 Grace R. Pidwill
Grace R. Pidwill Josie F. Gibson
Josie F. Gibson Joby Cole
Joby Cole Stephen A. Renshaw
Stephen A. Renshaw Simon J. Foster
Simon J. Foster- 1Department of Molecular Biology and Biotechnology, University of Sheffield, Sheffield, United Kingdom
- 2Florey Institute, University of Sheffield, Sheffield, United Kingdom
- 3The Bateson Centre, University of Sheffield, Sheffield, United Kingdom
- 4Department of Infection, Immunity and Cardiovascular Disease, Medical School, University of Sheffield, Sheffield, United Kingdom
Staphylococcus aureus is a member of the human commensal microflora that exists, apparently benignly, at multiple sites on the host. However, as an opportunist pathogen it can also cause a range of serious diseases. This requires an ability to circumvent the innate immune system to establish an infection. Professional phagocytes, primarily macrophages and neutrophils, are key innate immune cells which interact with S. aureus, acting as gatekeepers to contain and resolve infection. Recent studies have highlighted the important roles of macrophages during S. aureus infections, using a wide array of killing mechanisms. In defense, S. aureus has evolved multiple strategies to survive within, manipulate and escape from macrophages, allowing them to not only subvert but also exploit this key element of our immune system. Macrophage-S. aureus interactions are multifaceted and have direct roles in infection outcome. In depth understanding of these host-pathogen interactions may be useful for future therapeutic developments. This review examines macrophage interactions with S. aureus throughout all stages of infection, with special emphasis on mechanisms that determine infection outcome.
Introduction
Staphylococcus aureus is a Gram-positive commensal bacterium frequently found in the upper respiratory tract (1, 2), alongside various other locations on the human host (3). S. aureus is part of the normal microbiota, colonizing 40% of new-born babies and 50% of adults intermittently or permanently, normally without any ill-effects (1, 4). Despite this, S. aureus can become pathogenic, with colonization an important reservoir for infection (5).
Human diseases caused by S. aureus range from minor skin infections to life threatening bacteremia and meningitis. S. aureus is one of the most frequent causes of nosocomial and community-acquired pneumonia, skin and soft-tissue infections or bloodstream infections (6). Serious S. aureus infections cause approximately 20,000 deaths a year in the US, and 5,000 in the EU, costing an estimated €380 million in EU health costs (7, 8). A contributing factor to the high mortality rate of S. aureus infections is increasing antimicrobial resistance. Methicillin Resistant Staphylococcus aureus (MRSA) bacteremia has a high mortality rate: 30% to 40% (9–12). S. aureus resistance to antibiotics is widespread in both community and nosocomial-acquired infection. Some S. aureus strains have even developed resistance to the last-resort antibiotic vancomycin (13) and vaccine candidates have thus far been unsuccessful (14, 15). S. aureus infections represent a significant risk to human health, highlighting the pressing need for alternative prophylaxis and treatments.
The immune response to S. aureus infection is complex. Infection occurs when S. aureus breaches host external barriers, for example through a tissue injury. In most cases, an efficient immune response is mounted, involving innate immune cell recruitment and eventual clearance of infection. Macrophages, as antigen presenting cells, also activate the adaptive immune response. As such, phagocytes play a vital role in locating, restricting and destroying S. aureus.
Macrophage interactions with S. aureus are of particular interest. Macrophages are responsible for phagocytic uptake of the majority of invading bacteria and employ a multitude of bacterial killing mechanisms to effectively kill S. aureus. Despite this, some S. aureus are able to survive within macrophages - a source for intracellular persistence which eventually enables further bacterial dissemination (16–18). S. aureus can survive within mature macrophage phagosomes (16, 19, 20), as well as cause uncontrollable infection within monocyte-derived macrophages (MDMs) (18). Furthermore, S. aureus can evade and manipulate macrophages, using many strategies to impede macrophage recruitment, phagocytosis and degrative abilities (21–25). Understanding these complex host-pathogen interactions may provide promising new therapeutic targets, which are urgently required due to rising S. aureus antibiotic resistance.
This review examines macrophage interactions with S. aureus, from the role of macrophages in S. aureus infection dynamics to specific macrophage-S. aureus interactions, including macrophage recruitment, phagocytosis, macrophage polarization, bacterial killing mechanisms and nutrient restriction (Figure 1).
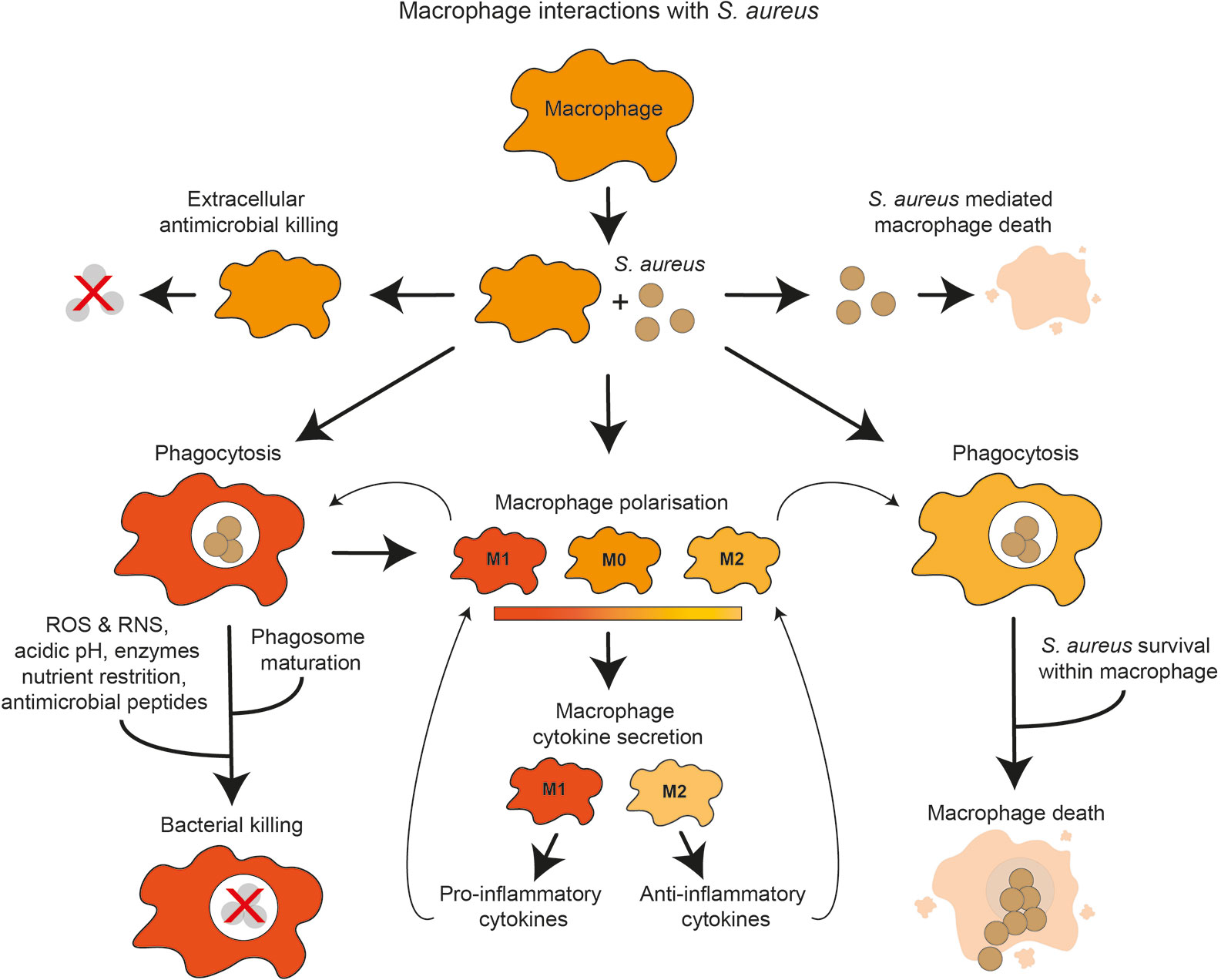
Figure 1 Overview of macrophage interactions with S. aureus. Presence of S. aureus influences macrophage polarization and cytokine secretion toward either pro-inflammatory or anti-inflammatory. S. aureus may be subject to extracellular or intracellular killing by macrophages. Macrophages may be killed by extracellular S. aureus factors. S. aureus phagocytosis by macrophages may lead to intracellular killing, which may destroy the bacteria using antimicrobial mechanisms. Alternatively, S. aureus may evade these mechanisms, proliferate within the macrophage and cause macrophage death.
What Are Macrophages?
Macrophages are professional phagocytes, able to engulf microorganisms and trigger responses leading to microbial death. Macrophages, like their close relatives neutrophils, both of which are professional phagocytes and are derived from myeloid precursor cells (22, 26), are an important part of the innate immune response. However, each phagocyte has multiple differences in cellular properties and functions. Both macrophages and neutrophils sense and migrate toward sites of infection and can phagocytose and kill invading pathogens. However, macrophages, as antigen presenting cells, also play a key role in activation of the adaptive immune response by presenting antigens of phagocytosed pathogens (27). Neutrophils are commonly the first immune cell to reach an infected site, and may be more bactericidal, whereas monocytes (which may differentiate into macrophages) are typically attracted later on (28). In comparison to neutrophils, macrophages are adapted to be much less reactive, which may be to avoid attacking self-antigens or stimulating unwanted immune responses due to being resident within tissues for a longer lifespan (29). Neutrophils are derived from within the bone marrow and have a very short lifespan which is thought to limit stimulation of unnecessary inflammation (28). In contrast, macrophages may live for weeks to months (30, 31) and are found within tissues through the body, termed tissue resident macrophages.
There are different cell lineage sources which give rise to tissue-resident macrophages. Traditionally, it was thought that macrophages develop only from circulating monocytes, the precursor cells for some macrophages and dendritic cells. Monocytes represent around 10% of leukocytes in humans, while tissue-resident macrophages represent another 10% to 15% (although this may increase following inflammatory stimulus) (26, 32). Monocytes develop from hematopoietic stem cells in the bone marrow (26) and circulate in the blood for 1 to 2 days, after which they die unless recruited to tissues for differentiation (26, 33, 34). However, many tissue-resident macrophages self-renew within the tissue (35). Self-renewing macrophages are derived from embryonic-origin cells which are seeded to sites of the body before birth (36–38), with examples including liver Kupffer cells, Langerhans skin cells and brain microglia (35, 39–41). Other macrophage populations develop from the macrophage and dendritic cell precursor (MDP) cell, a precursor to monocytes (42).
Once macrophages have differentiated according to their tissue, they develop distinct transcriptional profiles and are named according to tissue location (43). The properties of varied tissue-resident cells have been extensively reviewed (44, 45). However, macrophage function remains similar regardless of tissue location: (i) coordinating tissue development, (ii) tissue homeostasis through clearing apoptotic/senescent cells, (iii) acting as sentinels which survey and monitor changes in the tissue, and (iv) responding to pathogens in infection (26).
Kupffer cells are the largest group of tissue-resident macrophages in the body, making up 80% to 90% of the total population (46, 47). They display a unique phenotype characterized by downregulation of CR3, expression of liver-specific lectin CLEC4F and tissue-specific complement receptor CRIg (48, 49). Through a variety of receptors, Kupffer cells filter blood and mediate clearance of waste products and non–self-antigens (48, 50, 51). The close proximity of Kupffer cells to sinusoids also facilitates best access to pathogens arriving in the liver (46).
As mentioned above, Langerhans cells (LCs) are also self-renewing, although if they are exhausted by, for example, UV radiation, they are replaced by bone-marrow-derived precursor cells (52). LCs develop dendritic cell (DC) characteristics in the epidermis, and as such share attributes with both DCs and macrophages (53). Similar to tissue-resident macrophages, LCs self-renew and have a long half-life (approximately 2 months), however, like DCs, LCs can travel to lymph nodes (52, 53). Their presence at the barrier of the skin suggests a role as immune sentinels (54).
Macrophage diversity enables tissue-specific phenotypes which help macrophages to perform their function. However, macrophages are unified in their phagocytic and innate immune functions, allowing bridging of the innate and adaptive responses.
The Key Role of Macrophages in S. aureus Infection Outcome
A wide range of diseases are caused by S. aureus, from minor skin infections to life-threatening diseases, for example bacteremia and endocarditis. Numerous S. aureus infections of humans are associated with abscess formation (55) and in murine bacteremia infection models, kidney abscess formation is a key outcome (56, 57). Macrophages have a central role in S. aureus infection dynamics. Murine blood infection begins with hematogenous transit of extracellular S. aureus, which are rapidly phagocytosed in the liver by Kupffer cells. More than 90% of S. aureus are sequestered by the liver (58) - the majority of bacteria are then effectively killed. A small number of bacteria can survive intracellularly, ultimately escaping to form microabcesses in the liver. Extracellular S. aureus may also disseminate to seed kidney abscesses (59, 60).
The importance of macrophages in S. aureus infection is highlighted when macrophages are depleted in animal infection models. Mice lacking macrophages have increased bacterial burden and mortality following S. aureus sepsis (61). Similarly, in murine airway infection, macrophages are required for clearance of S. aureus, since loss of alveolar macrophages inhibited killing of bacteria at 5 hpi (62), significantly enhanced mortality (63), and increased bacterial load in the lungs (64). In zebrafish, macrophages phagocytose the majority of the initial bacterial inoculum and, similar to mice, loss of macrophages leads to increased S. aureus susceptibility (65, 66). Phagocytes are a known intracellular niche for S. aureus, allowing bacterial survival and eventual escape, allowing dissemination throughout the host (67–69). Human monocyte-derived macrophages (MDMs) also permit intracellular S. aureus survival and bacterial escape (16, 18). Despite this, macrophages efficiently phagocytose and degrade most S. aureus, with just a small proportion of bacteria surviving to potentially lead to dissemination throughout the host (59). Thus, the intraphagocyte niche represents a population bottleneck for S. aureus (70), as demonstrated for other intracellular pathogens including Salmonella enterica and Bacillus anthracis (71, 72). Micro-abscesses in the liver are formed from surviving bacterial cells which escape from macrophages. It has been demonstrated that S. aureus abscesses are formed by single, or very small numbers of bacteria (69, 70), leading to the emergence of clonal populations within abscesses. Depletion of macrophages causes loss of clonality whereas depletion of neutrophils does not (59), indicating that macrophages are the key phagocyte responsible for the emergence of clonality. Kupffer cells are especially instrumental as an intraphagocyte niche leading to the emergence of clonality in S. aureus murine sepsis infection, largely due to their key role in filtering blood (59, 61).
Extracellular bacteria, which have escaped macrophages can also seed infection at distant sites through the bloodstream. After staphylococcal cells survive and multiply inside Kupffer cells, the bacteria can escape into the peritoneal cavity where they are phagocytosed by peritoneal macrophages, which provide another intracellular niche, promoting dissemination to peritoneal organs (60). Cycles of macrophage phagocytosis and bacterial escape can allow S. aureus to survive intracellularly over time (73). Although macrophages are crucial for initial infection dynamics, neutrophils are thought to be significant for dissemination. Extracellular bacteria in the bloodstream may be phagocytosed by neutrophils, which can act as Trojan horses enabling spread to other organs, including the kidneys (59, 68). Together, these studies highlight the importance of macrophages in controlling the initial bacterial sepsis inoculum specifically in restricting early infection stages, and macrophage involvement in S. aureus infection features, including formation of a population bottleneck, clonal abscess formation and eventual dissemination.
Phagocytosis of S. aureus by Macrophages
As described above, macrophages are an important host defense against S. aureus infection, but in order to effectively eliminate S. aureus, macrophages must first locate and phagocytose the invading bacteria.
Recruitment of Macrophages to S. aureus Infection Sites
Phagocyte recruitment to S. aureus is coordinated through responding to host immune effectors released in response to S. aureus, or signals derived from S. aureus itself. Initial host responses to S. aureus are initiated by cells found at infected sites, often epithelial cells at mucosal surfaces. Epithelial cells sense invading S. aureus via pathogen recognition receptors (PRRs) which can recognize many staphylococcal molecules, including lipoproteins, lipoteichoic acid (LTA), phenol soluble modulins, protein A, toxins, and peptidoglycan (PGN) (74). Epithelial PRR signaling leads to phagocyte recruitment and activation by inducing pro-inflammatory cytokine and chemokine production; including granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein 3α (MIP-3α), IL-6, IL-1β, and IL-8 (75–78). Additionally, formylated peptides produced by S. aureus directly act as chemoattractants for macrophages (79) and S. aureus molecules activate the complement cascade (80), leading to release of strong phagocyte chemoattractant, C5a.
Macrophage recruitment has been demonstrated in S. aureus murine studies. MCP-1 is important for macrophage activation and clearance of S. aureus infection (81). Following S. aureus brain infection in mice, gene expression of multiple pro-inflammatory cytokines and chemokines are upregulated, leading to macrophage recruitment (82). In peritoneal infection, particulate S. aureus cell envelope promotes phagocyte recruitment by inducing chemotactic cytokine production (83). Of note, some macrophages subtypes, including Kupffer cells, are tissue-resident which may be recruited to local infection sites (84), whereas monocyte-derived macrophages are recruited to sites of infection from circulation in the blood (85). S. aureus also has strategies to prevent immune cell recruitment, such as expressing chemotaxis inhibitory protein of Staphylococcus aureus (CHIPS), which blocks phagocyte binding to activated complement proteins or formylated peptides excreted by S. aureus (86, 87). After recruitment to sites of infection, macrophages become activated and produce cytokines to enhance the immune response; discussed in the macrophage functional changes in response to S. aureus infection section.
Phagocytosis
Macrophages utilize micropinocytosis, macropinocytosis, receptor-mediated endocytosis and phagocytosis to ingest particles, fluids and molecules. Micropinocytosis is used for non-specific uptake of fluid and small molecules, while macropinocytosis can non-specifically engulf larger volumes of extracellular fluid and larger particles, including bacterial cells (88–90). Receptor-mediated endocytosis is the selective uptake of macromolecules bound to surface receptors. Receptor-mediated endocytosis is clathrin-dependent, micropinocytosis can involve clathrin pathways, but clathrin is not essential (88), while phagocytosis and macropinocytosis are actin-dependent (91). Phagocytosis is receptor-mediated targeted uptake of particles larger than 0.5 µm, and represents the primary pathway used by macrophages to internalize S. aureus (88). The physical state of bacterial cells is important for S. aureus phagocytosis, with particulate rather than soluble cell wall required to stimulate an efficient phagocyte immune response (83). S. aureus phagocytosis events occur following engagement of multiple receptors on the macrophage surface, including scavenger receptors (SRs), complement receptors and Fc receptors (Figure 2). The actin cytoskeleton at the cell membrane forms a phagocytic cup which extends to surround the extracellular bacterial cells and contracts to close the cup, forming a bacteria-containing phagosome within the phagocytic cell (91).
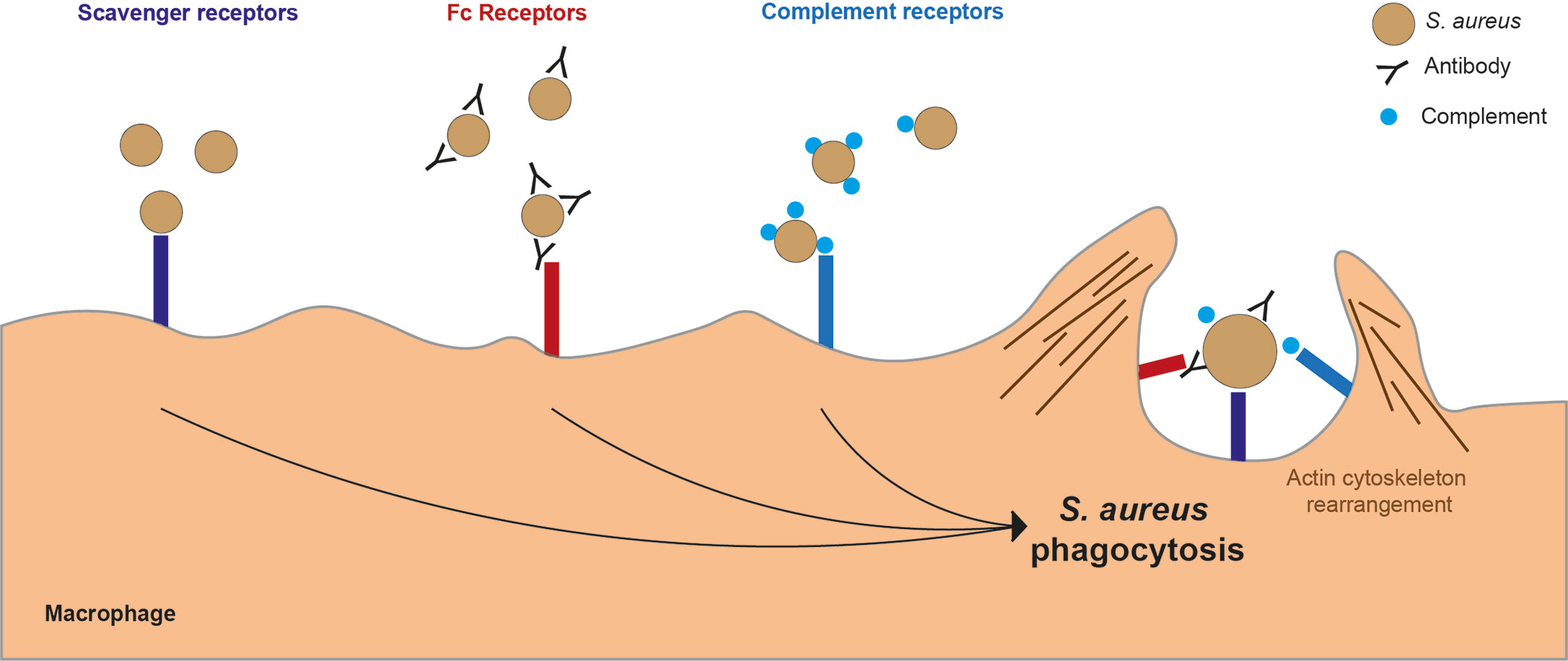
Figure 2 Key macrophage receptors used in phagocytosis of S. aureus. There are several receptors on the surface of a macrophages which can bind to S. aureus leading to phagocytosis. Scavenger receptors bind directly to S. aureus. Fc receptors bind to the Fc region of antibodies which have bound to S. aureus. Complement receptors bind to complement proteins which act as opsonins and are bound to S. aureus.
Scavenger Receptors
The SRs are a diverse group of receptors which recognize a wide range of pathogenic molecules, for example, proteins, polysaccharides, lipids, CpG motifs and lipoteichoic acid (LTA). SRs are grouped into classes based on what they bind, with S. aureus known to interact with multiple SR classes (92, 93). Macrophage SRs can bind to LTAs found on surface of Gram-positive bacteria, including S. aureus (94), leading to increased macrophage phagocytosis in an opsonin-independent manner (95). Scavenger receptor A (SR-A) contributes to Kupffer cell phagocytosis of S. aureus through mannose-binding lectin (a member of the C-type lectin family, which also binds to bacterial cells and activates the complement cascade), increasing SR-A expression on Kupffer cells (96). As well as increasing SR expression, mannose-binding lectin is also involved in opsonin-dependent S. aureus phagocytosis by phagocytes (97). Surfactant protein A (SP-A), like mannose-binding lectin, is a member of the C-type lectin family. Addition of SP-A to alveolar macrophages (AMs) increases S. aureus phagocytosis, potentially by upregulation of SR-A expression, as demonstrated for S. pneumoniae (98). SP-A can also act as an opsonin, binding to both S. aureus and SP-A receptors on macrophages. Interestingly, macrophages lacking SP-A receptors upregulate SR-A, promoting non-opsonic phagocytosis (99). Macrophage receptor with collagenous structure (MARCO) is another SR involved in macrophage phagocytosis of S. aureus, and is especially important in AM and Kupffer cell phagocytosis of S. aureus (100, 101). AMs from SR-A and MARCO knock-out mice showed reduced phagocytosis of S. aureus (102). Interestingly, the role of SRs in S. aureus infection appears to be dependent on the type of infection. Mice deficient in three different SRs (SR-A, CD36, and MARCO) were protected in peritoneal infection, but adversely effected in pulmonary infection (103). Furthermore, the importance of SRs appears to be dependent on S. aureus strain, with some strains showing no change in phagocytosis when SR binding is inhibited in human MDMs (104). Therefore, it is difficult to define a single role of SRs in S. aureus infection. However, it is clear that SRs are involved in non-opsonized S. aureus phagocytosis, and may play an important role in controlling lung infection.
Complement Receptors
The complement cascade is part of the innate immune system which targets pathogens, mediated by multiple complement proteins. The key complement component is C3 which, when cleaved by C3 convertase, generates important complement effector components to mediate three main activities: pathogens can be directly targeted with the formation of a membrane attack complex to cause cell lysis, complement proteins can promote recruitment of phagocytes to the infection site and complement proteins can act as opsonins to promote phagocytosis of coated pathogens.
Multiple S. aureus cell surface molecules activate the complement cascade in human sera (80), with changes in complement component levels observed in patients with S. aureus bacteremia (105). Furthermore, human serum studies show that mannose-binding lectin promotes complement activation in response to S. aureus (106), while depletion of complement is detrimental in S. aureus murine bacteremia or septic arthritis infections (107). A mouse model of S. aureus septic arthritis showed that deficiency in C3 increases susceptibility to infection, potentially through decreased peritoneal macrophage phagocytosis (108). The complement components used as opsonins are C3b and iC3b, these can bind phagocyte complement receptors CR1, or CR3 and CR4, respectively. Macrophage-expressed complement receptors, CR3 and CR4, promote binding and internalization of iC3b opsonized S. aureus (109). A therapeutic use of antibody complexes which interact with erythrocyte CR1 and S. aureus have been developed leading to enhanced bacterial degradation by macrophages (110).
S. aureus expresses multiple virulence factors to target complement components. To inhibit complement activation, S. aureus secretes extracellular fibrinogen-binding protein (Efb), which binds to C3, blocking complement cascade effects including opsonization (111). To interfere with C3 convertases, S. aureus expresses staphylococcal complement inhibitor (SCIN) (112). Although SCIN is a human-specific virulence factor, a modified version used in animal models indicated that targeting complement is important for host adaptation (113). S. aureus also blocks complement opsonization. A secreted protein, Staphylococcus aureus binder of IgG (Sbi), has multiple functions including binding C3b, and acting to inhibit complement activation and opsonin-mediated macrophage phagocytosis (114). Similarly, the S. aureus protein, extracellular complement binding protein (Ecb) is used to inhibit C3b interactions with CR1 (115). Another role of complement activation is immune cell recruitment, where complement component C5a is a chemoattractant. S. aureus reduces phagocyte recruitment, using CHIPS, which binds to C5a (86, 87). Together, these bacterial defenses act to reduce complement-aided phagocytosis of S. aureus. The large number of virulence factors targeting complement highlights the importance of complement-mediated immunity against S. aureus.
Fc Receptors
Fc receptors on the surfaces of phagocytes bind to the Fc region of antibodies. Invading pathogens opsonized with antibodies are more readily engulfed by phagocytes. Macrophages express Fcγ receptors, which bind IgG antibodies, triggering phagocytosis (116–118). Antibodies against S. aureus are detected in human sera in both healthy individuals and patients with S. aureus infection (119). There are specific IgG antibodies against staphylococcal α-hemolysin in the human population which are present from a young age and increase in prevalence during infection (120). There are differences in IgG antibody levels present dependent on S. aureus colonization of individuals, with colonization associated with higher IgG antibody titers (121).
S. aureus expresses virulence factors which inhibit antibody-mediated phagocytosis. Protein A (SpA) and Sbi interact with the Fc region of human IgG antibodies (122, 123). This inhibits the normal ability of the Fc region of IgG to bind to Fc receptors on phagocyte membranes, which has been thought to hide S. aureus from antibody-mediated phagocytosis. Despite this, it has been demonstrated that S. aureus strains with more protein A, and therefore more bound IgG, were not phagocytosed less by alveolar macrophages in mice (124). Furthermore, phagocytosis by neutrophils was actually higher for clinical strains with greater IgG binding than for commensal strains (125). These unexpected results could be due to differences between strains, or may be due to the lack of significant changes in the rate of phagocytosis caused by opsonin (126), suggesting that antibody opsonization is not essential for adequate S. aureus phagocytosis. Another virulence factor S. aureus uses to target antibodies is staphylokinase (SAK), which triggers degradation of IgG, as well as C3b on the bacterial cell surface (127). Since S. aureus has multiple strategies to target antibodies, it is likely beneficial for the bacteria to inhibit antibody binding, although whether this is to specifically protect against antibody mediated-phagocytosis is unclear.
Collectively, the presence of scavenger, complement and Fc receptors gives phagocytes their unique phagocytic capabilities. For example, if an Fc receptor is expressed on a non-phagocytic cell, that cell gains the ability to phagocytose in a similar manner to phagocytes (128). Many studies examine individual receptors in isolation to simplify their characterization, however it is important to note that in reality, all these receptors work simultaneously together to coordinate phagocytic engulfment of targets, including S. aureus.
Macrophage Functional Changes in Response to S. aureus
Upon interaction with S. aureus, macrophages may become activated and create a positive immune response to control infection, for example, by promoting phagocytosis and releasing pro-inflammatory cytokines. However, in some cases macrophage responses may be manipulated by S. aureus, leading to ineffective or even detrimental host responses (Figure 1). Macrophages respond to stimuli such as cytokines in their local environment which alter macrophage functions. Under homeostatic conditions, tissue macrophages are efficient at tissue repair and healing, often characterized as ‘M2’, with increased arginase metabolism (129, 130). In response to danger, for example infection, macrophages can become pro-inflammatory and efficient at pathogen killing, often characterized as ‘M1’ with enhanced nitric oxide (NO) production (130).
The M1 (pro-inflammatory) and M2 (anti-inflammatory) macrophage classifications are used widely in research and are referred to in this text. However, it is important to note that the M1 and M2 characterizations are based on in vitro studies which hypothetically represent two points on a continuum upon which macrophages lie. Furthermore, M1 and M2 definitions have been inaccurately associated with classical and alternatively polarized macrophages, respectively (131). These in vitro descriptions may not always correlate to in vivo macrophage phenotypes, which varies dependent on cell origin and microenvironment, and multiple stimuli in the in vivo environment may change over time, for example during infection progression (132).
Macrophage interactions with S. aureus are dependent on the type of pro- or anti- inflammatory immune response elicited (Figure 3). Macrophages actively phagocytose planktonic (single bacterial cells) S. aureus, but are less able to phagocytose biofilm-associated bacteria (133). This has been extended to keratinocytes, where S. aureus biofilms elicit a lesser inflammatory response than planktonic bacteria (134). Furthermore, adequate abscess formation in response to S. aureus dermal mouse infection requires M1 macrophages, whereas the presence of M2 macrophages was associated with uncontrolled bacterial spread (135). Changes in macrophage polarization are due, in part, to variations in macrophage stimulation in different S. aureus infection scenarios. M1 or M2 polarization leads macrophages to respond to S. aureus differently, promoting pro- or anti- inflammatory responses, respectively (136).
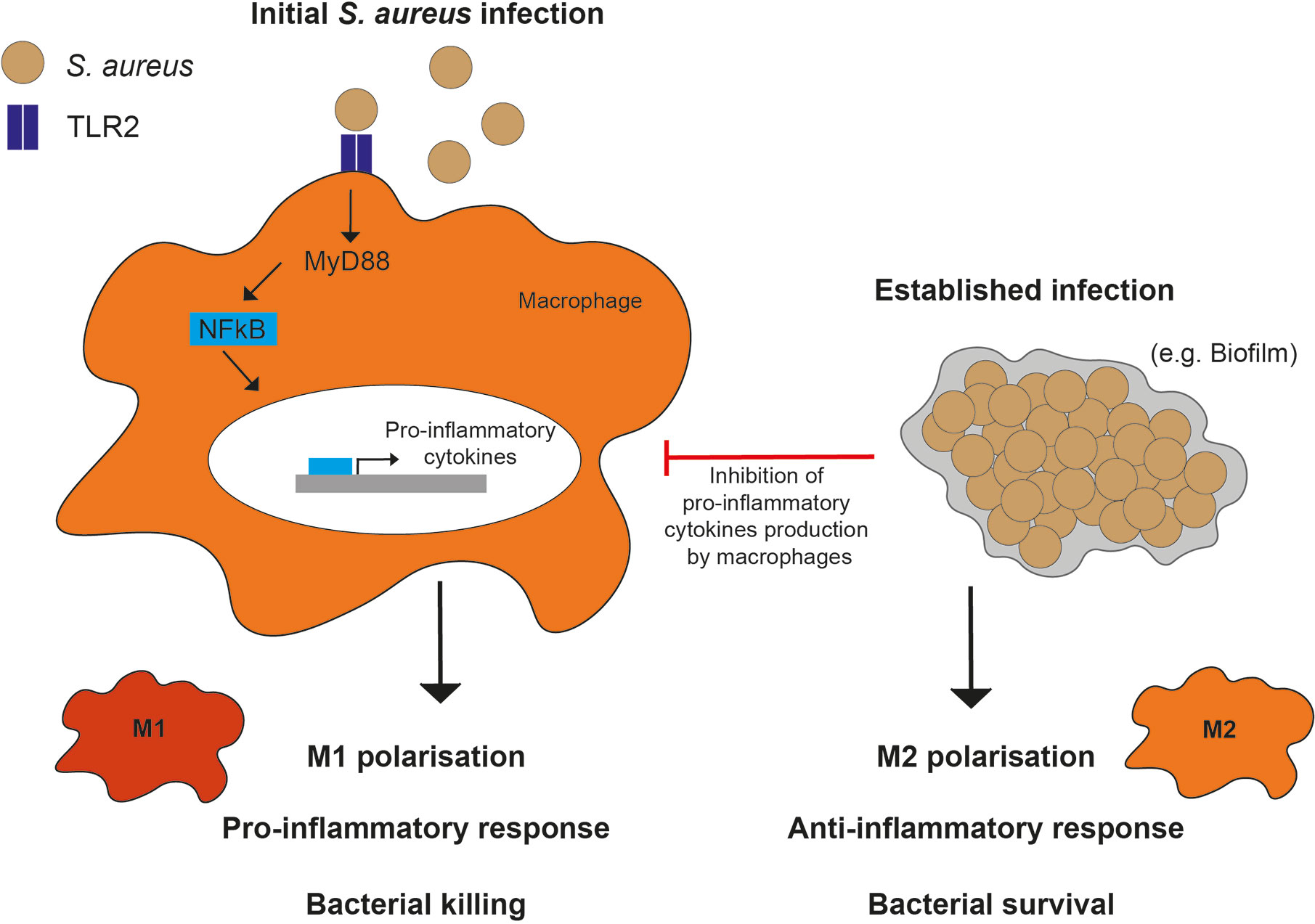
Figure 3 Macrophage polarization responses to S. aureus. M1 polarization in response to planktonic (or initial) infections, occurs through TLR2, MyD88, and NF- κB signaling resulting in a pro-inflammatory phenotype and cytokine production. In comparison, M2 polarization in response to established infections, such as biofilms, occurs through inhibition of macrophage pro-inflammatory cytokine production.
Pro-inflammatory Macrophage Polarization
In some S. aureus infections, a robust pro-inflammatory macrophage response can lead to efficient phagocytosis of S. aureus after sensing bacterial components. After initial S. aureus infection, AMs undergo M1 polarization and secrete pro-inflammatory cytokines (137). M1 macrophages phagocytose and kill intracellular pathogens, generate reactive oxygen species (ROS), nitric oxide (NO) and pro-inflammatory cytokines and can express class II major histocompatibility complex molecules (MHC-II) (138). Macrophages are also capable of longer-term memory in response to recurrent S. aureus infection. In localized skin infections, prior infection reduced subsequent infection severity by priming macrophages toward pro-inflammatory phenotypes (139).
Toll-like receptors (TLRs) recognize bacterial components, signaling through MYD88 innate immune signal transduction adaptor and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) to upregulate inflammatory gene expression, including pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β. TLR2 is particularly important in S. aureus infections. In peritoneal macrophages, TLR2 recognizes S. aureus PGN, leading to both MYD88 and NF-κB signaling (140). In addition, TLR2 to detects S. aureus lipoproteins, shown in keratinocytes where it induces NF- κB activity, lipoprotein activation of TLR2 which was similarly observed in J774 macrophages, leading to pro-inflammatory cytokine production (141, 142). Loss of MYD88 from macrophages inhibited production of TNF-α after exposure to S. aureus cell wall (143). Similarly, loss of TLR2 led to reduced pro-inflammatory cytokine expression in peritoneal macrophages infected with S. aureus (144). Mice deficient in TLR2 or MYD88 have an increased susceptibility to S. aureus infection, as well as a reduction or loss of macrophage expression of pro-inflammatory cytokines TNF-α and IL-6 (140, 145). TLR2 can also be recruited to S. aureus-containing phagosomes in macrophages, initiating cytokine production following bacterial degradation (146). CD14 is a co-receptor for TLR2 and, together, they act to promote a pro-inflammatory response, including by M1 polarization of macrophages (147–149). S. aureus PGN and LTA bind to CD14 and cause TLR2-mediated activation of NF-κB in HEK cells (150). Studies on S. aureus and TLR2 signaling have mainly focused on leukocytes, but S. aureus may promote alternate inflammatory responses in other cell types. For example, LTA stimulation of TLR2 on endothelial cells may promote an anti-inflammatory response (151).
As with other aspects of the immune system, TLR2 and NF-κB signaling may be undermined in S. aureus infection. Activity of c-Jun N-terminal kinase (JNK) has been associated with TLR2 in S. aureus infection, with JNK mediating cell responses to stress. TLR2 signaling through the JNK pathway may be required for macrophage phagocytosis of S. aureus (152), however, TLR2-activated JNK signaling in response to S. aureus reduces macrophage superoxide generation and enables prolonged survival within the phagosome (153). Similarly, loss of TLR2 in infected peritoneal macrophages was associated with reduced S. aureus catalase and superoxide dismutase activity (144). S. aureus strains lacking lipoproteins can escape immune recognition by TLR2 (154). Additionally, NF-κB activation is required for macrophage phagocytosis of S. aureus, since inhibition of NF-κB blocks bacterial uptake (155). NF-κB activation is reduced in S. aureus-stimulated macrophages by activation of a macrophage receptor involved in phagocytosis of apoptotic cells (MerTK), which leads to a reduced inflammatory response to staphylococcal LTA (156). Overall, these studies indicate that pro-inflammatory mediators TLR2 and NF-κB are important in the macrophage response to S. aureus and are a target of subversion.
S. aureus has further strategies to manipulate macrophage polarization to limit pro-inflammatory responses. Protein kinase B (Akt1) signaling induced by S. aureus was shown to decrease macrophage M1 polarization, with mice deficient in Akt1 having improved bacterial clearance. Akt1-deficient macrophages have increased pro-inflammatory cytokine expression and NF-κB activity (157). S. aureus induction of macrophage polarization is also modulated by microRNAs. MicroRNA-155 is involved in Akt1-mediated macrophage polarization (157), while microRNA-24, a regulator of macrophage polarization, has reduced expression during S. aureus infection (158).
Anti-inflammatory Macrophage Polarization
In certain S. aureus infections, for example in established biofilm infections, an anti-inflammatory response occurs, promoting continued bacterial survival within the host. M2 polarized macrophages and reduced phagocytosis are found in chronic rhinosinusitis, a condition associated with S. aureus colonization (159). In mice, S. aureus biofilms prevent phagocytosis by macrophages, as well as reduce inflammation through attenuation of pro-inflammatory host responses, favoring an M2 macrophage phenotype (133). In a rat S. aureus biofilm periprosthetic joint infection model, an increase in the number of M2 macrophages is observed (160). Additionally, AMs are more likely to become an M2 phenotype in S. aureus infections at later time-points in infection (137). Together these reports suggest that established S. aureus infections promote M2 polarization.
Antibodies may facilitate S. aureus-mediated M2 polarization in chronic rhinosinusitis, whereby bacterial virulence factors cause an increased production of IgE, which in turn promotes M2 polarization (159, 161). Furthermore, biofilm secretion of cyclic di-AMP promotes anti-inflammatory cytokine release from macrophages (162), and S. aureus virulence factor secretion from biofilms reduces macrophage phagocytosis (163). S. aureus expresses clumping factor A (ClfA) to reduce phagocytosis and subsequent pro-inflammatory response, and this is suggested to be due to immuno-modulation (164).
TLR2, MYD88 and NF-κB signaling are also involved in S. aureus biofilm infections and are targeted by S. aureus to manipulate the macrophage response. In early control of cranial biofilm infection spread, TLR2 is associated with macrophage IL-1β pro-inflammatory cytokine production, but this signaling was insufficient to clear infection (165), perhaps due to established infection manipulation of the macrophage response. Interestingly, addition of IL1-β to led to increased bacterial growth of biofilm, but not planktonic, S. aureus, suggesting that biofilms react to host cytokines to promote survival (166). Catheter-associated biofilm infections in MYD88-deficient mice have increased bacterial burden and dissemination, reduced expression of pro-inflammatory cytokines, and an increased number of M2 macrophages (167). This knowledge has led to production of biofilm treatments which promote a M1, rather than M2 macrophage polarization. Addition of M1 macrophages to the site of an in vivo biofilm, led to reduced bacterial burden (168). Remarkably, a therapeutic approach which promotes pro-inflammatory monocyte polarization lead to clearance of established biofilms in mice (169).
Cytokines in Macrophage Polarization
Macrophages are able to sense cytokines released in the local environment, including cytokines released by nearby activated macrophages in response to S. aureus infection. Following binding of cytokines to receptors, the action of the Janus kinase (JAK) and signal transducers and activators of transcription (STAT) signaling pathway mediate transcriptional changes (170). The JAK/STAT pathway is important for activation of macrophages, induction of inflammatory responses, and inhibition of apoptosis.
Exposure of MDMs to S. aureus alters the expression of 624 genes, with JAK/STAT signaling changed in early infection (171). JAK/STAT signaling is induced by PGN, leading to phagosome maturation in macrophages containing S. aureus (172). Interestingly, in murine influenza and MRSA co-infection, STAT2 is important in macrophage polarization, where STAT2-deficient mice had improved bacterial burden, potentially caused by an increased number of M1 macrophages (173). Human MDMs with an established S. aureus infection harbored viable intracellular bacteria within vesicles, however, MDM apoptosis or necrosis was not observed until S. aureus escaped to the cytosol (18). Further to this, addition of isolated S. aureus PGN can increase anti-apoptotic signals in infected macrophages, likely through the JAK/STAT and NF-κB signaling pathways (174). Macrophages which have phagocytosed S. aureus have increased expression of anti-apoptotic genes, enabling continued intracellular bacterial survival (175). To induce this, S. aureus upregulates macrophage myeloid cell leukemia-1 (MCL-1) expression, an anti-apoptotic gene which enhances anti-inflammatory cytokine release (176). In contrast to these macrophage studies, the presence of S. aureus increases apoptosis in neutrophils (177). Therefore, macrophages may be a prime target for subversion and intracellular persistence.
Interferon-beta (IFN-β) is a cytokine with roles in antimicrobial defense of infected cells, as well as innate and adaptive immunity (178). S. aureus can induce a strong IFN-β response in airway infection models, where protein A stimulates IFN-β production, likely via TLR9 or NOD2 signaling (179, 180). However, dependent on the S. aureus strain used, there is diversity in the IFN response induced (181). Following other routes of infection, S. aureus induces variable IFN-β production by macrophages, though IFN-β production or treatment has been shown to be beneficial for the host during S. aureus infection. S. aureus resistance to macrophage degradation causes the reduced IFN-β production, which is lower than that induced by comparable pathogens (182). This suggests a lack of sufficient IFN-β induction is detrimental to the host. IFN-β production by macrophages is inhibited by TLR2 signaling during S. aureus infection. TLR8, an intracellular TLR, senses S. aureus RNA in infected macrophages and monocytes, leading to IFN-β production via MYD88 signaling (183). TLR2 is a key sensor of S. aureus and therefore the antagonistic role of TLR2 and TLR8 signaling may ultimately reduce macrophage IFN-β production.
IL-1β is another pro-inflammatory cytokine with important roles in controlling S. aureus infection. In a brain abscess S. aureus infection, mice deficient in IL-1β (or TNF-α) were subject to significantly enhanced mortality and greater bacterial burden when compared to wild-type mice (82). In a sub-cutaneous model, mice deficient in MYD88 or IL-1R had significantly bigger lesions and bacterial burden, with IL-1R activation required for neutrophil recruitment to S. aureus-infected sites (184). Similarly, mice deficient in IL-1β had larger lesion size, greater colony forming units (CFUs) and reduced neutrophil attraction following in vivo cutaneous challenge (185). Supplementation of IL-1β KO mice with recombinant IL-1β restored the mice’s ability to control infection and clear S. aureus (185). In contrast, in murine airway S. aureus infection, IL1-β is associated with immunopathology (186), and addition of recombinant IL- β reduced bacterial clearance (187). Interestingly, activated platelets which release IL-1β act to enhance macrophage phagocytosis and killing of S. aureus, suggesting both platelets and IL1-β have an important role in the phagocyte response (188).
Mechanisms Used by Macrophages to Kill S. aureus
Once macrophages are activated, have located and phagocytosed S. aureus, the macrophage’s powerful degradative processes are used to kill the bacteria. Macrophages have a range of mechanisms to destroy phagocytosed pathogens (Figure 4), including release of reactive oxygen species (ROS), reactive nitrogen species (RNS), enzymes and antimicrobial peptides, as well as acidification of the phagolysosome, nutrient restriction, and autophagy. In addition, macrophages can target extracellular bacteria with extracellular traps.
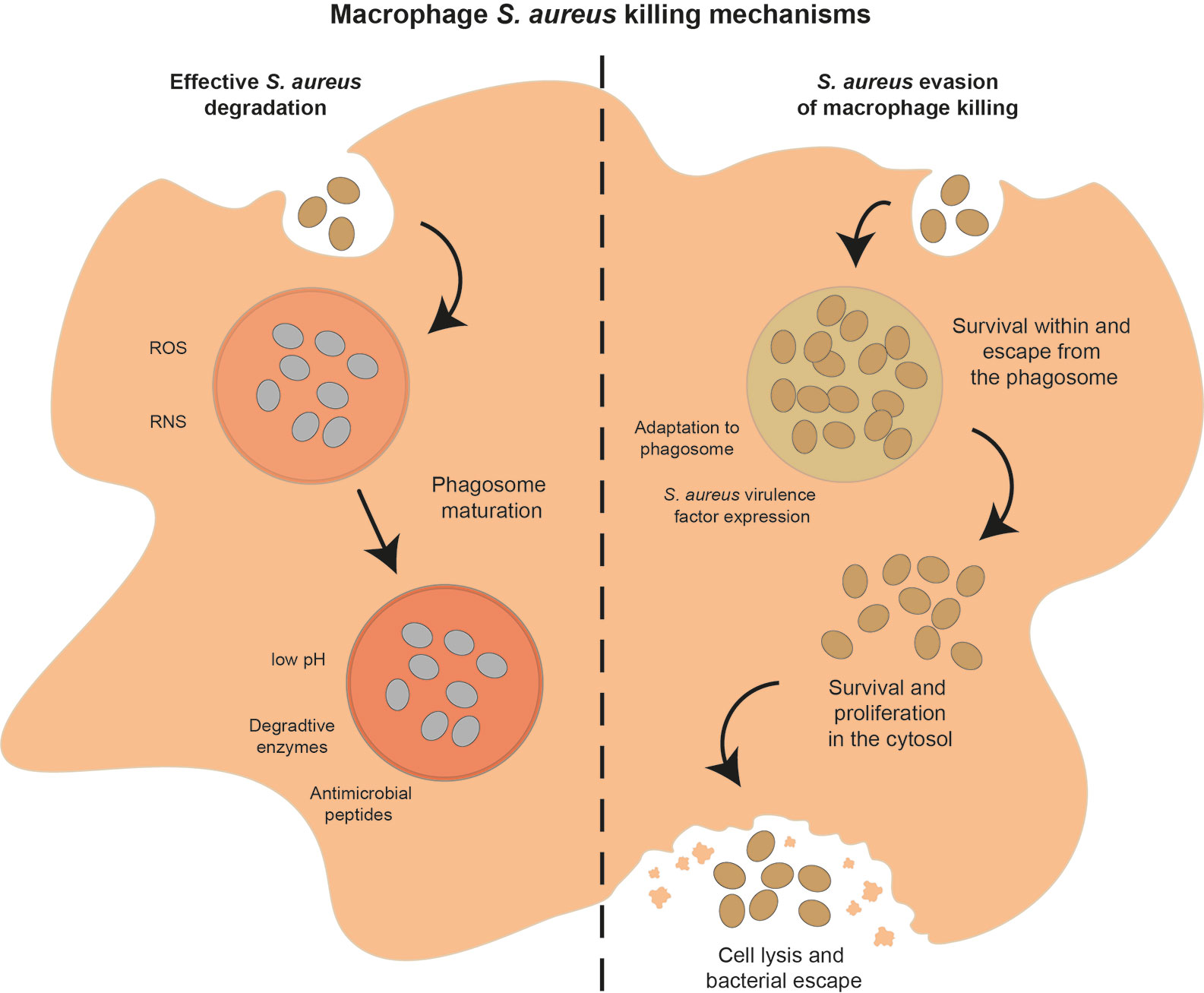
Figure 4 Potential outcomes of the interaction between macrophages and S. aureus. After phagocytosis, macrophages can successfully control and degrade S. aureus (left hand side of figure) using a range of mechanisms, including ROS and RNS soon after phagocytosis, phagosome acidification, nutrient restriction, release of degradative enzymes and AMPs as the phagosome matures. Alternatively, S. aureus can evade macrophage killing mechanisms (right hand side of figure) by adapting to the phagosome environment, expressing a range of virulence factors, or escape from the phagosome and survival in the cytosol, leading eventually to macrophage cell lysis and S. aureus dissemination.
Macrophage Production of ROS and RNS
NADPH oxidase (NOX2) is an enzyme located on the phagosome membrane, assembly of the oxidase is induced which then allows it to catalyze superoxide production and subsequent ROS, termed the oxidative burst. Superoxide can be converted into a variety of different ROS (see Figure 5), all of which are toxic to some degree. ROS production is considered the key killing mechanism for both macrophages and neutrophils (189), and is important for clearance of S. aureus (61, 190).
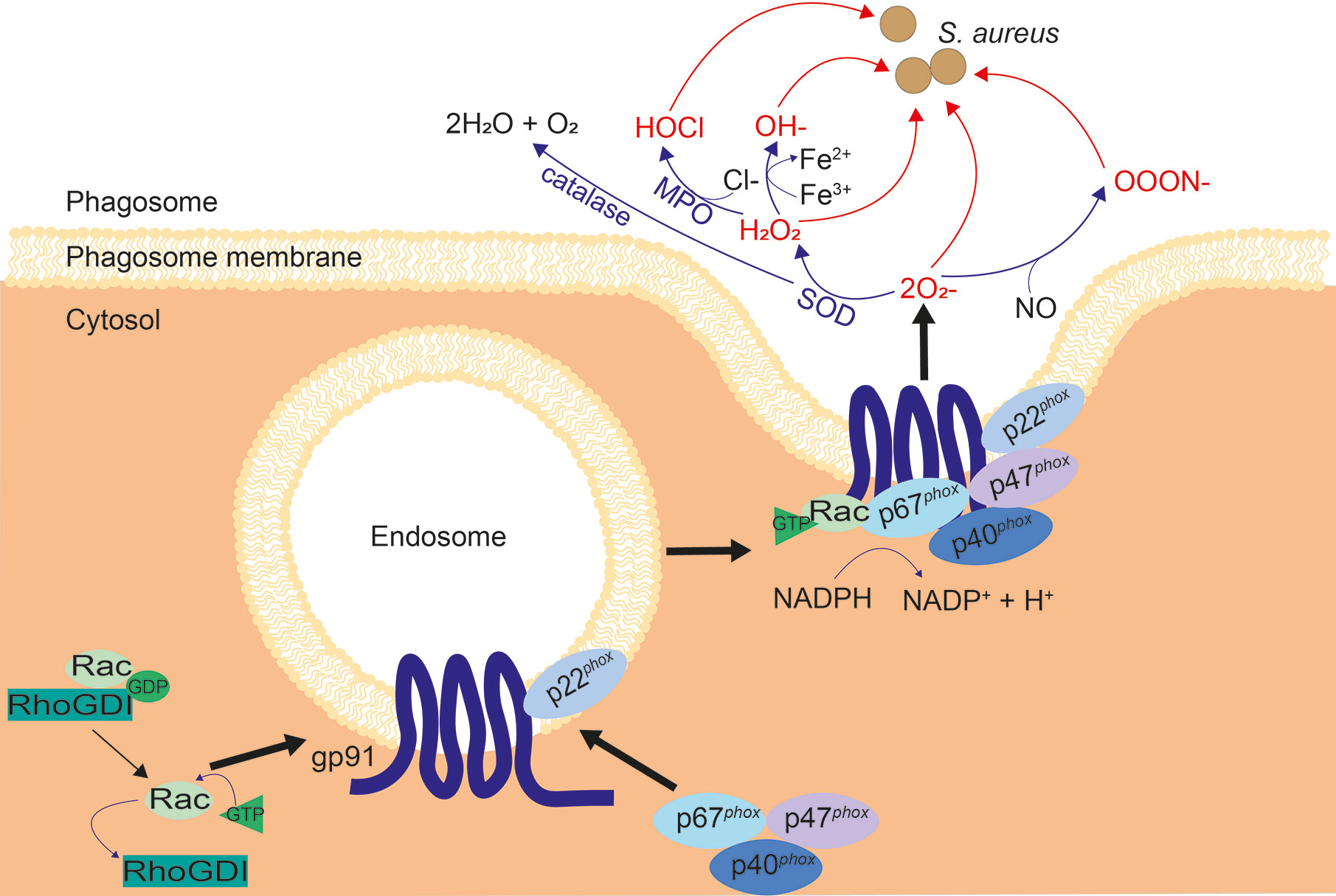
Figure 5 Assembly of NOX2 and subsequent ROS cascade. When inactive, NOX2 components gp91phox and p22phox are located on vesicles, while inactive Rac and p67phox, p47phox and p40phox exist in the cytosol. Upon activation, the cytosolic subunits are localized to phagocytic cups on the endosome membrane, to bind to gp91 and p22phox. Inhibition of Rac by RhoGDI is reversed, allowing GTP binding and recruitment of Rac to the NOX2 complex. The NOX2 vesicle merges with the membrane of the phagosome and produces superoxide. Superoxide is converted to other ROS: H2O2 by superoxide dismutase (SOD) and OOON− by interaction with nitric oxide (NO). H2O2 is converted into HOCl by myeloperoxidase (MPO), OH− by the Fenton reaction, and H2O + O2 by catalase, as shown.
NOX2 is activated by signals from phagocytic receptors, such as FcγR and macrophage-1 antigen (Mac-1) (22, 191), resulting in electron transfer from reduced NADPH in the cytosol to phagosomal oxygen. Ras-related C3 botulinum toxin substrate (Rac), a small GTPase, is necessary for best operation of NOX2 (192, 193). Rho GDP-dissociation inhibitor (RhoGDI) inhibits Rac, stabilizing the active, GDP-bound form until inhibition is reversed upon NOX2 activation (194, 195). NOX2 has 5 main components (see Figure 5), two of which are membrane-spanning: gp91phox and p22phox, while three are cytosolic: p40phox, p47phox, and p67phox (196). Gp91phox and p22phox are located on Rab11-positive recycling endosomes, Rab5-positive early endosomes and the plasma membrane (197). When NOX2 is activated, the three cytosolic components are recruited to bind gp91phox and p22phox at the vesicle membrane via phagocytic cups, Rac recruits GTP and binds to p67phox, and the NOX2 machinery fuses with the nascent phagosomal membrane, producing superoxide. Superoxide production occurs almost immediately, even before phagosomes are sealed, implying that the NOX2 assembly is fast (198, 199).
Although superoxide itself is able to destroy bacteria, it is extremely volatile and degrades into hydrogen peroxide (H2O2), or interacts with nitric oxide (NO) to produce peroxynitrite (ONOO−) (189, 200, 201) (Figure 5). When iron or other catalytic metals are present in the phagolysosome, possibly due to release from phagosomal proteins, H2O2 and can react to form a hydroxyl radical (OH−); a process known as the Fenton reaction (202–204). Myeloperoxidase (MPO) is the enzyme that catalyzes hypochlorous acid (HOCl) formation from H2O2 and chloride. This is abundant in neutrophils, although other phagocytes including macrophages express it (201, 205). Hypochlorous acid is thought to contribute to microbicidal activity induced by H2O2, however, hypochlorous acid is not critical for antimicrobial activity. This is demonstrated by the fact that patients deficient in MPO have similar susceptibilities to bacterial infections as healthy individuals (61, 206, 207). Chronic granulomatous disease (CGD) patients have mutations in one of the subunits of NOX2, resulting in an inability to make ROS. CGD patients are significantly more susceptible to S. aureus infection (208). CGD is most commonly due to defects in the genes for gp91phox or p47phox, with only 5% of CGD cases due to mutations in genes coding for p22phox, p40phox and p67phox (208–214). Macrophages are implicated in CGD bacterial diseases, since a characteristic of CGD is hepatic abscesses, suggesting the importance of Kupffer cells in control of microbes (215).
In addition to ROS, phagocytes produce RNS. Production of NO radicals is catalyzed by inducible nitric oxide synthase (iNOS) (216). iNOS is only expressed in response to inflammatory stimuli, with IFN-γ being the key cytokine required for iNOS induction in macrophages (216, 217). Upon reaction of NO with superoxide, peroxynitrite is formed, which is toxic to phagocytosed microbes’ proteins and DNA (218, 219). However, mice deficient in iNOS do not suffer a significant increase in intracellular S. aureus upon infection, while mice with NOX2 deleted (cybb−/−) had significantly increased intracellular burden and hence greater mortality (61, 220, 221). This underlines the importance of NOX2 in defense against S. aureus.
There is limited data on the concentration of ROS within the phagosome, much of it relating to neutrophils. Macrophage oxidative burst peaks approximately 30 min post-phagocytosis, although it is maintained for over 60 min (222, 223). In the neutrophil phagosome, the concentrations of and H2O2 are estimated to be 25 and 2 µM, respectively. However, in the absence of MPO, these concentrations are higher: over 100 µM and 30 µM, respectively (224). This is important, due to macrophages possessing lower concentrations of MPO than neutrophils (205). Macrophages have been estimated to produce 50 µM of and 1 to 4 µM H2O2 at neutral pH (225). These concentrations were determined using computer modeling to approximate the speed NOX2 can produce , the volume of the phagosome, the rate of spontaneous dismutation into H2O2, and the frequency of H2O2 diffusion across the phagosome membrane into the cytoplasm (224, 225). There are margins for error at each stage of these calculations, particularly as this assumes homogeneity within the phagosome. In vitro measurement of macrophage ROS may be more accurate. However, as these concentrations of ROS are very small and the oxidative burst occurs rapidly, there are difficulties in accurately measuring this.
ROS are also produced by the mitochondria. Mitochondrial ROS (mROS) are, in most cases, the by-product of oxidative phosphorylation. However, more recent studies have demonstrated mROS act as a microbial defense mechanism within macrophages (226, 227). When macrophages were treated with histone deacetylase inhibitors (to test possible downregulation of host immune responses) alongside infection with either Salmonella or E. coli, intracellular bacterial clearance was enhanced via upregulation of mitochondrial ROS, an effect which was reversible upon inhibition of mitochondrial function (226). Furthermore, signaling through Toll-like receptors, specifically TLR1, TLR2, and TLR4, in macrophages leads to recruitment of mitochondria to the phagosome and an alteration in mROS (227, 228). Additionally, when mitochondria were induced to express catalase, Salmonella clearance was decreased (227). Likewise, infection of macrophages with S. aureus triggered production of mROS, primarily H2O2, which was delivered to the bacterial-containing phagosome by mitochondria-derived vesicles, contributing to bacterial killing (228). This was determined to be induced by endoplasmic reticulum stress, dependent on TLR signaling and mitochondrial superoxide dismutase 2 (228). Moreover, mitochondria associate to the membrane of S. aureus-containing macrophage phagosomes to increase mROS production and activate caspase-1, leading to acidification of the phagosome. However, expression of alpha-hemolysin by S. aureus was able to counteract these effects (229). Furthermore, S. aureus counteracts recruitment of mitochondria to the macrophage phagosome membrane in a caspase-11–dependent manner, with caspase-11 deletion in mice enabling mitochondrial association with S. aureus vacuoles, increased mROS and improved bacterial clearance (230).
S. aureus Response to ROS
ROS can damage biomolecules including essential enzymes and DNA (225). However, bacteria have evolved mechanisms to withstand ROS and RNS. Staphylococcal peroxidase inhibitor (SPIN) is secreted by S. aureus. SPIN attaches to and incapacitates human MPO (231). Structural analysis revealed that SPIN acts as a “molecular plug”, occupying the active site of MPO and thus refusing entry to the H2O2 substrate (231). Expression of SPIN was maximal within a phagosome, which is the location of MPO, and S. aureus mutants deficient in SPIN have reduced survival following phagocytosis when compared to wild-type S. aureus (231), this was demonstrated with neutrophils but is likely to occur in macrophages.
S. aureus possesses two superoxide dismutases which incapacitate superoxide radicals, superoxide dismutase A (SodA) and superoxide dismutase M (SodM) (232). Some studies have identified SodA and SodM as important for S. aureus virulence (232, 233), while others show only marginal effects (234, 235). Manganese ions act as a co-factor for SodA and SodM, upregulating superoxide dismutase activity without affecting transcription, and S. aureus is more susceptible to manganese starvation in the absence of these proteins (232, 236). SodA is valuable in resisting superoxide stress in the presence of manganese, while SodM is crucial when in manganese-scarce environments (236). SodM is not present in other staphylococci, and this role of inhibiting host ROS during manganese restriction may explain why S. aureus has acquired a second superoxide dismutase.
Resistance to oxidative stress in S. aureus is mediated, in part, by transcriptional regulators. Peroxide regulator (PerR) is an important regulator which controls a regulon of many antioxidant genes. In particular, alkylhydroperoxide reductase (AhpC) and catalase (KatA) are involved in resisting peroxides and H2O2 respectively (237). The genes encoding these two proteins are regulated in a compensatory manner: mutation in ahpC enhanced (rather than reduced) H2O2 resistance, as katA is upregulated by removal of PerR repression (237). AhpC was similarly able to compensate for katA mutation. Deletion of both katA and ahpC caused a significant growth defect, with S. aureus unable to remove intra- or extracellular H2O2, meaning H2O2 accumulated to toxic levels in the media (237). S. aureus mutants lacking two component regulator staphylococcal respiratory response AB (SrrAB) were more susceptible to H2O2, with katA and ahpC transcriptionally downregulated (238). Susceptibility to H2O2 was reversed by iron sequestration or perR repressor gene deletion (238). Another study showed that the msaABCR operon of S. aureus regulates expression of genes involved in oxidative stress (239). Staphyloxanthin, a carotenoid pigment, is a strong antioxidant which is regulated by cold shock protein (CspA), alongside the organic hydroperoxide resistance gene which defends specifically against oxidative stress caused by organic hydroperoxides. This implies involvement of ROS resistance genes in persistence of S. aureus (239).
A transposon screen found there were five S. aureus regulons which are crucial for NO resistance (240). Flavohemoglobin (Hmp) is necessary for resistance to NO in some bacterial species, because it acts as a denitrosylase, removing NO (241). This is strictly controlled, as Hmp expression in the absence of NO leads to enhanced oxidative stress (242). Nitrite-sensitive repressor (NsrR), is the NO-sensing transcriptional regulator of Hmp used by many bacteria to detect and react to NO (242–245). S. aureus does not possess NsrR, instead, the two-component regulator SrrAB controls Hmp (243). Additionally, modifications to S. aureus metabolism may increase bacterial NO resistance. Infection of RAW 264.7 cells with a S. aureus TCA cycle mutant had reduced NO production and iNOS activity when compared to wild-type S. aureus (246).
S. aureus can take advantage of host signaling in order to escape oxidative killing. In wild-type mice, S. aureus phagocytosis by macrophages led to JNK activation in a TLR2-dependent manner; JNK activation caused inhibition in superoxide production, impairing the ROS cascade and prolonging survival of the bacteria. When TLR2-deficient mice were used, the macrophages were more able to readily kill S. aureus (153). TLR2 expression is higher in S. aureus-infected macrophages (144), and S. aureus were more able to escape killing by peritoneal macrophages when anti-TLR2 antibodies were used (247).
S. aureus produces lipoic acid, which also restricts ROS and RNS production by macrophages, to enhance bacterial survival (248). Lipoic acid is a metabolic cofactor which is synthesized by the lipoic acid synthetase (LipA), which limits macrophage activation by reducing TLR1 and TLR2 activation by bacterial products (249). A S. aureus lipA deletion mutant caused significantly more TLR2-dependent pro-inflammatory cytokine production (249). Exogenous lipoic acid can reduce neutrophil oxidative burst through radical binding as well as recycling antioxidants, inhibiting NF-κB transport into the nucleus, and reducing production of inflammatory cytokines (250–254). Macrophages which were recruited to the site of infection with the lipA mutant produced significantly greater amounts of ROS and RNS than those attracted to sites infected with wild-type S. aureus (248); in this case, ROS and RNS (but not mitochondrial ROS) were important for controlling S. aureus lipA infection (248). This suggests that lipoic acid production by S. aureus promotes persistence of the bacteria.
Macrophage Phagosomal Acidification
Acidification of the phagosome is another key mechanism involved in killing phagocytosed bacteria. A low phagosomal pH may directly affect S. aureus survival, since bacterial growth is reduced at pH 4.5 (255). Additionally, acidification has an important impact on phagosomal enzymes, for example cathepsins, which have optimal efficacy at low pH. Phagosomal enzymes are discussed in detail in the enzymes section below.
Macrophage phagosome acidification is generated by an influx of protons (H+) into the phagosome by vacuolar-type proton transporting ATPase (v-ATPase), which is present in phagosome membranes (256). The action of v-ATPase reduces the pH of endosomes and lysosomes to ~6 and ~4.5, respectively (257). Fusion of endosomes and lysosomes, which are enriched with v-ATPase, is an important part of phagosome maturation, the continued delivery of v-ATPase causes increasing acidification throughout sequential stages of phagosome maturation (258). In addition to this, the permeability of the phagosome to protons is important in maintenance of low pH, therefore as phagosomes mature, proton permeability is decreased to preserve acidification (259). However, phagosome acidification commences before lysosomal fusion events occur, demonstrating that v-ATPase is also present at an earlier stage is phagosome maturation (256). Indeed, v-ATPase is found on the plasma membrane of phagocytes where it is used to maintain cytosolic pH (260, 261). The v-ATPase present in plasma membranes are likely internalized during phagocytosis and responsible for acidification at very early timepoints of phagocytosis, with additional v-ATPase delivered during phagosomal maturation leading to increased acidification.
Phagosomal acidification is well documented in S. aureus infection. In S. aureus-infected murine peritoneal macrophages, the phagosomal pH is reduced to 5.7 to 6 within 6 to 8 min of infection, and this is dependent on v-ATPase (256, 259). Indeed, the average phagolysosomal pH of RAW 264.7 cells infected with S. aureus was measured as 5.43 12 hours post-infection (262). S. aureus phagocytosed by Kupffer cells is trafficked to an acidified phagosome, as demonstrated in intravital imaging of murine infections (263). Another study shows that S. aureus peptidoglycan can induce macrophage phagosome maturation through JAK-STAT signaling (172). Low pH is also important in efficiently killing S. aureus in neutrophils (264). Non-professional phagocytes, including epithelial cells and endothelial cells, are also shown to traffic S. aureus to an acidic phagosome (19, 20, 265, 266). Phagosome maturation proteins are involved in S. aureus degradation. For example, copper metabolism gene MURR1 domain (COMMD) proteins regulate both intracellular trafficking and transcription factors. Kupffer cells effectively kill S. aureus, where phagosomes mature in a COMMD10-dependent manner, required for phagosome acidification and optimal bacterial killing (267).
S. aureus can adapt to the acidic phagosome, with recent studies suggesting that exposure to acidification may even promote intracellular bacterial survival. S. aureus can survive and replicate within mature acidic phagosomes, as demonstrated using murine macrophages and human MDMs (16). S. aureus can survive and replicate within murine AMs, and inhibiting phagosome acidification caused a small drop in bacterial survival (268). Similarly, THP-1 cells were also used to show that inhibiting phagosome acidification reduced S. aureus survival, where exposure to low pH was shown to induce virulence factor expression (269). THP-1 cells which are deficient in phagosomal acidification had improved bacterial killing of S. aureus strain USA300, although not the Newman strain (270). Phagosomal acidification has even been proposed to be requisite for S. aureus intracellular survival, the bacterial GraXRS regulatory system is used to sense low pH, where S. aureus promotes adaptive responses enabling bacterial growth within the phagosome, shown to be required for bacterial survival within murine Kupffer cells in vivo (262).
Other studies show that macrophages with phagosomes containing S. aureus do not acidify appropriately. Reduced acidification of the phagosome was observed in THP-1 cells when infected with S. aureus, in comparison to E. coli or S. pneumoniae, and the authors suggest that reduced acidification may precede bacterial escape (73). S. aureus has also been shown to reside within non-acidified vesicles in epithelial cells (271). Presence of other material in the phagosome with S. aureus reduced the acidification of Kupffer cell phagosomes, promoting S. aureus survival (272). Whether phagosomes containing S. aureus properly acidify, leading to beneficial or detrimental effects on the host, likely depends on multiple factors; cell types, bacterial strains, timepoints and phagosomal markers studied, as well as the antagonistic roles of ROS production and proton influx discussed below.
There is evidence that the actions of phagosomal NOX2 and v-ATPase are antagonistic. In the early stages of phagosomal maturation, ROS production by NOX2 may buffer acidification through rapid consumption of protons. The oxidative burst is therefore intrinsically linked to phagolysosome acidification (73, 273), and, as such, oxidation can delay phagosomal maturation (223). In neutrophils, NOX2-dependent reduction of phagosome acidification is caused by proton consumption, as well as decreased v-ATPase recruitment to the phagosome and increased membrane permeability to protons (274). Caspase-1 limits the antagonism of NOX2 and v-ATPase in macrophages infected with S. aureus by regulating NOX2 activity (through cleavage of NOX2 components) to promote phagosomal acidification (275). Interestingly, phagosomes of pro-inflammatory M1-like human macrophages acidify less in comparison to anti-inflammatory M2-like macrophages, due to sustained NOX2 retention on the phagosome and associated proton consumption by the ROS produced (223). Since proteolytic enzymes are less functional at higher pH, the antagonistic effects of NOX2 activity on pH may reduce the degradative capacity of the phagosome. It has been hypothesized this ensures ROS-mediated destruction of microbes before subsequent degradation of microbial products (22). Antigen presenting cells present antigens to the adaptive immune system. In macrophages and dendritic cells, increased NOX2 activity is associated with reduced proteolysis (273, 276–278), meaning antigens are retained longer for improved presentation to adaptive immune cells (279). There appear to be multiple mechanisms causing NOX2 and v-ATPase antagonism, which differ between cell types, likely due to their different roles. As limited studies use macrophages, which have important roles in antigen presentation, there remain many unanswered questions.
The Role of Macrophage Enzymes in Controlling S. aureus Infection
Mature phagosomes may contain hydrolytic enzymes that kill bacteria efficiently. These include proteases, lipases, phosphatases and glycosidases. These enzymes have optimal efficacy in acidic conditions (280, 281). The acidification of mature phagosomes is discussed above.
The phagosome of macrophages can contain lysozyme, which is an enzyme that cleaves bacterial peptidoglycan. S. aureus is resistant to lysozyme due to acetylation of PGN by O-acetyltransferase (OatA) (282). PGN acetylation may also reduce activation of the NLRP3 inflammasome, avoiding induction of IL-1β (283). IL-1β is produced by phagocytes in response to inflammasome activation and is a key weapon in the arsenal of the immune system against S. aureus (82). The NLRP3 inflammasome is activated by exposure to phagocytosed PGN (283). In order to trigger this response, PGN must be partially digested by lysozyme. Thus, the ability of OatA to induce resistance to lysozyme suppresses activation of the NLRP3 inflammasome and subsequent IL-1β induction, demonstrated both in vitro and in vivo (282, 283). Underlining the importance of the inflammasome, mice deficient in inflammasome component apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), failed to induce IL-1β expression and suffered increased lesion size, increased CFUs and decreased neutrophil attraction upon challenge with S. aureus (185). Interestingly, the route of S. aureus infection influences the role of the inflammasome. S. aureus is known to commandeer the NLRP3 inflammasome during lung infection to aggravate pathology (284), while inflammasome activation during skin and soft tissue infection leads to clearance of the bacteria (285). For further discussion of inflammasome involvement in S. aureus infection, see (286).
Cathepsins are proteases found in the lysosomal compartment which are highly expressed in macrophages. Cathepsin D-deficient mice were more susceptible to infection with intracellular pathogen Listeria monocytogenes, which survived phagosomal killing significantly more than in wild-type mice (287). Cathepsin D is thought to act by degrading secreted bacterial virulence factors (287). Cathepsin G secreted from neutrophils damages S. aureus biofilms (288). Cathepsins have been shown to be involved in macrophage S. aureus engulfment and killing, with cathepsin L indicated as an inducer of non-oxidative killing, and cathepsin K important in induction of IL-6 production (289). The method of cathepsin-mediated S. aureus killing is thought to be direct proteolytic damage (289). In addition to modulating IL-6 production from macrophages, cathepsins can also influence IL-1β production (290, 291). This has been demonstrated in a bone marrow-derived macrophage model of Mycobacterium tuberculosis infection, whereby cathepsin release was critical for inflammasome activation and IL-1β production (292), providing evidence that this may be a common mechanism to control intracellular bacteria.
A further example of macrophage antimicrobial enzymes is phospholipases, which influence immunomodulatory compounds and attack the membrane of microbes. For example, the group IIA secreted phospholipase A2 (IIA-sPLA2) has strong antimicrobial activity against bacteria, especially Gram-positives (293, 294). IIA-sPLA2 mediates S. aureus cell membrane and cell wall damage, leading to bacterial cell death (293). Specifically, IIA-sPLA2 targets phosphatidylglycerol in the bacterial cell membrane, with the strong positive charge of PLA2 binding efficiently to the negative charge of bacteria (295). A S. aureus mutant deficient in wall teichoic acid (WTA) was around 100-fold more resistant to IIA-sPLA2 killing, likely caused by reduced access to the cell surface for PLA2 binding (296). Interestingly, one study found that S. aureus degradation was only successful when IIA-sPLA2 was accompanied by neutrophil NOX2 activity, independent of MPO (297). Since macrophages, unlike neutrophils, produce IIA-sPLA2, these complementary oxygen-dependent and -independent killing mechanisms may play a role in macrophage-mediated S. aureus degradation.
Antimicrobial Peptides in Macrophage Defense Against S. aureus
Antimicrobial peptides (AMP) tend to be positively charged and damage the membrane of pathogens. In order to defend itself against AMPs, S. aureus modifies its cell membrane by reducing the negative charge to repel cationic AMPs, thus minimizing electrostatic interactions. Negatively-charged lipids in the cytoplasmic membrane have positively-charged lysine added to them, catalyzed by enzyme multiple peptide resistance factor (MprF), with a similar effect carried out by addition of D-alanine onto cell WTAs, produced by the dlt operon gene products (298–300). S. aureus which have accumulated extra copies of the dlt operon possess teichoic acids with more D-alanine, and hence a greater positive surface charge and lesser susceptibility to binding and damage by cationic AMPs (300). Mutants that are more susceptible to AMPs display teichoic acids that lack D-alanine, when compared to wild-type bacteria (300, 301), meaning they were more attractive to cationic AMPs, including human defensin HNP1-3 (300). Through this mechanism, MprF was found to enable resistance to defensins and protegrins (299). S. aureus with an MprF deficiency were significantly attenuated in mice and killed with considerably more efficiency by human neutrophils, as well as displaying an inability to grow within macrophages (262, 299). S. aureus has also been shown to counteract the activities of AMPs by integrating lysyl-phosphatidylglycerol in S. aureus cell membranes, and expressing the AMP transporter VraFG, which promotes resistance to cationic AMPs (301).
AMP hepcidin is released by macrophages (and neutrophils) in vitro and in vivo upon microbial detection via TLR-4 in order to limit iron availability (302). Furthermore, cytokines including TNF-α, IFN-γ, IL-1, and IL-6 also induce iron modulation (303–309). For example, upon detection of bacteria, IL-6 is stimulated to directly induce expression of hepcidin, leading to hepcidin binding ferroportin, an iron transporter, which causes ferroportin degradation. Degradation of ferroportin reduces the concentration of circulating iron, although it may increase intracellular iron which may have a beneficial effect on intracellular bacteria (310–312). However, other studies show that hepcidin mRNA was induced in RAW 264.7 macrophage-like cells when stimulated with IFN-γ and mycobacteria, but not when the stimulating cytokine was either IL-6 or IL-1β (313).
Calprotectin, an AMP present in monocytes, neutrophils and early macrophages (314, 315), sequesters metal ions to reduce their bioavailability. This has been particularly well-documented for iron, manganese and zinc (316–318). In fact, calprotectin is able to use this sequestration of metal ions to successfully inhibit growth of S. aureus in a mouse abscess model (317, 319). The S. aureus manganese transporters MntH and MntABC have been shown to work synergistically to overcome manganese scavenging by calprotectin (320). To overcome zinc scavenging, S. aureus expresses two zinc transporters and the metallophore staphylopine (321). Furthermore, bone marrow derived macrophages (BMDM) which were primed with calprotectin were induced to produce IL-6, CXCL1 and TNF-α, while BMDMs without calprotectin had a significantly reduced pro-inflammatory response (322).
Cathelicidins have multiple functions, including inducing antimicrobial action, guiding immune cell differentiation toward proinflammatory effects and steering chemotaxis (323). Cathelicidins opsonize bacteria to significantly enhance phagocytosis of S. aureus by macrophages in vitro by up to 10-fold (324). Cathelicidin fowlcidin-1 induces expression of pro-inflammatory cytokines in order to activate macrophages protecting mice from death in a normally lethal intraperitoneal MRSA infection (325). Furthermore, cathelicidin LL-37 improves macrophage killing of S. aureus, with LL-37 endocytosis by macrophages correlated with enhanced ROS production and lysosomal fusion (326).
Metal Accumulation and Restriction in the Phagosome
Metal ions are essential for bacterial metabolic activity, reproduction and oxidative stress defense (327). However, metal ions are also involved in production of ROS and RNS (328). Immune cells reduce availability of metal ions and alter the metabolic use of metal ions, termed ‘nutritional immunity’ (204, 328). Nutritional immunity studies show limiting availability to metal ions inhibits bacterial growth (329).
The role of metals in phagocytic microbial control has been extensively reviewed (330). Briefly, metal ions iron and manganese are restricted from the phagosome, while copper and zinc are used to overwhelm microbes with toxicity (330).
Although metal ions are essential and contribute to the functionality of many bacterial enzymes, high concentrations can be toxic to bacteria by enabling ROS production, as well as possessing high-affinity to metal-binding portions of proteins which can lead to bacterial enzymes binding excess metal ions, interfering with enzyme function (331). For example, copper has been described to be toxic to microbes by replacing iron ions in essential enzymes, as well as facilitating the production of hydroxyl radicals (332).
Manganese sequestration was found to be crucial for maximal inhibition of S. aureus growth in vitro (319). Manganese acquisition is essential for S. aureus survival (333–335), and is important for oxidative stress resistance due to acting as a cofactor for superoxide dismutase enzymes (334, 336). S. aureus with mutations in manganese transporters MntC or MntE were unable to resist methyl viologen (which interacts with electron donors to produce superoxide) likely due to an inability of the superoxide dismutases to function properly in the absence of manganese (334, 335). Similarly, mutations in manganese transporters MntABC and MntH resulted in S. aureus with increased sensitivity to methyl viologen, which was reversed by manganese supplementation (337).
Iron is essential for the functioning of many vital bacterial enzymes. However, when present in abundant quantities, iron catalyzes the generation of hydroxyl radicals via the Fenton reaction (204). Macrophages control iron homeostasis in part via NO-facilitated nuclear factor erythroid 2–related factor 2 transcription factor activation which upregulates the iron exporter ferroportin-1 (338). Macrophages with the gene for iNOS deleted had significantly higher concentrations of iron due to less expression of ferroportin-1, and this iron was able to be harnessed by intracellular Salmonella (338). S. aureus overcomes iron restriction by production of siderophores, which are able to competitively bind to iron to prevent sequestration by iron-binding host molecules such as lactoferrin and transferrin. In fact, both staphyloferrin A and staphyloferrin B have been shown to displace iron from transferrin (339, 340). Staphylococcal iron-regulated transporter (SirABC) is a transporter of S. aureus staphyloferrin B, and has been found to be expressed in response to oxidative and nitrative stress, providing protection from oxidative killing (341). These effects underline the importance of iron in macrophage antimicrobial defense.
Host cytokines are involved in regulating metal ion homeostasis in phagocytes. A number of cytokines identified as particularly important in defense against S. aureus, IL-1, IL-6, IL-10, and TNF-α, can act to make iron less available in monocytes and macrophages (82, 328). Unfortunately, this can have the unintended side-effect of anemia in the host. Accumulation of iron was correlated with reduced expression of pro-inflammatory cytokines TNF-α, IL-12, and IFN-γ, leading to an inability to control intracellular bacteria. This effect was reversed upon addition of an iron chelator (338). Host expression of GM-CSF activates the sequestration of zinc, leading to enhancement of H+ channels in the phagosome membrane, and induces NOX2 to produce ROS (342). Pro-inflammatory cytokine IFN-γ has been shown to upregulate expression of copper transporter Ctr1. This stimulates copper influx, which was found to be necessary for efficient bactericidal activity (343).
Nutrients in Control of S. aureus Infection
Nutrients, such as fatty acids and amino acids, are important for S. aureus survival. Additionally, fatty acids can also be antimicrobial. The host environment can be unfavorable for bacterial growth, as nutrients are restricted. Therefore, bacterial metabolism, essential compound scavenging, and defense against antimicrobial fatty acids is associated with S. aureus survival during infection.
Amino acid availability is critical for S. aureus growth, in fact many staphylococcal strains isolated from human skin are auxotrophic for multiple amino acids (344). In bovine mastitis infections, at least seven amino acids were required for S. aureus growth (345). Following exposure to H2O2, S. aureus amino acid metabolism is altered, likely with increased amino acid consumption promoting bacterial survival (346). Also, amino acid catabolism enables S. aureus survival within abscesses, where glucose supply is limited (347). In macrophages, S. aureus may induce host cell autophagy to increase metabolite availability to support intracellular proliferation (348). S. aureus is able to incorporate exogenous fatty acids into bacterial membranes (349). Host low-density lipoprotein (LDL) can be used as a fatty acid supply by S. aureus, removing the need for bacterial synthesis of fatty acids (350). Indeed, S. aureus uses host derived fatty acids when available, which is associated with higher levels of staphyloxanthin; thus saving energy in fatty acid synthesis and allowing virulence factor expression (351). Alternatively, S. aureus may obtain nutrients from the extracellular milieu via macrophage macropinocytosis, inhibition of which reduced S. aureus intracellular replication (352). Macrophage micropinocytosis occurs constitutively (353), indicating a potential route of nutrition for intracellular S. aureus.
In macrophages, the role of host lipids in infection with intracellular pathogens has been comprehensively reviewed; highlighting how fatty acids and their derivatives can have positive and negative consequences for pathogens, and that lipid metabolism changes with macrophage polarization (354). Antimicrobial fatty acid production by HeLa cells is protective against S. aureus infection (355). Leukocytes may also generate bactericidal fatty acids against S. aureus biofilms (356). Multiple unsaturated fatty acids are bactericidal against S. aureus, including linolenic acid and arachidonic acid, and fatty acid efficacy increases with greater unsaturation (357). Therefore, poly-unsaturated fatty acids (PUFAs) have greater antibacterial properties. Mice fed high levels of PUFAs had increased survival and reduced bacterial burden, along with an improved neutrophil response, following S. aureus sepsis infection (358). PUFA bactericidal effects against S. aureus were suggested to occur through a mechanism involving ROS (359). Arachidonic acid is a PUFA released at the same time as the oxidative burst in phagocytes, contributing to S. aureus killing. Arachidonic acid is oxidized to create electrophiles which are toxic to S. aureus, which is likely exasperated by ROS produced during the oxidative burst (360). Fatty acid cis-6-hexadecenoic acid is found on the skin and inhibits S. aureus survival, so, S. aureus increases defense gene expression (361).
In response to unsaturated fatty acids, S. aureus expression of genes involved in membrane stability and metabolism is increased as part of the stress response, indicating that fatty acids disrupt both bacterial lipid membranes and bacterial metabolism (362). S. aureus can increase resistance to fatty acids by reducing exogenous fatty acid incorporation into lipid membranes (363). S. aureus also uses fatty acid modifying enzyme (FAME) to inactivate fatty acids in abscesses (364). When host fatty acids are incorporated into the membrane of S. aureus, expression of the T7SS is increased, leading to virulence factor export (365). Furthermore, S. aureus expresses fatty acid resistance genes which confer resistance against linolenic acid and arachidonic acid (366). Studies on the role of macrophage unsaturated fatty acids and PUFAs on S. aureus are lacking, perhaps due to focus on the major role of fatty acids on the skin. However, further research may be beneficial due to fatty acid presence in biofilms, as well as fatty acids being associated with the phagocyte oxidative burst.
Macrophage Autophagy in S. aureus Infection
Macroautophagy (autophagy) is the cellular lysosomal self-degradation of damaged or unwanted components; however, autophagy components can be used to target pathogens for degradation. In recent publications, macrophage autophagy machinery has been revealed as an important host target which S. aureus is able to manipulate (367, 368). Autophagy proteins may be present at multiple stages of S. aureus infection, from phagosomes and autophagosomes, to targeting cytosolic bacteria. Autophagy machinery is more abundant in S. aureus infected murine macrophages (369). S. aureus can manipulate autophagy to promote survival and subsequent escape from within phagocytic cells in an Agr-dependent manner, potentially enabling persistence in sepsis infections (367). Similarly, high expression of vancomycin resistance-associated sensor/regulator also induces autophagy to a greater extent and is associated with increased intracellular survival in macrophages (368). Therefore, the extent autophagy may benefit S. aureus appears to be dependent on the virulence of individual strains. In a murine lung infection model, inhibiting autophagy with drug treatments reduces bacterial burden in the lung (370), again indicating that autophagy is beneficial for S. aureus. In agreement, in diabetic settings associated with increased autophagy, a larger number of autophagosomes containing S. aureus are observed, and blocking lysosome fusion to autophagosomes promotes S. aureus survival in macrophages (371). In bovine macrophages, S. aureus infection increased the number of autophagosomes, leading to increased bacterial survival, which also suggests later stages of the autophagy pathway are blocked (372). Together these data suggest that S. aureus resides within an autophagic vesicle within macrophages, possibly by blocking autophagy pathway advancement, thereby inhibiting macrophage-mediated killing. The role of autophagy in S. aureus-infected neutrophils is less clear, although it seems that the involvement of different autophagic machinery involved at early and late autophagy stages may lead to alternative bacterial outcomes (65, 373). Autophagy also represents a potential therapeutic target in S. aureus infection, whereby selenium may promote autophagy within macrophages to an extent that overcomes the bacterial block of the autophagy pathway (374). The involvement of autophagy in macrophage-S. aureus interactions is clearly demonstrated, but whether it directly affects infection outcome has yet to be examined in detail and remains an interesting area with possible therapeutic potential.
Macrophage Apoptosis-Associated Killing Is Deficient in S. aureus Infections
Macrophage apoptosis-associated bacterial killing is important for the clearance of a number of pathogens such as M. tuberculosis (375) and, in particular, S. pneumoniae (376, 377). It is suggested that macrophage phagocytic ability outpacing bactericidal activity leads to permeabilization of the phagolysosome, leading to cathepsin D release, which causes a reduction in anti-apoptotic Mcl-1 expression and, eventually, macrophage apoptosis (378). Interestingly, this mechanism is not observed following phagocytosis of S. aureus. Indeed, phagocytosis of S. aureus is associated with upregulation of both B cell lymphoma 2 gene (BCL2) and Mcl-1 (175), leading to decreased apoptosis. It has also been proposed that S. aureus inhibition of phagolysosome acidification and maturation circumvents apoptosis, enabling persistence in macrophages, although the exact mechanism remains unclear (73). Since persisting intracellular S. aureus can be found in the cytoplasm, further studies have suggested that S. aureus escapes the phagolysosome, which was associated with increased antiapoptotic host cell proteins (379). This is in stark contrast to extracellular S. aureus, which actively promotes macrophage apoptosis by releasing α-hemolysin or Panton-Valentine leucocidin (PVL) toxins (380–382). Detailed discussion of S. aureus virulence factors is outside the scope of this review (22, 24, 383, 384).
Macrophage Extracellular Traps and S. aureus
Extracellular traps (ET) are protrusions of chromatin, histone proteins, DNA, proteases and AMPs, that ensnare bacteria and form an important part of the immune response to infection (385), first described in association with neutrophils (386). Neutrophil extracellular traps (NET) vary in their formation (fast or slow) (387, 388) and composition (chromatin or mitochondrial DNA) (389). Original descriptions of NETs demonstrated formation over 3 hours by the destruction of the nuclear membrane leading to death of the neutrophil. More recently, certain NETs were shown to form within 60 min by extrusion of vesicles containing chromatin in a rapid and oxidant independent mechanism (387). Moreover, NETs can be formed of mitochondrial DNA rather than chromatin, in a mechanism that is independent of cell death but was associated with increased survival of neutrophils (389). This allows the neutrophil to continue to contribute to the host immune response. NET formation can be induced in response to a number of different stimuli such as LPS, IL-8, complement factor C5a, and bacteria including S. aureus (386, 387, 389).
There is a growing body of evidence that many different innate immune cells are capable of producing ETs to control bacteria, including eosinophils (390), mast cells (391) and macrophages (392). Macrophage ETs (MET) play a role in host defense. Bovine monocyte-derived macrophages form METs in response to Mannheimiae haemolytica and to its leukotoxin (LKT) (392). Interestingly LKT did not induce MET formation by bovine alveolar macrophages, suggesting that macrophage differentiation determines the ability to trigger MET formation. Additionally, MET formation was demonstrated by bovine macrophages in response to Histophilus somni (393). E. coli also induced MET formation in RAW 264.7 macrophages, which was NADPH oxidase-dependent (392). Similarly, METs were induced in J774 cells in response to E. coli and Candida albicans, with authors suggesting that the role of METs is to slow dissemination of microbes (394). However, phagocytosis and MET formation have been observed to coincide for control and clearance of C. albicans (395).
MET formation can be stimulated by the use of statins. The pre-treatment of human and murine macrophages with statins is associated with increased S. aureus killing (396). In neutrophils the proposed mechanism for this enhanced clearance was a significant increase in NET formation, leading to increased S. aureus entrapment. A similar result was observed in PMA-stimulated RAW 264.7 macrophages. Initially, NETosis was considered to culminate in cell death, but emerging evidence has shown in neutrophils NETs can be independent of cell death (389). However, the formation of METs appears to trigger a form of cell death as the macrophage exhibits loss of membrane integrity (396, 397), and this may be associated with caspase-1 activity (398), although data on this are sparse. This is further evidence that MET formation may act to slow the dissemination of infection and allow neighboring macrophages to phagocytose bacteria (394).
Pathogens have evolved mechanisms to overcome ETs. For instance, Streptococcus pneumoniae evades NETs by producing endonuclease EndA, which degrades the DNA in the NET (399). Similarly, S. aureus secretes nuclease and adenosine synthase, leading to the conversion of NETs to deoxyadenosine and in turn triggering caspase-3–mediated cell death to cause non-inflammatory macrophage apoptosis (400–402). This effectively leads to the removal of phagocytic cells from the site of infection allowing abscess formation.
Extracellular Vesicles From S. aureus and Macrophages
Extracellular vesicles (EV) are known to be secreted by a number of different Gram-positive bacteria, including S. aureus (403). These bacterial EVs have been shown to contain a variety of virulence factors and provoke significant immune responses. Indeed, one of the first descriptions of S. aureus EV demonstrated they could contain β-lactamases, which enabled surrounding bacteria to withstand ampicillin (404). EVs also trigger apoptosis. S. aureus EVs deliver virulence factors such as α-hemolysin to macrophages, leading to NLRP3 inflammasome-induced pyroptosis (host cell death) (405), which can be inhibited by fosfomycin (406). In a murine model, EVs were shown to cause atopic dermatitis-like inflammation in the skin (407). EVs secreted by S. aureus can also cause mastitis (408). Furthermore, it has been postulated that EVs could be a target for vaccine development (409). The use of statins in a murine survival model decreased macrophage responses to S. aureus EVs, suggesting a possible novel therapeutic approach (410).
In addition to extracellular vesicles (EV) secreted by S. aureus to subvert the host, there are numerous examples of immune cells releasing EVs. These can vary in size and content and have been isolated from several different cell types, including macrophages. EVs are primarily thought to act as communicating mechanisms allowing an orchestration of immune responses and are an important part of the junction between the adaptive and innate immune responses (411). Indeed, EV from macrophage infected with M. bovis modulated T lymphocytes responses (412). When macrophages were infected with M. tuberculosis, the content of the EV changed to confer decreased inflammatory cytokine release and decrease lung mycobacterial load (413). Furthermore, during infection with hepatitis C virus, macrophages secreted EVs that inhibited viral replication (414). AM-derived EVs are suggested to play a role in the pathogenesis of acute lung injury by encapsulating TNF-α (415). The majority of studies have been termed EVs “microvesicles” due to their size, but more recently, larger “macrolets” containing IL-6 have been described which are capable of engulfing and killing E. coli following macrophage LPS stimulation (416). The full role of S. aureus-infected macrophage EVs on host-pathogen interactions and their interplay with the adaptive immune response merits further studies as possible targets for therapeutic approaches.
Therapeutic Approaches to S. aureus Infection
Antimicrobials
S. aureus has acquired resistance to a wide range of antibiotics, which is an expanding problem for the treatment of human infections. In fact, the specter of antimicrobial resistant (AMR) S. aureus has been described as a pandemic (417), with global incidence rising (418–421). Resistance most commonly arises due to horizontal gene transfer from resistant bacteria, however, mutation of the S. aureus chromosome and mobile genetic elements may also lead to resistance (417, 422). Antibiotic resistance is a particular threat to modern medicine, with multiple procedures dependent on antibiotic use (6). Last resort antibiotics used to treat MRSA are often expensive, less efficacious, and more likely to cause severe side effects (422).
The intracellular nature of S. aureus impedes antibiotic activity, as many antibiotics cannot access the intraphagocyte niche (423, 424). Methods to combat this have included development of intracellular antimicrobials (425), nanoparticles which can distribute antimicrobials to infected macrophages (426, 427) and active targeting of macrophages to induce receptor-mediated endocytosis, releasing singlet oxygen to kill intracellular S. aureus (428). Furthermore, therapeutic nanoparticles which favored pro-inflammatory macrophage polarization enabled clearance of S. aureus biofilms in vivo (169). The antimicrobial protein plectasin, can kill S. aureus inside THP-1 macrophages in vitro, or inside peritoneal macrophages in vivo, however, plectasin is significantly more effective against extracellular bacteria (425). A nanogel which preferentially targets macrophages uses bacterial enzymes to initiate release of antibiotic (vancomycin), inhibiting MRSA growth at sites of infection in vivo (426). After treatment with the nanogel, zebrafish embryos infected with MRSA survived to significantly higher levels with no visible (GFP-expressing) bacteria 9 h post-infection. Similarly, macrophages treated with nanogel had significantly reduced CFUs recovered from S. aureus infected RAW 264.7 macrophages (426). Also, conjugation of penicillin G to squalene enabled antibiotic endocytosis into J774 macrophages, whereby S. aureus was significantly less able to survive intracellularly (427).
Vaccines
Decades of work have been devoted to production of a S. aureus vaccine, however, none has yet been approved (14, 15). A key difficulty in producing a S. aureus vaccine is that it must provide broad immunity, since the bacteria can cause a wide range of infections in a variety of tissues. S. aureus was traditionally believed to be extracellular; however, it is now recognized as a facultative intracellular pathogen. This may partially explain the lack of an effective S. aureus vaccine, especially with S. aureus able to exist within immune cells. Indeed, since S. aureus exploits macrophages during infection, vaccine design inducing successful macrophage defenses against S. aureus could be valuable.
Attempts have been made to produce whole cell or live/killed vaccines against S. aureus, but these have failed to produce effective immunity (14, 15). Vaccines have been targeted against S. aureus polysaccharide, with initial positive results in animal studies and partial protection in early human trials (429, 430). Other targets include surface polysaccharide poly-N-acetylglucosamine (431, 432), surface proteins such as iron surface determinant (Isd) A or IsdB (433, 434), clumping factor (Clf) A or ClfB (435–437) and fibronectin binding protein (FnBP) (438). These vaccines led to partial immune protection, but overall they were not successful (439). A limiting factor may be that the proteins used are not essential components of S. aureus (440). To combat this, research groups have tried combining multiple antigens into a single vaccine. Newer approaches include targeting S. aureus molecules which stimulate varied immune responses, to mimic the different immune responses observed with natural S. aureus infection. It is now thought that approaches which induce Th1/Th17 responses may be more effective, although this is thought to ideally be best when combined with induction of opsonophagocytic antibody generation (439, 441). Many of the aforementioned vaccine studies investigated whether the treatment was able to induce opsonophagocytic killing by phagocytes (432, 433, 437, 438). However, it has also been suggested that vaccines which neutralize S. aureus toxins, rather than aiming to induce opsonophagocytic killing, may be more effective (15).
Differences in animal and human responses to vaccines hinders their production. Some studies have found that, despite promising results in animal models, human trials showed no protective immunity (433, 439). This suggests that positive animal trials do not correlate with positive human immune responses, which may be in part to the differences between the human and murine/rabbit immune system. One of the limitations of S. aureus mouse models is that a much higher dose of the bacteria is required to initiate infection when compared to the estimated human infective dose. Co-injection of mice with commensal bacteria alongside a dose of S. aureus more comparable to natural human infection led to increased CFUs, and decreased survival of the mice (272). This phenomenon was labeled “augmentation.” As S. aureus exists in a polymicrobial environment, this model is likely closer to that of natural infection. It is possible that using this augmentation model in murine models to better represent human infection would improve the assessment of therapeutic efficacy against S. aureus.
Future Perspectives
S. aureus is a highly successful pathogen due to a wide variety of virulence factors and immune evasion strategies (22). Macrophages play a crucial role in the control of S. aureus infection as macrophage depletion in mice led to increased susceptibility to S. aureus (61, 272). However, macrophages do not always eliminate staphylococci, which can use the macrophages as a reservoir for persistence, causing continued infection. Therefore, it is important to further characterize the mechanisms used by S. aureus to overcome macrophage killing and manipulate the host cell as these may present novel therapeutic adjuncts preventing dissemination and persistence of infection.
It remains unclear which antimicrobial strategy above all others is responsible for killing the majority of S. aureus. As detailed above, macrophage killing mechanisms, including ROS, RNS, phagosome acidification, antimicrobial enzymes and AMPs, nutritional immunity and autophagy contribute to S. aureus clearance and it is therefore likely through a combination of these mechanisms. NOX2-dependent ROS is seemingly critical, as CGD patients are particularly susceptible to S. aureus infection (208). S. aureus appears to require exposure to an acidic environment for intracellular survival, again suggesting NOX2-dependent ROS rather than downstream phagosomal maturation is most critical for bacterial killing. Further studies are required to confirm which ROS, within the macrophage phagosome, are necessary to overcome S. aureus infection to fully understand the ROS killing capacity. Since NOX2-dependent ROS appears to be vital for bacterial killing, enhancement of macrophage NOX2 activity may be useful as a therapeutic target.
In addition to further characterizing the killing mechanism, a greater understanding of the strategies used by S. aureus to evade the host is required to prevent dissemination of infection. To date, much effort has gone into evaluating the role of neutrophils in S. aureus infection, while macrophages, despite being a source of bacterial persistence, have been far less studied. The role of macrophages in controlling infection highlights these cells as an important target for investigation and exploitation. Indeed, studies targeting macrophages during S. aureus infection show beneficial outcomes (426–428).
Finally, in light of the rising antimicrobial resistance, determining the optimal antibiotic strategies to control S. aureus infections, and use of novel agents or combinations to provide synergistic activity merit further studies. The use of immunomodulation and preventative approaches to the peri-operative patient, if fruitful, would lead to significant decreases in the public health burden posed by S. aureus.
Author Contributions
All authors contributed to the writing and editing of the article and GP and JG created the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Medical Research Council (MRC) grant MR/R001111/1 and was supported in part by AMR cross-council funding from the MRC to the SHIELD consortium “Optimising Innate Host Defence to Combat Antimicrobial Resistance” MRNO2995X/1.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wertheim HFL, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, et al. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis (2005) 5:751–62. doi: 10.1016/S1473-3099(05)70295-4
2. Gorwitz RJ, Kruszon-Moran D, McAllister SK, McQuillan G, McDougal LK, Fosheim GE, et al. Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001-2004. J Infect Dis (2008) 197:1226–34. doi: 10.1086/533494
3. Albrecht VS, Limbago BM, Moran GJ, Krishnadasan A, Gorwitz RJ, McDougal LK, et al. Staphylococcus aureus colonization and strain type at various body sites among patients with a closed abscess and uninfected controls at U.S. emergency departments. J Clin Microbiol (2015) 53:3478–84. doi: 10.1128/JCM.01371-15
4. Peacock SJ, Justice A, Griffiths D, de Silva GDI, Kantzanou MN, Crook D, et al. Determinants of acquisition and carriage of Staphylococcus aureus in infancy. J Clin Microbiol (2003) 41:5718–25. doi: 10.1128/jcm.41.12.5718-5725.2003
5. von Eiff C, Becker K, Machka K, Stammer H, Peters G. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study group. N Engl J Med (2001) 344:11–6. doi: 10.1056/NEJM200101043440102
6. Prestinaci F, Pezzotti P, Pantosti A. Antimicrobial resistance: a global multifaceted phenomenon. Pathog Glob Health (2015) 109:309–18. doi: 10.1179/2047773215Y.0000000030
7. Kourtis AP, Hatfield K, Baggs J, Mu Y, See I, Epson E, et al. Vital signs: epidemiology and recent trends in methicillin-resistant and in methicillin-susceptible Staphylococcus aureus bloodstream infections - United States. MMWR Morb Mortal Wkly Rep (2019) 68:214–9. doi: 10.15585/mmwr.mm6809e1
8. Köck R, Becker K, Cookson B, van Gemert-Pijnen JE, Harbarth S, Kluytmans J, et al. Methicillin-resistant Staphylococcus aureus (MRSA): burden of disease and control challenges in Europe. Euro Surveill (2010) 15:19688. doi: 10.2807/ese.15.41.19688-en
9. Kim T, Chong YP, Park K-H, Bang KM, Park S-J, Kim S-H, et al. Clinical and microbiological factors associated with early patient mortality from methicillin-resistant Staphylococcus aureus bacteremia. Korean J Intern Med (2019) 34:184–94. doi: 10.3904/kjim.2016.351
10. Gasch O, Ayats J, Angeles Dominguez M, Tubau F, Liñares J, Peña C, et al. Epidemiology of methicillin-resistant Staphylococcus aureus (MRSA) bloodstream infection: secular trends over 19 years at a university hospital. Med (Baltimore) (2011) 90:319–27. doi: 10.1097/MD.0b013e31822f0b54
11. Paul M, Kariv G, Goldberg E, Raskin M, Shaked H, Hazzan R, et al. Importance of appropriate empirical antibiotic therapy for methicillin-resistant Staphylococcus aureus bacteraemia. J Antimicrob Chemother (2010) 65:2658–65. doi: 10.1093/jac/dkq373
12. Lam JC, Gregson DB, Robinson S, Somayaji R, Conly JM, Parkins MD. Epidemiology and outcome determinants of Staphylococcus aureus bacteremia revisited: a population-based study. Infection (2019) 47:961–71. doi: 10.1007/s15010-019-01330-5
13. McGuinness WA, Malachowa N, DeLeo FR. Vancomycin resistance in Staphylococcus aureus. Yale J Biol Med (2017) 90:269–81.
14. DeDent A, Kim HK, Missiakas D, Schneewind O. Exploring Staphylococcus aureus pathways to disease for vaccine development. Semin Immunopathol (2012) 34:317–33. doi: 10.1007/s00281-011-0299-z
15. Miller LS, Fowler VG, Shukla SK, Rose WE, Proctor RA. Development of a vaccine against Staphylococcus aureus invasive infections: Evidence based on human immunity, genetics and bacterial evasion mechanisms. FEMS Microbiol Rev (2020) 44:123–53. doi: 10.1093/femsre/fuz030
16. Flannagan RS, Heit B, Heinrichs DE. Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell Microbiol (2016) 18:514–35. doi: 10.1111/cmi.12527
17. DuMont AL, Yoong P, Surewaard BGJ, Benson MA, Nijland R, van Strijp JAG, et al. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect Immun (2013) 81:1830–41. doi: 10.1128/IAI.00095-13
18. Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, et al. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS One (2008) 3:e1409. doi: 10.1371/journal.pone.0001409
19. Lam T-T, Giese B, Chikkaballi D, Kuhn A, Wolber W, Pane-Farre J, et al. Phagolysosomal integrity is generally maintained after Staphylococcus aureus invasion of nonprofessional phagocytes but is modulated by strain 6850. Infect Immun (2010) 78:3392–403. doi: 10.1128/IAI.00012-10
20. Grosz M, Kolter J, Paprotka K, Winkler A-C, Schafer D, Chatterjee SS, et al. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin alpha. Cell Microbiol (2014) 16:451–65. doi: 10.1111/cmi.12233
21. Cole J, Aberdein J, Jubrail J, Dockrell DH. The role of macrophages in the innate immune response to Streptococcus pneumoniae and Staphylococcus aureus: mechanisms and contrasts. Adv Microb Physiol (2014) 65:125–202. doi: 10.1016/bs.ampbs.2014.08.004
22. Flannagan RS, Heit B, Heinrichs DE. Antimicrobial mechanisms of macrophages and the immune evasion strategies of Staphylococcus aureus. Pathogens (2015) 4:826–68. doi: 10.3390/pathogens4040826
23. Lacey KA, Geoghegan JA, McLoughlin RM. The role of Staphylococcus aureus virulence factors in skin infection and their potential as vaccine antigens. Pathogens (2016) 5:22. doi: 10.3390/pathogens5010022
24. Jenkins A, Diep BA, Mai TT, Vo NH, Warrener P, Suzich J, et al. Differential expression and roles of Staphylococcus aureus virulence determinants during colonization and disease. MBio (2015) 6:e02272–14. doi: 10.1128/mBio.02272-14
25. Richardson AR. Virulence and metabolism. Microbiol Spectr (2019) 7:2. doi: 10.1128/microbiolspec.GPP3-0011-2018
26. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol (2014) 5:514. doi: 10.3389/fimmu.2014.00514
27. Silva MT, Correia-Neves M. Neutrophils and macrophages: the main partners of phagocyte cell systems. Front Immunol (2012) 3:174. doi: 10.3389/fimmu.2012.00174
28. McCracken JM, Allen L-AH. Regulation of human neutrophil apoptosis and lifespan in health and disease. J Cell Death (2014) 7:15–23. doi: 10.4137/JCD.S11038
29. Davies LC, Taylor PR. Tissue-resident macrophages: then and now. Immunology (2015) 144:541–8. doi: 10.1111/imm.12451
30. Parihar A, Eubank TD, Doseff AI. Monocytes and macrophages regulate immunity through dynamic networks of survival and cell death. J Innate Immun (2010) 2:204–15. doi: 10.1159/000296507
31. van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med (1968) 128:415–35. doi: 10.1084/jem.128.3.415
32. Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol (2009) 27:669–92. doi: 10.1146/annurev.immunol.021908.132557
33. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol (2013) 14:986–95. doi: 10.1038/ni.2705
34. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med (2017) 214:1913–23. doi: 10.1084/jem.20170355
35. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci (2007) 10:1538–43. doi: 10.1038/nn2014
36. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity (2014) 41:21–35. doi: 10.1016/j.immuni.2014.06.013
37. Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science (2013) 342:1242974. doi: 10.1126/science.1242974
38. Honold L, Nahrendorf M. Resident and monocyte-derived macrophages in cardiovascular disease. Circ Res (2018) 122:113–27. doi: 10.1161/CIRCRESAHA.117.311071
39. Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol (2012) 13:753–60. doi: 10.1038/ni.2360
40. Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte-derived macrophages and resident Kupffer cells display different ontogeny and functions in acute liver injury. J Immunol (2014) 193:344–53. doi: 10.4049/jimmunol.1400574
41. Ikarashi M, Nakashima H, Kinoshita M, Sato A, Nakashima M, Miyazaki H, et al. Distinct development and functions of resident and recruited liver Kupffer cells/macrophages. J Leukoc Biol (2013) 94:1325–36. doi: 10.1189/jlb.0313144
42. Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, et al. In vivo analysis of dendritic cell development and homeostasis. Science (2009) 324:392–7. doi: 10.1126/science.1170540
43. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol (2012) 13:1118–28. doi: 10.1038/ni.2419
44. Guilliams M, Scott CL. Does niche competition determine the origin of tissue-resident macrophages? Nat Rev Immunol (2017) 17:451–60. doi: 10.1038/nri.2017.42
45. Okabe Y. Molecular control of the identity of tissue-resident macrophages. Int Immunol (2018) 30:485–91. doi: 10.1093/intimm/dxy019
46. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol (2013) 3:785–97. doi: 10.1002/cphy.c120026
47. Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int (2006) 26:1175–86. doi: 10.1111/j.1478-3231.2006.01342.x
48. Gordon S, Plüddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol (2017) 15:53. doi: 10.1186/s12915-017-0392-4
49. Yang C-Y, Chen J-B, Tsai T-F, Tsai Y-C, Tsai C-Y, Liang P-H, et al. CLEC4F is an inducible C-type lectin in F4/80-positive cells and is involved in alpha-galactosylceramide presentation in liver. PLoS One (2013) 8:e65070. doi: 10.1371/journal.pone.0065070
50. Herrmann M, Schäfer C, Heiss A, Gräber S, Kinkeldey A, Büscher A, et al. Clearance of fetuin-A–containing calciprotein particles is mediated by scavenger receptor-A. Circ Res (2012) 111:575–84. doi: 10.1161/CIRCRESAHA.111.261479
51. Martinez-Pomares L. The mannose receptor. J Leukoc Biol (2012) 92:1177–86. doi: 10.1189/jlb.0512231
52. Merad M, Manz MG, Karsunky H, Wagers A, Peters W, Charo I, et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol (2002) 3:1135–41. doi: 10.1038/ni852
53. Doebel T, Voisin B, Nagao K. Langerhans cells - the macrophage in dendritic cell clothing. Trends Immunol (2017) 38:817–28. doi: 10.1016/j.it.2017.06.008
54. West HC, Bennett CL. Redefining the role of Langerhans cells as immune regulators within the skin. Front Immunol (2017) 8:1941. doi: 10.3389/fimmu.2017.01941
55. Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VG. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev (2015) 28:603–61. doi: 10.1128/CMR.00134-14
56. Kim HK, Missiakas D, Schneewind O. Mouse models for infectious diseases caused by Staphylococcus aureus. J Immunol Methods (2014) 410:88. doi: 10.1016/J.JIM.2014.04.007
57. Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J (2009) 23:3393–404. doi: 10.1096/fj.09-135467
58. Rogers DE. Studies on bacteriemia. I. Mechanisms relating to the persistence of bacteriemia in rabbits following the intravenous injection of staphylococci. J Exp Med (1956) 103:713. doi: 10.1084/JEM.103.6.713
59. Pollitt EJG, Szkuta PT, Burns N, Foster SJ. Staphylococcus aureus infection dynamics. PLoS Pathog (2018) 14:e1007112. doi: 10.1371/journal.ppat.1007112
60. Jorch SK, Surewaard BG, Hossain M, Peiseler M, Deppermann C, Deng J, et al. Peritoneal GATA6+ macrophages function as a portal for Staphylococcus aureus dissemination. J Clin Invest (2019) 129:4643–56. doi: 10.1172/JCI127286
61. Surewaard BGJ, Deniset JF, Zemp FJ, Amrein M, Otto M, Conly J, et al. Identification and treatment of the Staphylococcus aureus reservoir in vivo. J Exp Med (2016) 213:1141–51. doi: 10.1084/jem.20160334
62. Yajjala VK, Thomas VC, Bauer C, Scherr TD, Fischer KJ, Fey PD, et al. Resistance to acute macrophage killing promotes airway fitness of prevalent community-acquired Staphylococcus aureus strains. J Immunol (2016) 196:4196–203. doi: 10.4049/jimmunol.1600081
63. Martin FJ, Parker D, Harfenist BS, Soong G. Prince A. Participation of CD11c(+) leukocytes in methicillin-resistant Staphylococcus aureus clearance from the lung. Infect Immun (2011) 79:1898–904. doi: 10.1128/IAI.01299-10
64. Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, et al. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog (2015) 11:e1004820. doi: 10.1371/journal.ppat.1004820
65. Prajsnar TK, Serba JJ, Dekker BM, Gibson JF, Masud S, Fleming A, et al. The autophagic response to Staphylococcus aureus provides an intracellular niche in neutrophils. Autophagy (2020) 15:1–15. doi: 10.1080/15548627.2020.1739443
66. Prajsnar TK, Cunliffe VT, Foster SJ. Renshaw S a. A novel vertebrate model of Staphylococcus aureus infection reveals phagocyte-dependent resistance of zebrafish to non-host specialized pathogens. Cell Microbiol (2008) 10:2312–25. doi: 10.1111/j.1462-5822.2008.01213.x
67. Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol (2000) 164:3713–22. doi: 10.4049/jimmunol.164.7.3713
68. Thwaites GE, Gant V. Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat Rev Microbiol (2011) 9:215–22. doi: 10.1038/nrmicro2508
69. Prajsnar TK, Hamilton R, Garcia-Lara J, McVicker G, Williams A, Boots M, et al. A privileged intraphagocyte niche is responsible for disseminated infection of Staphylococcus aureus in a zebrafish model. Cell Microbiol (2012) 14:1600–19. doi: 10.1111/j.1462-5822.2012.01826.x
70. McVicker G, Prajsnar TK, Williams A, Wagner NL, Boots M, Renshaw SA, et al. Clonal expansion during Staphylococcus aureus infection dynamics reveals the effect of antibiotic intervention. PLoS Pathog (2014) 10:e1003959. doi: 10.1371/journal.ppat.1003959
71. Grant AJ, Restif O, McKinley TJ, Sheppard M, Maskell DJ, Mastroeni P. Modelling within-Host Spatiotemporal Dynamics of Invasive Bacterial Disease. PLoS Biol (2008) 6:e74. doi: 10.1371/journal.pbio.0060074
72. Plaut RD, Kelly VK, Lee GM, Stibitz S, Merkel TJ. Dissemination bottleneck in a murine model of inhalational anthrax. Infect Immun (2012) 80:3189–93. doi: 10.1128/IAI.00515-12
73. Jubrail J, Morris P, Bewley MA, Stoneham S, Johnston SA, Foster SJ, et al. Inability to sustain intraphagolysosomal killing of Staphylococcus aureus predisposes to bacterial persistence in macrophages. Cell Microbiol (2016) 18:80–96. doi: 10.1111/cmi.12485
74. Fournier B, Philpott DJ. Recognition of Staphylococcus aureus by the innate immune system. Clin Microbiol Rev (2005) 18:521–40. doi: 10.1128/CMR.18.3.521-540.2005
75. Gómez MI, Prince A. Airway epithelial cell signaling in response to bacterial pathogens. Pediatr Pulmonol (2008) 43:11–9. doi: 10.1002/ppul.20735
76. Peterson ML, Ault K, Kremer MJ, Klingelhutz AJ, Davis CC, Squier CA, et al. The innate immune system is activated by stimulation of vaginal epithelial cells with Staphylococcus aureus and toxic shock syndrome toxin 1. Infect Immun (2005) 73:2164–74. doi: 10.1128/IAI.73.4.2164-2174.2005
77. Kumar A, Zhang J, Yu F-SX. Innate immune response of corneal epithelial cells to Staphylococcus aureus infection: role of peptidoglycan in stimulating proinflammatory cytokine secretion. Invest Opthalmol Vis Sci (2004) 45:3513. doi: 10.1167/iovs.04-0467
78. Al Alam D, Escorte S, Puchelle E, Guenounou M, Gangloff SC. 037 Comparative chemokine secretory profiles in normal and CF polarized epithelial cells following S. aureus infection. Rev Mal Respir (2005) 22:862. doi: 10.1016/S0761-8425(05)92449-2
79. Rot A, Henderson LE, Leonard EJ. Staphylococcus aureus-derived chemoattractant activity for human monocytes. J Leukoc Biol (1986) 40:43–53. doi: 10.1002/jlb.40.1.43
80. Wilkinson BJ, Kim Y, Peterson PK, Quie PG, Michael AF. Activation of complement by cell surface components of Staphylococcus aureus. Infect Immun (1978) 20:388. doi: 10.1128/IAI.20.2.388-392.1978
81. Nandi A, Bishayi B. Murine macrophage response from peritoneal cavity requires signals mediated by chemokine receptor CCR-2 during Staphylococcus aureus infection. Immunol Res (2016) 64:213–32. doi: 10.1007/s12026-015-8739-9
82. Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-α play a pivotal role in the host immune response in a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuropathol Exp Neurol (2004) 63:381–96. doi: 10.1093/jnen/63.4.381
83. Kim B, Jiang T, Bae J-H, Yun HS, Jang SH, Kim JH, et al. In Staphylococcus aureus, the particulate state of the cell envelope is required for the efficient induction of host defense responses. Infect Immun (2019) 87:e00674–19. doi: 10.1128/IAI.00674-19
84. Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, et al. Kupffer cell heterogeneity: Functional properties of bone marrow-derived and sessile hepatic macrophages. Blood (2007) 110:4077–85. doi: 10.1182/blood-2007-02-073841
85. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol (2011) 11:762–74. doi: 10.1038/nri3070
86. Postma B, Poppelier MJ, van Galen JC, Prossnitz ER, van Strijp JAG, de Haas CJC, et al. Chemotaxis inhibitory protein of Staphylococcus aureus binds specifically to the C5a and formylated peptide receptor. J Immunol (2004) 172:6994–7001. doi: 10.4049/jimmunol.172.11.6994
87. de Haas CJC, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJB, Heezius ECJM, et al. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med (2004) 199:687–95. doi: 10.1084/jem.20031636
88. Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol (1999) 17:593–623. doi: 10.1146/annurev.immunol.17.1.593
89. Alpuche-Aranda CM, Racoosin EL, Swanson JA, Miller SI. Salmonella stimulate macrophage macropinocytosis and persist within spacious phagosomes. J Exp Med (1994) 179:601. doi: 10.1084/JEM.179.2.601
90. Watarai M, Derre I, Kirby J, Growney JD, Dietrich WF, Isberg RR. Legionella pneumophila is internalized by a macropinocytotic uptake pathway controlled by the dot/icm system and the mouse lgn1 locus. J Exp Med (2001) 194:1081. doi: 10.1084/JEM.194.8.1081
91. Swanson JA. Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol (2008) 9:639–49. doi: 10.1038/nrm2447
92. Plüddemann A, Mukhopadhyay S, Gordon S. The interaction of macrophage receptors with bacterial ligands. Expert Rev Mol Med (2006) 8:1–25. doi: 10.1017/S1462399406000159
93. Plüddemann A, Neyen C, Gordon S. Macrophage scavenger receptors and host-derived ligands. Methods (2007) 43:207–17. doi: 10.1016/J.YMETH.2007.06.004
94. Dunne DW, Resnick D, Greenberg J, Krieger M, Joiner KA. The type I macrophage scavenger receptor binds to Gram-positive bacteria and recognizes lipoteichoic acid. Proc Natl Acad Sci USA (1994) 91:1863–7. doi: 10.1073/pnas.91.5.1863
95. Thomas CA, Li Y, Kodama T, Suzuki H, Silverstein SC, El Khoury J. Protection from lethal Gram-positive infection by macrophage scavenger receptor–dependent phagocytosis. J Exp Med (2000) 191:147–56. doi: 10.1084/jem.191.1.147
96. Ono K, Nishitani C, Mitsuzawa H, Shimizu T, Sano H, Suzuki H, et al. Mannose-binding lectin augments the uptake of lipid a, Staphylococcus aureus , and Escherichia coli by Kupffer cells through increased cell surface expression of scavenger receptor A. J Immunol (2006) 177:5517–23. doi: 10.4049/jimmunol.177.8.5517
97. Shi L, Takahashi K, Dundee J, Shahroor-Karni S, Thiel S, Jensenius JC, et al. Mannose-binding lectin-deficient mice are susceptible to infection with Staphylococcus aureus. J Exp Med (2004) 199:1379–90. doi: 10.1084/jem.20032207
98. Kuronuma K, Sano H, Kato K, Kudo K, Hyakushima N, Yokota S, et al. Pulmonary surfactant protein A augments the phagocytosis of Streptococcus pneumoniae by alveolar macrophages through a casein kinase 2-dependent increase of cell surface localization of scavenger receptor A. J Biol Chem (2004) 279:21421–30. doi: 10.1074/jbc.M312490200
99. Sever-Chroneos Z, Krupa A, Davis J, Hasan M, Yang C-H, Szeliga J, et al. (SP-A)-mediated clearance of Staphylococcus aureus involves binding of SP-A to the staphylococcal adhesin Eap and the macrophage receptors SP-A receptor 210 and scavenger receptor class A. J Biol Chem (2011) 286:4854–70. doi: 10.1074/JBC.M110.125567
100. Arredouani MS, Palecanda A, Koziel H, Huang Y-C, Imrich A, Sulahian TH, et al. MARCO is the major binding receptor for unopsonized particles and bacteria on human alveolar macrophages. J Immunol (2005) 175:6058–64. doi: 10.4049/jimmunol.175.9.6058
101. van der Laan LJ, Döpp EA, Haworth R, Pikkarainen T, Kangas M, Elomaa O, et al. Regulation and functional involvement of macrophage scavenger receptor MARCO in clearance of bacteria in vivo. J Immunol (1999) 162:939–47.
102. Zhou H, Imrich A, Kobzik L. Characterization of immortalized MARCO and SR-AI/II-deficient murine alveolar macrophage cell lines. Part Fibre Toxicol (2008) 5:7. doi: 10.1186/1743-8977-5-7
103. Blanchet C, Jouvion G, Fitting C, Cavaillon J-M, Adib-Conquy M. Protective or deleterious role of scavenger receptors SR-A and CD36 on host resistance to Staphylococcus aureus depends on the site of infection. PLoS One (2014) 9:e87927. doi: 10.1371/JOURNAL.PONE.0087927
104. DeLoid GM, Sulahian TH, Imrich A, Kobzik L. Heterogeneity in macrophage phagocytosis of Staphylococcus aureus strains: high-throughput scanning cytometry-based analysis. PLoS One (2009) 4:e6209. doi: 10.1371/journal.pone.0006209
105. Hallett AF, Cooper R. Complement activation in Staphylococcus aureus bacteraemia. Clin Exp Immunol (1980) 40:306.
106. Neth O, Jack DL, Johnson M, Klein NJ, Turner MW. Enhancement of complement activation and opsonophagocytosis by complexes of mannose-binding lectin with mannose-binding lectin-associated serine protease after binding to Staphylococcus aureus. J Immunol (2002) 169:4430–6. doi: 10.4049/jimmunol.169.8.4430
107. Sakiniene E, Bremell T, Tarkowski A. Complement depletion aggravates Staphylococcus aureus septicaemia and septic arthritis. Clin Exp Immunol (1999) 115:95–102. doi: 10.1046/j.1365-2249.1999.00771.x
108. Na M, Jarneborn A, Ali A, Welin A, Magnusson M, Stokowska A, et al. Deficiency of the complement component 3 but not factor b aggravates Staphylococcus aureus septic arthritis in mice. Infect Immun (2016) 84:930–9. doi: 10.1128/IAI.01520-15
109. Lukácsi S, Nagy-Baló Z, Erdei A, Sándor N, Bajtay Z. The role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in complement-mediated phagocytosis and podosome formation by human phagocytes. Immunol Lett (2017) 189:64–72. doi: 10.1016/j.imlet.2017.05.014
110. Gyimesi E, Bankovich AJ, Schuman TA, Goldberg JB, Lindorfer MA, Taylor RP. Staphylococcus aureus bound to complement receptor 1 on human erythrocytes by bispecific monoclonal antibodies is phagocytosed by acceptor macrophages. Immunol Lett (2004) 95:185–92. doi: 10.1016/j.imlet.2004.07.007
111. Lee LYL, Höök M, Haviland D, Wetsel RA, Yonter EO, Syribeys P, et al. Inhibition of complement activation by a secreted Staphylococcus aureus protein. J Infect Dis (2004) 190:571–9. doi: 10.1086/422259
112. Rooijakkers SHM, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, et al. Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat Immunol (2005) 6:920–7. doi: 10.1038/ni1235
113. de Jong NWM, Vrieling M, Garcia BL, Koop G, Brettmann M, PC A, et al. Identification of a staphylococcal complement inhibitor with broad host specificity in equid Staphylococcus aureus strains. J Biol Chem (2018) 293:4468–77. doi: 10.1074/JBC.RA117.000599
114. Haupt K, Reuter M, van den Elsen J, Burman J, Hälbich S, Richter J, et al. The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement factor H and C3b. PLoS Pathog (2008) 4:e1000250. doi: 10.1371/journal.ppat.1000250
115. Amdahl H, Haapasalo K, Tan L, Meri T, Kuusela PI, van SJA, et al. Staphylococcal protein Ecb impairs complement receptor-1 mediated recognition of opsonized bacteria. PLoS One (2017) 12:e0172675. doi: 10.1371/JOURNAL.PONE.0172675
116. Dorrington KJ. Properties of the Fc receptor on macrophages and monocytes. Immunol Commun (1976) 5:263–80. doi: 10.3109/08820137609044280
117. Mellman I, Koch T, Healey G, Hunziker W, Lewis V, Plutner H, et al. Structure and function of Fc receptors on macrophages and lymphocytes. J Cell Sci (1988) 1988:45–65. doi: 10.1242/jcs.1988.Supplement_9.3
118. Fitzer-Attas CJ, Lowry M, Crowley MT, Finn AJ, Meng F, DeFranco AL, et al. Fcγ Receptor–Mediated Phagocytosis in Macrophages Lacking the Src Family Tyrosine Kinases Hck, Fgr, and Lyn. J Exp Med (2000) 191:669. doi: 10.1084/JEM.191.4.669
119. Christensson B, Hedstrom SÅ, Kronvall G. Detection of staphylococcus aureus antibodies in patrients with S. aureus infections and in normal persons, using solid phase radioimmunoassay. Acta Pathol Microbiol Scand Ser B Microbiol (1982) 90B:251–5. doi: 10.1111/j.1699-0463.1982.tb00113.x
120. Wu Y, Liu X, Akhgar A, Li JJ, Mok H, Sellman BR, et al. Prevalence of IgG and neutralizing antibodies against Staphylococcus aureus alpha-toxin in healthy human subjects and diverse patient populations. Infect Immun (2017) 86:e00671–17. doi: 10.1128/IAI.00671-17
121. Kloppot P, Selle M, Kohler C, Stentzel S, Fuchs S, Liebscher V, et al. Microarray-based identification of human antibodies against Staphylococcus aureus antigens. Proteomics - Clin Appl (2015) 9:1003–11. doi: 10.1002/prca.201400123
122. Forsgren A, Sjöquist J. “Protein A” from S. aureus I. Pseudo-immune reaction with human gamma-globulin. J Immunol (1966) 97:822–7.
123. Zhang L, Jacobsson K, Vasi J, Lindberg M, Frykberg L. A second IgG-binding protein in Staphylococcus aureus. Microbiology (1998) 144:985–91. doi: 10.1099/00221287-144-4-985
124. Hsieh S, Goldstein E, Lippert W, Margulies L. Effect of Protein A on the antistaphylococcal defense mechanisms of the murine lung. J Infect Dis (1978) 138:754–9. doi: 10.1093/infdis/138.6.754
125. Vega B, Garcia JD, Hernandez F. Immunoglobulin Fc receptors in clinical strains of Staphylococcus aureus do not confer resistance to Phagocytosis in an in vitro assay. Rev Inst Med Trop Sao Paulo (1999) 41:143–5. doi: 10.1590/S0036-46651999000300001
126. Lee DA, Hoidal JR, Garlich DJ, Clawson CC, Quie PG, Peterson PK. Opsonin-independent phagocytosis of surface-adherent bacteria by human alveolar macrophages. J Leukoc Biol (1984) 36:689–701. doi: 10.1002/jlb.36.6.689
127. Rooijakkers SHM, van Wamel WJB, Ruyken M, van Kessel KPM, van Strijp JAG. Anti-opsonic properties of staphylokinase. Microbes Infect (2005) 7:476–84. doi: 10.1016/j.micinf.2004.12.014
128. Downey GP, Botelho RJ, Butler JR, Moltyaner Y, Chien P, Schreiber AD, et al. Phagosomal maturation, acidification, and inhibition of bacterial growth in nonphagocytic cells transfected with FcγRIIA receptors. J Biol Chem (1999) 274:28436–44. doi: 10.1074/jbc.274.40.28436
129. Buscher K, Ehinger E, Gupta P, Pramod AB, Wolf D, Tweet G, et al. Natural variation of macrophage activation as disease-relevant phenotype predictive of inflammation and cancer survival. Nat Commun (2017) 8:16041. doi: 10.1038/ncomms16041
130. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (2000) 161:6878–84. doi: 10.4049/jimmunol.164.12.6166
131. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS–) vs. alternatively activated macrophages. Front Immunol (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
132. Nahrendorf M, Swirski FK. Abandoning M1/M2 for a network model of macrophage function. Circ Res (2016) 119:414. doi: 10.1161/CIRCRESAHA.116.309194
133. Thurlow LR, Hanke ML, Fritz T, Angle A, Aldrich A, Williams SH, et al. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol (2011) 186:6585–96. doi: 10.4049/jimmunol.1002794
134. Secor PR, James GA, Fleckman P, Olerud JE, McInnerney K, Stewart PS. Staphylococcus aureus biofilm and planktonic cultures differentially impact gene expression, MAPK phosphorylation, and cytokine production in human keratinocytes. BMC Microbiol (2011) 11:143. doi: 10.1186/1471-2180-11-143
135. Asai A, Tsuda Y, Kobayashi M, Hanafusa T, Herndon DN, Suzuki F. Pathogenic role of macrophages in intradermal infection of methicillin-resistant Staphylococcus aureus in thermally injured mice. Infect Immun (2010) 78:4311–9. doi: 10.1128/IAI.00642-10
136. Werz O, Gerstmeier J, Libreros S, De la Rosa X, Werner M, Norris PC, et al. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat Commun (2018) 9:59. doi: 10.1038/s41467-017-02538-5
137. Brann KR, Fullerton MS, Onyilagha FI, Prince AA, Kurten RC, Rom JS, et al. Infection of Primary Human Alveolar Macrophages Alters Staphylococcus aureus Toxin Production and Activity. Infect Immun (2019) 87:e00167–19. doi: 10.1128/IAI.00167-19
138. Bosedasgupta S, Pieters J. Inflammatory stimuli reprogram macrophage phagocytosis to macropinocytosis for the rapid elimination of pathogens. PLoS Pathog (2014) 10:e1003879. doi: 10.1371/journal.ppat.1003879
139. Chan LC, Rossetti M, Miller LS, Filler SG, Johnson CW, Lee HK, et al. Protective immunity in recurrent Staphylococcus aureus infection reflects localized immune signatures and macrophage-conferred memory. Proc Natl Acad Sci (2018) 115:E11111–9. doi: 10.1073/pnas.1808353115
140. Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity (1999) 11:443–51. doi: 10.1016/s1074-7613(00)80119-3
141. Schäffler H, Demircioglu DD, Kühner D, Menz S, Bender A, Autenrieth IB, et al. NOD2 stimulation by Staphylococcus aureus-derived peptidoglycan is boosted by Toll-like receptor 2 costimulation with lipoproteins in dendritic cells. Infect Immun (2014) 82:4681–8. doi: 10.1128/IAI.02043-14
142. Hashimoto M, Tawaratsumida K, Kariya H, Aoyama K, Tamura T, Suda Y. Lipoprotein is a predominant Toll-like receptor 2 ligand in Staphylococcus aureus cell wall components. Int Immunol (2006) 18:355–62. doi: 10.1093/intimm/dxh374
143. Takeuchi O, Takeda K, Hoshino K, Adachi O, Ogawa T, Akira S. Cellular responses to bacterial cell wall components are mediated through MyD88-dependent signaling cascades. Int Immunol (2000) 12:113–7. doi: 10.1093/intimm/12.1.113
144. Bishayi B, Bandyopadhyay D, Majhi A, Adhikary R. Possible role of Toll-like receptor-2 in the intracellular survival of Staphylococcus aureus in murine peritoneal macrophages: involvement of cytokines and anti-oxidant enzymes. Scand J Immunol (2014) 80:127–43. doi: 10.1111/sji.12195
145. Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol (2000) 165:5392–6. doi: 10.4049/jimmunol.165.10.5392
146. Ip WKE, Sokolovska A, Charriere GM, Boyer L, Dejardin S, Cappillino MP, et al. Phagocytosis and phagosome acidification are required for pathogen processing and MyD88-dependent responses to Staphylococcus aureus. J Immunol (2010) 184:7071–81. doi: 10.4049/jimmunol.1000110
147. Janot L, Secher T, Torres D, Maillet I, Pfeilschifter J, Quesniaux VFJ, et al. CD14 works with Toll-like receptor 2 to contribute to recognition and control of Listeria monocytogenes infection. J Infect Dis (2008) 198:115–24. doi: 10.1086/588815
148. Raby A-C, Holst B, Le Bouder E, Diaz C, Ferran E, Conraux L, et al. Targeting the TLR co-receptor CD14 with TLR2-derived peptides modulates immune responses to pathogens. Sci Transl Med (2013) 5:185ra64. doi: 10.1126/scitranslmed.3005544
149. da Silva TA, Zorzetto-Fernandes ALV, Cecílio NT, Sardinha-Silva A, Fernandes FF, Roque-Barreira MC. CD14 is critical for TLR2-mediated M1 macrophage activation triggered by N-glycan recognition. Sci Rep (2017) 7:7083. doi: 10.1038/s41598-017-07397-0
150. Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by Toll-like receptor 2. J Biol Chem (1999) 274:17406–9. doi: 10.1074/jbc.274.25.17406
151. Gillrie MR, Zbytnuik L, McAvoy E, Kapadia R, Lee K, Waterhouse CCM, et al. Divergent roles of Toll-like receptor 2 in response to lipoteichoic acid and. Staphylococcus aureus vivo Eur J Immunol (2010) 40:1639–50. doi: 10.1002/eji.200939929
152. Fang L, Wu H-M, Ding P-S, Liu R-Y. TLR2 mediates phagocytosis and autophagy through JNK signaling pathway in Staphylococcus aureus-stimulated RAW264.7 cells. Cell Signal (2014) 26:806–14. doi: 10.1016/J.CELLSIG.2013.12.016
153. Watanabe I, Ichiki M, Shiratsuchi A, Nakanishi Y. TLR2-mediated survival of Staphylococcus aureus in macrophages: a novel bacterial strategy against host innate immunity. J Immunol (2007) 178:4917–25. doi: 10.4049/jimmunol.178.8.4917
154. Bubeck Wardenburg J, Williams WA, Missiakas D. Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc Natl Acad Sci USA (2006) 103:13831–6. doi: 10.1073/pnas.0603072103
155. Zhu F, Yue W, Wang Y. The Nuclear Factor Kappa B (NF-κB) activation is required for phagocytosis of Staphylococcus aureus by RAW 264.7 cells. Exp Cell Res (2014) 327:256–63. doi: 10.1016/J.YEXCR.2014.04.018
156. Zhang B, Wu H, Fang L, Ding P, Xu K, Yang Q, et al. MerTK does not mediate phagocytosis of Staphylococcus aureus but attenuates inflammation induced by staphylococcal lipoteichoic acid through blocking NF-κb activation. Inflammation (2017) 40:1543–52. doi: 10.1007/s10753-017-0595-4
157. Xu F, Kang Y, Zhang H, Piao Z, Yin H, Diao R, et al. Akt1-mediated regulation of macrophage polarization in a murine model of Staphylococcus aureus pulmonary infection. J Infect Dis (2013) 208:528–38. doi: 10.1093/infdis/jit177
158. Jingjing Z, Nan Z, Wei W, Qinghe G, Weijuan W, Peng W, et al. MicroRNA-24 modulates Staphylococcus aureus-induced macrophage polarization by suppressing CHI3L1. Inflammation (2017) 40:995–1005. doi: 10.1007/s10753-017-0543-3
159. Krysko O, Holtappels G, Zhang N, Kubica M, Deswarte K, Derycke L, et al. Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy (2011) 66:396–403. doi: 10.1111/j.1398-9995.2010.02498.x
160. Peng K-T, Hsieh C-C, Huang T-Y, Chen P-C, Shih H-N, Lee MS, et al. Staphylococcus aureus biofilm elicits the expansion, activation and polarization of myeloid-derived suppressor cells in vivo and. vitro PLoS One (2017) 12:e0183271. doi: 10.1371/journal.pone.0183271
161. Van Zele T, Gevaert P, Watelet J-B, Claeys G, Holtappels G, Claeys C, et al. Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis. J Allergy Clin Immunol (2004) 114:981–3. doi: 10.1016/j.jaci.2004.07.013
162. Gries CM, Bruger EL, Moormeier DE, Scherr TD, Waters CM, Kielian T. Cyclic di-AMP released from Staphylococcus aureus biofilm induces a macrophage type I interferon response. Infect Immun (2016) 84:3564–74. doi: 10.1128/IAI.00447-16
163. Scherr TD, Hanke ML, Huang O, James DBA, Horswill AR, Bayles KW, et al. Staphylococcus aureus biofilms induce macrophage dysfunction through leukocidin AB and alpha-toxin. MBio (2015) 6:e01021–15. doi: 10.1128/mBio.01021-15
164. Palmqvist N, Patti J, Tarkowski A, Josefsson E. Expression of staphylococcal clumping factor A impedes macrophage phagocytosis. Microbes Infect (2004) 6:188–95. doi: 10.1016/J.MICINF.2003.11.005
165. Aldrich AL, Heim CE, Shi W, Fallet RW, Duan B, Kielian T. TLR2 and caspase-1 signaling are critical for bacterial containment but not clearance during craniotomy-associated biofilm infection. J Neuroinflamm (2020) 17:114. doi: 10.1186/s12974-020-01793-6
166. McLaughlin R, Hoogewerf A. Interleukin-1beta-induced growth enhancement of Staphylococcus aureus occurs in biofilm but not planktonic cultures. Microb Pathog (2006) 41:67–79. doi: 10.1016/J.MICPATH.2006.04.005
167. Hanke ML, Angle A, Kielian T. MyD88-dependent signaling influences fibrosis and alternative macrophage activation during Staphylococcus aureus biofilm infection. PLoS One (2012) 7:e42476. doi: 10.1371/JOURNAL.PONE.0042476
168. Hanke ML, Heim CE, Angle A, Sanderson SD, Kielian T. Targeting macrophage activation for the prevention and treatment of Staphylococcus aureus biofilm infections. J Immunol (2013) 190:2159–68. doi: 10.4049/jimmunol.1202348
169. Yamada KJ, Heim CE, Xi X, Attri KS, Wang D, Zhang W, et al. Monocyte metabolic reprogramming promotes pro-inflammatory activity and Staphylococcus aureus biofilm clearance. PLoS Pathog (2020) 16:e1008354. doi: 10.1371/journal.ppat.1008354
170. Harrison DA. The Jak/STAT pathway. Cold Spring Harb Perspect Biol (2012) 4:a011205. doi: 10.1101/cshperspect.a011205
171. Sun A, Zhang H, Pang F, Niu G, Chen J, Chen F, et al. Essential genes of the macrophage response to Staphylococcus aureus exposure. Cell Mol Biol Lett (2018) 23:25. doi: 10.1186/s11658-018-0090-4
172. Zhu F, Zhou Y, Jiang C, Zhang X. Role of JAK-STAT signaling in maturation of phagosomes containing Staphylococcus aureus. Sci Rep (2015) 5:14854. doi: 10.1038/srep14854
173. Gopal R, Lee B, McHugh KJ, Rich HE, Ramanan K, Mandalapu S, et al. STAT2 signaling regulates macrophage phenotype during influenza and bacterial super-infection. Front Immunol (2018) 9:2151. doi: 10.3389/fimmu.2018.02151
174. Wang Z, Zhu F. The expression profiles of immune genes in Mus musculus macrophages during Staphylococcus aureus infection. PLoS One (2018) 13:e0190490. doi: 10.1371/journal.pone.0190490
175. Koziel J, Maciag-Gudowska A, Mikolajczyk T, Bzowska M, Sturdevant DE, Whitney AR, et al. Phagocytosis of Staphylococcus aureus by macrophages exerts cytoprotective effects manifested by the upregulation of antiapoptotic factors. PLoS One (2009) 4:e5210. doi: 10.1371/journal.pone.0005210
176. Koziel J, Kmiecik K, Chmiest D, Maresz K, Mizgalska D, Maciag-Gudowska A, et al. The role of Mcl-1 in S. aureus-induced cytoprotection of infected macrophages. Mediators Inflamm (2013) 2013:1–12. doi: 10.1155/2013/427021
177. Kobayashi SD, Braughton KR, Whitney AR, Voyich JM, Schwan TG, Musser JM, et al. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc Natl Acad Sci (2003) 100:10948–53. doi: 10.1073/pnas.1833375100
178. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14:36–49. doi: 10.1038/nri3581
179. Parker D, Planet PJ, Soong G, Narechania A, Prince A. Induction of type I interferon signaling determines the relative pathogenicity of Staphylococcus aureus strains. PLoS Pathog (2014) 10:e1003951. doi: 10.1371/journal.ppat.1003951
180. Martin FJ, Gomez MI, Wetzel DM, Memmi G, O’Seaghdha M, Soong G, et al. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J Clin Invest (2009) 119:1931–9. doi: 10.1172/jci35879
181. Peignier A, Planet PJ, Parker D. Differential induction of type I and III interferons by Staphylococcus aureus. Infect Immun (2020) 88:e00352–20. doi: 10.1128/IAI.00352-20
182. Kaplan A, Ma J, Kyme P, Wolf AJ, Becker CA, Tseng CW, et al. Failure to induce interferon-beta production during Staphylococcus aureus infection contributes to pathogenicity. J Immunol (2012) 189:4537. doi: 10.4049/JIMMUNOL.1201111
183. Bergstrøm B, Aune MH, Awuh JA, Kojen JF, Blix KJ, Ryan L, et al. TLR8 senses Staphylococcus aureus RNA in human primary monocytes and macrophages and induces IFN-β production via a TAK1-IKKβ-IRF5 signaling pathway. J Immunol (2015) 195:1100–11. doi: 10.4049/jimmunol.1403176
184. Miller LS, O’Connell RM, Gutierrez MA, Pietras EM, Shahangian A, Gross CE, et al. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity (2006) 24:79–91. doi: 10.1016/j.immuni.2005.11.011
185. Miller LS, Pietras EM, Uricchio LH, Hirano K, Rao S, Lin H, et al. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol (2007) 179:6933–42. doi: 10.4049/jimmunol.179.10.6933
186. Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1β causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol (2005) 32:311–8. doi: 10.1165/rcmb.2004-0309OC
187. Pires S, Parker D. IL-1β activation in response to Staphylococcus aureus lung infection requires inflammasome-dependent and independent mechanisms. Eur J Immunol (2018) 48:1707–16. doi: 10.1002/eji.201847556
188. Ali RA, Wuescher LM, Dona KR, Worth RG. Platelets mediate host defense against Staphylococcus aureus through direct bactericidal activity and by enhancing macrophage activities. J Immunol (2017) 198:344–51. doi: 10.4049/jimmunol.1601178
189. Dupre-Crochet S, Erard M, Nubetae O. ROS production in phagocytes: why, when, and where? J Leukoc Biol (2013) 94:657–70. doi: 10.1189/jlb.1012544
190. Guerra FE, Borgogna TR, Patel DM, Sward EW, Voyich JM. Epic immune battles of history: neutrophils vs. Staphylococcus aureus. Front Cell Infect Microbiol (2017) 7:286. doi: 10.3389/fcimb.2017.00286
191. Tian W, Li XJ, Stull ND, Ming W, Suh C-I, Bissonnette SA, et al. Fc gamma R-stimulated activation of the NADPH oxidase: phosphoinositide-binding protein p40phox regulates NADPH oxidase activity after enzyme assembly on the phagosome. Blood (2008) 112:3867–77. doi: 10.1182/blood-2007-11-126029
192. Abo A, Pick E, Hall A, Totty N, Teahan CG, Segal AW. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature (1991) 353:668–70. doi: 10.1038/353668a0
193. Knaus UG, Heyworth PG, Evans T, Curnutte JT, Bokoch GM. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science (1991) 254:1512–5. doi: 10.1126/science.1660188
194. Di-Poï N, Fauré J, Grizot S, Molnár G, Pick E, Dagher MC. Mechanism of NADPH oxidase activation by the Rac/Rho-GDI complex. Biochemistry (2001) 40:10014–22. doi: 10.1021/bi010289c
195. Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J (2005) 390:1–9. doi: 10.1042/BJ20050104
196. Segal AW. The NADPH oxidase and chronic granulomatous disease. Mol Med Today (1996) 2:129–35. doi: 10.1016/1357-4310(96)88723-5
197. Casbon A-J, Allen L-AH, Dunn KW, Dinauer MC. Macrophage NADPH oxidase flavocytochrome B localizes to the plasma membrane and Rab11-positive recycling endosomes. J Immunol (2009) 182:2325–39. doi: 10.4049/jimmunol.0803476
198. Anderson KE, Chessa TAM, Davidson K, Henderson RB, Walker S, Tolmachova T, et al. PtdIns3P and Rac direct the assembly of the NADPH oxidase on a novel, pre-phagosomal compartment during FcR-mediated phagocytosis in primary mouse neutrophils. Blood (2010) 116:4978–89. doi: 10.1182/blood-2010-03-275602
199. Schlam D, Bohdanowicz M, Chatgilialoglu A, Steinberg BE, Ueyama T, Du G, et al. Diacylglycerol kinases terminate diacylglycerol signaling during the respiratory burst leading to heterogeneous phagosomal NADPH oxidase activation. J Biol Chem (2013) 288:23090–104. doi: 10.1074/jbc.M113.457606
200. Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Med (Baltimore) (2000) 79:170–200. doi: 10.1097/00005792-200005000-00004
201. Fang FC. Antimicrobial actions of reactive oxygen species. MBio (2011) 2:e00141–11. doi: 10.1128/mBio.00141-11
202. Kuhns DB. Assessment of neutrophil function. Clin Immunol (2008), 1461–70. doi: 10.1016/B978-0-323-04404-2.10099-5
203. Lam GY, Huang J, Brumell JH. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin Immunopathol (2010) 32:415–30. doi: 10.1007/s00281-010-0221-0
204. Cassat JE, Skaar EP. Iron in infection and immunity. Cell Host Microbe (2013) 13:509–19. doi: 10.1016/j.chom.2013.04.010
205. Rodrigues MR, Rodriguez D, Russo M, Campa A. Macrophage activation includes high intracellular myeloperoxidase activity. Biochem Biophys Res Commun (2002) 292:869–73. doi: 10.1006/bbrc.2002.6724
206. Lanza F. Clinical manifestation of myeloperoxidase deficiency. J Mol Med (1998) 76:676–81. doi: 10.1007/s001090050267
207. Lehrer RI, Cline MJ. Leukocyte myeloperoxidase deficiency and disseminated candidiasis: the role of myeloperoxidase in resistance to Candida infection. J Clin Invest (1969) 48:1478–88. doi: 10.1172/JCI106114
208. Goldblatt D. Recent advances in chronic granulomatous disease. J Infect (2014) 69:S32–5. doi: 10.1016/j.jinf.2014.07.013
209. Nunoi H, Rotrosen D, Gallin JI, Malech HL. Two forms of autosomal chronic granulomatous disease lack distinct neutrophil cytosol factors. Science (1988) 242:1298–301. doi: 10.1126/science.2848319
210. Clark RA, Malech HL, Gallin JI, Nunoi H, Volpp BD, Pearson DW, et al. Genetic variants of chronic granulomatous disease: prevalence of deficiencies of two cytosolic components of the NADPH oxidase system. N Engl J Med (1989) 321:647–52. doi: 10.1056/NEJM198909073211005
211. Curnutte JT, Scott PJ, Mayo LA. Cytosolic components of the respiratory burst oxidase: resolution of four components, two of which are missing in complementing types of chronic granulomatous disease. Proc Natl Acad Sci USA (1989) 86:825–9. doi: 10.1073/pnas.86.3.825
212. Teahan C, Rowe P, Parker P, Totty N, Segal AW. The X-linked chronic granulomatous disease gene codes for the beta-chain of cytochrome b-245. Nature (1987) 327:720–1. doi: 10.1038/327720a0
213. Segal AW. Absence of both cytochrome b-245 subunits from neutrophils in X-linked chronic granulomatous disease. Nature (1987) 326:88–91. doi: 10.1038/326088a0
214. Winkelstein JA, Marino MC, Johnston RBJ, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Med (Baltimore) (2000) 79:155–69. doi: 10.1097/00005792-200005000-00003
215. Lublin M, Bartlett DL, Danforth DN, Kauffman H, Gallin JI, Malech HL, et al. Hepatic abscess in patients with chronic granulomatous disease. Ann Surg (2002) 235:383–91. doi: 10.1097/00000658-200203000-00010
216. Bogdan C, Rollinghoff M, Diefenbach A. The role of nitric oxide in innate immunity. Immunol Rev (2000) 173:17–26. doi: 10.1034/j.1600-065x.2000.917307.x
217. Weinberg JB, Misukonis MA, Shami PJ, Mason SN, Sauls DL, Dittman WA, et al. Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood (1995) 86:1184–95. doi: 10.1182/blood.V86.3.1184.1184
218. Richardson AR, Payne EC, Younger N, Karlinsey JE, Thomas VC, Becker LA, et al. Multiple targets of nitric oxide in the tricarboxylic acid cycle of Salmonella enterica serovar typhimurium. Cell Host Microbe (2011) 10:33–43. doi: 10.1016/j.chom.2011.06.004
219. Richardson AR, Soliven KC, Castor ME, Barnes PD, Libby SJ, Fang FC. The Base Excision Repair system of Salmonella enterica serovar typhimurium counteracts DNA damage by host nitric oxide. PLoS Pathog (2009) 5:e1000451. doi: 10.1371/journal.ppat.1000451
220. Pizzolla A, Hultqvist M, Nilson B, Grimm MJ, Eneljung T, Jonsson I-M, et al. Reactive oxygen species produced by the NADPH oxidase 2 complex in monocytes protect mice from bacterial infections. J Immunol (2012) 188:5003–11. doi: 10.4049/jimmunol.1103430
221. Reeves EP, Lu H, Jacobs HL, Messina CGM, Bolsover S, Gabella G, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature (2002) 416:291–7. doi: 10.1038/416291a
222. VanderVen B, Yates RM, Russell DG. Intraphagosomal measurement of the magnitude and duration of the oxidative burst. Traffic (2009) 10:372–8. doi: 10.1111/j.1600-0854.2009.00877.x
223. Canton J, Khezri R, Glogauer M, Grinstein S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol Biol Cell (2014) 25:3330–41. doi: 10.1091/mbc.E14-05-0967
224. Winterbourn CC, Hampton MB, Livesey JH, Kettle AJ. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. J Biol Chem (2006) 281:39860–9. doi: 10.1074/jbc.M605898200
225. Slauch JM. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol Microbiol (2011) 80:580–3. doi: 10.1111/j.1365-2958.2011.07612.x
226. Ariffin JK, das Gupta K, Kapetanovic R, Iyer A, Reid RC, Fairlie DP, et al. Histone deacetylase inhibitors promote mitochondrial reactive oxygen species production and bacterial clearance by human macrophages. Antimicrob Agents Chemother (2015) 60:1521–9. doi: 10.1128/AAC.01876-15
227. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature (2011) 472:476–80. doi: 10.1038/nature09973
228. Abuaita BH, Schultz TL, O’Riordan MX. Mitochondria-derived vesicles deliver antimicrobial reactive oxygen species to control phagosome-localized Staphylococcus aureus. Cell Host Microbe (2018) 24:625–36.e5. doi: 10.1016/j.chom.2018.10.005
229. Cohen TS, Boland ML, Boland BB, Takahashi V, Tovchigrechko A, Lee Y, et al. S. aureus evades macrophage killing through NLRP3-dependent effects on mitochondrial trafficking. Cell Rep (2018) 22:2431–41. doi: 10.1016/j.celrep.2018.02.027
230. Krause K, Daily K, Estfanous S, Hamilton K, Badr A, Abu Khweek A, et al. Caspase-11 counteracts mitochondrial ROS-mediated clearance of Staphylococcus aureus in macrophages. EMBO Rep (2019) 20:e48109. doi: 10.15252/embr.201948109
231. de Jong NWM, Ramyar KX, Guerra FE, Nijland R, Fevre C, Voyich JM, et al. Immune evasion by a staphylococcal inhibitor of myeloperoxidase. Proc Natl Acad Sci USA (2017) 114:9439–44. doi: 10.1073/pnas.1707032114
232. Karavolos MH, Horsburgh MJ, Ingham E, Foster SJ. Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology (2003) 149:2749–58. doi: 10.1099/mic.0.26353-0
233. Das D, Saha SS, Bishayi B. Intracellular survival of Staphylococcus aureus: correlating production of catalase and superoxide dismutase with levels of inflammatory cytokines. Inflammation Res (2008) 57:340–9. doi: 10.1007/s00011-007-7206-z
234. Clements MO, Watson SP, Foster SJ. Characterization of the major superoxide dismutase of Staphylococcus aureus and its role in starvation survival, stress resistance, and pathogenicity. J Bacteriol (1999) 181:3898–903. doi: 10.1128/JB.181.13.3898-3903.1999
235. Schneider WP, Ho SK, Christine J, Yao M, Marra A, Hromockyj AE. Virulence gene identification by differential fluorescence induction analysis of Staphylococcus aureus gene expression during infection-simulating culture. Infect Immun (2002) 70:1326–33. doi: 10.1128/iai.70.3.1326-1333.2002
236. Garcia YM, Barwinska-Sendra A, Tarrant E, Skaar EP, Waldron KJ, Kehl-Fie TE. A superoxide dismutase capable of functioning with iron or manganese promotes the resistance of Staphylococcus aureus to calprotectin and nutritional immunity. PLoS Pathog (2017) 13:e1006125. doi: 10.1371/journal.ppat.1006125
237. Cosgrove K, Coutts G, Jonsson I-M, Tarkowski A, Kokai-Kun JF, Mond JJ, et al. Catalase (KatA) and alkyl hydroperoxide reductase (AhpC) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J Bacteriol (2007) 189:1025–35. doi: 10.1128/JB.01524-06
238. Mashruwala AA, Boyd JM. The Staphylococcus aureus SrrAB regulatory system modulates hydrogen peroxide resistance factors, which imparts protection to aconitase during aerobic growth. PLoS One (2017) 12:e0170283. doi: 10.1371/journal.pone.0170283
239. Pandey S, Sahukhal GS, Elasri MO. The msaABCR operon regulates the response to oxidative stress in Staphylococcus aureus. J Bacteriol (2019) 201:e00417–19. doi: 10.1128/JB.00417-19
240. Grosser MR, Weiss A, Shaw LN, Richardson AR. Regulatory requirements for Staphylococcus aureus nitric oxide resistance. J Bacteriol (2016) 198:2043–55. doi: 10.1128/JB.00229-16
241. Nobre LS, Gonçalves VL, Saraiva LM. Flavohemoglobin of Staphylococcus aureus. Methods Enzymol (2008) 436:203–16. doi: 10.1016/S0076-6879(08)36011-X
242. Bang I-S, Liu L, Vazquez-Torres A, Crouch M-L, Stamler JS, Fang FC. Maintenance of nitric oxide and redox homeostasis by the Salmonella flavohemoglobin hmp. J Biol Chem (2006) 281:28039–47. doi: 10.1074/jbc.M605174200
243. Kinkel TL, Roux CM, Dunman PM, Fang FC. The Staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. MBio (2013) 4:e00696–13. doi: 10.1128/mBio.00696-13
244. Rodionov DA, Dubchak IL, Arkin AP, Alm EJ, Gelfand MS. Dissimilatory metabolism of nitrogen oxides in bacteria: comparative reconstruction of transcriptional networks. PLoS Comput Biol (2005) 1:e55. doi: 10.1371/journal.pcbi.0010055
245. Gill SR, Fouts DE, Archer GL, Mongodin EF, Deboy RT, Ravel J, et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol (2005) 187:2426–38. doi: 10.1128/JB.187.7.2426-2438.2005
246. Massilamany C, Gangaplara A, Gardner DJ, Musser JM, Steffen D, Somerville GA, et al. TCA cycle inactivation in Staphylococcus aureus alters nitric oxide production in RAW 264.7 cells. Mol Cell Biochem (2011) 355:75–82. doi: 10.1007/s11010-011-0840-3
247. Nandi A, Bishayi B. Intracellularly survived Staphylococcus aureus after phagocytosis are more virulent in inducing cytotoxicity in fresh murine peritoneal macrophages utilizing TLR-2 as a possible target. Microb Pathog (2016) 97:131–47. doi: 10.1016/j.micpath.2016.06.007
248. Grayczyk JP, Alonzo F3. Staphylococcus aureus lipoic acid synthesis limits macrophage reactive oxygen and nitrogen species production to promote survival during infection. Infect Immun (2019) 87:e00344–19. doi: 10.1128/IAI.00344-19
249. Grayczyk JP, Harvey CJ, Laczkovich I, Alonzo F3. A lipoylated metabolic protein released by Staphylococcus aureus suppresses macrophage activation. Cell Host Microbe (2017) 22:678–87.e9. doi: 10.1016/j.chom.2017.09.004
250. Zhang WJ, Frei B. Alpha-lipoic acid inhibits TNF-alpha-induced NF-kappaB activation and adhesion molecule expression in human aortic endothelial cells. FASEB J (2001) 15:2423–32. doi: 10.1096/fj.01-0260com
251. Packer L. alpha-Lipoic acid: a metabolic antioxidant which regulates NF-kappa B signal transduction and protects against oxidative injury. Drug Metab Rev (1998) 30:245–75. doi: 10.3109/03602539808996311
252. Kim H-S, Kim H-J, Park K-G, Kim Y-N, Kwon T-K, Park J-Y, et al. Alpha-lipoic acid inhibits matrix metalloproteinase-9 expression by inhibiting NF-kappaB transcriptional activity. Exp Mol Med (2007) 39:106–13. doi: 10.1038/emm.2007.12
253. Packer L, Witt EH, Tritschler HJ. alpha-Lipoic acid as a biological antioxidant. Free Radic Biol Med (1995) 19:227–50. doi: 10.1016/0891-5849(95)00017-r
254. Zhang WJ, Wei H, Hagen T, Frei B. Alpha-lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc Natl Acad Sci USA (2007) 104:4077–82. doi: 10.1073/pnas.0700305104
255. Bore E, Langsrud S, Langsrud Ø, Rode T, Holck A. Acid-shock Responses in Staphylococcus Aureus Investigated by Global Gene Expression Analysis. Microbiology (2007) 153:2289–303. doi: 10.1099/MIC.0.2007/005942-0
256. Lukacs G, Rotstein O, Grinstrein S. Phagosomal acidification is mediated by a vacuolar-type H+-ATPase in murine macrophages. J Biol Chem (1990) 265:21099–107.
257. Maxson ME, Grinstein S. The vacuolar-type H+-ATPase at a glance – more than a proton pump. J Cell Sci (2014) 127:4987–93. doi: 10.1242/JCS.158550
258. Sun-Wada G-H, Tabata H, Kawamura N, Aoyama M, Wada Y. Direct recruitment of H+-ATPase from lysosomes for phagosomal acidification. J Cell Sci (2009) 122:2504–13. doi: 10.1242/JCS.050443
259. Lukacs G, Rotstein O, Grinstein S. Determinants of the phagosomal pH in macrophages. J Biol Chem (1991) 266:24540–8.
260. Brisseau GF, Grinstein S, Hackam DJ, Nordström T, Manolson MF, Khine AA, et al. Interleukin-1 increases vacuolar-type H-ATPase activity in murine peritoneal macrophages. J Biol Chem (1996) 271:2005–11. doi: 10.1074/JBC.271.4.2005
261. Swallow C, Grinstein S, Sudsbury R, Rotstein O. Modulation of the macrophage respiratory burst by an acidic environment: the critical role of cytoplasmic pH regulation by proton extrusion pumps. Surgery (1990) 108:363–8.
262. Flannagan RS, Kuiack RC, McGavin MJ, Heinrichs DE. Staphylococcus aureus uses the GraXRS regulatory system to sense and adapt to the acidified phagolysosome in macrophages. MBio (2018) 9:e01143–18. doi: 10.1128/mBio.01143-18
263. Surewaard B, Kubes P. Measurement of bacterial capture and phagosome maturation of Kupffer cells by intravital microscopy. Methods (2017) 128:12–9. doi: 10.1016/J.YMETH.2017.05.004
264. Styrt B, Klempner M. Effects of pH on killing of Staphylococcus aureus and Escherichia coli by constituents of the neutrophil phagolysosome. J Med Microbiol (1988) 25:101–7. doi: 10.1099/00222615-25-2-101
265. Jarry TM, Cheung AL. Staphylococcus aureus escapes more efficiently from the phagosome of a cystic fibrosis bronchial epithelial cell line than from its normal counterpart. Infect Immun (2006) 74:2568–77. doi: 10.1128/IAI.74.5.2568-2577.2006
266. Lowy FD, Fant J, Higgins LL, Ogawa SK, Hatcher VB. Staphylococcus aureus-human endothelial cell interactions. J Ultrastruct Mol Struct Res (1988) 98:137–46. doi: 10.1016/s0889-1605(88)80906-6
267. Ben Shlomo S, Mouhadeb O, Cohen K, Varol C, Gluck N. COMMD10-Guided Phagolysosomal Maturation Promotes Clearance of Staphylococcus aureus in Macrophages. iScience (2019) 14:147. doi: 10.1016/J.ISCI.2019.03.024
268. Lacoma A, Cano V, Moranta D, Regueiro V, Domínguez-Villanueva D, Laabei M, et al. Investigating intracellular persistence of Staphylococcus aureus within a murine alveolar macrophage cell line. Virulence (2017) 8:1761–75. doi: 10.1080/21505594.2017.1361089
269. Tranchemontagne ZR, Camire RB, O’Donnell VJ, Baugh J, Burkholder KM. Staphylococcus aureus strain USA300 perturbs acquisition of lysosomal enzymes and requires phagosomal acidification for survival inside macrophages. Infect Immun (2016) 84:241–53. doi: 10.1128/IAI.00704-15
270. Sedlyarov V, Eichner R, Girardi E, Essletzbichler P, Goldmann U, Nunes-Hasler P, et al. The bicarbonate transporter SLC4A7 plays a key role in macrophage phagosome acidification. Cell Host Microbe (2018) 23:766–74.e5. doi: 10.1016/J.CHOM.2018.04.013
271. Kahl BC, Goulian M, van Wamel W, Herrmann M, Simon SM, Kaplan G, et al. Staphylococcus aureus RN6390 replicates and induces apoptosis in a pulmonary epithelial cell line. Infect Immun (2000) 68:5385–92. doi: 10.1128/IAI.68.9.5385-5392.2000
272. Boldock E, Surewaard BGJ, Shamarina D, Renshaw S, Foster S. Human skin commensals augment Staphylococcus aureus pathogenesis. Nat Microbiol (2018) 3:881–90. doi: 10.1038/s41564-018-0198-3
273. Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell (2006) 126:205–18. doi: 10.1016/j.cell.2006.05.035
274. Jankowski A, Scott C, Grinstein S. Determinants of the phagosomal pH in neutrophils. J Biol Chem (2002) 277:6059–66. doi: 10.1074/JBC.M110059200
275. Sokolovska A, Becker CE, Ip WKE, Rathinam VAK, Brudner M, Paquette N, et al. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nat Immunol (2013) 14:543–53. doi: 10.1038/ni.2595
276. Rybicka JM, Balce DR, Khan MF, Krohn RM, Yates RM. NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes. Proc Natl Acad Sci USA (2010) 107:10496. doi: 10.1073/PNAS.0914867107
277. Rybicka JM, Balce DR, Chaudhuri S, Allan ERO, Yates RM. Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner. EMBO J (2012) 31:932–44. doi: 10.1038/emboj.2011.440
278. Mantegazza AR, Savina A, Vermeulen M, Pérez L, Geffner J, Hermine O, et al. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood (2008) 112:4712–22. doi: 10.1182/blood-2008-01-134791
279. Delamarre L, Pack M, Chang H, Mellman I, Trombetta E. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science (2005) 307:1630–4. doi: 10.1126/SCIENCE.1108003
280. Steinberg BE, Huynh KK, Brodovitch A, Jabs S, Stauber T, Jentsch TJ, et al. A cation counterflux supports lysosomal acidification. J Cell Biol (2010) 189:1171–86. doi: 10.1083/jcb.200911083
281. Schroder BA, Wrocklage C, Hasilik A, Saftig P. The proteome of lysosomes. Proteomics (2010) 10:4053–76. doi: 10.1002/pmic.201000196
282. Bera A, Biswas R, Herbert S, Gotz F. The presence of peptidoglycan O-acetyltransferase in various staphylococcal species correlates with lysozyme resistance and pathogenicity. Infect Immun (2006) 74:4598–604. doi: 10.1128/IAI.00301-06
283. Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe (2010) 7:38–49. doi: 10.1016/j.chom.2009.12.008
284. Kebaier C, Chamberland RR, Allen IC, Gao X, Broglie PM, Hall JD, et al. Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J Infect Dis (2012) 205:807–17. doi: 10.1093/infdis/jir846
285. Muñoz-Planillo R, Franchi L, Miller LS, Núñez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol (2009) 183:3942–8. doi: 10.4049/jimmunol.0900729
286. Melehani JH, Duncan JA. Inflammasome activation can mediate tissue-specific pathogenesis or protection in Staphylococcus aureus infection. Curr Top Microbiol Immunol (2016) 397:257–82. doi: 10.1007/978-3-319-41171-2_13
287. del Cerro-Vadillo E, Madrazo-Toca F, Carrasco-Marín E, Fernandez-Prieto L, Beck C, Leyva-Cobián F, et al. Cutting edge: a novel nonoxidative phagosomal mechanism exerted by cathepsin-D controls Listeria monocytogenes intracellular growth. J Immunol (2006) 176:1321–5. doi: 10.4049/jimmunol.176.3.1321
288. Kavanaugh JS, Leidal KG, Nauseef WM, Horswill AR. Cathepsin G degrades Staphylococcus aureus biofilms. J Infect Dis (2020) 6:12. doi: 10.1093/infdis/jiaa612
289. Müller S, Faulhaber A, Sieber C, Pfeifer D, Hochberg T, Gansz M, et al. The endolysosomal cysteine cathepsins L and K are involved in macrophage-mediated clearance of Staphylococcus aureus and the concomitant cytokine induction. FASEB J (2014) 28:162–75. doi: 10.1096/fj.13-232272
290. Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple cathepsins promote pro-IL-1β synthesis and NLRP3-mediated IL-1β activation. J Immunol (2015) 195:1685–97. doi: 10.4049/jimmunol.1500509
291. Chevriaux A, Pilot T, Derangère V, Simonin H, Martine P, Chalmin F, et al. Cathepsin B is required for NLRP3 inflammasome activation in macrophages, through NLRP3 interaction. Front Cell Dev Biol (2020) 8:167. doi: 10.3389/fcell.2020.00167
292. Amaral EP, Riteau N, Moayeri M, Maier N, Mayer-Barber KD, Pereira RM, et al. Lysosomal cathepsin release is required for NLRP3-inflammasome activation by Mycobacterium tuberculosis in infected macrophages. Front Immunol (2018) 9:1427. doi: 10.3389/fimmu.2018.01427
293. Weinrauch Y, Elsbach P, Madsen LM, Foreman A, Weiss J. The potent anti-Staphylococcus aureus activity of a sterile rabbit inflammatory fluid is due to a 14-kD phospholipase A2. J Clin Invest (1996) 97:250–7. doi: 10.1172/JCI118399
294. Wu Y, Raymond B, Goossens PL, Njamkepo E, Guiso N, Paya M, et al. Type-IIA secreted phospholipase A2 is an endogenous antibiotic-like protein of the host. Biochimie (2010) 92:583–7. doi: 10.1016/j.biochi.2010.01.024
295. Hunt CL, Nauseef WM, Weiss JP. Effect of D-alanylation of (lipo)teichoic acids of Staphylococcus aureus on host secretory phospholipase A2 action before and after phagocytosis by human neutrophils. J Immunol (2006) 176:4987–94. doi: 10.4049/jimmunol.176.8.4987
296. Koprivnjak T, Weidenmaier C, Peschel A, Weiss JP. Wall teichoic acid deficiency in Staphylococcus aureus confers selective resistance to mammalian group IIA phospholipase A(2) and human beta-defensin 3. Infect Immun (2008) 76:2169–76. doi: 10.1128/IAI.01705-07
297. Femling JK, Nauseef WM, Weiss JP. Synergy between extracellular group IIA phospholipase A2 and phagocyte NADPH oxidase in digestion of phospholipids of Staphylococcus aureus ingested by human neutrophils. J Immunol (2005) 175:4653–61. doi: 10.4049/jimmunol.175.7.4653
298. Kristian SA, Durr M, Van Strijp JAG, Neumeister B, Peschel A. MprF-mediated lysinylation of phospholipids in Staphylococcus aureus leads to protection against oxygen-independent neutrophil killing. Infect Immun (2003) 71:546–9. doi: 10.1128/iai.71.1.546-549.2003
299. Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J Exp Med (2001) 193:1067–76. doi: 10.1084/jem.193.9.1067
300. Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gotz F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem (1999) 274:8405–10. doi: 10.1074/jbc.274.13.8405
301. Li M, Cha DJ, Lai Y, Villaruz AE, Sturdevant DE, Otto M. The antimicrobial peptide-sensing system aps of Staphylococcus aureus. Mol Microbiol (2007) 66:1136–47. doi: 10.1111/j.1365-2958.2007.05986.x
302. Peyssonnaux C, Zinkernagel AS, Datta V, Lauth X, Johnson RS, Nizet V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood (2006) 107:3727–32. doi: 10.1182/blood-2005-06-2259
303. Weiss G. Modification of iron regulation by the inflammatory response. Best Pract Res Clin Haematol (2005) 18:183–201. doi: 10.1016/j.beha.2004.09.001
304. Nairz M, Schroll A, Sonnweber T, Weiss G. The struggle for iron - a metal at the host-pathogen interface. Cell Microbiol (2010) 12:1691–702. doi: 10.1111/j.1462-5822.2010.01529.x
305. Byrd TF, Horwitz MA. Interferon gamma-activated human monocytes downregulate transferrin receptors and inhibit the intracellular multiplication of Legionella pneumophila by limiting the availability of iron. J Clin Invest (1989) 83:1457–65. doi: 10.1172/JCI114038
306. Olakanmi O, Schlesinger LS, Ahmed A, Britigan BE. Intraphagosomal Mycobacterium tuberculosis acquires iron from both extracellular transferrin and intracellular iron pools. Impact of interferon-gamma and hemochromatosis. J Biol Chem (2002) 277:49727–34. doi: 10.1074/jbc.M209768200
307. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science (2004) 306:2090–3. doi: 10.1126/science.1104742
308. Ludwiczek S, Aigner E, Theurl I, Weiss G. Cytokine-mediated regulation of iron transport in human monocytic cells. Blood (2003) 101:4148–54. doi: 10.1182/blood-2002-08-2459
309. Sow FB, Alvarez GR, Gross RP, Satoskar AR, Schlesinger LS, Zwilling BS, et al. Role of STAT1, NF-kappaB, and C/EBPbeta in the macrophage transcriptional regulation of hepcidin by mycobacterial infection and IFN-gamma. J Leukoc Biol (2009) 86:1247–58. doi: 10.1189/jlb.1208719
310. Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood (2006) 108:3204–9. doi: 10.1182/blood-2006-06-027631
311. Chlosta S, Fishman DS, Harrington L, Johnson EE, Knutson MD, Wessling-Resnick M, et al. The iron efflux protein ferroportin regulates the intracellular growth of Salmonella enterica. Infect Immun (2006) 74:3065–7. doi: 10.1128/IAI.74.5.3065-3067.2006
312. Paradkar PN, De Domenico I, Durchfort N, Zohn I, Kaplan J, Ward DM. Iron depletion limits intracellular bacterial growth in macrophages. Blood (2008) 112:866–74. doi: 10.1182/blood-2007-12-126854
313. Sow FB, Florence WC, Satoskar AR, Schlesinger LS, Zwilling BS, Lafuse WP. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J Leukoc Biol (2007) 82:934–45. doi: 10.1189/jlb.0407216
314. Pepper RJ, Hamour S, Chavele K-M, Todd SK, Rasmussen N, Flint S, et al. Leukocyte and serum S100A8/S100A9 expression reflects disease activity in ANCA-associated vasculitis and glomerulonephritis. Kidney Int (2013) 83:1150–8. doi: 10.1038/ki.2013.2
315. Nacken W, Roth J, Sorg C, Kerkhoff C. S100A9/S100A8: Myeloid representatives of the S100 protein family as prominent players in innate immunity. Microsc Res Tech (2003) 60:569–80. doi: 10.1002/jemt.10299
316. Zygiel EM, Nelson CE, Brewer LK, Oglesby-Sherrouse AG, Nolan EM. The human innate immune protein calprotectin induces iron starvation responses in Pseudomonas aeruginosa. J Biol Chem (2019) 294:3549–62. doi: 10.1074/jbc.RA118.006819
317. Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science (2008) 319:962–5. doi: 10.1126/science.1152449
318. Liu JZ, Jellbauer S, Poe AJ, Ton V, Pesciaroli M, Kehl-Fie TE, et al. Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe (2012) 11:227–39. doi: 10.1016/j.chom.2012.01.017
319. Damo SM, Kehl-Fie TE, Sugitani N, Holt ME, Rathi S, Murphy WJ, et al. Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc Natl Acad Sci USA (2013) 110:3841–6. doi: 10.1073/pnas.1220341110
320. Radin JN, Zhu J, Brazel EB, McDevitt CA, Kehl-Fie TE. Synergy between nutritional immunity and independent host defenses contributes to the importance of the MntABC manganese transporter during Staphylococcus aureus infection. Infect Immun (2019) 87:e00642–18. doi: 10.1128/IAI.00642-18
321. Grim KP, San Francisco B, Radin JN, Brazel EB, Kelliher JL, Parraga Solorzano PK, et al. The metallophore staphylopine enables Staphylococcus aureus to compete with the host for zinc and overcome nutritional immunity. MBio (2017) 8:e01281–17. doi: 10.1128/mBio.01281-17
322. Pepper RJ, Wang H-H, Rajakaruna GK, Papakrivopoulou E, Vogl T, Pusey CD, et al. S100A8/A9 (calprotectin) is critical for development of glomerulonephritis and promotes inflammatory leukocyte-renal cell interactions. Am J Pathol (2015) 185:1264–74. doi: 10.1016/j.ajpath.2015.01.015
323. van Harten RM, van Woudenbergh E, van Dijk A, Haagsman HP. Cathelicidins: immunomodulatory antimicrobials. Vaccines (2018) 6:63. doi: 10.3390/vaccines6030063
324. Lishko VK, Moreno B, Podolnikova NP, Ugarova TP. Identification of human cathelicidin peptide LL-37 as a ligand for macrophage Integrin α(M)β(2) (Mac-1, CD11b/CD18) that promotes phagocytosis by opsonizing bacteria. Res Rep Biochem (2016) 2016:39–55. doi: 10.2147/rrbc.s107070
325. Bommineni YR, Pham GH, Sunkara LT, Achanta M, Zhang G. Immune regulatory activities of fowlicidin-1, a cathelicidin host defense peptide. Mol Immunol (2014) 59:55–63. doi: 10.1016/j.molimm.2014.01.004
326. Tang X, Basavarajappa D, Haeggström JZ, Wan M. P2X7 receptor regulates internalization of antimicrobial peptide LL-37 by human macrophages that promotes intracellular pathogen clearance. J Immunol (2015) 195:1191–201. doi: 10.4049/jimmunol.1402845
327. Cassat JE, Skaar EP. Metal ion acquisition in Staphylococcus aureus: overcoming nutritional immunity. Semin Immunopathol (2012) 34:215–35. doi: 10.1007/s00281-011-0294-4
328. Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev (2015) 264:182–203. doi: 10.1111/imr.12266
329. Weinberg ED. Iron and susceptibility to infectious disease. Science (1974) 184:952–6. doi: 10.1126/science.184.4140.952
330. Sheldon JR, Skaar EP. Metals as phagocyte antimicrobial effectors. Curr Opin Immunol (2019) 60:1–9. doi: 10.1016/j.coi.2019.04.002
331. Palmer LD, Skaar EP. Transition metals and virulence in bacteria. Annu Rev Genet (2016) 50:67–91. doi: 10.1146/annurev-genet-120215-035146
332. Samanovic MI, Ding C, Thiele DJ, Darwin KH. Copper in microbial pathogenesis: meddling with the metal. Cell Host Microbe (2012) 11:106–15. doi: 10.1016/j.chom.2012.01.009
333. Coady A, Xu M, Phung Q, Cheung TK, Bakalarski C, Alexander MK, et al. The Staphylococcus aureus ABC-type manganese transporter MntABC is critical for reinitiation of bacterial replication following exposure to phagocytic oxidative burst. PLoS One (2015) 10:e0138350. doi: 10.1371/journal.pone.0138350
334. Handke LD, Gribenko AV, Timofeyeva Y, Scully IL, Anderson AS. MntC-dependent manganese transport is essential for Staphylococcus aureus oxidative stress resistance and virulence. mSphere (2018) 3:e00336–18. doi: 10.1128/mSphere.00336-18
335. Grunenwald CM, Choby JE, Juttukonda LJ, Beavers WN, Weiss A, Torres VJ, et al. Manganese detoxification by MntE is critical for resistance to oxidative stress and virulence of Staphylococcus aureus. MBio (2019) 10:e02915–18. doi: 10.1128/mBio.02915-18
336. Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol (2012) 10:525–37. doi: 10.1038/nrmicro2836
337. Horsburgh MJ, Wharton SJ, Cox AG, Ingham E, Peacock S, Foster SJ. MntR modulates expression of the PerR regulon and superoxide resistance in Staphylococcus aureus through control of manganese uptake. Mol Microbiol (2002) 44:1269–86. doi: 10.1046/j.1365-2958.2002.02944.x
338. Nairz M, Schleicher U, Schroll A, Sonnweber T, Theurl I, Ludwiczek S, et al. Nitric oxide-mediated regulation of ferroportin-1 controls macrophage iron homeostasis and immune function in Salmonella infection. J Exp Med (2013) 210:855–73. doi: 10.1084/jem.20121946
339. Modun B, Evans RW, Joannou CL, Williams P. Receptor-mediated recognition and uptake of iron from human transferrin by Staphylococcus aureus and Staphylococcus epidermidis. Infect Immun (1998) 66:3591–6. doi: 10.1128/IAI.66.8.3591-3596.1998
340. Beasley FC, Marolda CL, Cheung J, Buac S, Heinrichs DE. Staphylococcus aureus transporters Hts, Sir, and Sst capture iron liberated from human transferrin by Staphyloferrin A, Staphyloferrin B, and catecholamine stress hormones, respectively, and contribute to virulence. Infect Immun (2011) 79:2345–55. doi: 10.1128/IAI.00117-11
341. Nobre LS, Saraiva LM. Role of the siderophore transporter SirABC in the Staphylococcus aureus resistance to oxidative stress. Curr Microbiol (2014) 69:164–8. doi: 10.1007/s00284-014-0567-y
342. Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, Deepe GSJ. Granulocyte macrophage-colony stimulating factor induced Zn sequestration enhances macrophage superoxide and limits intracellular pathogen survival. Immunity (2013) 39:697–710. doi: 10.1016/j.immuni.2013.09.006
343. White C, Lee J, Kambe T, Fritsche K, Petris MJ. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J Biol Chem (2009) 284:33949–56. doi: 10.1074/jbc.M109.070201
344. Emmett M, Kloos WE. Amino acid requirements of staphylococci isolated from human skin. Can J Microbiol (1975) 21:729–33. doi: 10.1139/m75-107
345. Lincoln RA, Leigh JA, Jones NC. The amino acid requirements of Staphylococcus aureus isolated from cases of bovine mastitis. Vet Microbiol (1995) 45:275–9. doi: 10.1016/0378-1135(95)00041-8
346. Murphy GR, Dunstan RH, Macdonald MM, Gottfries J, Roberts TK. Alterations in amino acid metabolism during growth by Staphylococcus aureus following exposure to H2O2 - a multifactorial approach. Heliyon (2018) 4:e00620. doi: 10.1016/j.heliyon.2018.e00620
347. Halsey CR, Lei S, Wax JK, Lehman MK, Nuxoll AS, Steinke L, et al. Amino acid catabolism in Staphylococcus aureus and the function of carbon catabolite repression. MBio (2017) 8:e01434–16. doi: 10.1128/mBio.01434-16
348. Bravo-Santano N, Ellis JK, Mateos LM, Calle Y, Keun HC, Behrends V, et al. Intracellular Staphylococcus aureus modulates host central carbon metabolism to activate autophagy. mSphere (2018) 3:e00374–18. doi: 10.1128/mSphere.00374-18
349. Parsons JB, Frank MW, Jackson P, Subramanian C, Rock CO. Incorporation of extracellular fatty acids by a fatty acid kinase-dependent pathway in Staphylococcus aureus. Mol Microbiol (2014) 92:234–45. doi: 10.1111/mmi.12556
350. Delekta PC, Shook JC, Lydic TA, Mulks MH, Hammer ND. Staphylococcus aureus utilizes host-derived lipoprotein particles as sources of fatty acids. J Bacteriol (2018) 200:e00728–17. doi: 10.1128/JB.00728-17
351. Sen S, Sirobhushanam S, Johnson SR, Song Y, Tefft R, Gatto C, et al. Growth-environment dependent modulation of Staphylococcus aureus branched-chain to straight-chain fatty acid ratio and incorporation of unsaturated fatty acids. PLoS One (2016) 11:e0165300. doi: 10.1371/journal.pone.0165300
352. Flannagan RS, Heinrichs DE. Macrophage-driven nutrient delivery to phagosomal Staphylococcus aureus supports bacterial growth. EMBO Rep (2020) 21:e50348. doi: 10.15252/embr.202050348
353. Canton J, Schlam D, Breuer C, Gütschow M, Glogauer M, Grinstein S. Calcium-sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat Commun (2016) 7:11284. doi: 10.1038/ncomms11284
354. Teng O, Ang CKE, Guan XL. Macrophage-bacteria interactions-a lipid-centric relationship. Front Immunol (2017) 8:1836. doi: 10.3389/fimmu.2017.01836
355. Bravo-Santano N, Ellis JK, Calle Y, Keun HC, Behrends V, Letek M. Intracellular Staphylococcus aureus elicits the production of host very long-chain saturated fatty acids with antimicrobial activity. Metabolites (2019) 9:148. doi: 10.3390/metabo9070148
356. Dye ES, Kapral FA. Characterization of a bactericidal lipid developing within staphylococcal abscesses. Infect Immun (1981) 32:98–104. doi: 10.1128/IAI.32.1.98-104.1981
357. Butcher GW, King G, Dyke KGH. Sensitivity of Staphylococcus aureus to unsaturated fatty acids. J Gen Microbiol (1976) 94:290–6. doi: 10.1099/00221287-94-2-290
358. Svahn SL, Grahnemo L, Pálsdóttir V, Nookaew I, Wendt K, Gabrielsson B, et al. Dietary polyunsaturated fatty acids increase survival and decrease bacterial load during septic Staphylococcus aureus infection and improve neutrophil function in mice. Infect Immun (2015) 83:514–21. doi: 10.1128/IAI.02349-14
359. Knapp HR, Melly MA. Bactericidal effects of polyunsaturated fatty acids. J Infect Dis (1986) 154:84–94. doi: 10.1093/infdis/154.1.84
360. Beavers WN, Monteith AJ, Amarnath V, Mernaugh RL, Roberts LJ, Chazin WJ, et al. Arachidonic acid kills Staphylococcus aureus through a lipid peroxidation mechanism. MBio (2019) 10:e01333–19. doi: 10.1128/mBio.01333-19
361. Neumann Y, Ohlsen K, Donat S, Engelmann S, Kusch H, Albrecht D, et al. The effect of skin fatty acids on Staphylococcus aureus. Arch Microbiol (2015) 197:245–67. doi: 10.1007/s00203-014-1048-1
362. Kenny JG, Ward D, Josefsson E, Jonsson I-M, Hinds J, Rees HH, et al. The Staphylococcus aureus response to unsaturated long chain free fatty acids: survival mechanisms and virulence implications. PLoS One (2009) 4:e4344. doi: 10.1371/journal.pone.0004344
363. Krute CN, Ridder MJ, Seawell NA, Bose JL. Inactivation of the exogenous fatty acid utilization pathway leads to increased resistance to unsaturated fatty acids in Staphylococcus aureus. Microbiology (2019) 165:197–207. doi: 10.1099/mic.0.000757
364. Mortensen JE, Shryock TR, Kapral FA. Modification of bactericidal fatty acids by an enzyme of Staphylococcus aureus. J Med Microbiol (1992) 36:293–8. doi: 10.1099/00222615-36-4-293
365. Lopez MS, Tan IS, Yan D, Kang J, McCreary M, Modrusan Z, et al. Host-derived fatty acids activate type VII secretion in Staphylococcus aureus. Proc Natl Acad Sci (2017) 114:11223–8. doi: 10.1073/PNAS.1700627114
366. Alnaseri H, Arsic B, Schneider JET, Kaiser JC, Scinocca ZC, Heinrichs DE, et al. Inducible expression of a resistance-nodulation-division-type efflux pump in Staphylococcus aureus provides resistance to linoleic and arachidonic acids. J Bacteriol (2015) 197:1893–905. doi: 10.1128/JB.02607-14
367. O’Keeffe KM, Wilk MM, Leech JM, Murphy AG, Laabei M, Monk IR, et al. Manipulation of autophagy in phagocytes facilitates Staphylococcus aureus bloodstream infection. Infect Immun (2015) 83:3445. doi: 10.1128/IAI.00358-15
368. Dai Y, Gao C, Chen L, Chang W, Yu W, Ma X, et al. Heterogeneous vancomycin-intermediate Staphylococcus aureus uses the VraSR regulatory system to modulate autophagy for increased intracellular survival in macrophage-like cell line raw264.7. Front Microbiol (2019) 10:1222 doi: 10.3389/fmicb.2019.01222
369. Wu H-M, Wang J, Zhang B, Fang L, Xu K, Liu R-Y. CpG-ODN promotes phagocytosis and autophagy through JNK/P38 signal pathway in Staphylococcus aureus-stimulated macrophage. Life Sci (2016) 161:51–9. doi: 10.1016/j.lfs.2016.07.016
370. Zhu Y, Li H, Ding S, Wang Y. Autophagy inhibition promotes phagocytosis of macrophage and protects mice from methicillin-resistant Staphylococcus aureus pneumonia. J Cell Biochem (2018) 119:4808–14. doi: 10.1002/jcb.26677
371. Xie X, Yang C, Duan C, Chen H, Zeng T, Huang S, et al. Advanced glycation end products reduce macrophage-mediated killing of Staphylococcus aureus by ARL8 upregulation and inhibition of autolysosome formation. Eur J Immunol (2020) 50:1174–86. doi: 10.1002/eji.201948477
372. Cai J, Li J, Zhou Y, Wang J, Li J, Cui L, et al. Staphylococcus aureus facilitates its survival in bovine macrophages by blocking autophagic flux. J Cell Mol Med (2020) 24:3460–8. doi: 10.1111/jcmm.15027
373. Gibson JF, Prajsnar TK, Hill CJ, Tooke AK, Serba JJ, Tonge RD, et al. Neutrophils use selective autophagy receptor Sqstm1/p62 to target Staphylococcus aureus for degradation in vivo in zebrafish. Autophagy (2020) 19:1–10. doi: 10.1080/15548627.2020.1765521
374. Zang H, Qian S, Li J, Zhou Y, Zhu Q, Cui L, et al. The effect of selenium on the autophagy of macrophage infected by Staphylococcus aureus. Int Immunopharmacol (2020) 83:106406. doi: 10.1016/j.intimp.2020.106406
375. Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, et al. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun (1997) 65:298–304. doi: 10.1128/IAI.65.1.298-304.1997
376. Preston JA, Bewley MA, Marriott HM, McGarry Houghton A, Mohasin M, Jubrail J, et al. Alveolar macrophage apoptosis-associated bacterial killing helps prevent murine pneumonia. Am J Respir Crit Care Med (2019) 200:84–97. doi: 10.1164/rccm.201804-0646OC
377. Aberdein JD, Cole J, Bewley MA, Marriott HM, Dockrell DH. Alveolar macrophages in pulmonary host defence the unrecognized role of apoptosis as a mechanism of intracellular bacterial killing. Clin Exp Immunol (2013) 174:193–202. doi: 10.1111/cei.12170
378. Bewley MA, Marriott HM, Tulone C, Francis SE, Mitchell TJ, Read RC, et al. A cardinal role for cathepsin D in co-ordinating the host-mediated apoptosis of macrophages and killing of pneumococci. PLoS Pathog (2011) 7:e1001262. doi: 10.1371/journal.ppat.1001262
379. Palma Medina LM, Becker A-K, Michalik S, Yedavally H, Raineri EJM, Hildebrandt P, et al. Metabolic cross-talk between human bronchial epithelial cells and internalized Staphylococcus aureus as a driver for infection. Mol Cell Proteomics (2019) 18:892–908. doi: 10.1074/mcp.RA118.001138
380. Holzinger D, Gieldon L, Mysore V, Nippe N, Taxman DJ, Duncan JA, et al. Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J Leukoc Biol (2012) 92:1069–81. doi: 10.1189/jlb.0112014
381. Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, et al. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS One (2009) 4:e7446. doi: 10.1371/journal.pone.0007446
382. Zhang X, Hu X, Rao X. Apoptosis induced by Staphylococcus aureus toxins. Microbiol Res (2017) 205:19–24. doi: 10.1016/j.micres.2017.08.006
383. Otto M. Staphylococcus aureus toxins. Curr Opin Microbiol (2014) 17:32–7. doi: 10.1016/j.mib.2013.11.004
384. Pietrocola G, Nobile G, Rindi S, Speziale P. Staphylococcus aureus manipulates innate immunity through own and host-expressed proteases. Front Cell Infect Microbiol (2017) 7:166. doi: 10.3389/fcimb.2017.00166
385. von Köckritz-Blickwede M, Nizet V. Innate immunity turned inside-out: antimicrobial defense by phagocyte extracellular traps. J Mol Med (2009) 87:775–83. doi: 10.1007/s00109-009-0481-0
386. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
387. Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
388. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176:231–41. doi: 10.1083/jcb.200606027
389. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
390. Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med (2008) 14:949–53. doi: 10.1038/nm.1855
391. von Köckritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood (2008) 111:3070–80. doi: 10.1182/blood-2007-07-104018
392. Aulik NA, Hellenbrand KM, Czuprynski CJ. Mannheimia haemolytica and its leukotoxin cause macrophage extracellular trap formation by bovine macrophages. Infect Immun (2012) 80:1923–33. doi: 10.1128/IAI.06120-11
393. Hellenbrand KM, Forsythe KM, Rivera-Rivas JJ, Czuprynski CJ, Aulik NA. Histophilus somni causes extracellular trap formation by bovine neutrophils and macrophages. Microb Pathog (2013) 54:67–75. doi: 10.1016/j.micpath.2012.09.007
394. Liu P, Wu X, Liao C, Liu X, Du J, Shi H, et al. Escherichia coli and Candida albicans induced macrophage extracellular trap-like structures with limited microbicidal activity. PLoS One (2014) 9:e90042. doi: 10.1371/journal.pone.0090042
395. Loureiro A, Pais C, Sampaio P. Relevance of macrophage extracellular traps in C. albicans killing. Front Immunol (2019) 10:2767. doi: 10.3389/fimmu.2019.02767
396. Chow OA, von Köckritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, et al. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe (2010) 8:445–54. doi: 10.1016/j.chom.2010.10.005
397. Nakazawa D, Shida H, Kusunoki Y, Miyoshi A, Nishio S, Tomaru U, et al. The responses of macrophages in interaction with neutrophils that undergo NETosis. J Autoimmun (2016) 67:19–28. doi: 10.1016/j.jaut.2015.08.018
398. Webster SJ, Daigneault M, Bewley MA, Preston JA, Marriott HM, Walmsley SR, et al. Distinct cell death programs in monocytes regulate innate responses following challenge with common causes of invasive bacterial disease. J Immunol (2010) 185:2968–79. doi: 10.4049/jimmunol.1000805
399. Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol (2006) 16:401–7. doi: 10.1016/j.cub.2006.01.056
400. Thammavongsa V, Missiakas DM, Schneewind O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science (2013) 342:863–6. doi: 10.1126/science.1242255
401. Berends ETM, Horswill AR, Haste NM, Monestier M, Nizet V, von Köckritz-Blickwede M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun (2010) 2:576–86. doi: 10.1159/000319909
402. Winstel V, Schneewind O, Missiakas D. Staphylococcus aureus exploits the host apoptotic pathway to persist during infection. MBio (2019) 10:e02270–19. doi: 10.1128/mBio.02270-19
403. Brown L, Wolf JM, Prados-Rosales R, Casadevall A. Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat Rev Microbiol (2015) 13:620–30. doi: 10.1038/nrmicro3480
404. Lee J, Lee E-Y, Kim S-H, Kim D-K, Park K-S, Kim KP, et al. Staphylococcus aureus extracellular vesicles carry biologically active β-lactamase. Antimicrob Agents Chemother (2013) 57:2589–95. doi: 10.1128/AAC.00522-12
405. Wang X, Eagen WJ, Lee JC. Orchestration of human macrophage NLRP3 inflammasome activation by Staphylococcus aureus extracellular vesicles. Proc Natl Acad Sci (2020) 117:3174–84. doi: 10.1073/pnas.1915829117
406. An Y, Wang Y, Zhan J, Tang X, Shen K, Shen F, et al. Fosfomycin protects mice from Staphylococcus aureus pneumonia caused by α-hemolysin in extracellular vesicles by inhibiting MAPK-regulated NLRP3 inflammasomes. Front Cell Infect Microbiol (2019) 9:253. doi: 10.3389/fcimb.2019.00253
407. Hong S-W, Kim M-R, Lee E-Y, Kim JH, Kim Y-S, Jeon SG, et al. Extracellular vesicles derived from Staphylococcus aureus induce atopic dermatitis-like skin inflammation. Allergy (2011) 66:351–9. doi: 10.1111/j.1398-9995.2010.02483.x
408. Tartaglia NR, Breyne K, Meyer E, Cauty C, Jardin J, Chrétien D, et al. Staphylococcus aureus extracellular vesicles elicit an immunostimulatory response in vivo on the murine mammary gland. Front Cell Infect Microbiol (2018) 8:277. doi: 10.3389/fcimb.2018.00277
409. Wang X, Thompson CD, Weidenmaier C, Lee JC. Release of Staphylococcus aureus extracellular vesicles and their application as a vaccine platform. Nat Commun (2018) 9:1379. doi: 10.1038/s41467-018-03847-z
410. Burns EM, Smelser LK, Then JE, Stankiewicz TE, Kushdilian M, McDowell SA, et al. Short term statin treatment improves survival and differentially regulates macrophage-mediated responses to Staphylococcus aureus. Curr Pharm Biotechnol (2013) 14:233–41. doi: 10.2174/138920113805219395
411. Groot Kormelink T, Mol S, de Jong EC, Wauben MHM. The role of extracellular vesicles when innate meets adaptive. Semin Immunopathol (2018) 40:439–52. doi: 10.1007/s00281-018-0681-1
412. Giri PK, Schorey JS. Exosomes derived from M. Bovis BCG infected macrophages activate antigen-specific CD4+ and CD8+ T cells in vitro and in vivo. PLoS One (2008) 3:e2461. doi: 10.1371/journal.pone.0002461
413. García-Martínez M, Vázquez-Flores L, Álvarez-Jiménez VD, Castañeda-Casimiro J, Ibáñez-Hernández M, Sánchez-Torres LE, et al. Extracellular vesicles released by J774A.1 macrophages reduce the bacterial load in macrophages and in an experimental mouse model of tuberculosis. Int J Nanomed (2019) 14:6707–19. doi: 10.2147/IJN.S203507
414. Cai C, Koch B, Morikawa K, Suda G, Sakamoto N, Rueschenbaum S, et al. Macrophage-derived extracellular vesicles induce long-lasting immunity against hepatitis C virus which is blunted by polyunsaturated fatty acids. Front Immunol (2018) 9:723. doi: 10.3389/fimmu.2018.00723
415. Soni S, Wilson MR, O’Dea KP, Yoshida M, Katbeh U, Woods SJ, et al. Alveolar macrophage-derived microvesicles mediate acute lung injury. Thorax (2016) 71:1020–9. doi: 10.1136/thoraxjnl-2015-208032
416. Ding W, Rivera OC, Kelleher SL, Soybel DI. Macrolets: outsized extracellular vesicles released from lipopolysaccharide-stimulated macrophages that trap and kill Escherichia coli. iScience (2020) 23:101135. doi: 10.1016/j.isci.2020.101135
417. Grundmann H, Aires-de-Sousa M, Boyce J, Tiemersma E. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet (2006) 368:874–85. doi: 10.1016/S0140-6736(06)68853-3
418. Kaplan SL, Hulten KG, Gonzalez BE, Hammerman WA, Lamberth L, Versalovic J, et al. Three-year surveillance of community-acquired Staphylococcus aureus infections in children. Clin Infect Dis (2005) 40:1785–91. doi: 10.1086/430312
419. Laupland KB, Ross T, Gregson DB. Staphylococcus aureus bloodstream infections: risk factors, outcomes, and the influence of methicillin resistance in Calgary, Canada, 2000-2006. J Infect Dis (2008) 198:336–43. doi: 10.1086/589717
420. Hersh AL, Chambers HF, Maselli JH, Gonzales R. National trends in ambulatory visits and antibiotic prescribing for skin and soft-tissue infections. Arch Intern Med (2008) 168:1585–91. doi: 10.1001/archinte.168.14.1585
421. Hope R, Livermore DM, Brick G, Lillie M, Reynolds R. Non-susceptibility trends among staphylococci from bacteraemias in the UK and Ireland, 2001-06. J Antimicrob Chemother (2008) 62:ii65–74. doi: 10.1093/jac/dkn353
422. Chambers HF, Deleo FR. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol (2009) 7:629–41. doi: 10.1038/nrmicro2200
423. Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature (2015) 527:323–8. doi: 10.1038/nature16057
424. Watson K, Russell CD, Baillie JK, Dhaliwal K, Fitzgerald JR, Mitchell TJ, et al. Developing novel host-based therapies targeting microbicidal responses in macrophages and neutrophils to combat bacterial antimicrobial resistance. Front Immunol (2020) 11:786. doi: 10.3389/fimmu.2020.00786
425. Brinch KS, Sandberg A, Baudoux P, Van Bambeke F, Tulkens PM, Frimodt-Møller N, et al. Plectasin shows intracellular activity against Staphylococcus aureus in human THP-1 monocytes and in a mouse peritonitis model. Antimicrob Agents Chemother (2009) 53:4801–8. doi: 10.1128/AAC.00685-09
426. Xiong M-H, Li Y-J, Bao Y, Yang X-Z, Hu B, Wang J. Bacteria-responsive multifunctional nanogel for targeted antibiotic delivery. Adv Mater (2012) 24:6175–80. doi: 10.1002/adma.201202847
427. Sémiramoth N, Di Meo C, Zouhiri F, Saïd-Hassane F, Valetti S, Gorges R, et al. Self-assembled squalenoylated penicillin bioconjugates: an original approach for the treatment of intracellular infections. ACS Nano (2012) 6:3820–31. doi: 10.1021/nn204928v
428. Cai Q, Fei Y, An H-W, Zhao X-X, Ma Y, Cong Y, et al. Macrophage-instructed intracellular Staphylococcus aureus killing by targeting photodynamic dimers. ACS Appl Mater Interf (2018) 10:9197–202. doi: 10.1021/acsami.7b19056
429. Fattom A, Schneerson R, Szu SC, Vann WF, Shiloach J, Karakawa WW, et al. Synthesis and immunologic properties in mice of vaccines composed of Staphylococcus aureus type 5 and type 8 capsular polysaccharides conjugated to Pseudomonas aeruginosa exotoxin A. Infect Immun (1990) 58:2367–74. doi: 10.1128/IAI.58.7.2367-2374.1990
430. Shinefield H, Black S, Fattom A, Horwith G, Rasgon S, Ordonez J, et al. Use of a Staphylococcus aureus conjugate vaccine in patients receiving hemodialysis. N Engl J Med (2002) 346:491–6. doi: 10.1056/NEJMoa011297
431. McKenney D, Pouliot KL, Wang Y, Murthy V, Ulrich M, Döring G, et al. Broadly protective vaccine for Staphylococcus aureus based on an in vivo-expressed antigen. Science (1999) 284:1523–7. doi: 10.1126/science.284.5419.1523
432. Maira-Litrán T, Kropec A, Goldmann DA, Pier GB. Comparative opsonic and protective activities of Staphylococcus aureus conjugate vaccines containing native or deacetylated staphylococcal poly-N-acetyl-beta-(1-6)-glucosamine. Infect Immun (2005) 73:6752–62. doi: 10.1128/IAI.73.10.6752-6762.2005
433. Stranger-Jones YK, Bae T, Schneewind O. Vaccine assembly from surface proteins of Staphylococcus aureus. Proc Natl Acad Sci USA (2006) 103:16942–7. doi: 10.1073/pnas.0606863103
434. Kuklin NA, Clark DJ, Secore S, Cook J, Cope LD, McNeely T, et al. A novel Staphylococcus aureus vaccine: iron surface determinant B induces rapid antibody responses in rhesus macaques and specific increased survival in a murine S. aureus sepsis model. Infect Immun (2006) 74:2215–23. doi: 10.1128/IAI.74.4.2215-2223.2006
435. Josefsson E, Hartford O, O’Brien L, Patti JM, Foster T. Protection against experimental Staphylococcus aureus arthritis by vaccination with clumping factor A, a novel virulence determinant. J Infect Dis (2001) 184:1572–80. doi: 10.1086/324430
436. Schaffer AC, Solinga RM, Cocchiaro J, Portoles M, Kiser KB, Risley A, et al. Immunization with Staphylococcus aureus clumping factor B, a major determinant in nasal carriage, reduces nasal colonization in a murine model. Infect Immun (2006) 74:2145–53. doi: 10.1128/IAI.74.4.2145-2153.2006
437. Li X, Wang X, Thompson CD, Park S, Park WB, Lee JC. Preclinical efficacy of clumping factor A in prevention of Staphylococcus aureus infection. MBio (2016) 7:e02232–15. doi: 10.1128/mBio.02232-15
438. Zhou H, Xiong Z-Y, Li H-P, Zheng Y-L, Jiang Y-Q. An immunogenicity study of a newly fusion protein Cna-FnBP vaccinated against Staphylococcus aureus infections in a mice model. Vaccine (2006) 24:4830–7. doi: 10.1016/j.vaccine.2006.03.020
439. Daum RS, Spellberg B. Progress toward a Staphylococcus aureus vaccine. Clin Infect Dis (2012) 54:560–7. doi: 10.1093/cid/cir828
440. Mazmanian SK, Ton-That H, Schneewind O. Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol Microbiol (2001) 40:1049–57. doi: 10.1046/j.1365-2958.2001.02411.x
Keywords: macrophage, Staphylococcus, phagocytosis, immunity, immune evasion
Citation: Pidwill GR, Gibson JF, Cole J, Renshaw SA and Foster SJ (2021) The Role of Macrophages in Staphylococcus aureus Infection. Front. Immunol. 11:620339. doi: 10.3389/fimmu.2020.620339
Received: 22 October 2020; Accepted: 02 December 2020;
Published: 19 January 2021.
Edited by:
Rachel McLoughlin, Trinity College Dublin, IrelandReviewed by:
Dane Parker, The State University of New Jersey, United StatesDavid Heinrichs, Western University, Canada
Copyright © 2021 Pidwill, Gibson, Cole, Renshaw and Foster. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon J. Foster, cy5mb3N0ZXJAc2hlZmZpZWxkLmFjLnVr
†These authors share first authorship