
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 28 January 2021
Sec. Molecular Innate Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.622326
This article is part of the Research TopicDiagnostic and Therapeutic Applications of Pentraxin and Pentraxin-Associated ProteinsView all 20 articles
 Helena Enocsson1*
Helena Enocsson1* Birgitta Gullstrand2
Birgitta Gullstrand2 Maija-Leena Eloranta3
Maija-Leena Eloranta3 Jonas Wetterö1
Jonas Wetterö1 Dag Leonard3
Dag Leonard3 Lars Rönnblom3
Lars Rönnblom3 Anders A. Bengtsson2
Anders A. Bengtsson2 Christopher Sjöwall1
Christopher Sjöwall1Objectives: Patients with systemic lupus erythematosus (SLE) often display modest elevations of C-reactive protein (CRP) despite raised disease activity and increased interleukin (IL-) 6. We asked to what extent IL-6 levels, the CRP polymorphism rs1205, and the type I interferon (IFN) gene signature affects the basal CRP levels in patients with SLE during a quiescent phase of the disease.
Methods: CRP and IL-6 were analyzed in plasma from 57 patients meeting established classification criteria for SLE. The CRP polymorphism rs1205 was assessed and gene expression analyzed including four type I IFN-regulated genes (IGS).
Results: CRP was increased in patients with detectable IL-6 levels (p=0.001) and decreased among IGS-positive subjects (p=0.033). A multiple linear regression model revealed IL-6 to have a positive association with CRP levels, whereas both IGS-positivity and CRP genotype (rs1205) AA/GA were negatively associated with CRP-levels.
Conclusion: Our data offer an explanation to the modest CRP levels seen in viral infections and IFN-α driven autoimmunity and corroborate prior observations showing an IFN-α dependent downregulation of CRP. The latter observation, together with the fact that the CRP-lowering polymorphism rs1205 is overrepresented in human SLE, could explain low basal CRP and inadequate CRP-responses among patients with active SLE.
The acute-phase protein C-reactive protein (CRP) is a key actor in the clearance of bacteria and dying cells. Its pentameric structure encompasses an effector face with affinity for C1q and Fcγ-receptors and a recognition face with the ability to bind phosphocholine on e.g., dying cells and pathogens, but also nuclear constituents exposed during apoptosis (i.e., snRNP and histones) (1, 2). These qualities enable CRP to contribute to efficient clearance of cell remnants and immune complexes by complement activation/modulation, opsonization, and phagocytosis, biological processes considered dysfunctional in systemic lupus erythematosus (SLE) (3, 4). Indeed, protective effects of CRP in the disease process have been demonstrated in animal models of lupus (5–7).
Due to increased CRP production from hepatocytes upon interleukin (IL-) 6 stimulation, CRP is widely used to monitor infectious and inflammatory conditions (8). With few exceptions, CRP reflects ongoing inflammation/tissue damage, seemingly without discrimination of the underlying trigger. However, whereas bacterial infections generally result in impressive CRP levels, the response in many viral infections is typically less pronounced (9). Similarly, modest CRP-responses are recorded in autoimmune diseases characterized by increased type I interferon (IFN) activity, e.g., SLE (8), and lack of correlation between IL-6 and CRP has been demonstrated in patients with SLE (10). Type I IFNs are strong activators of the anti-viral immune response, but may also contribute to autoantibody production in several autoimmune conditions (11).
IFN-α, the most studied type I IFN in autoimmunity, has previously been evaluated in relation to regulation of pentraxins (12–15). We have previously demonstrated an inhibitory effect of IFN-α in IL-6/IL-1β-induced CRP gene transcription and protein production from hepatocytes (12). Thus, the presence of IFN-α per se could contribute to modest CRP levels during IFN-α associated disease flares of type I IFN-driven autoimmunity as well as in viral infections. Furthermore, a combined effect of the CRP-lowering polymorphism of the CRP gene (rs1205) (16) and detectable IFN-α levels resulted in inability of CRP to reflect inflammatory activity among SLE patients (13). However, circulating IFN-α is difficult to detect with standard methods despite evidence of an increased gene transcription of type I IFN-regulated genes. Thus, a selection of type I IFN-regulated genes is typically assessed as a marker of an increased type I IFN activity, i.e., the type I IFN gene signature (IGS).
The aim of this study was to confirm and extend the knowledge from previous in vivo and in vitro studies showing an impact of type I IFNs (12, 13, 15) and rs1205 (13) on CRP levels among patients with SLE. Herein, the objective was to evaluate the potential impact of the IGS and rs1205 in patients unbiased from high disease activity.
All patients (Table 1) were followed within the frame of an observational research program at the University Hospital in Linköping (17). Each participant (n=57) was classified with SLE according to the 1982 American College of Rheumatology and/or the 2012 Systemic Lupus International Collaborating Clinics criteria (18, 19).

Table 1 Clinical characteristics of the 57 included patients with systemic lupus erythematosus (SLE).
The patients donated peripheral blood and the disease activity, defined by SLE disease activity index 2000 (SLEDAI-2K), was recorded from their closest regular visit to rheumatologist. All patients were considered to have low disease activity but could be serologically active and clinically quiescent at sampling (20, 21). Each participant gave written informed consent, and the study protocol was approved by the regional ethics board in Linköping.
Venous blood was drawn, and plasma was prepared and blood lipids were analyzed at the local Clinical Chemistry unit, including triglycerides, total cholesterol, high-density lipoprotein (HDL), low-density lipoprotein (LDL), non-HDL, IL-6, and CRP. We utilized a high sensitivity method for CRP with 0.15 mg/L as limit of quantification (LOQ). For IL-6, LOQ was 1.5 ng/L.
The type I IFN gene signature (IGS) was based on gene expression of IFI27, IFI44, IFI44L, and RSAD2 (22). Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized whole blood by density gradient centrifugation and lysed by RLT-buffer. Total RNA was extracted using the RNeasy Mini Kit (Qiagen GmbH, Hilden, Germany). Quantification and purity of the RNA was assessed on a DS-11 spectrophotometer (DeNovix Inc. Wilmington, DE, USA). Total RNA was reverse transcribed using iScript™ cDNA synthesis kit (Bio Rad, Hercules, California, USA) to obtain cDNA for RT-qPCR. The RT-qPCR was performed by preheating for 10 min at 95°C, followed by 40 cycles of 95°C 15 s and 60°C for 60 s using StepOnePlus (Applied Biosystems, CA, USA). TaqMan™ Gene Expression assay (FAM) (Applied Biosystems, CA, USA) were used with the following primers from Thermo Fisher Scientific, IFI44L: Hs00199115_m1, RSAD2: Hs00369813_m1, IFI27: Hs00271467_m1, IFI44: Hs00197427_m1, GAPDH: Hs03929097_g1.
Fold‐change of gene expression was determined by the relative quantification method (ΔΔCt) after normalization to the housekeeping gene (GAPDH). A subsequent log-transformation was performed to achieve comparability between genes. IGS-score was expressed as mean of log-transformed fold change in relation to a control sample (consisting of a pooled sample from healthy control donors) of the four genes (detailed in Supplementary Data 1). The patient distribution revealed two groups regarding the IGS; a cut-off of 0.5 was applied to separate IGS-positive patients from IGS-negative.
DNA was extracted from peripheral blood using Qiagen Blood Midi Kit (Qiagen) and genotyping was performed using Illumina Infinium Global Screening Array-Multi Disease version3 (Illumina Inc. California, USA) at the SNP&SEQ Technology Platform at Uppsala University. The genotyping is detailed in Supplementary Data 2.
CRP levels were not normally distributed why Mann-Whitney U test, Kruskal-Wallis test, or Spearman’s correlation analysis were applied. CRP and IL-6 were log-transformed (10log) prior to linear regression analysis with CRP or IL-6 as dependent variables. A two-sided p-value of <0.05 was considered statistically significant.
Levels of CRP correlated significantly with IL-6 (rho=0.415, p=0.001; Figure 1A) and with age (rho=0.292, p=0.027). No correlation was observed between CRP levels and IGS-score (Figure 1B). Since 28 patients had non-detectable IL-6, and 21 were judged IGS-negative, IL-6 and IGS were further tested as binary variables. CRP levels were significantly higher among those with detectable IL-6 (p=0.001; Figure 1C) and lower among IGS-positive subjects (p=0.033; Figure 1D). The CRP genotype (rs1205) was not significantly associated with CRP, neither based on number of rare alleles, nor based on presence or absence of one or two rare alleles (binary variables; Figure 1E).
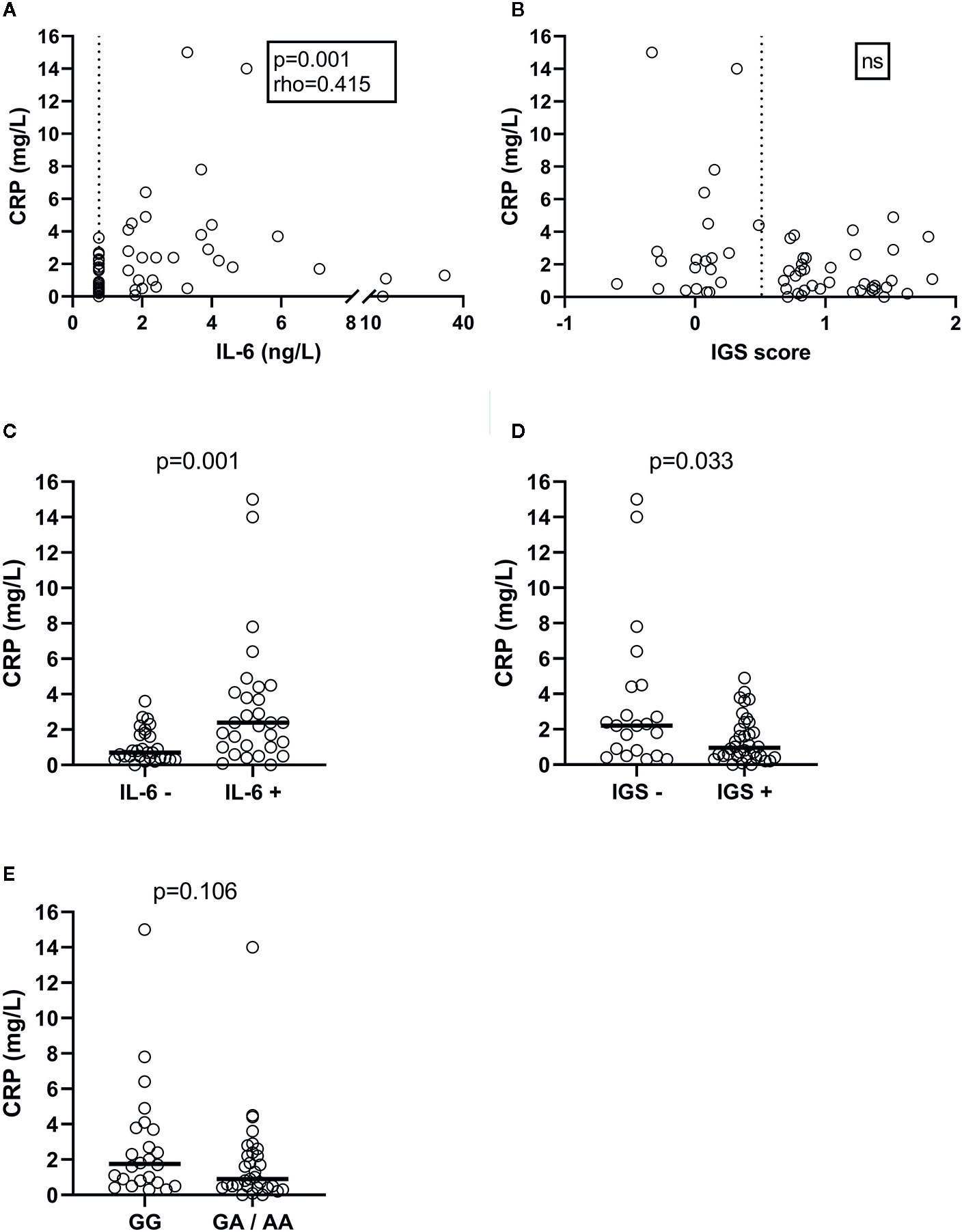
Figure 1 Plasma CRP levels in SLE patients, and the correlation and association with plasma IL-6 concentrations (A), the IGS score measured in peripheral blood mononuclear cells (B), detectable plasma IL-6 levels (C), IGS positivity (D) and CRP rs1205 genotype (E). Dashed lines represent cut-off levels for IL-6 detection and IGS positivity, respectively.
No significant correlations between CRP and corticosteroid dose, SLEDAI-2K or blood lipids were observed. Neither did we observe any differences in CRP related to sex, tobacco smoking, or use of antimalarials, corticosteroids, mycophenolate mofetil, methotrexate or use of any immunosuppressive agent (disease-modifying anti-rheumatic drugs except antimalarials, n=28). Finally, CRP levels did not associate with disease phenotypes (i.e., fulfilled classification criteria).
To evaluate impact of CRP genotype rs1205, IL-6, and IGS-positivity, these variables were included in a linear regression model with 10log CRP as the dependent variable. Age was further added as an independent variable due to its correlation with CRP.
A stepwise analysis revealed IGS-positivity (p=0.017) and alleles AA/AG of rs1205 (p=0.041) to be associated with low levels of CRP. Age and IL-6 levels did not associate with CRP in the regression analysis (Table 2). However, a regression analysis with IL-6 as binary variable resulted in a significant association of both IL-6 (p=0.012), CRP genotype (AA/AG) (p=0.030), and IGS-positivity (p=0.030) with CRP levels (Table 2 and visualized in Figure 2).
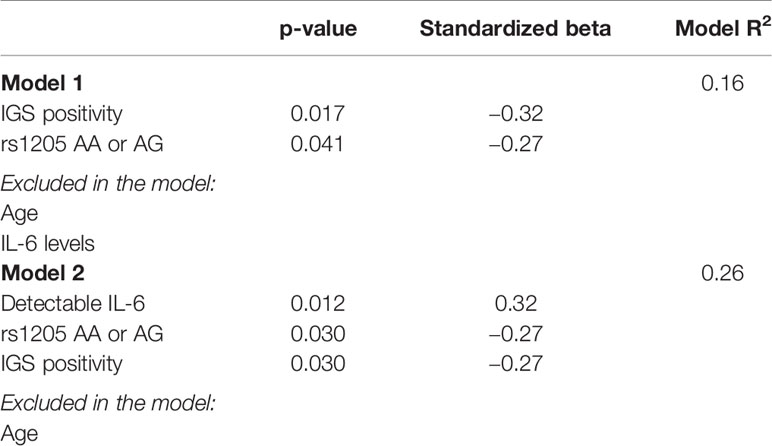
Table 2 Variables associated with CRP levels (10log CRP) in stepwise linear regression analyses.
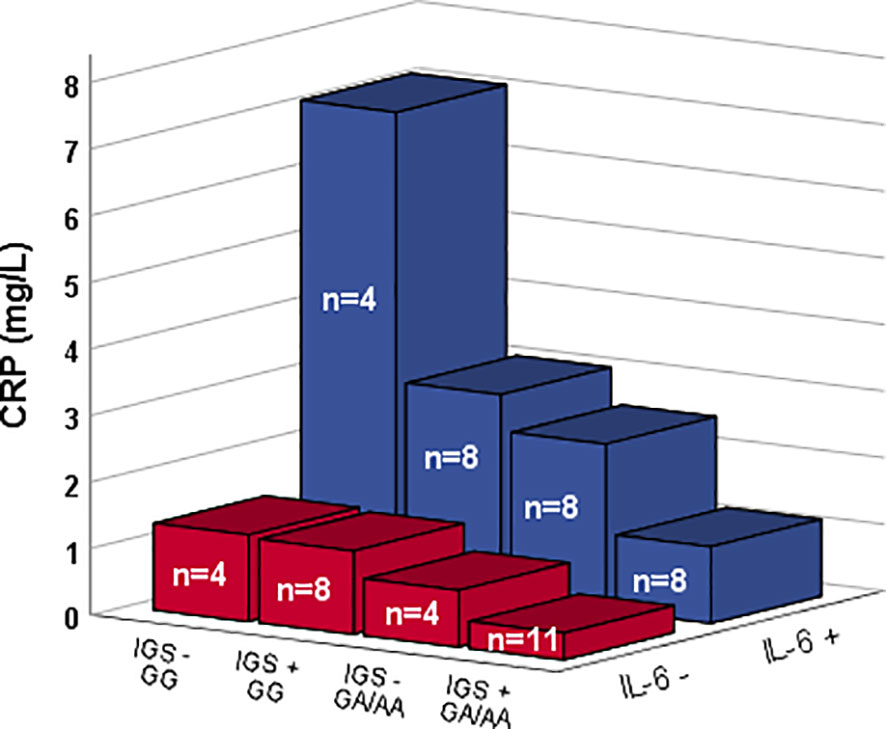
Figure 2 CRP levels among patients with and without detectable IL-6 levels (Z-axis) along with different CRP genotype (rs1205) and type I IFN gene signature (IGS) status (X-axis). Bars show median values.
We observed no associations between IGS-status and CRP genotype, IL-6 (binary), or CRP genotype as determined by chi2 (Fisher’s exact test). A regression analysis with 10log IL-6 as the dependent variable and CRP genotype and IGS-status as dependent variables did not reach statistical significance, indicating that the effect of IGS and CRP genotype on CRP levels were independent of IL-6.
The present study demonstrates that low CRP levels coincide with the CRP polymorphism rs1205 and the presence of an activated type I IFN system, as determined by the IGS-score. Our observations therefore add further support to an important role of type I IFNs in the regulation of circulating CRP via an inhibitory effect by IFN-α on CRP production (12, 15). Furthermore, we previously demonstrated an association between CRP and SLE disease activity when patients with the rs1205 minor allele and detectable serum IFN-α were excluded (13). Thus, it is plausible that our findings together with the fact that the CRP-lowering polymorphism rs1205 is overrepresented among SLE patients (16), explain the low basal CRP and inadequate CRP-responses in those with active SLE.
The extensive use of CRP as a biomarker motivate research aiming to unravel its regulation in different inflammatory conditions. Indeed, viral infections generally result in lower CRP-responses compared with bacterial infections (9), and SLE patients rarely mount a CRP-response that corresponds to their inflammatory activity (10, 23) or circulating IL-6 levels (10).
IL-6 is the main inducer of CRP-production, and it is therefore not surprising with a positive impact of IL-6 on circulating CRP (24). Herein, it was however evident that the CRP polymorphism, as well as type I IFN activity, coincides with a limited ability of IL-6 to induce a CRP-response. In fact, none of the IL-6-positive patients with the rs1205 minor allele and IGS-positivity reached a higher CRP level than 3.6 mg/L, i.e., the highest CRP found among IL-6-negative subjects regardless of rs1205 genotype or IGS-status.
Hypothetically, other polymorphisms or presence of soluble receptors for e.g., IL-6 and IFN-α could also impact CRP levels. As alternative explanations to the muted CRP response in SLE, increased consumption of CRP has been put forward. Increased complement consumption, due to inflammation and increased cell death, is a well-known feature of SLE flares (25). CRP has similar functions in the clearance of debris and could potentially be utilized and eliminated at a higher rate in patients with SLE. However, the elimination rate of CRP in SLE patients seems unaltered compared with healthy controls (26). Furthermore, anti-CRP autoantibodies, which are more frequently found in SLE, are not directed toward native circulating CRP, but toward epitopes that are exposed upon CRP dissociation on surfaces (27).
Limitations of the present study encompassed a relatively small number of patients and the lack of a replication cohort as well as a control group with unrelated inflammatory disease. The patients included had no known concomitant infections and low/stable disease activity, which reduce the potential impact of specific SLE-manifestations and comorbidities affecting the CRP-response. During bacterial infections, SLE patients usually present with an adequate CRP-response (28) which may be due to the massive increase of IL-6 that overrides the inhibitory effect of type I IFNs and/or genetic variants of CRP. In addition, bacterial infections could result in a shift of the immune activation with reduced type I IFN production. CRP itself has furthermore been shown to inhibit type I IFN production (29, 30) and it is tempting to speculate in a reciprocal regulation between pentraxins and type I IFNs. Finally, also leukocyte secretion/release of the closely related protein pentraxin-3 (PTX3) appear to be regulated by IFN-α in vitro (14).
Given the essential biological functions of pentraxins, such as CRP, in facilitating silent removal of cellular debris via Fc-receptors and by interacting with the complement system (1, 4) it is perhaps not surprising that administration of CRP to a murine lupus model prevent and reverse ongoing nephritis (5, 7). The crucial role of type I IFNs in SLE was once again demonstrated by the encouraging results of the phase 3 trial investigating the anti-IFN α/β receptor anifrolumab in patients with active SLE (31). These observations, together with our results, suggests that the consequences of CRP gene polymorphisms and IL-6/type I IFN interactions in SLE deserves further attention.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Regional Ethics Review Board in Linköping, Sweden. The patients/participants provided their written informed consent to participate in this study.
HE and CS conceived the original idea and project planning. HE, BG, JW, AAB, and CS contributed to the study design. CS collected clinical patient data. HE, BG, and M-LE carried out the laboratory work. M-LE, DL, and LR planned and supervised the genotyping. HE, BG, and CS analyzed the data. HE, LR, and CS drafted the manuscript. All authors contributed to the article and approved the submitted version.
This study was funded by grants from the Swedish Research Council for Medicine and Health, the Swedish Society of Medicine and the Ingegerd Johansson donation, the Swedish Rheumatism Association, the Region Östergötland (ALF grants), the King Gustaf V’s 80-year Anniversary Foundation, the King Gustaf V and Queen Victoria’s Freemasons’ Foundation, the Gustafsson, Selander, Alfred Österlund, Anna-Greta Crafoord and the Greta and Johan Kock’s Foundations.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to participating patients and to study nurse Marianne Petersson, Linköping. Pascal Pucholt and Johanna Sandling, Uppsala University, are acknowledged for genotype data extraction. Genotyping was performed by the SNP&SEQ Technology Platform in Uppsala (www.genotyping.se). The facility is part of the National Genomics Infrastructure supported by the Swedish Research Council for Infrastructures and Science for Life Laboratory, Sweden. The SNP&SEQ Technology Platform is supported by the Knut and Alice Wallenberg Foundation.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.622326/full#supplementary-material
1. Lu J, Mold C, Du Clos TW, Sun PD. Pentraxins and Fc Receptor-Mediated Immune Responses. Front Immunol (2018) 9:2607. doi: 10.3389/fimmu.2018.02607
2. Pathak A, Agrawal A. Evolution of C-Reactive Protein. Front Immunol (2019) 10:943. doi: 10.3389/fimmu.2019.00943
3. Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev (2010) 6(5):280–9. doi: 10.1038/nrrheum.2010.46
4. Ma YJ, Garred P. Pentraxins in Complement Activation and Regulation. Front Immunol (2018) 9:3046. doi: 10.3389/fimmu.2018.03046
5. Du Clos TW, Zlock LT, Hicks PS, Mold C. Decreased autoantibody levels and enhanced survival of (NZB x NZW) F1 mice treated with C-reactive protein. Clin Immunol Immunopathol (1994) 70(1):22–7. doi: 10.1006/clin.1994.1005
6. Szalai AJ, Weaver CT, McCrory MA, van Ginkel FW, Reiman RM, Kearney JF, et al. Delayed lupus onset in (NZB x NZW)F1 mice expressing a human C-reactive protein transgene. Arthritis Rheum (2003) 48(6):1602–11. doi: 10.1002/art.11026
7. Rodriguez W, Mold C, Marnell LL, Hutt J, Silverman GJ, Tran D, et al. Prevention and reversal of nephritis in MRL/lpr mice with a single injection of C-reactive protein. Arthritis Rheum (2006) 54(1):325–35. doi: 10.1002/art.21556
8. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med (1999) 340(6):448–54. doi: 10.1056/NEJM199902113400607
9. Nakayama T, Sonoda S, Urano T, Yamada T, Okada M. Monitoring both serum amyloid protein A and C-reactive protein as inflammatory markers in infectious diseases. Clin Chem (1993) 39(2):293–7. doi: 10.1093/clinchem/39.2.293
10. Gabay C, Roux-Lombard P, de Moerloose P, Dayer JM, Vischer T, Guerne PA. Absence of correlation between interleukin 6 and C-reactive protein blood levels in systemic lupus erythematosus compared with rheumatoid arthritis. J Rheumatol (1993) 20(5):815–21.
11. Crow MK, Ronnblom L. Type I interferons in host defence and inflammatory diseases. Lupus Sci Med (2019) 6(1):e000336. doi: 10.1136/lupus-2019-000336
12. Enocsson H, Sjöwall C, Skogh T, Eloranta ML, Rönnblom L, Wetterö J. Interferon-alpha mediates suppression of C-reactive protein: explanation for muted C-reactive protein response in lupus flares? Arthritis Rheumatol (2009) 60(12):3755–60. doi: 10.1002/art.25042
13. Enocsson H, Sjöwall C, Kastbom A, Skogh T, Eloranta ML, Rönnblom L, et al. Association of serum C-reactive protein levels with lupus disease activity in the absence of measurable interferon-alpha and a C-reactive protein gene variant. Arthritis Rheumatol (2014) 66(6):1568–73. doi: 10.1002/art.38408
14. Wirestam L, Enocsson H, Skogh T, Eloranta ML, Ronnblom L, Sjowall C, et al. Interferon-alpha coincides with suppressed levels of pentraxin-3 (PTX3) in systemic lupus erythematosus and regulates leucocyte PTX3 in vitro. Clin Exp Immunol (2017) 189(1):83–91. doi: 10.1111/cei.12957
15. Vaez A, Jansen R, Prins BP, Hottenga JJ, de Geus EJ, Boomsma DI, et al. In Silico Post Genome-Wide Association Studies Analysis of C-Reactive Protein Loci Suggests an Important Role for Interferons. Circ Cardiovasc Genet (2015) 8(3):487–97. doi: 10.1161/CIRCGENETICS.114.000714
16. Russell AI, Cunninghame Graham DS, Shepherd C, Roberton CA, Whittaker J, Meeks J, et al. Polymorphism at the C-reactive protein locus influences gene expression and predisposes to systemic lupus erythematosus. Hum Mol Genet (2004) 13(1):137–47. doi: 10.1093/hmg/ddh021
17. Ighe A, Dahlstrom O, Skogh T, Sjowall C. Application of the 2012 Systemic Lupus International Collaborating Clinics classification criteria to patients in a regional Swedish systemic lupus erythematosus register. Arthritis Res Ther (2015) 17:3. doi: 10.1186/s13075-015-0521-9
18. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum (1982) 25(11):1271–7. doi: 10.1002/art.1780251101
19. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum (2012) 64(8):2677–86. doi: 10.1002/art.34473
20. Steiman AJ, Gladman DD, Ibanez D, Urowitz MB. Prolonged serologically active clinically quiescent systemic lupus erythematosus: frequency and outcome. J Rheumatol (2010) 37(9):1822–7. doi: 10.3899/jrheum.100007
21. Svensson C, Eriksson P, Zachrisson H, Sjöwall C. High-frequency ultrasound of multiple arterial areas reveals increased intima media thickness, vessel wall appearance and atherosclerotic plaques in systemic lupus erythematosus. Front Med (2020) 7:581336. doi: 10.3389/fmed.2020.581336
22. Strauss R, Rose T, Flint SM, Klotsche J, Haupl T, Peck-Radosavljevic M, et al. Type I interferon as a biomarker in autoimmunity and viral infection: a leukocyte subset-specific analysis unveils hidden diagnostic options. J Mol Med (Berl) (2017) 95(7):753–65. doi: 10.1007/s00109-017-1515-7
23. Rezaieyazdi Z, Sahebari M, Hatef MR, Abbasi B, Rafatpanah H, Afshari JT, et al. Is there any correlation between high sensitive CRP and disease activity in systemic lupus erythematosus? Lupus (2011) 20(14):1494–500. doi: 10.1177/0961203311418706
24. Agrawal A, Cha-Molstad H, Samols D, Kushner I. Transactivation of C-reactive protein by IL-6 requires synergistic interaction of CCAAT/enhancer binding protein beta (C/EBP beta) and Rel p50. J Immunol (2001) 166(4):2378–84. doi: 10.4049/jimmunol.166.4.2378
25. Leffler J, Bengtsson AA, Blom AM. The complement system in systemic lupus erythematosus: an update. Ann Rheumatic Dis (2014) 73(9):1601–6. doi: 10.1136/annrheumdis-2014-205287
26. Vigushin DM, Pepys MB, Hawkins PN. Metabolic and scintigraphic studies of radioiodinated human C-reactive protein in health and disease. J Clin Invest (1993) 91(4):1351–7. doi: 10.1172/JCI116336
27. Sjöwall C, Wetterö J. Pathogenic implications for autoantibodies against C-reactive protein and other acute phase proteins. Clin Chim Acta (2007) 378(1-2):13–23. doi: 10.1016/j.cca.2006.12.002
28. Becker GJ, Waldburger M, Hughes GR, Pepys MB. Value of serum C-reactive protein measurement in the investigation of fever in systemic lupus erythematosus. Ann Rheum Dis (1980) 39(1):50–2. doi: 10.1136/ard.39.1.50
29. Mold C, Du Clos TW. C-reactive protein inhibits plasmacytoid dendritic cell interferon responses to autoantibody immune complexes. Arthritis Rheum (2013) 65(7):1891–901. doi: 10.1002/art.37968
30. Svanberg C, Enocsson H, Martinsson K, Potempa LA, Rajab I, Wetterö J, et al editors. Pentameric, but Not Monomeric C-reactive Protein, Limits the SnRNP-immune Complex Triggered Type I Interferon Response: Implications for Lupus Pathogenesis. Arthritis Rheumatol (2019) 71 Supplement:10.
Keywords: C-reactive protein, type I interferons, systemic lupus erythematosus, inflammation, biomarker, pentraxins, interferon, gene variants
Citation: Enocsson H, Gullstrand B, Eloranta M-L, Wetterö J, Leonard D, Rönnblom L, Bengtsson AA and Sjöwall C (2021) C-Reactive Protein Levels in Systemic Lupus Erythematosus Are Modulated by the Interferon Gene Signature and CRP Gene Polymorphism rs1205. Front. Immunol. 11:622326. doi: 10.3389/fimmu.2020.622326
Received: 28 October 2020; Accepted: 17 December 2020;
Published: 28 January 2021.
Edited by:
Kenji Daigo, Nippon Medical School, JapanReviewed by:
Marta E. Alarcon-Riquelme, Junta de Andalucía de Genómica e Investigación Oncológica (GENYO), SpainCopyright © 2021 Enocsson, Gullstrand, Eloranta, Wetterö, Leonard, Rönnblom, Bengtsson and Sjöwall. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helena Enocsson, SGVsZW5hLmVub2Nzc29uQGxpdS5zZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.