 Anna Villa
Anna Villa Valentina Capo
Valentina Capo Maria Carmina Castiello
Maria Carmina Castiello- 1San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), IRCCS San Raffaele Scientific Institute, Milan, Italy
- 2Istituto di Ricerca Genetica e Biomedica, Consiglio Nazionale delle Ricerche (IRGB-CNR), Milan, Italy
Genetic defects in recombination activating genes (RAG) 1 and 2 cause a broad spectrum of severe immune defects ranging from early severe and repeated infections to inflammation and autoimmune manifestations. A correlation between in vitro recombination activity and immune phenotype has been described. Hematopoietic cell transplantation is the treatment of care; however, the availability of next generation sequencing and whole genome sequencing has allowed the identification of novel genetic RAG variants in immunodeficient patients at various ages, raising therapeutic questions. This review addresses the recent advances of novel therapeutic approaches for RAG deficiency. As conventional myeloablative conditioning regimens are associated with acute toxicities and transplanted-related mortality, innovative minimal conditioning regimens based on the use of monoclonal antibodies are now emerging and show promising results. To overcome shortage of compatible donors, gene therapy has been developed in various RAG preclinical models. Overall, the transplantation of autologous gene corrected hematopoietic precursors and the use of non-genotoxic conditioning will open a new era, offering a cure to an increasing number of RAG patients regardless of donor availability and severity of clinical conditions.
Introduction
Effective adaptive immunity relies on the ability of T and B lymphocytes to express the vast majority of antigen receptors. During differentiation, T and B cells assemble T cell antigen receptor (TCR) and B cell receptor (BCR) respectively, by a complex process named V(D)J recombination that recognizes each segment of V, D and J flanked by recombination signal sequences (RSSs) (1). The recombination activating genes (RAG) 1 and 2, the first players of this molecular process, form a complex and introduce a DNA double strand break (DSB) in the RSSs giving rise to a diverse repertoire of antigen specific receptors. Null mutations in RAG genes cause an arrest at DN3 and pre-B1 stage of T and B cell development respectively (2). The T- B- natural killer (NK)+ severe combined immunodeficiency (SCID) phenotype is characterized by repeated severe infections caused by common viral pathogens and opportunistic pathogens that lead to death in the absence of hematopoietic stem cell transplantation (HSCT), which represents the treatment of choice (3).
Missense mutations impairing RAG functions while permitting occasional recombination activity lead to the generation and expansion of oligoclonal T cell population. Extensive molecular studies have defined the biochemical effect of amino acid changes on the recombination activity, providing evidence that residual activity sustained at least by one allele can lead to a peculiar immune phenotype named Omenn syndrome (OS) (4, 5). These patients present severe erythroderma, lymphadenopathy with hepatosplenomegaly, colitis, repeated infections and inflammatory pneumonitis. Activated oligoclonal and autoreactive T cells circulate in the peripheral blood and tend to migrate to the gut and skin mainly contributing to tissue damage that correlates with the severity of the disease (6, 7). IgM, IgA and IgG are usually absent or barely detectable in the serum but elevated IgE levels are found despite lack of circulating B cells (8, 9). Recently, next generation sequence identified new forms of RAG-SCID presenting with a milder phenotype than the classical signs of Omenn syndrome (10, 11). The description of these cases has further broadened the spectrum of clinical manifestations caused by RAG mutations, posing diagnostic and therapeutic questions (12, 13).
Leaky or atypical SCID (AS) patients harbor missense mutations and in the majority of these patients the diagnosis is delayed, with RAG mutations identified in childhood (median 5 years) or even in adolescent or adult individuals (10, 13, 14). Of note, while autoimmune manifestations are rare in null SCID, cytopenia and autoimmune hemolytic anemia have been frequently reported in AS patients (15) and in some cases vasculitis resulting in digital necrosis have been reported (16). Other RAG-associated phenotypes include autoimmune cytopenia and oligoclonal expansion of TCR expressing γδ T cells in disseminated CMV infection (17, 18) or specific antibody and autoantibody production (19). Furthermore, Schuetz et al. reported granuloma formation in internal organs, skin, and mucous membranes in three unrelated females with severe viral infections and B cells lymphoma (20). Presence of the rubella virus vaccine strain has been demonstrated in the granulomas of some of these patients (21). This condition referred as “combined immunodeficiency with granuloma and/or autoimmunity” (CID-G/AI) may associate with autoimmune cytopenia and other autoimmune manifestations including myopathy and nephrotic syndrome (15). Finally, biallelic RAG mutations have been found in patients with idiopathic CD4 T cell lymphopenia (22), IgA deficiency (23, 24), hyper IgM syndrome (25) and impaired antibody production against polysaccharide antigens (24).
Extensive studies in mouse models have contributed to understand the in vivo effect of amino acid changes and the impact of RAG defects on the immune dysregulation (5, 11). Based on the evolutionary conservation of amino acid change identified in various clinical conditions, several groups have reproduced hypomorphic mutations in mouse models [reviewed in (11)]: the Rag2R229Q/R229Q mouse that fully recapitulates the clinical manifestations of OS (26); the spontaneous mouse mutant carrying a homozygous point mutation (R972Q) in the Rag1 gene (27); the Rag1S723C/S723C showing profound B cell lymphopenia in the presence of significant serum levels of immunoglobulins and activated oligoclonal T cells (28). Novel mutants carrying mutations at C-terminal domain of RAG1 and reproducing amino acid changes found in patients with CID-G/AI have been generated by gene editing (F971L, R972Q and R972W) corresponding to the human mutations (F974L, R975Q and R975W) (29). These models, associated with analysis of RAG patients, have allowed to dissect the defective mechanisms of central and peripheral tolerance and the contributive role of environment to the disease severity (7, 30). Parallel studies have identified the broad Th1/Th2/Th17 inflammatory signature, highlighting the complex scenario and its clinical implication (7). Therefore, these models have been exploited to assess the efficacy and safety of novel therapeutic strategies. Recent advances in conditioning regimens and availability of donor cells source have dramatically improved the outcome of HSCT that in case of null RAG forms was significantly worse than other forms of SCID (31).
In RAG SCID, engraftment of donor cells requires myeloablative regimen, that eliminates arrested precursors fully occupying bone marrow and thymic niches. However, chemotherapy may cause severe organ damage, worsening the infections and limiting access to transplant. Conventional gene addition or gene editing of autologous stem cells represent promising technologies that might overcome the limited number of available donors and offer a cure to all patients. Conditioning regimens based on monoclonal antibodies (mAb) specifically targeting hematopoietic stem and progenitor cells (HSPCs) are an attractive alternative to conventional myeloablation, in order to obtain depletion while preserving hematopoietic tissue homeostasis (32) (Figure 1). Overall, the development of these novel therapies will pave the way toward a new scenario of treatment to all RAG patients offering a cure regardless of the severity of the disease and age of treatment.
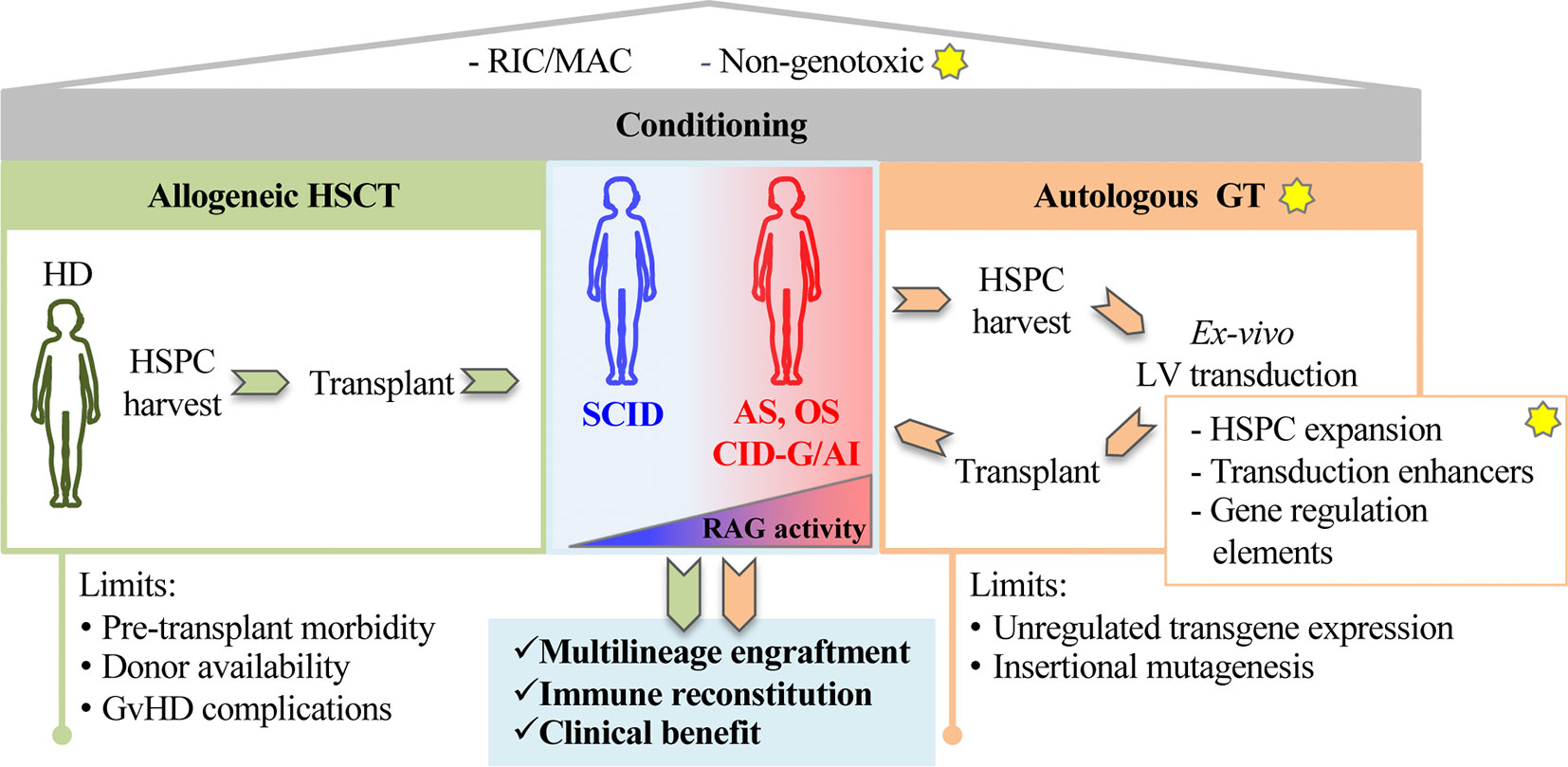
Figure 1 The figure shows principles of stem cell based gene therapy for recombination activating genes (RAG) disorders. The yellow stars indicate proposed innovative steps for the treatment of RAG-severe combined immunodeficiency (SCID). A preparative regimen as Reduced Intensity Conditioning (RIC) or myeloablative conditioning (MAC) is required to deplete endogenous hematopoietic precursors in RAG-deficient niches. Non-genotoxic compounds (anti-cKit mAb, anti-CD45 SAP) recently tested in preclinical models of RAG1 deficiencies represent a step forward toward new and safer form of conditioning. Patients carrying null RAG defect (SCID) or hypomorphic RAG defects (AS, OS, CID-G/AI) undergo conventional allogeneic hematopoietic stem cell transplantation (HSCT) (left green panel). Alternatively, autologous gene therapy (GT, right orange panel) CD34+ cells, isolated from bone marrow or mobilized peripheral blood, cultured and transduced in GMP conditions, are reinfused in the patient. New methods and reagents aimed at expanding HSPCs, enhancing transduction levels and regulating gene expression are being developed to boost GT efficiency. Limits of allogeneic HSCT and autologous GT are indicated at the bottom of the figure.
Conventional Stem Cell Transplantation
Hematopoietic stem cell transplantation is the only curative treatment available for RAG deficiencies up to date (Table 1). Similarly to other SCIDs, highest survival is observed in patients transplanted before onset of infection and early in life (3.5 months of age or younger), regardless of donor or conditioning (43, 44). In this scenario, newborn screening is fundamental to expedite early treatment (45, 46).
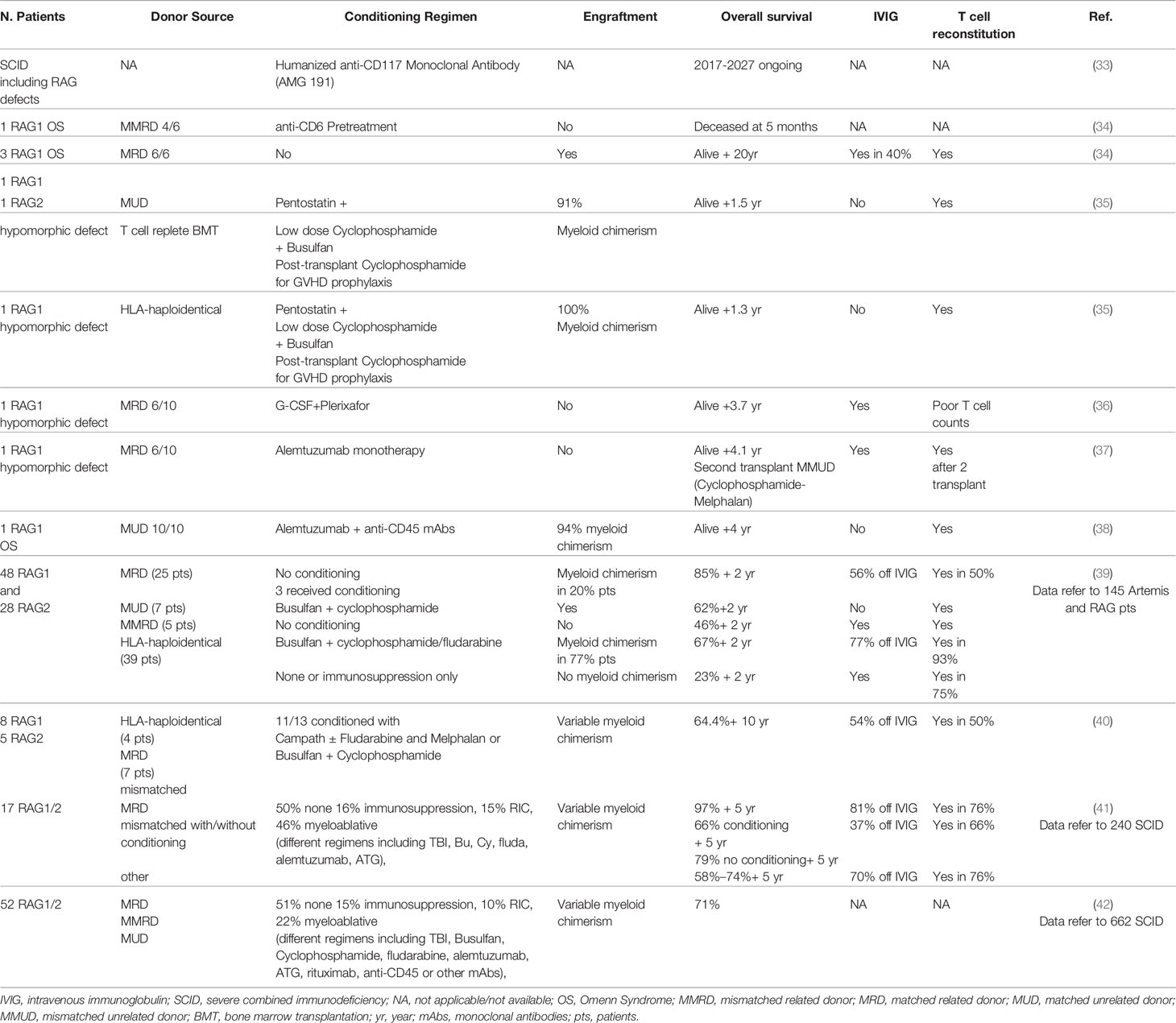
Table 1 Main hematopoietic stem cell transplantation (HSCT) approaches and outcomes in recombination activating genes (RAG) patients.
A study conducted in three main centers reported an 88% survival without recurrent infections in HLA-identical grafts without conditioning (39). Remarkably, half of the patients still required intravenous immunoglobulin (IVIG) and had incomplete T cell reconstitution, indicating that pre-transplant conditioning is necessary to eliminate immature progenitors. Conversely, patients receiving T-cell-depleted HLA-haploidentical family donors after myeloablative conditioning (busulfan combined with cyclophosphamide/fludarabine), had significant lower survival (63%) but showed higher T-cell reconstitution without the need for IVIG. Chronic GvHD and autoimmunity remained the most frequent late effects, observed in 24% of patients (39). The retrospective study from the Primary Immune Deficiency Treatment Consortium (PIDTC) confirmed poorer immune reconstitution in absence of conditioning, with ~80% overall survival (42).
Recently, a smaller cohort of 11 RAG patients receiving matched or haploidentical graft after pre-HSCT chemotherapy, showed similar overall survival (64.4%). Consistently, conditioning was associated with poorer overall survival and higher long-term side effects but better immune reconstitution (40). This observation has been reported in other SCIDs, as described in a large multicenter study on 240 SCID patients (including 17 RAG patients) (44).
Overall, myeloablative treatment can cause severe complications and mortality, while absence of pre-transplant conditioning may lead to poor immune reconstitution, due to competition with immature progenitors in bone marrow and thymic niches of RAG patients.
Approaching Non-Genotoxic Conditioning for RAG Deficiencies
Although current conditioning regimens may cause acute and chronic toxicities, lymphoid progenitors limit proper engraftment of donor cells (11, 47–49), highlighting the urgent need for safe transplant protocols (Table 1). Biologic approaches based on mAbs specifically targeting HSPC while sparing non-hematopoietic cells are emerging as attractive conditionings for safely improving HSCT outcome. HSPC-depleting mAb as the anti-CD117 ACK2 (c-kit antagonist) allowed increased chimerism in Rag2-/-γc-/- mice, but not in immunocompetent mice (50) and X-linked chronic granulomatous disease (X-CGD) mice (51). Conversely, ACK2 synergistically acted with low-dose irradiation or CD47 blockade to allow higher engraftment in X-CGD (51) or immunocompetent mice (52), respectively. These data paved the way for the clinical trial using anti-CD117 antibody currently ongoing to treat SCID patients (NCT02963064) (33), which provides the proof of concept that a humanized mAb can safely clear human hematopoietic stem cells (HSC) niches facilitating donor cell engraftment in two T-B-NK+ SCID patients (with Artemis mutations) (53).
In the same direction, CD45 mAbs have been tested in a RAG1-deficient patient with immune dysregulation showing promising results (38). Alternative reduced intensity conditioning, including mAb combined with or without chemotherapy, has been tested in few RAG patients. The outcome of this treatment was variable, and some patients developed post-transplant severe complications (34–37, 54).
Antibody-Drug Conjugates (ADCs), extensively applied in cancer therapy, have recently proposed as non-myeloablative agents. CD117-ADC was exploited to deplete host HSPCs while preserving host immunity in immunocompetent mice (55), in MHC-mismatched allotransplantation (56) and in hemophilia A gene therapy (GT) mice (57). Encouraging results in non-human primates (58, 59) support ADC applicability in the clinical setting. In parallel, anti-CD45 mAb coupled with Saporin (CD45-SAP), a ribosome inactivating protein lacking the cell-entry domain and toxic only upon receptor-mediated internalization, has been exploited to efficiently deplete HSPCs enabling multilineage engraftment with minimal organ toxicity in immunocompetent mice (60). Due to CD45 expression pattern, CD45-SAP can be a good candidate in CID-G/AI patients to target autoreactive T cells, improving HSCT outcome and immune recovery. To this end, the efficacy of this compound was tested in null and hypomorhic Rag1 models, achieving multilineage engraftment and robust immune reconstitution while preserving thymic epithelial cell homeostasis. A synergistic effect on myeloid chimerism and immune recovery was achieved when CD45-SAP was combined with low-dose of irradiation (61). However, mild transient hepatotoxicity secondary to CD45-SAP (60, 61) or upon single saporin injection (62) or radiolabeled anti-CD45 antibody (63–65) have been observed posing clinical concerns.
Overall, while these preclinical models indicate ADCs as safer conditional regimens than conventional chemotherapy, further investigation is needed before moving to the clinical setting. In particular, future studies assessing the dosage, safety, and efficacy of other HSC-depleting ADCs, alone or in combination with other mAb-based conditioning agents, are needed to bring ADC conditioning in the clinical arena.
Gene Therapy of RAG Defects
The clinical spectrum of autoimmunity and hyper inflammation due to RAG mutations highlights the clinical need to offer a cure to patients without suitable donors or in critical clinical conditions.
Various groups have developed novel strategies, based on the hypothesis that gene corrected cells should acquire a selective advantage and overcome lymphocyte differentiation block. However, RAG tight regulation during cell cycle and expression level may constrain the clinical feasibility of GT. Ectopic or dysregulated RAG expression may lead to genotoxicity or immune dysregulation (66, 67). In the past, stable immune reconstitution in Rag1-/- mice was achieved using retroviral gene transfer of human RAG1 cDNA, but high transgene copy number was associated with a risk of lymphoproliferation (66). Based on this evidence, preclinical Rag1-/- models have been generated using self-inactivating (SIN) lentiviral vectors (LVs) carrying the human codon-optimized RAG1 cDNA (67–69) (Table 2). Different promoters have been tested to drive RAG expression: the human elongation factor 1α (EFS) promoter, the enhancer–promoter of the spleen-focus-forming virus (SFFV) and the ubiquitously acting chromatin opening element from the human HNRPA2B1-CBX3 locus (A2UCOE) (67, 73). Increased number and improved function of T cells were observed in the cohort of Rag1-/- mice transplanted with Lineage negative (Lin-) cells transduced with SFFV.coRAG1 and A2UCOE.coRAG1, with respect to EFS.coRAG1. However, poor B cell reconstitution was achieved with all promoters (68, 73). Despite reduced B cell number, GT mice showed increased production of IgM, IgG and IgA in the serum, antigen-specific antibody production upon challenges and polyclonal Vβ TCR repertoire. A parallel study using the same vectors reported contrasting results, showing inflammation, tissue cellular infiltrate and circulating anti-double strand DNA, resembling Omenn clinical features (67). Various factors may account for these discrepancies, including suboptimal transgene expression, partial immune reconstitution and immune dysregulation (73, 74). To improve LV titer and transduction efficiency, the Staal group switched to the CCL backbone, widely used in the clinics (69). They compared small and large scale production of the four SIN LVs carrying the following promoters: the phosphoglycerate kinase 1 promoter (PGK), the MND myeloproliferative sarcoma virus enhancer, the ubiquitous chromatin opening element (UCOE), Cbx3.MND (a tandem combination of UCOE and the MND promoter). The comparison identified the MND promoter as the optimal vector, able to reach a sufficient expression of the RAG1 transgene (with a vector copy number ~1) in order to obtain stable immune reconstitution of GT Rag1-/- treated mice. Despite low B cell number, gene therapy treated mice had normal Ig levels in the serum and showed a normalization of T cell specific antigen response. While polyclonal Vβ repertoire was restored, T cell counts in peripheral blood reached 30% of normal levels (69). Remarkably, in case of low RAG1 expression (mainly driven by PGK and UCOE promoters) 4 out of 9 GT mice developed skin erythroderma and wasting syndrome leading to death resembling observation previously reported in literature, further confirming the importance of achieving sufficient transgene expression. However, as previously discussed, irradiation and the expression of human RAG1 in mouse system may contribute to suboptimal immune reconstitution.
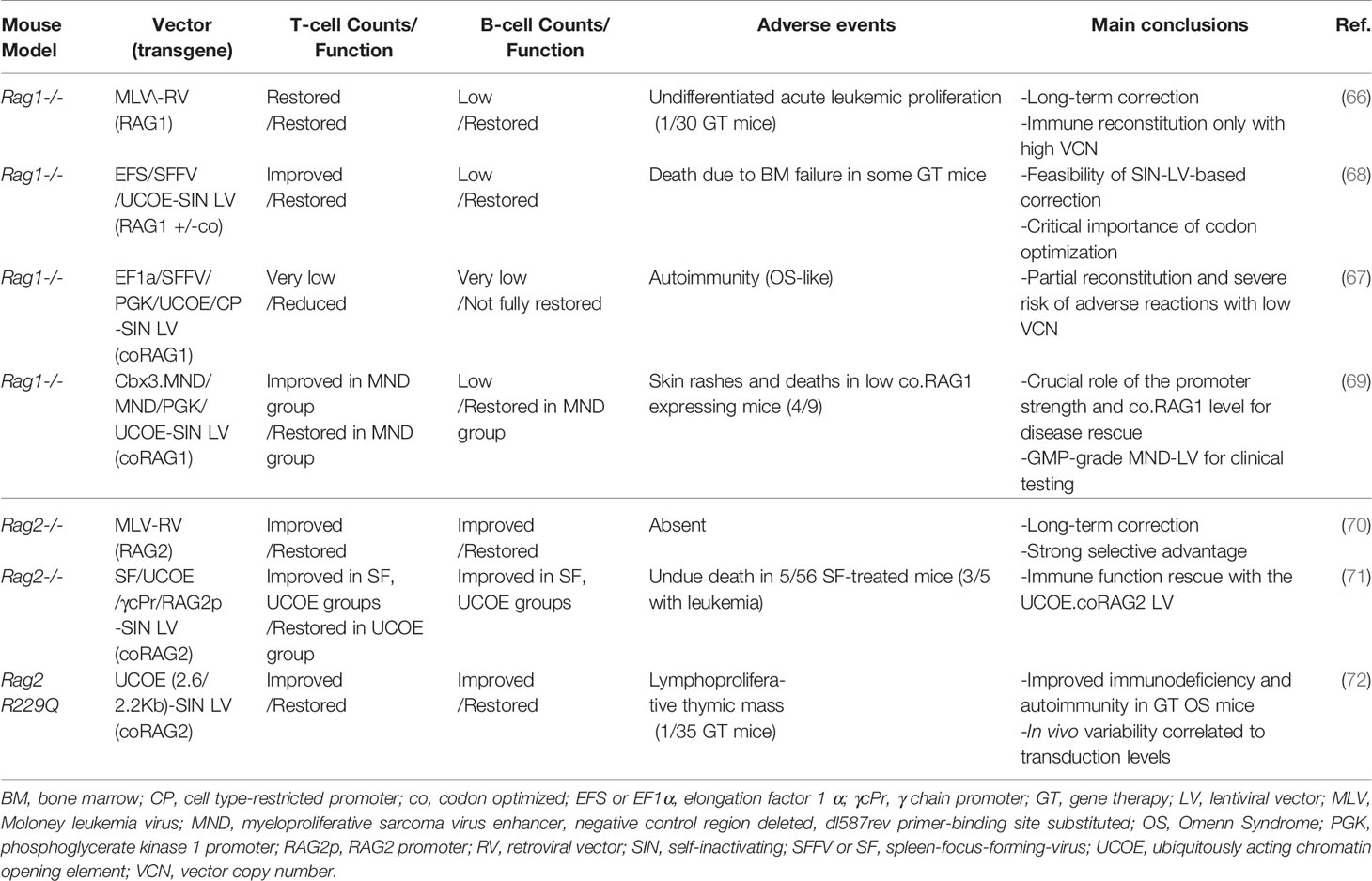
Table 2 Gene therapy preclinical studies in Rag1 and Rag2 mouse models.
Different LVs were also tested in CD34+ cells from RAG1 patients to test their ability to induce the differentiation of functional B and T cells in vivo in NSG mice. In a first report, Rag1 expression driven by EF1α allowed the differentiation of functional B cells in a minority of transplanted mice, while no information on T cells are reported (75). Recently, LV carrying MND promoter was used to transduce CD34+ cells from a hypomorphic RAG1 patient with residual B cells and no T cells, transplanted in a single NSG mouse. CD34+ GT cells could differentiate in polyclonal B cells and T cells, despite the low vector copy number, in line with the stronger MND promoter activity (69).
GT studies in hypomorphic Rag1 models will be instrumental to understand the efficacy of GT in atypical SCID or OS, which represent the majority of RAG patients. Finally, safety tests and long-term follow up of GT treated animals are required to further validate the use of RAG1 GT in the clinical setting.
With regard to RAG2, preliminary studies using a retroviral vector carrying human RAG2 cDNA showed stable immune reconstitution in the absence of detectable toxicity (70). Because of the genotoxicity of retroviral vectors (76, 77), a SIN LV carrying human codon optimized RAG2 cDNA driven by UCOE promoter has been developed showing promising data in the preclinical model of Rag2-/- mice in terms of immune reconstitution. GT Rag2-/- mice presented polyclonal Vβ TCR repertoire, increase of naïve T cells and redistribution of T cell subpopulation. Reduced B cell counts were accompanied by normal levels of IgM, IgG subclasses and antigen-specific antibodies production upon T-dependent and independent antigens (71).
Based on these promising results, the UCOE-RAG2co LV has been tested in the mouse model of OS, the Rag2R229Q/R229Q mutant (26). While GT OS mice showed decreased absolute T and B absolute counts as compared to wild-type mice, significant change in T cell distribution with a dramatic increase in naïve T cells and reduction in effector/memory T cells was obtained (72). Treated thymic displayed improvement in the structure with appearance of medullary compartment containing mature TEC expressing AIRE. Consistently, spectratyping indicated a polyclonal T cell repertoire. Finally, gene corrected mice responded properly to in vivo challenges. Remarkably, treated OS animals did not show cellular infiltration in the skin and gut indicating a resolution of Omenn clinical signs. Taken together, these data indicate the feasibility of lentiviral GT for RAG2 deficiencies, even in the context of residual recombination activity and in inflammatory conditions.
Conclusions and Future Directions
Autologous gene corrected stem cell transplant represents the next therapeutic step to treat RAG patients without suitable donors. Preclinical RAG models have provided instrumental data unrevealing advantages and drawbacks of novel cell-based therapies and non-genotoxic conditioning posing the basis for the future development of clinical GT trials.
HSPC expansion protocols assuring maintenance of stemness potential are currently being tested when limited donor cells are available, especially in case of cord blood donors (78, 79). Notably, these protocols can be exploited to decrease the burden of CD34+ cell harvest in very young patients in gene therapy and editing settings (80). Conventional GT approach has shown promising results in murine models, highlighting at the same time the need for physiological level of RAG expression while maintaining low vector copy number. Preclinical GT studies are now in progress to implement transgene expression. To this end, the recent description of the effect of immunomodulatory compounds is particularly relevant, leading to increased transduction levels in long-term HSC while preserving engraftment potential (81–83). Cyclosporines A and H have been recently demonstrated improve transduction without altering HSCP subpopulation composition nor the cell-cycle status (81, 82). Prostaglandin E2 (PGE2) allows enhanced transduction efficiency in one hit protocol thus limiting HSPC manipulation (83, 84). Consistently, PGE2 has been recently applied in a GT phase I/II clinical trial to treat mucopolysaccharidosis type I, Hurler Syndrome (85).
Although currently still challenging, genome editing at a specific locus is now emerging as a new potential technique that allows to insert the corrective sequence downstream its endogenous promoter thus maintaining the physiological expression of the gene of interest (86–89). The development of engineered nucleases, including Zinc Finger Nucleases (ZFNs), transcription activator like effector nucleases (TALEN) and clustered regularly interspaced short palindromic repeats (CRISPR-Cas9), associated with improvement in the efficiency of homology directed repair in HSCs (90) will allow to obtain rapid advances for the future transfer of this technique to the clinical setting.
Author Contribution
AV, VC, and MC designed and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was sponsored in part by EU H2020 grant RECOMB (755170-2), Telethon Core grant E2 project, RF-PE- 2016-02363691 and PRIN 2017 5XHBPN.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Schatz DG, Swanson PC. V(D)J Recombination: Mechanisms of Initiation. Annu Rev Genet (2011) 45:167–202. doi: 10.1146/annurev-genet-110410-132552
2. Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG mutations in human B cell-negative SCID. Sci (80- ) (1996) 274:97–9. doi: 10.1126/science.274.5284.97
3. Fischer A, Notarangelo LD, Neven B, Cavazzana M, Puck JM. Severe combined immunodeficiencies and related disorders. Nat Rev Dis Prim (2015) 1:15061. doi: 10.1038/nrdp.2015.61
4. Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, et al. Partial V(D)J recombination activity leads to omenn syndrome. Cell (1998) 93:885–96. doi: 10.1016/S0092-8674(00)81448-8
5. Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: Biochemistry and clinical implications. Nat Rev Immunol (2016) 16:234–46. doi: 10.1038/nri.2016.28
6. Rigoni R, Fontana E, Guglielmetti S, Fosso B, D’Erchia AM, Maina V, et al. Intestinal microbiota sustains inflammation and autoimmunity induced by hypomorphic RAG defects. J Exp Med (2016) 213:355–75. doi: 10.1084/jem.20151116
7. Rigoni R, Fontana E, Dobbs K, Marrella V, Taverniti V, Maina V, et al. Cutaneous barrier leakage and gut inflammation drive skin disease in Omenn syndrome. J Allergy Clin Immunol (2020) S0091-6749(20):30492–9. doi: 10.1016/j.jaci.2020.04.005
8. Cassani B, Poliani PL, Marrella V, Schena F, Sauer AV, Ravanini M, et al. Homeostatic expansion of autoreactive immunoglobulin-secreting cells in the Rag2 mouse model of Omenn syndrome. J Exp Med (2010) 207:1525–40. doi: 10.1084/jem.20091928
9. Walter JE, Rucci F, Patrizi L, Recher M, Regenass S, Paganini T, et al. Expansion of immunoglobulin-secreting cells and defects in B cell tolerance in Rag-dependent immunodeficiency. J Exp Med (2010) 207:1541–54. doi: 10.1084/jem.20091927
10. Delmonte OM, Villa A, Notarangelo LD. Immune dysregulation in patients with RAG deficiency and other forms of combined immune deficiency. Blood (2020) 135:610–9. doi: 10.1182/BLOOD.2019000923
11. Villa A, Notarangelo LD. RAG gene defects at the verge of immunodeficiency and immune dysregulation. Immunol Rev (2019) 287:73–90. doi: 10.1111/imr.12713
12. Tirosh I, Yamazaki Y, Frugoni F, Ververs FA, Allenspach EJ, Zhang Y, et al. Recombination activity of human recombination-activating gene 2 (RAG2) mutations and correlation with clinical phenotype. J Allergy Clin Immunol (2019) 143:726–35. doi: 10.1016/j.jaci.2018.04.027
13. Farmer JR, Foldvari Z, Ujhazi B, De Ravin SS, Chen K, Bleesing JJH, et al. Outcomes and Treatment Strategies for Autoimmunity and Hyperinflammation in Patients with RAG Deficiency. J Allergy Clin Immunol Pract (2019) 7:1970–85. doi: 10.1016/j.jaip.2019.02.038
14. Bulkhi AA, Dasso JF, Schuetz C, Walter JE. Approaches to patients with variants in RAG genes: from diagnosis to timely treatment. Expert Rev Clin Immunol (2019) 15:1033–46. doi: 10.1080/1744666X.2020.1670060
15. Delmonte OM, Schuetz C, Notarangelo LD. RAG Deficiency: Two Genes, Many Diseases. J Clin Immunol (2018) 38:646–55. doi: 10.1007/s10875-018-0537-4
16. Henderson LA, Frugoni F, Hopkins G, De Boer H, Pai SY, Lee YN, et al. Expanding the spectrum of recombination-activating gene 1 deficiency: A family with early-onset autoimmunity. J Allergy Clin Immunol (2013) 132(4):969–71. doi: 10.1016/j.jaci.2013.06.032
17. De Villartay JP, Lim A, Al-Mousa H, Dupont S, Déchanet-Merville J, Coumau-Gatbois E, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest (2005) 115:3291–9. doi: 10.1172/JCI25178
18. Ehl S, Schwarz K, Enders A, Duffner U, Pannicke U, Kühr J, et al. A variant of SCID with specific immune responses and predominance of γδ T cells. J Clin Invest (2005) 115:3140–8. doi: 10.1172/JCI25221
19. Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest (2015) 125:4135–48. doi: 10.1172/JCI80477
20. Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med (2008) 358:2030–8. doi: 10.1056/NEJMoa073966
21. Neven B, Pérot P, Bruneau J, Pasquet M, Ramirez M, Diana JS, et al. Cutaneous and visceral chronic granulomatous disease triggered by a rubella virus vaccine strain in children with primary immunodeficiencies. Clin Infect Dis (2017) 64:83–6. doi: 10.1093/cid/ciw675
22. Kuijpers TW, IJspeert H, Van Leeuwen EMM, Jansen MH, Hazenberg MD, Weijer KC, et al. Idiopathic CD4+ T lymphopenia without autoimmunity or granulomatous disease in the slipstream of RAG mutations. Blood (2011) 117:5892–6. doi: 10.1182/blood-2011-01-329052
23. Kato T, Crestani E, Kamae C, Honma K, Yokosuka T, Ikegawa T, et al. RAG1 Deficiency May Present Clinically as Selective IgA Deficiency. J Clin Immunol (2015) 35:280–8. doi: 10.1007/s10875-015-0146-4
24. Geier CB, Piller A, Linder A, Sauerwein KMT, Eibl MM, Wolf HM. Leaky RAG deficiency in adult patients with impaired antibody production against bacterial polysaccharide antigens. PloS One (2015) 10(7):e0133220. doi: 10.1371/journal.pone.0133220
25. Chou J, Hanna-Wakim R, Tirosh I, Kane J, Fraulino D, Lee YN, et al. A novel homozygous mutation in recombination activating gene 2 in 2 relatives with different clinical phenotypes: Omenn syndrome and hyper-IgM syndrome. J Allergy Clin Immunol (2012) 130:1414–6. doi: 10.1016/j.jaci.2012.06.012
26. Marrella V, Poliani PL, Casati A, Rucci F, Frascoli L, Gougeon ML, et al. A hypomorphic R229Q Rag2 mouse mutant recapitulates human Omenn syndrome. J Clin Invest (2007) 117:1260–9. doi: 10.1172/JCI30928
27. Khiong K, Murakami M, Kitabayashi C, Ueda N, Sawa SI, Sakamoto A, et al. Homeostatically proliferating CD4+ T cells are involved in the pathogenesis of an Omenn syndrome murine model. J Clin Invest (2007) 117:1270–81. doi: 10.1172/JCI30513
28. Giblin W, Chatterji M, Westfield G, Masud T, Theisen B, Cheng HL, et al. Leaky severe combined immunodeficiency and aberrant DNA rearrangements due to a hypomorphic RAG1 mutation. Blood (2009) 113:2965–75. doi: 10.1182/blood-2008-07-165167
29. Ott de Bruin LM, Bosticardo M, Barbieri A, Lin SG, Rowe JH, Poliani PL, et al. Hypomorphic Rag1 mutations alter the preimmune repertoire at early stages of lymphoid development. Blood (2018) 132:281–92. doi: 10.1182/blood-2017-12-820985
30. Rigoni R, Grassi F, Villa A, Cassani B. RAGs and BUGS: An alliance for autoimmunity. Gut Microbes (2016) 7:503–11. doi: 10.1080/19490976.2016.1228517
31. Gennery AR. The challenges presented by haematopoietic stem cell transplantation in children with primary immunodeficiency. Br Med Bull (2020) 135 (1):4–15. doi: 10.1093/bmb/ldaa017
32. Lum SH, Hoenig M, Gennery AR, Slatter MA. Conditioning Regimens for Hematopoietic Cell Transplantation in Primary Immunodeficiency. Curr Allergy Asthma Rep (2019) 19(11):52. doi: 10.1007/s11882-019-0883-1
33. AMG 191 Antibody Targeting Conditioning in SCID Patients . ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT02963064 (Accessed September 3, 2020).
34. Patel NC, Chinen J, Rosenblatt HM, Hanson IC, Brown BS, Paul ME, et al. Long-term outcomes of nonconditioned patients with severe combined immunodeficiency transplanted with HLA-identical or haploidentical bone marrow depleted of T cells with anti-CD6 mAb. J Allergy Clin Immunol (2008) 122:1185–93. doi: 10.1016/j.jaci.2008.10.030
35. Dimitrova D, Gea-Banacloche J, Steinberg SM, Sadler JL, Hicks SN, Carroll E, et al. Prospective Study of a Novel, Radiation-Free, Reduced-Intensity Bone Marrow Transplantation Platform for Primary Immunodeficiency Diseases. Biol Blood Marrow Transplant (2020) 26:94–106. doi: 10.1016/j.bbmt.2019.08.018
36. Dvorak CC, Horn BN, Puck JM, Czechowicz A, Shizuru JA, Ko RM. Cowan MJ. A trial of plerixafor adjunctive therapy in allogeneic hematopoietic cell transplantation with minimal conditioning for severe combined immunodeficiency. Pediatr Transplant (2014) 18:602–8. doi: 10.1111/petr.12309
37. Dvorak CC, Horn BN, Puck JM, Adams S, Veys P, Czechowicz A. Cowan MJ. A trial of alemtuzumab adjunctive therapy in allogeneic hematopoietic cell transplantation with minimal conditioning for severe combined immunodeficiency. Pediatr Transplant (2014) 18:609–16. doi: 10.1111/petr.12310
38. Straathof KC, Rao K, Eyrich M, Hale G, Bird P, Berrie E, et al. Haemopoietic stem-cell transplantation with antibody-based minimal-intensity conditioning: a phase 1/2 study. Lancet (2009) 374:912–20. doi: 10.1016/S0140-6736(09)60945-4
39. Schuetz C, Neven B, Dvorak CC, Leroy S, Ege MJ, Pannicke U, et al. SCID patients with ARTEMIS vs RAG deficiencies following HCT: Increased risk of late toxicity in ARTEMIS-deficient SCID. Blood (2014) 123:281–9. doi: 10.1182/blood-2013-01-476432
40. Abd Hamid IJ, Slatter MA, McKendrick F, Pearce MS, Gennery AR. Long-Term Health Outcome and Quality of Life Post-HSCT for IL7Rα-, Artemis-, RAG1- and RAG2-Deficient Severe Combined Immunodeficiency: a Single Center Report. J Clin Immunol (2018) 38:727–32. doi: 10.1007/s10875-018-0540-9
41. Hacein-Bey-Abina S, Pai SY, Gaspar HB, Armant M, Berry CC, Blanche S, et al. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med (2014) 371:1407–17. doi: 10.1056/NEJMoa1404588
42. Haddad E, Logan BR, Griffith LM, Buckley RH, Parrott RE, Prockop SE, et al. SCID genotype and 6-month posttransplant CD4 count predict survival and immune recovery. Blood (2018) 132:1737–49. doi: 10.1182/blood-2018-03-840702
43. Heimall J, Puck J, Buckley R, Fleisher TA, Gennery AR, Neven B, et al. Current Knowledge and Priorities for Future Research in Late Effects after Hematopoietic Stem Cell Transplantation (HCT) for Severe Combined Immunodeficiency Patients: A Consensus Statement from the Second Pediatric Blood and Marrow Transplant Consortium. Biol Blood Marrow Transplant (2017) 23:379–87. doi: 10.1016/j.bbmt.2016.12.619
44. Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med (2014) 371:434–46. doi: 10.1056/NEJMoa1401177
45. Castagnoli R, Delmonte OM, Calzoni E, Notarangelo LD. Hematopoietic Stem Cell Transplantation in Primary Immunodeficiency Diseases: Current Status and Future Perspectives. Front Pediatr (2019) 7:295. doi: 10.3389/fped.2019.00295
46. van der Burg M, Mahlaoui N, Gaspar HB, Pai SY. Universal Newborn Screening for Severe Combined Immunodeficiency (SCID). Front Pediatr (2019) 7:373. doi: 10.3389/fped.2019.00373
47. Severe N, Karabacak NM, Gustafsson K, Baryawno N, Courties G, Kfoury Y, et al. Stress-Induced Changes in Bone Marrow Stromal Cell Populations Revealed through Single-Cell Protein Expression Mapping. Cell Stem Cell (2019) 25:570–583.e7. doi: 10.1016/j.stem.2019.06.003
48. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol (2016) 18:607–18. doi: 10.1038/ncb3346
49. Liu A, Vosshenrich CAJ, Lagresle-Peyrou C, Malassis-Seris M, Hue C, Fischer A, et al. Competition within the early B-cell compartment conditions B-cell reconstitution after hematopoietic stem cell transplantation in nonirradiated recipients. Blood (2006) 108:1123–8. doi: 10.1182/blood-2006-01-0061
50. Czechowicz A, Kraft D, Weissman IL, Bhattacharya D. Efficient transplantation via antibody-based clearance of hematopoietic stem cell niches. Sci (80- ) (2007) 318:1296–9. doi: 10.1126/science.1149726
51. Xue X, Pech NK, Shelley WC, Srour EF, Yoder MC, Dinauer MC. Antibody targeting KIT as pretransplantation conditioning in immunocompetent mice. Blood (2010) 116:5419–22. doi: 10.1182/blood-2010-07-295949
52. Chhabra A, Ring AM, Weiskopf K, Schnorr PJ, Gordon S, Le AC, et al. Hematopoietic stem cell transplantation in immunocompetent hosts without radiation or chemotherapy. Sci Transl Med (2016) 8(351):351ra105. doi: 10.1126/scitranslmed.aae0501
53. Agarwal R, Dvorak CC, Prohaska S, Long-Boyle J, Kwon H-S, Brown J(M, et al. Toxicity-Free Hematopoietic Stem Cell Engraftment Achieved with Anti-CD117 Monoclonal Antibody Conditioning. Biol Blood Marrow Transplant (2019) 25:S92. doi: 10.1016/j.bbmt.2018.12.172
54. Haines HL, Bleesing JJ, Davies SM, Hornung L, Jordan MB, Marsh RA, et al. Outcomes of Donor Lymphocyte Infusion for Treatment of Mixed Donor Chimerism after a Reduced-Intensity Preparative Regimen for Pediatric Patients with Nonmalignant Diseases. Biol Blood Marrow Transplant (2015) 21:288–92. doi: 10.1016/j.bbmt.2014.10.010
55. Czechowicz A, Palchaudhuri R, Scheck A, Hu Y, Hoggatt J, Saez B, et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat Commun (2019) 10:617. doi: 10.1038/s41467-018-08201-x
56. Li Z, Czechowicz A, Scheck A, Rossi DJ, Murphy PM. Hematopoietic chimerism and donor-specific skin allograft tolerance after non-genotoxic CD117 antibody-drug-conjugate conditioning in MHC-mismatched allotransplantation. Nat Commun (2019) 10(1):616. doi: 10.1038/s41467-018-08202-w
57. Gao C, Schroeder JA, Xue F, Jing W, Cai Y, Scheck A, et al. Nongenotoxic antibody-drug conjugate conditioning enables safe and effective platelet gene therapy of hemophilia A mice. Blood Adv (2019) 3:2700–11. doi: 10.1182/bloodadvances.2019000516
58. Pearse BR, McDonough SM, Proctor JL, Panwar R, Sarma GN, Kien L, et al. A CD117-Amanitin Antibody Drug Conjugate (ADC) Effectively Depletes Human and Non-Human Primate Hematopoietic Stem and Progenitor Cells (HSPCs): targeted Non-Genotoxic Conditioning for Bone Marrow Transplant. Biol Blood Marrow Transplant (2019) 25:S29–30. doi: 10.1016/j.bbmt.2018.12.101
59. Tisdale JF, Donahue RE, Uchida N, Pearse BR, McDonough SM, Proctor JL, et al. A Single Dose of CD117 Antibody Drug Conjugate Enables Autologous Gene-Modified Hematopoietic Stem Cell Transplant (Gene Therapy) in Nonhuman Primates. Blood (2019) 134:610–0. doi: 10.1182/blood-2019-125968
60. Palchaudhuri R, Saez B, Hoggatt J, Schajnovitz A, Sykes DB, Tate TA, et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat Biotechnol (2016) 34:738–45. doi: 10.1038/nbt.3584
61. Castiello MC, Bosticardo M, Sacchetti N, Calzoni E, Fontana E, Yamazaki Y, et al. Efficacy and safety of anti-CD45–saporin as conditioning agent for RAG deficiency. J Allergy Clin Immunol (2020) S0091-6749(20)30629–1. doi: 10.1016/j.jaci.2020.04.033
62. Polito L, Bortolotti M, Mercatelli D, Battelli MG, Bolognesi A. Saporin-S6: A useful tool in cancer therapy. Toxins (Basel) (2013) 5:1698–722. doi: 10.3390/toxins5101698
63. Burtner CR, Chandrasekaran D, Santos EB, Beard BC, Adair JE, Hamlin DK, et al. 211Astatine-Conjugated Monoclonal CD45 Antibody-Based Nonmyeloablative Conditioning for Stem Cell Gene Therapy. Hum Gene Ther (2015) 26:399–406. doi: 10.1089/hum.2015.021
64. Nakamae H, Wilbur DS, Hamlin DK, Thakar MS, Santos EB, Fisher DR, et al. Biodistributions, myelosuppression, and toxicities in mice treated with an anti-CD45 antibody labeled with the α-emitting radionuclides bismuth-213 or astatine-211. Cancer Res (2009) 69:2408–15. doi: 10.1158/0008-5472.CAN-08-4363
65. Chen Y, Kornblit B, Hamlin DK, Sale GE, Santos EB, Wilbur DS, et al. Durable donor engraftment after radioimmunotherapy using α-emitter astatine-211-labeled anti-CD45 antibody for conditioning in allogeneic hematopoietic cell transplantation. Blood (2012) 119:1130–8. doi: 10.1182/blood-2011-09-380436
66. Lagresle-Peyrou C, Yates F, Malassis-Séris M, Hue C, Morillon E, Garrigue A, et al. Long-term immune reconstitution in RAG-1-deficient mice treated by retroviral gene therapy: A balance between efficiency and toxicity. Blood (2006) 107:63–72. doi: 10.1182/blood-2005-05-2032
67. Van Til NP, Sarwari R, Visser TP, Hauer J, Lagresle-Peyrou C, Van Der Velden G, et al. Recombination-activating gene 1 (Rag1)-deficient mice with severe combined immunodeficiency treated with lentiviral gene therapy demonstrate autoimmune Omenn-like syndrome. J Allergy Clin Immunol (2014) 133:1116–23. doi: 10.1016/j.jaci.2013.10.009
68. Pike-Overzet K, Rodijk M, Ng YY, Baert MRM, Lagresle-Peyrou C, Schambach A, et al. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia (2011) 25:1471–83. doi: 10.1038/leu.2011.106
69. Garcia-Perez L, van Eggermond M, van Roon L, Vloemans SA, Cordes M, Schambach A, et al. Successful Preclinical Development of Gene Therapy for Recombinase-Activating Gene-1-Deficient SCID. Mol Ther - Methods Clin Dev (2020) 17:666–82. doi: 10.1016/j.omtm.2020.03.016
70. Yates F, Malassis-Séris M, Stockholm D, Bouneaud C, Larousserie F, Noguiez-Hellin P, et al. Gene therapy of RAG-2-/- mice: Sustained correction of the immunodeficiency. Blood (2002) 100:3942–9. doi: 10.1182/blood-2002-03-0782
71. Van Til NP, De Boer H, Mashamba N, Wabik A, Huston M, Visser TP, et al. Correction of murine rag2 severe combined immunodeficiency by lentiviral gene therapy using a codon-optimized RAG2 therapeutic transgene. Mol Ther (2012) 20:1968–80. doi: 10.1038/mt.2012.110
72. Capo V, Castiello MC, Fontana E, Penna S, Bosticardo M, Draghici E, et al. Efficacy of lentivirus-mediated gene therapy in an Omenn syndrome recombination-activating gene 2 mouse model is not hindered by inflammation and immune dysregulation. J Allergy Clin Immunol (2018) 142:928–941.e8. doi: 10.1016/j.jaci.2017.11.015
73. Pike-Overzet K, Baum C, Bredius RGM, Cavazzana M, Driessen GJ, Fibbe WE, et al. Successful RAG1-SCID gene therapy depends on the level of RAG1 expression. J Allergy Clin Immunol (2014) 134:242–3. doi: 10.1016/j.jaci.2014.04.033
74. Van Til NP, Cortes P, Danos O, Cassani B, Poliani PL, Villa A, et al. Reply. J Allergy Clin Immunol (2014) 134:243–4. doi: 10.1016/j.jaci.2014.04.032
75. Lagresle-Peyrou C, Benjelloun F, Hue C, Andre-Schmutz I, Bonhomme D, Forveille M, et al. Restoration of human B-cell differentiation Into NOD-SCID mice engrafted with gene-corrected CD34+ cells isolated from artemis or RAG1-deficient patients. Mol Ther (2008) 16:396–403. doi: 10.1038/sj.mt.6300353
76. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-Associated Clonal T Cell Proliferation in Two Patients after Gene Therapy for SCID-X1. Sci (80- ) (2003) 302:415–9. doi: 10.1126/science.1088547
77. Pike-Overzet K, De Ridder D, Weerkamp F, Baert MRM, Verstegen MM, Brugman MH, et al. Gene therapy: Is IL2RG oncogenic in T-cell development? Nature (2006) 443(7109):E5; discussion E6-7. doi: 10.1038/nature05218
78. Cohen S, Roy J, Lachance S, Marinier A, Delisle J-S, Roy D-C, et al. Single UM171 Expanded Cord Blood Permits Transplantation of Better HLA Matched Cords with Excellent Gvhd Relapse Free Survival. Blood (2018) 132:4658–8. doi: 10.1182/BLOOD-2018-99-110188
79. Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Sci (80- ) (2010) 329:1345–8. doi: 10.1126/science.1191536
80. Capo V, Penna S, Merelli I, Barcella M, Scala S, Basso-Ricci L, et al. Expanded circulating hematopoietic stem/progenitor cells as novel cell source for the treatment of TCIRG1 osteopetrosis. Haematologica (2020). doi: 10.3324/haematol.2019.238261. haematol.2019.238261.
81. Petrillo C, Calabria A, Piras F, Capotondo A, Spinozzi G, Cuccovillo I, et al. Assessing the Impact of Cyclosporin A on Lentiviral Transduction and Preservation of Human Hematopoietic Stem Cells in Clinically Relevant Ex Vivo Gene Therapy Settings. Hum Gene Ther (2019) 30:1133–46. doi: 10.1089/hum.2019.016
82. Petrillo C, Thorne LG, Unali G, Schiroli G, Giordano AMS, Piras F, et al. Cyclosporine H Overcomes Innate Immune Restrictions to Improve Lentiviral Transduction and Gene Editing In Human Hematopoietic Stem Cells. Cell Stem Cell (2018) 23:820–32. doi: 10.1016/j.stem.2018.10.008
83. Zonari E, Desantis G, Petrillo C, Boccalatte FE, Lidonnici MR, Kajaste-Rudnitski A, et al. Efficient Ex Vivo Engineering and Expansion of Highly Purified Human Hematopoietic Stem and Progenitor Cell Populations for Gene Therapy. Stem Cell Rep (2017) 8:977–90. doi: 10.1016/j.stemcr.2017.02.010
84. Heffner GC, Bonner M, Christiansen L, Pierciey FJ, Campbell D, Smurnyy Y, et al. Prostaglandin E2 Increases Lentiviral Vector Transduction Efficiency of Adult Human Hematopoietic Stem and Progenitor Cells. Mol Ther (2018) 26:320–8. doi: 10.1016/j.ymthe.2017.09.025
85. Gentner B, Bernardo ME, Zonari E, Tucci F, Fumagalli F, Redaelli D, et al. Ex-Vivo Gene Therapy for Hurler Disease: Initial Results from a Phase I/II Clinical Study. Mol Ther (2019) 27:abstr. 2. doi: 10.1016/j.ymthe.2019.04.004
86. Genovese P, Schiroli G, Escobar G, Di Tomaso T, Firrito C, Calabria A, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature (2014) 510:235–40. doi: 10.1038/nature13420
87. Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature (2016) 539:384–9. doi: 10.1038/nature20134
88. De Ravin SS, Li L, Wu X, Choi U, Allen C, Koontz S, et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci Transl Med (2017) 9:eaah3480. doi: 10.1126/scitranslmed.aah3480
89. Schiroli G, Ferrari S, Conway A, Jacob A, Capo V, Albano L, et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci Transl Med (2017) 9(411):eaan0820. doi: 10.1126/scitranslmed.aan0820
Keywords: RAG genes, gene therapy, non-genotoxic conditioning, severe combined immunodeficiency, hematopoietic stem cell transplantation, Omenn syndrome, leaky SCID
Citation: Villa A, Capo V and Castiello MC (2020) Innovative Cell-Based Therapies and Conditioning to Cure RAG Deficiency. Front. Immunol. 11:607926. doi: 10.3389/fimmu.2020.607926
Received: 18 September 2020; Accepted: 26 October 2020;
Published: 19 November 2020.
Edited by:
Claudio Pignata, University of Naples Federico II, ItalyReviewed by:
Andrew R. Gennery, Newcastle University, United KingdomAlexandra Freeman, National Institutes of Health (NIH), United States
Copyright © 2020 Villa, Capo and Castiello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Villa, dmlsbGEuYW5uYUBoc3IuaXQ=