolerance and Trained Immunity. Cell Metab (2019) 29(1):211–20.e215. doi: 10.1016/j.cmet.2018.09.003
244. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab (2016) 24(1):158–66. doi: 10.1016/j.cmet.2016.06.004
245. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature (2011) 472(7344):476–80. doi: 10.1038/nature09973
246. van der Heijden C, Noz MP, Joosten LAB, Netea MG, Riksen NP, Keating ST. Epigenetics and Trained Immunity. Antioxid Redox Signaling (2018) 29(11):1023–40. doi: 10.1089/ars.2017.7310
247. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science (2014) 345(6204):1251086. doi: 10.1126/science.1251086
248. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab (2016) 24(6):807–19. doi: 10.1016/j.cmet.2016.10.008
249. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol (2017) 18(9):985–94. doi: 10.1038/ni.3796
250. Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, et al. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics (2012) 11(5):100–7. doi: 10.1074/mcp.M111.015875
251. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature (2019) 574(7779):575–80. doi: 10.1038/s41586-019-1678-1
252. Ifrim DC, Quintin J, Joosten LA, Jacobs C, Jansen T, Jacobs L, et al. Trained immunity or tolerance: opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin Vaccine Immunol (2014) 21(4):534–45. doi: 10.1128/CVI.00688-13
253. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest (2016) 126(1):23–31. doi: 10.1172/JCI82224
254. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis (2013) 13(3):260–8. doi: 10.1016/S1473-3099(13)70001-X
255. van der Poll T, Opal SM. Host-pathogen interactions in sepsis. Lancet Infect Dis (2008) 8(1):32–43. doi: 10.1016/S1473-3099(07)70265-7
256. Volk HD, Reinke P, Docke WD. Clinical aspects: from systemic inflammation to ‘immunoparalysis’. Chem Immunol (2000) 74:162–77. doi: 10.1159/000058753
257. Farrugia M, Baron B. The Role of Toll-Like Receptors in Autoimmune Diseases through Failure of the Self-Recognition Mechanism. Int J Inflam (2017) 2017:8391230. doi: 10.1155/2017/8391230
258. Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature (2009) 459(7247):717–21. doi: 10.1038/nature07968
259. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature (2011) 470(7332):115–9. doi: 10.1038/nature09671
260. Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature (2009) 459(7247):712–6. doi: 10.1038/nature07969
261. Schnack L, Sohrabi Y, Lagache SMM, Kahles F, Bruemmer D, Waltenberger J, et al. Mechanisms of Trained Innate Immunity in oxLDL Primed Human Coronary Smooth Muscle Cells. Front Immunol (2019) 10:13. doi: 10.3389/fimmu.2019.00013
262. Jasani B, Navabi H, Adams M. Ampligen: a potential toll-like 3 receptor adjuvant for immunotherapy of cancer. Vaccine (2009) 27(25-26):3401–4. doi: 10.1016/j.vaccine.2009.01.071
263. Jin Y, Mu Y, Zhang S, Li P, Wang F. Preparation and evaluation of the adjuvant effect of curdlan sulfate in improving the efficacy of dendritic cell-based vaccine for antitumor immunotherapy. Int J Biol Macromol (2020) 146:273–84. doi: 10.1016/j.ijbiomac.2019.12.256
264. Breckpot K, Aerts-Toegaert C, Heirman C, Peeters U, Beyaert R, Aerts JL, et al. Attenuated expression of A20 markedly increases the efficacy of double-stranded RNA-activated dendritic cells as an anti-cancer vaccine. J Immunol (2009) 182(2):860–70. doi: 10.4049/jimmunol.182.2.860
265. Sato-Kaneko F, Yao S, Ahmadi A, Zhang SS, Hosoya T, Kaneda MM, et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight (2017) 2(18):e93397. doi: 10.1172/jci.insight.93397
266. Wang E, Feng Y, Zhang M, Zou L, Li Y, Buys ES, et al. Toll-like receptor 4 signaling confers cardiac protection against ischemic injury via inducible nitric oxide synthase- and soluble guanylate cyclase-dependent mechanisms. Anesthesiology (2011) 114(3):603–13. doi: 10.1097/ALN.0b013e31820a4d5b
267. Markowski P, Boehm O, Goelz L, Haesner AL, Ehrentraut H, Bauerfeld K, et al. Pre-conditioning with synthetic CpG-oligonucleotides attenuates myocardial ischemia/reperfusion injury via IL-10 up-regulation. Basic Res Cardiol (2013) 108(5). doi: 10.1007/s00395-013-0376-7
268. Rego A, Viana SD, Ribeiro CA, Rodrigues-Santos P, Pereira FC. Monophosphoryl Lipid-A: A Promising Tool for Alzheimer’s Disease Toll. J Alzheimers Dis (2016) 52(4):1189–202. doi: 10.3233/JAD-151183
269. Michaud JP, Halle M, Lampron A, Theriault P, Prefontaine P, Filali M, et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc Natl Acad Sci U S A (2013) 110(5):1941–6. doi: 10.1073/pnas.1215165110
270. Hamada A, Torre C, Drancourt M, Ghigo E. Trained Immunity Carried by Non-immune Cells. Front Microbiol (2018) 9:3225. doi: 10.3389/fmicb.2018.03225
271. Llewellyn A, Foey A. Probiotic Modulation of Innate Cell Pathogen Sensing and Signaling Events. Nutrients (2017) 9(10):1156. doi: 10.3390/nu9101156
272. O’Neill LAJ, Netea MG. BCG-induced trained immunity: can it offer protection against COVID-19? Nat Rev Immunol (2020) 20(6):335–7. doi: 10.1038/s41577-020-0337-y
273. Geller A, Yan J. Could the Induction of Trained Immunity by beta-Glucan Serve as a Defense Against COVID-19? Front Immunol (2020) 11:1782. doi: 10.3389/fimmu.2020.01782
274. Poulas K, Farsalinos K, Zanidis C. Activation of TLR7 and Innate Immunity as an Efficient Method Against COVID-19 Pandemic: Imiquimod as a Potential Therapy. Front Immunol (2020) 11:1373. doi: 10.3389/fimmu.2020.01373
275. Anwar MA, Shah M, Kim J, Choi S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med Res Rev (2019) 39(3):1053–90. doi: 10.1002/med.21553
276. Sanchez-Ramon S, Conejero L, Netea MG, Sancho D, Palomares O, Subiza JL. Trained Immunity-Based Vaccines: A New Paradigm for the Development of Broad-Spectrum Anti-infectious Formulations. Front Immunol (2018) 9:2936. doi: 10.3389/fimmu.2018.02936
Keywords: trained immunity, immunomodulators, Toll-like receptors (TLRs), TLR agonists, nosocomial infections, immunosuppression, antibiotic resistance, vaccine adjuvant
Citation: Owen AM, Fults JB, Patil NK, Hernandez A and Bohannon JK (2021) TLR Agonists as Mediators of Trained Immunity: Mechanistic Insight and Immunotherapeutic Potential to Combat Infection. Front. Immunol. 11:622614. doi: 10.3389/fimmu.2020.622614
Received: 28 October 2020; Accepted: 24 December 2020;
Published: 18 February 2021.
Edited by:
Jose Luis Subiza, Inmunotek SL, SpainReviewed by:
Belén De Andrés, Instituto de Salud Carlos III (ISCIII), SpainMengji Lu, Essen University Hospital, Germany
Copyright © 2021 Owen, Fults, Patil, Hernandez and Bohannon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julia K. Bohannon, anVsaWEuay5ib2hhbm5vbkB2dW1jLm9yZw==