 Matthew Clark1,2
Matthew Clark1,2 Charles J. Kroger
Charles J. Kroger Qi Ke
Qi Ke Roland M. Tisch
Roland M. Tisch- 1Department of Microbiology and Immunology, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 2Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
T cell receptor (TCR) signaling influences multiple aspects of CD4+ and CD8+ T cell immunobiology including thymic development, peripheral homeostasis, effector subset differentiation/function, and memory formation. Additional T cell signaling cues triggered by co-stimulatory molecules and cytokines also affect TCR signaling duration, as well as accessory pathways that further shape a T cell response. Type 1 diabetes (T1D) is a T cell-driven autoimmune disease targeting the insulin producing β cells in the pancreas. Evidence indicates that dysregulated TCR signaling events in T1D impact the efficacy of central and peripheral tolerance-inducing mechanisms. In this review, we will discuss how the strength and nature of TCR signaling events influence the development of self-reactive T cells and drive the progression of T1D through effects on T cell gene expression, lineage commitment, and maintenance of pathogenic anti-self T cell effector function.
Introduction
T cell-driven autoimmune diseases are heterogeneous and complex, typically leading to chronic organ-specific inflammation and tissue damage. Type 1 diabetes (T1D) is a prominent example of a T cell-mediated autoimmune disease. T1D is characterized by the destruction and/or dysregulation of the insulin producing β cells found in the pancreatic islets of Langerhans (1–5). Typically, β cell autoimmunity progresses for a number of years prior to clinical diagnosis of T1D (6, 7). The autoimmune response is heterogeneous marked by the disparate onset between childhood and adult diabetes (7–22). Inflammation of the islets, termed insulitis, involves infiltration of CD4+ and CD8+ T cells, B cells, dendritic cells (DC) and macrophages, as well as production of islet-specific autoantibodies (7, 9–18, 23, 24). When 80%–90% of β cell mass is rendered nonfunctional, hyperglycemia is established, and overt diabetes diagnosed. Indirect evidence indicates that human T1D is driven by T cells (9–18). For instance, the strongest genetic risk factor for T1D is associated with specific alleles of HLA class II and class I molecules (25–30). CD4+ and particularly CD8+ T cells are typically detected infiltrating the islets of T1D subjects (13, 25–28). Furthermore, the more aggressive childhood versus adult T1D onset is characterized by an expanded effector T cell (Teff) response to β cell autoantigens, such as proinsulin and insulin (11). Moreover, in non-obese diabetic (NOD) mice, a spontaneous model of T1D, CD4+ and CD8+ T cells are essential for β cell destruction, and respond to similar β cell autoantigens (5, 20, 31, 32). The NOD mouse model has been integral for studying the immunology of T1D, and investigating immunotherapies. The diabetogenic response in NOD mice, however, is considered to represent only one scenario of the heterogenous disease process in human T1D. For instance, examples exist suggesting that human T1D can progress in a T cell-independent manner. Nevertheless, CD4+ and CD8+ T cells are thought to be the primary drivers of β cell autoimmunity in most cases of human T1D (13, 33, 34).
The tissue-specificity and progression of T cell-mediated autoimmunity is dictated in part by the repertoire of T cell receptors (TCR) expressed by pathogenic Teff (18). The TCR repertoire in general is inherently self-reactive, and T cells recognizing self-peptides are detected in healthy individuals (11, 35–38). Typically, autoreactive T cells are tightly regulated by both central and peripheral tolerance-inducing mechanisms that prevent autoimmune-mediated pathology. However, in susceptible individuals, aberrant self-tolerance allows for the development, expansion and differentiation of autoreactive Teff that mediate pathology. In T1D numerous genetic variants are associated with disease susceptibility and resistance (27–30, 39–44). Notably, several of these insulin-dependent diabetes mellitus (IDDM) loci contain candidate genes that are involved with various aspects T cell immunobiology (45–50). The result is preferential skewing largely toward differentiation and expansion of β cell-specific type 1 CD4+ (e.g. T helper 1 (Th1)) and CD8+ (e.g. type 1 CD8+ (Tc1)) Teff, marked by expression of IFNγ and other proinflammatory cytokines (11, 51–53). Additional Teff subsets have been associated with driving β cell autoimmunity (51, 54–56). For example, the frequency of peripheral blood T follicular helper cells (Tfh) in human T1D patients correlates with increasing autoantibody production and reductions in C-peptide levels (57, 58). Furthermore, the IL2/IL21 genomic region has also been identified as a risk factor in genome-wide association studies of human T1D subjects (41). Interestingly, clinical responsiveness of T1D patients to Abatacept treatment, which entails blockade of the CD80 and CD86 costimulatory molecules, directly correlates with a reduced pool of functional Tfh (59). These studies highlight Tfh as a key predictor of T1D disease progression (59). In addition, NOD mice fail to develop diabetes in the absence of IL-21, further suggesting that Tfh as well as Th17 are important contributors to β cell autoimmunity (56, 60–66).
Aberrant immunoregulation also contributes to the differentiation and expansion of pathogenic Teff in T1D (67, 68). In general, both natural and induced immunoregulatory CD4+ T cells expressing the forkhead box P3 protein transcription factor (nFoxp3+Treg and iFoxp3+Treg, respectively) play a critical role in suppressing autoimmunity (69–76). In human and NOD mouse T1D, reports have described aberrant maintenance, fitness and/or function of the Foxp3+Treg pool (54, 77–91). In addition, intrinsic defects within human and murine T1D Teff promote resistance to Foxp3+Treg-mediated suppression (77, 92).
The nature of a Teff response is influenced by multiple stimuli including TCR signal strength and duration, and/or the availability of co-stimulatory molecules and cytokines provided by antigen presenting cells (APC) (93, 94). Typically, strong TCR signaling is associated with a Th1 and Tc1 response regulated by the transcription factor T-bet. Since islet resident T cells largely exhibit a type 1 phenotype, this suggests that TCR signaling events favor differentiation of proinflammatory Teff (51, 52). In this review, we will discuss how key TCR signaling events in human T1D patients and the NOD mouse alters T cell development in the thymus that favors an autoreactive TCR repertoire, and how dysregulation of TCR signaling in the periphery imprints a proinflammatory phenotype in β cell-specific Teff that drives pancreatic islet damage.
Thymic Origins of T Cell Receptor-Driven β Cell-Specific Autoimmunity
Thymic Selection Events Shape the Anti-Self T Cell Receptor Repertoire
TCR signaling plays a pinnacle role in regulating T cell homeostasis, activation, expansion and effector function upon recognition of cognate foreign- or self-antigens. The specificity and properties of the TCR repertoire are established via selection events ongoing in the thymus (95–100). Positive selection occurring in the thymus cortex establishes a functional pool of TCR that bind self-peptide-MHC class I or II complexes. Cortical thymic epithelial cells (cTEC) mediate positive selection by presenting alternatively processed self-peptides to double positive thymocytes (DP), characterized in part by expression of the TCR, and both CD4 and CD8 co-receptors. Sufficient binding of TCR to self-peptide-MHC class I or class II molecules results in signaling events that promote DP survival and differentiation into CD8+ or CD4+ single positive thymocytes (SP), respectively. In the absence of TCR stimulation, DP undergo apoptosis. Since self-peptides drive positive selection, all functional TCR exhibit some degree of self-reactivity.
Positively selected SP migrate from the thymic cortex into the medulla to undergo negative selection. The thymic medulla is populated by medullary TEC (mTEC), DC, and B cells which present peripheral tissue-specific antigens (TSA) (101–103). mTEC express the transcription factors autoimmune regulator (AIRE) and forebrain expressed zinc finger 2 (Fezf2), which drive expression of a spectrum of TSA, including β cell-expressed proteins such as insulin (104–109). Thymic DC on the other hand acquire TSA and associated peptides through various mechanisms, including uptake of apoptotic mTEC, and trogocytosis of surface pMHC from mTEC (110–115). DC and B cells that traffick into the thymus also ferry TSA acquired from the periphery (116–120). SP expressing TCR with increased affinity/avidity for a given TSA-derived peptide are negatively selected and undergo apoptosis. A fraction of CD4+ SP expressing high affinity TCR for self-peptide, however, survive negative selection by differentiating into nFoxp3+Treg. SP that exhibit a low affinity for a TSA-derived peptide survive negative selection and egress from the thymus into the periphery (121). Noteworthy is that the efficiency of negative selection is limited early in ontogeny, thereby providing a discrete window of increased development of autoreactive T cells with enhanced affinity/avidity (122, 123). For instance, β cell autoimmunity develops in immunodeficient NOD.scid recipients reconstituted with T cells derived from transplanted thymic lobes of newborn but not 10 day or older NOD mice (124). Interestingly, autoimmunity has been reported in immunodeficient children with congenital athymia that receive an infant thymus transplant (125). The lack of structural organization of the medulla coupled with a largely immature mTEC pool expressing reduced TSA are thought to contribute to the limited efficacy of negative selection early in life.
Factors That Impact the Specificity of the Anti-Self T Cell Receptor Repertoire Pool
The breadth and diversity of the TCR repertoire established during positive and negative selection reflects the TSA expressed and self-peptides processed and presented by cTEC and mTEC (98–100). Effective deletion of self-reactive SP for instance, is tightly linked to thymic TSA expression. In mice deficient of AIRE and Fezf2, mTEC fail to present a sufficiently broad constellation of TSA. Consequently, SP expressing anti-self TCR with increased affinity escape to the periphery and drive systemic autoimmunity (126). AIRE-deficiency also results in the failure of SP expressing TSA-specific TCR to differentiate into nFoxp3+Treg (127, 128). Humans with AIRE mutations develop Autoimmune Polyglandular Syndrome type-1 (APS-1) marked by multi-organ autoimmunity that includes T1D, Addison’s disease, and hypoparathyroidism (129, 130). Notably, the level of mTEC expression of insulin, a critical β cell autoantigen, can have a significant impact on the progression of T1D. Non-autoimmune prone C57BL/6 (B6) mice develop diabetes when mTEC lack insulin expression (131). Similarly, thymic insulin gene expression in humans is strongly associated with T1D susceptibility. Humans that have decreased variable number of tandem repeats (VNTRs) upstream of the insulin gene exhibit reduced thymic insulin expression, and increased T1D susceptibility (132, 133).
The efficiency of processing of self-antigens by cTEC and mTEC is also thought to play a key role in establishing the anti-self TCR repertoire (98, 100). Diabetes is prevented in NOD mice lacking thymus serine specific protease (TSSP) and Cathespin L (CatL), which are expressed by cTEC to generate MHC class II peptides (134–137). Lack of diabetes in TSSP- and CatL-deficient NOD mice correlates with an altered TCR repertoire expressed by CD4+ T cells, as well as increased thymic nFoxp3+Treg development. A modest decrease in diabetes is also seen in NOD mice lacking Cathespin S, which is involved in mTEC processing of MHC class II peptides (137). It is notable that TCR diversity is limited in CD4+CD25- and CD4+CD25+ thymocytes from NOD (H2g7) versus B6 mice congenic for the H2g7 haplotype (138). Altered thymic antigen processing contributing to human T1D and myasthenia gravis is suggested by risk variants of TSSP and the CatL homolog CTSL2 gene encoding CatV (139–141).
Finally, intrinsic properties of alleles of MHC molecules affect the repertoire of self-peptides presented in the thymus. As noted above the strongest genetic association with human T1D susceptibility is linked to specific alleles of the DQB1 gene; namely HLA-DQ8 (DQB1*03:02) and HLA-DQ2 (DQB1*02:01) (25–27). In NOD mice expression of the unique IAg7 class II molecule is needed for the development of β cell autoimmunity (142, 143). Notably, the DQ2/8 and IAg7 molecules share a common polymorphism at position 57 on the β chain which encodes a non-aspartic acid residue (144). This polymorphism favors the binding of peptides with a hydrophobic P9 anchor residue. In addition, IAg7-peptide complexes exhibit reduced surface half-life relative to other IA alleles (145). Therefore, T1D-associated MHC alleles are expected to promote sub-optimal presentation of self-peptides which enhances escape of β cell-specific SP with increased TCR affinity, while limiting the TCR repertoire of thymic Foxp3+Treg.
In sum, thymic selection and central tolerance involve a complex set of events in which the expression and presentation of TSA-derived peptides impact: 1) the properties of the TCR repertoire expressed by T cells specific for self and foreign antigen, as well as 2) the development of protective nFoxp3+Treg. Dysregulation of these events can lead to an elevated peripheral frequency of autoreactive T cells expressing TCR with increased affinity, thereby enhancing susceptibility for pathogenic autoimmunity (Figure 1). The latter is further enhanced via peripheral processing and post-translational modification of islet antigens. De novo islet antigens such as hybrid insulin peptides (HIP) not presented in the thymus, stimulate the expansion of diabetogenic Teff in NOD mice (146–148). Interestingly, some HIP induce stronger TCR signaling than native proinsulin, suggesting low affinity/avidity autoreactive T cells can be readily activated by novel islet antigens (146, 147).
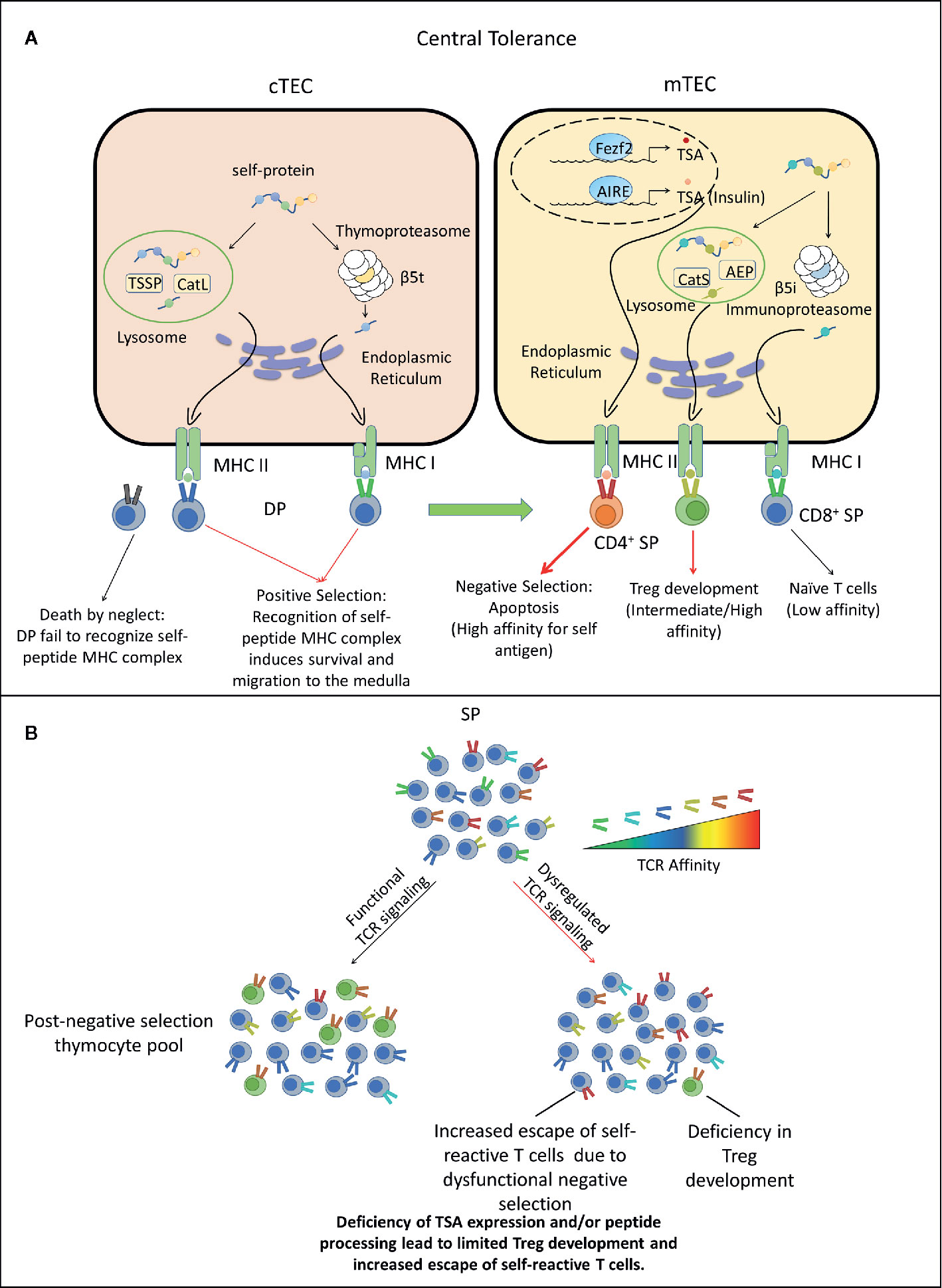
Figure 1 Dysregulated central tolerance impacts T cell receptor (TCR) signaling and establishes a T cell repertoire with increased type 1 diabetes (T1D) susceptibility. (A) In the thymus cortex, double positive thymocytes (DP) engage cortical thymic epithelial cell (cTEC) through TCR/peptide-MHC complexes (pMHC) interactions. Self-peptides are processed via the thymoproteasome and lysosomal proteases and displayed by MHC I and II molecules expressed by cTEC. DP recognizing these self-peptides receive survival signals, differentiate into CD4+ or CD8+ single positive thymocytes (SP) and migrate to the medulla. Alternatively, DP thymocytes incapable of transmitting TCR mediated survival signals die due to neglect. In the medulla, SP interact with mTEC or DC. Medullary TEC (mTEC) express a variety of tissue-specific antigens (TSA) such as insulin under the control of transcription factors autoimmune regulator (AIRE) and forebrain expressed zinc finger 2 (Fezf2). TSA are processed and presented on the MHC of mTEC, which can be acquired by DC by various mechanisms. SP with high TCR affinity for self-antigens are deleted through apoptosis, whereas SP exhibiting low TCR self-reactivity become naïve T cells. A fraction of CD4+ SP thymocytes with intermediate to high affinity differentiate into nFoxp3+Treg. Surviving SP exit the thymus and comprise the peripheral TCR repertoire. Red thick arrow: high TCR/pMHC affinity. Red thin arrow: intermediate TCR/pMHC affinity. Black arrow: low TCR/pMHC affinity. (B) Deficiency in self-peptide expression or processing by cTEC reduces TCR repertoire diversity and potentially increases susceptibility to β cell autoimmunity. Reduced expression and presentation of β cell-specific peptides by mTEC and other thymic APC limits: 1) negative selection leading to escape of an increased frequency of β cell-specific T cells with increased affinity/avidity, as well as impaired development of β cell-specific Foxp3+Treg. AEP, Asparaginyl endopeptidase.
Factors That Influence T Cell Receptor Signaling Strength During Type 1 Diabetes
Efficient T Cell Activation Is Dependent on Signals 1, 2, and 3
TCR signaling involves a series of phosphorylation events initiated by the tyrosine kinase LCK-ZAP70 axis. LCK and ZAP70 activity is then propagated by secondary messengers including the PLCγ1, PI3K, MAPK, and Ca++ flux pathways (149–152). Over time, these signaling events culminate in epigenetic reorganization and gene transcription (93, 149, 153). Intrinsic and extrinsic factors regulate the nature, strength and functional outcome of TCR signaling in peripheral T cells. Engagement of the TCR with the peptide-MHC complexes (pMHC) forms an immune synapse (IS) between T cells and APC, which serves to regulate TCR signaling intensity and duration (154, 155). The IS facilitates interactions with the TCR and pMHC resulting in “signal 1” (154, 155). Optimal activation and differentiation of naïve T cells, as well as maintenance of Teff subsets, and differentiation of long-lived memory T cells (Tmem), however, requires additional signals (150). This includes “signal 2” delivered by T cell co-stimulatory molecules such as CD4, CD8, CD28, ICOS, and CD40L, and “signal 3” mediated by cytokines derived by T cells and APC (152).
Anti-Self T Cell Receptor Signaling Is Required for T Cell Homeostasis
Preventing self-reactive TCR from exceeding the required signaling threshold between naïve T cell quiescence versus differentiation of pathogenic Teff after self-peptide recognition is a critical step to maintaining peripheral self-tolerance (151). This is particularly important since T cells continuously sample self-antigens via brief, low affinity TCR-pMHC interactions resulting in limited TCR-mediated phosphorylation events (156–158). This tonic TCR signaling is required for T cell survival and homeostasis. For instance, CD8+ and CD4+ T cells transferred into MHC class I and II deficient hosts, respectively, undergo accelerated apoptosis (159–162). Recent thymic emigrant (RTE) T cells also require tonic TCR signaling to fully develop into mature naïve T cells in the periphery. Therefore, the immune system exploits the intrinsic self-reactivity of TCR generated in the thymus to establish a balance between T cell homeostasis and unwanted autoimmunity through regulated T cell activation thresholds. This regulation is achieved partly through signaling events that are analog versus digital in nature (163). The proximal TCR-mediated phosphorylation of ZAP70 and upregulated gene expression of the transcription factor Nur77 are examples of analog signals that develop in a graded manner, directly reflecting the strength of TCR stimulus (163–165). In contrast, digital signaling involves events that are activated once a threshold is achieved with no intermediate state. Activation of NF-κB, a transcription factor driving T cell activation and function, is an example of TCR-mediated digital signaling (166). TCR analog events including Ca++-stimulated activation of IRF4 enable tonic signaling to occur below the threshold needed for efficient T cell activation (163, 167). In this way, despite low TCR reactivity to self, T cells sustain homeostatic gene expression without initiating an autoimmune response.
Multiple Factors Influence T Cell Receptor Signaling Strength and Type 1 Diabetes Development
The strength of TCR signaling delivered upon recognition of a cognate, self-peptide-MHC complex is governed by a number of parameters including: TCR affinity, the frequency of pMHC, and the duration of the TCR-pMHC interaction (168). For any given antigen, the polyclonal TCR repertoire exhibits a spectrum of affinities (169). T cells expressing high affinity TCR can be activated at a relatively low pMHC frequency and vice versa. As alluded to above, the majority of autoreactive T cells are believed to express TCR with relatively low affinity that in turn permits escape of thymic negative selection. Accordingly, a high frequency of pMHC is required to activate this autoreactive T cell pool. On the other hand, inefficient thymic negative selection that is believed to occur in NOD mice, results in an autoreactive T cell pool expressing TCR with increased affinity (170–173). Therefore, a reduced frequency of pMHC is sufficient to stimulate and overcome the threshold for efficient activation of autoreactive T cells. The frequency of pMHC expressed by APC is dictated by a number of factors including the stability of the complex, which is in part influenced by peptide binding affinity and intrinsic properties of the MHC molecule. As noted above the IAg7 molecule has been reported to exhibit reduced stability which would be expected to limit T cell stimulation in the periphery (145). However, under proinflammatory conditions, APC upregulate antigen processing and presentation machinery as well as the expression level of co-stimulatory and MHC molecules that can compensate for the limited stability of IAg7 (174). One interesting scenario is that IAg7 instability reduces induction of T cell exhaustion, particularly during chronic antigen stimulation that occurs in the islets. Sustained TCR signaling promotes the formation of exhausted T cells (Tex), which serves to limit a given T cell response (175, 176). Levels of peptide-IAg7 complexes may be sufficient to stimulate Teff reactivity without driving exhaustion, thereby propagating chronic β cell autoimmunity.
Enhancing TCR signaling events in the periphery can break self-tolerance leading to autoimmunity. Polymorphisms in the protein tyrosine phosphatase, non-receptor type 22 (PTPN22) is associated with increased susceptibility to T1D and other autoimmune diseases (45, 46, 177–179). The role of PTPN22 in autoimmunity is complex, particularly since this tyrosine phosphatase is also expressed in B cells and DC, which contribute to the disease process (180, 181). In T cells, PTPN22 downregulates TCR signaling at multiple points including dephosphorylation and inactivation of the proximal kinases such as LCK and ZAP70 (179). PTPN22-deficient T cells exhibit increased LCK activation and Ca++ signaling (179). Furthermore, in NOD mice expressing the knock-in human PTPN22 risk variant, Teff and Tmem populations are increased, analogous to that seen in human subjects (182). This and other studies with a loss-in-function phenotype associated with the human PTPN22 risk variant consistently show increased T cell activation (179, 182). In this scenario, T cells expressing low affinity self-reactive TCR more readily overcome signaling thresholds needed for activation, expansion, and/or differentiation. Similarly, gene variants of PTPN2, another tyrosine phosphatase, have been associated with a T1D susceptibility gene (183). Human T1D risk variants have been identified that impact PTPN2 mRNA stability, protein structure, and expression levels (50, 184). β cell autoimmunity is exacerbated in NOD mice that selectively lack PTPN2 expression in T cells (185). Interestingly, enhanced disease progression is marked by an increased frequency of CD4+ Th1 and CD8+ Tc1 cells, as well as Tfh (185).
The strength of TCR signaling is also impacted by the CD28-CTLA-4 axis, which plays a role in T1D. A number of variants encoding CTLA-4 are associated with increased human T1D risk (47, 48, 186). Normally, CTLA-4 is upregulated shortly after T cell activation, competes with CD28 for binding to CD80 and CD86, and downregulates co-stimulatory “signal 2” (187, 188). The latter is achieved by recruitment of the phosphatases SHP-2 and PP2A to the cytoplasmic tail of CTLA-4. Activation of SHP-2 then inhibits LCK mediated phosphorylation of ZAP70 and activation of PI3K signaling pathways to block TCR signaling events. T1D risk variants are associated with reduced CTLA4 mRNA levels and altered mRNA splicing resulting in a soluble isoform of CTLA-4 (47, 48, 186). Limited expression of CTLA-4 on the surface of conventional T cells (e.g. FoxP3- T cells) is expected to readily promote expansion of Teff. Indeed, cancer immunotherapy trials have recently reported that CTLA-4 blockade induces T1D onset in some subjects (187). Furthermore, induced deletion of CTLA-4 in adult mice results in increased Teff numbers (189).
In addition to having a critical role in Teff subset differentiation, cytokine-mediated “signal 3” contributes to TCR signaling (152). IL-18 and IL-21 are particularly important in T1D. Deficiency in either IL-18 or IL-21 prevents the onset of diabetes in NOD mice (60, 190). IL-18 is known to stabilize Th1 responses, and in tandem with the TCR signaling related molecule Themis, enhances TCR signaling to self-antigens promoting T cell proliferation (190, 191). IL-21 is a key driver of B cell responses, but also enhances T cell proliferation of islet specific CD8+ T cells (63, 65, 192–196). Interestingly, IL-21 can mitigate development of Tex via STAT3 activation and the induction of the transcription factor BATF. TCR-induced IRF4 and IL-21 driven BATF cooperate to induce the expression of Blimp-1 that maintains Teff reactivity and blocks induction of exhaustion. IL-21 signaling has also been found to decrease expression of the Tex programmed cell death protein-1 (PD-1) (196). Therefore, IL-21 may help sustain β cell-specific T cell responses long-term through STAT3-regulated events, despite chronic TCR signaling. Interestingly, an intrinsic polymorphism increases STAT3 activation which is associated with severe multi-organ autoimmunity including T1D in humans (197). Approaches to attenuate pathogenic “signal 3” TCR events have been attempted in the clinic to alter T1D progression (198, 199). Specifically, an anti-IL-21 antibody therapy is currently being evaluated (NCT02443155) for efficacy in β cell preservation in newly diagnosed T1D patients.
Impact of T Cell Receptor Signaling on the Molecular Landscape of T Cells
T Cell Receptor Signaling and Regulation of Epigenetic Events
TCR signaling affects epigenetic and transcriptional regulation. A number of transcription factors are governed by TCR signaling and cytokine stimulation (Table 1), which shape the molecular landscape of naïve T cells transitioning into Teff, as well as the viability and expansion of Teff and differentiation of Tmem (200, 201). T cell activation induces changes in DNA methylation and acetylation, creating broad and lasting genetic modifications (200, 201). The most prominent markers denoting alterations in gene transcription are histone H3 lysine 27 trimethylation (H3K27Me3), and histone H3 lysine 27 acetylation (H3K27Ac). Methylation of histones such as H3K27Me3 have classically been associated with a closed chromatin state preventing gene transcription. On the other hand, acetylation markers such as H3K27Ac, correlate with an open chromatin state permissive for transcription. Accordingly, histone methyltransferases and deacetylases suppress gene expression, whereas histone acetyltransferases and demethylases promote gene expression. Enhancer of zeste homolog 2 (EZH2), the functional unit of the polycomb repressive complex 2 (PRC2), is a histone methylase that plays a key role in regulating various aspects of T cell immunobiology such a Foxp3+Treg stability (202, 203). Aberrant chromatin landscapes following T cell activation have been noted in numerous autoimmune diseases including rheumatoid arthritis, systemic lupus erythematosus, Grave’s disease, and T1D (204). Teff isolated from NOD mice display a unique chromatin structure conferring expression of T1D associated genetic loci (205).
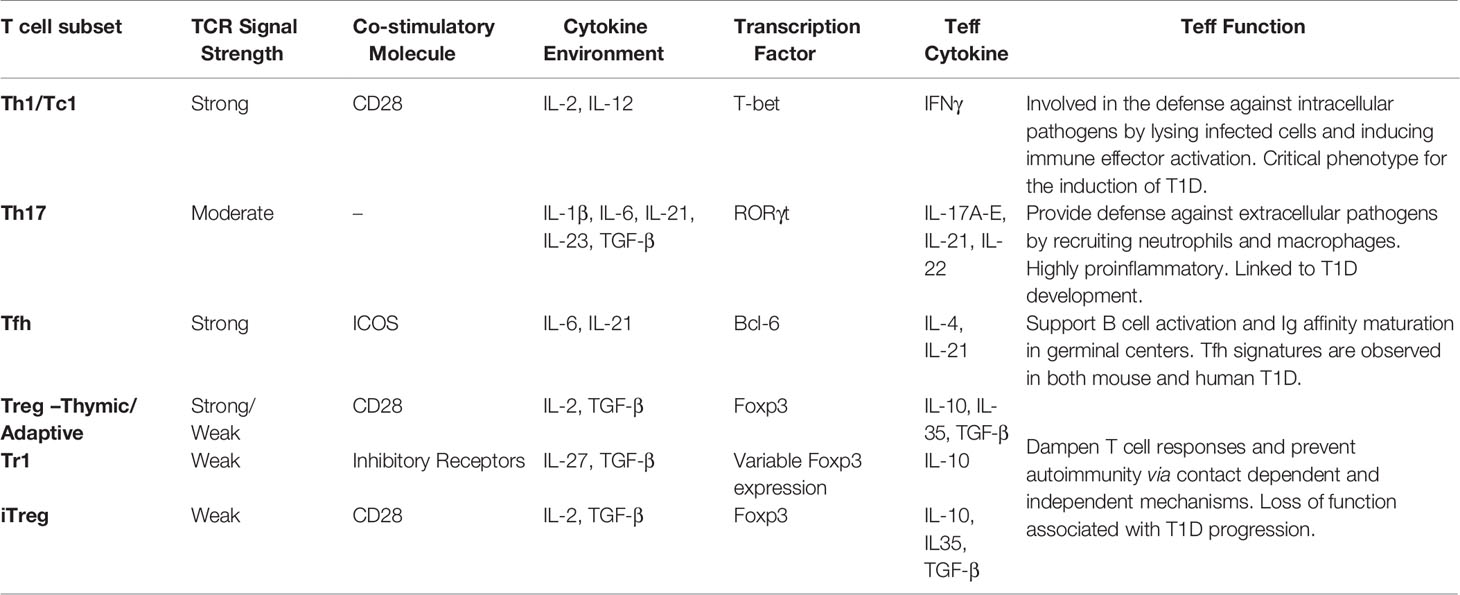
Table 1 T cell receptor (TCR) signals influencing T cell subset differentiation and Teff function in type 1 diabetes (T1D).
The role of TCR signaling influencing epigenetic and transcriptional events is aptly seen with the transcription factor T-bet (206). T-bet expression is required for the development of β cell-specific pathogenic Teff in NOD mice (207). Expression of T-bet is regulated in an analog fashion directly reflecting the strength of TCR signaling (163, 208). High expression levels of T-bet drive the differentiation of short-lived type 1 Teff. T-bet has also been implicated in the differentiation of Tmem (209, 210). Intermediate TCR-induced expression of T-bet is needed for the generation of long-lived Tmem populations (209, 210). In addition to functioning as a transcription factor, T-bet remodels chromatin via recruiting histone demethylases such as JMJD3 (211). Here, T-bet induces transcriptionally permissive histone changes in promoters regulating expression of genes such as CXCR3 and IFNγ (211). Thus, the magnitude of TCR signaling establishes expression levels of T-bet, which affects the phenotypic fate and migration of Teff. Similarly, reduced H3K27Me3 deposition is observed after TCR signaling (202). Here PI3K/AKT-mediated phosphorylation results in dissociation of EZH2 from promoter regions (202). This allows recently activated transcription factors to gain access to promoters exhibiting an euchromatin state primed for transcription. Notably the genetic changes established during activation of naïve T cells are preserved allowing for rapid recall responses of Teff and Tmem (212).
Signals 2 and 3 Contribute to Shaping the T Cell Molecular Landscape
TCR signaling alone is typically insufficient to induce widespread chromatin changes and establish a given transcriptional profile. In addition to “signal 1”, co-stimulation of T cells via “signal 2” related molecules is needed to induce the full complement of gene expression during activation. For example, expression of IL-2 fails to occur when T cells are activated by TCR-alone (213). Co-stimulation by TCR and CD28 results in the acetylation and demethylation of the IL-2 promoter driving IL-2 expression (213). Interestingly, CD28 expression in NOD mice is critical to preserving self-tolerance during T1D (214). Here, reduced CD28 signaling is believed to contribute to poor IL-2 production compromising Foxp3+Treg function (214). Additionally, polymorphisms in NOD Il2 can result in decreased IL-2 transcription also limiting the function and fitness of Foxp3+Treg (79, 215). Together, these findings suggest T cell activation signals work in concert to induce gene expression and maintain self-tolerance.
Cytokines produced by APC, as well as T cells in an autocrine or paracrine fashion, activate the STAT family of transcription factors that regulate Teff differentiation (Table 1) (209, 216). Specifically, cytokine mediated signaling and transcription through STATs can further upregulate expression of other T cell transcription factors, which both enhances and stabilizes the respective Teff phenotype (209, 216). Conversely the absence of essential cytokines can negatively impact the T cell response. In NOD mice, reduced T cell secretion of IL-2 generates a local milieu thought to promote chromatin remodeling and transcription that favors Teff by compromising the fitness and function of Foxp3+Treg (71, 85, 217–221). Similarly, in human T1D, polymorphisms in CD25 result in reduced FOXP3+ Treg sensitivity to local IL-2 (68). A diminished FOXP3+ Treg pool then further enhances the pathogenic β cell-specific Teff response.
The Influence of T Cell Receptor Signal Strength on Expanded Effector T Cell Differentiation and Type 1 Diabetes Progression
T Cell Receptor Signaling Strength and CD4+ Subset Differentiation
TCR signaling activates a common set of pathways, but the magnitude and quality of the events influence the outcome of Teff differentiation (93, 163, 222–224). Upon appropriate stimulation, naïve CD4+ T cells have the plasticity to differentiate into a number of distinct subsets based on pMHC frequency and/or cytokine environment (93, 222–224). Teff subsets have a signature transcription factor and secreted cytokine profile that establishes effector function (Table 1). Furthermore, specific Teff subsets require unique synergy of “signal 1, 2, and 3” for optimal differentiation and maintenance of Teff subsets. Lineage commitment is a pivotal aspect of autoimmunity; in multiple sclerosis (MS) for instance, increased disease severity is associated with Th17 reactivity (225–227). As previously mentioned, Teff in T1D largely exhibit Th1 and Tc1 phenotypes, although a role for Th17 and Tfh cells have been observed (51, 54, 64, 228, 229).
As noted above the strength of signal 1 can influence the differentiation of CD4+ T cells, dictating commitment toward Treg versus proinfammatory Teff (93, 222–224, 230). At low levels of TCR signaling, differentiation of various subsets of Treg, such as iFoxp3+Treg is favored (231, 232). In the case of iFoxp3+Treg for example, reduced signal 1 facilitates activity of the PTEN phosphatase, which blocks PI3K/AKT-mediated phosphorylation events, and in turn enables FoxP3 transcription driven via NF-κB, NFAT, and Foxo1 (233–236). Strong TCR signaling, however, antagonizes PTEN expression and activity, permitting PI3K/AKT signaling that inhibits FoxP3 transcription.
On the other hand, heightened signal 1 favors the differentiation of proinflammatory Teff such as Th1 cells. At relatively high TCR signaling threshold, T-bet expression is increased, which regulates expression of IFNγ and IL-12rβ chain (206, 237, 238). In an autocrine fashion IFNγ induces activation of STAT1, which then serves as a feed-forward loop promoting further T-bet transcription (224). IFNγ also acts on local APC to promote secretion of IL-12, which further promotes Th1 differentiation via STAT4 and increased T-bet and IFNγ expression (206, 237). T-bet and STAT4 also induce expression of the transcription factor Runx3, which enhances IFNγ expression and stabilizes the Th1 phenotype (206, 237, 239). Interestingly, islet-specific T cells from T1D patients have been found in vitro to have irregular T cell-APC immune synapse formation which favors low affinity TCR signaling. However, the increased antigenic density of the islets may promote robust TCR signaling required for progression of autoimmunity (240).
Strength of TCR signaling has also been reported to play a role in the differentiation of Th1 versus Tfh cells (93, 241, 242). Increased and chronic TCR signaling favors Tfh over Th1 differentiation (93). Elevated constitutive TCR signaling, along with T-bet, induces expression of Bcl-6 and the IL-2 receptor. At relatively reduced levels of TCR stimulation, IL-2 receptor activation of STAT5 induces IL-12 receptor and Blimp1 (241, 243, 244). These events lead to the suppression of Bcl-6, thereby permitting Th1 cell differentiation. However, increased TCR signaling uncouples the IL-2 receptor from the STAT5 signaling pathway, so that Bcl-6 is not suppressed, and Tfh cell differentiation progresses. Furthermore, signal 2 in Tfh cells through CD28 and ICOS play fundamental roles in preserving the Tfh phenotype during chronic immune responses such as murine and human T1D (241). One such mechanism is the chronic activation of ICOS on Tfh cells induces PI3K/AKT signaling that serves to repress the transcription factor Foxo1 which is a repressor of Bcl-6 expression and the Tfh lineage.
Co-stimulatory signals also can negatively regulate maintenance of the Th17 phenotype (245, 246). Previous studies have demonstrated that Th17 subset differentiation is impaired in the presence of combined TCR and CD28 signaling, resulting in: 1) activation of multiple negative regulators such as Bcl-6 and SOCS3, and 2) reduced sensitivity to the lineage skewing cytokines IL-1β and IL-2 (245, 246). In the absence of CD28 signaling, Th17 production of IL-17 is maximal whereas in the presence of CD28 signals T cells are diverted to a “Th1-like” IFNγ high producing population (246). These results suggest strong TCR signaling events skew toward Th1 lineage fates, and weaker TCR signals accompanied by the cytokine milieu result in Th17 commitment. Importantly, a role for Th17 cells has been associated with T1D (51, 66, 228). Th17 cells have been detected in the NOD mouse pancreas, and administration of blocking IL-17 antibodies reduced T1D onset in NOD mice (66). Overall, Th subset require distinct signaling conditions for specific lineage commitment.
T Cell Receptor Signaling Strength and CD8+ Effector T Cell Differentiation
Analogous to CD4+ T cells, strength of signal 1 impacts the quality and magnitude of the CD8+ Teff response (163). Strong TCR signaling events in human and murine CD8+ T cells promote activation of inducible T cell kinase (ITK) that regulates the CD8+ T cell response by influencing the magnitude of Ca++ signaling. ITK activation controls the expression of transcription factors such as Blimp-1, T-bet and IRF4 that are needed to generate terminally differentiated CD8+ Teff capable of cytotoxic function (163, 247). Lack of Blimp-1, T-bet, or IRF4 upregulation impairs the function and expansion of short-lived Teff (163, 167, 248–255). Other properties which influence the pathogenicity of CD8+ Teff in the context of autoimmunity are also impacted by the strength of signal 1. Ovalbumin (OVA)-specific OT-1 CD8+ T cells primed with high but not low affinity altered peptide ligands (APLs) induce diabetes upon transfer into transgenic mice expressing OVA by β cells. The pathogenicity of these CD8+ Teff in part correlates with upregulation of the integrin very late antigen-4 (VLA-4), which facilitates islet infiltration (256).
The Role of T Cell Receptor Signaling in the Maintenance of Effector T Cell Function and Type 1 Diabetes Progression
T Cell Receptor Signaling in Effector T Cell Function and Memory T Cell Development
TCR signaling is critical for the maintenance of T cell viability, effector functions, and tissue residency (163). Inhibition of TCR signaling in NOD mice by FK506 treatment or blockade of the CD4 and CD8 co-receptors by monoclonal antibodies both prevent the progression and induce remission of T1D (257–259). Here, the diminution of TCR signaling in islet resident T cells promotes islet egress and trafficking back into circulation (257–259). This results from reduced IFNγ production by islet Teff, which in turn reduces the production of CXCL9/10 causing a failure in islet T cell retention (257–259). These findings highlight how TCR signaling directly affects Teff cytokine production and tissue residency critical to the progression of T1D.
The long-term survival and function of T cells is also a requirement for the development of T1D (260, 261). Typically, long-lived T cell responses are carried out by Tmem. An increased precursor frequency, rapid cytokine production, and the expression of unique trafficking molecules characterize Tmem responses (209, 216). The precise role of Tmem in autoimmunity is difficult to define since anti-self-reactivity is chronic due to the constitutive presentation of autoantigens. However, it is likely that a Tmem-like phenotype is necessary to sustain persistent autoreactivity. Indeed, GAD65-specific CD4+ T cells marked by IFNγ secretion and a capacity to infiltrate and destroy human islets transplanted in mice are detected over numerous years in human T1D patients (262–264).
TCR signaling plays a fundamental role in the development of Tmem cells (150). Both strong and weak TCR signals induce Tmem (208). Studies suggest that the quality and duration of TCR signaling coupled with the activation of specific transcription factors such as IRF4 influence Tmem formation (150, 209, 216). Increasing TCR signaling strength directly induces IRF4 expression with high IRF4 levels resulting in Teff and intermediate IRF4 expression establishing Tmem (167, 247, 255). Mice deficient in IRF4 expression fail to develop fully functional Tmem, and lack of IRF4 expression results in Tex (265, 266). Strikingly, NOD mice deficient in IRF4 fail to develop T1D and show dysfunctional T cell responses (267). Maintenance of long-lived Tmem responses has been tied to the pro-memory transcription factor TCF1 (268–270). High expression of IRF4 inhibits TCF1 expression showing that TCR signaling controls the maintenance of Teff responses (265). Recent work has suggested the long-lived β cell-specific responses are maintained via TCF1 which sustains the autoimmune response in human T1D (260). Furthermore, transplantation of pancreatic islets into diabetic mice leads to recurrent T1D and transplanted islet destruction (271). These results indicate a long-lasting autoreactive T cell population exists after self-antigen clearance.
T Cell Receptor Signaling and the Formation of Exhausted T Cell and Type 1 Diabetes
The clearance of antigen also appears to be an important step in the development of Tmem. Failure to clear antigen in a timely fashion leads to a state of chronic inflammation which negatively impacts the formation of Tmem in studies comparing acute versus chronic lymphocytic choriomeningitis virus (LCMV) infection (272). Sustained TCR signaling initially favors Teff development, but then leads to the formation of Tex (175, 176, 188). This continued signaling and proinflammatory cytokine production can suppress the expression of pro-memory genes such as Foxo1 (273). Tex have been reported in several immune models of infection, cancer, and autoimmunity with both CD4+ and CD8+ T cells susceptible to developing an exhausted phenotype as chronic TCR signaling is a hallmark of these conditions (175, 176, 188).
A key exhaustion marker that has been associated with a Tex phenotype in multiple diseases is PD-1 (175, 176, 188, 272). Interestingly, functional inhibition of this molecule has significant consequences on autoimmunity (175, 176, 272, 274). Phenotypically, Tex exhibit a reduced Teff capacity due to the upregulation of molecules associated with dampening TCR signaling events (275). PD-1 is upregulated rapidly in an analog fashion following TCR stimulation. Chronic TCR signaling, however, results in permanent upregulation of PD-1 expression, and T cell dysfunction (175, 176, 272, 275). Inhibition of TCR signaling occurs upon PD-1 binding to PDL-1 or PDL-2 expressed by APC or the targeted tissues (275). Ligand binding results in phosphorylation of PD-1 by LCK, and recruitment of the tyrosine phosphatase SHP-2, which in turn negatively regulates phosphorylation of CD28 (275, 276). The latter limits Zap70 and PI3K signaling to inhibit TCR-dependent Teff functions (275, 276). This continued expression of PD-1 then drives T cells into a terminally exhausted state (175, 176, 272, 275).
The PD-1-PDL-1/2 axis is a prominent regulatory pathway in autoimmune diseases such as T1D (274, 277–281). PD-1 deficient NOD mice develop aggressive T1D, as do NOD mice treated with an anti-PD-1 antibody (277, 278). A subset of human cancer patients receiving check-point blockade including anti-PD1 antibody have also been reported to develop autoimmune diseases including T1D (187). In addition to PD-1, there are a number of other factors associated with the Tex phenotype including cell surface proteins such as CTLA-4, LAG3, and TIM-3 which further dampen TCR signaling and cellular metabolism suppressing Teff function (175, 176, 188, 272). NOD mice deficient in or receiving blocking antibodies to these molecules results in acceleration of T1D similar to the disruption of PD-1 function (282–284). A diminished frequency of Tex correlates with the progression of human β cell autoimmunity (279, 285). The latter is consistent with T1D subjects exhibiting an increase in Tfh, which produce IL-21. Notably, IL-21 blocks the induction of T cell exhaustion (196). Interestingly, anti-CD3 therapy has been found to induce a Tex cell phenotype in both NOD mice and human T1D patients (280, 281, 286). These results indicate that inducing and maintaining Tex are a key component of T1D progression. How TCR signaling and the cytokine milieu during T1D limits the transcriptional development of terminally exhausted autoreactive T cells continues to be an area of study.
Conclusion
The disease process of T1D is complex and heterogenous, influenced by genetic and ill-defined environmental factors. Multiple immune defects contribute to a failure of β cell-specific self-tolerance which impacts the function of various immune effector cells, including T cells. Genetic variants associated with increased T1D susceptibility have been linked to the generation of self-reactive TCR and dysregulated TCR signaling events. Evidence indicates that alterations in signals 1, 2, and 3 lead to impaired central and peripheral tolerance inducing mechanisms (Figure 2B). The latter culminates in β cell autoantigen-induced T cell activation, expansion, and subset differentiation driven by epigenetic and transcriptional outcomes that promote a proinflammatory response (Figure 2A). We propose that signal 1 is the dominant TCR signaling event driving disease progression, consistent with the fact that the strongest genetic association exists between specific HLA alleles and T1D susceptibility. The greatest impact of signal 1 is expected to be in the thymus, which establishes the repertoire specificity, affinity, and frequency of the anti-self TCR pool, as well as development of nFoxp3+Treg. On the other hand, dysregulation of signals 2 and/or 3 serve to modify signaling outcomes in peripheral β cell-specific T cells that favor differentiation and maintenance of pathogenic Teff and Tmem, while limiting terminal Tex cell development and a protective FOXP3+Treg response. The degree of dysregulation within the respective TCR signaling pathways may influence the heterogeneity observed in the pace of disease progression, the frequency of T cell infiltration of the islets, and the age of T1D onset in humans.
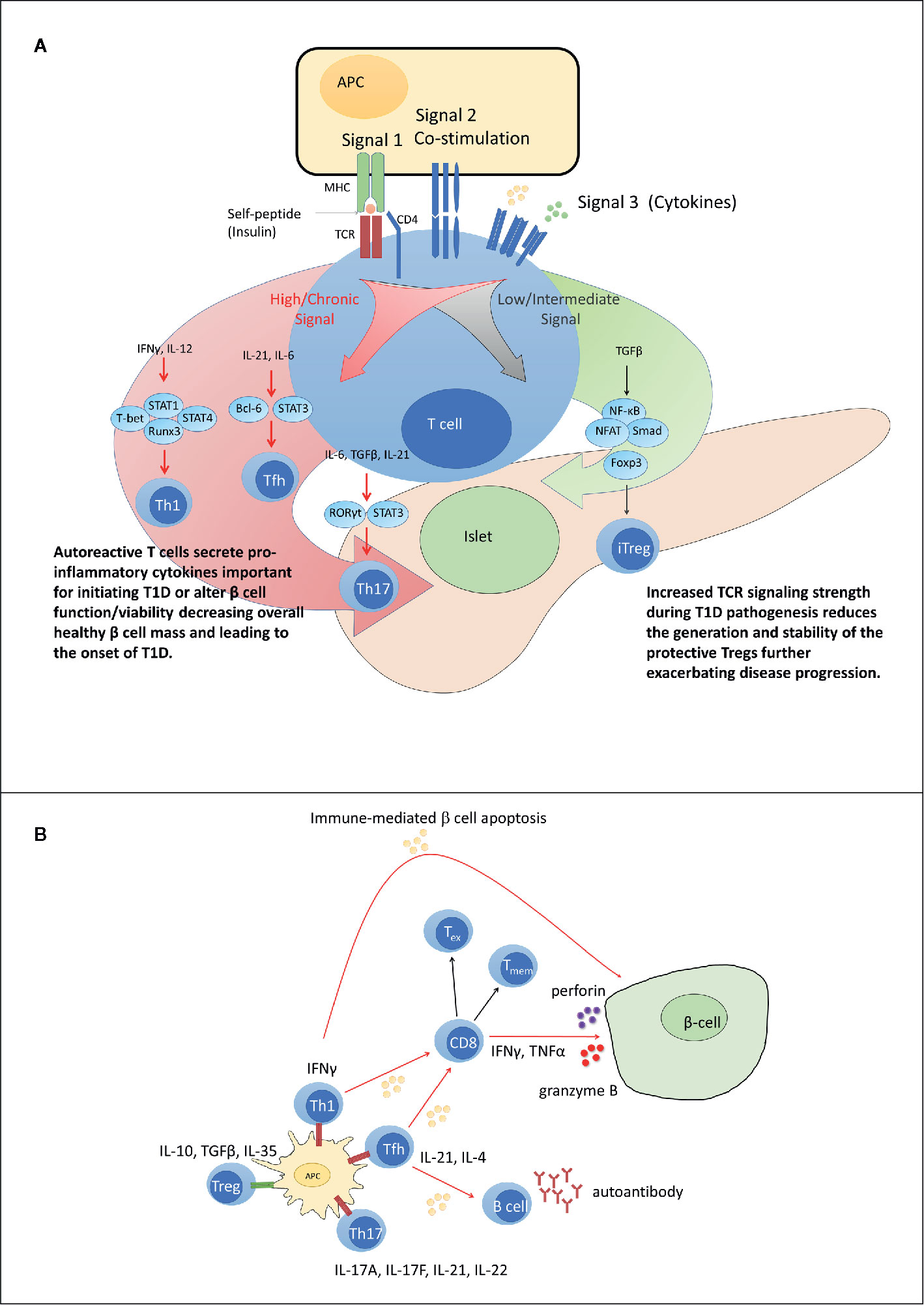
Figure 2 Dysregulated peripheral tolerance and the role of T cell receptor (TCR) signaling in type 1 diabetes (T1D) pathogenesis. (A) Upon interacting with antigen presenting cell (APC), peripheral T cells differentiate into distinct subsets through activation of specific sets of transcription factors based on TCR signal strength (signal 1), co-stimulatory molecule engagement (signal 2) and the cytokine environment (signal 3). Low TCR signaling favors differentiation of adaptive regulatory T cell subsets such as iFoxp3+Treg. Strong and/or persistent TCR signaling favors differentiation of Th1, Tfh, and Th17 subsets. Increased TCR signaling in T1D aids in disrupting the balance between pro inflammatory and regulatory immune responses. (B) In T1D, increased affinity for and/or prolonged interaction with β cell peptide-MHC complexes results in elevated TCR signaling. Elevated TCR signaling coupled with signals derived via co-stimulation and proinflammatory cytokines, promotes differentiation of pathogenic Teff such as IFNγ-producing Th1 cells and cytolytic Tc1 cells, that migrate into the islets and mediate β cell destruction. Chronic TCR signaling normally leads to T cell exhaustion and dampened Teff function. However, elevated levels of IL-21 rescue Teff from terminal exhaustion to maintain β cell destruction. Tmem contribute to maintenance of β cell autoimmunity by providing a source of chronic proinflammatory cytokines upon antigen stimulation. Red TCR (high TCR signal). Green TCR (low TCR signal).
Although TCR signaling defects are associated with mediating β cell autoimmunity, a better understanding of those alterations may also provide clues on how to design effective individualized treatments. The targeting of one or multiple TCR signaling events offers one strategy to modulate the differentiation and activity of autoreactive Teff for the purpose of T1D prevention and treatment. Indeed, varying degrees of efficacy have been seen in preclinical and clinical studies targeting TCR signaling pathways via pharmacological inhibitors, β cell-derived peptides and APLs, recombinant cytokines, monoclonal antibodies and fusion molecules (287–289). Future work is needed to focus on the targeted delivery of TCR modulating therapeutic agents directly to autoreactive T cells, thereby selectively dampening the autoimmune responses and preserving protective acquired immunity.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institutes of Health grants R01DK100256, R01AI139475, R01AI141631, R21AI115752 (RT), and T32AI007273 (MC), and the American Diabetes Association 1-18-PDF-108 (MC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Clark M, Kroger CJ, Tisch RM. Type 1 Diabetes: A Chronic Anti-Self-Inflammatory Response. Front Immunol (2017) 8:1898:1898. doi: 10.3389/fimmu.2017.01898
2. Bach JF. Insulin-dependent diabetes mellitus as an autoimmune disease. Endocr Rev (1994) 15(4):516–42. doi: 10.1210/edrv-15-4-516
3. Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell (1996) 85(3):291–7. doi: 10.1016/s0092-8674(00)81106-x
4. Eisenbarth GS. Type 1 diabetes: molecular, cellular and clinical immunology. Adv Exp Med Biol (2004) 552:306–10.
5. Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol (2005) 23:447–85. doi: 10.1146/annurev.immunol.23.021704.115643
6. von Herrath M, Sanda S, Herold K. Type 1 diabetes as a relapsing-remitting disease? Nat Rev Immunol (2007) 7(12):988–94. doi: 10.1038/nri2192
7. Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, et al. Insulitis and beta-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes (2016) 65(3):719–31. doi: 10.2337/db15-0779
8. Arif S, Leete P, Nguyen V, Marks K, Nor NM, Estorninho M, et al. Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes (2014) 63(11):3835–45. doi: 10.2337/db14-0365
9. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med (2012) 209(1):51–60. doi: 10.1084/jem.20111187
10. Atkinson MA, von Herrath M, Powers AC, Clare-Salzler M. Current concepts on the pathogenesis of type 1 diabetes–considerations for attempts to prevent and reverse the disease. Diabetes Care (2015) 38(6):979–88. doi: 10.2337/dc15-0144
11. Arif S, Gibson VB, Nguyen V, Bingley PJ, Todd JA, Guy C, et al. beta-cell specific T-lymphocyte response has a distinct inflammatory phenotype in children with Type 1 diabetes compared with adults. Diabetes Med (2017) 34(3):419–25. doi: 10.1111/dme.13153
12. Heninger AK, Eugster A, Kuehn D, Buettner F, Kuhn M, Lindner A, et al. A divergent population of autoantigen-responsive CD4+ T cells in infants prior to beta cell autoimmunity. Sci Transl Med (2017) 9(378):1–11. doi: 10.1126/scitranslmed.aaf8848
13. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol (2009) 155(2):173–81. doi: 10.1111/j.1365-2249.2008.03860.x
14. Coppieters KT, Roep BO, von Herrath MG. Beta cells under attack: toward a better understanding of type 1 diabetes immunopathology. Semin Immunopathol (2011) 33(1):1–7. doi: 10.1007/s00281-010-0236-6
15. Richardson SJ, Willcox A, Bone AJ, Morgan NG, Foulis AK. Immunopathology of the human pancreas in type-I diabetes. Semin Immunopathol (2011) 33(1):9–21. doi: 10.1007/s00281-010-0205-0
16. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med (2016) 22(12):1482–7. doi: 10.1038/nm.4203
17. Michels AW, Landry LG, McDaniel KA, Yu L, Campbell-Thompson M, Kwok WW, et al. Islet-Derived CD4 T Cells Targeting Proinsulin in Human Autoimmune Diabetes. Diabetes (2017) 66(3):722–34. doi: 10.2337/db16-1025
18. Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest (2017) 127(8):2881–91. doi: 10.1172/JCI94549
19. Kent SC, Mannering S II, Michels AW, Babon JAB. Deciphering the Pathogenesis of Human Type 1 Diabetes (T1D) by Interrogating T Cells from the “Scene of the Crime”. Curr Diabetes Rep (2017) 17(10):95. doi: 10.1007/s11892-017-0915-y
20. Gomez-Tourino I, Arif S, Eichmann M, Peakman M. T cells in type 1 diabetes: Instructors, regulators and effectors: A comprehensive review. J Autoimmun (2016) 66:7–16. doi: 10.1016/j.jaut.2015.08.012
21. Atkinson MA, Gianani R. The pancreas in human type 1 diabetes: providing new answers to age-old questions. Curr Opin Endocrinol Diabetes Obes (2009) 16(4):279–85. doi: 10.1097/MED.0b013e32832e06ba
22. Campbell-Thompson M. Organ donor specimens: What can they tell us about type 1 diabetes? Pediatr Diabetes (2015) 16(5):320–30. doi: 10.1111/pedi.12286
23. Yu L, Rewers M, Gianani R, Kawasaki E, Zhang Y, Verge C, et al. Antiislet autoantibodies usually develop sequentially rather than simultaneously. J Clin Endocrinol Metab (1996) 81(12):4264–7. doi: 10.1210/jcem.81.12.8954025
24. Bonifacio E, Scirpoli M, Kredel K, Fuchtenbusch M, Ziegler AG. Early autoantibody responses in prediabetes are IgG1 dominated and suggest antigen-specific regulation. J Immunol (1999) 163(1):525–32.
25. Nerup J, Platz P, Andersen OO, Christy M, Lyngsoe J, Poulsen JE, et al. HL-A antigens and diabetes mellitus. Lancet (1974) 2(7885):864–6. doi: 10.1016/s0140-6736(74)91201-x
26. Singal DP, Blajchman MA. Histocompatibility (HL-A) antigens, lymphocytotoxic antibodies and tissue antibodies in patients with diabetes mellitus. Diabetes (1973) 22(6):429–32. doi: 10.2337/diab.22.6.429
27. Noble JA, Erlich HA. Genetics of type 1 diabetes. Cold Spring Harb Perspect Med (2012) 2(1):a007732. doi: 10.1101/cshperspect.a007732
28. Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med (2009) 360(16):1646–54. doi: 10.1056/NEJMra0808284
29. Pociot F, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G, et al. Genetics of type 1 diabetes: what’s next? Diabetes (2010) 59(7):1561–71. doi: 10.2337/db10-0076
30. Pociot F, McDermott MF. Genetics of type 1 diabetes mellitus. Genes Immun (2002) 3(5):235–49. doi: 10.1038/sj.gene.6363875
31. Mallone R, Brezar V, Boitard C. T cell recognition of autoantigens in human type 1 diabetes: clinical perspectives. Clin Dev Immunol (2011) 2011:513210. doi: 10.1155/2011/513210
32. Roep BO, Peakman M. Antigen targets of type 1 diabetes autoimmunity. Cold Spring Harb Perspect Med (2012) 2(4):a007781. doi: 10.1101/cshperspect.a007781
33. In’t Veld P. Insulitis in human type 1 diabetes: The quest for an elusive lesion. Islets (2011) 3(4):131–8. doi: 10.4161/isl.3.4.15728
34. Skog O, Korsgren S, Melhus A, Korsgren O. Revisiting the notion of type 1 diabetes being a T-cell-mediated autoimmune disease. Curr Opin Endocrinol Diabetes Obes (2013) 20(2):118–23. doi: 10.1097/MED.0b013e32835edb89
35. Monti P, Scirpoli M, Rigamonti A, Mayr A, Jaeger A, Bonfanti R, et al. Evidence for in vivo primed and expanded autoreactive T cells as a specific feature of patients with type 1 diabetes. J Immunol (2007) 179(9):5785–92. doi: 10.4049/jimmunol.179.9.5785
36. Skowera A, Ladell K, McLaren JE, Dolton G, Matthews KK, Gostick E, et al. beta-cell-specific CD8 T cell phenotype in type 1 diabetes reflects chronic autoantigen exposure. Diabetes (2015) 64(3):916–25. doi: 10.2337/db14-0332
37. Viglietta V, Kent SC, Orban T, Hafler DA. GAD65-reactive T cells are activated in patients with autoimmune type 1a diabetes. J Clin Invest (2002) 109(7):895–903. doi: 10.1172/JCI14114
38. Velthuis JH, Unger WW, Abreu JR, Duinkerken G, Franken K, Peakman M, et al. Simultaneous detection of circulating autoreactive CD8+ T-cells specific for different islet cell-associated epitopes using combinatorial MHC multimers. Diabetes (2010) 59(7):1721–30. doi: 10.2337/db09-1486
39. Atkinson MA. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb Perspect Med (2012) 2(11):1–18. doi: 10.1101/cshperspect.a007641
40. Katsarou A, Gudbjornsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers (2017) 3:17016. doi: 10.1038/nrdp.2017.16
41. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. and C. Type 1 Diabetes Genetics. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet (2009) 41(6):703–7. doi: 10.1038/ng.381
42. Concannon P, Erlich HA, Julier C, Morahan G, Nerup J, Pociot F, et al. and C. Type 1 Diabetes Genetics. Type 1 diabetes: evidence for susceptibility loci from four genome-wide linkage scans in 1,435 multiplex families. Diabetes (2005) 54(10):2995–3001. doi: 10.2337/diabetes.54.10.2995
43. Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet (2015) 47(4):381–6. doi: 10.1038/ng.3245
44. Jerram ST, Leslie RD. The Genetic Architecture of Type 1 Diabetes. Genes (Basel) (2017) 8(8):1–22. doi: 10.3390/genes8080209
45. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet (2004) 36(4):337–8. doi: 10.1038/ng1323
46. Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JM, et al. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes (2004) 53(11):3020–3. doi: 10.2337/diabetes.53.11.3020
47. Nistico L, Buzzetti R, Pritchard LE, Van der Auwera B, Giovannini C, Bosi E, et al. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Hum Mol Genet (1996) 5(7):1075–80. doi: 10.1093/hmg/5.7.1075
48. Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature (2003) 423(6939):506–11. doi: 10.1038/nature01621
49. Morahan G, Huang D, Ymer S II, Cancilla MR, Stephen K, Dabadghao P, et al. Linkage disequilibrium of a type 1 diabetes susceptibility locus with a regulatory IL12B allele. Nat Genet (2001) 27(2):218–21. doi: 10.1038/84872
50. Long SA, Cerosaletti K, Wan JY, Ho JC, Tatum M, Wei S, et al. An autoimmune-associated variant in PTPN2 reveals an impairment of IL-2R signaling in CD4(+) T cells. Genes Immun (2011) 12(2):116–25. doi: 10.1038/gene.2010.54
51. Walker LS, von Herrath M. CD4 T cell differentiation in type 1 diabetes. Clin Exp Immunol (2016) 183(1):16–29. doi: 10.1111/cei.12672
52. Yoon JW, Jun HS. Autoimmune destruction of pancreatic beta cells. Am J Ther (2005) 12(6):580–91. doi: 10.1097/01.mjt.0000178767.67857.63
53. Arif S, Tree T II, Astill TP, Tremble JM, Bishop AJ, Dayan CM, et al. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest (2004) 113(3):451–63. doi: 10.1172/JCI19585
54. Ferraro A, Socci C, Stabilini A, Valle A, Monti P, Piemonti L, et al. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes (2011) 60(11):2903–13. doi: 10.2337/db11-0090
55. Marwaha AK, Crome SQ, Panagiotopoulos C, Berg KB, Qin H, Ouyang Q, et al. Cutting edge: Increased IL-17-secreting T cells in children with new-onset type 1 diabetes. J Immunol (2010) 185(7):3814–8. doi: 10.4049/jimmunol.1001860
56. Ferreira RC, Simons HZ, Thompson WS, Cutler AJ, Dopico XC, Smyth DJ, et al. IL-21 production by CD4+ effector T cells and frequency of circulating follicular helper T cells are increased in type 1 diabetes patients. Diabetologia (2015) 58(4):781–90. doi: 10.1007/s00125-015-3509-8
57. Xu X, Shi Y, Cai Y, Zhang Q, Yang F, Chen H, et al. Inhibition of increased circulating Tfh cell by anti-CD20 monoclonal antibody in patients with type 1 diabetes. PloS One (2013) 8(11):e79858. doi: 10.1371/journal.pone.0079858
58. Ahmed S, Cerosaletti K, James E, Long SA, Mannering S, Speake C, et al. Standardizing T-Cell Biomarkers in Type 1 Diabetes: Challenges and Recent Advances. Diabetes (2019) 68(7):1366–79. doi: 10.2337/db19-0119
59. Edner NM, Heuts F, Thomas N, Wang CJ, Petersone L, Kenefeck R, et al. Follicular helper T cell profiles predict response to costimulation blockade in type 1 diabetes. Nat Immunol (2020) 21(10):1244–55. doi: 10.1038/s41590-020-0744-z
60. Sutherland AP, Van Belle T, Wurster AL, Suto A, Michaud M, Zhang D, et al. Interleukin-21 is required for the development of type 1 diabetes in NOD mice. Diabetes (2009) 58(5):1144–55. doi: 10.2337/db08-0882
61. Liu SM, King C. IL-21-producing Th cells in immunity and autoimmunity. J Immunol (2013) 191(7):3501–6. doi: 10.4049/jimmunol.1301454
62. McGuire HM, Walters S, Vogelzang A, Lee CM, Webster KE, Sprent J, et al. Interleukin-21 is critically required in autoimmune and allogeneic responses to islet tissue in murine models. Diabetes (2011) 60(3):867–75. doi: 10.2337/db10-1157
63. Spolski R, Kashyap M, Robinson C, Yu Z, Leonard WJ. IL-21 signaling is critical for the development of type I diabetes in the NOD mouse. Proc Natl Acad Sci U S A (2008) 105(37):14028–33. doi: 10.1073/pnas.0804358105
64. Kenefeck R, Wang CJ, Kapadi T, Wardzinski L, Attridge K, Clough LE, et al. Follicular helper T cell signature in type 1 diabetes. J Clin Invest (2015) 125(1):292–303. doi: 10.1172/JCI76238
65. Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol (2005) 5(11):853–65. doi: 10.1038/nri1714
66. Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, et al. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes (2009) 58(6):1302–11. doi: 10.2337/db08-1113
67. Hull CM, Peakman M, Tree T II. Regulatory T cell dysfunction in type 1 diabetes: what’s broken and how can we fix it? Diabetologia (2017) 60(10):1839–50. doi: 10.1007/s00125-017-4377-1
68. Hulme MA, Wasserfall CH, Atkinson MA, Brusko TM. Central role for interleukin-2 in type 1 diabetes. Diabetes (2012) 61(1):14–22. doi: 10.2337/db11-1213
69. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27(1):68–73. doi: 10.1038/83784
70. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27(1):18–20. doi: 10.1038/83707
71. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
72. Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet (2002) 39(8):537–45. doi: 10.1136/jmg.39.8.537
73. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
74. Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol (2005) 6(4):331–7. doi: 10.1038/ni1179
75. Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med (2007) 13(3):108–16. doi: 10.1016/j.molmed.2007.01.003
76. Schmitt EG, Williams CB. Generation and function of induced regulatory T cells. Front Immunol (2013) 4:152:152. doi: 10.3389/fimmu.2013.00152
77. Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol (2008) 181(10):7350–5. doi: 10.4049/jimmunol.181.10.7350
78. D’Alise AM, Auyeung V, Feuerer M, Nishio J, Fontenot J, Benoist C, et al. The defect in T-cell regulation in NOD mice is an effect on the T-cell effectors. Proc Natl Acad Sci U S A (2008) 105(50):19857–62. doi: 10.1073/pnas.0810713105
79. Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity (2008) 28(5):687–97. doi: 10.1016/j.immuni.2008.03.016
80. Visperas A, Vignali DA. Are Regulatory T Cells Defective in Type 1 Diabetes and Can We Fix Them? J Immunol (2016) 197(10):3762–70. doi: 10.4049/jimmunol.1601118
81. Bailey-Bucktrout SL, Bluestone JA. Regulatory T cells: stability revisited. Trends Immunol (2011) 32(7):301–6. doi: 10.1016/j.it.2011.04.002
82. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
83. Bluestone JA, Trotta E, Xu D. The therapeutic potential of regulatory T cells for the treatment of autoimmune disease. Expert Opin Ther Targ (2015) 19(8):1091–103. doi: 10.1517/14728222.2015.1037282
84. Garg G, Tyler JR, Yang JH, Cutler AJ, Downes K, Pekalski M, et al. Type 1 diabetes-associated IL2RA variation lowers IL-2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol (2012) 188(9):4644–53. doi: 10.4049/jimmunol.1100272
85. Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA, Atkinson MA. Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes (2005) 54(5):1407–14. doi: 10.2337/diabetes.54.5.1407
86. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree T II. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes (2005) 54(1):92–9. doi: 10.2337/diabetes.54.1.92
87. Attridge K, Wang CJ, Wardzinski L, Kenefeck R, Chamberlain JL, Manzotti C, et al. IL-21 inhibits T cell IL-2 production and impairs Treg homeostasis. Blood (2012) 119(20):4656–64. doi: 10.1182/blood-2011-10-388546
88. Goudy KS, Wang B, Tisch R. Gene gun-mediated DNA vaccination enhances antigen-specific immunotherapy at a late preclinical stage of type 1 diabetes in nonobese diabetic mice. Clin Immunol (2008) 129(1):49–57. doi: 10.1016/j.clim.2008.06.001
89. Johnson MC, Garland AL, Nicolson SC, Li C, Samulski RJ, Wang B, et al. beta-cell-specific IL-2 therapy increases islet Foxp3+Treg and suppresses type 1 diabetes in NOD mice. Diabetes (2013) 62(11):3775–84. doi: 10.2337/db13-0669
90. Flores RR, Zhou L, Robbins PD. Expression of IL-2 in beta cells by AAV8 gene transfer in pre-diabetic NOD mice prevents diabetes through activation of FoxP3-positive regulatory T cells. Gene Ther (2014) 21(8):715–22. doi: 10.1038/gt.2014.45
91. Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes (2010) 59(2):407–15. doi: 10.2337/db09-0694
92. Putnam AL, Vendrame F, Dotta F, Gottlieb PA. CD4+CD25high regulatory T cells in human autoimmune diabetes. J Autoimmun (2005) 24(1):55–62. doi: 10.1016/j.jaut.2004.11.004
93. Bhattacharyya ND, Feng CG. Regulation of T Helper Cell Fate by TCR Signal Strength. Front Immunol (2020) 11:624:624. doi: 10.3389/fimmu.2020.00624
94. Gascoigne NR, Rybakin V, Acuto O, Brzostek J. TCR Signal Strength and T Cell Development. Annu Rev Cell Dev Biol (2016) 32:327–48. doi: 10.1146/annurev-cellbio-111315-125324
95. Kisielow P. How does the immune system learn to distinguish between good and evil? The first definitive studies of T cell central tolerance and positive selection. Immunogenetics (2019) 71(8-9):513–8. doi: 10.1007/s00251-019-01127-8
96. Hogquist KA, Baldwin TA, Jameson SC. Central tolerance: learning self-control in the thymus. Nat Rev Immunol (2005) 5(10):772–82. doi: 10.1038/nri1707
97. Klein L, Robey EA, Hsieh CS. Central CD4(+) T cell tolerance: deletion versus regulatory T cell differentiation. Nat Rev Immunol (2019) 19(1):7–18. doi: 10.1038/s41577-018-0083-6
98. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol (2014) 14(6):377–91. doi: 10.1038/nri3667
99. Takaba H, Takayanagi H. The Mechanisms of T Cell Selection in the Thymus. Trends Immunol (2017) 38(11):805–16. doi: 10.1016/j.it.2017.07.010
100. Breed ER, Lee ST, Hogquist KA. Directing T cell fate: How thymic antigen presenting cells coordinate thymocyte selection. Semin Cell Dev Biol (2018) 84:2–10. doi: 10.1016/j.semcdb.2017.07.045
101. Yamano T, Steinert M, Klein L. Thymic B Cells and Central T Cell Tolerance. Front Immunol (2015) 6:376:376. doi: 10.3389/fimmu.2015.00376
102. Gotter J, Brors B, Hergenhahn M, Kyewski B. Medullary epithelial cells of the human thymus express a highly diverse selection of tissue-specific genes colocalized in chromosomal clusters. J Exp Med (2004) 199(2):155–66. doi: 10.1084/jem.20031677
103. Wang HX, Pan W, Zheng L, Zhong XP, Tan L, Liang Z, et al. Thymic Epithelial Cells Contribute to Thymopoiesis and T Cell Development. Front Immunol (2019) 10:3099:3099. doi: 10.3389/fimmu.2019.03099
104. Anderson MS, Su MA. AIRE expands: new roles in immune tolerance and beyond. Nat Rev Immunol (2016) 16(4):247–58. doi: 10.1038/nri.2016.9
105. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science (2002) 298(5597):1395–401. doi: 10.1126/science.1075958
106. Derbinski J, Schulte A, Kyewski B, Klein L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol (2001) 2(11):1032–9. doi: 10.1038/ni723
107. Takaba H, Morishita Y, Tomofuji Y, Danks L, Nitta T, Komatsu N, et al. Fezf2 Orchestrates a Thymic Program of Self-Antigen Expression for Immune Tolerance. Cell (2015) 163(4):975–87. doi: 10.1016/j.cell.2015.10.013
109. Abramson J, Anderson G. Thymic Epithelial Cells. Annu Rev Immunol (2017) 35:85–118. doi: 10.1146/annurev-immunol-051116-052320
110. Lei Y, Ripen AM, Ishimaru N, Ohigashi I, Nagasawa T, Jeker LT, et al. Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory T cell development. J Exp Med (2011) 208(2):383–94. doi: 10.1084/jem.20102327
111. Kroger CJ, Spidale NA, Wang B, Tisch R. Thymic Dendritic Cell Subsets Display Distinct Efficiencies and Mechanisms of Intercellular MHC Transfer. J Immunol (2017) 198(1):249–56. doi: 10.4049/jimmunol.1601516
112. Koble C, Kyewski B. The thymic medulla: a unique microenvironment for intercellular self-antigen transfer. J Exp Med (2009) 206(7):1505–13. doi: 10.1084/jem.20082449
113. Millet V, Naquet P, Guinamard RR. Intercellular MHC transfer between thymic epithelial and dendritic cells. Eur J Immunol (2008) 38(5):1257–63. doi: 10.1002/eji.200737982
114. Gallegos AM, Bevan MJ. Central tolerance to tissue-specific antigens mediated by direct and indirect antigen presentation. J Exp Med (2004) 200(8):1039–49. doi: 10.1084/jem.20041457
115. Nakayama M. Antigen Presentation by MHC-Dressed Cells. Front Immunol (2014) 5:672:672. doi: 10.3389/fimmu.2014.00672
116. Yamano T, Nedjic J, Hinterberger M, Steinert M, Koser S, Pinto S, et al. Thymic B Cells Are Licensed to Present Self Antigens for Central T Cell Tolerance Induction. Immunity (2015) 42(6):1048–61. doi: 10.1016/j.immuni.2015.05.013
117. Bonasio R, Scimone ML, Schaerli P, Grabie N, Lichtman AH, von Andrian UH. Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat Immunol (2006) 7(10):1092–100. doi: 10.1038/ni1385
118. Hadeiba H, Lahl K, Edalati A, Oderup C, Habtezion A, Pachynski R, et al. Plasmacytoid dendritic cells transport peripheral antigens to the thymus to promote central tolerance. Immunity (2012) 36(3):438–50. doi: 10.1016/j.immuni.2012.01.017
119. Atibalentja DF, Byersdorfer CA, Unanue ER. Thymus-blood protein interactions are highly effective in negative selection and regulatory T cell induction. J Immunol (2009) 183(12):7909–18. doi: 10.4049/jimmunol.0902632
120. Atibalentja DF, Murphy KM, Unanue ER. Functional redundancy between thymic CD8alpha+ and Sirpalpha+ conventional dendritic cells in presentation of blood-derived lysozyme by MHC class II proteins. J Immunol (2011) 186(3):1421–31. doi: 10.4049/jimmunol.1002587
121. Savage PA, Klawon DEJ, Miller CH. Regulatory T Cell Development. Annu Rev Immunol (2020) 38:421–53. doi: 10.1146/annurev-immunol-100219-020937
122. Kroger CJ, Wang B, Tisch R. Temporal increase in thymocyte negative selection parallels enhanced thymic SIRPalpha+ DC function. Eur J Immunol (2016) 46(10):2352–62. doi: 10.1002/eji.201646354
123. Guerau-de-Arellano M, Martinic M, Benoist C, Mathis D. Neonatal tolerance revisited: a perinatal window for Aire control of autoimmunity. J Exp Med (2009) 206(6):1245–52. doi: 10.1084/jem.20090300
124. He Q, Morillon YM2, Spidale NA, Kroger CJ, Liu B, Sartor RB, et al. Thymic development of autoreactive T cells in NOD mice is regulated in an age-dependent manner. J Immunol (2013) 191(12):5858–66. doi: 10.4049/jimmunol.1302273
125. Markert ML, Devlin BH, Alexieff MJ, Li J, McCarthy EA, Gupton SE, et al. Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants. Blood (2007) 109(10):4539–47. doi: 10.1182/blood-2006-10-048652
126. Ramsey C, Winqvist O, Puhakka L, Halonen M, Moro A, Kampe O, et al. Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Hum Mol Genet (2002) 11(4):397–409. doi: 10.1093/hmg/11.4.397
127. Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science (2015) 348(6234):589–94. doi: 10.1126/science.aaa7017
128. Aricha R, Feferman T, Scott HS, Souroujon MC, Berrih-Aknin S, Fuchs S. The susceptibility of Aire(-/-) mice to experimental myasthenia gravis involves alterations in regulatory T cells. J Autoimmun (2011) 36(1):16–24. doi: 10.1016/j.jaut.2010.09.007
129. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional cloning of the APECED gene. Nat Genet (1997) 17(4):393–8. doi: 10.1038/ng1297-393
130. Finnish-German AC. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet (1997) 17(4):399–403. doi: 10.1038/ng1297-399
131. Fan Y, Rudert WA, Grupillo M, He J, Sisino G, Trucco M. Thymus-specific deletion of insulin induces autoimmune diabetes. EMBO J (2009) 28(18):2812–24. doi: 10.1038/emboj.2009.212
132. Pugliese A, Zeller M, Fernandez A Jr, Zalcberg LJ, Bartlett RJ, Ricordi C, et al. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet (1997) 15(3):293–7. doi: 10.1038/ng0397-293
133. Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet (1997) 15(3):289–92. doi: 10.1038/ng0397-289
134. Viret C, Leung-Theung-Long S, Serre L, Lamare C, Vignali DA, Malissen B, et al. Thymus-specific serine protease controls autoreactive CD4 T cell development and autoimmune diabetes in mice. J Clin Invest (2011) 121(5):1810–21. doi: 10.1172/JCI43314
135. Viret C, Mahiddine K, Baker RL, Haskins K, Guerder S. The T Cell Repertoire-Diversifying Enzyme TSSP Contributes to Thymic Selection of Diabetogenic CD4 T Cell Specificities Reactive to ChgA and IAPP Autoantigens. J Immunol (2015) 195(5):1964–73. doi: 10.4049/jimmunol.1401683
136. Maehr R, Mintern JD, Herman AE, Lennon-Dumenil AM, Mathis D, Benoist C, et al. Cathepsin L is essential for onset of autoimmune diabetes in NOD mice. J Clin Invest (2005) 115(10):2934–43. doi: 10.1172/JCI25485
137. Hsing LC, Kirk EA, McMillen TS, Hsiao SH, Caldwell M, Houston B, et al. Roles for cathepsins S, L, and B in insulitis and diabetes in the NOD mouse. J Autoimmun (2010) 34(2):96–104. doi: 10.1016/j.jaut.2009.07.003
138. Ferreira C, Singh Y, Furmanski AL, Wong FS, Garden OA, Dyson J. Non-obese diabetic mice select a low-diversity repertoire of natural regulatory T cells. Proc Natl Acad Sci U S A (2009) 106(20):8320–5. doi: 10.1073/pnas.0808493106
139. Lie BA, Akselsen HE, Bowlus CL, Gruen JR, Thorsby E, Undlien DE. Polymorphisms in the gene encoding thymus-specific serine protease in the extended HLA complex: a potential candidate gene for autoimmune and HLA-associated diseases. Genes Immun (2002) 3(5):306–12. doi: 10.1038/sj.gene.6363858
140. Viken MK, Blomhoff A, Olsson M, Akselsen HE, Pociot F, Nerup J, et al. Reproducible association with type 1 diabetes in the extended class I region of the major histocompatibility complex. Genes Immun (2009) 10(4):323–33. doi: 10.1038/gene.2009.13
141. Viken MK, Sollid HD, Joner G, Dahl-Jorgensen K, Ronningen KS, Undlien DE, et al. Polymorphisms in the cathepsin L2 (CTSL2) gene show association with type 1 diabetes and early-onset myasthenia gravis. Hum Immunol (2007) 68(9):748–55. doi: 10.1016/j.humimm.2007.05.009
142. Miyazaki T, Uno M, Uehira M, Kikutani H, Kishimoto T, Kimoto M, et al. Direct evidence for the contribution of the unique I-ANOD to the development of insulitis in non-obese diabetic mice. Nature (1990) 345(6277):722–4. doi: 10.1038/345722a0
143. Singer SM, Tisch R, Yang XD, McDevitt HO. An Abd transgene prevents diabetes in nonobese diabetic mice by inducing regulatory T cells. Proc Natl Acad Sci U S A (1993) 90(20):9566–70. doi: 10.1073/pnas.90.20.9566
144. Acha-Orbea H, McDevitt HO. The first external domain of the nonobese diabetic mouse class II I-A beta chain is unique. Proc Natl Acad Sci U S A (1987) 84(8):2435–9. doi: 10.1073/pnas.84.8.2435
145. Carrasco-Marin E, Shimizu J, Kanagawa O, Unanue ER. The class II MHC I-Ag7 molecules from non-obese diabetic mice are poor peptide binders. J Immunol (1996) 156(2):450–8.
146. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science (2016) 351(6274):711–4. doi: 10.1126/science.aad2791
147. Baker RL, Jamison BL, Wiles TA, Lindsay RS, Barbour G, Bradley B, et al. CD4 T Cells Reactive to Hybrid Insulin Peptides are Indicators of Disease Activity in the NOD Mouse. Diabetes (2018) 67(9):1836–46. doi: 10.2337/db18-0200
148. Baker RL, Rihanek M, Hohenstein AC, Nakayama M, Michels A, Gottlieb PA, et al. Hybrid Insulin Peptides Are Autoantigens in Type 1 Diabetes. Diabetes (2019) 68(9):1830–40. doi: 10.2337/db19-0128
149. Hwang JR, Byeon Y, Kim D, Park SG. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp Mol Med (2020) 52(5):750–61. doi: 10.1038/s12276-020-0435-8
150. Daniels MA, Teixeiro E. TCR Signaling in T Cell Memory. Front Immunol (2015) 6:1–10. doi: 10.3389/fimmu.2015.00617
151. Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signalling. Nat Rev Immunol (2018) 18(8):485–97. doi: 10.1038/s41577-018-0020-8
152. Cantrell D. Signaling in lymphocyte activation. Cold Spring Harb Perspect Biol (2015) 7(6):1–13. doi: 10.1101/cshperspect.a018788
153. Tough DF, Rioja I, Modis LK, Prinjha RK. Epigenetic Regulation of T Cell Memory: Recalling Therapeutic Implications. Trends Immunol (2020) 41(1):29–45. doi: 10.1016/j.it.2019.11.008
154. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science (1999) 285(5425):221–7. doi: 10.1126/science.285.5425.221
155. Davis DM, Dustin ML. What is the importance of the immunological synapse? Trends Immunol (2004) 25(6):323–7. doi: 10.1016/j.it.2004.03.007
156. Myers DR, Zikherman J, Roose JP. Tonic Signals: Why Do Lymphocytes Bother? Trends Immunol (2017) 38(11):844–57. doi: 10.1016/j.it.2017.06.010
157. van Oers NS, Killeen N, Weiss A. ZAP-70 is constitutively associated with tyrosine-phosphorylated TCR zeta in murine thymocytes and lymph node T cells. Immunity (1994) 1(8):675–85. doi: 10.1016/1074-7613(94)90038-8
158. van Oers NS, Tao W, Watts JD, Johnson P, Aebersold R, Teh HS. Constitutive tyrosine phosphorylation of the T-cell receptor (TCR) zeta subunit: regulation of TCR-associated protein tyrosine kinase activity by TCR zeta. Mol Cell Biol (1993) 13(9):5771–80. doi: 10.1128/mcb.13.9.5771
159. Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science (1997) 276(5321):2057–62. doi: 10.1126/science.276.5321.2057
160. Kirberg J, Berns A, von Boehmer H. Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex-encoded molecules. J Exp Med (1997) 186(8):1269–75. doi: 10.1084/jem.186.8.1269
161. Rooke R, Waltzinger C, Benoist C, Mathis D. Targeted complementation of MHC class II deficiency by intrathymic delivery of recombinant adenoviruses. Immunity (1997) 7(1):123–34. doi: 10.1016/s1074-7613(00)80515-4
162. Takeda S, Rodewald HR, Arakawa H, Bluethmann H, Shimizu T. MHC class II molecules are not required for survival of newly generated CD4+ T cells, but affect their long-term life span. Immunity (1996) 5(3):217–28. doi: 10.1016/s1074-7613(00)80317-9
163. Conley JM, Gallagher MP, Berg LJ. T Cells and Gene Regulation: The Switching On and Turning Up of Genes after T Cell Receptor Stimulation in CD8 T Cells. Front Immunol (2016) 7:76:76. doi: 10.3389/fimmu.2016.00076
164. Ashouri JF, Weiss A. Endogenous Nur77 Is a Specific Indicator of Antigen Receptor Signaling in Human T and B Cells. J Immunol (2017) 198(2):657–68. doi: 10.4049/jimmunol.1601301
165. Au-Yeung BB, Zikherman J, Mueller JL, Ashouri JF, Matloubian M, Cheng DA, et al. A sharp T-cell antigen receptor signaling threshold for T-cell proliferation. Proc Natl Acad Sci U S A (2014) 111(35):E3679–88. doi: 10.1073/pnas.1413726111
166. Kingeter LM, Paul S, Maynard SK, Cartwright NG, Schaefer BC. Cutting edge: TCR ligation triggers digital activation of NF-kappaB. J Immunol (2010) 185(8):4520–4. doi: 10.4049/jimmunol.1001051
167. Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, et al. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol (2013) 14(11):1155–65. doi: 10.1038/ni.2710
168. Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol (2013) 13(4):257–69. doi: 10.1038/nri3403
169. Blanchfield JL, Shorter SK, Evavold BD. Monitoring the Dynamics of T Cell Clonal Diversity Using Recombinant Peptide:MHC Technology. Front Immunol (2013) 4:170:170. doi: 10.3389/fimmu.2013.00170
170. Liston A, Lesage S, Gray DH, O’Reilly LA, Strasser A, Fahrer AM, et al. Generalized resistance to thymic deletion in the NOD mouse; a polygenic trait characterized by defective induction of Bim. Immunity (2004) 21(6):817–30. doi: 10.1016/j.immuni.2004.10.014
171. Kishimoto H, Sprent J. A defect in central tolerance in NOD mice. Nat Immunol (2001) 2(11):1025–31. doi: 10.1038/ni726
172. Mingueneau M, Jiang W, Feuerer M, Mathis D, Benoist C. Thymic negative selection is functional in NOD mice. J Exp Med (2012) 209(3):623–37. doi: 10.1084/jem.20112593
173. Zucchelli S, Holler P, Yamagata T, Roy M, Benoist C, Mathis D. Defective central tolerance induction in NOD mice: genomics and genetics. Immunity (2005) 22(3):385–96. doi: 10.1016/j.immuni.2005.01.015
174. Mbongue JC, Nieves HA, Torrez TW, Langridge WH. The Role of Dendritic Cell Maturation in the Induction of Insulin-Dependent Diabetes Mellitus. Front Immunol (2017) 8:327:327. doi: 10.3389/fimmu.2017.00327
176. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
177. Bottini N, Vang T, Cucca F, Mustelin T. Role of PTPN22 in type 1 diabetes and other autoimmune diseases. Semin Immunol (2006) 18(4):207–13. doi: 10.1016/j.smim.2006.03.008
178. Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet (2005) 37(12):1317–9. doi: 10.1038/ng1673
179. Hasegawa K, Martin F, Huang G, Tumas D, Diehl L, Chan AC. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science (2004) 303(5658):685–9. doi: 10.1126/science.1092138
180. Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol (2007) 179(7):4704–10. doi: 10.4049/jimmunol.179.7.4704
181. Clarke F, Purvis HA, Sanchez-Blanco C, Gutierrez-Martinez E, Cornish GH, Zamoyska R, et al. The protein tyrosine phosphatase PTPN22 negatively regulates presentation of immune complex derived antigens. Sci Rep (2018) 8(1):12692. doi: 10.1038/s41598-018-31179-x
182. Lin X, Pelletier S, Gingras S, Rigaud S, Maine CJ, Marquardt K, et al. CRISPR-Cas9-Mediated Modification of the NOD Mouse Genome With Ptpn22R619W Mutation Increases Autoimmune Diabetes. Diabetes (2016) 65(8):2134–8. doi: 10.2337/db16-0061
183. Sharp RC, Abdulrahim M, Naser ES, Naser SA. Genetic Variations of PTPN2 and PTPN22: Role in the Pathogenesis of Type 1 Diabetes and Crohn’s Disease. Front Cell Infect Microbiol (2015) 5:95:95. doi: 10.3389/fcimb.2015.00095
184. Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet (2007) 39(7):857–64. doi: 10.1038/ng2068
185. Wiede F, Brodnicki TC, Goh PK, Leong YA, Jones GW, Yu D, et al. T-Cell-Specific PTPN2 Deficiency in NOD Mice Accelerates the Development of Type 1 Diabetes and Autoimmune Comorbidities. Diabetes (2019) 68(6):1251–66. doi: 10.2337/db18-1362
186. Anjos S, Nguyen A, Ounissi-Benkalha H, Tessier MC, Polychronakos C. A common autoimmunity predisposing signal peptide variant of the cytotoxic T-lymphocyte antigen 4 results in inefficient glycosylation of the susceptibility allele. J Biol Chem (2002) 277(48):46478–86. doi: 10.1074/jbc.M206894200
187. Stamatouli AM, Quandt Z, Perdigoto AL, Clark PL, Kluger H, Weiss SA, et al. Collateral Damage: Insulin-Dependent Diabetes Induced With Checkpoint Inhibitors. Diabetes (2018) 67(8):1471–80. doi: 10.2337/dbi18-0002
188. Attanasio J, Wherry EJ. Costimulatory and Coinhibitory Receptor Pathways in Infectious Disease. Immunity (2016) 44(5):1052–68. doi: 10.1016/j.immuni.2016.04.022
189. Klocke K, Sakaguchi S, Holmdahl R, Wing K. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc Natl Acad Sci U S A (2016) 113(17):E2383–92. doi: 10.1073/pnas.1603892113
190. Marleau AM, Sarvetnick NE. IL-18 is required for self-reactive T cell expansion in NOD mice. J Autoimmun (2011) 36(3-4):263–77. doi: 10.1016/j.jaut.2011.02.005
191. Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol (2001) 19:423–74. doi: 10.1146/annurev.immunol.19.1.423
192. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
193. Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med (1996) 184(5):2049–53. doi: 10.1084/jem.184.5.2049
194. Noorchashm H, Lieu YK, Noorchashm N, Rostami SY, Greeley SA, Schlachterman A, et al. I-Ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell tolerance to islet beta cells of nonobese diabetic mice. J Immunol (1999) 163(2):743–50.
195. Frohlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, et al. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science (2009) 324(5934):1576–80. doi: 10.1126/science.1172815
196. Yi JS, Du M, Zajac AJ. A vital role for interleukin-21 in the control of a chronic viral infection. Science (2009) 324(5934):1572–6. doi: 10.1126/science.1175194
197. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet (2014) 46(8):812–4. doi: 10.1038/ng.3040
198. Nepom GT, Ehlers M, Mandrup-Poulsen T. Anti-cytokine therapies in T1D: Concepts and strategies. Clin Immunol (2013) 149(3):279–85. doi: 10.1016/j.clim.2013.02.003
199. Lu J, Liu J, Li L, Lan Y, Liang Y. Cytokines in type 1 diabetes: mechanisms of action and immunotherapeutic targets. Clin Transl Immunol (2020) 9(3):e1122. doi: 10.1002/cti2.1122
200. Weng NP, Araki Y, Subedi K. The molecular basis of the memory T cell response: differential gene expression and its epigenetic regulation. Nat Rev Immunol (2012) 12(4):306–15. doi: 10.1038/nri3173
201. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol (2018) 18(5):340–56. doi: 10.1038/nri.2017.146
202. He S, Liu Y, Meng L, Sun H, Wang Y, Ji Y, et al. Ezh2 phosphorylation state determines its capacity to maintain CD8(+) T memory precursors for antitumor immunity. Nat Commun (2017) 8(1):2125. doi: 10.1038/s41467-017-02187-8
203. DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity (2015) 42(2):227–38. doi: 10.1016/j.immuni.2015.01.007
204. Jerram ST, Dang MN, Leslie RD. The Role of Epigenetics in Type 1 Diabetes. Curr Diabetes Rep (2017) 17(10):89. doi: 10.1007/s11892-017-0916-x
205. Fasolino M, Goldman N, Wang W, Cattau B, Zhou Y, Petrovic J, et al. Genetic Variation in Type 1 Diabetes Reconfigures the 3D Chromatin Organization of T Cells and Alters Gene Expression. Immunity (2020) 52(2):257–274 e11. doi: 10.1016/j.immuni.2020.01.003
206. Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol (2013) 13(11):777–89. doi: 10.1038/nri3536
207. Esensten JH, Lee MR, Glimcher LH, Bluestone JA. T-bet-deficient NOD mice are protected from diabetes due to defects in both T cell and innate immune system function. J Immunol (2009) 183(1):75–82. doi: 10.4049/jimmunol.0804154
208. Knudson KM, Goplen NP, Cunningham CA, Daniels MA, Teixeiro E. Low-affinity T cells are programmed to maintain normal primary responses but are impaired in their recall to low-affinity ligands. Cell Rep (2013) 4(3):554–65. doi: 10.1016/j.celrep.2013.07.008
209. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol (2012) 12(11):749–61. doi: 10.1038/nri3307
210. Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, et al. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity (2011) 35(4):633–46. doi: 10.1016/j.immuni.2011.08.016
211. Miller SA, Huang AC, Miazgowicz MM, Brassil MM, Weinmann AS. Coordinated but physically separable interaction with H3K27-demethylase and H3K4-methyltransferase activities are required for T-box protein-mediated activation of developmental gene expression. Genes Dev (2008) 22(21):2980–93. doi: 10.1101/gad.1689708
212. Youngblood B, Hale JS, Ahmed R. T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology (2013) 139(3):277–84. doi: 10.1111/imm.12074
213. Thomas RM, Gao L, Wells AD. Signals from CD28 induce stable epigenetic modification of the IL-2 promoter. J Immunol (2005) 174(8):4639–46. doi: 10.4049/jimmunol.174.8.4639
214. Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity (2000) 12(4):431–40. doi: 10.1016/s1074-7613(00)80195-8
215. Denny P, Lord CJ, Hill NJ, Goy JV, Levy ER, Podolin PL, et al. Mapping of the IDDM locus Idd3 to a 0.35-cM interval containing the interleukin-2 gene. Diabetes (1997) 46(4):695–700. doi: 10.2337/diab.46.4.695
216. Seder RA, Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat Immunol (2003) 4(9):835–42. doi: 10.1038/ni969
217. Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet (2007) 39(9):1074–82. doi: 10.1038/ng2102
218. Maier LM, Anderson DE, Severson CA, Baecher-Allan C, Healy B, Liu DV, et al. Soluble IL-2RA levels in multiple sclerosis subjects and the effect of soluble IL-2RA on immune responses. J Immunol (2009) 182(3):1541–7. doi: 10.4049/jimmunol.182.3.1541
219. Giordano C, Panto F, Caruso C, Modica MA, Zambito AM, Sapienza N, et al. Interleukin 2 and soluble interleukin 2-receptor secretion defect in vitro in newly diagnosed type I diabetic patients. Diabetes (1989) 38(3):310–5. doi: 10.2337/diab.38.3.310
220. Brusko T, Wasserfall C, McGrail K, Schatz R, Viener HL, Schatz D, et al. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes (2007) 56(3):604–12. doi: 10.2337/db06-1248
221. Qu HQ, Verlaan DJ, Ge B, Lu Y, Lam KC, Grabs R, et al. A cis-acting regulatory variant in the IL2RA locus. J Immunol (2009) 183(8):5158–62. doi: 10.4049/jimmunol.0901337
222. van Panhuys N. TCR Signal Strength Alters T-DC Activation and Interaction Times and Directs the Outcome of Differentiation. Front Immunol (2016) 7:6:6. doi: 10.3389/fimmu.2016.00006
223. Zhu J, Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev (2010) 238(1):247–62. doi: 10.1111/j.1600-065X.2010.00951.x
224. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
225. Babaloo Z, Aliparasti MR, Babaiea F, Almasi S, Baradaran B, Farhoudi M. The role of Th17 cells in patients with relapsing-remitting multiple sclerosis: interleukin-17A and interleukin-17F serum levels. Immunol Lett (2015) 164(2):76–80. doi: 10.1016/j.imlet.2015.01.001
226. Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A (2008) 105(26):9041–6. doi: 10.1073/pnas.0802218105
227. Moser T, Akgun K, Proschmann U, Sellner J, Ziemssen T. The role of TH17 cells in multiple sclerosis: Therapeutic implications. Autoimmun Rev (2020) 19(10):102647. doi: 10.1016/j.autrev.2020.102647
228. Kuriya G, Uchida T, Akazawa S, Kobayashi M, Nakamura K, Satoh T, et al. Double deficiency in IL-17 and IFN-gamma signalling significantly suppresses the development of diabetes in the NOD mouse. Diabetologia (2013) 56(8):1773–80. doi: 10.1007/s00125-013-2935-8
229. Heuts F, Edner NM, Walker LS. Follicular T Helper Cells: A New Marker of Type 1 Diabetes Risk? Diabetes (2017) 66(2):258–60. doi: 10.2337/dbi16-0062
230. Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol (1997) 15:297–322. doi: 10.1146/annurev.immunol.15.1.297
231. Li MO, Rudensky AY. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol (2016) 16(4):220–33. doi: 10.1038/nri.2016.26
232. Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J Exp Med (2010) 207(8):1701–11. doi: 10.1084/jem.20091999
233. Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity (2009) 31(6):921–31. doi: 10.1016/j.immuni.2009.09.022
234. Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene M II, et al. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity (2009) 31(6):932–40. doi: 10.1016/j.immuni.2009.10.006
235. Vaeth M, Schliesser U, Muller G, Reissig S, Satoh K, Tuettenberg A, et al. Dependence on nuclear factor of activated T-cells (NFAT) levels discriminates conventional T cells from Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A (2012) 109(40):16258–63. doi: 10.1073/pnas.1203870109
236. Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature (2012) 491(7425):554–9. doi: 10.1038/nature11581
237. Kallies A, Good-Jacobson KL. Transcription Factor T-bet Orchestrates Lineage Development and Function in the Immune System. Trends Immunol (2017) 38(4):287–97. doi: 10.1016/j.it.2017.02.003
238. Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol (2002) 3(6):549–57. doi: 10.1038/ni794
239. Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol (2007) 8(2):145–53. doi: 10.1038/ni1424
240. Schubert DA, Gordo S, Sabatino JJ Jr., Vardhana S, Gagnon E, Sethi DK, et al. Self-reactive human CD4 T cell clones form unusual immunological synapses. J Exp Med (2012) 209(2):335–52. doi: 10.1084/jem.20111485
241. Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity (2019) 50(5):1132–48. doi: 10.1016/j.immuni.2019.04.011
242. Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol (2009) 10(4):375–84. doi: 10.1038/ni.1704
243. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med (2012) 209(2):243–50. doi: 10.1084/jem.20111174
244. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325(5943):1006–10. doi: 10.1126/science.1175870
245. Bhaumik S, Basu R. Cellular and Molecular Dynamics of Th17 Differentiation and its Developmental Plasticity in the Intestinal Immune Response. Front Immunol (2017) 8:254:254. doi: 10.3389/fimmu.2017.00254
246. Bouguermouh S, Fortin G, Baba N, Rubio M, Sarfati M. CD28 co-stimulation down regulates Th17 development. PloS One (2009) 4(3):e5087. doi: 10.1371/journal.pone.0005087
247. Nayar R, Enos M, Prince A, Shin H, Hemmers S, Jiang JK, et al. TCR signaling via Tec kinase ITK and interferon regulatory factor 4 (IRF4) regulates CD8+ T-cell differentiation. Proc Natl Acad Sci U S A (2012) 109(41):E2794–802. doi: 10.1073/pnas.1205742109
248. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity (2007) 27(2):281–95. doi: 10.1016/j.immuni.2007.07.010
249. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol (2005) 6(12):1236–44. doi: 10.1038/ni1268
250. Kalia V, Sarkar S. Regulation of Effector and Memory CD8 T Cell Differentiation by IL-2-A Balancing Act. Front Immunol (2018) 9:2987:2987. doi: 10.3389/fimmu.2018.02987
251. Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, et al. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity (2009) 31(2):296–308. doi: 10.1016/j.immuni.2009.05.014
252. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity (2010) 32(1):79–90. doi: 10.1016/j.immuni.2009.11.012
253. Boulet S, Daudelin JF, Labrecque N. IL-2 induction of Blimp-1 is a key in vivo signal for CD8+ short-lived effector T cell differentiation. J Immunol (2014) 193(4):1847–54. doi: 10.4049/jimmunol.1302365
254. Nayar R, Schutten E, Bautista B, Daniels K, Prince AL, Enos M, et al. Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J Immunol (2014) 192(12):5881–93. doi: 10.4049/jimmunol.1303187
255. Yao S, Buzo BF, Pham D, Jiang L, Taparowsky EJ, Kaplan MH, et al. Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity (2013) 39(5):833–45. doi: 10.1016/j.immuni.2013.10.007
256. King CG, Koehli S, Hausmann B, Schmaler M, Zehn D, Palmer E. T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity (2012) 37(4):709–20. doi: 10.1016/j.immuni.2012.06.021
257. Yi Z, Diz R, Martin AJ, Morillon YM, Kline DE, Li L, et al. Long-term remission of diabetes in NOD mice is induced by nondepleting anti-CD4 and anti-CD8 antibodies. Diabetes (2012) 61(11):2871–80. doi: 10.2337/db12-0098
258. Morillon YM2, Lessey-Morillon EC, Clark M, Zhang R, Wang B, Burridge K, et al. Antibody Binding to CD4 Induces Rac GTPase Activation and Alters T Cell Migration. J Immunol (2016) 197(9):3504–11. doi: 10.4049/jimmunol.1501600
259. Martin AJ, Clark M, Gojanovich G, Manzoor F, Miller K, Kline DE, et al. Anti-coreceptor therapy drives selective T cell egress by suppressing inflammation-dependent chemotactic cues. JCI Insight (2016) 1(17):e87636. doi: 10.1172/jci.insight.87636
260. Abdelsamed HA, Zebley CC, Nguyen H, Rutishauser RL, Fan Y, Ghoneim HE, et al. Beta cell-specific CD8(+) T cells maintain stem cell memory-associated epigenetic programs during type 1 diabetes. Nat Immunol (2020) 21(5):578–87. doi: 10.1038/s41590-020-0633-5
261. Spanier JA, Sahli NL, Wilson JC, Martinov T, Dileepan T, Burrack AL, et al. Increased Effector Memory Insulin-Specific CD4(+) T Cells Correlate With Insulin Autoantibodies in Patients With Recent-Onset Type 1 Diabetes. Diabetes (2017) 66(12):3051–60. doi: 10.2337/db17-0666
262. Laughlin E, Burke G, Pugliese A, Falk B, Nepom G. Recurrence of autoreactive antigen-specific CD4+ T cells in autoimmune diabetes after pancreas transplantation. Clin Immunol (2008) 128(1):23–30. doi: 10.1016/j.clim.2008.03.459
263. Vendrame F, Pileggi A, Laughlin E, Allende G, Martin-Pagola A, Molano RD, et al. Recurrence of type 1 diabetes after simultaneous pancreas-kidney transplantation, despite immunosuppression, is associated with autoantibodies and pathogenic autoreactive CD4 T-cells. Diabetes (2010) 59(4):947–57. doi: 10.2337/db09-0498
264. Burke GW3, Vendrame F, Virdi SK, Ciancio G, Chen L, Ruiz P, et al. Lessons From Pancreas Transplantation in Type 1 Diabetes: Recurrence of Islet Autoimmunity. Curr Diabetes Rep (2015) 15(12):121. doi: 10.1007/s11892-015-0691-5
265. Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, et al. Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity (2017) 47(6):1129–1141 e5. doi: 10.1016/j.immuni.2017.11.021
266. Wu J, Zhang H, Shi X, Xiao X, Fan Y, Minze LJ, et al. Ablation of Transcription Factor IRF4 Promotes Transplant Acceptance by Driving Allogenic CD4(+) T Cell Dysfunction. Immunity (2017) 47(6):1114–1128 e6. doi: 10.1016/j.immuni.2017.11.003
267. Akazawa S, Kobayashi M, Kuriya G, Horie I, Yu L, Yamasaki H, et al. Haploinsufficiency of interferon regulatory factor 4 strongly protects against autoimmune diabetes in NOD mice. Diabetologia (2015) 58(11):2606–14. doi: 10.1007/s00125-015-3724-3
268. Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity (2019) 51(5):840–855 e5. doi: 10.1016/j.immuni.2019.09.013
269. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity (2016) 45(2):415–27. doi: 10.1016/j.immuni.2016.07.021
270. Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol (2016) 1(6):1–27. doi: 10.1126/sciimmunol.aai8593
271. Burrack AL, Martinov T, Fife BT. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front Endocrinol (Lausanne) (2017) 8:343:343. doi: 10.3389/fendo.2017.00343
272. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol (2009) 10(1):29–37. doi: 10.1038/ni.1679
273. Utzschneider DT, Delpoux A, Wieland D, Huang X, Lai CY, Hofmann M, et al. Active Maintenance of T Cell Memory in Acute and Chronic Viral Infection Depends on Continuous Expression of FOXO1. Cell Rep (2018) 22(13):3454–67. doi: 10.1016/j.celrep.2018.03.020
274. Linsley PS, Long SA. Enforcing the checkpoints: harnessing T-cell exhaustion for therapy of T1D. Curr Opin Endocrinol Diabetes Obes (2019) 26(4):213–8. doi: 10.1097/MED.0000000000000488
275. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
276. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355(6332):1428–33. doi: 10.1126/science.aaf1292
277. Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med (2003) 198(1):63–9. doi: 10.1084/jem.20022125
278. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A (2005) 102(33):11823–8. doi: 10.1073/pnas.0505497102
279. Wiedeman AE, Muir VS, Rosasco MG, DeBerg HA, Presnell S, Haas B, et al. Autoreactive CD8+ T cell exhaustion distinguishes subjects with slow type 1 diabetes progression. J Clin Invest (2020) 130(1):480–90. doi: 10.1172/JCI126595
280. Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, Bour-Jordan H, et al. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J Exp Med (2006) 203(12):2737–47. doi: 10.1084/jem.20061577
281. Long SA, Thorpe J, DeBerg HA, Gersuk V, Eddy J, Harris KM, et al. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci Immunol (2016) 1(5):1–23. doi: 10.1126/sciimmunol.aai7793
282. Bettini M, Szymczak-Workman AL, Forbes K, Castellaw AH, Selby M, Pan X, et al. Cutting edge: accelerated autoimmune diabetes in the absence of LAG-3. J Immunol (2011) 187(7):3493–8. doi: 10.4049/jimmunol.1100714
283. Luhder F, Hoglund P, Allison JP, Benoist C, Mathis D. Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) regulates the unfolding of autoimmune diabetes. J Exp Med (1998) 187(3):427–32. doi: 10.1084/jem.187.3.427
284. Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol (2003) 4(11):1093–101. doi: 10.1038/ni987
285. McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature (2015) 523(7562):612–6. doi: 10.1038/nature14468
286. Long SA, Thorpe J, Herold KC, Ehlers M, Sanda S, Lim N, et al. Remodeling T cell compartments during anti-CD3 immunotherapy of type 1 diabetes. Cell Immunol (2017) 319:3–9. doi: 10.1016/j.cellimm.2017.07.007
287. Schloot NC, Cohen IR. DiaPep277(R) and immune intervention for treatment of type 1 diabetes. Clin Immunol (2013) 149(3):307–16. doi: 10.1016/j.clim.2013.09.001
288. Kroger CJ, Clark M, Ke Q, Tisch RM. Therapies to Suppress beta Cell Autoimmunity in Type 1 Diabetes. Front Immunol (2018) 9:1891:1891. doi: 10.3389/fimmu.2018.01891
Keywords: autoimmunity, diabetes, immunoregulation, T cell receptor (TCR) signaling, T cell differentiation
Citation: Clark M, Kroger CJ, Ke Q and Tisch RM (2021) The Role of T Cell Receptor Signaling in the Development of Type 1 Diabetes. Front. Immunol. 11:615371. doi: 10.3389/fimmu.2020.615371
Received: 08 October 2020; Accepted: 15 December 2020;
Published: 02 February 2021.
Edited by:
Urs Christen, Goethe University Frankfurt, GermanyReviewed by:
Helen Thomas, University of Melbourne, AustraliaGerald Nepom, Benaroya Research Institute, United States
Karen Cerosaletti, Benaroya Research Institute, United States
Copyright © 2021 Clark, Kroger, Ke and Tisch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roland M. Tisch, cm10aXNjaEBtZWQudW5jLmVkdQ==