 Elise M. N. Ferré
Elise M. N. Ferré Michail S. Lionakis
Michail S. Lionakis- Fungal Pathogenesis Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, United States
Autoimmune-polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), a monogenic disorder caused by biallelic mutations in the AIRE gene, has historically been defined by the development of chronic mucocutaneous candidiasis together with autoimmune endocrinopathies, primarily hypoparathyroidism and adrenal insufficiency. Recent work has drawn attention to the development of life-threatening non-endocrine manifestations such as autoimmune pneumonitis, which has previously been poorly recognized and under-reported. In this review, we present the clinical, radiographic, autoantibody, and pulmonary function abnormalities associated with APECED pneumonitis, we highlight the cellular and molecular basis of the autoimmune attack in the AIRE-deficient lung, and we provide a diagnostic and a therapeutic roadmap for patients with APECED pneumonitis. Beyond APECED, we discuss the relevance and potential broader applicability of these findings to other interstitial lung diseases seen in secondary AIRE deficiency states such as thymoma and RAG deficiency or in common polygenic autoimmune disorders such as idiopathic Sjögren’s syndrome.
Introduction
Autoimmune-polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), also known as autoimmune polyglandular syndrome type-1 (APS-1), is a rare disorder resulting from biallelic mutations in the autoimmune regulator (AIRE) gene. AIRE is a thymus-enriched transcription regulator integral for enforcing central immune tolerance. AIRE-deficiency leads to multiorgan system autoimmunity and susceptibility to chronic mucocutaneous candidiasis (CMC). Diagnosis relies on developing two (“diagnostic dyad”) out of any three “classic triad” manifestations of CMC, hypoparathyroidism, and adrenal insufficiency. Development of a diagnostic dyad raises suspicion for APECED, which is then confirmed by AIRE gene sequencing. Detection of type I interferon (IFN-α/IFN-ω) autoantibodies is sensitive and specific for APECED and is useful for diagnosis (1). While the classic triad is quite characteristic for APECED, exclusive reliance on the classic triad manifestations results in delayed clinical diagnosis as a variety of non-triad non-endocrine manifestations develop often before reaching a classic diagnostic dyad (2). To that end, we have proposed inclusion of an adjunct triad of early-onset manifestations, namely APECED rash, intestinal dysfunction, and enamel hypoplasia, into expanded diagnostic criteria which would reduce the time to clinical diagnosis by half (3). Establishing an earlier diagnosis is important as it can enable screening for life-threatening endocrinopathies and prompt recognition and treatment of non-endocrine autoimmune manifestations such as hepatitis (4) or pneumonitis (5).
With regard to pneumonitis, prior studies had suggested it to be an uncommon manifestation of APECED (prevalence in all previously-published work, ~2%). A small number of affected patients (2.7–4.5%) had been described among Turkish, Russian, and Indian APECED cohorts. Importantly, the foundational APECED cohort descriptions in Finns, Sardinians, or Iranian Jews do not highlight pneumonitis nor is it a prominent feature in the literature among APECED patients from the British Isles (6–25). In contrast, in a prospective observational natural history study at the NIH, we diagnosed >40% of consecutively-enrolled APECED patients with autoimmune pneumonitis; notably, pneumonitis symptoms presented early in life, often before developing a classic diagnostic dyad (5).
Definition and Clinical Presentation of Autoimmune-Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy Pneumonitis
APECED pneumonitis presents clinically with chronic respiratory symptoms lasting >4 weeks with accompanying radiographic abnormalities of interstitial lung disease (ILD) and/or bronchiectasis. Affected patients most commonly present with daily cough with or without sputum production, and frequently report nocturnal bouts of cough (60%) awakening them from sleep. Less frequently, dyspnea on exertion (57%), pleuritic chest pain (48%), wheezing (43%), and fevers (29%) occur (5). Importantly, a small proportion of patients (<5–10%) is asymptomatic early in the course of pneumonitis (5).
Non-contrast computed tomography (CT) of the chest reveals abnormalities consistent with ILD and/or bronchiectasis. Specifically, ground-glass opacities (GGO) or mosaicism and bronchiectasis are the most common abnormalities; they are seen, either alone or in combination, in all patients with APECED pneumonitis, including those without respiratory symptoms and negative lung-targeted autoantibodies (see below) (5). Additional less common radiographic findings include a tree-in-bud pattern, nodular opacities, and mucus plugging. Taken together, non-contrast chest CT imaging is the most sensitive screening tool for APECED pneumonitis.
In keeping with these chronic symptoms and radiographic abnormalities, APECED pneumonitis leads to abnormal pulmonary function (5, 26, 27). Indeed, affected patients display decreased diffusing capacity of the lungs for carbon monoxide with or without a ventilatory defect by spirometry presenting as obstructive, restrictive, or a mixed pattern of both. A 6 min walk test typically shows decreased walk distance and oxygen desaturation (5).
Progression of Untreated Pneumonitis Causes Morbidity and Mortality
Through the course of our study, we encountered patients across the spectrum of pneumonitis severity which allowed us to characterize the temporal progression of clinical and radiographic features of APECED pneumonitis. Early-stage disease manifests with dry cough associated with GGO and/or a tree-in-bud pattern without bronchiectasis (Figure 1). Without immunosuppression, pneumonitis progresses to bronchiectasis-associated structural lung disease presenting with productive cough and bacterial airway colonization. Late-stage untreated pneumonitis features progressively worsening bronchiectasis-associated structural lung disease with development of recurrent infections by Gram-negative bacteria, Gram-positive bacteria, or nontuberculous mycobacteria (NTM) leading to hypoxemia requiring home oxygen therapy (5).
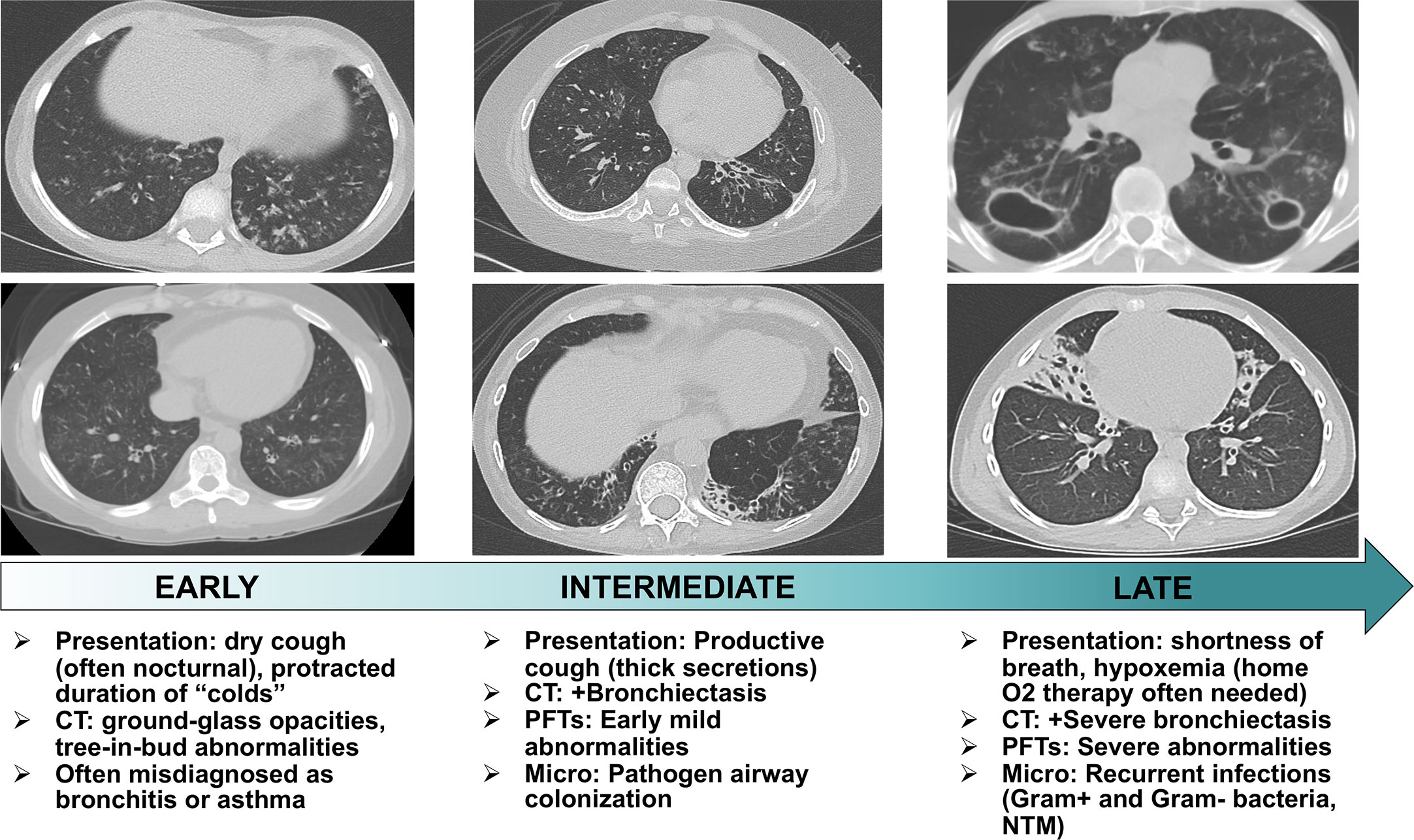
Figure 1 Stages of temporal progression of APECED-associated pneumonitis. Flow chart summarizing the temporal progression of symptoms, radiographic and pulmonary function test abnormalities, and microbiological findings in patients with APECED pneumonitis. CT, computed tomography; PFT, pulmonary function tests; Micro, microbiological findings; NTM, nontuberculous mycobacteria.
The few clinical cases previously described in the literature corroborate our study observations. DeLuca et al. and Alimohammadi et al. reported a Sicilian child who first developed productive cough and recurrent lower respiratory tract infections at the age of 5 years. The patient’s pneumonitis progressed over time with development of a severe obstructive defect, bronchiectasis, chronic airway colonization with Burkholderia, and hypoxemia requiring daily oxygen supplementation at the age of 14 years. The patient succumbed to pneumonitis complications when 18 years-old (26, 27). Alimohammadi and colleagues described three additional patients who developed chronic cough in childhood and progressed clinically with recurrent lower respiratory tract infections, an obstructive ventilatory defect, and radiographic evidence of bronchiectasis and/or GGO. One of the patients was oxygen-dependent by 19 years and another died at 37 years from respiratory failure (27).
Therefore, disease progression from symptom onset to end-stage lung disease is highly variable as demonstrated by the aforementioned cases. Similarly, in our recent study we reported a 54-year-old man who developed chronic cough when 5 years-old and progressed over 40 years to eventually develop cavitary pulmonary NTM infection complicated by bronchopulmonary fistula and empyema, chronic hypoxemia requiring daily supplemental oxygen, and death at 56 years. His case stands in contrast to a 14-year-old boy who rapidly progressed from cough onset at 7 years to home oxygen therapy at 11 years and death at 14 years (5).
Therefore, timely diagnosis is necessary to ensure early initiation of immunomodulation in order to arrest progression to bronchiectasis-associated structural lung disease. However, this can be challenging to achieve as symptoms frequently begin in early life and often before the patient develops a classic diagnostic dyad that would raise suspicion for APECED. Even patients with confirmed APECED typically experience delays in pneumonitis diagnosis due to the poor characterization of the entity in the previously-published literature. Consequently, patients are often misdiagnosed with asthma or bronchitis resulting in treatment delays thereby increasing the risk of developing structural lung disease and associated morbidity and mortality. For this reason, we recommend that all APECED patients, regardless of symptoms, undergo periodic screening with chest CT to achieve early diagnosis of APECED pneumonitis (5). Moreover, a high index of suspicion for APECED is required by pediatricians and pulmonologists in children who develop chronic respiratory symptoms in the setting of CMC and/or autoimmune manifestations within the classic and/or adjunct diagnostic criteria of APECED.
Pathogenesis of Autoimmune-Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy Pneumonitis
AIRE Genetics and Non-AIRE Modifiers may Impact Pneumonitis Prevalence
APECED is caused by biallelic AIRE mutations (28, 29). In our genotype-phenotype analysis, we found an association between carrying the c.967_979del13 mutation in homozygosity with decreased time to development of pneumonitis (5). Autosomal dominant (AD) AIRE mutations in the first plant homeodomain (PHD1) zinc finger domain and in the SAND domain have been described to cause organ-specific autoimmune disease resulting in milder phenotypes with reduced penetrance (30–32). While CMC, endocrinopathies and non-endocrine manifestations such as pernicious anemia, nail dystrophy, vitiligo and alopecia have been reported, autoimmune pneumonitis has thus far not been reported in those carrying AD mutations in AIRE. The enrichment of the c.967_979del13 mutation in American and British cohorts may explain the differences in prevalence among Americans and British. Alternatively, or in parallel, non-AIRE genetic modifiers (33), differential pulmonary microbiome, environmental factors, and/or our unbiased enrollment coupled with a uniform prospective evaluation in all patients regardless of symptoms may contribute to the increased prevalence of pneumonitis among Americans. Future enrollment and uniform multidisciplinary evaluation of European and additional American patients in our and other institutions will be essential to validate our findings.
Thymic Escape of Autoreactive Lymphocytes
AIRE is expressed in thymic medullary epithelial cells (mTECs) where it facilitates the negative selection of self-reactive T-lymphocytes. As a transcription regulator, AIRE promotes the expression of peripheral tissue-restricted antigens on mTECs and the clonal deletion of self-reactive T-lymphocytes; in the AIRE-deficient state, these cells escape in the periphery and are both necessary and sufficient to cause tissue-specific autoimmunity as shown by lymphocyte depletion and adoptive transfer experiments in mice (2, 34–38).
AIRE-deficiency also impairs B-lymphocyte tolerance (39), which contributes to the development of autoimmunity in some, but not all, tissues (40). AIRE-deficient humans and mice produce a broad repertoire of high-affinity autoantibodies (1, 41–44), although these autoantibodies have not demonstrated direct pathogenicity via serum transfer studies in mice (37, 40). Instead, B-lymphocytes appear to contribute to autoimmune inflammation through priming effector T-lymphocytes (40).
Several tissue-specific autoantibodies correlate with the development of organ-specific disease in APECED (38, 45–47). Among these, autoantibodies against bactericidal/permeability-increasing fold-containing family B member 1 (BPIFB1) and the potassium channel regulator KCNRG have been associated with development of APECED pneumonitis (3, 21, 27, 48). We corroborated this finding in our cohort where both autoantibodies were highly specific for pneumonitis and significantly associated with the time to development of pneumonitis (5). Autoantibodies against BPIFB1 were more sensitive compared to those against KCNRG (5). Although the majority (76%) of affected patients carried at least one of these lung-targeted autoantibodies in serum and/or bronchoalveolar lavage (BAL), a quarter of patients with pneumonitis were negative for both autoantibodies. Therefore, while identification of autoantibodies in patient serum may aid as a screening modality of pneumonitis, such testing alone does not suffice to rule out pneumonitis in all individuals, further underscoring the importance of universal screening via chest CT imaging. Importantly, these data also underscore the need for future research aimed to identify the lung autoantigens that might be the target of autoimmune attack in patients with APECED pneumonitis who do not carry BPIFB1 or KCNRG autoantibodies.
Autoimmune-Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy Pneumonitis Features a Characteristic Compartmentalized Immunopathology
We performed bronchoscopies in APECED patients with untreated pneumonitis and obtained BAL fluid and endobronchial and transbronchial tissue biopsies for immunological and histological analyses in comparison to healthy volunteer specimens obtained in bronchoscopy. A characteristic compartmentalized immune response was noted, which carries significant diagnostic value. In the airways, an enrichment of neutrophils was seen in the absence of bacterial or other lung infection. In agreement, we observed a significant increase of neutrophil-targeted CXC chemokines in the BAL (CXCL1, CXCL2, IL-8), although the cellular source of these chemokines remains unknown (Figure 2). BAL neutrophils exhibited an activated phenotype evidenced by increased expression of the extracellular epitope of the NADPH oxidase b558, of primary, secondary, and tertiary granule contents (CD18, CD63, CD66b), and of CD45, and decreased CD16 expression. Both myeloperoxidase (MPO) and matrix metallopeptidase-9 (MMP-9), products of activated neutrophils, and lactate dehydrogenase (LDH), a surrogate marker of tissue injury, were markedly increased in the BAL fluid of patients with pneumonitis (5). Thus, activated neutrophils appear to contribute to airway tissue injury and may instigate bronchiectasis as postulated in patients with cystic fibrosis and non-cystic fibrosis bronchiectasis (49, 50).
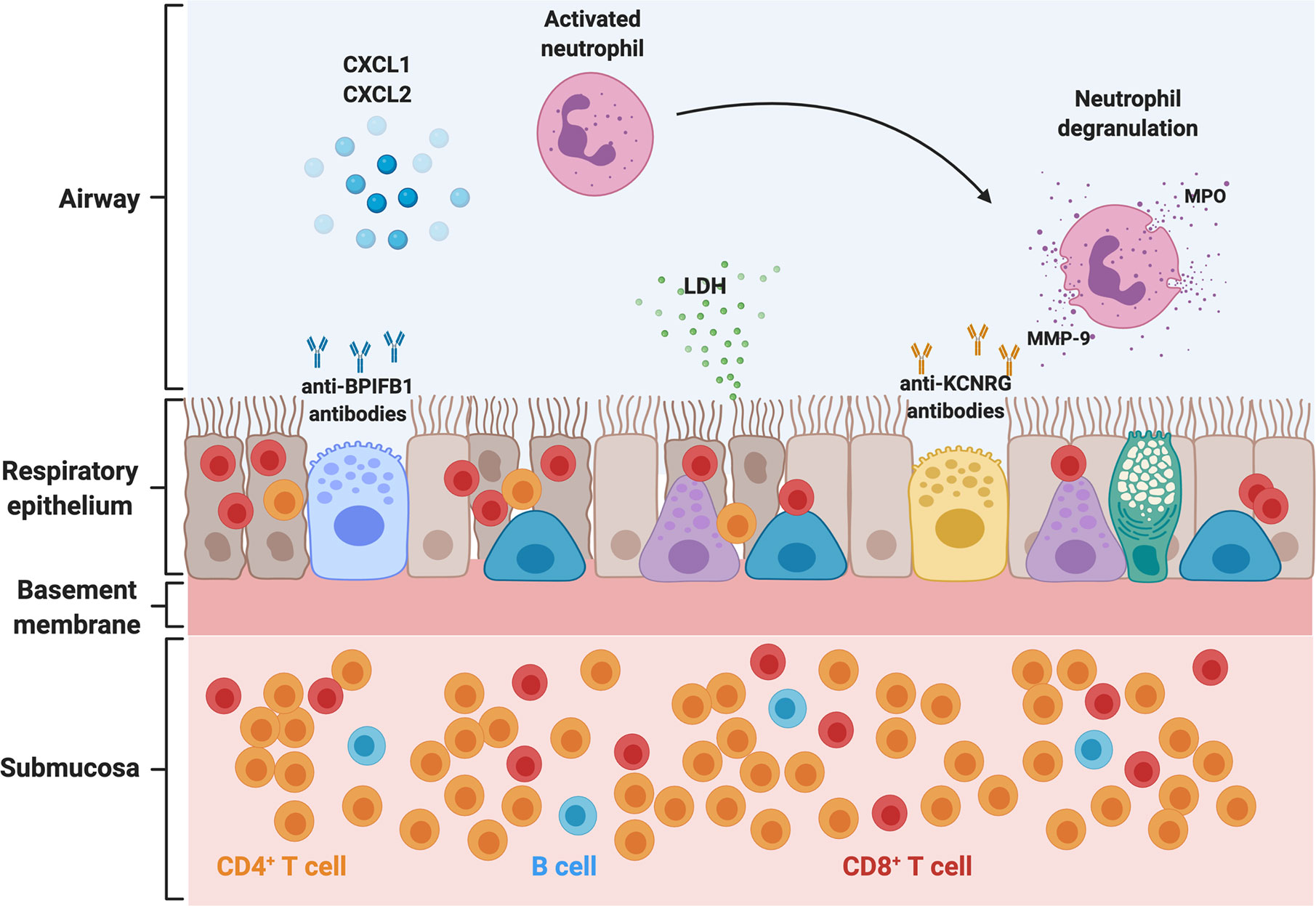
Figure 2 Pathogenesis of APECED-associated pneumonitis. Schematic representation of the abnormalities in the airway, respiratory epithelium, and submucosal tissue in the setting of APECED pneumonitis. T- and B-lymphocytes infiltrate the respiratory tissue. CD4+ T-lymphocytes predominate in the submucosal tissue and peribronchiolar/bronchiolar areas (not depicted), while CD8+ T-lymphocytes display a predominantly intraepithelial distribution. Neutrophils predominate in the airways where they accumulate through the release of CXC chemokines such as CXCL1, CXCL2, and IL-8. Recruited neutrophils acquire an activated phenotype and release MPO and MMP-9 into the airway, which further exacerbates tissue injury, as seen with release of LDH within the airways. Chronic epithelial irritation results in a thickened basement membrane. KCNRG and the BPIFB1 have been identified as bronchial autoantigens targeted by autoimmunity in APECED pneumonitis, and autoantibodies against these targets can be detected in the bronchoalveolar lavage and serum (not depicted) of patients with APECED pneumonitis. BPIFB1, bactericidal/permeability-increasing fold containing family B member 1; LDH, lactate dehydrogenase; MPO, myeloperoxidase; MMP-9, matrix metallopeptidase 9; CXCL1, C-X-C chemokine ligand 1; CXCL2, C-X-C chemokine ligand 2.
In contrast to the neutrophilic response in the airways, histological examination of endobronchial and deeper lung tissue biopsies demonstrated a chronic inflammatory infiltrate consistent with prior literature describing lymphocytic peribronchiolar inflammation in few patients (Figure 2) (5, 26, 27, 51). Endobronchial biopsies from patients with APECED pneumonitis displayed a thickened basement membrane with submucosal and intraepithelial lymphocytosis composed predominately of T-lymphocytes with fewer B-lymphocytes. CD4+ T-lymphocytes predominated in the submucosa whereas CD8+ T-lymphocytes were enriched within the intraepithelial compartment (Figure 2) (5). No eosinophils or neutrophils were observed infiltrating the tissue. Deeper lung biopsies unveiled lymphocytic or lymphoplasmacytic bronchiolitis and/or peribronchiolar inflammation dominated by CD4+ and CD8+ T-lymphocytes, with mild-to-moderate fibrosis noted in some patients. As with endobronchial biopsy specimens, CD8+ T-lymphocytes predominated within the bronchiolar epithelium while CD4+ T-lymphocytes were prominent in the submucosal bronchiolar tissue. Notably, whereas infiltration of B-lymphocytes was less prominent on endobronchial biopsy specimens, deep peribronchial tissue examination demonstrated marked B-lymphocyte infiltration with development of lymphoid nodules and primary follicles, some of which showed germinal center formation (5).
The mouse model of Aire-deficiency recapitulated the immunological characteristics of autoimmune pneumonitis of patients. Specifically, Aire-/- mice exhibited airway neutrophilia with increased neutrophil-targeted CXC chemokines in the absence of an infectious challenge. Moreover, the lung parenchyma of Aire-/- mice featured similar histological abnormalities consisting of intraepithelial, submucosal, peribronchiolar and interstitial infiltration composed of T- and B-lymphocytes with B-lymphocyte aggregates observed deeper in the lung tissue (5).
Collectively, APECED pneumonitis features a characteristic pattern of compartmentalized immunopathology consisting of activated neutrophils in the airways with lymphocytic inflammation within the lung parenchyma. This information has important diagnostic value. For example, the presence of neutrophils in the BAL or even in induced sputum examination in an APECED patient with pulmonary symptoms and radiographic abnormalities should raise suspicion for pneumonitis in the absence of pneumonia. Endobronchial biopsies, which we favor as the preferred modality for making a histological diagnosis of pneumonitis, allow for demonstration of intraepithelial and submucosal lymphocytosis, which together with the airway neutrophil expansion provide a high degree of probability for the diagnosis of APECED pneumonitis, especially when combined with BPIFB1- and/or KCNRG-targeted autoantibody positivity.
Combination Lymphocyte-Directed Immunomodulation Remits Pneumonitis
Previous reports of various immunomodulatory treatments had demonstrated mixed results with one patient responding to T-lymphocyte immunomodulation with azathioprine (27) while other patients required multiple different T-lymphocyte therapies with mixed results (21, 27). Data in the Aire-deficient mouse from our group and others would suggest that a T-lymphocyte depletion approach such as with the CD52-targeting alemtuzumab would remit APECED pneumonitis (5, 37); however, the risk of opportunistic infections makes such T-cell depleting strategies difficult to implement for the lifelong management of pneumonitis (52, 53). Thus, we elected a combination of T-lymphocyte modulation with azathioprine [or mycophenolate mofetil in patients with thiopurine methyltransferase (TPMT) mutations] together with B cell-targeting rituximab to capitalize on the beneficial effects of B-lymphocyte deficiency observed in mice (5). This regimen is used successfully to treat granulomatous and lymphocytic interstitial lung disease (GLILD) seen in combined variable immunodeficiency (CVID) (54).
Combination T and B lymphocyte-directed therapy resulted in resolution of respiratory symptoms in all symptomatic patients within 1 month. Those who had recurrent pulmonary infections secondary to their bronchiectasis before onset of immunomodulatory treatment did not develop infection recurrences after therapy initiation, indicating that the hyper-inflammatory milieu within the untreated airways is permissive for pathogen overgrowth. Immunomodulatory treatment was accompanied by marked improvement of radiographic abnormalities of GGO, tree-in-bud pattern, nodular opacities, and mucus plugging. Improvement was also noted in pulmonary function abnormalities with increased 6 min walk distance and resolution of oxygen desaturation (5). Lymphocyte immunophenotyping showed no changes in CD3+, CD4+, and CD8+ T-lymphocyte numbers in blood and an expected decline in CD19+ B-lymphocytes. Titers of BPIFB1 and KCNRG autoantibodies did not decline despite clinical and radiographic remission of pneumonitis, further suggesting that the pathogenic role of B-cells might be conferred via priming of T-cells in the lung tissue, rather than through autoantibody production. This early treatment study of five consecutive patients (5) with pneumonitis and treatment of 6 additional patients with similar results (manuscript in preparation) indicate that combination T and B lymphocyte-directed therapy can remit clinical symptoms and radiographic and functional abnormalities in APECED pneumonitis. Importantly, early initiation of treatment, preferably before the establishment of irreversible bronchiectatic abnormalities, is desirable to avoid the long-term pulmonary complications and morbidity and mortality associated with untreated pneumonitis.
Autoimmune-Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy Pneumonitis Shares Immunological Features With Interstitial Lung Diseases Associated With Secondary Autoimmune Regulator-Deficiency States
Conditions associated with documented secondary AIRE-deficiency in the thymus such as thymoma (55) and inherited RAG deficiency due to hypomorphic RAG mutations that cause delayed onset combined immunodeficiency with granulomas and/or autoimmunity (CID-G/AI) feature autoimmunity and display broad-spectrum autoantibodies against cytokines and tissue autoantigens (56–58) similar to APECED patients. A subset of these patients develops lung disease, which had previously been poorly-characterized (59, 60). We hypothesized that the lung disease seen in patients with thymoma or hypomorphic RAG mutations share similar features with APECED pneumonitis. Indeed, thymoma-associated autoimmune lung disease exhibits a similar compartmentalized immunopathology with airway neutrophil expansion and intraepithelial, submucosal, and peri-bronchiolar lymphocytic inflammation as seen in APECED pneumonitis (5). A smaller proportion of these patients carry autoantibodies against BPIFB1 and KCNRG compared to patients with APECED pneumonitis (5), pointing to additional yet-unidentified lung autoantigens in these diseases. Notably, the similarities between autoimmune lung disease seen in the setting of these secondary AIRE-deficiency states and APECED suggest common pathogenetic mechanisms and imply that the lymphocyte-targeted immunomodulatory regimen that is effective in APECED pneumonitis might also remit ILD in patients with thymoma (manuscript in preparation) and may serve as a bridge to hematopoietic stem cell transplantation in patients with ILD in the setting of hypomorphic RAG mutations with CID-G/AI.
Beyond primary and secondary AIRE-deficiency states, ILD with a similar compartmentalized immunopathology consisting of airway neutrophil expansion and lymphocytic bronchiolitis develops among a subset of patients with certain polygenic autoimmune diseases such as Sjögren’s syndrome (SS), ulcerative colitis (UC), systemic lupus erythematosus (SLE), and dermatomyositis (DM) (61–64). Future research is required to determine whether, based on the shared pathologic features of these ILDs with APECED pneumonitis, these ILDs may also be responsive to the lymphocyte-directed therapy that is effective in APECED pneumonitis and GLILD. In addition, whether other primary immune dysregulatory disorders that manifest with ILD such as STAT3 gain-of-function (GOF), CTLA4 haploinsufficiency, and LRBA deficiency share common immunopathological mechanisms with APECED pneumonitis merits future investigation (65–68).
Conclusion
Herein, we highlighted clinical, radiographic, pulmonary function, autoantibody, immunological, and histological abnormalities of APECED pneumonitis, a previously-unrecognized manifestation of AIRE-deficiency that causes significant morbidity and mortality when untreated. Periodic screening with chest CT and bronchoscopic performance of endobronchial biopsies to reveal the characteristic compartmentalized immunopathology of pneumonitis have important implications for early diagnosis and initiation of lymphocyte-directed immunomodulation that can remit pneumonitis and prevent irreversible pulmonary complications. The common immunological and histological features between APECED pneumonitis and ILDs seen in secondary AIRE-deficiency states (thymoma, RAG deficiency), and certain polygenic autoimmune disorders (SS, UC, SLE, DM) suggest that the pathogenesis of autoimmune lung disease is shared among disorders of central immune tolerance and show promise for the potential efficacy of a similar lymphocyte-directed immunomodulatory regimen for these common ILDs.
Author Contributions
EF conducted the literature review and wrote the initial draft of the manuscript. ML revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases, National Institutes of Health, USA.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the patients and their families for their participation in our study. The authors also thank Drs. Kenneth Olivier and Kevin Fennelly for their contributions to our understanding of APECED-associated pneumonitis.
References
1. Meager A, Visvalingam K, Peterson P, Moll K, Murumagi A, Krohn K, et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med (2006) 3:e289. doi: 10.1371/journal.pmed.0030289
2. Constantine GM, Lionakis MS. Lessons from primary immunodeficiencies: Autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol Rev (2019) 287:103–20. doi: 10.1111/imr.12714
3. Ferre EM, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight (2016) 1. doi: 10.1172/jci.insight.88782
4. Chascsa DM, Ferre EMN, Hadjiyannis Y, Alao H, Natarajan M, Quinones M, et al. APECED-Associated Hepatitis: Clinical, Biochemical, Histological and Treatment Data from a Large Predominantly American Cohort. Hepatology (2020). doi: 10.1002/hep.31421
5. Ferre EMN, Break TJ, Burbelo PD, Allgauer M, Kleiner DE, Jin D, et al. Lymphocyte-driven regional immunopathology in pneumonitis caused by impaired central immune tolerance. Sci Transl Med (2019) 11. doi: 10.1126/scitranslmed.aav5597
6. Cetani F, Barbesino G, Borsari S, Pardi E, Cianferotti L, Pinchera A, et al. A novel mutation of the autoimmune regulator gene in an Italian kindred with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, acting in a dominant fashion and strongly cosegregating with hypothyroid autoimmune thyroiditis. J Clin Endocrinol Metab (2001) 86:4747–52. doi: 10.1210/jcem.86.10.7884
7. Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med (1990) 322:1829–36. doi: 10.1056/NEJM199006283222601
8. Perheentupa J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab (2006) 91:2843–50. doi: 10.1210/jc.2005-2611
9. Wolff AS, Erichsen MM, Meager A, Magitta NF, Myhre AG, Bollerslev J, et al. Autoimmune polyendocrine syndrome type 1 in Norway: phenotypic variation, autoantibodies, and novel mutations in the autoimmune regulator gene. J Clin Endocrinol Metab (2007) 92:595–603. doi: 10.1210/jc.2006-1873
10. Dominguez M, Crushell E, Ilmarinen T, Mcgovern E, Collins S, Chang B, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in the Irish population. J Pediatr Endocrinol Metab (2006) 19:1343–52. doi: 10.1515/JPEM.2006.19.11.1343
11. Meloni A, Willcox N, Meager A, Atzeni M, Wolff AS, Husebye ES, et al. Autoimmune polyendocrine syndrome type 1: an extensive longitudinal study in Sardinian patients. J Clin Endocrinol Metab (2012) 97:1114–24. doi: 10.1210/jc.2011-2461
12. N FM, Pura M, A SBW, Vanuga P, Meager A, M.K. P, et al. Autoimmune polyendocrine syndrome type I in Slovakia: relevance of screening patients with autoimmune Addison’s disease. Eur J Endocrinol (2008) 158:705–9. doi: 10.1530/EJE-07-0843
13. Zlotogora J, Shapiro MS. Polyglandular autoimmune syndrome type I among Iranian Jews. J Med Genet (1992) 29:824–6. doi: 10.1136/jmg.29.11.824
14. Orlova EM, Bukina AM, Kuznetsova ES, Kareva MA, Zakharova EU, Peterkova VA, et al. Autoimmune polyglandular syndrome type 1 in Russian patients: clinical variants and autoimmune regulator mutations. Horm Res Paediatr (2010) 73:449–57. doi: 10.1159/000313585
15. Pearce SH, Cheetham T, Imrie H, Vaidya B, Barnes ND, Bilous RW, et al. A common and recurrent 13-bp deletion in the autoimmune regulator gene in British kindreds with autoimmune polyendocrinopathy type 1. Am J Hum Genet (1998) 63:1675–84. doi: 10.1086/302145
16. Myhre AG, Halonen M, Eskelin P, Ekwall O, Hedstrand H, Rorsman F, et al. Autoimmune polyendocrine syndrome type 1 (APS I) in Norway. Clin Endocrinol (Oxf) (2001) 54:211–7. doi: 10.1046/j.1365-2265.2001.01201.x
17. Podkrajsek KT, Bratanic N, Krzisnik C, Battelino T. Autoimmune regulator-1 messenger ribonucleic acid analysis in a novel intronic mutation and two additional novel AIRE gene mutations in a cohort of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients. J Clin Endocrinol Metab (2005) 90:4930–5. doi: 10.1210/jc.2005-0418
18. Stolarski B, Pronicka E, Korniszewski L, Pollak A, Kostrzewa G, Rowinska E, et al. Molecular background of polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome in a Polish population: novel AIRE mutations and an estimate of disease prevalence. Clin Genet (2006) 70:348–54. doi: 10.1111/j.1399-0004.2006.00690.x
19. Valenzise M, Fierabracci A, Cappa M, Porcelli P, Barcellona R, De Luca F, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy: report of seven additional sicilian patients and overview of the overall series from sicily. Horm Res Paediatr (2014) 82:127–32. doi: 10.1159/000363537
20. Bruserud O, Oftedal BE, Landegren N, Erichsen MM, Bratland E, Lima K, et al. A Longitudinal Follow-up of Autoimmune Polyendocrine Syndrome Type 1. J Clin Endocrinol Metab (2016) 101:2975–83. doi: 10.1210/jc.2016-1821
21. Popler J, Alimohammadi M, Kampe O, Dalin F, Dishop MK, Barker JM, et al. Autoimmune polyendocrine syndrome type 1: Utility of KCNRG autoantibodies as a marker of active pulmonary disease and successful treatment with rituximab. Pediatr Pulmonol (2012) 47:84–7. doi: 10.1002/ppul.21520
22. Friedman TC, Thomas PM, Fleisher TA, Feuillan P, Parker RI, Cassorla F, et al. Frequent occurrence of asplenism and cholelithiasis in patients with autoimmune polyglandular disease type I. Am J Med (1991) 91:625–30. doi: 10.1016/0002-9343(91)90215-J
23. Huibregtse KE, Wolfgram P, Winer KK, Connor EL. Polyglandular autoimmune syndrome type I - a novel AIRE mutation in a North American patient. J Pediatr Endocrinol Metab (2014) 27:1257–60. doi: 10.1515/jpem-2013-0328
24. Neufeld M, Maclaren NK, Blizzard RM. Two types of autoimmune Addison’s disease associated with different polyglandular autoimmune (PGA) syndromes. Medicine (Baltimore) (1981) 60:355–62. doi: 10.1097/00005792-198109000-00003
25. Orlova EM, Sozaeva LS, Kareva MA, Oftedal BE, Wolff ASB, Breivik L, et al. Expanding the Phenotypic and Genotypic Landscape of Autoimmune Polyendocrine Syndrome Type 1. J Clin Endocrinol Metab (2017) 102:3546–56. doi: 10.1210/jc.2017-00139
26. De Luca F, Valenzise M, Alaggio R, Arrigo T, Crisafulli G, Salzano G, et al. Sicilian family with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) and lethal lung disease in one of the affected brothers. Eur J Pediatr (2008) 167:1283–8. doi: 10.1007/s00431-008-0668-3
27. Alimohammadi M, Dubois N, Skoldberg F, Hallgren A, Tardivel I, Hedstrand H, et al. Pulmonary autoimmunity as a feature of autoimmune polyendocrine syndrome type 1 and identification of KCNRG as a bronchial autoantigen. Proc Natl Acad Sci U S A (2009) 106:4396–401. doi: 10.1073/pnas.0809986106
28. Aaltonen J, Horelli-Kuitunen N, Fan JB, Bjorses P, Perheentupa J, Myers R, et al. High-resolution physical and transcriptional mapping of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy locus on chromosome 21q22.3 by FISH. Genome Res (1997) 7:820–9. doi: 10.1101/gr.7.8.820
29. Bjorses P, Aaltonen J, Horelli-Kuitunen N, Yaspo ML, Peltonen L. Gene defect behind APECED: a new clue to autoimmunity. Hum Mol Genet (1998) 7:1547–53. doi: 10.1093/hmg/7.10.1547
30. Oftedal BE, Hellesen A, Erichsen MM, Bratland E, Vardi A, Perheentupa J, et al. Dominant Mutations in the Autoimmune Regulator AIRE Are Associated with Common Organ-Specific Autoimmune Diseases. Immunity (2015) 42:1185–96. doi: 10.1016/j.immuni.2015.04.021
31. Abbott JK, Huoh YS, Reynolds PR, Yu L, Rewers M, Reddy M, et al. Dominant-negative loss of function arises from a second, more frequent variant within the SAND domain of autoimmune regulator (AIRE). J Autoimmun (2018) 88:114–20. doi: 10.1016/j.jaut.2017.10.010
32. Waterfield M, Khan IS, Cortez JT, Fan U, Metzger T, Greer A, et al. The transcriptional regulator Aire coopts the repressive ATF7ip-MBD1 complex for the induction of immunotolerance. Nat Immunol (2014) 15:258–65. doi: 10.1038/ni.2820
33. Proekt I, Miller CN, Jeanne M, Fasano KJ, Moon JJ, Lowell CA, et al. LYN- and AIRE-mediated tolerance checkpoint defects synergize to trigger organ-specific autoimmunity. J Clin Invest (2016) 126:3758–71. doi: 10.1172/JCI84440
34. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science (2002) 298:1395–401. doi: 10.1126/science.1075958
35. Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The cellular mechanism of Aire control of T cell tolerance. Immunity (2005) 23:227–39. doi: 10.1016/j.immuni.2005.07.005
36. Mathis D, Benoist C. Aire. Annu Rev Immunol (2009) 27:287–312. doi: 10.1146/annurev.immunol.25.022106.141532
37. Devoss JJ, Shum AK, Johannes KP, Lu W, Krawisz AK, Wang P, et al. Effector mechanisms of the autoimmune syndrome in the murine model of autoimmune polyglandular syndrome type 1. J Immunol (2008) 181:4072–9. doi: 10.4049/jimmunol.181.6.4072
38. Vazquez SE, Ferre EM, Scheel DW, Sunshine S, Miao B, Mandel-Brehm C, et al. Identification of novel, clinically correlated autoantigens in the monogenic autoimmune syndrome APS1 by proteome-wide PhIP-Seq. Elife (2020) 9. doi: 10.7554/eLife.55053
39. Sng J, Ayoglu B, Chen JW, Schickel JN, Ferre EMN, Glauzy S, et al. AIRE expression controls the peripheral selection of autoreactive B cells. Sci Immunol (2019) 4. doi: 10.1126/sciimmunol.aav6778
40. Gavanescu I, Benoist C, Mathis D. B cells are required for Aire-deficient mice to develop multi-organ autoinflammation: A therapeutic approach for APECED patients. Proc Natl Acad Sci U S A (2008) 105:13009–14. doi: 10.1073/pnas.0806874105
41. Meyer S, Woodward M, Hertel C, Vlaicu P, Haque Y, Karner J, et al. AIRE-Deficient Patients Harbor Unique High-Affinity Disease-Ameliorating Autoantibodies. Cell (2016) 166:582–95. doi: 10.1016/j.cell.2016.06.024
42. Kisand K, Link M, Wolff AS, Meager A, Tserel L, Org T, et al. Interferon autoantibodies associated with AIRE deficiency decrease the expression of IFN-stimulated genes. Blood (2008) 112:2657–66. doi: 10.1182/blood-2008-03-144634
43. Landegren N, Sharon D, Freyhult E, Hallgren A, Eriksson D, Edqvist PH, et al. Proteome-wide survey of the autoimmune target repertoire in autoimmune polyendocrine syndrome type 1. Sci Rep (2016) 6:20104. doi: 10.1038/srep20104
44. Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med (2010) 207:291–7. doi: 10.1084/jem.20091983
45. Betterle C, Dal Pra C, Mantero F, Zanchetta R. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev (2002) 23:327–64. doi: 10.1210/edrv.23.3.0466
46. Yu L, Brewer KW, Gates S, Wu A, Wang T, Babu SR, et al. DRB1*04 and DQ alleles: expression of 21-hydroxylase autoantibodies and risk of progression to Addison’s disease. J Clin Endocrinol Metab (1999) 84:328–35. doi: 10.1210/jcem.84.1.5414
47. Winqvist O, Karlsson FA, Kampe O. 21-Hydroxylase, a major autoantigen in idiopathic Addison’s disease. Lancet (1992) 339:1559–62. doi: 10.1016/0140-6736(92)91829-W
48. Shum AK, Alimohammadi M, Tan CL, Cheng MH, Metzger TC, Law CS, et al. BPIFB1 is a lung-specific autoantigen associated with interstitial lung disease. Sci Transl Med (2013) 5:206ra139. doi: 10.1126/scitranslmed.3006998
49. Guan WJ, Gao YH, Xu G, Lin ZY, Tang Y, Gu YY, et al. Sputum matrix metalloproteinase-8 and -9 and tissue inhibitor of metalloproteinase-1 in bronchiectasis: clinical correlates and prognostic implications. Respirology (2015) 20:1073–81. doi: 10.1111/resp.12582
50. Bergin DA, Hurley K, Mehta A, Cox S, Ryan D, O’neill SJ, et al. Airway inflammatory markers in individuals with cystic fibrosis and non-cystic fibrosis bronchiectasis. J Inflammation Res (2013) 6:1–11. doi: 10.2147/JIR.S40081
51. Shum AK, Devoss J, Tan CL, Hou Y, Johannes K, O’gorman CS, et al. Identification of an autoantigen demonstrates a link between interstitial lung disease and a defect in central tolerance. Sci Transl Med (2009) 1:9ra20. doi: 10.1126/scitranslmed.3000284
52. Martin SI, Marty FM, Fiumara K, Treon SP, Gribben JG, Baden LR. Infectious complications associated with alemtuzumab use for lymphoproliferative disorders. Clin Infect Dis (2006) 43:16–24. doi: 10.1086/504811
53. Maus MV, Lionakis MS. Infections associated with the new ‘nibs and mabs’ and cellular therapies. Curr Opin Infect Dis (2020) 33:281–9. doi: 10.1097/QCO.0000000000000656
54. Chase NM, Verbsky JW, Hintermeyer MK, Waukau JK, Tomita-Mitchell A, Casper JT, et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol (2013) 33:30–9. doi: 10.1007/s10875-012-9755-3
55. Wolff AS, Karner J, Owe JF, Oftedal BE, Gilhus NE, Erichsen MM, et al. Clinical and serologic parallels to APS-I in patients with thymomas and autoantigen transcripts in their tumors. J Immunol (2014) 193:3880–90. doi: 10.4049/jimmunol.1401068
56. Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest (2015) 125:4135–48. doi: 10.1172/JCI80477
57. De Ravin SS, Cowen EW, Zarember KA, Whiting-Theobald NL, Kuhns DB, Sandler NG, et al. Hypomorphic Rag mutations can cause destructive midline granulomatous disease. Blood (2010) 116:1263–71. doi: 10.1182/blood-2010-02-267583
58. Delmonte OM, Schuetz C, Notarangelo LD. RAG Deficiency: Two Genes, Many Diseases. J Clin Immunol (2018) 38:646–55. doi: 10.1007/s10875-018-0537-4
59. Maiolo C, Fuso L, Benedetto RT, Boniello V, Basso S, Granieri AM, et al. A case of nonspecific interstitial pneumonia associated with thymoma. Sarcoidosis Vasc Diffuse Lung Dis (2003) 20:75–6.
60. Gonlugur U, Sahin E, Yildiz E, Gonlugur TE. Early autoimmune complications after thymomectomy in a patient with interstitial lung disease. Case report. Acta Microbiol Immunol Hung (2006) 53:105–11. doi: 10.1556/AMicr.53.2006.1.8
61. Dong X, Gao YL, Lu Y, Zheng Y. Characteristics of primary Sjogren’s syndrome related lymphocytic interstitial pneumonia. Clin Rheumatol (2020) 1:1–12. doi: 10.1007/s10067-020-05236-8
62. Cheema GS, Quismorio FP Jr. Interstitial lung disease in systemic lupus erythematosus. Curr Opin Pulm Med (2000) 6:424–9. doi: 10.1097/00063198-200009000-00007
63. Papiris SA, Maniati M, Constantopoulos SH, Roussos C, Moutsopoulos HM, Skopouli FN. Lung involvement in primary Sjogren’s syndrome is mainly related to the small airway disease. Ann Rheum Dis (1999) 58:61–4. doi: 10.1136/ard.58.1.61
64. Douglas WW, Tazelaar HD, Hartman TE, Hartman RP, Decker PA, Schroeder DR, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med (2001) 164:1182–5. doi: 10.1164/ajrccm.164.7.2103110
65. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood (2015) 125:591–9. doi: 10.1182/blood-2014-09-602763
66. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (2014) 345:1623–7. doi: 10.1126/science.1255904
67. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med (2014) 20:1410–6. doi: 10.1038/nm.3746
Keywords: Autoimmune-polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), autoimmune polyglandular syndrome type-1 (APS-1), autoimmune regulator (AIRE), pneumonitis, interstitial lung disease, bronchiectasis
Citation: Ferré EMN and Lionakis MS (2021) An AIREless Breath: Pneumonitis Caused by Impaired Central Immune Tolerance. Front. Immunol. 11:609253. doi: 10.3389/fimmu.2020.609253
Received: 22 September 2020; Accepted: 01 December 2020;
Published: 27 January 2021.
Edited by:
John R. Hurst, University College London, United KingdomReviewed by:
Tadashi Ariga, Hokkaido University, JapanEleonora Gambineri, University of Florence, Italy
Copyright © 2021 Ferré and Lionakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michail S. Lionakis, bGlvbmFraXNtQG1haWwubmloLmdvdg==