
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 16 December 2020
Sec. Primary Immunodeficiencies
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.612703
This article is part of the Research Topic Advances in Primary Immunodeficiencies in India View all 12 articles
 Priyanka Madhav Kambli1
Priyanka Madhav Kambli1 Umair Ahmed Bargir1
Umair Ahmed Bargir1 Reetika Malik Yadav1
Reetika Malik Yadav1 Maya Ravishankar Gupta1
Maya Ravishankar Gupta1 Aparna Dhondi Dalvi1
Aparna Dhondi Dalvi1 Gouri Hule1
Gouri Hule1 Madhura Kelkar1Sneha Sawant-Desai1
Madhura Kelkar1Sneha Sawant-Desai1 Priyanka Setia1
Priyanka Setia1 Neha Jodhawat1Nayana Nambiar1Amruta Dhawale1Pallavi Gaikwad1Shweta Shinde1Prasad Taur2Vijaya Gowri2
Neha Jodhawat1Nayana Nambiar1Amruta Dhawale1Pallavi Gaikwad1Shweta Shinde1Prasad Taur2Vijaya Gowri2 Ambreen Pandrowala3Anju Gupta4
Ambreen Pandrowala3Anju Gupta4 Vibhu Joshi4Madhubala Sharma4
Vibhu Joshi4Madhubala Sharma4 Kanika Arora4
Kanika Arora4 Rakesh Kumar Pilania4
Rakesh Kumar Pilania4 Himanshi Chaudhary4
Himanshi Chaudhary4 Amita Agarwal5Shobita Katiyar5Sagar Bhattad6
Amita Agarwal5Shobita Katiyar5Sagar Bhattad6 Stalin Ramprakash7Raghuram CP7Ananthvikas Jayaram8Vinod Gornale9
Stalin Ramprakash7Raghuram CP7Ananthvikas Jayaram8Vinod Gornale9 Revathi Raj10Ramya Uppuluri10
Revathi Raj10Ramya Uppuluri10 Meena Sivasankaran11Deenadayalan Munirathnam11Harsha Prasad Lashkari12Manas Kalra13Anupam Sachdeva13Avinash Sharma14Sarath Balaji15
Meena Sivasankaran11Deenadayalan Munirathnam11Harsha Prasad Lashkari12Manas Kalra13Anupam Sachdeva13Avinash Sharma14Sarath Balaji15 Geeta Madathil Govindraj16Sunil Karande17Ruchi Nanavati18
Geeta Madathil Govindraj16Sunil Karande17Ruchi Nanavati18 Mamta Manglani19Girish Subramanyam20Abhilasha Sampagar21Indumathi CK22Parinitha Gutha23Swati Kanakia24Shiv Prasad Mundada25
Mamta Manglani19Girish Subramanyam20Abhilasha Sampagar21Indumathi CK22Parinitha Gutha23Swati Kanakia24Shiv Prasad Mundada25 Vidya Krishna26Sheela Nampoothiri27Sandeep Nemani28
Vidya Krishna26Sheela Nampoothiri27Sandeep Nemani28 Amit Rawat4Mukesh Desai2
Amit Rawat4Mukesh Desai2 Manisha Madkaikar1*
Manisha Madkaikar1*Leukocyte adhesion deficiency (LAD) syndrome is a group of inborn errors of immunity characterized by a defect in the cascade of the activation and adhesion leading to the failure of leukocyte to migrate to the site of tissue injury. Three different types of LAD have been described. The most common subtype is LAD type 1 (LAD1) caused due to defects in the ITGβ2 gene. LAD type 2 (LAD2) is caused by mutations in the SLC35C1 gene leading to a generalized loss of expression of fucosylated glycans on the cell surface and LAD type 3 (LAD3) is caused by mutations in the FERMT3 gene resulting in platelet function defects along with immunodeficiency. There is a paucity of data available from India on LAD syndromes. The present study is a retrospective analysis of patients with LAD collated from 28 different centers across India. For LAD1, the diagnosis was based on clinical features and flow cytometric expression of CD18 on peripheral blood leukocytes and molecular confirmation by Sanger sequencing. For patients with LAD3 diagnosis was largely based on clinical manifestations and identification of the pathogenic mutation in the FERMT3 gene by next-generation Sequencing. Of the total 132 cases diagnosed with LAD, 127 were LAD1 and 5 were LAD3. The majority of our patients (83%) had CD18 expression less than 2% on neutrophils (LAD1°) and presented within the first three months of life with omphalitis, skin and soft tissue infections, delayed umbilical cord detachment, otitis media, and sepsis. The patients with CD18 expression of more than 30% (LAD1+) presented later in life with skin ulcers being the commonest manifestation. Bleeding manifestations were common in patients with LAD3. Persistent neutrophilic leukocytosis was the characteristic finding in all patients. 35 novel mutations were detected in the ITGβ2 gene, and 4 novel mutations were detected in the FERMT3 gene. The study thus presents one of the largest cohorts of patients from India with LAD, focusing on clinical features, immunological characteristics, and molecular spectrum.
Leukocyte adhesion deficiency (LAD) is a rare phagocytic disorder characterized by a defect in the trafficking of leukocytes from the blood vessels to the site of tissue injury (1–4). These patients usually present in infancy with delayed separation of the umbilical cord, omphalitis, and necrotic infections of the skin and mucosal surfaces (5). The absence of pus and persistent marked neutrophilic leukocytosis are the hallmarks of LAD. Three types of LAD have been described, with LAD type 1 (LAD1) being the most common form. LAD1 is caused due to defect in the ITGβ2 gene encoding the common beta subunit of β2 integrins (CD18) (5–9). β2 integrins form a heterodimer by non-covalently binding to the different subunits including, αL (CD11a), αM (CD11b), αX (CD11c), and αD (CD11d) (1, 4, 9). In a setting of strong clinical suspicion, the immunological workup for diagnosis of LAD1 involves studying the flow cytometric expression of CD18 and CD11 on leukocytes followed by molecular confirmation. Depending on the CD18 expression on neutrophils LAD1 patients are classified into severe (CD18 expression <2%), moderate (2%–30%), and mild (>30%) (1, 2, 5, 10). LAD type 2 (LAD2) is caused due to mutations in the SLC35C1 gene leading to defective expression of cell surface fucosylated glycan structures (11, 12). These patients suffer from recurrent bacterial infections, severe mental, and growth retardation characterized by distinct facial characteristics (12). Flow cytometry demonstrates the absence of SLeX (CD15a) expression on cell-surface glycoproteins, along with deficiency of H antigen on erythroid cells, resulting in the Bombay phenotype (13). LAD type 3 (LAD3) is caused by a mutation in the FERMT3 gene that encodes protein kindlin-3 which plays a crucial in integrin activation (14–16). These patients also have severe recurrent bacterial infections, persistent leukocytosis, and delayed umbilical cord fall with a platelet aggregation defect that results in severe bleeding manifestation (17). Though individual case reports and small case series are available from India (18–23), there is a paucity of data on the clinical, immunological, and molecular spectrum in LAD. In this study, we report a retrospective cohort study of 132 patients LAD patients from 28 different centers of India.
Patients with a clinical suspicion of LAD referred to the Indian Council of Medical Research-National Institute of Immunohaematology (ICMR-NIIH) and other tertiary care centers in India between 1990 and 2020 were retrospectively analyzed in this study. The clinical and laboratory information about the age of presentation, age at diagnosis, site of infections, organisms isolated, umbilical cord complications, family history, consanguinity, complete blood count (CBC), immunological investigations were collected from the data available. The study was approved by the institutional ethics committee of ICMR-NIIH.
As a part of the diagnostic workup of LAD the flow cytometric expression of CD18, CD11 markers on leukocytes was assessed. Based on the CD18 expression on neutrophils, LAD1 was sub-classified into three phenotypes viz. severe (LAD1°) with CD18 expression <2% moderate (LAD1-) phenotype with CD18 expression 2%–30%, and mild (LAD1+) phenotype with CD18 expression ≥30%. CD11a expression data were available for 89 patients. We analyzed our data by looking at different parameters like median fluorescence intensity (MFI) and stain index (SI) index on different populations of leukocytes via, neutrophils, lymphocytes, and monocytes.
Molecular confirmation was performed was done using Sanger sequencing for LAD1 and next-generation sequencing (NGS) for LAD3. The candidate variants identified by NGS were confirmed by Sanger sequencing in the index and family members.
Graph pad prism version 5.03 statistical software was used to perform statistical analysis. The descriptive variables were expressed as percentage counts and the median-interquartile range (IQR) were used. The groups in this study were compared using the one-way ANOVA. The test was performed at a 95% confidence interval (95% CI), and p<0.05 was considered statistically significant. Kaplan–Meier evaluation was used to predict the survival probabilities of the patients.
In this study, we analyzed a total of 127 cases from 125 families with LAD1 and 5 cases from 4 families with LAD3. LAD2 were not reported in our cohort. The clinical and demographic features of the patients are as shown in Table 1. Consanguinity was seen in 51% of the patients. Male preponderance was seen in our cohort (62%). The genetic diagnosis was available in 80% of cases (n=105) cases. White blood count (WBC) and absolute neutrophil count (ANC) were noted in all the cases with a median of 53 x103/μl (14–167 x103/all) and 36 x103/μl (22–137x103/μl), respectively. It was observed that the ANC was higher in LAD1° 40 × 103/μl (11–136× 103/μl) cases as compared to LAD1- 25 × 103/μl (16–74× 103/μl), LAD1+ 27 × 103/μl (10–91× 103/μl), and LAD3 21 × 103/μl (10–38× 103/μl).
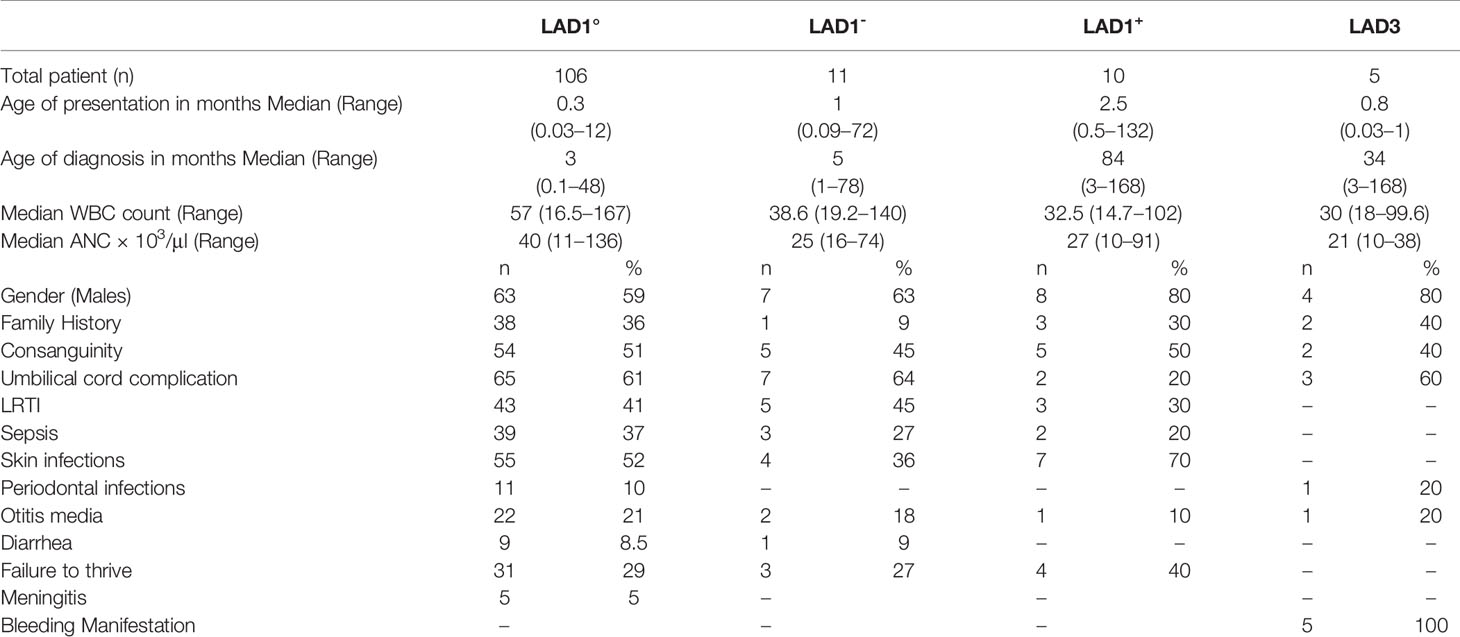
Table 1 Clinical characteristics of patients with leukocyte adhesion deficiency (LAD).
On the basis of absent or abnormal CD18 expression on the surface of neutrophils, three distinct phenotypes were observed in our cohort. The commonest being LAD1° seen in 83% (n=106) individuals, followed by LAD1- phenotype seen in 9% (n=11) of the cases and LAD1+ in 8% (n=10). The mean CD18 expression in LAD1- and LAD1+ patients was 9.6 ± 5% (3%–18%) and 68 ± 24% (32%–99%), respectively. The median fluorescence intensity was noted in 76/127 LAD1 patients. Figure S1, presents the percentage, MFI, and SI on neutrophils, monocytes, and lymphocytes in LAD1°, LAD1-, and LAD1+ cases. Although the expression of CD18 was >30% in LAD1+ cases, the CD11a was significantly reduced in 80% (8/10) of the cases. The median age of presentation was 0.3 months (0.03–12 months) for the LAD1° patients, 1 month (0.09–72 months) for LAD1- and 2.5 months (0.5–132 months) for patients with LAD1+. The median age of diagnosis was 3 months (0.1–48 months) for LAD1°, 5 months (1–78 months) for LAD1- and 84 months (3–168 months) for LAD1+.
Umbilical cord related complications like omphalitis (64%) and delayed separation (62%) were the most common manifestation seen in the LAD1° and LAD1- cases. Other frequent infections included lower respiratory tract infection (LRTI) in 41% (43/106), sepsis in 37%. Necrotic skin ulcer was the most common infection in LAD1+ which may mimic pyoderma gangrenosum. The perianal region was the commonest site in LAD1- cases (27%). Infectious organisms were isolated from 69 cases comprising predominantly bacterial infections including Pseudomonas aeruginosa (n=28), Staphylococcus aureus (n=17), and Klebsiella pneumonia (n=11), and fungal infections were noted in 7 patient. Also, unusual organisms like Proteus sp., Citrobacter sp., Stingomonas paucimobilis, and Acinetobacter baumani were noted in a few patients.
Direct Sanger sequencing of the ITGβ2 gene revealed 57 disease-causing variants in 105 patients (Table S2), including 30 patients we have previously reported (18). These mutations were clustered mostly in exons 6 (22%) and exon 7 (11%). The spectrum of mutation has been shown in Figure 1. The frequency of mutation c.533C>T (p.Pro178Leu) and c.817G>A (p.Gly273Arg) was high. The majority of the patients (n=95) had homozygous mutations, while compound heterozygous mutations were identified in only 10 patients. These compound heterozygous mutations were seen only LAD1° cases. Missense mutations (40%) were the most common mutations identified in our cohort followed by nonsense (21%), splice site (19%), and frameshift (19%). 54% of the mutations were located in exon 5–9, a highly conserved region of the extracellular domain of CD18 followed by cysteine-rich repeat region (CRR) domain (32%), Mid region (7%), plexins, semaphorins, and integrins domain (PSI) domain (5%) and transmembrane domain (TM) region (2%). Missense and nonsense mutations were frequently seen in LAD1° and LAD1- patients. On the other hand, splice site mutations affected almost 60% of LAD1+ patients.
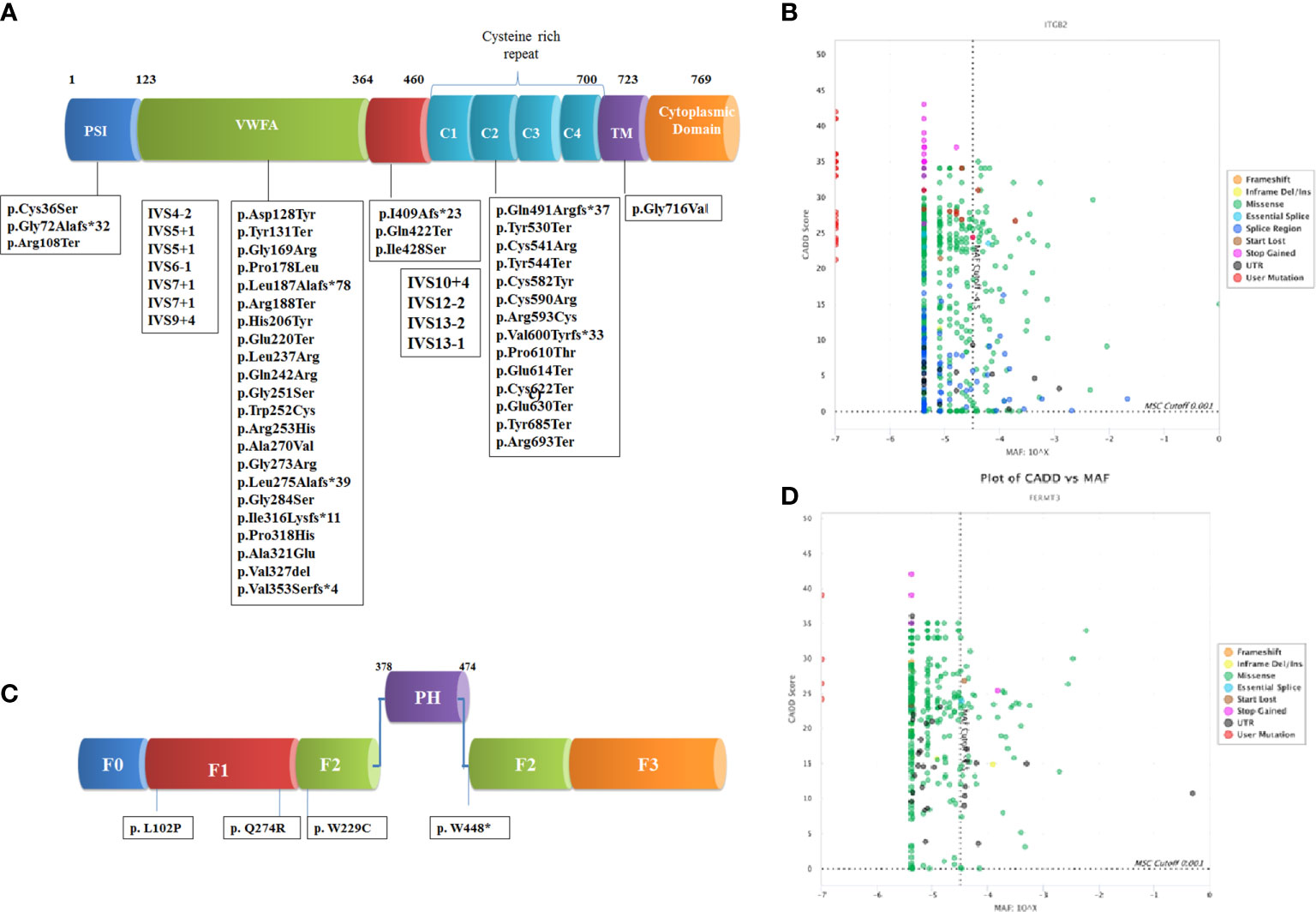
Figure 1 Schematic representation of domain wise distribution of the mutations identified in our LADpatients (A) β2 integrin (B) kindlin-3 protein. Combined annotation–dependent depletion (CADD) and Minor allele frequency (MAF) scores of the variants reported in gnomAD and novel variants identified in our cohort for the genes (C) ITGβ2 and (D) FERMT3 using PopViz software (24). The mutation significance cutoff (MSC) with 99% confidence interval is shown in dotted line.
Recurrent infections and severe bleeding manifestation were seen in all patients within 1 month of life. The median age of diagnosis was 34 months (3–168 months). Omphalitis was seen in 3/5 patients. CD18 expression on the surface of the leukocytes was normal in all patients. The molecular diagnosis using NGS technology identified four novel pathogenic variants in the FERMT3 gene in five LAD3 patients which were conserved across the species. Out of these 3 were missense mutations and one was nonsense mutations.
Follow up data were available for 124 patients with the median follow up duration of 7 months (0.033–216 months), 6 months (2–120 months), 182 months (5–276 months), and 60 months (11–192 months) for LAD1°, LAD1-, LAD1+, and LAD3, respectively. Out of these, 81% of patients expired due to severe infections in absence of hematopoietic stem cell transplant (HSCT). The majority of them died within the first year of life (n=70). The overall survival in our LAD1 cohort is only 14% while that of LAD3 is 83%. Mortality was higher in LAD1° patients with only 6% survival beyond 2 years as compared to 16% of the LAD1- and 60% of the LAD1+ patients in our cohort (Figure 2). Twelve patients underwent HSCT. Of these, ten patients underwent stem cell transplant (SCT) from HLA-matched related donors, whereas two patients received graft from matched unrelated donor. Three patients expired due to severe graph versus host disease (GVHD) and secondary complications.
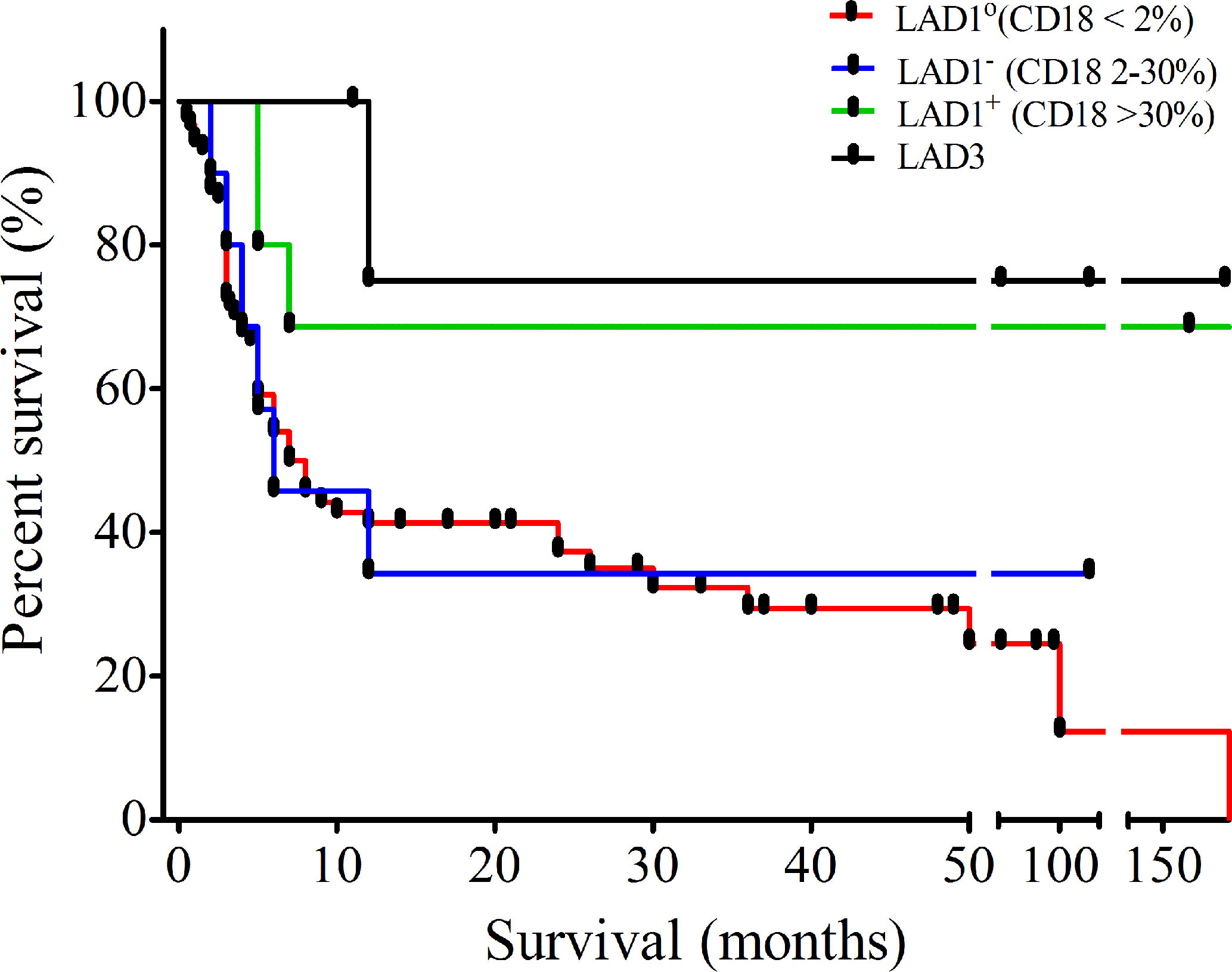
Figure 2 Outcome of leukocyte adhesion deficiency (LAD) patients: Kaplan-Meier curve showing survival of patients diagnosed with LAD in our cohort.
Leukocyte adhesion deficiency is a rare phagocytic disorder associated with defective neutrophil recruitment, rolling, and adhesion (1, 25–27). To the best of our knowledge, this is the largest comprehensive study on clinical, immunological, and molecular findings of LAD from India. Most of our patients presented with recurrent bacterial infections with a history of umbilical cord related complications. The infective spectra was similar to other reported cohorts with LRTI and sepsis being the commonest (5, 8, 9, 28–37). Delayed umbilical cord fall was seen in only 66% of the cases. Periodontal infections including gingivitis and oral ulcers are reported in 24% of severe and 52% of moderate LAD1 patients (5). In our cohort, it was observed in 10% of LAD1° patients. This might be because of under-diagnosis of mild LAD1 phenotype. Late- onset autoimmune complications have been reported previously (38). However, it was not observed in our patients as most of them expired in infancy.
The degree of severity of infections in LAD1° and LAD1- patients was high with poor survival of 8% and 33% (beyond 24 months), respectively. On the other hand, patients with LAD1+ had a more variable clinical phenotype with a significant difference between the age of onset and diagnosis. We observed that the mortality rate was high in LAD1° as well as LAD1- compared to LAD1+.
The clinical suspicion was strengthened with the presence of marked neutrophilic leukocytosis observed in all patients with total WBC count >25 x 103/μl in 86% cases and ANC of >15 x 103/μl in 82% of the cases. The median WBC count was higher in LAD1° as compared to the other subtypes and LAD3 patients, however, there was no significant correlation (r<0.1) between the WBC count and CD18 expression on neutrophil for the entire LAD1 cohort and the three phenotype as seen in the earlier studies (Figure S2).
The diagnosis of LAD1 often relies on the percentage of positive neutrophils expressing CD18. For severe forms of LAD1 where the expression is <2% diagnosis is easy and reliable. The expression may vary from patient to patient despite the same underlying disease-causing mutation (5, 30). It was observed that positive predictive value (PPV) of the assay significantly increased from 98.51 to 100% when the MFI of patients and healthy controls are compared (p<0.001) (data not shown). It is known that α- subunit of LFA-1 cannot be efficiently expressed unless it first associates with the β subunit. Previous studies have also reported that the expression of CD11a is abnormal in all the patients of LAD1 irrespective of CD18 expression (7, 32, 37). In P20 and P28, the CD18 expression was 50 & 90% and CD11a expression was 70 & 50%, respectively; SI observed was comparatively below laboratory lower limits obtained from the SI of healthy controls (SI of CD18/CD11a: P20- 1.81/1.59 & P28- 1.61/1.75). This concurs that the addition of the CD11a marker to the assay may increase the diagnostic accuracy (32).
We identified 57 mutations in 105 patients of which 35 were novel suggesting heterogeneity in the mutation spectrum for LAD1. 51% (29/57) of the mutations were identified in the VWFA domain followed by 16% (n=9) in cysteine-rich region and 3% (n=2) PSI domain, 4% (n=2) mid-region and 2% (n=1) in TM. The common mutation identified included c.533C>T (n=9), c.817G>A (n=9), c.751G>A (n=5), c.1224+4A>G (n=5), and c.2077C>T (n=5) in different domains. Out of the total mutations identified in the VWFA domain, 89% of them resulted in absent expression of CD18 on PMNs causing severe infection in LAD1° and LAD1- cases.
50% of LAD1+ had a splice site mutation. These patients presented later in life with recurrent skin lesions like pyoderma gangrenosum. All of them had the same c.1224+4A>G mutation and 4 of these patients have been reported by us earlier (39). In contrast to other patients with pyoderma gangrenosum, there is a paucity of neutrophils in the dermis of the skin lesions in patients with LAD1+ and they also show only partial and temporary response to steroids. Though the pathogenesis of these inflammatory lesions is not clear, partial expression of CD18 resulting in an aberrant oscillation of integrins on the neutrophil surface and Th17 mediated aberrant inflammatory response may be responsible for these inflammatory skin lesions (40, 41).
LAD3 was diagnosed in 5 patients classically presenting with recurrent infection of skin, ear, and mucosal surfaces, and bleeding from gums and skin. There was a significant variation in the age of diagnosis and presentation in these patients. Unfortunately, platelet aggregation studies were not available for these cases. Unlike LAD1, the surface expression of CD18/CD11 expression on leukocytes was normal in these patients. This disorder has mostly been reported in patients of Arab Maltese, Turkish, or African American origin (15, 16, 20, 42, 43).
The overall outcome in our cohort was poor with 81% mortality. The time taken for the patients from the diagnosis to treatment is critical and many patients are lost before they reach the stage of transplant. HSCT was possible in only 12 cases as most of the patients included in the study were from the last decade when limited transplantation facilities were available in India. However, with the increase in the number of HSCT centers, this scenario may change in the near future. Recent advancements in gene therapy for LAD1 may change the course of management (30).
This study describes the clinical and molecular spectrum of a large cohort of patients of LAD from India. It highlights the importance of analyzing MFI and SI of CD11a along with CD18 for accurate diagnosis of LAD1. It reports a large number of previously unreported mutations in the ITGβ2 and FERMT3 gene. Knowledge of the nature and frequency of these mutations is not only important for providing accurate diagnosis and genetic counseling to the families but will also help in the future for planning gene editing and gene therapy strategies for these rare genetic disorders.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by the Indian Council of Medical Research National Institute of Immunohematology. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
PK analyzed the data and wrote the manuscript. UB and RM helped in procuring the clinical details and follow-up of the patients. MD, AR, PT, VG, AP, AG, KA RKP, HC, PK, AA, SunK, SagB, SR, RCP, VinG, RR, RU, MSi, DM, HP, MKa, AnuS, AviS, SarB, GG, ShoK, RN, MamM, GS, AbhS, ICK, ParG, SwaK, SPM, VK, ShN, and SNem supervised the management and follow up of the patients. PK, MG, AD, GH, PS, MKe, SS, NJ, NN, AmrD, PalG, ShwS, ShoK, and AJ performed the laboratory investigations for the different cases. PK, PK, MG, MSh, and VJ were involved in the molecular analysis of the different patients. MM supervised the study and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Support from Indian Council of Medical Research to NIIH is gratefully acknowledged.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge the Foundation for Primary Immunodeficiency (FPID) for providing support for establishing FPID centers across India, which has been beneficial in increased awareness, diagnosis, and management.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.612703/full#supplementary-material
Supplementary Figure S1 | The percentage expression, median fluorescence intensity (MFI), stain index (SI- ratio of intensity of stain/unstain) of CD18 and CD11a on a) Neutrophils b) Lymphocytes, and c) monocytes.
Supplementary Figure S2 | Correlation between WBC count and CD18 expression on neutrophils a) all LAD patients b) LAD1° c) LAD1- d) LAD1+ e) LAD3.
Supplementary Table S1 | Clinical summary of all the patients diagnosed with LAD.
Supplementary Table S2 | Mutation spectrum of ITGβ2 gene and FERMT3 genes.
1. Roos D, Law SKA. Hematologically important mutations: Leukocyte adhesion deficiency. Blood Cells Mol Dis (2001) 27:1000–4. doi: 10.1006/bcmd.2001.0473
2. Harris ES, Weyrich AS, Zimmerman GA. Lessons from rare maladies: Leukocyte adhesion deficiency syndromes. Curr Opin Hematol (2013) 20:16–25. doi: 10.1097/MOH.0b013e32835a0091
3. Etzioni A. Defects in the leukocyte adhesion cascade. Clin Rev Allergy Immunol (2010) 38:54–60. doi: 10.1007/s12016-009-8132-3
4. Bednarczyk M, Stege H, Grabbe S, Bros M. β2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease. Int J Mol Sci (2020) 21:1–43. doi: 10.3390/ijms21041402
5. Almarza Novoa E, Kasbekar S, Thrasher AJ, Kohn DB, Sevilla J, Nguyen T, et al. Leukocyte adhesion deficiency-I: A comprehensive review of all published cases. J Allergy Clin Immunol Pract (2018) 6:1418–1420.e10. doi: 10.1016/j.jaip.2017.12.008
6. Yassaee VR, Hashemi-Gorji F, Boosaliki S, Parvaneh N. Mutation spectra of the ITGB2 gene in Iranian families with leukocyte adhesion deficiency type 1. Hum Immunol (2016) 77:191–5. doi: 10.1016/j.humimm.2015.11.019
7. Wolach B, Gavrieli R, Wolach O, Stauber T, Abuzaitoun O, Kuperman A, et al. Leucocyte adhesion deficiency—A multicentre national experience. Eur J Clin Invest (2019) 49:e13047. doi: 10.1111/eci.13047
8. Sun B, Chen Q, Dong X, Liu D, Hou J, Wang W, et al. Report of a Chinese Cohort with Leukocyte Adhesion Deficiency-I and Four Novel Mutations. J Clin Immunol (2019) 39(3)309–15. doi: 10.1007/s10875-019-00617-4
9. Gorjipour H, Chavoshzadeh Z, Fahimzad A, Darougar S. Leukocyte Adhesion Deficiency Type 1 : A Case Series and Review of the Literature. EMJ Allergy Immunol (2019) 4(1):95–100.
10. van de Vijver E, Maddalena A, Sanal Ö, Holland SM, Uzel G, Madkaikar M, et al. Hematologically important mutations: Leukocyte adhesion deficiency (first update). Blood Cells Mol Dis (2012) 48:53–61. doi: 10.1016/j.bcmd.2011.10.004
11. Dauber A, Ercan A, Lee J, James P, Jacobs PP, Ashline DJ, et al. Congenital disorder of fucosylation type 2c (LADII) presenting with short stature and developmental delay with minimal adhesion defect. Hum Mol Genet (2014) 23:2880–7. doi: 10.1093/hmg/ddu001
12. Knapp KM, Luu R, Baerenfaenger M, Zijlstra F, Wessels HJCT, Jenkins D, et al. Biallelic variants in SLC35C1 as a cause of isolated short stature with intellectual disability. J Hum Genet (2020) 65:743–50. doi: 10.1038/s10038-020-0764-4
13. Phillips ML, Schwartz BR, Etzioni A, Bayer R, Ochs HD, Paulson JC, et al. Neutrophil adhesion in leukocyte adhesion deficiency syndrome type 2. J Clin Invest (1995) 96:2898–906. doi: 10.1172/JCI118361
14. Fagerholm SC, Guenther C, Asens ML, Savinko T, Uotila LM. Beta2-Integins and interacting proteins in leukocyte trafficking, immune supression, and immunodeficiency disease. Front Immunol (2019) 10:254. doi: 10.3389/fimmu.2019.00254
15. McDowall A, Svensson L, Stanley P, Patzak I, Chakravarty P, Howarth K, et al. Two mutations in the KINDLIN3 gene of a new leukocyte adhesion deficiency III patient reveal distinct effects on leukocyte function in vitro. Blood (2010) 115:4834–42. doi: 10.1182/blood-2009-08-238709
16. Kuijpers TW, Van De Vijver E, Weterman MAJ, De Boer M, Tool ATJ, Van Den Berg TK, et al. LAD-1/variant syndrome is caused by mutations in FERMT3. Blood (2009) 113:4740–6. doi: 10.1182/blood-2008-10-182154
17. Alon R, Etzioni A. LAD-III, a novel group of leukocyte integrin activation deficiencies. Trends Immunol (2003) 24:561–6. doi: 10.1016/j.it.2003.08.001
18. Madkaikar M, Italia K, Gupta M, Chavan S, Mishra A, Rao M, et al. Molecular characterization of leukocyte adhesion deficiency-I in Indian patients: Identification of 9 novel mutations. Blood Cells Mol Dis (2015) 54:217–23. doi: 10.1016/j.bcmd.2015.01.012
19. Deshpande P, Kathirvel K, Alex AA, Korula A, George B, Shaji RV, et al. Leukocyte Adhesion Deficiency-I: Clinical and Molecular Characterization in an Indian Population. Indian J Pediatr (2016) 83:799–804. doi: 10.1007/s12098-016-2051-0
20. Mishra A, Gupta M, Dalvi A, Ghosh K, Madkaikar M. Rapid flow cytometric prenatal diagnosis of primary immunodeficiency (PID) Disorders. J Clin Immunol (2014) 34:316–22. doi: 10.1007/s10875-014-9993-7
21. Madkaikar M, Currimbhoy Z, Gupta M, Desai M, Rao M. Clinical profile of leukocyte adhesion deficiency type I. Indian Pediatr (2012) 49:43–5. doi: 10.1007/s13312-012-0005-9
22. Das J, Sharma A, Jindal A, Aggarwal V, Rawat A. Leukocyte adhesion defect: Where do we stand circa 2019? Genes Dis (2020) 7:107–14. doi: 10.1016/j.gendis.2019.07.012
23. Madkaikar MR, Gupta M, Rao M, Ghosh K. Prenatal diagnosis of LAD-I on cord blood by flowcytometry. Indian J Pediatr (2012) 79:1605–9. doi: 10.1007/s12098-012-0737-5
24. Zhang P, Bigio B, Rapaport F, Zhang SY, Casanova JL, Abel L, et al. PopViz: A webserver for visualizing minor allele frequencies and damage prediction scores of human genetic variations. Bioinformatics (2018) 34:4307–9. doi: 10.1093/bioinformatics/bty536
25. Anderson DC, Children T, Springer TA, Ph D. LEUKOCYTE ADHESION DEFICIENCY : An Inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med (1987) 38:175–94. doi: 10.1146/annurev.me.38.020187.001135
26. Tan S. The leucocyte β2 (CD18) integrins: the structure, functional regulation and signalling properties. Biosci Rep (2012) 32:241–69. doi: 10.1042/bsr20110101
27. Arnaout MA. Leukocyte Adhesion Molecules Deficiency: Its Structural Basis, Pathophysiology and Implications for Modulating the Inflammatory Response. Immunol Rev (1990) 114:145–80. doi: 10.1111/j.1600-065X.1990.tb00564.x
28. Aghamohammadi A, Chavoshzadeh Z, Ghalehbaghi B, Mamishi S, Ashrafi F, Parvaneh N, et al. Characterization of 11 New Cases of Leukocyte Adhesion Deficiency Type 1 with Seven Novel Mutations in the ITGB2 Gene. J Clin Immunol (2010) 30:756–60. doi: 10.1007/s10875-010-9433-2
29. Bleesing JH, Hinze CH, Lucky AW, Marsh RA, Passo MH, Bove KE. Leukocyte Adhesion Deficiency Type 1 Presenting with Recurrent Pyoderma Gangrenosum and Flaccid Scarring. Pediatr Dermatol (2010) 27:500–3. doi: 10.1111/j.1525-1470.2010.01260.x
30. Hu J, Zhang Q, Zheng H, Chang H, Xian Y, Nie N, et al. Novel mutations in the β2 integrin gene (ITGB2) in a moderate leukocyte adhesion defect type 1 patient. Arch Iran Med (2018) 21:296–301.
31. Wright AH, Douglass WA, Taylor GM, Lau Y -L, Higgins D, Davies KA, et al. Molecular characterization of leukocyte adhesion deficiency in six patients. Eur J Immunol (1995) 25:717–22. doi: 10.1002/eji.1830250313
32. Levy-Mendelovich S, Rechavi E, Abuzaitoun O, Vernitsky H, Simon AJ, Lev A, et al. Highlighting the problematic reliance on CD18 for diagnosing leukocyte adhesion deficiency type 1. Immunol Res (2016) 64:476–82. doi: 10.1007/s12026-015-8706-5
33. Cox DP, Weathers DR. Leukocyte adhesion deficiency type 1: an important consideration in the clinical differential diagnosis of prepubertal periodontitis. A case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endodontol (2008) 105:86–90. doi: 10.1016/j.tripleo.2007.02.026
34. Wardlaw AJ, Hibbs ML, Stacker SA, Springer TA. Distinct mutations in two patients with leukocyte adhesion deficiency and their functional correlates. J Exp Med (1990) 172:335–45. doi: 10.1084/jem.172.1.335
35. Teimourian S, De Boer M, Roos D, Isaian A, Bemanian MH, Lashkary S, et al. Genetic Analysis of 13 Iranian Families With Leukocyte Adhesion Deficiency Type 1. J Pediatr Hematol Oncol (2018) 00:1–4. doi: 10.1097/MPH.0000000000001221
36. Wada T, Tone Y, Shibata F, Toma T, Yachie A. Delayed Wound Healing in Leukocyte Adhesion Deficiency Type 1. J Pediatr (2011) 158:342. doi: 10.1016/j.jpeds.2010.07.057
37. Cabanillas D, Regairaz L, Deswarte C, García M, Richard ME, Casanova JL, et al. Leukocyte Adhesion Deficiency Type 1 (LAD1) with Expressed but Nonfunctional CD11/CD18. J Clin Immunol (2016) 36:627–30. doi: 10.1007/s10875-016-0322-1
38. De Rose DU, Giliani S, Dora L, Lougaris V, Lanfranchi A, Martire B, et al. Long term outcome of eight patients with type 1 Leukocyte Adhesion Deficiency (LAD-1): Not only infections, but high risk of autoimmune complications. Clin Immunol (2018) 191:75–80. doi: 10.1016/j.clim.2018.03.005
39. Madkaikar M, Italia K, Gupta M, Desai M, Aggarwal A, Singh S, et al. Leukocyte Adhesion Deficiency-I with a Novel Intronic Mutation Presenting with Pyoderma Gangrenosum- Like Lesions. J Clin Immunol (2015) 35:431–4. doi: 10.1007/s10875-015-0155-3
40. Shaya S, Kindzehkii AL, Minor J, Moore EC, Todd RF, Petty HR. Aberrant integrin (CR4; α(x)β2; CD11c/CD18) oscillations on neutrophils in a mild form of pyoderma gangrenosum. J Invest Dermatol (1998) 111:154–8. doi: 10.1046/j.1523-1747.1998.00255.x
41. Adachi Y, Kindzelskii AL, Cookingham G, Shaya S, Moore EC, Todd RF, et al. Aberrant neutrophil trafficking and metabolic oscillations in severe pyoderma gangrenesum. J Invest Dermatol (1998) 111:259–68. doi: 10.1046/j.1523-1747.1998.00311.x
42. Aygun D, Nepesov S, Gershoni R, Camcıoglu Y. Leukocyte Adhesion Deficiency III: Report of Two Siblings. Pediatr Neonatol (2017) 58:99–100. doi: 10.1016/j.pedneo.2016.07.006
Keywords: Leukocyte Adhesion deficiency, CD18, CD11, FERMT3, ITGβ2
Citation: Kambli PM, Bargir UA, Yadav RM, Gupta MR, Dalvi AD, Hule G, Kelkar M, Sawant-Desai S, Setia P, Jodhawat N, Nambiar N, Dhawale A, Gaikwad P, Shinde S, Taur P, Gowri V, Pandrowala A, Gupta A, Joshi V, Sharma M, Arora K, Pilania RK, Chaudhary H, Agarwal A, Katiyar S, Bhattad S, Ramprakash S, CP R, Jayaram A, Gornale V, Raj R, Uppuluri R, Sivasankaran M, Munirathnam D, Lashkari HP, Kalra M, Sachdeva A, Sharma A, Balaji S, Govindraj GM, Karande S, Nanavati R, Manglani M, Subramanyam G, Sampagar A, CK I, Gutha P, Kanakia S, Mundada SP, Krishna V, Nampoothiri S, Nemani S, Rawat A, Desai M and Madkaikar M (2020) Clinical and Genetic Spectrum of a Large Cohort of Patients With Leukocyte Adhesion Deficiency Type 1 and 3: A Multicentric Study From India. Front. Immunol. 11:612703. doi: 10.3389/fimmu.2020.612703
Received: 30 September 2020; Accepted: 09 November 2020;
Published: 16 December 2020.
Edited by:
Sudhir Gupta, University of California, Irvine, United StatesCopyright © 2020 Kambli, Bargir, Yadav, Gupta, Dalvi, Hule, Kelkar, Sawant-Desai, Setia, Jodhawat, Nambiar, Dhawale, Gaikwad, Shinde, Taur, Gowri, Pandrowala, Gupta, Joshi, Sharma, Arora, Pilania, Chaudhary, Agarwal, Katiyar, Bhattad, Ramprakash, CP, Jayaram, Gornale, Raj, Uppuluri, Sivasankaran, Munirathnam, Lashkari, Kalra, Sachdeva, Sharma, Balaji, Govindraj, Karande, Nanavati, Manglani, Subramanyam, Sampagar, CK, Gutha, Kanakia, Mundada, Krishna, Nampoothiri, Nemani, Rawat, Desai and Madkaikar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manisha Madkaikar, bWFka2Fpa2FybWFuaXNoYUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.