 Katerina Pyrillou†
Katerina Pyrillou† Laura C. Burzynski
Laura C. Burzynski Murray C. H. Clarke
Murray C. H. Clarke
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 December 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.613170
This article is part of the Research Topic NLRP3 Inflammasome: Regulatory Mechanisms, Role in Health and Disease and Therapeutic Potential View all 25 articles
Cytokines activate or inhibit immune cell behavior and are thus integral to all immune responses. IL-1α and IL-1β are powerful apical cytokines that instigate multiple downstream processes to affect both innate and adaptive immunity. Multiple studies show that IL-1β is typically activated in macrophages after inflammasome sensing of infection or danger, leading to caspase-1 processing of IL-1β and its release. However, many alternative mechanisms activate IL-1α and IL-1β in atypical cell types, and IL-1 function is also important for homeostatic processes that maintain a physiological state. This review focuses on the less studied, yet arguably more interesting biology of IL-1. We detail the production by, and effects of IL-1 on specific innate and adaptive immune cells, report how IL-1 is required for barrier function at multiple sites, and discuss how perturbation of IL-1 pathways can drive disease. Thus, although IL-1 is primarily studied for driving inflammation after release from macrophages, it is clear that it has a multifaceted role that extends far beyond this, with various unconventional effects of IL-1 vital for health. However, much is still unknown, and a detailed understanding of cell-type and context-dependent actions of IL-1 is required to truly understand this enigmatic cytokine, and safely deploy therapeutics for the betterment of human health.
IL-1α and IL-1β are the most studied members of the IL-1 superfamily, and although both ligate the same receptor (IL-1R1), and therefore induce identical downstream signaling, activation pathways of the two cytokines differ. Both IL-1α and IL-1β are expressed as proforms that require proteolytic processing for maximum cytokine activity (1), with removal of the N-terminus (N-term) leading to unmasking of key residues and/or a conformational change that enables the signature C-terminal beta-trefoil motif to interact with the receptor. The study of IL-1 activation has historically focused on processing of IL-1β by caspase-1 (casp-1) (2) and IL-1α by calpain (3). However, as summarized in Figure 1A, pro-IL-1 processing and activation can be mediated by caspases other than casp-1 and a range of other tissue and/or cell-type specific proteases. Together, this allows IL-1 activation to be controlled in specialized environments across the body (1). This review focuses on alternative modes of IL-1 activation and the more unconventional effects IL-1 can have in health and disease. Throughout the review use of the term “IL-1” relates to effects that can be driven by either IL-1α or IL-1β, or where experiments could not determine the form responsible (e.g. use of an Il1r1 knockout), whilst use of “IL-1α” or “IL-1β” refers to processes that were driven by a specific form.
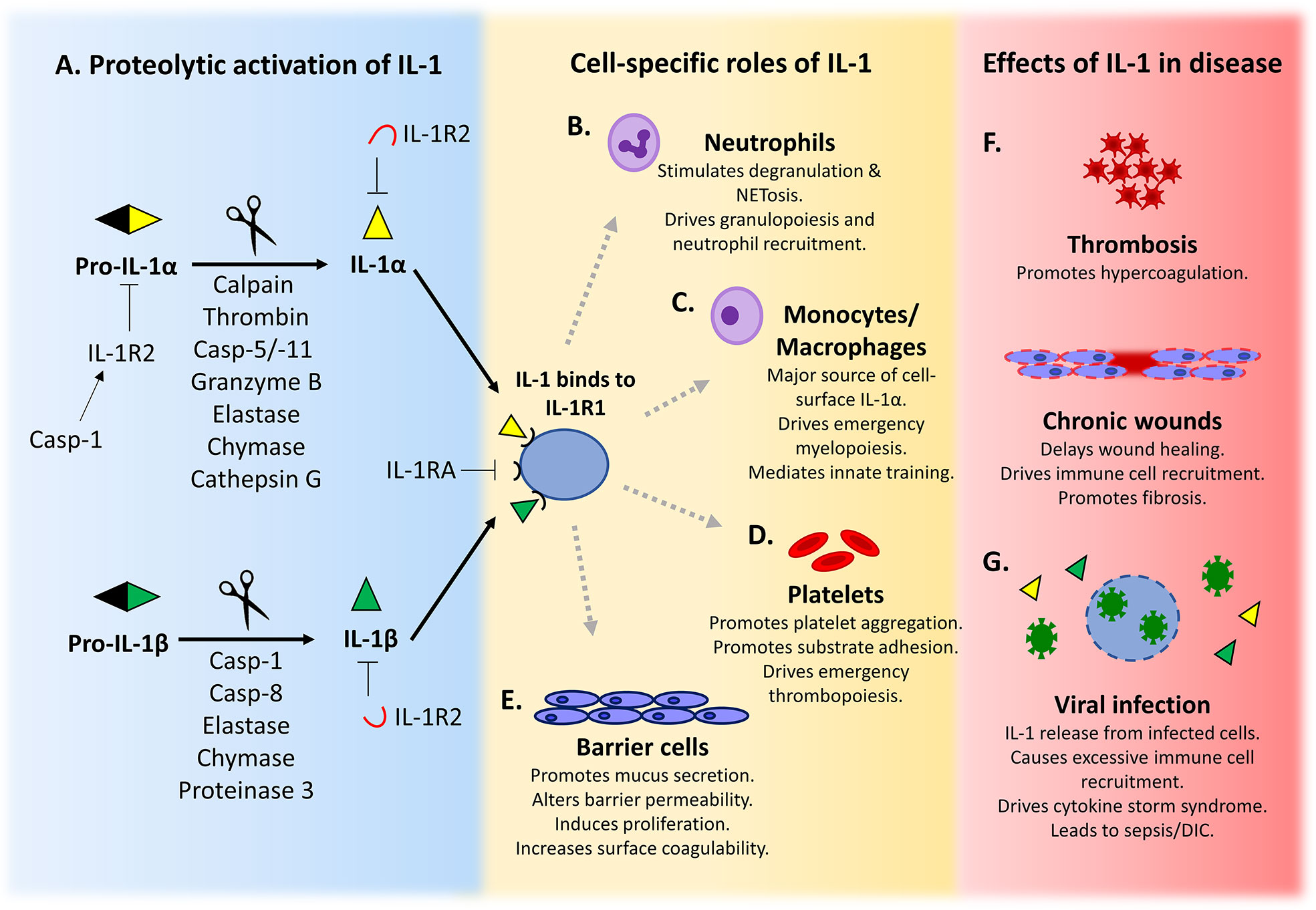
Figure 1 IL-1 is activated after proteolysis and has a variety of cell-specific roles that maintain homeostasis. (A) Pro-IL-1α/β is activated after cleavage by a diverse range of proteases. Active IL-1 binds to its signalling receptor IL-1R1 and elicits downstream signalling. IL-1 signalling is inhibited by binding to soluble or cell surface decoy receptor IL-1R2, or by competition from the IL-1 receptor antagonist (IL-1RA) for IL-1R1. (B–E) The production and response to IL-1 by specific cell types is important for the maintenance of cell function and homeostasis. (F, G) Dysregulation of IL-1 signalling can exacerbate or drive development of disease.
Typical IL-1β activation is mediated by casp-1 cleavage after inflammasome formation. Activation of inflammasomes is extensively reviewed elsewhere (1, 4). Briefly, inflammasomes are intracellular multiprotein complexes that assemble in response to pathogen-associated molecular patterns (PAMPs) (e.g. bacterial LPS, viral dsRNA) or danger-associated molecular patterns (DAMPs) (e.g. uric acid crystals, ATP), as well as other environmental factors (5). The NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome is the best studied and unique in its ability to form in response to a wide range of stimuli. Canonical activation occurs after the sensing of factors by NLRP3, causing assembly of NLRP3, ASC and casp-1 into a complex that results in the activation of casp-1. Active casp-1 cleaves pro-IL-1β and pro-IL-18 to active cytokines and cleaves Gasdermin D (GSDMD) to a form able to generate pores in the plasma membrane, both releasing cytokines and inducing pyroptotic cell death by loss of plasma membrane integrity (6, 7). Furthermore, IL-1α is also able to be released from cells via GSDMD pores (8), but it is unclear if proteolytic processing of pro-IL-1α is required prior to release.
Non-canonical inflammasome activation typically occurs in response to intracellular bacteria or internalized LPS. Depending on the cell type, non-canonical activation of inflammasomes occurs via a one- or two-step process. Macrophages require two-steps, with an initial engagement of a pathogen recognition receptor by PAMPs (e.g. LPS binding to toll-like receptor 4 (TLR4)) leading to NF-κB-mediated upregulation of inflammasome components (e.g. NLRP3 and pro-casp-1) and type I interferon (IFN) signaling-mediated upregulation of pro-casp-5 (in humans) or casp-11 (in mice), complementing the constitutively expressed human pro-casp-4. In the second step, these non-canonical caspases bind to intracellular LPS via the Lipid A domain, leading to their activation (9–11) and the subsequent cleavage of GSDMD. The now active N-term of GSDMD forms pores in the plasma membrane, to drive pyroptosis, and in turn triggers NLRP3 inflammasome activation (4, 12). In the event of infection with cytosol-invading bacteria such as Salmonella typhimurium or Shigella flexneri, where the Lipid A moiety is not accessible, interferon-induced guanylate-binding proteins (GBPs) are critical for LPS recognition and activation of non-canonical caspases, due to their association with the vacuole containing the phagocytosed pathogen. The GBPs coating the vacuole either recruit the caspase to the vacuole to create a platform for LPS detection and casp-4 activation (13), or mediate vacuolar rupture that exposes LPS to the cytosol (14). GBP1 initiates the formation of the platform, followed by GBP2, GBP4 and GBP3, eventually controlling casp-4 recruitment and activation (15). In contrast, non-canonical inflammasome activation in human monocytes occurs in a single step, with internalized free LPS directly activating ready-made casp-4/5 (16), negating the need for IFN signaling.
Recent developments identified an alternative pathway of non-canonical inflammasome activation involving caspase-8 (casp-8). Casp-8 is best known for transmitting pro-apoptotic signals downstream of death receptor signaling during extrinsic apoptosis (17). However, casp-8 is also able to promote both the upregulation of pro-IL-1β and its activation by direct processing at the same site targeted by casp-1 (18, 19). This occurs in dendritic cells (DCs) exposed to fungi, with Dectin-1 signaling causing formation of a non-canonical casp-8 inflammasome that upregulates and matures IL-1β (20). Similarly, bacterial infection inside macrophages also induces casp-8-dependent IL-1β secretion (21, 22). However, as casp-8 can also mediate casp-1 activation (21, 22), it is likely that pro-IL-1β is processed by both casp-1 and casp-8. Stimulation of DCs and macrophages with cellular stressors (e.g. chemotherapeutics) also causes direct cleavage of pro-IL-1β by casp-8, and casp-8-mediated NLRP3 inflammasome activation (23). However, casp-8 can also induce IL-1β maturation completely independent of casp-1. In DCs where casp-1 proteolytic activity is inhibited by casp-1 mutation, resulting in no GSDMD activation or pyroptosis, casp-8 is recruited to the NLRP3 inflammasome, which enhances casp-8 activity and drives IL-1β release, albeit in a delayed fashion due to either reduced processing or release (24). Casp-8 can also mediate IL-1β release completely independent of the NLRP3 inflammasome after engagement of DC Fas with Fas ligand (FasL) on invariant natural killer (NK) T cells (25). Finally, casp-8 can mediate an alternative form of NLRP3-dependent cell death coined incomplete pyroptosis, which is driven by gasdermin E pores in the absence of casp-1/11 (26). Although activation of these non-canonical pathways is often accompanied by pyroptosis and IL-1α release via GSDMD (4) or GSDME (26) pores, the exact mechanism for IL-1α release remains unclear.
The historic view is that IL-1α does not require proteolytic cleavage for full activity. However, these conclusions were drawn from studies that did not directly compare the activity of pro- and cleaved IL-1α (27), or used recombinant forms of IL-1α that were likely denatured during the purification process (i.e. by using HPLC) (28). Further work has shown that mature IL-1α has much higher cytokine activity than pro-IL-1α (29–31), with the calpain cleaved form having a ~50-fold higher affinity for IL-1R1 than the pro-form (30). In addition, cleavage of pro-IL-1α is regulated by its binding to a cytosolic form of the decoy receptor IL-1R2, which prevents calpain cleavage. However, after inflammasome activation casp-1 cleaves IL-1R2, which releases pro-IL-1α and allows calpain cleavage to the mature form (30).
Pro-IL-1α can also be cleaved by granzyme B (a cytotoxic T and NK cell protease), neutrophil elastase and mast-cell chymase, which confers bioactivity similar to calpain and up to a ~10-fold increase in activity over pro-IL-1α (29). This was found to be important in persistent inflammatory lung conditions such as cystic fibrosis, as patient bronchoalveolar lavage fluids could process IL-1α to a mature form. Interestingly, neutrophil elastase and mast cell chymase can also cleave IL-1β, IL-18, and IL-33 (32–34), perhaps revealing historic processes that could activate the ancestral IL-1 ligand before gene duplications formed the IL-1 family (35).
Thrombin, the key protease of coagulation, is also able to directly cleave and activate pro-IL-1α (36). Pro-IL-1α is cleaved by thrombin at a (K)PRS motif that is highly conserved across disparate mammalian species, suggesting functional importance. IL-1α cytokine activity after cleavage by either thrombin or calpain is equivalent, suggesting that removal of the N-term is critical for IL-1α activity, as is seen for IL-1β. The co-localization of tissue factor (TF) (a thrombin activator) and pro-IL-1α in the epidermis means that following injury thrombin generated during hemostasis can rapidly activate IL-1α, leading to inflammation and recruitment of immune cells that can safeguard against potential infection.
In addition to responding to intracellular LPS, casp-5 and -11 can also directly process and activate IL-1α (31). Again, pro-IL-1α cleavage occurs at an Asp residue that is highly conserved between different species, and this processing partially controls the release of mature IL-1α from macrophages after both canonical and non-canonical inflammasome activation (31). Importantly, CASP5 expression is increased in senescent human fibroblasts, and release of IL-1α, which drives the senescence-associated secretory phenotype (SASP), and SASP factors (e.g. IL-6/8, monocyte chemoattractant protein-1) is reduced without casp-5 (31). Furthermore, in an in vivo model of hepatocyte senescence, reducing casp-11 in senescent cells leads to their accumulation, which is caused by a reduced SASP failing to recruit immune cells. Together this suggests that casp-5/11 plays a key role in regulating IL-1α activation and release in both myeloid and senescent cells.
All the proteases described above cleave pro-IL-1α within the same target region located between the N-term propiece and the C-term cytokine domain. Together, this means that cleavage of IL-1α by any protease results in an active cytokine that only differs by a few amino acids and has comparable biologically activity. IL-1α activation can therefore be affected in a broad range of scenarios, allowing it to act as a versatile universal danger signal.
IL-1 was historically studied under many names, including leukocyte endogenous mediator, hematopoietin 1, endogenous pyrogen, lymphocyte activating factor, catabolin and osteoclast activating factor (37)—underscoring the pleiotropic effects of this widely expressed cytokine. In multi-cellular organisms cells exist in specialized niches that require distinct environmental cues to maintain homeostasis and functionality, much of which is controlled by soluble signaling factors such as cytokines and growth factors. Both IL-1α and IL-1β are active at very low concentrations, are tightly regulated and have important roles that extend beyond typical inflammation. Cell type-specific examples of both IL-1α and IL-1β production and response are discussed in this section.
IL-1 signaling is vital for effective innate immunity, with most innate immune cells able to produce IL-1 and almost all mesenchymal/tissue cells able to respond to it (38). Innate responses typically occur after sensing of DAMPs or PAMPs by tissue resident immune cells (e.g. macrophages), which leads to upregulation of pro-IL-1α/β and other cytokines (e.g. TNFα). If the insult is severe enough, leaked factors such as ATP drive inflammasome activation and large-scale release of mature IL-1α/β. Importantly, as pro-IL-1α is constitutively expressed by many tissue cells, necrosis can release fully active IL-1α that may instigate a low level responses able to resolve an insult before full inflammasome activation and potential collateral tissue damage. IL-1-mediated processes important for innate responses include cytokine secretion, upregulation of adhesion, MHC and/or co-stimulatory molecules and induction of vascular leakage, ultimately leading to the recruitment, activation and instruction of immune cells (39).
Neutrophils are the most abundant white blood cell in the circulation. They are rapidly recruited to sites of injury or infection where they phagocytose microbes and undergo degranulation to release bactericidal reactive oxygen species and proteases (40). Because of a short life span (~23–38 h), neutrophils require continuous replacement and robust mechanisms to control circulating numbers (41). Granulopoiesis and release of mature neutrophils from the bone marrow can be driven by IL-17 from Th17 cells (42), via granulocyte-colony stimulating factor. As Th17 differentiation is regulated by IL-1 (43) and Th17 cells produce IL-17 after exposure to IL-1 and IL-23, this gives IL-1 an indirect role in granulopoiesis (Figure 1B) (44, 45). More generally, infection and inflammation trigger neutrophilia via IL-1-induced proliferation of hematopoietic stem cells, a process known as emergency granulopoiesis (46).
IL-1 does not directly recruit neutrophils (47), rather it causes recruitment to sites of inflammation by upregulating neutrophil chemoattractants such as CXC- and CCL- chemokines in other cell types (48). For example, IL-1-dependent production of the chemokine IL-8 (CXCL8) occurs in endothelial cells (49), fibroblasts, and keratinocytes (50). IL-1 can also induce production of neutrophil chemotactic lipids including leukotriene B4 (51) and platelet activating factor (52). Recruitment of mature neutrophils to sites of infection or injury can also be driven by Th17 cell IL-17, which induces release of the neutrophil chemokines CXCL1, CXCL2, CXCL5, and CXCL8 from local endothelial and epithelial cells (53). Th17 cells also directly recruit neutrophils by production of CXCL8 (54). After administration of hydrocarbon oils to the peritoneal cavity, locally released IL-1α drives production of CXCL5 that recruits neutrophils (55).
IL-1 also drives calcium-dependent degranulation of neutrophils (56), with the exteriorized proteases (e.g. cathepsin G, elastase, and proteinase 3) subsequently able to cleave and activate multiple IL-1 family members (e.g. IL-1α, IL-1β, IL-33, IL-36) (57, 58). Indeed, it has been suggested that neutrophil proteases may be more effective at processing IL-1 family members than directly killing microbes (58), suggesting the ancestral purpose of these proteases was to activate cytokines. Proteinase 3 can also process IL-1β inside neutrophils lacking NF-κB signaling (59), which could serve as a host defense mechanism against pathogens able to evade NF-κB dependent innate immunity (60). Activated neutrophils release a meshwork of chromatin and proteases that form extracellular fibers, known as neutrophil extracellular traps (NETs), which trap and kill bacteria (61). Formation of NETs often leads to a lytic form of cell death called NETosis, but NETs can be released without cell death, known as vital NETosis (62). Interestingly, NET extrusion and NETosis are dependent on GSDMD pore formation that occurs during non-canonical inflammasome activation by intracellular LPS or bacteria (63, 64). NET formation also occurs in response to exogenous IL-1 (65, 66). In atherosclerotic plaques, cholesterol efflux-driven NLRP3 inflammasome activation in myeloid cells induces subsequent IL-1R1-dependent neutrophil accumulation and NET formation (67). Conversely, NET formation can also elicit IL-1 signaling, with the NET-associated proteases elastase and cathepsin G able to cleave and activate IL-1α (68, 69). Indeed, in diabetic wounds NET overproduction triggers macrophage NLRP3 inflammasome activation and IL-1β release, which sustains inflammation and impairs healing (70).
Finally, myelopoiesis following myocardial infarction is driven by neutrophil-derived IL-1β. Neutrophils recruited to the infarcted tissue release the alarmins S100A8 and S100A9 that bind to TLR4 on naïve neutrophils and induce NLRP3 inflammasome-dependent IL-1β secretion, which subsequently drives IL-1R1-dependent granulopoiesis in the bone marrow (71). Together, IL-1 signaling is either directly or indirectly capable of inducing neutrophil production, recruitment, degranulation, and NETosis. In turn, neutrophil proteases are able to activate IL-1 family members, which could act as a means to rapidly amplify inflammation at sites of injury or infection.
The main functions of a macrophage are to phagocytose cell debris and foreign bodies and release cytokines that orchestrate immune responses. Phagocytosis is fundamental for host defense, as it combines microbial killing with innate immune activation and the presentation of antigens to T cells. Macrophage phagosome maturation depends on progressive acidification, which activates pH-dependent proteases (e.g. cathepsins) that degrade the contents (72). Interestingly, mechanisms for macrophage cytokine release and phagocytosis overlap, with gram-positive bacteria within phagosomes inducing NLRP3 inflammasome activation. This leads to active casp-1 accumulation on phagosomes, where it controls acidification via the NADPH oxidase NOX2 (73, 74). To evade digestion in the phagosome, Staphylococcus aureus has evolved a mechanism that allows it to sequester mitochondria away from phagosomes, thereby preventing mitochondrial reactive oxygen species generation, local casp-1 activation, and subsequent phagosome acidification (75).
Macrophages can produce IL-1 in a variety of ways, as detailed above. However, macrophages appear unique in their ability to present IL-1α on the cell surface (Figure 1C) (76, 77). Cell surface (csIL-1α) is induced de-novo after TLR ligation and associates with the membrane via IL-1R2 and a glycosylphosphatidylinositol (GPI)-anchored protein, with trafficking to the membrane specifically inhibited by IFNγ (77). Because csIL-1α is the less active pro-form tethered via the N-term (77), it is both activated and released after cleavage, for example by thrombin (36), permitting wider ranging effects. However, because IL-33 causes macrophage activation and differentiation and IL-33 signaling via ST2 needs IL-1 receptor accessory protein (IL-1AcP) as a co-receptor (78, 79), this signaling complex likely sequesters IL-1AcP away from any IL-1R1 and reduces the potential for IL-1 signaling by macrophages.
Macrophages change phenotype in response to the microenvironment, typically becoming polarized to M1 (classically activated) or M2 (alternatively activated) subtypes. NLRP3 and IL-1β expression is reduced in M2 cells (80), and inhibition of NLRP3 inflammasome activation drives M2 polarization (81, 82). In keeping with this, NLRP3 inflammasome activation in Treponema pallidum infected macrophages induces M1 polarization and IL-1β release (83). In contrast, NLRP3 activation can also cause M2 polarization via upregulation of IL-4 in an inflammasome-independent process (84). Together, although NLRP3-controlled M1/M2 polarization is clearly important, the exact picture is yet to be elucidated.
Platelets are key for hemostasis in response to vascular injury, but also form pathogenic thrombi. Activated platelets change shape, aggregate, degranulate, and upregulate receptors (85), but also release bioactive molecules that cause inflammation and modulate immune responses. Activation of platelets increases cell surface IL-1α (86, 87), which can be cleaved and released from the surface by thrombin (36). During stroke or brain injury, IL-1α released from activated platelets is important for driving cerebrovascular inflammation via activation of the brain endothelium, which enhances adhesion molecule and CXCL1 expression (Figure 1D) (88). Platelet microparticle-associated IL-1β also promotes platelet-neutrophil aggregation in injured lung microvasculature during sickle cell disease (89). Additionally, platelets are able to license NLRP3 inflammasome activation and IL-1 production in innate immune cells, particularly monocytes. This licensing is via an unknown soluble platelet factor, independent of contact, and is not IL-1 or an inflammasome component (90). Platelets also express IL-1R1, and treatment with exogenous IL-1β can activate platelets and enhance adhesion to substrates (91). Platelets can also respond to the IL-1β they release in an autocrine signaling loop after LPS stimulation (92).
Platelet count and thrombopoiesis are both tightly regulated. Normal thrombopoiesis occurs after thrombopoietin induces caspase activation in megakaryocytes, causing compartmentalized apoptosis and pro-platelet formation (93, 94). However, emergency thrombopoiesis also occurs after acute platelet loss, with IL-1α signaling causing alternative platelet production via megakaryocyte rupture (95). Indeed, mice with thrombin-resistant pro-IL-1α do not rapidly recover platelet count after platelet depletion, suggesting thrombin activation of IL-1α is the critical mechanism for this form of emergency thrombopoiesis (36). Platelets are at the interplay of hemostasis and inflammation and together it is clear that their role in host defense goes much deeper than their ability to cross link fibrin during hemostasis.
An appreciation of the effects of IL-1 on the adaptive immune system dates to the 1970s, when IL-1 was originally named lymphocyte activating factor) to account for its lymphoproliferative activity (96–98). Indeed, T cell expansion under the influence of either IL-1α or IL-1β is demonstrated for a variety of T helper (Th) cell subsets such as Th1 (99, 100), Th2 (100, 101), and Th17 (100), in a process that is antigen and/or T cell receptor (TCR)-dependent. These observations are partly explained by a pro-survival role of IL-1 on the cells (100); however, other studies show no pro-survival effects of IL-1 downstream signaling on CD4 T cells (102). Similar observations were noted with Granzyme B+ CD8+ T cells, where IL-1β regulates expansion and effector function (103). IL-1α also drives Th cell differentiation, in particular Th17, but also synergizes with TNF to enable IL-12-driven Th1 differentiation (99). Th17 lineage commitment in both human (104, 105) and mouse systems (43) is conferred by IL-1-driven upregulation of the transcription factor Rorc (106). Production of signature cytokines by the majority of CD4 effector T cells is also augmented by other IL-1 family members (e.g. IL-18, IL-33 and IL-36), with Th9 (107, 108) and Th17 (106) cells directly responding to IL-1, which bypasses the requirement for TCR engagement and CD28 co-stimulation (100). Indeed, the tri-fold effects of IL-1 on T cell proliferation, differentiation and cytokine secretion in the absence of TCR stimulation is suggestive of the negative effects dysregulated IL-1 might have on adaptive immunity (as summarized in Figure 2A). This is further compounded by the inability of Tregs to effectively suppress IL-1-driven T cell proliferation and cytokine production (102). Thus, IL-1-driven activation of effector T cells with reduced Treg suppression could breach tolerance and drive autoimmunity.
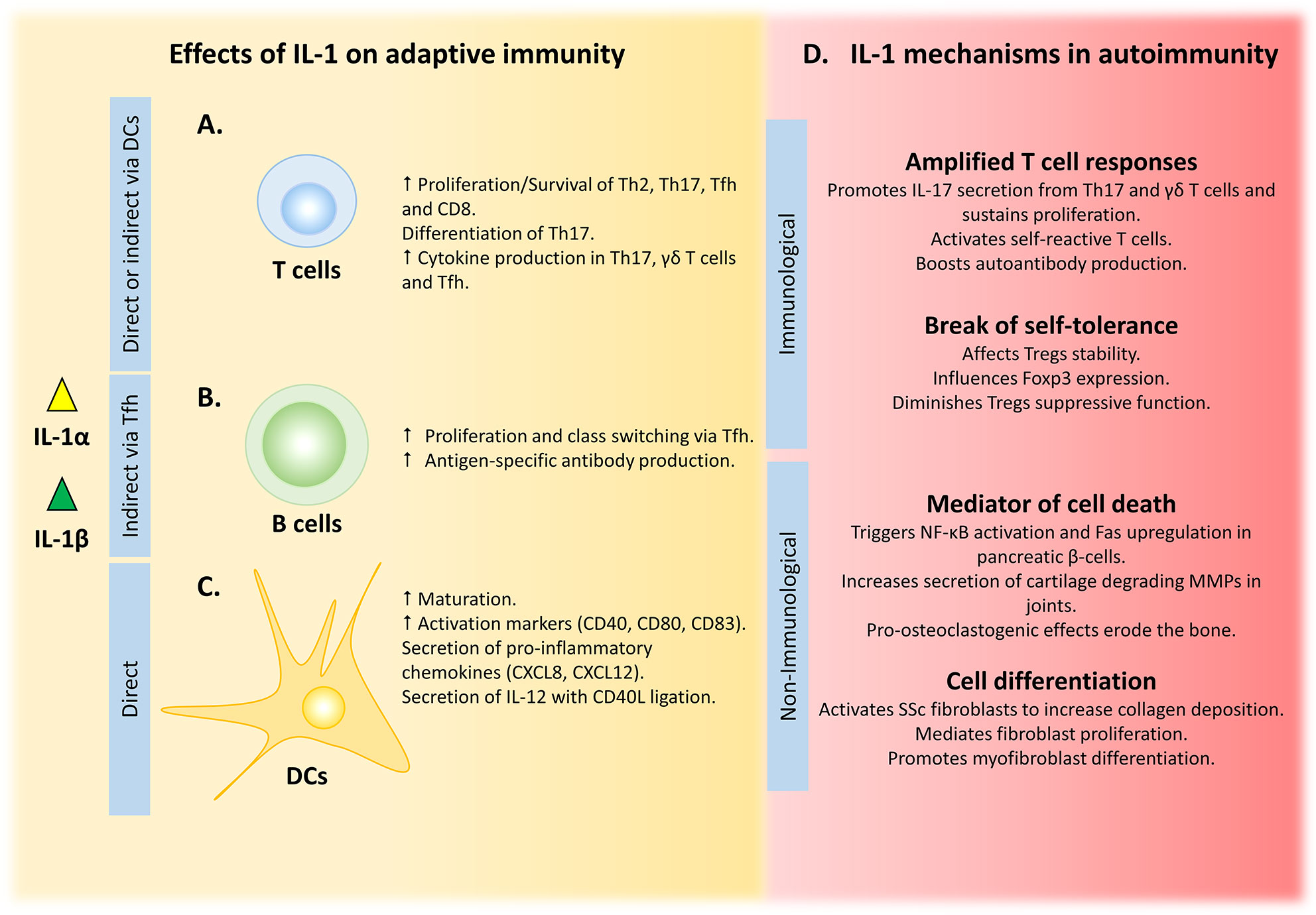
Figure 2 IL-1 influences adaptive immunity by multiple mechanisms, with dysregulation leading to disease. (A) IL-1 can shape T cell responses either by direct binding to T cell IL-1R1 or indirectly via IL-1-primed DCs. (B) Amplified B cell responses are driven by IL-1-activated Tfh cells. (C) Maturation, activation and cytokine secretion by DCs is directly driven by IL-1 signalling. (D) Uncontrolled IL-1 amplifies T cell responses and can break self-tolerance to drive autoimmunity, while its non-immunological role in tissue damage and remodelling can exacerbate these conditions.
It is not totally clear whether the effects of IL-1 on T cells are direct or indirect. Direct regulation implies expression of IL-1 receptor 1 (IL-1R1) on T cells, and although some evidence supports this it is not currently conclusive. Competitive binding experiments using radiolabeled IL-1 demonstrates that Th2 cells, but not Th1, bind IL-1 (109), which is supported by flow cytometry for IL-1R1 expression on murine Th2 cells (110). However, circulating human Th17 precursor cells (CCR6+ CD161+ CD4+) do not express Il1r1 mRNA (111), and a subset of Th2 cells with the ability to express IL-17 (CRTH2+ CD4+) contain very low levels of IL-1R1 protein (112). In contrast, bona fide Th17 cells show clear Il1r1 transcription (43) and expression of functional IL-1R1 on the cell surface (106). Furthermore, the presence of IL-1R1 on naive or memory CD4 T cells isolated from human blood determines Th17 fate and subsequent secretion of IL-17, with or without TCR triggering (113). Importantly, formation of an active IL-1 signaling complex requires IL-1R accessory protein (IL-1RAcP), and therefore the expression pattern of this co-receptor in conjunction with IL-1R1 is required to determine which T cells respond to IL-1 (114, 115). CD4 and CD8 T cells express cell surface IL-1RAcP, with higher abundance on regulatory CD4+ CD127low CD25hi cells that also express high levels of forkhead box P3 (FoxP3) (116). IL-1RAcP is also stably expressed on Th2 cells, where it acts as a co-receptor for ST2 that mediates T helper type 2 reactions in response to IL-33 (117, 118), suggesting that Th2 cells would respond to IL-1 if IL-1R1 is co-expressed.
T follicular helper (Tfh) cells also express IL-1R1 transcript and protein, and functionally respond to IL-1 (119), suggesting they must also express IL-1RAcP. Tfh cells provide growth and differentiation signals (i.e. IL-4, IL-21 and ICOS) to germinal center (GC) B cells, which drives affinity maturation, somatic hypermutation and long-lived humoral immunity (Figure 2B) (120–122). Indeed, administration of IL-1β during immunization substitutes for adjuvant-induced Tfh expansion and cytokine release (specifically IL-4 and IL-21). Moreover, IL-1β augments adjuvant-driven immunization by enhancing antigen-specific antibody production. In contrast, IL-1RA (Anakinra) inhibits these responses, showing the importance of IL-1 signaling in Tfh cells (119). The need of IL-1β for a productive GC and antibody response after immunization (123) suggests that driving IL-1-dependent Tfh-mediated B cell activation might be beneficial for enhancing vaccine efficacy, either by direct IL-1 administration or by using agents that induce or release IL-1 (e.g. alum and necrotic cell death, respectively). However, GC activation has to be tightly regulated, because loss of control could breach B cell tolerance and allow self-reactive clones to escape the GC, ultimately causing systemic autoimmune disease (124, 125).
Tregs maintain immune homeostasis by suppressing unwanted immune responses via multiple mechanisms (126, 127), including regulation of IL-1 availability via expression of IL-1 antagonists. Within the GC, Treg subtypes can restrain humoral responses by inhibiting secretion of the B cell activating cytokines IL-4 and IL-21 from activated Tfh (128, 129). T follicular regulatory (Tfr) cells in particular express high levels of IL-1R2 and IL-1RA, which suppresses IL-1-driven GC activation by inhibiting Tfh activation and secretion of IL-4 and IL-21 (119). Thus, in cases of IL-1-mediated GC dysregulation, IL-1 antagonists may be useful in controlling these responses and restoring protective immunity. However, non-physiological upregulation of IL-1R2 represents a maladaptive mechanism utilized by Tregs infiltrating colorectal, non-small cell lung or breast cancers that are more aggressive with poor prognosis (130, 131). Supported by evidence showing that DC-derived IL-1β-dependent priming of CD8+ T cells augments their tumoricidal properties and that Nlrp3-/- or Casp-1-/- DCs sub-optimally prime T cells (132), these data suggest that IL-1β (or potentially IL-1α) depletion from the tumor microenvironment could confer immunosuppression and poor tumor control, and thus IL-1R2 expression on intra-tumoral Tregs could be utilized as a prognostic marker.
In addition to the direct effects of IL-1 on T cells, their function can also be indirectly influenced by interaction with IL-1-primed DCs (Figure 2C). Both IL-1α or IL-1β-pulsed DCs enhance T cell-dependent cytokine secretion from CD4 and CD8 T cells, with IFN-γ secretion being consistently augmented (133–135). Although IL-1β is not as strong an inducer of DC maturation as LPS, it still potently upregulates activation markers (e.g. CD40, CD80 and CD83) and induces secretion of pro-inflammatory mediators (e.g. CXCL8 and CXCL12) (133). In contrast, IL-1β induces minimal secretion of the major polarizing cytokine IL-12p70, but in combination with CD40L both expression of Il12b and release of IL-12p70 is increased (134, 136). In addition, antigen endocytosis, which is diminished during DC maturation, is decreased by IL-1β to a similar degree as LPS (137). What is clear is that the role of IL-1 on adaptive immunity is multifaceted and cannot be summarized in a single conclusion. A multitude of normal immune responses depend on IL-1 signaling, including T cell proliferation, expansion and differentiation, as well as humoral responses to T cell-dependent antigens. Moreover, IL-1 synergizes with CD40L to potentiate activation and maturation of DCs, and the subsequent adaptive immune response. Together, these processes are critical for host defense, and dysregulation of the equilibrium leads to either an ineffective adaptive reaction or an overwrought maladaptive response that drives autoimmune pathology.
Epithelial cells form a highly specialized physical barrier against the external environment, and thus are exposed to a myriad of PAMPs, damage-associated molecular patterns (DAMPs), and other environmental factors that induce inflammation. Although inflammasomes are primarily studied in myeloid cells they are also essential for barrier defense in non-myeloid cells, including specialized epithelial cells. Indeed, IL-1 is constitutively expressed in many epithelial cells, where it helps defend against pathogens and injury (Figure 1E) (138). However, loss of control can lead to inappropriate activation, chronic inflammation and disease (139).
Keratinocytes secrete pro-IL-1α that can be processed by thrombin activated during hemostasis after wounding. This connection between the coagulation and immune systems allows rapid IL-1α activation and immune cell recruitment that can safeguard against potential infection after breach of the epidermal barrier (36). In response to UVB irradiation (140) or analogs of viral double stranded RNA (141) keratinocytes activate the NLRP3 inflammasome and secrete IL-1β. The IL-1 released from keratinocytes after viral infection induces expression of anti-viral interferon-stimulated genes in local fibroblasts and endothelial cells. However, this is not observed in mouse models, suggesting human skin has an alternative IL-1-driven anti-viral system to prevent viruses that can evade detection by pattern recognition receptors (142).
The gastrointestinal tract epithelium is highly specialized throughout its length and must respond appropriately to its local environment to maintain homeostasis. In the stomach, IL-1-mediated reduction of gastric acid production (143–145) and prostaglandin synthesis (146) by parietal epithelial cells inhibits neutrophil infiltration, which protects the gastric lining from non-steroidal anti-inflammatory drug-induced gastropathy (146) and ulceration (147). In the duodenum, IL-1 signaling causes epithelial goblet cells to increase mucus secretion (148, 149), and IL-1 can also confer protection to disrupted mucosal layers during Helicobacter pylori infection (150). In contrast, elevated IL-1 signaling can also contribute to gut epithelium dysfunction by increasing tight junction permeability (151, 152) and inhibiting effective absorption (153–155). Finally, IL-1 signaling in colonic epithelial cells plays a direct pro-tumorigenic role by regulating the early proliferation and survival of colorectal cancer cells, independent of inflammation (156).
The respiratory epithelium comprises basal, ciliated, and secretory epithelial cells responsible for homeostatic regulation of lung fluid, clearance of inhaled agents, recruitment of immune cells, and regulation of airway smooth muscle function (157). Epithelial IL-1 signaling regulates mucin secretion and airway surface liquid metabolism, resulting in enhanced mucociliary clearance during inflammation (158). In cystic fibrosis, where CFTR-mediated fluid secretion is impaired, IL-1 drives secretion of epithelial mucin, but not fluid, causing mucus hyperconcentration (159). IL-1 and TNFα also promote the regeneration of alveolar epithelial cells by stimulating proliferation of type 2 epithelial progenitor cells (160). Indeed, IL-1 and TNFα released during influenza infection promotes repair of damaged alveoli (161).
Endothelial cells are critical for blood vessel formation, coagulation, regulation of vascular tone, and inflammation (162). Heme from ruptured red blood cells can activate the NLRP3 inflammasome and cause IL-1β release from endothelial cells, and thus inflammation can occur in response to sterile hemolysis (163). Endothelial cells activated by IL-1 express TF on their surface, which initiates the coagulation cascade (69, 164, 165). IL-1 also causes endothelial cells to reduce tight junction integrity (166–168) and increase expression of adhesion molecules, such as ICAM (169) and PECAM1 (170), facilitating diapedesis of leukocytes. In contrast, IL-1 reduces transcellular diapedesis through endothelial cells (167). Finally, IL-1 also acts as a potent proangiogenic signal via endothelial cell-derived vascular endothelial growth factor (VEGF) (171). For example, during tumorigenesis, IL-1 from recruited myeloid cells causes endothelial cells to upregulate VEGF and other proangiogenic factors, which promotes an inflammatory microenvironment that supports tumor angiogenesis (172, 173). In these models IL-1 inhibition restricted tumor growth and angiogenesis (174). Together, IL-1 signaling is essential to many specialized epithelial cell processes and is vital for effective barrier defense; however, dysregulation can lead to excessive inflammation and tumorigenesis.
Immune responses are necessary for the maintenance of homeostasis; for example, to maintain an environment that is inhospitable for invading pathogens (e.g. mucous membranes), or in contrast, training the immune system to tolerate bacteria (e.g. gut microbiota). However, balance is everything, with loss of homeostasis leading to chronic inflammation and/or inappropriate adaptive immunity, which both exacerbates existing disease and drives emergence of new pathology. IL-1 signaling is central to many immune responses, and thus blocking IL-1 therapeutically is important in a wide spectrum of diseases (175). This section focuses on some of the more unconventional effects of IL-1 in health and disease.
Skin wounding occurs when the epidermal barrier is disrupted by cutting, excessive force, chemicals, or extreme temperature. Wounding triggers a series of events critical to the healing process: hemostasis, inflammation, proliferation, and remodeling (176). Initially, hemostasis is activated after vessel wall injury to allow local thrombus formation that stymies blood loss (177). Subsequent inflammation at the wound site is integral for removal of damaged tissue and recruitment of immune cells that coordinate repair (176).
Epidermal IL-1α colocalizes with TF, a potent thrombin activator. Thus, epidermal injury results in generation of active thrombin, which cleaves and activates pro-IL-1α (36). Indeed, genetically modified mice with thrombin-resistant pro-IL-1α recruit fewer neutrophils and macrophages to the granulation tissue under the wound and show delayed healing (36). Furthermore, excisional wounds in Nlrp3-/- and Casp1-/- mice contain less IL-1β and TNFα, recruit fewer neutrophils and macrophages, and also show delayed healing that can be partially rescued by addition of exogenous IL-1β (178). In keeping with this, application of the NLRP3 activator ATP accelerates wound closure in wild type mice (179). Staphylococcus aureus infection of wounds upregulates keratinocyte IL-1 that drives neutrophil recruitment via IL-1R1 signaling (180), while NLRP3 activation is also seen after sterile burn injuries, with Nlrp3-/- mice showing reduced macrophage infiltration and impaired healing after chemical burns (181). In contrast, excessive IL-1 can lead to chronic non-resolving wounds (Figure 1F). For example, sustained NLRP3 activation and elevated IL-1β is found in non-healing diabetic wounds (182), with inflammasome inhibition improving healing via increased angiogenesis and reduced inflammatory macrophages (182, 183). Additionally, excessive neutrophil NETs cause NLRP3-dependent IL-1β release from macrophages (70), with NET digestion reducing inflammasome activation and macrophage infiltration, leading to diabetic wound healing (70). IL-1 signaling also promotes fibrosis in the wound, with IL-1 inhibition reducing scarring and improving healing in deep tissue (184) and diabetic wounds (185). Together, this shows that while IL-1 signaling in the wound is necessary for effective wound healing, it must be tightly regulated to prevent pathological chronic inflammation and/or scarring.
Hemostasis is essential to prevent injury-related blood loss; however pathological thrombosis occurs when anticoagulant mechanisms are unable to limit excessive activation of the hemostatic pathway, leading to vessel occlusion and downstream ischemia. Excessive IL-1 signaling can cause thrombosis (Figure 1F), with the CANTOS study finding that IL-1β inhibition with Canakinumab reduces secondary atherothrombotic events in patients with residual inflammatory risk, which was likely due to modulation of procoagulant factors and reduced leukocyte recruitment to the plaque (186). Additionally, increased NLRP3-dependent IL-1β maturation and venous thrombosis is seen in mice deficient for the vasculoprotective enzyme CD39, which is reversed by IL-1 inhibition (187). Thrombosis relies on the interplay between blood cells, plasma proteins and the vessel wall. Neutrophils drive thrombosis by producing TF and NETs, which act as scaffolds for thrombus stabilization (188, 189), which can induce coagulopathy during sepsis (190), acute respiratory distress syndrome (ARDS) (191), and coronary artery thrombosis (192). ST-elevation myocardial infarction (STEMI) patients with elevated inflammatory markers (e.g. hsCRP) have increased circulating IL-1β and NET-associated TF, while mouse models of thrombosis have reduced NET-associated TF and delayed thrombotic occlusion when IL-1β is blocked (193). Similarly, IL-1β or NLRP3 inhibition attenuates NET-associated thrombosis in mouse models of breast cancer, supporting IL-1β as the driver of this mechanism (194). Additionally, as NET-associated proteases can activate IL-1α, this can also promote IL-1R1-dependent TF expression on the endothelial cell surface (69). Macrophages can also release TF after inflammasome activation by bacteria during sepsis, with GSDMD-dependent pyroptosis and pore formation allowing externalization of TF that triggers systemic coagulation and lethality (195, 196). Overall, wounding is a complex mechanism that requires rapid, tightly controlled, hemostasis and inflammation for effective healing. IL-1 is widely expressed in the epidermis and the vessel wall and can be activated by many proteases, and is therefore integral to the wounding response.
Trained immunity refers to a response in innate immune cells after encountering a pathogen that is more adaptive-like and confers a degree of protection against secondary exposure to an un-related infection (197). Establishment of trained immunity is mostly driven by epigenetic remodeling and metabolic changes that occur after the first exposure (198, 199). Because the epigenetic imprinting is locus-specific and allows access to transcription factors, it can alter cell identity and prime specific functions. Similarly, changes in cell metabolism also establish new cell functions (199). However, cell metabolism can affect epigenetic reprogramming that further alters the cells metabolic status, and thus these processes are inter-dependent. IL-1 family agonists are implicated in trained innate immunity, with macrophage-derived IL-18 playing a non-redundant role in training NK cells’ anti-tumor activity via secretion of IFN-γ (200–202). Moreover, IL-1β participates in induction of trained immunity in response to Bacille Calmette-Guerin (BCG) vaccination, where increased IL-1β and TNFα from peripheral monocytes correlates with enriched histone H3K4 tri-methylation at Il6, Tnf, and Tlr4 promoters after in vitro re-stimulation with M. tuberculosis, heat-killed S. aureus or C. albicans (203).
Epigenetic changes after training via cytokines can be mediated by altered metabolism that leads to accumulation of metabolic products such as glucose, lactate (204), succinate, itaconate (205), mevalonate (206) and fumarate (207). In particular, the transition from oxidative phosphorylation to aerobic glycolysis is a determining step towards trained immunity, and is associated with H3K4me3 and H3K27Ac modifications in the presence of glucose, lactate (204) and mevalonate (206). Interestingly, mevalonate accumulation in patients with hyper IgD syndrome (HIDS) that lack mevalonate kinase is associated with increased IL-1β production by LPS-stimulated monocytes (206). Similarly, glycolysis-driven accumulation of succinate in response to LPS induces macrophage IL-1β expression, which occurs via HIF-1α binding to the Il1b promoter (208).
Trained immunity is also established at a systemic/central level after exposure of bone marrow hemopoietic stem and progenitor cells (HSPCs) to pathogen-derived products, such as β-glucan, BCG, or Western diet (WD) feeding. β-glucan induces bone marrow IL-1β production that acts on HSPCs, leading to preferential myelopoiesis and metabolism that favors glycolysis and cholesterol synthesis (209). BCG vaccination also causes HSPCs to skew toward myelopoiesis and epigenetic reprogramming of macrophages, such that they upregulate Ifng, Tnf, and Il1b upon re-stimulation with M. tuberculosis (210). Indeed, BCG vaccination increases circulating IL-1β, which inversely correlates with viremia after a secondary yellow fever vaccination (211). Together, this suggests that IL-1β contributes to both training process and direct anti-mycobacterial response. Interestingly, diet can also drive trained immunity, with WD fed mice exhibiting myelopoiesis skewed towards the monocytic lineage and upregulation of genes associated with hematopoiesis, metabolism, immune cell differentiation and leukocyte activation. This WD-dependent reprogramming was long lasting (4 weeks) and was dependent on NLRP3 (212).
However, despite beneficial effects of trained immunity on host defense, the long-lasting reprogramming of innate immune cells can become maladaptive and contribute to pathology. Human monocytes trained ex vivo with oxidized low density lipoprotein (oxLDL) or β-glucan produce pro-atherogenic cytokines and increase foam cell formation upon re-stimulation (213). Furthermore, chronic systemic exposure to IL-1 can lead to haematopoietic stem cell exhaustion that compromises blood cell homeostasis and reduces the ability to endure replicative challenges such as transplantation (214). Under such circumstances metabolites such as itaconate, which limits aerobic glycolysis and induces anti-inflammatory and anti-oxidative programs (215–217), might be utilized to counteract innate training and re-instate immune tolerance (205).
In the brain, microglia are able to undergo priming in response to injury similar to immune training seen in macrophages in the peripheral immune system (218). IL-1 is constitutively expressed at very low levels in the normal brain but appears to have a role in some physiological processes, including regulation of sleep and synaptic plasticity (219, 220). Following neurotoxic injury, upregulation of IL-1α and IL-1β occurs rapidly in the brain (219), and facilitates recruitment of leukocytes across the blood brain barrier (221). Microglia are able to retain memory of an inflammatory state via histone H3 modifications, which may contribute to hyper-reactive IL-1 expression in response to subsequent inflammatory stimuli, which is seen in many neurodegenerative pathologies (218).
Although IL-1 is critical for host defense, it can also contribute to autoimmune disease via its ability to amplify T cell responses, shift the balance for immune tolerance and its direct action on non-immune cells that induces inflammation and tissue damage (102, 222) (Figure 2D). Indeed, each of the above mechanisms, either alone or in combination, underlie the pathophysiology of common autoimmune diseases.
IL-17-producing Th17 and γδ T cells are causal of several mouse autoimmune syndromes, such as experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis (44, 223, 224). IL-17 production in these cell types is stimulated by IL-1, suggesting that excess IL-1 would exacerbate these conditions. Indeed, both Il1r1-/- and Il1a/b-/- mice are resistant to the development of EAE due to lower Th17 cell proliferation, less IL-17 and lower self-reactive T cell activation, leading to less autoantibodies (44, 225). Similarly, IL-1β and IL-23-treated γδ T cells drive EAE by increased secretion of IL-17 directly into the brain milieu and by indirect augmentation of IL-17 production by αβ T cells (223).
As demonstrated in the Deficiency of Interleukin-1 Receptor antagonist (DIRA) syndrome, IL-1 antagonism is critical for the amelioration of IL-17-driven diseases. DIRA is an autoinflammatory condition associated with hyperactivation of IL-1 signaling due to loss-of-function mutations in IL-1RA, with severe manifestations in skin and bone. As such, DIRA is very responsive to IL-1 blockade by administration of recombinant IL-1RA (Anakinra) (226). Furthermore, IL-1 antagonists (i.e. IL-1RA and SIGIRR) delay onset and reduce severity of mouse EAE (225, 227). Interestingly, classic drugs used to manage multiple sclerosis (MS) (i.e. IFNβ) and its clinical relapse (i.e. steroids) associate with increased systemic levels of IL-1RA and IL-1R2 (228–230). In addition, monitoring the IL-1 to IL-1RA ratio in cerebrospinal fluid and/or lesions in MS patients may predict susceptibility of relapse-onset MS, hinting towards the use of IL-1 as a biomarker (231).
In keeping, loss of NLRP3 inflammasome components (i.e. NLRP3, ASC, caspases-1 or -11) confers some protection from EAE (232–235). However, inflammasome-independent IL-1β production has also been demonstrated as a result of trans-cellular interactions between effector CD4 T cells and DCs. Thus, CD4 T cell-derived TNF induces DC expression of pro-IL-1β, which is subsequently activated by casp-8 after T cell FasL ligation of DC Fas, leading to IL-1β release and induction of EAE (236).
Tregs are critical for maintenance of peripheral tolerance through their ability to suppress inappropriate activation of effector T cells. Foxp3 is essential for the suppressive functions of Tregs, and thus its expression maintains physiological T cell responses. Foxp3 expression can be reduced in highly inflammatory tissue microenvironments, especially in the presence of IL-1 and IL-6, which can convert Tregs into effector T cells that produce IFN-γ and IL-17 (237–239). Indeed, this confirms observations that IL-1β reduces susceptibility of CD4 effector/memory T cells to Treg suppression due to expansion of IFN-γ producing effector CD25+ cells (240). This is more pronounced in the autoimmune-prone NOD mice, where splenocyte IL-1β production is enhanced and IL-1β neutralization restores Treg suppression and normalizes IFN-γ secretion, altogether suggesting that breach of tolerance can be driven by IL-1β (240).
Besides its direct immunomodulatory effects, IL-1 can also act on non-immune cells to either alter their function or direct them towards apoptosis, thus contributing to the progression of several autoimmune and metabolic syndromes. Tissue damaging effects of IL-1 have been identified in the development of Type 1 and Type 2 Diabetes Mellitus (T1/T2DM) as well as rheumatoid arthritis (RA), while in systemic sclerosis (SSc) IL-1 has been shown to mediate pathological tissue remodeling.
Pancreatic inflammation and β-cell deregulation and loss are determining factors in the pathogenesis of both T1/T2DM (241, 242), and IL-1 can interfere with all these processes. Pancreatic β-cells express high levels or IL-1R1 and respond to IL-1 by upregulating inflammatory cytokines and chemokines (243, 244), which in turn recruit macrophages (245) and T cells (246). In addition, IL-1β has direct cytotoxic effects on β-cells (247) via canonical NF-κB activation and upregulation of Fas, in response to glucose, ultimately contributing to insulin resistance and development of T2DM (248). This IL-1β-mediated glucotoxicity is also relevant to T1DM, where islet resident macrophages secrete IL-1β upon stimulation with LPS and TNF (249), leading to β-cell production of cytotoxic molecules (e.g. inducible nitric oxide synthase) (250). In addition, β-cell apoptosis can also be triggered by IL-1β and IFN-γ activation of the non-canonical NF-κB pathway (251). Indeed, glucotoxicity and cell loss is abrogated by IL-1RA, improving β-cell secretory function and glycemic control (248, 252).
RA is characterized by progressive destruction of the articular joints due to IL-1β-dependent degradation of both bone and cartilage (253). BALB/c mice lacking IL-1RA spontaneously develop an inflammatory arthropathy that shares features with human RA, such as inflammatory cell infiltrates, fibrin clots, bone erosion, increased IL-1β in the affected joints and autoantibodies in serum (254). IL-1 signaling in synovial cells and chondrocytes causes upregulation and secretion of matrix metalloproteinases that degrade cartilage (255–257). In addition, IL-1 and TNF, when in the presence of T cell-derived RANKL, exert pro-osteoclastogenic effects that result in bone erosion and further joint damage (258), suggesting that IL-1 could be a druggable target for ameliorating symptoms and impeding disease progression. Indeed, neutralization of IL-1 with antibodies or natural antagonists (i.e. IL-1RA or IL-1R2) diminish local inflammation and protect the joint from bone erosion, which led to the approval of Anakinra (IL-1RA) as an effective therapeutic for management of RA (259–261).
SSc is an idiopathic autoimmune syndrome that exhibits fibrosis in the skin and other organs like the heart and lungs, due to fibroblast activation and deposition of extracellular matrix (262). IL-1α and IL-1β regulate IL-6 and PDGF-A expression on SSc fibroblasts, promoting collagen deposition and proliferation (263, 264) and differentiation into myofibroblasts (265–267). In addition, inflammasome activation in fibroblasts leading to IL-1β production acts in an autocrine manner to trigger expression of mIR-155, which in turn regulates collagen deposition in SSc (268–270).
The novel coronavirus SARS-CoV-2 is the causative agent of the acute respiratory disease COVID-19, which has caused a global pandemic (271). Symptoms of COVID-19 range from mild to severe, with viral pneumonia leading to ARDS, sepsis/disseminated intravascular coagulation (DIC) and/or multi-organ failure identified as the major causes of death (272). A key stage of the virus lifecycle is release of newly replicated virons (273), which escape via the host cell’s secretory pathways or by cell lysis (274). Egress of coronaviruses depends on the coronavirus envelope (E) protein, which acts as a viroporin that forms pores in the plasma membrane, causing lysis and subsequent release of DAMPs (275–277). For example, respiratory syncytial virus-infected cells release IL-1α that activates neighboring cells to promote leukocyte recruitment (278) and interferon-mediated anti-viral mechanisms (Figure 1G) (142). However, excessive cell lysis and DAMP release can trigger an over-the-top innate immune response and overt production of cytokines, known as a cytokine storm, which can spill over into the circulation and cause sepsis (279). Severe SARS-CoV-2 infection causes a systemic cytokine storm and ARDS, with accompanying thrombosis (271, 280, 281). IL-1 inhibition improves pathologies associated with cytokine storms, including sepsis and DIC (282–284), and thus maybe beneficial for treating COVID-19. Indeed, Anakinra (IL-1RA) appears to dampen markers of systemic inflammation and improve ARDS in COVID-19 patients. Importantly, Anakinra has a short half-life (3h) that allows rapid discontinuation of treatment if needed, in contrast to Canakinumab (half-life 26 d) (284, 285). At the time of writing there were 15 registered clinical trials examining IL-1 blockade by Anakinra for COVID-19. However, patient selection, dosing, and outcome measures are not harmonized between studies (286), and only 11 were randomized control trials. Finally, the multi-center RECOVERY trial has recently shown that the corticosteroid dexamethasone reduces deaths by one-third in ventilated COVID-19 patients (287), reinforcing the use of anti-inflammatory agents. Interestingly, dexamethasone is long known to profoundly inhibit IL-1 production (288, 289), again signifying a likely role for this apical cytokine in human health and disease. Together, there is existing evidence that blockade of inflammatory pathways, including IL-1, is beneficial for reducing symptoms of cytokine storm in COVID-19. Further clinical trials will be essential to determine the full extent of this benefit.
KP and LB contributed equally. KP, LB, and MC conceived and wrote the review. All authors contributed to the article and approved the submitted version.
This work was funded by British Heart Foundation Grants FS/09/005/26845, FS/13/3/30038, FS/18/19/33371, and RG/16/8/32388 to MC, the BHF Cambridge Centre for Research Excellence RE/13/6/30180, and the Cambridge NIHR Biomedical Research Centre.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Afonina IS, Müller C, Martin SJ, Beyaert R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity (2015) 42:991–1004. doi: 10.1016/j.immuni.2015.06.003
2. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature (1992) 356:768–74. doi: 10.1038/356768a0
3. Kobayashi Y, Yamamoto K, Saido T, Kawasaki H, Oppenheim JJ, Matsushima K. Identification of calcium-activated neutral protease as a processing enzyme of human interleukin 1 alpha. Proc Natl Acad Sci U S A (1990) 87:5548–52. doi: 10.1073/pnas.87.14.5548
4. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
5. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
6. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A (2016) 113:7858–63. doi: 10.1073/pnas.1607769113
7. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity (2016) 44:833–46. doi: 10.1016/j.immuni.2016.01.012
8. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity (2018) 48:35–44.e6. doi: 10.1016/j.immuni.2017.11.013
9. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science (80) (2013) 341:1250–3. doi: 10.1126/science.1240988
10. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514:187–92. doi: 10.1038/nature13683
11. Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol (2015) 45:2918–26. doi: 10.1002/eji.201545655
12. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526:666–71. doi: 10.1038/nature15541
13. Santos JC, Boucher D, Schneider LK, Demarco B, Dilucca M, Shkarina K, et al. Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun (2020) 11:1–15. doi: 10.1038/s41467-020-16889-z
14. Meunier E, Dick MS, Dreier RF, Schürmann N, Broz DK, Warming S, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature (2014) 509:366–70. doi: 10.1038/nature13157
15. Wandel MP, Kim BH, Park ES, Boyle KB, Nayak K, Lagrange B, et al. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat Immunol (2020) 21:880–91. doi: 10.1038/s41590-020-0697-2
16. Viganò E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat Commun (2015) 6:1–13. doi: 10.1038/ncomms9761
17. van Raam BJ, Salvesen GS. Proliferative versus apoptotic functions of caspase-8 Hetero or homo: the caspase-8 dimer controls cell fate. Biochim Biophys Acta (2012) 1824:113–22. doi: 10.1016/j.bbapap.2011.06.005
18. Maelfait J, Beyaert R. Non-apoptotic functions of caspase-8. Biochem Pharmacol (2008) 76:1365–73. doi: 10.1016/j.bcp.2008.07.034
19. Feltham R, Vince JE, Lawlor KE. Caspase-8: Not so silently deadly. Clin Transl Immunol (2017) 6:e124. doi: 10.1038/cti.2016.83
20. Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, Van Der Vlist M, Boekhout T, et al. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat Immunol (2012) 13:246–54. doi: 10.1038/ni.2222
21. Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE. Salmonella Infection Induces Recruitment of Caspase-8 to the Inflammasome To Modulate IL-1β Production. J Immunol (2013) 191:5239–46. doi: 10.4049/jimmunol.1301581
22. Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci U S A (2014) 111:7391–6. doi: 10.1073/pnas.1403477111
23. Antonopoulos C, El Sanadi C, Kaiser WJ, Mocarski ES, Dubyak GR. Proapoptotic Chemotherapeutic Drugs Induce Noncanonical Processing and Release of IL-1β via Caspase-8 in Dendritic Cells. J Immunol (2013) 191:4789–803. doi: 10.4049/jimmunol.1300645
24. Schneider KS, Groß CJ, Dreier RF, Saller BS, Mishra R, Gorka O, et al. The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Rep (2017) 21:3846–59. doi: 10.1016/j.celrep.2017.12.018
25. Donado CA, Cao AB, Simmons DP, Croker BA, Brennan PJ, Brenner MB. A Two-Cell Model for IL-1β Release Mediated by Death-Receptor Signaling. Cell Rep (2020) 31:107466. doi: 10.1016/j.celrep.2020.03.030
26. Aizawa E, Karasawa T, Watanabe S, Komada T, Kimura H, Kamata R, et al. GSDME-Dependent Incomplete Pyroptosis Permits Selective IL-1α Release under Caspase-1 Inhibition. iScience (2020) 23:101070. doi: 10.1016/j.isci.2020.101070
27. March CJ, Mosley B, Larsen A, Cerretti DP, Braedt G, Price V, et al. Cloning, sequence and expression of two distinct human interleukin-1 complementary DNAs. Nature (1985) 315:641–7. doi: 10.1038/315641a0
28. Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, et al. The Interleukin-1alpha Precursor is Biologically Active and is Likely a Key Alarmin in the IL-1 Family of Cytokines. Front Immunol (2013) 4:391. doi: 10.3389/fimmu.2013.00391
29. Afonina IS, Tynan GA, Logue SE, Cullen SP, Bots M, Lüthi AU, et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1α. Mol Cell (2011) 44:265–78. doi: 10.1016/j.molcel.2011.07.037
30. Zheng Y, Humphry M, Maguire JJ, Bennett MR, Clarke MC. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity (2013) 38:285–95. doi: 10.1016/j.immuni.2013.01.008
31. Wiggins KA, Parry AJ, Cassidy LD, Humphry M, Webster SJ, Goodall JC, et al. IL-1α cleavage by inflammatory caspases of the noncanonical inflammasome controls the senescence-associated secretory phenotype. Aging Cell (2019) 18:e12946. doi: 10.1111/acel.12946
32. Mizutani H, Schechter N, Lazarus G, Black RA, Kupper TS. Rapid and specific conversion of precursor interleukin 1 beta (IL-1 beta) to an active IL-1 species by human mast cell chymase. J Exp Med (1991) 174:821–5. doi: 10.1084/jem.174.4.821
33. Lefrançais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard J-P, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci U S A (2012) 109:1673–8. doi: 10.1073/pnas.1115884109
34. Omoto Y, Tokime K, Yamanaka K, Habe K, Morioka T, Kurokawa I, et al. Human mast cell chymase cleaves pro-IL-18 and generates a novel and biologically active IL-18 fragment. J Immunol (2006) 177:8315–9. doi: 10.4049/jimmunol.177.12.8315
35. Rivers-Auty J, Daniels MJD, Colliver I, Robertson DL, Brough D. Redefining the ancestral origins of the interleukin-1 superfamily. Nat Commun (2018) 9:1156. doi: 10.1038/s41467-018-03362-1
36. Burzynski LC, Humphry M, Pyrillou K, Wiggins KA, Chan JNE, Figg N, et al. The Coagulation and Immune Systems Are Directly Linked through the Activation of Interleukin-1α by Thrombin. Immunity (2019) 50:1033–1042.e6. doi: 10.1016/j.immuni.2019.03.003
37. Sims JE, Smith DE. The IL-1 family: Regulators of immunity. Nat Rev Immunol (2010) 10:89–102. doi: 10.1038/nri2691
38. Garlanda C, Dinarello CA, Mantovani A. The Interleukin-1 Family: Back to the Future. Immunity (2013) 39:1003–18. doi: 10.1016/j.immuni.2013.11.010
39. Janeway. CA. How the immune system works to protect the host from infection: a personal view. Proc Natl Acad Sci U S A (2001) 98:7461–8. doi: 10.1073/pnas.131202998
40. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol (2010) 31:318–24. doi: 10.1016/j.it.2010.05.006
41. Lahoz-Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, et al. Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood (2016) 127:3431–8. doi: 10.1182/blood-2016-03-700336
42. von Vietinghoff S, Ley K. IL-17A controls IL-17F production and maintains blood neutrophil counts in mice. J Immunol (2009) 183:4136. doi: 10.4049/jimmunol.0990072
43. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity (2009) 30:576–87. doi: 10.1016/j.immuni.2009.02.007
44. Sutton C, Brereton C, Keogh B, Mills KHGG, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med (2006) 203:1685–91. doi: 10.1084/jem.20060285
45. Mills KH, Dungan LS, Jones SA, Harris J. The role of inflammasome-derived IL-1 in driving IL-17 responses. J Leukoc Biol (2013) 93:489–97. doi: 10.1189/jlb.1012543
46. Ueda Y, Cain DW, Kuraoka M, Kondo M, Kelsoe G. IL-1R Type I-Dependent Hemopoietic Stem Cell Proliferation Is Necessary for Inflammatory Granulopoiesis and Reactive Neutrophilia. J Immunol (2009) 182:6477. doi: 10.4049/jimmunol.0803961
47. Yoshimura T, Matsushima K, Oppenheim JJ, Leonard EJ. Neutrophil chemotactic factor produced by lipopolysaccharide (LPS)-stimulated human blood mononuclear leukocytes: partial characterization and separation from interleukin 1 (IL 1). J Immunol (1987) 139:788.
48. Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol (2011) 32:452–60. doi: 10.1016/j.it.2011.06.008
49. Sica A, Matsushima K, Van Damme J, Wang JM, Polentarutti N, Dejana E, et al. IL-1 transcriptionally activates the neutrophil chemotactic factor/IL-8 gene in endothelial cells. Immunology (1990) 69:548–53.
50. Larsen CG, Anderson AO, Oppenheim JJ, Matsushima K. Production of interleukin-8 by human dermal fibroblasts and keratinocytes in response to interleukin-1 or tumour necrosis factor. Immunology (1989) 68:31–6.
51. Oliveira SH, Canetti C, Ribeiro RA, Cunha FQ. Neutrophil migration induced by IL-1beta depends upon LTB4 released by macrophages and upon TNF-alpha and IL-1beta released by mast cells. Inflammation (2008) 31:36–46. doi: 10.1007/s10753-007-9047-x
52. Biancone L, Tetta C, Turello E, Montrucchio G, Iorio EL, Servillo L, et al. Platelet-activating factor biosynthesis by cultured mesangial cells is modulated by proteinase inhibitors. J Am Soc Nephrol (1992) 2:1251.
53. Zenobia C, Hajishengallis G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol 2000 (2015) 69:142–59. doi: 10.1111/prd.12083
54. Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, Costantini C, et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood (2010) 115:335–43. doi: 10.1182/blood-2009-04-216085
55. Lee PY, Kumagai Y, Xu Y, Li Y, Barker T, Liu C, et al. IL-1α Modulates Neutrophil Recruitment in Chronic Inflammation Induced by Hydrocarbon Oil. J Immunol (2011) 186:1747. doi: 10.4049/jimmunol.1001328
56. Smith RJ, Bowman BJ, Speziale SC. Interleukin-1 stimulates granule exocytosis from human neutrophils. Int J Immunopharmacol (1986) 8:33–40. doi: 10.1016/0192-0561(86)90070-6
57. Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, et al. Converting enzyme-independent release of tumor necrosis factor α and IL-1β from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci (1999) 96:6261. doi: 10.1073/pnas.96.11.6261
58. Clancy DM, Sullivan GP, Moran HBT, Henry CM, Reeves EP, McElvaney NG, et al. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep (2018) 22:2937–50. doi: 10.1016/j.celrep.2018.02.062
59. Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell (2007) 130:918–31. doi: 10.1016/j.cell.2007.07.009
60. Orth K, Xu Z, Mudgett MB, Bao ZQ, Palmer LE, Bliska JB, et al. Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science (80) (2000) 290:1594–7. doi: 10.1126/science.290.5496.1594
61. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (80) (2004) 303:1532. doi: 10.1126/science.1092385
62. Yipp BG, Kubes P. NETosis: how vital is it? Blood (2013) 122:2784–94. doi: 10.1182/blood-2013-04-457671
63. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol (2018) 3:eaar6676. doi: 10.1126/sciimmunol.aar6676
64. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol (2018) 3:eaar6689. doi: 10.1126/sciimmunol.aar6689
65. Meher AK, Spinosa M, Davis JP, Pope N, Laubach VE, Su G, et al. Novel Role of IL (Interleukin)-1β in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler Thromb Vasc Biol (2018) 38:843–53. doi: 10.1161/ATVBAHA.117.309897
66. Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, et al. Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PLoS One (2011) 6:e29318–8. doi: 10.1371/journal.pone.0029318
67. Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation (2018) 138:898–912. doi: 10.1161/circulationaha.117.032636
68. Clancy DM, Henry CM, Sullivan GP, Martin SJ. Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J (2017) 284:1712–25. doi: 10.1111/febs.14075
69. Folco EJ, Mawson TL, Vromman A, Bernardes-Souza B, Franck G, Persson O, et al. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1α and Cathepsin G. Arter Thromb Vasc Biol (2018) 38:1901–12. doi: 10.1161/atvbaha.118.311150
70. Liu D, Yang P, Gao M, Yu T, Shi Y, Zhang M, et al. NLRP3 activation induced by neutrophil extracellular traps sustains inflammatory response in the diabetic wound. Clin Sci (2019) 133:565–82. doi: 10.1042/cs20180600
71. Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation (2020) 141:1080–94. doi: 10.1161/circulationaha.119.043833
72. Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol (1999) 17:593–623. doi: 10.1146/annurev.immunol.17.1.593
73. Sokolovska A, Becker CE, Ip WK, Rathinam VA, Brudner M, Paquette N, et al. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nat Immunol (2013) 14:543–53. doi: 10.1038/ni.2595
74. Yoon SJ, Jo DH, Park SH, Park JY, Lee YK, Lee MS, et al. Thioredoxin-Interacting Protein Promotes Phagosomal Acidification Upon Exposure to Escherichia coli Through Inflammasome-Mediated Caspase-1 Activation in Macrophages. Front Immunol (2019) 10:2636. doi: 10.3389/fimmu.2019.02636
75. Cohen TS, Boland ML, Boland BB, Takahashi V, Tovchigrechko A, Lee Y, et al. S. aureus Evades Macrophage Killing through NLRP3-Dependent Effects on Mitochondrial Trafficking. Cell Rep (2018) 22:2431–41. doi: 10.1016/j.celrep.2018.02.027
76. Kurt-Jones EA, Beller DI, Mizel SB, Unanue ER. Identification of a membrane-associated interleukin 1 in macrophages. Proc Natl Acad Sci (1985) 82:1204. doi: 10.1073/pnas.82.4.1204
77. Chan JNE, Humphry M, Kitt L, Krzyzanska D, Filbey KJ, Bennett MR, et al. Cell surface IL-1α trafficking is specifically inhibited by interferon-γ, and associates with the membrane via IL-1R2 and GPI anchors. Eur J Immunol (2020) 50:1663–75. doi: 10.1002/eji.201948521
78. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol (2009) 183:6469–77. doi: 10.4049/jimmunol.0901575
79. Palmer G, Lipsky BP, Smithgall MD, Meininger D, Siu S, Talabot-Ayer D, et al. The IL-1 receptor accessory protein (AcP) is required for IL-33 signaling and soluble AcP enhances the ability of soluble ST2 to inhibit IL-33. Cytokine (2008) 42:358–64. doi: 10.1016/j.cyto.2008.03.008
80. Awad F, Assrawi E, Jumeau C, Georgin-Lavialle S, Cobret L, Duquesnoy P, et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS One (2017) 12:e0175336. doi: 10.1371/journal.pone.0175336
81. Qing L, Fu J, Wu P, Zhou Z, Yu F, Tang J. Metformin induces the M2 macrophage polarization to accelerate the wound healing via regulating AMPK/mTOR/NLRP3 inflammasome singling pathway. Am J Transl Res (2019) 11:655–68.
82. Zhao P, Zhou W, Zhang Y, Li J, Zhao Y, Pan L, et al. Aminooxyacetic acid attenuates post-infarct cardiac dysfunction by balancing macrophage polarization through modulating macrophage metabolism in mice. J Cell Mol Med (2020) 24:2593–609. doi: 10.1111/jcmm.14972
83. Lin LR, Liu W, Zhu XZ, Chen YY, Gao ZX, Gao K, et al. Treponema pallidum promotes macrophage polarization and activates the NLRP3 inflammasome pathway to induce interleukin-1β production. BMC Immunol (2018) 19:28. doi: 10.1186/s12865-018-0265-9
84. Liu Y, Gao X, Miao Y, Wang Y, Wang H, Cheng Z, et al. NLRP3 regulates macrophage M2 polarization through up-regulation of IL-4 in asthma. Biochem J (2018) 475:1995–2008. doi: 10.1042/BCJ20180086
85. Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol (2010) 30:2341–9. doi: 10.1161/ATVBAHA.110.207522
86. Kaplanski G, Porat R, Aiura K, Erban JK, Gelfand JA, Dinarello CA. Activated platelets induce endothelial secretion of interleukin-8 in vitro via an interleukin-1-mediated event. Blood (1993) 81:2492–5.
87. Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science (80) (2010) 327:580–3. doi: 10.1126/science.1181928
88. Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM, Rothwell NJ. Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood (2010) 115:3632–9. doi: 10.1182/blood-2009-11-252643
89. Vats R, Brzoska T, Bennewitz MF, Jimenez MA, Pradhan-Sundd T, Tutuncuoglu E, et al. Platelet Extracellular Vesicles Drive Inflammasome-IL-1β-Dependent Lung Injury in Sickle Cell Disease. Am J Respir Crit Care Med (2020) 201:33–46. doi: 10.1164/rccm.201807-1370OC
90. Rolfes V, Ribeiro LS, Hawwari I, Böttcher L, Rosero N, Maasewerd S, et al. Platelets Fuel the Inflammasome Activation of Innate Immune Cells. Cell Rep (2020) 31:107615. doi: 10.1016/j.celrep.2020.107615
91. Beaulieu LM, Lin E, Mick E, Koupenova M, Weinberg EO, Kramer CD, et al. Interleukin 1 receptor 1 and interleukin 1β regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler Thromb Vasc Biol (2014) 34:552–64. doi: 10.1161/ATVBAHA.113.302700
92. Brown GT, Narayanan P, Li W, Silverstein RL, McIntyre TM. Lipopolysaccharide stimulates platelets through an IL-1β autocrine loop. J Immunol (2013) 191:5196–203. doi: 10.4049/jimmunol.1300354
93. Clarke MCH, Savill J, Jones DB, Noble BS, Brown SB. Compartmentalized megakaryocyte death generates functional platelets committed to caspase-independent death. J Cell Biol (2003) 160:577–87. doi: 10.1083/jcb.200210111
94. De Botton S, Sabri S, Daugas E, Zermati Y, Guidotti JE, Hermine O, et al. Platelet formation is the consequence of caspase activation within megakaryocytes. Blood (2002) 100:1310–7. doi: 10.1182/blood-2002-03-0686
95. Nishimura S, Nagasaki M, Kunishima S, Sawaguchi A, Sakata A, Sakaguchi H, et al. IL-1alpha induces thrombopoiesis through megakaryocyte rupture in response to acute platelet needs. J Cell Biol (2015) 209:453–66. doi: 10.1083/jcb.201410052
96. Gery I, Handschumacher RE. Potentiation of the T lymphocyte response to mitogens. III. Properties of the mediator(s) from adherent cells. Cell Immunol (1974) 11:162–9. doi: 10.1016/0008-8749(74)90016-1
97. Aarden LA, Brunner TK, Cerottini J-C, Dayer J-M, de Weck AL, Dinarello CA, et al. Revised Nomenclature for Antigen-Nonspecific T Cell Proliferation and Helper Factors. J Immunol (1979) 123:2928–9.
98. Smith KA, Lachman LB, Oppenheim JJ, Favata MF. The functional relationship of the interleukins. J Exp Med (1980) 151:1551–6. doi: 10.1084/jem.151.6.1551
99. Shibuya K, Robinson D, Zonin F, Hartley SB, Macatonia SE, Somoza C, et al. IL-1 alpha and TNF-alpha are required for IL-12-induced development of Th1 cells producing high levels of IFN-gamma in BALB/c but not C57BL/6 mice. J Immunol (1998) 160:1708–16.
100. Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, et al. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A (2009) 106:7119–24. doi: 10.1073/pnas.0902745106
101. Huber M, Beuscher HU, Rohwer P, Kurrle R, Rollinghoff M, Lowoff M. Costimulation via TCR and IL-1 reveals a novel autocrine pathway of T helper type 2 cell proliferation. Immunobiology (1998) 160:4242 LP –4247. doi: 10.1016/s0165-2478(97)87026-5
102. Schenten D, Nish SA, Yu S, Yan X, Lee HK, Brodsky I, et al. Signaling through the adaptor molecule MyD88 in CD4+ T cells is required to overcome suppression by regulatory T cells. Immunity (2014) 40:78–90. doi: 10.1016/j.immuni.2013.10.023
103. Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, et al. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J Exp Med (2013) 210:491–502. doi: 10.1084/jem.20122006
104. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol (2007) 8:942–9. doi: 10.1038/ni1496
105. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol (2007) 8:950–7. doi: 10.1038/ni1497
106. Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci U S A (2009) 106:13463–8. doi: 10.1073/pnas.0906988106
107. Horka H, Staudt V, Klein M, Taube C, Reuter S, Dehzad N, et al. The Tick Salivary Protein Sialostatin L Inhibits the Th9-Derived Production of the Asthma-Promoting Cytokine IL-9 and Is Effective in the Prevention of Experimental Asthma. J Immunol (2012) 188:2669–76. doi: 10.4049/jimmunol.1100529
108. Végran F, Berger H, Boidot R, Mignot G, Bruchard M, Dosset M, et al. The transcription factor IRF1 dictates the IL-21-dependent anticancer functions of T H9 cells. Nat Immunol (2014) 15:758–66. doi: 10.1038/ni.2925
109. Lichtman AH, Chin J, Schmidt JA, Abbas AK. Role of interleukin 1 in the activation of T lymphocytes. Proc Natl Acad Sci U S A (1988) 85:9699–703. doi: 10.1073/pnas.85.24.9699
110. Taylor-Robinson AW, Phillips RS. Expression of the IL-1 receptor discriminates Th2 from Th1 cloned CD4+ T cells specific for Plasmodium chabaudi. Immunology (1994) 81:216–21.
111. Cosmi L, Maggi L, Santarlasci V, Capone M, Cardilicchia E, Frosali F, et al. Identification of a novel subset of human circulating memory CD4+ T cells that produce both IL-17A and IL-4. J Allergy Clin Immunol (2010) 125:222–30. doi: 10.1016/j.jaci.2009.10.012
112. Wang YH, Voo KS, Liu B, Chen CY, Uygungil B, Spoede W, et al. A novel subset of CD4+ TH2 memory/ effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J Exp Med (2010) 207:2479–91. doi: 10.1084/jem.20101376
113. Lee W-WW, Kang SW, Choi J, Lee S-HH, Shah K, Eynon EE, et al. Regulating human Th17 cells via differential expression of IL-1 receptor. Blood (2010) 115:530–40. doi: 10.1182/blood-2009-08-236521
114. Wesche H, Korherr C, Kracht M, Falk W, Resch K, Martin MU. The interleukin-1 receptor accessory protein (IL-1RAcP) is essential for IL-1-induced activation of interleukin-1 receptor-associated kinase (IRAK) and stress-activated protein kinases (SAP kinases). J Biol Chem (1997) 272:7727–31. doi: 10.1074/jbc.272.12.7727
115. Korherr C, Hofmeister R, Wesche H, Falk W. A critical role for interleukin-1 receptor accessory protein in interleukin-1 signaling. Eur J Immunol (1997) 27:262–7. doi: 10.1002/eji.1830270139
116. Mansur IG, Schiavon V, Giustiniani J, Bagot M, Bensussan A, Marie-Cardine A. Engagement of IL-1 receptor accessory protein (IL-1RAcP) with the monoclonal antibody AY19 provides co-activating signals and prolongs the CD2-induced proliferation of peripheral blood lymphocytes. Immunol Lett (2011) 139:52–7. doi: 10.1016/j.imlet.2011.04.015
117. Xu D, Chan WL, Leung BP, Huang FP, Wheeler R, Piedrafita D, et al. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J Exp Med (1998) 187:787–94. doi: 10.1084/jem.187.5.787
118. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
119. Ritvo P-GG, Churlaud G, Quiniou V, Florez L, Brimaud F, Fourcade G, et al. T fr cells lack IL-2Rα but express decoy IL-1R2 and IL-1Ra and suppress the IL-1–dependent activation of T fh cells. Sci Immunol (2017) 2:eaan0368. doi: 10.1126/sciimmunol.aan0368
120. Vinuesa CG, Linterman MA, Yu D, Maclennan ICM. Follicular Helper T Cells. Annu Rev Immunol (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
121. Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, et al. T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature (2015) 517:214–8. doi: 10.1038/nature13803
122. Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, et al. Germinal centers: Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science (80) (2014) 345:1058–62. doi: 10.1126/science.1257861
123. Muñoz-Wolf N, Lavelle EC. A Guide to IL-1 family cytokines in adjuvanticity. FEBS J (2018) 285:2377–401. doi: 10.1111/febs.14467
124. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435:452–8. doi: 10.1038/nature03555
125. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, et al. Persistent Antigen and Germinal Center B Cells Sustain T Follicular Helper Cell Responses and Phenotype. Immunity (2013) 38:596–605. doi: 10.1016/j.immuni.2012.11.020
126. Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med (2007) 13:108–16. doi: 10.1016/j.molmed.2007.01.003
127. Sakaguchi S, Wing K, Onishi A, Prieto-Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int Immunol (2009) 21:1105–11. doi: 10.1093/intimm/dxp095
128. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17:975–82. doi: 10.1038/nm.2425
129. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med (2011) 17:983–8. doi: 10.1038/nm.2426
130. Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity (2016) 45:1122–34. doi: 10.1016/j.immuni.2016.10.032
131. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity (2016) 45:1135–47. doi: 10.1016/j.immuni.2016.10.021
132. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1|[beta]||[ndash]|dependent adaptive immunity against tumors. Nat Med Publ (2009) 15:1170. doi: 10.1038/nm.2028. online 20 Sept 2009; | doi101038/nm2028
133. Michelini S, Sarajlic M, Duschl A, Horejs-Hoeck J. IL-1β induces expression of costimulatory molecules and cytokines but not immune feedback regulators in dendritic cells. Hum Immunol (2018) 79:610–5. doi: 10.1016/j.humimm.2018.06.002
134. Luft T, Jefford M, Luetjens P, Hochrein H, Masterman K-A, Maliszewski C, et al. IL-1β Enhances CD40 Ligand-Mediated Cytokine Secretion by Human Dendritic Cells (DC): A Mechanism for T Cell-Independent DC Activation. J Immunol (2002) 168:713–22. doi: 10.4049/jimmunol.168.2.713
135. Matjeka T, Summerfield V, Noursadeghi M, Chain BM. Chemical toxicity to keratinocytes triggers dendritic cell activation via an IL-1α path. J Allergy Clin Immunol (2012) 129:247–50. doi: 10.1016/j.jaci.2011.08.018
136. Wesa AK, Galy A. IL-1β induces dendritic cells to produce IL-12. Int Immunol (2001) 13:1053–61. doi: 10.1093/intimm/13.8.1053
137. Sallusto F, Cella M, Danieli C, Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: Downregulation by cytokines and bacterial products. J Exp Med (1995) 182:389–400. doi: 10.1084/jem.182.2.389
138. Anttila HS, Reitamo S, Erkko P, Miettinen A, Didierjean L, Saurat JH. Membrane and cytosolic interleukin-1 alpha and beta in normal human epidermal cells: variability of epitope exposure in immunohistochemistry. J Invest Dermatol (1990) 95:31–8. doi: 10.1111/1523-1747.ep12873278
139. Palazon-Riquelme P, Lopez-Castejon G. The inflammasomes, immune guardians at defence barriers. Immunology (2018) 155:320–30. doi: 10.1111/imm.12989
140. Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol (2007) 17:1140–5. doi: 10.1016/j.cub.2007.05.074
141. Dai X, Tohyama M, Murakami M, Sayama K. Epidermal keratinocytes sense dsRNA via the NLRP3 inflammasome, mediating interleukin (IL)-1β and IL-18 release. Exp Dermatol (2017) 26:904–11. doi: 10.1111/exd.13334
142. Orzalli MH, Smith A, Jurado KA, Iwasaki A, Garlick JA, Kagan JC. An Antiviral Branch of the IL-1 Signaling Pathway Restricts Immune-Evasive Virus Replication. Mol Cell (2018) 71:825–40. doi: 10.1016/j.molcel.2018.07.009
143. Beales IL, Calam J. Interleukin 1 beta and tumour necrosis factor alpha inhibit acid secretion in cultured rabbit parietal cells by multiple pathways. Gut (1998) 42:227–34. doi: 10.1136/gut.42.2.227
144. Wallace JL, Cucala M, Mugridge K, Parente L. Secretagogue-specific effects of interleukin-1 on gastric acid secretion. Am J Physiol (1991) 261:G559–64. doi: 10.1152/ajpgi.1991.261.4.G559
145. Uehara A, Okumura T, Sekiya C, Okamura K, Takasugi Y, Namiki M. Interleukin-1 inhibits the secretion of gastric acid in rats: possible involvement of prostaglandin. Biochem Biophys Res Commun (1989) 162:1578–84. doi: 10.1016/0006-291x(89)90855-3
146. Wallace JL, Keenan CM, Cucala M, Mugridge KG, Parente L. Mechanisms underlying the protective effects of interleukin 1 in experimental nonsteroidal anti-inflammatory drug gastropathy. Gastroenterology (1992) 102:1176–85.
147. Wallace JL, Keenan CM, Mugridge KG, Parente L. Reduction of the severity of experimental gastric and duodenal ulceration by interleukin-1 beta. Eur J Pharmacol (1990) 186:279–84. doi: 10.1016/0014-2999(90)90444-b
148. Cohan VL, Scott AL, Dinarello CA, Prendergast RA. Interleukin-1 is a mucus secretagogue. Cell Immunol (1991) 136:425–34. doi: 10.1016/0008-8749(91)90364-h
149. Han V, Resau J, Prendergast R, Scott A, Levy DA. Interleukin-1 induces mucus secretion from mouse intestinal explants. Int Arch Allergy Appl Immunol (1987) 82:364–5. doi: 10.1159/000234228
150. Takahashi S, Nakamura E, Okabe S. Effects of cytokines, without and with Helicobacter pylori components, on mucus secretion by cultured gastric epithelial cells. Dig Dis Sci (1998) 43:2301–8. doi: 10.1023/a:1026635110099
151. Al-Sadi R, Ye D, Dokladny K, Ma TY. Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction permeability. J Immunol (2008) 180:5653–61. doi: 10.4049/jimmunol.180.8.5653
152. Al-Sadi R, Ye D, Said HM, Ma TY. Cellular and molecular mechanism of interleukin-1β modulation of Caco-2 intestinal epithelial tight junction barrier. J Cell Mol Med (2011) 15:970–82. doi: 10.1111/j.1582-4934.2010.01065.x
153. Viñuales C, Gascón S, Barranquero C, Osada J, Rodríguez-Yoldi MJ. Inhibitory effect of IL-1β on galactose intestinal absorption in rabbits. Cell Physiol Biochem (2012) 30:173–86. doi: 10.1159/000339056
154. García-Barrios A, Guillén N, Gascón S, Osada J, Vazquez CM, Miguel-Carrasco JL, et al. Nitric oxide involved in the IL-1β-induced inhibition of fructose intestinal transport. J Cell Biochem (2010) 111:1321–9. doi: 10.1002/jcb.22859
155. Rodríguez-Yoldi MJ, Gascón S, Barranquero C, García-Barrios A, Osada J. Involvement of intracellular signaling in the IL-1β inhibitory effect on fructose intestinal absorption. J Cell Physiol (2015) 230:896–902. doi: 10.1002/jcp.24820
156. Dmitrieva-Posocco O, Dzutsev A, Posocco DF, Hou V, Yuan W, Thovarai V, et al. Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity (2019) 50:166–80.e7. doi: 10.1016/j.immuni.2018.11.015
157. Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology (2003) 8:432–46. doi: 10.1046/j.1440-1843.2003.00493.x
158. Gray T, Coakley R, Hirsh A, Thornton D, Kirkham S, Koo JS, et al. Regulation of MUC5AC mucin secretion and airway surface liquid metabolism by IL-1beta in human bronchial epithelia. Am J Physiol Lung Cell Mol Physiol (2004) 286:L320–30. doi: 10.1152/ajplung.00440.2002
159. Chen G, Sun L, Kato T, Okuda K, Martino MB, Abzhanova A, et al. IL-1β dominates the promucin secretory cytokine profile in cystic fibrosis. J Clin Invest (2019) 129:4433–50. doi: 10.1172/jci125669
160. Katsura H, Kobayashi Y, Tata PR, Hogan BLM. IL-1 and TNFα Contribute to the Inflammatory Niche to Enhance Alveolar Regeneration. Stem Cell Rep (2019) 12:657–66. doi: 10.1016/j.stemcr.2019.02.013
161. Guo XJ, Thomas PG. New fronts emerge in the influenza cytokine storm. Semin Immunopathol (2017) 39:541–50. doi: 10.1007/s00281-017-0636-y
162. Michiels C. Endothelial cell functions. J Cell Physiol (2003) 196:430–43. doi: 10.1002/jcp.10333
163. Erdei J, Tóth A, Balogh E, Nyakundi BB, Bányai E, Ryffel B, et al. Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxid Med Cell Longev (2018) 2018:4310816. doi: 10.1155/2018/4310816
164. Bevilacqua MP, Pober JS, Majeau GR, Cotran RS, Gimbrone MA Jr. Interleukin 1 (IL-1) induces biosynthesis and cell surface expression of procoagulant activity in human vascular endothelial cells. J Exp Med (1984) 160:618–23. doi: 10.1084/jem.160.2.618
165. Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA Jr. Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J Clin Invest (1985) 76:2003–11. doi: 10.1172/jci112200
166. Woodfin A, Reichel CA, Khandoga A, Corada M, Voisin MB, Scheiermann C, et al. JAM-A mediates neutrophil transmigration in a stimulus-specific manner in vivo: evidence for sequential roles for JAM-A and PECAM-1 in neutrophil transmigration. Blood (2007) 110:1848–56. doi: 10.1182/blood-2006-09-047431
167. Ferreira AM, McNeil CJ, Stallaert KM, Rogers KA, Sandig M. Interleukin-1beta reduces transcellular monocyte diapedesis and compromises endothelial adherens junction integrity. Microcirculation (2005) 12:563–79. doi: 10.1080/10739680500253493
168. Labus J, Wöltje K, Stolte KN, Häckel S, Kim KS, Hildmann A, et al. IL-1β promotes transendothelial migration of PBMCs by upregulation of the FN/α(5)β(1) signalling pathway in immortalised human brain microvascular endothelial cells. Exp Cell Res (2018) 373:99–111. doi: 10.1016/j.yexcr.2018.10.002
169. Ebisawa M, Bochner BS, Georas SN, Schleimer RP. Eosinophil transendothelial migration induced by cytokines. I. Role of endothelial and eosinophil adhesion molecules in IL-1 beta-induced transendothelial migration. J Immunol (1992) 149:4021–8.
170. Dangerfield J, Larbi KY, Huang MT, Dewar A, Nourshargh S. PECAM-1 (CD31) homophilic interaction up-regulates alpha6beta1 on transmigrated neutrophils in vivo and plays a functional role in the ability of alpha6 integrins to mediate leukocyte migration through the perivascular basement membrane. J Exp Med (2002) 196:1201–11. doi: 10.1084/jem.20020324
171. Carmi Y, Voronov E, Dotan S, Lahat N, Rahat MA, Fogel M, et al. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J Immunol (2009) 183:4705–14. doi: 10.4049/jimmunol.0901511
172. Carmi Y, Dotan S, Rider P, Kaplanov I, White MR, Baron R, et al. The role of IL-1β in the early tumor cell-induced angiogenic response. J Immunol (2013) 190:3500–9. doi: 10.4049/jimmunol.1202769
173. Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A (2003) 100:2645–50. doi: 10.1073/pnas.0437939100
174. Bar D, Apte RN, Voronov E, Dinarello CA, Cohen S. A continuous delivery system of IL-1 receptor antagonist reduces angiogenesis and inhibits tumor development. FASEB J (2004) 18:161–3. doi: 10.1096/fj.03-0483fje
175. Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov (2012) 11:633–52. doi: 10.1038/nrd3800
176. Zhao R, Liang H, Clarke E, Jackson C, Xue M. Inflammation in Chronic Wounds. Int J Mol Sci (2016) 17:2085. doi: 10.3390/ijms17122085
177. Lippi G, Franchini M, Targher G. Arterial thrombus formation in cardiovascular disease. Nat Rev Cardiol (2011) 8:502–12. doi: 10.1038/nrcardio.2011.91
178. Weinheimer-Haus EM, Mirza RE, Koh TJ. Nod-like receptor protein-3 inflammasome plays an important role during early stages of wound healing. PLoS One (2015) 10:e0119106. doi: 10.1371/journal.pone.0119106
179. Ito H, Kanbe A, Sakai H, Seishima M. Activation of NLRP3 signalling accelerates skin wound healing. Exp Dermatol (2018) 27:80–6. doi: 10.1111/exd.13441
180. Brauweiler AM, Goleva E, Leung DYM. Staphylococcus aureus Lipoteichoic Acid Damages the Skin Barrier through an IL-1-Mediated Pathway. J Invest Dermatol (2019) 139:1753–1761.e4. doi: 10.1016/j.jid.2019.02.006
181. Vinaik R, Abdullahi A, Barayan D, Jeschke MG. NLRP3 inflammasome activity is required for wound healing after burns. Transl Res (2020) 217:47–60. doi: 10.1016/j.trsl.2019.11.002
182. Mirza RE, Fang MM, Weinheimer-Haus EM, Ennis WJ, Koh TJ. Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice. Diabetes (2014) 63:1103–14. doi: 10.2337/db13-0927
183. Bitto A, Altavilla D, Pizzino G, Irrera N, Pallio G, Colonna MR, et al. Inhibition of inflammasome activation improves the impaired pattern of healing in genetically diabetic mice. Br J Pharmacol (2014) 171:2300–7. doi: 10.1111/bph.12557
184. Thomay AA, Daley JM, Sabo E, Worth PJ, Shelton LJ, Harty MW, et al. Disruption of interleukin-1 signaling improves the quality of wound healing. Am J Pathol (2009) 174:2129–36. doi: 10.2353/ajpath.2009.080765
185. Perrault DP, Bramos A, Xu X, Shi S, Wong AK. Local Administration of Interleukin-1 Receptor Antagonist Improves Diabetic Wound Healing. Ann Plast Surg (2018) 80:S317–s321. doi: 10.1097/sap.0000000000001417
186. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
187. Yadav V, Chi L, Zhao R, Tourdot BE, Yalavarthi S, Jacobs BN, et al. Ectonucleotidase tri(di)phosphohydrolase-1 (ENTPD-1) disrupts inflammasome/interleukin 1β-driven venous thrombosis. J Clin Invest (2019) 129:2872–7. doi: 10.1172/jci124804
188. Kambas K, Mitroulis I, Apostolidou E, Girod A, Chrysanthopoulou A, Pneumatikos I, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS One (2012) 7:e45427. doi: 10.1371/journal.pone.0045427
189. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
190. Kambas K, Mitroulis I, Ritis K. The emerging role of neutrophils in thrombosis-the journey of TF through NETs. Front Immunol (2012) 3:385. doi: 10.3389/fimmu.2012.00385
191. Kambas K, Markiewski MM, Pneumatikos IA, Rafail SS, Theodorou V, Konstantonis D, et al. C5a and TNF-alpha up-regulate the expression of tissue factor in intra-alveolar neutrophils of patients with the acute respiratory distress syndrome. J Immunol (2008) 180:7368–75. doi: 10.4049/jimmunol.180.11.7368
192. Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Hear J (2015) 36:1405–14. doi: 10.1093/eurheartj/ehv007
193. Liberale L, Holy EW, Akhmedov A, Bonetti NR, Nietlispach F, Matter CM, et al. Interleukin-1β Mediates Arterial Thrombus Formation via NET-Associated Tissue Factor. J Clin Med (2019) 8:2072. doi: 10.3390/jcm8122072
194. Gomes T, Várady CBS, Lourenço AL, Mizurini DM, Rondon AMR, Leal AC, et al. IL-1β Blockade Attenuates Thrombosis in a Neutrophil Extracellular Trap-Dependent Breast Cancer Model. Front Immunol (2019) 10:2088. doi: 10.3389/fimmu.2019.02088
195. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity (2019) 50:1401–1411.e4. doi: 10.1016/j.immuni.2019.04.003
196. Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity (2019) 51:983–96. doi: 10.1016/j.immuni.2019.11.005
197. Quintin J, Cheng SC, van der Meer JWM, Netea MG. Innate immune memory: Towards a better understanding of host defense mechanisms. Curr Opin Immunol (2014) 29:1–7. doi: 10.1016/j.coi.2014.02.006
198. Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science (80) (2014) 345:1251086. doi: 10.1126/science.1251086
199. Arts RJW, Joosten LAB, Netea MG. Immunometabolic circuits in trained immunity. Semin Immunol (2016) 28:425–30. doi: 10.1016/j.smim.2016.09.002
200. Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med (2012) 209:2351–65. doi: 10.1084/jem.20120944
201. Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A (2009) 106:1915–9. doi: 10.1073/pnas.0813192106
202. van den Boorn JG, Jakobs C, Hagen C, Renn M, Luiten RM, Melief CJM, et al. Inflammasome-Dependent Induction of Adaptive NK Cell Memory. Immunity (2016) 44:1406–21. doi: 10.1016/j.immuni.2016.05.008
203. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Ifrim DC, Saeed S, et al. Bacille Calmette-Guérin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A (2012) 109:17537–42. doi: 10.1073/pnas.1202870109
204. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. MTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (80) (2014) 345:1250684. doi: 10.1126/science.1250684
205. Domínguez-Andrés J, Novakovic B, Li Y, Scicluna BP, Gresnigt MS, Arts RJW, et al. The Itaconate Pathway Is a Central Regulatory Node Linking Innate Immune Tolerance and Trained Immunity. Cell Metab (2019) 29:211–220.e5. doi: 10.1016/j.cmet.2018.09.003
206. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell (2018) 172:135–146.e9. doi: 10.1016/j.cell.2017.11.025
207. Arts RJW, Novakovic B, ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab (2016) 24:807–19. doi: 10.1016/j.cmet.2016.10.008
208. Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature (2013) 496:238–42. doi: 10.1038/nature11986
209. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell (2018) 172:147–61.e12. doi: 10.1016/j.cell.2017.11.034
210. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell (2018) 172:176–90.e19. doi: 10.1016/j.cell.2017.12.031
211. Arts RJW, Moorlag SJCFM, Novakovic B, Li Y, Wang SY, Oosting M, et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe (2018) 23:89–100.e5. doi: 10.1016/j.chom.2017.12.010
212. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell (2018) 172:162–175.e14. doi: 10.1016/j.cell.2017.12.013
213. Bekkering S, Quintin J, Joosten LAB, Van Der Meer JWM, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887
214. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol (2016) 18:607–18. doi: 10.1038/ncb3346
215. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature (2018) 556:113–7. doi: 10.1038/nature25986
216. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab (2016) 24:158–66. doi: 10.1016/j.cmet.2016.06.004
217. O’Neill LAJ, Artyomov MN. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol (2019) 19:273–81. doi: 10.1038/s41577-019-0128-5
218. Haley MJ, Brough D, Quintin J, Allan SM. Microglial Priming as Trained Immunity in the Brain. Neuroscience (2019) 405:47–54. doi: 10.1016/j.neuroscience.2017.12.039
219. Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol (2005) 5:629–40. doi: 10.1038/nri1664
220. Vitkovic L, Bockaert J, Jacque C. “Inflammatory” cytokines’ neuromodulators in normal brain? J Neurochem (2000) 74:457–71. doi: 10.1046/j.1471-4159.2000.740457.x
221. Ching S, He L, Lai W, Quan N. IL-1 type I receptor plays a key role in mediating the recruitment of leukocytes into the central nervous system. Brain Behav Immun (2005) 19:127–37. doi: 10.1016/j.bbi.2004.06.001
222. Jain A, Song R, Wakeland EK, Pasare C. T cell-intrinsic IL-1R signaling licenses effector cytokine production by memory CD4 T cells. Nat Commun (2018) 9:1–13. doi: 10.1038/s41467-018-05489-7
223. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 Induce Innate IL-17 Production from γδ T Cells, Amplifying Th17 Responses and Autoimmunity. Immunity (2009) 31:331–41. doi: 10.1016/j.immuni.2009.08.001
224. Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci U S A (2003) 100:5986–90. doi: 10.1073/pnas.1035999100
225. Matsuki T, Nakae S, Sudo K, Horai R, Iwakura Y. Abnormal T cell activation caused by the imbalance of the IL-1/IL-1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int Immunol (2006) 18:399–407. doi: 10.1093/intimm/dxh379
226. Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An Autoinflammatory Disease with Deficiency of the Interleukin-1–Receptor Antagonist. N Engl J Med (2009) 360:2426–37. doi: 10.1056/nejmoa0807865
227. Gulen MF, Kang Z, Bulek K, Youzhong W, Kim TW, Chen Y, et al. The Receptor SIGIRR Suppresses Th17 Cell Proliferation via Inhibition of the Interleukin-1 Receptor Pathway and mTOR Kinase Activation. Immunity (2010) 32:54–66. doi: 10.1016/j.immuni.2009.12.003
228. Comabella M, Julià E, Tintoré M, Brieva L, Téllez N, Río J, et al. Induction of serum soluble tumor necrosis factor receptor II (sTNF-RII) and interleukin-1 receptor antagonist (IL-1ra) by interferon beta-1b in patients with progressive multiple sclerosis. J Neurol (2008) 255:1136–41. doi: 10.1007/s00415-008-0855-1
229. Nicoletti F, Patti F, Di Marco R, Zaccone P, Nicoletti A, Meroni PL, et al. Circulating serum levels of IL-1ra in patients with relapsing remitting multiple sclerosis are normal during remission phases but significantly increased either during exacerbations or in response to IFN-β treatment. Cytokine (1996) 8:395–400. doi: 10.1006/cyto.1996.0054
230. Dujmovic I, Mangano K, Pekmezovic T, Quattrocchi C, Mesaros S, Stojsavljevic N, et al. The analysis of IL-1 beta and its naturally occurring inhibitors in multiple sclerosis: The elevation of IL-1 receptor antagonist and IL-1 receptor type II after steroid therapy. J Neuroimmunol (2009) 207:101–6. doi: 10.1016/j.jneuroim.2008.11.004
231. De Jong BA, Huizinga TWJ, Bollen ELEM, Uitdehaag BMJ, Bosma GPT, Van Buchem MA, et al. Production of IL-1β and IL-1Ra as risk factors for susceptibility and progression of relapse-onset multiple sclerosis. J Neuroimmunol (2002) 126:172–9. doi: 10.1016/S0165-5728(02)00056-5
232. Jha S, Srivastava SY, Brickey WJ, Iocca H, Toews A, Morrison JP, et al. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci (2010) 30:15811–20. doi: 10.1523/JNEUROSCI.4088-10.2010
233. Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, et al. NLRP3 Plays a Critical Role in the Development of Experimental Autoimmune Encephalomyelitis by Mediating Th1 and Th17 Responses. J Immunol (2010) 185:974–81. doi: 10.4049/jimmunol.0904145
234. Hisahara S, Yuan J, Momoi T, Okano H, Miura M. Caspase-11 mediates oligodendrocyte cell death and pathogenesis of autoimmune-mediated demyelination. J Exp Med (2001) 193:111–22. doi: 10.1084/jem.193.1.111
235. Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, et al. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol (1999) 163:2403–9.
236. Jain A, Irizarry-Caro RA, McDaniel MM, Chawla AS, Carroll KR, Overcast GR, et al. T cells instruct myeloid cells to produce inflammasome-independent IL-1β and cause autoimmunity. Nat Immunol (2020) 21:69. doi: 10.1038/s41590-019-0559-y
237. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular Antagonism and Plasticity of Regulatory and Inflammatory T Cell Programs. Immunity (2008) 29:44–56. doi: 10.1016/j.immuni.2008.05.007
238. Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature (2007) 445:766–70. doi: 10.1038/nature05479
239. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol (2009) 10:1000–7. doi: 10.1038/ni.1774
240. O’Sullivan BJ, Thomas HE, Pai S, Santamaria P, Iwakura Y, Steptoe RJ, et al. IL-1β Breaks Tolerance through Expansion of CD25 + Effector T Cells. J Immunol (2006) 176:7278–87. doi: 10.4049/jimmunol.176.12.7278
241. Atkinson MA, Eisenbarth GS. Type 1 diabetes: New perspectives on disease pathogenesis and treatment. Lancet (2001) 358:221–9. doi: 10.1016/S0140-6736(01)05415-0
242. Donath MY, Dinarello CA, Mandrup-Poulsen T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat Rev Immunol (2019) 19:734–46. doi: 10.1038/s41577-019-0213-9
243. Böni-Schnetzler M, Boller S, Debray S, Bouzakri K, Meier DT, Prazak R, et al. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology (2009) 150:5218–29. doi: 10.1210/en.2009-0543
244. Donath MY, Størling J, Berchtold LA, Billestrup N, Mandrup-Poulsen T. Cytokines and β-cell biology: From concept to clinical translation. Endocr Rev (2008) 29:334–50. doi: 10.1210/er.2007-0033
245. Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes (2007) 56:2356–70. doi: 10.2337/db06-1650
246. Dotson AL, Novikova L, Stehno-Bittel L, Benedict SH. Elimination of T cell reactivity to pancreatic β cells and partial preservation of β cell activity by peptide blockade of LFA-1: ICAM-1 interaction in the NOD mouse model. Clin Immunol (2013) 148:149–61. doi: 10.1016/j.clim.2013.04.016
247. Bendtzen K, Mandrup-Poulsen T, Nerup J, Nielsen JH, Dinarello CA, Svenson M. Cytotoxicity of human pI 7 interleukin-1 for pancreatic islets of Langerhans. Science (80) (1986) 232:1545–7. doi: 10.1126/science.3086977
248. Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, et al. Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J Clin Invest (2002) 110:851–60. doi: 10.1172/jci15318
249. Arnush M, Scarim AL, Heitmeier MR, Kelly CB, Corbett JA. Potential role of resident islet macrophage activation in the initiation of autoimmune diabetes. J Immunol (1998) 160:2684–91.
250. Corbett JA, Kwon G, Misko TP, Rodi CP, McDaniel ML. Tyrosine kinase involvement in IL-1β-induced expression of iNOS by β- cells purified from islets of Langerhans. Am J Physiol Cell Physiol (1994) 267:48–54. doi: 10.1152/ajpcell.1994.267.1.c48
251. Meyerovich K, Fukaya M, Terra LF, Ortis F, Eizirik DL, Cardozo AK. The non-canonical NF-κB pathway is induced by cytokines in pancreatic beta cells and contributes to cell death and proinflammatory responses in vitro. Diabetologia (2016) 59:512–21. doi: 10.1007/s00125-015-3817-z
252. Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med (2007) 356:1517–26. doi: 10.1056/NEJMoa065213
253. Abramson SB, Amin A. Blocking the effects of IL-1 in rheumatoid arthritis protects bone and cartilage. Rheumatology (2002) 41:972–80. doi: 10.1093/rheumatology/41.9.972
254. Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin I receptor antagonist-deficient mice. J Exp Med (2000) 191:313–20. doi: 10.1084/jem.191.2.313
255. Bau B, Gebhard PM, Haag J, Knorr T, Bartnik E, Aigner T. Relative messenger RNA expression profiling of collagenases and aggrecanases in human articular chondrocytes in vivo and in vitro. Arthritis Rheum (2002) 46:2648–57. doi: 10.1002/art.10531
256. Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor κB: Differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum (2000) 43:801–11. doi: 10.1002/1529-0131(200004)43:4<801::AID-ANR10>3.0.CO;2-4
257. Fan Z, Söder S, Oehler S, Fundel K, Aigner T. Activation of interleukin-1 signaling cascades in normal and osteoarthritis articular cartilage. Am J Pathol (2007) 171:938–46. doi: 10.2353/ajpath.2007.061083
258. Theill LE, Boyle WJ, Penninger JM. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol (2002) 20:795–823. doi: 10.1146/annurev.immunol.20.100301.064753
259. Bessis N, Guéry L, Mantovani A, Vecchi A, Sims JE, Fradelizi D, et al. The type II decoy receptor of IL-1 inhibits murine collagen-induced arthritis. Eur J Immunol (2000) 30:867–75. doi: 10.1002/1521-4141(200003)30:3<867::AID-IMMU867>3.0.CO;2-M
260. Makarov SS, Olsen JC, Johnston WN, Anderle SK, Brown RR, Baldwin AS, et al. Suppression of experimental arthritis by gene transfer of interleukin 1 receptor antagonist cDNA. Proc Natl Acad Sci U S A (1996) 93:402–6. doi: 10.1073/pnas.93.1.402
261. Fleischmann R, Tesser J, Schiff MH, Schechtman J, Burmester GR, Bennett R, et al. Safety of extended treatment with anakinra in patients with rheumatoid arthritis. Ann Rheum Dis (2006) 65:1006–12. doi: 10.1136/ard.2005.048371
262. Xu D, Mu R, Wei X. The Roles of IL-1 Family Cytokines in the Pathogenesis of Systemic Sclerosis. Front Immunol (2019) 10:2025. doi: 10.3389/fimmu.2019.02025
263. Kawaguchi Y, Hara M, Wright TM. Endogenous IL-1α from systemic sclerosis fibroblasts induces IL-6 and PDGF-A. J Clin Invest (1999) 103:1253–60. doi: 10.1172/JCI4304
264. Maleszewska M, Moonen JRAJ, Huijkman N, van de Sluis B, Krenning G, Harmsen MC. IL-1β and TGFβ2 synergistically induce endothelial to mesenchymal transition in an NFκB-dependent manner. Immunobiology (2013) 218:443–54. doi: 10.1016/j.imbio.2012.05.026
265. Kawaguchi Y, McCarthy SA, Watkins SC, Wright TM. Autocrine activation by interleukin 1α induces the fibrogenic phenotype of systemic sclerosis fibroblasts. J Rheumatol (2004) 31:1946–54.
266. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat Med (2012) 18:1028–40. doi: 10.1038/nm.2807
267. Kirk TZ, Mayes MD. IL-1 rescues scleroderma myofibroblasts from serum-starvation-induced cell death. Biochem Biophys Res Commun (1999) 255:129–32. doi: 10.1006/bbrc.1999.0155
268. Artlett CM, Sassi-Gaha S, Hope JL, Feghali-Bostwick CA, Katsikis PD. Mir-155 is overexpressed in systemic sclerosis fibroblasts and is required for NLRP3 inflammasome-mediated collagen synthesis during fibrosis. Arthritis Res Ther (2017) 19:144. doi: 10.1186/s13075-017-1331-z
269. Artlett CM, Sassi-Gaha S, Rieger JL, Boesteanu AC, Feghali-Bostwick CA, Katsikis PD. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum (2011) 63:3563–74. doi: 10.1002/art.30568
270. Pottier N, Maurin T, Chevalier B, Puisségur MP, Lebrigand K, Robbe-Sermesant K, et al. Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: Implication in epithelial-mesenchymal interactions. PLoS One (2009) 4:e6718. doi: 10.1371/journal.pone.0006718
271. Coperchini F, Chiovato L, Croce L, Magri F, Rotondi M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev (2020) 53:25–32. doi: 10.1016/j.cytogfr.2020.05.003
272. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intens Care Med (2020) 46:846–8. doi: 10.1007/s00134-020-05991-x
273. Wang IH, Burckhardt CJ, Yakimovich A, Greber UF. Imaging, Tracking and Computational Analyses of Virus Entry and Egress with the Cytoskeleton. Viruses (2018) 10:166. doi: 10.3390/v10040166
274. Ravindran MS, Bagchi P, Cunningham CN, Tsai B. Opportunistic intruders: how viruses orchestrate ER functions to infect cells. Nat Rev Microbiol (2016) 14:407–20. doi: 10.1038/nrmicro.2016.60
275. Schoeman D, Fielding BC. Coronavirus envelope protein: current knowledge. Virol J (2019) 16:69. doi: 10.1186/s12985-019-1182-0
276. Nieva JL, Madan V, Carrasco L. Viroporins: structure and biological functions. Nat Rev Microbiol (2012) 10:563–74. doi: 10.1038/nrmicro2820
277. Liao Y, Lescar J, Tam JP, Liu DX. Expression of SARS-coronavirus envelope protein in Escherichia coli cells alters membrane permeability. Biochem Biophys Res Commun (2004) 325:374–80. doi: 10.1016/j.bbrc.2004.10.050
278. Chang CH, Huang Y, Anderson R. Activation of vascular endothelial cells by IL-1alpha released by epithelial cells infected with respiratory syncytial virus. Cell Immunol (2003) 221:37–41. doi: 10.1016/s0008-8749(03)00058-3
279. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev (2012) 76:16–32. doi: 10.1128/mmbr.05015-11
280. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
281. McGonagle D, O’Donnell JS, Sharif K, Emery P, Bridgewood C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol (2020) 2:e437–e45. doi: 10.1016/S2665-9913(20)30121-1
282. Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 Receptor Blockade Is Associated With Reduced Mortality in Sepsis Patients With Features of Macrophage Activation Syndrome: Reanalysis of a Prior Phase III Trial. Crit Care Med (2016) 44:275–81. doi: 10.1097/ccm.0000000000001402
283. Fisher CJ, Slotman GJ, Opal SM, Pribble JP, Bone RC, Emmanuel G, et al. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: a randomized, open-label, placebo-controlled multicenter trial. Crit Care Med (1994) 22:12–21. doi: 10.1097/00003246-199401000-00008
284. Rajasekaran S, Kruse K, Kovey K, Davis AT, Hassan NE, Ndika AN, et al. Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children*. Pediatr Crit Care Med (2014) 15:401–8. doi: 10.1097/pcc.0000000000000078
285. Cavalli G, Dinarello CA. Anakinra Therapy for Non-cancer Inflammatory Diseases. Front Pharmacol (2018) 9:1157. doi: 10.3389/fphar.2018.01157
286. King A, Vail A, O’Leary C, Hannan C, Brough D, Patel H, et al. Anakinra in COVID-19: important considerations for clinical trials. Lancet Rheumatol (2020) 2:e379–81. doi: 10.1016/S2665-9913(20)30160-0
287. Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, et al. Dexamethasone in Hospitalized Patients with Covid-19 — Preliminary Report. N Engl J Med (2020) NEJMoa2021436. doi: 10.1056/nejmoa2021436
288. Kern JA, Lamb RJ, Reed JC, Daniele RP, Nowell PC. Dexamethasone inhibition of interleukin 1 beta production by human monocytes. Posttranscriptional mechanisms. J Clin Invest (1988) 81:237–44. doi: 10.1172/jci113301
Keywords: cytokines, interleukin-1, inflammasome, innate immunity, inflammation, adaptive immunity
Citation: Pyrillou K, Burzynski LC and Clarke MCH (2020) Alternative Pathways of IL-1 Activation, and Its Role in Health and Disease. Front. Immunol. 11:613170. doi: 10.3389/fimmu.2020.613170
Received: 01 October 2020; Accepted: 16 November 2020;
Published: 18 December 2020.
Edited by:
Kuo-Feng Hua, National Ilan University, TaiwanReviewed by:
Luiz Eduardo Baggio Savio, Federal University of Rio de Janeiro, BrazilCopyright © 2020 Pyrillou, Burzynski and Clarke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Murray C. H. Clarke, bWNoYzJAY2FtLmFjLnVr
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.