 Norman Fultang
Norman Fultang Xinyuan Li
Xinyuan Li Ting Li
Ting Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 14 January 2021
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.619253
This article is part of the Research Topic Immune Cell Lineage Reprogramming in Cancer View all 19 articles
Myeloid-derived Suppressor Cells (MDSCs) are a sub-population of leukocytes that are important for carcinogenesis and cancer immunotherapy. During carcinogenesis or severe infections, inflammatory mediators induce MDSCs via aberrant differentiation of myeloid precursors. Although several transcription factors, including C/EBPβ, STAT3, c-Rel, STAT5, and IRF8, have been reported to regulate MDSC differentiation, none of them are specifically expressed in MDSCs. How these lineage-non-specific transcription factors specify MDSC differentiation in a lineage-specific manner is unclear. The recent discovery of the c-Rel−C/EBPβ enhanceosome in MDSCs may help explain these context-dependent roles. In this review, we examine several transcriptional regulators of MDSC differentiation, and discuss the concept of non-modular regulation of MDSC signature gene expression by transcription factors such as c-Rel and C/EBPß.
Tumor immune evasion is an essential feature of tumorigenesis (1, 2). To successfully establish themselves within a host, tumor cells leverage biochemical signals and rogue immune cells to hide from and repress host immune responses (1–3). Immunotherapy, which can restore immune response and anti-cancer immunity, has revolutionized cancer therapy. However, rogue immunosuppressive cells, including tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), regulatory T-cells (Tregs), regulatory dendritic cells (RegDCs), cancer-associated fibroblasts, and myeloid-derived suppressor cells (MDSCs), still represent significant impediments to immunotherapy, contributing to therapy failure and poor clinical outcomes (4–8). Of these pro-tumoral cell types, MDSCs are perhaps the least well characterized.
MDSCs are a heterogenous population of immunosuppressive pro-tumoral leukocytes which arise as a result of defects in myelopoiesis (9). Under physiological conditions, progenitor myeloid cells differentiate into macrophages, dendritic cells or granulocytes. Under pathological conditions like cancer or chronic infections, aberrant myelopoiesis allows the accumulation and expansion of immature myeloid cells with strong immunosuppressive capabilities (10–16). While these cells possess many phenotypic and morphological hallmarks of anti-tumor myeloid-lineage cells like monocytes and neutrophils, they differ significantly in their activation programs and function to inhibit anti-tumor immunity by producing immunosuppressive factors like arginase, nitrogen species and reactive oxygen species, among others (10, 17–19). MDSCs are a significant obstacle to immunotherapies including checkpoint inhibitors; accumulation of MDSCs populations within circulating and tumor-infiltrating leukocytes have been observed in patients who fail to respond to checkpoint inhibitor therapy (18, 19).
There are two major subsets of MDSCs– granulocytic or polymorphonuclear MDSCs (G-MDSCs or PMN-MDSCs), which are phenotypically similar to granulocytes, and monocytic or mononuclear MDSCs (M-MDSCs), which are phenotypically similar to monocytes. PMN-MDSCs have a CD11b+Ly6G+Ly6Clo phenotype in mice and a CD11b+CD14−CD15+/CD66b+ phenotype in humans while M-MDSCs are identified as CD11b+Ly6G−Ly6Chi in mice, and CD11b+CD14+HLA-DR−/loCD15− in humans (20, 21). MDSC markers were recently reviewed here (21). A third mixed population of MDSCs, early-stage MDSC (e-MDSC), with phenotype Lin- (including CD3, CD14, CD15, CD19, and CD56) HLA-DR-CD33+ was recently proposed in humans (22). e-MDSCs also contain immature progenitor myeloid cells and their equivalent in mice is yet to be identified (22).
While a lot is known about the phenotypic and morphological delineations of MDSCs, the biochemical markers and effectors underlying their development and function are still poorly understood. As such, the identification of these drivers of pathological MDSC expansion and immunosuppressive activity has been the subject of intensive research in recent years. Recently identified MDSC effectors, mostly transcription factors (TFs) and apoptotic regulators, include IRF8 (23), STAT3 (23–26), C/EBPß (27, 28), S100A8/9 (29), TIPE2 (30, 31), GCN2 (32), among others (Table 1). Of all these regulators, C/EBPß has emerged as an essential “master” regulator of MDSC expansion and immunosuppressive activity. Most of the known MDSC regulators drive expansion and immunosuppressive activity in C/EBPß-dependent mechanisms. Additionally, C/EBPß deletion alone in myeloid cells was sufficient to halt MDSC generation and immunosuppressive activity (27). Recent evidence, however, suggests that c-Rel, a member of the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) family of transcription factors, regulates C/EBPß activity and expression in MDSCs (33). In this review we describe c-Rel and C/EBPß as master effectors of MDSC biology and highlight how a non-modular c-Rel-C/EBPß “enhanceosome” drives MDSC development and function in cancer.

Table 1 Known effectors or regulators of MDSC biology.
MDSCs arise when sustained pathologic inflammation induces an aberrant differentiation program in myeloid precursors giving rise to immunosuppressive cells (10–16). This is mediated by activation of complex transcriptional machinery within these cells by inflammatory cytokines including GM-CSF, IL-6, G-CSF, IL-1ß, PGE2, TNFα, and VEGF (10–16). Currently known transcriptional regulators of MDSC biology include STAT3, CEBP/β, STAT5, IRF8, S100A8/9, RB, TIPE2 and GCN2 (Table 1).
STAT3 is a key repressor of antitumor immunity (39, 40). It impairs antigen presentation and inhibits the production of immunostimulatory cytokines while promoting the expression of immunosuppressive molecules. It is highly active in most cancers where it promotes the production of inflammatory cytokines and growth factors like IL-6, IL-10, IL-23, LIF, VEGF, and HGF (39, 41). These molecules induce STAT3 activation in myeloid precursors which drives cell survival, transcription of immunosuppressive enzymes (ARG1 and iNOS), and aberrant differentiation into MDSCs. It also interacts with C/EBPß at promoter sites to regulate transcription (33, 34). Intriguingly, a decrease in MDSC STAT3 activity in the tumor environment is associated with differentiation into TAMs (42). Within myeloid precursors, STAT3 and STAT5 also inhibit IRF8, a crucial transcription factor for normal myeloid differentiation into monocytes and dendritic cells (23). IRF8 functions as a negative regulator of MDSCs and its downregulation is necessary for pathologic MDSC expansion (23).
S100A8/9 produced by tumors binds to RAGE receptors on myeloid precursors inducing activation of an NF-κB-C/EBPß-STAT3 axis (29). This promotes production of S100A8/9 in MDSCs and drives both expansion and chemotactic migration to tumor sites for immunosuppression. The MDSC-secreted S100A8/9 creates an autocrine feedback loop that exacerbates MDSC accumulation.
High reactive oxygen species (ROS) associated within tumor microenvironments and IL-6 induce TIPE2 in myeloid precursors (30, 31). Active TIPE2 promotes the expression of C/EBPß and STAT3 via the PI3K/AKT and MAPK/ERK pathways. This leads to MDSC accumulation and polarization into an immunosuppressive phenotype. In the absence of TIPE2 MDSCs became anti-tumoral indicating TIPE2 functions as a molecular polarity switch in MDSCs (30). GCN2 similarly functions as a polarity switch in MDSCs. It alters myeloid function by inducing C/EBPß and CREB-2/ATF4 which promote MDSC expansion and immunosuppressive activity (32). Epigenetic silencing of Rb by HDAC-2 in myeloid precursors also promotes accumulation of PMN-MDSCs (38).
C/EBPß appears to be an essential player among these effectors in MDSCs.
C/EBPß is the second member of the CCAAT/Enhancer Binding Protein (C/EBP) family of transcription factors (28). C/EBP proteins are basic-region-leucine zipper transcription factors which regulate both emergency and steady state myelopoiesis (35, 43–45). C/EBPα, the first member of the family, regulates steady state myelopoiesis. C/EBPα is highly expressed early identified n the myeloid differentiation process and is an essential molecular switch for the transition from common myeloid precursors to granulocyte macrophage progenitors (46). The role of other C/EBP family proteins, including C/EBPδ and CHOP, are less clear but they are all thought to similarly regulate myelopoiesis as well as modulate the activity of other C/EBP proteins (28). C/EBPδ regulates the expression of inflammatory cytokines including COX-2, iNOS, G-CSF, IL-1β, IL-6, and TNF-α, and has been implicated in MDSC expansion (47, 48). CHOP on the other hand, lacks DNA-binding activity but can form heterodimers with C/EBPß isoforms and other family members, regulating their activity (49). It has similarly been implicated in MDSC expansion via these regulatory events (50).
Within the context of MDSC development and function, C/EBPß (also known as IL6-DBP, CRP2, NF-IL6, NF-M or TCF5) is the most important C/EBP (Figure 1). It has three isoforms with diverse, context-dependent roles (28, 51). The first two, LAP and LAP*, contain both a DNA-binding domain and an activation domain. The third isoform, LIP, lacks an activation domain and attenuates transcriptional activity via heterodimerization with LAP/LAP* (35, 45, 52). C/EBPß controls emergency myelopoiesis, which is a characteristic feature of many solid tumors due to chronic tumor-induced inflammation (53–55). Deregulations of C/EBPß activity are thus a significant contributing factor to aberrant myelopoiesis and MDSC expansion under pathological conditions (27, 28).
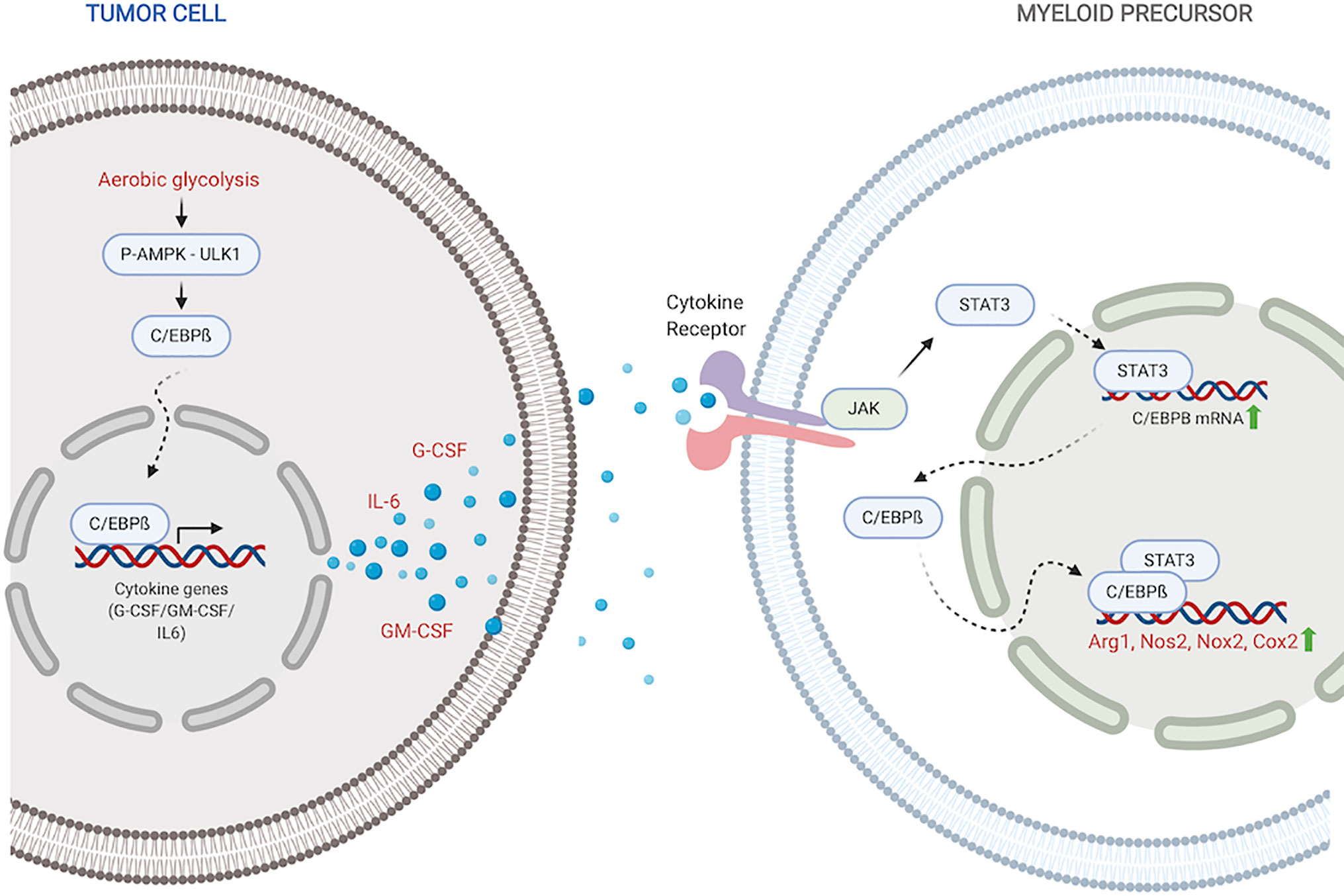
Figure 1 C/EBPß regulates MDSC expansion and function. Within the tumor, C/EBPß promotes transcription of inflammatory cytokines. Inflammatory cytokines then reciprocally induce C/EBPß in myeloid compartment which promotes transcription of immunosuppressive molecules. Created with BioRender.com.
Stimulation with inflammatory cytokines like G-CSF, GM-CSF and IL-6 drives an increase in C/EBPß expression and DNA-binding activity (27, 35, 56). Upregulated LAP and LAP* isoforms of C/EBPß function as mediators of cytokine-induced inflammatory response via transcriptional activation of inflammatory genes IL-6, TNF and G-CSF, exacerbating the response (45). Under pathological conditions, this sustained inflammatory activation promotes aberrant myeloid development and differentiation into immunosuppressive phenotypes (27, 35, 36). Following IL-6 stimulation, C/EBPß, in concert with STAT3, also promotes miR-21 and miR-181b, which induce NFI-A to promote MDSC accumulation in the bone marrow and spleen (34).
Within tumors, aerobic glycolysis, a hallmark of cancer, leads to an increase in LAP which promotes G-CSF+GM-CSF expression and secretion (37). Li et al. showed that in breast cancer cells, preferential activation of aerobic glycolysis over oxidative phosphorylation, inhibits AMPK-ULK1 and autophagy signaling, allowing stabilization and activity of LAP (37). Cytokines, induced by LAP, travel to the myeloid compartment where they promote expansion of MDSC precursors and direct their differentiation into suppressor cells. Within MDSCs, activated C/EBPß directly binds to and promotes the transcription of immunosuppressive enzymes including Arg1, Nos2, Nox2, and Cox2 (27, 36, 57). These enzymes are crucial members of the MDSC immunosuppressive machinery. Arg1 and Nos2 deplete environmental L-arginine, a crucial amino acid for T-cell survival and anti-tumor activity (58–61). Nox2 increases ROS which block T-cell activation and activity (62, 63). The COX-2-PGE2 cascade suppresses both dendritic and natural killer cell activity, while promoting the expression of immunity repressor PD-L1 (64, 65). It is also plausible that activated C/EBPß in myeloid precursors similarly induces the production of GM-CSF and IL-6 which drive MDSC accumulation and function in autocrine signaling mechanisms.
In macrophages, PI3Kγ activates C/EBPß, which serves as a critical polarization switch from an immunostimulatory to an immunosuppressive phenotype during tumor progression (66). This suggests C/EBPß could also regulate MDSC differentiation into TAMs in the tumor microenvironment.
Seminal work by Marigo et al. showed that C/EBPß deletion in all hematopoietic lineage cells was enough to halt MDSC genesis and completely abrogate their immunosuppressive activity on antigen activated T-cells (27). They also observed significant reduction in both Arg1 and Nos2 expression and activity. C/EBPß deletion potentiated adoptive T-cell therapy resulting in a complete cure for 60% of mice bearing subcutaneous fibrosarcoma. Their work and subsequent studies suggest C/EBPß is an essential mediator of MDSC development and activity (36, 67, 68).
Perhaps unsurprisingly, many studies into molecular effectors of MDSCs have focused on upstream regulators of C/EBPß. Of these recently found effectors, c-Rel, appears to be an essential regulatory partner for C/EBPß in MDSC.
c-Rel, is a member of the NF-κB family of TFs which regulate a variety of molecular processes from embryogenesis to hematopoiesis and inflammation (69, 70). Being a class 2 member of the family, it contains both an N-terminal Rel-homology domain (RHD) and a transactivation domain (TAD) (70, 71). c-Rel’s RHD mediates interactions with other proteins and transcriptional regulators at promoter sites where its TAD recognizes and binds to consensus GGGCTTTCC sequences (69, 72). These interactions, especially with other NF-κB members to form heterodimers, are essential for c-Rel transcriptional activity. c-Rel’s TAD also contains several serine residues which are readily phosphorylated, regulating c-Rel nuclear localization, transactivation and DNA binding activity (73–76).
c-Rel is an important regulator of immune cell function. It is crucial for normal B- and T- cell activation and proliferation (77–81). Upon lymphocyte activation, c-Rel induces IRF-4 in B-cells which promotes cell cycle progression and proliferation. IRF-4 has κB elements in its promoter region to which a c-Rel:p50 heterodimer binds. B-cell proliferation defects have been observed in c-Rel deficient mice (82). Similar defects in T-cell activation and proliferation following stimulation have been observed in c-Rel knockout mice (77).
c-Rel is a key regulator of autoimmunity via its role in promoting the generation of Th1, Th17 and Foxp3+ regulatory T cells (Tregs) (83–87). c-Rel is responsible for assembling a transcriptional enhanceosome including RelA, NFAT, SMAD and CREB that binds and transcribes Foxp3, a master regulator of Treg immunosuppression (84). c-Rel also directly regulates the expression of many proinflammatory cytokines via its context-dependent binding events at promoter sequences (79, 80, 88). Intriguingly, despite its significant roles in both inflammation and autoimmunity, the effects of c-Rel deficiency on immune homeostasis appear to be mostly minor (77).
Although previously thought to primarily function in the lymphoid compartment, mounting evidence suggests a significant role for c-Rel in myeloid cells. We recently showed that c-Rel regulates MDSC expansion and function in cancer (57). Both global and myeloid-specific c-Rel deletion blocked tumor growth and markedly decreased MDSC accumulation in melanoma and lymphoma mice models. The few MDSCs that were generated in the c-Rel knockout mice were defective in suppression when compared to MDSCs from Wild-type mice. c-Rel deletion also altered MDSC metabolism, reducing mitochondrial respiration and glycolysis, inducing a Warburg-like metabolic state. We also observed downregulation of signature MDSC genes in c-Rel knockout mice including Arg1, Nos2, and C/EBPß, key members of the MDSC immunosuppression machinery. There was also heightened inflammatory gene expression in c-Rel deficient MDSCs compared to wild type, a phenotype that was rescued by C/EBPß overexpression. This suggests that c-Rel’s effect in MDSCs is C/EBPß dependent.
Mechanistically, c-Rel directly regulates the transcription of these MDSC signature genes (57). Upon stimulation with GM-CSF and IL-6, c-Rel binds to the promoters of Arg1 and Cebpb where it forms a transcriptional complex with pSTAT3, C/EBPß and p65. ReChIP analyses showed that these factors all bind to the same promoter element, suggesting the formation of a single enhanceosome complex which drives MDSC biology. c-Rel-C/EBPß enhanceosomes have previously been identified as transcriptional regulators in hepatocytes (89, 90).
Enhanceosomes are high-order protein complexes, usually transcription factors, that bind cooperatively at a gene’s promoter or enhancer regions to activate transcription (91, 92). Many cis-regulatory elements, including promoters and enhancers, contain overlapping DNA binding sites for various transcription factors. This allows the formation of elaborate protein complexes which alter chromatin architecture and recruit the RNA polymerase transcription machinery, regulating gene expression as a functional, nucleoprotein unit (91, 92). These enhanceosome complexes effectively function as “on” and “off” transcriptional switches, specifying key developmental and cell lineage-determining gene regulation events (91, 92). Enhanceosomes could comprise any number of multifunctional transcriptional regulators in an almost limitless number of combinations, specifying the varied cell differentiation programs found in multicellular organisms. An increasing number of enhanceosomes are being described, shifting previously established transcription paradigms.
Fiedler et al. recently described a “Wnt enhanceosome” consisting of ChiLS, Runt/RUNX2, ARID1 and Groucho/TLE which is integrated by Pygo at TCF enhancers to drive Wnt signaling in Drosophila (93). Additionally, the Wnt enhanceosome could incorporate a number of factors in a lineage-dependent manner and be switched “off” by Notch. This allows context-dependent regulation of TCF/LEF target genes to simultaneously promote embryogenesis and development while preventing hyperproliferation and cancer. Pawlus et al. similarly described a multifactorial HIF enhanceosome comprising of HIF1, HIF2, RNA poll II and varied transcription factors at enhancer sites for HIF target genes (94). These context-dependent enhanceosomes help explain the dual oncogenic and tumor-suppressive role of HIF-mediate hypoxia. Scotto et al. also showed that multidrug resistance in cancer is governed by an MDR1 enhanceosome at the MDR1 promoter which can be activated by a variety of stimuli including differentiation agents like retinoic acid, UV radiation and chemotherapy (95). The MDR1 enhanceosome included NF-Y, Sp family transcription factors and histone acetyltransferase PCAF and could be targeted to reverse multidrug resistance.
The assembly and disassembly of enhanceosomes is essential for tight gene regulation in a cell. Because the assembly of a functional enhanceosome complex depends on several factors including local DNA conformation, protein availability and modifications, gene regulation via enhanceosomes can be very cell-specific. The absence of any one factor disrupts enhanceosome activity, preventing transactivation. In the case of MDSCs, enhanceosomes at regulatory sites for MDSC signature genes are compelling as key effectors of aberrant MDSC development under pathological conditions.
It is plausible that higher levels of active c-Rel and C/EBPß within the nucleus of pathologically activated myeloid cells drive the formation of altered enhanceosomes at regulatory regions for Arg1, Nos2, Nox2, Cebpb, and other MDSC genes. Previous work has identified enhanceosomes for several immunosuppressive mediators including Nos2, Arg1, and Nox2 that do not contain either C/EBPß or c-Rel (96–98). We recently showed abundant c-Rel and C/EBPß accumulation at the gene promoters of both Arg1 and C/EBPß following stimulation with GM-CSF and IL-6 (57). In this c-Rel-C/EBPß MDSC enhanceosome model, c-Rel is recruited first to the promoter site and in its absence, the enhanceosome fails to assemble. Following c-Rel binding, pSTAT3, p65 and C/EBPß are recruited to the promoter site to drive transcription and differentiation into immunosuppressive MDSCs (Figure 2).
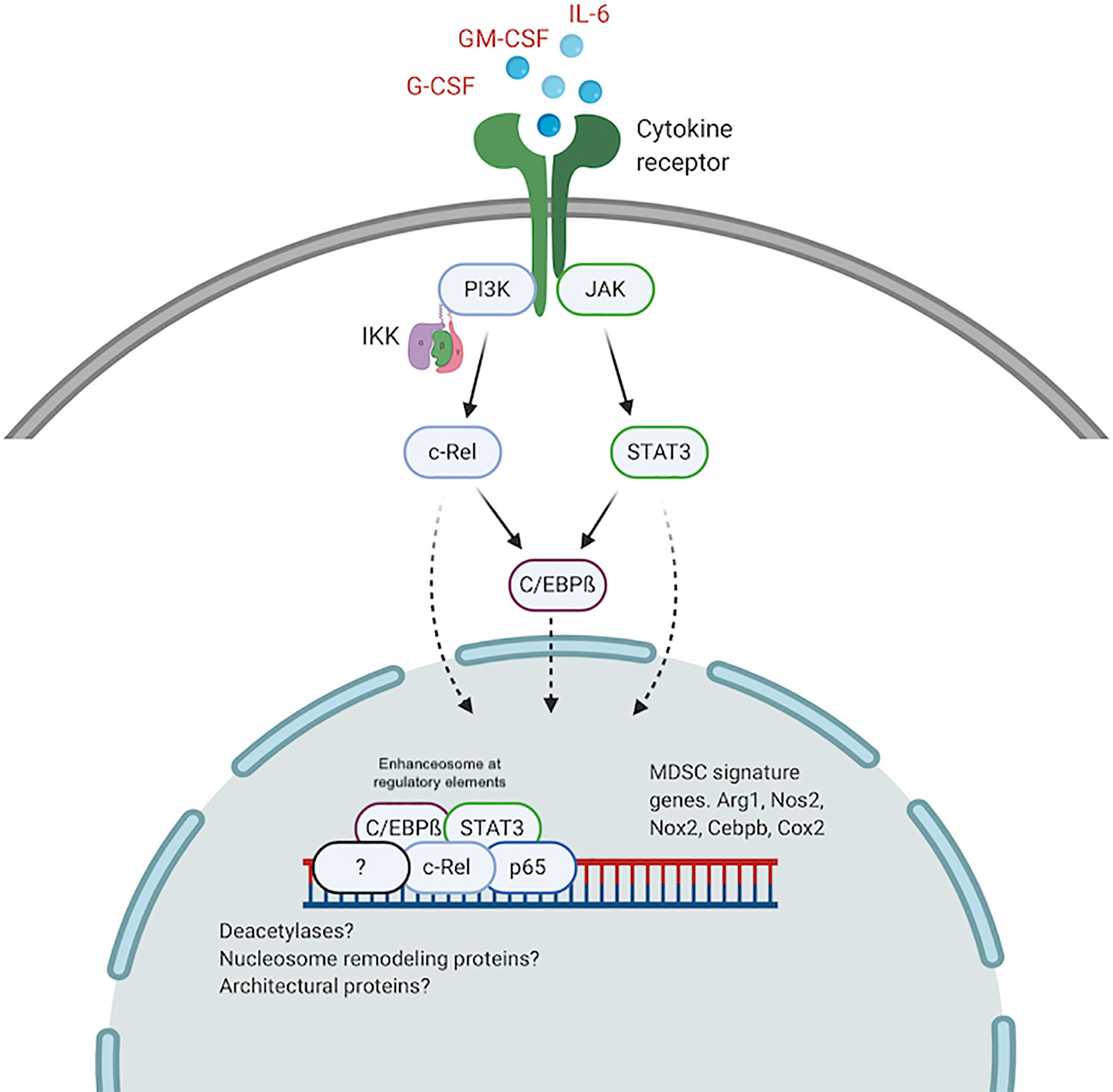
Figure 2 The c-Rel/C/EBPß enhanceosome in MDSCs. c-Rel and C/EBPß induced by tumor secreted cytokines, translocate to the nucleus and assemble an enhanceosome containing STAT3, p65 and other regulators at enhancer sites for immunosuppressive molecules. Created with BioRender.com.
A similar c-Rel-C/EBPß enhanceosome was previously described (89, 90). Cha-Molstad et al. showed that in hepatocytes, cytokine stimulation promotes c-Rel-mediated recruitment of C/EBPß and STAT3 to the CRP gene promoter to activate transcription (89). Intriguingly, c-Rel itself was not directly bound to the DNA sequence. c-Rel DNA binding activity is regulated by phosphorylation of the many serine residues within its TAD (73–76). Because we found c-Rel binding to DNA forming the MDSC enhanceosome, it is plausible highly active kinases within pathologically activated myeloid cells contribute to the formation of the MDSC-specific c-Rel enhanceosome. Other post-translational modifications, specific to myeloid cells under pathological activation, that modulate protein-protein interactions and protein-DNA interactions, might drive the formation of MDSC enhanceosomes. Other NF-κB proteins, including p50, have been reported to be involved in MDSC expansion following stimulation by tumor-derived PGE2 (99). We previously showed that c-Rel could bind p50 in MDSCs (57). P50 could similarly be incorporated into the MDSC enhanceosome during tumorigenesis to drive MDSC expansion and activity. The c-Rel-C/EBPß enhanceosome might also contain other nuclear proteins including co-regulators, deacetylases, architectural proteins like HMG I/Y and nucleosome remodeling proteins.
The c-Rel-C/EBPß enhanceosome is also a promising candidate as a biochemical marker for MDSCs. A significant constraint in MDSC research is the lack of reliable markers to characterize this highly heterogeneous cell population (22). Because yields are often low when isolating MDSCs, especially from in vivo systems, most studies lack functional validation of immunoregulatory activity. Improved biochemical markers, specific to MDSCs, would provide a simple validatable phenotype for MDSCs. The individual factors within the enhanceosome are not specific to MDSCs: C/EBPß is enriched in monocytes/macrophages (100, 101). c-Rel and p65 are pervasive regulators of B- and T- cell proliferation (77–81). pSTAT3 is a ubiquitous transcription factor within eukaryotic cells (25, 41). However, concurrent activation of all four, as well as other putative members of the enhanceosome, could be indicative of an MDSC phenotype. Monitoring assembly and activation of the c-Rel-C/EBPß enhanceosome could thus be a testable marker for MDSC activation and expansion.
This also provides an exciting therapeutic avenue. We showed that a small molecule inhibitor of c-Rel abrogated MDSC development and immunosuppression via disruption of the c-Rel complex (57). Similar approaches targeting individual members, aiming to disrupt their interactions in the MDSC enhanceosome, could have thrilling outcomes. Lee et al. showed that cerulenin, a small molecule inhibitor of the NF-κB enhanceosome in macrophages, might disrupt the assembly of the enhanceosome, suppressing pro-inflammatory activation and sepsis (102). Cerulenin specifically disrupted the p65-TonEBP-p300 complex without affecting their expression or DNA-binding. It had no detectable toxicity and animals could tolerate high doses for several weeks (103). Additionally, our c-Rel inhibitor enhanced the anti-tumor effect of anti-PD-1 antibodies suggesting combinatorial restoration of T cell function (via MDSC inhibition) and activation (via PD-1 inhibition) as a viable clinical strategy (57). The development of a novel class of enhanceosome inhibitors targeting MDSCs could represent an exciting approach to potentiate immunotherapy.
MDSCs are a product of sustained pathologic inflammation, which develop as a result of aberrant cytokine-mediated activation of complex transcriptional machinery in myeloid precursors (9, 10). They are involved in the pathogenesis of a host of human diseases from cancers to acute infections. In cancer, tumor-produced cytokines mediated by C/EBPß induce c-Rel and C/EBPß in the myeloid compartment, which drives the formation of a c-Rel-C/EBPß-pSTAT3-p65 MDSC enhanceosome. This enhanceosome promotes the transcription of immunosuppressive enzymes and other MDSC signature genes, guiding their differentiation into immunosuppressive cell populations. Because this putative enhanceosome is MDSC-specific, it can be targeted to repress MDSC expansion and immunosuppression. It is thus imperative to further characterize this enhanceosome and develop modalities to inhibit it. Additionally, further studies into other complex transcription programs underlying spatiotemporal gene regulation during aberrant myeloid cell differentiation are warranted. These would identify novel mechanisms and therapeutic targets, which could be blocked clinically to enhance the efficacy of immunotherapies like checkpoint blockade.
NF drafted the manuscript and designed the figures. XL and TI reviewed the manuscript structure and science. YC reviewed the manuscript structure, ideas and science. All authors contributed to the article and approved the submitted version.
This work was supported in part by grants from the National Institutes of Health (nos. R01-AI152195, R01-AI099216, R01-AI121166, R01-AI143676, and R01-AI136945 to YC); XL was partially supported by grant no. NIH-T32-DK007780.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AKT, Protein kinase B; ATF, Activating transcription factor; C/EBPβ, CCAAT-enhancer-binding protein ß; COX-2, Cyclooxygenase 2; CREB, cAMP response element-binding protein; ERK, Extracellular-signal-regulated kinase; G-CSF, Granulocyte colony-stimulating factor; GCN2, General control nonderepressible 2; GM-CSF, Granulocyte-macrophage colony-stimulating factor; HDAC2, Histone Deacetylase 2; HMG I/Y, High mobility group protein; IL-1ß/6/10/23, Interleukin -1ß/6/10/23; IRF8, Interferon Regulatory Factor 8; LAP/LAP*, Liver-enriched activating protein – C/EBPß isoforms; LIF, Leukemia inhibitory factor; LIP, Liver-enriched inhibitory protein; MAPK, Mitogen-activated protein kinase; MDSC, Myeloid-derived suppressor cell; NFAT, Nuclear factor of activated T-cells; NFI-A, Nuclear factor 1 A-type; NOX2, NADPH oxidase 2; PGE2, Prostaglandin E2; PI3K, Phosphoinositide 3-kinase; RAGE, Receptor for advanced glycation endproducts; RB, Retinoblastoma protein; ReCHIP, Re-Chromatin immunoprecipitation; S100A8/9, S100 Calcium Binding Protein A8; STAT3/5, Signal transducer and activator of transcription 3/5; TIPE2, TNF alpha induced protein 8 like 2; TNF, Tumor necrosis factor; VEGF, Vascular endothelial growth factor; c-Rel, Cellular Avian Reticuloendotheliosis Viral Oncogene Homolog; iNOS/NOS2, inducible NO synthase/Nitric Oxide Synthase 2.
1. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3:991–8. doi: 10.1038/ni1102-991
2. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol (2015) 35:S185–98. doi: 10.1016/j.semcancer.2015.03.004
3. Kim H-S, Skurk C, Thomas SR, Bialik A, Suhara T, Kureishi Y, et al. Regulation of angiogenesis by glycogen synthase kinase-3β. J Biol Chem (2002) 277:41888–96. doi: 10.1074/jbc.M206657200
4. Mantovani A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur J Immunol (2010) 40:3317–20. doi: 10.1002/eji.201041170
5. Quaranta V, Schmid MC. Macrophage-Mediated Subversion of Anti-Tumour Immunity. Cells (2019) 8:747. doi: 10.3390/cells8070747
6. Ha T-Y. The role of regulatory T cells in cancer. Immune Netw (2009) 9:209–35. doi: 10.4110/in.2009.9.6.209
7. Ma Y, Shurin GV, Peiyuan Z, Shurin MR. Dendritic Cells in the Cancer Microenvironment. J Cancer (2013) 4:36–44. doi: 10.7150/jca.5046
8. Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine (2019) 47:235–46. doi: 10.1016/j.ebiom.2019.08.025
9. Talmadge JE, Gabrilovich D II. History of myeloid-derived suppressor cells. Nat Rev Cancer (2013) 13:739–52. doi: 10.1038/nrc3581
10. Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget (2017) 8:3649. doi: 10.18632/oncotarget.12278
11. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell (2012) 21:822–35. doi: 10.1016/j.ccr.2012.04.025
12. Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol (1999) 162:5728–37.
13. Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1-bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat (2010) 123:39–49. doi: 10.1007/s10549-009-0622-8
14. Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+ Ly6C+ granulocytes. Proc Natl Acad Sci (2010) 107:21248–55. doi: 10.1073/pnas.1015855107
15. Waight JD, Hu Q, Miller A, Liu S, Abrams S II. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS One (2011) 6:e27690. doi: 10.1371/journal.pone.0027690
16. Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol (2010) 185.4:2273–84. doi: 10.4049/jimmunol.1000901
17. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202:931–9. doi: 10.1084/jem.20050715
18. Cassetta L, Bruderek K, Skrzeczynska-Moncznik J, Osiecka O, Hu X, Rundgren IM, et al. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J Immunother Cancer (2020) 8(2):e001223. doi: 10.1136/jitc-2020-001223
19. Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: Immune suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol (2018) 200:422–31. doi: 10.4049/jimmunol.1701019
20. Gabrilovich D II, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253–68. doi: 10.1038/nri3175
21. Bruger AM, Dorhoi A, Esendagli G, Barczyk-Kahlert K, van der Bruggen P, Lipoldova M, et al. How to measure the immunosuppressive activity of MDSC: assays, problems and potential solutions. Cancer Immunol Immunother (2019) 68(4):631–44. doi: 10.1007/s00262-018-2170-8
22. Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
23. Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest (2013) 123:4464–78. doi: 10.1172/JCI68189
24. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin J-P, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest (2010) 120:457–71. doi: 10.1172/JCI40483
25. Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol (2004) 172:464–74. doi: 10.4049/jimmunol.172.1.464
26. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol (2009) 182:5693–701. doi: 10.4049/jimmunol.0900092
27. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-Induced Tolerance and Immune Suppression Depend on the C/EBPβ Transcription Factor. Immunity (2010) 32:790–802. doi: 10.1016/j.immuni.2010.05.010
28. Wang W, Xia X, Mao L, Wang S. The CCAAT/Enhancer-Binding Protein Family: Its Roles in MDSC Expansion and Function. Front Immunol (2019) 10:1804. doi: 10.3389/fimmu.2019.01804
29. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol (2008) 181:4666–75. doi: 10.4049/jimmunol.181.7.4666
30. Yan D, Wang J, Sun H, Zamani A, Zhang H, Chen W, et al. TIPE2 specifies the functional polarization of myeloid-derived suppressor cells during tumorigenesis. J Exp Med (2020) 217. doi: 10.1084/jem.20182005
31. Yan DH, Wang JH, Wan XC. TIPE2 is a novel direct target of STAT3 in MDSC and inhibition of its expression on MDSC enhanced T cell activation in tumor. J Immunol (2017) 198:205.3–3.
32. Halaby MJ, Hezaveh K, Lamorte S, Ciudad MT, Kloetgen A, MacLeod BL, et al. GCN2 drives macrophage and MDSC function and immunosuppression in the tumor microenvironment. Sci Immunol (2019) 4. doi: 10.1126/sciimmunol.aax8189
33. Huang YH, Yang HY, Huang SW, Ou G, Hsu YF, Hsu MJ, et al. Interleukin-6 Induces Vascular Endothelial Growth Factor-C Expression via Src-FAK-STAT3 Signaling in Lymphatic Endothelial Cells. PLoS One (2016) 11:e0158839–e0158839. doi: 10.1371/journal.pone.0158839
34. McClure C, McPeak MB, Youssef D, Yao ZQ, McCall CE, El Gazzar M. Stat3 and C/EBPβ synergize to induce miR-21 and miR-181b expression during sepsis. Immunol Cell Biol (2017) 95:42–55. doi: 10.1038/icb.2016.63
35. Huber R, Pietsch D, Panterodt T, Brand K. Regulation of C/EBPβ and resulting functions in cells of the monocytic lineage. Cell Signal (2012) 24:1287–96. doi: 10.1016/j.cellsig.2012.02.007
36. Gao Y, Sun W, Shang W, Li Y, Zhang D, Wang T, et al. Lnc-C/EBPβ Negatively Regulates the Suppressive Function of Myeloid-Derived Suppressor Cells. Cancer Immunol Res (2018) 6:1352–63. doi: 10.1158/2326-6066.CIR-18-0108
37. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab (2018) 28:87–103.e6. doi: 10.1016/j.cmet.2018.04.022
38. Youn J-I, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol (2013) 14:211–20. doi: 10.1038/ni.2526
39. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol (2007) 7:41–51. doi: 10.1038/nri1995
40. Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med (2015) 7:314ra185. doi: 10.1126/scitranslmed.aac5272
41. Su Y-L, Banerjee S, White SV, Kortylewski M. STAT3 in Tumor-Associated Myeloid Cells: Multitasking to Disrupt Immunity. Int J Mol Sci (2018) 19:1803. doi: 10.3390/ijms19061803
42. Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity (2016) 44:303–15. doi: 10.1016/j.immuni.2016.01.014
43. Scott LM, Civin C II, Rorth P, Friedman AD. A novel temporal expression pattern of three C/EBP family members in differentiating myelomonocytic cells. Blood (1992) 80:1725–35. doi: 10.1182/blood.V80.7.1725.bloodjournal8071725
44. Bjerregaard MD, Jurlander J, Klausen P, Borregaard N, Cowland JB. The in vivo profile of transcription factors during neutrophil differentiation in human bone marrow. Blood (2003) 101:4322–32. doi: 10.1182/blood-2002-03-0835
45. Lekstrom-Himes J, Xanthopoulos KG. Biological Role of the CCAAT/Enhancer-binding Protein Family of Transcription Factors. J Biol Chem (1998) 273:28545–8. doi: 10.1074/jbc.273.44.28545
46. Jones LC, Lin ML, Chen SS, Krug U, Hofmann WK, Lee S, et al. Expression of C/EBPβ from the C/ebpα gene locus is sufficient for normal hematopoiesis in vivo. Blood (2002) 99:2032–6. doi: 10.1182/blood.V99.6.2032
47. Takata Y, Kitami Y, Yang ZH, Nakamura M, Okura T, Hiwada K. Vascular Inflammation Is Negatively Autoregulated by Interaction Between CCAAT/Enhancer-Binding Protein-δ and Peroxisome Proliferator-Activated Receptor-γ. Circ Res (2002) 91:427–33. doi: 10.1161/01.RES.0000031271.20771.4F
48. Min Y, Li J, Qu P, Lin PC. C/EBP-δ positively regulates MDSC expansion and endothelial VEGFR2 expression in tumor development. Oncotarget (2017) 8:50582–93. doi: 10.18632/oncotarget.16410
49. Ubeda M, Wang XZ, Zinszner H, Wu I, Habener JF, Ron D. Stress-induced binding of the transcriptional factor CHOP to a novel DNA control element. Mol Cell Biol (1996) 16:1479–89. doi: 10.1128/MCB.16.4.1479
50. Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, et al. The Stress-Response Sensor Chop Regulates the Function and Accumulation of Myeloid-Derived Suppressor Cells in Tumors. Immunity (2014) 41:389–401. doi: 10.1016/j.immuni.2014.08.015
51. Condamine T, Mastio J, Gabrilovich D II. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol (2015) 98:913–22. doi: 10.1189/jlb.4RI0515-204R
52. Descombes P, Schibler U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the sam mRNA. Cell (1991) 67:569–79. doi: 10.1016/0092-8674(91)90531-3
53. Lin W-W. & Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest (2007) 117:1175–83. doi: 10.1172/JCI31537
54. Wang K, Karin M. Tumor-Elicited Inflammation and Colorectal Cancer. Adv Cancer Res (2015) 128:173–96. doi: 10.1016/bs.acr.2015.04.014
55. Hirai H, Yokota A, Tamura A, Sato A, Maekawa T. Non-steady-state hematopoiesis regulated by the C/EBPβ transcription factor. Cancer Sci (2015) 106:797–802. doi: 10.1111/cas.12690
56. Tengku-Muhammad TS, Hughes TR, Ranki H, Cryer A, Ramji DP. DIFFERENTIAL REGULATION OF MACROPHAGE CCAAT-ENHANCER BINDING PROTEIN ISOFORMS BY LIPOPOLYSACCHARIDE AND CYTOKINES. Cytokine (2000) 12:1430–6. doi: 10.1006/cyto.2000.0711
57. Li T, Li X, Zamani A, Wang W, Lee CN, Li M, et al. c-Rel is a myeloid checkpoint for cancer immunotherapy. Nat Cancer (2020) 1:507–17. doi: 10.1038/s43018-020-0061-3
58. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res (2005) 65:3044–8. doi: 10.1158/0008-5472.CAN-04-4505
59. Arlauckas SP, Garren SB, Garris CS, Kohler RH, Oh J, Pittet MJ, et al. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics (2018) 8:5842–54. doi: 10.7150/thno.26888
60. Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J (1994) 298:249–58. doi: 10.1042/bj2980249
61. Ekmekcioglu S, Grimm EA, Roszik J. Targeting iNOS to increase efficacy of immunotherapies. Hum Vaccines Immunother (2017) 13:1105–8. doi: 10.1080/21645515.2016.1276682
62. Belikov AV, Schraven B, Simeoni L. T cells and reactive oxygen species. J Biomed Sci (2015) 22:85. doi: 10.1186/s12929-015-0194-3
63. Hubert MT, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, et al. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol (2010) 185:5247–58. doi: 10.4049/jimmunol.1001472
64. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci (2017) 114:1117–22. doi: 10.1073/pnas.1612920114
65. Kim S-H, Roszik J, Cho SN, Ogata D, Milton DR, Peng W, et al. The COX2 effector microsomal PGE2 synthase-1 is a regulator of immunosuppression in cutaneous melanoma. Clin Cancer Res (2019) 25:1650–63. doi: 10.1158/1078-0432.CCR-18-1163
66. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature (2016) 539:437–42. doi: 10.1038/nature19834
67. Dai J, Kumbhare A, Youssef D, Yao ZQ, McCall CE, El Gazzar M. Expression of C/EBPβ in myeloid progenitors during sepsis promotes immunosuppression. Mol Immunol (2017) 91:165–72. doi: 10.1016/j.molimm.2017.09.008
68. Dai J, Kumbhare A, Williams DA, Youssef D, Yao ZQ, McCall CE, et al. Nfia deletion in myeloid cells blocks expansion of myeloid-derived suppressor cells during sepsis. Innate Immun (2018) 24:54–65. doi: 10.1177/1753425917742956
69. Fullard N, Wilson CL, Oakley F. Roles of c-Rel signalling in inflammation and disease. Int J Biochem Cell Biol (2012) 44:851–60. doi: 10.1016/j.biocel.2012.02.017
70. Gilmore TD, Gerondakis S. The c-Rel Transcription Factor in Development and Disease. Genes Cancer (2011) 2:695–711. doi: 10.1177/1947601911421925
71. Leeman JR, Weniger MA, Barth TF, Gilmore TD. Deletion analysis and alternative splicing define a transactivation inhibitory domain in human oncoprotein REL. Oncogene (2008) 27:6770–81. doi: 10.1038/onc.2008.284
72. Bunting K, Rao S, Hardy K, Woltring D, Denyer GS, Wang J, et al. Genome-wide analysis of gene expression in T cells to identify targets of the NF-kappa B transcription factor c-Rel. J Immunol (2007) 178:7097–109. doi: 10.4049/jimmunol.178.11.7097
73. Starczynowski DT, Trautmann H, Pott C, Harder L, Arnold N, Africa JA, et al. Mutation of an IKK phosphorylation site within the transactivation domain of REL in two patients with B-cell lymphoma enhances REL’s in vitro transforming activity. Oncogene (2007) 26:2685–94. doi: 10.1038/sj.onc.1210089
74. Starczynowski DT, Reynolds JG, Gilmore TD. Mutations of tumor necrosis factor α -responsive serine residues within the C-terminal transactivation domain of human transcription factor REL enhance its in vitro transforming ability. Oncogene (2005) 24:7355–68. doi: 10.1038/sj.onc.1208902
75. Yu S-H, Chiang W-C, Shih H-M, Wu K-J. Stimulation of c-Rel transcriptional activity by PKA catalytic subunit beta. J Mol Med Berl Ger (2004) 82:621–8. doi: 10.1007/s00109-004-0559-7
76. Martin AG, San-Antonio B, Fresno M. Regulation of Nuclear Factor κB Transactivation IMPLICATION OF PHOSPHATIDYLINOSITOL 3-KINASE AND PROTEIN KINASE C ζ IN c-Rel ACTIVATION BY TUMOR NECROSIS FACTOR α. J Biol Chem (2001) 276:15840–9. doi: 10.1074/jbc.M011313200
77. Köntgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev (1995) 9:1965–77. doi: 10.1101/gad.9.16.1965
78. Grumont RJ, Rourke IJ, O’Reilly LA, Strasser A, Miyake K, Sha W, et al. B Lymphocytes Differentially Use the Rel and Nuclear Factor κB1 (NF-κB1) Transcription Factors to Regulate Cell Cycle Progression and Apoptosis in Quiescent and Mitogen-activated Cells. J Exp Med (1998) 187:663–74. doi: 10.1084/jem.187.5.663
79. Tumang JR, Owyang A, Andjelic S, Jin Z, Hardy RR, Liou ML, et al. c-Rel is essential for B lymphocyte survival and cell cycle progression. Eur J Immunol (1998) 28:4299–312. doi: 10.1002/(SICI)1521-4141(199812)28:12<4299::AID-IMMU4299>3.0.CO;2-Y
80. Liou H-C, Smith KA. The roles of c-rel and interleukin-2 in tolerance: a molecular explanation of self–nonself discrimination. Immunol Cell Biol (2011) 89:27–32. doi: 10.1038/icb.2010.120
81. Strasser A, Grumont RJ, Stanley ML, Gerondakis S. The transcriptional regulator Rel is essential for antigen receptor-mediated stimulation of mature T cells but dispensable for positive and negative selection of thymocytes and T cell apoptosis. Eur J Immunol (1999) 29:928–35. doi: 10.1002/(SICI)1521-4141(199903)29:03<928::AID-IMMU928>3.0.CO;2-P
82. Gugasyan R, Grumont R, Grossmann M, Nakamura Y, Pohl T, Nesic D, et al. Rel/NF-kappaB transcription factors: key mediators of B-cell activation. Immunol Rev (2000) 176:134–40. doi: 10.1034/j.1600-065X.2000.00615.x
83. Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med (2009) 206:3001–14. doi: 10.1084/jem.20091411
84. Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, et al. Development of Foxp3+ Regulatory T Cells Is Driven by the c-Rel Enhanceosome. Immunity (2009) 31:932–40. doi: 10.1016/j.immuni.2009.10.006
85. Vang KB, Yang J, Pagán AJ, Li LX, Wang J, Green JM, et al. Cutting Edge: CD28 and c-Rel–Dependent Pathways Initiate Regulatory T Cell Development. J Immunol (2010) 184:4074–7. doi: 10.4049/jimmunol.0903933
86. Long M, Park S-G, Strickland I, Hayden MS, Ghosh S. Nuclear Factor-κB Modulates Regulatory T Cell Development by Directly Regulating Expression of Foxp3 Transcription Factor. Immunity (2009) 31:921–31. doi: 10.1016/j.immuni.2009.09.022
87. Visekruna A, Huber M, Hellhund A, Bothur E, Reinhard K, Bollig N, et al. c-Rel is crucial for the induction of Foxp3+ regulatory CD4+ T cells but not TH17 cells. Eur J Immunol (2010) 40:671–6. doi: 10.1002/eji.200940260
88. Grigoriadis G, Zhan Y, Grumont RJ, Metcalf D, Handman E, Cheers C, et al. The Rel subunit of NF-kappaB-like transcription factors is a positive and negative regulator of macrophage gene expression: distinct roles for Rel in different macrophage populations. EMBO J (1996) 15:7099–107. doi: 10.1002/j.1460-2075.1996.tb01101.x
89. Cha-Molstad H, Young DP, Kushner I, Samols D. The interaction of C-Rel with C/EBPbeta enhances C/EBPbeta binding to the C-reactive protein gene promoter. Mol Immunol (2007) 44:2933–42. doi: 10.1016/j.molimm.2007.01.015
90. Agrawal A, Samols D, Kushner I. Transcription factor c-Rel enhances C-reactive protein expression by facilitating the binding of C/EBPβ to the promoter. Mol Immunol (2003) 40:373–80. doi: 10.1016/S0161-5890(03)00148-2
91. Joughin BA, Cheung E, Karuturi RK, Saez-Rodriguez J, Lauffenburger DA, Liu ET. Chapter 4 - Cellular Regulatory Networks. In: Liu ET, Lauffenburger DA, editors. Systems Biomedicine. Cambridge, Massachusetts:Academic Press (2010). p. 57–108. doi: 10.1016/B978-0-12-372550-9.00004-3
92. Merika M, Thanos D. Enhanceosomes. Curr Opin Genet Dev (2001) 11:205–8. doi: 10.1016/S0959-437X(00)00180-5
93. Fiedler M, Graeb M, Mieszczanek J, Rutherford TJ, Johnson CM, Bienz M. An ancient Pygo-dependent Wnt enhanceosome integrated by Chip/LDB-SSDP. Elife (2015) 4:e09073. doi: 10.7554/eLife.09073
94. Pawlus MR, Hu CJ. Enhanceosomes as integrators of hypoxia inducible factor (HIF) and other transcription factors in the hypoxic transcriptional response. Cell Signal (2013) 25(9):1895–903. doi: 10.1016/j.cellsig.2013.05.018
96. Carvajal IM, Baron RM, Perrella MA. High-mobility group-I/Y proteins: Potential role in the pathophysiology of critical illnesses. Crit Care Med (2002) 30:S36. doi: 10.1097/00003246-200201001-00005
97. Li Y, Cifuentes-Pagano E, DeVallance ER, de Jesus DS, Sahoo S, Meijles DN, et al. NADPH oxidase 2 inhibitors CPP11G and CPP11H attenuate endothelial cell inflammation & vessel dysfunction and restore mouse hind-limb flow. Redox Biol (2019) 22:101143. doi: 10.1016/j.redox.2019.101143
98. Qiu H, Hu C, Yoon S, Natarajan K, Swanson MJ, Hinnebusch AG. An Array of Coactivators Is Required for Optimal Recruitment of TATA Binding Protein and RNA Polymerase II by Promoter-Bound Gcn4p. Mol Cell Biol (2004) 24:4104–17. doi: 10.1128/MCB.24.10.4104-4117.2004
99. Porta C, Consonni FM, Morlacchi S, Sangaletti S, Bleve A, Totaro MG, et al. Tumor-derived prostaglandin E2 promotes p50 NF-κB-dependent differentiation of monocytic MDSC. Cancer Res (2020) 80(13): 2874–88. doi: 10.1158/0008-5472.CAN-19-2843
100. Tamura A, Hirai H, Yokota A, Kamio N, Sato A, Shoji T, et al. C/EBPβ is required for survival of Ly6C– monocytes. Blood (2017) 130:1809–18. doi: 10.1182/blood-2017-03-772962
101. Pope RM, Lovis R, Mungre S, Perlman H, Koch AE, Haines III GK. C/EBPβ in Rheumatoid Arthritis: Correlation with Inflammation, Not Disease Specificity1. Clin Immunol (1999) 91:271–82. doi: 10.1006/clim.1999.4723
102. Lee HH, Sanada S, An SM, Ye BJ, Lee JH, Seo YK, et al. LPS-induced NFκB enhanceosome requires TonEBP/NFAT5 without DNA binding. Sci Rep (2016) 6:24921. doi: 10.1038/srep24921
Keywords: myeloid-derived suppressor cell, immunosuppression, enhanceosome, aberrant myelopoiesis, tumor immunobiology
Citation: Fultang N, Li X, Li T and Chen YH (2021) Myeloid-Derived Suppressor Cell Differentiation in Cancer: Transcriptional Regulators and Enhanceosome-Mediated Mechanisms. Front. Immunol. 11:619253. doi: 10.3389/fimmu.2020.619253
Received: 19 October 2020; Accepted: 30 November 2020;
Published: 14 January 2021.
Edited by:
Lewis Z. Shi, University of Alabama at Birmingham, United StatesReviewed by:
Katherine Chiappinelli, George Washington University, United StatesCopyright © 2021 Fultang, Li, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youhai H. Chen, eWhjQHBlbm5tZWRpY2luZS51cGVubi5lZHU=; Norman Fultang, RnVsdGFuZ25AcGVubm1lZGljaW5lLnVwZW5uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.