 Jean-Marie Berthelot
Jean-Marie Berthelot Frédéric Lioté2
Frédéric Lioté2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol. , 01 December 2020
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.607069
This article is part of the Research Topic Sensing DNA in Antiviral Innate Immunity View all 14 articles
Upon recognition of microbial DNA or self-DNA, the cyclic-GMP-AMP synthase (cGAS) of the host catalyzes the production of the cyclic dinucleotide cGAMP. cGAMP is the main activator of STING, stimulator of interferon genes, leading to interferon synthesis through the STING-TBK1-IRF3 pathway. STING is also a hub for activation of NF-κB and autophagy. The present review details the striking similarities between T and B cell responses in severe coronavirus disease 2019 (COVID-19) and both animal or human models of STING gain of function (SAVI syndromes: STING-associated vasculopathy with onset in infancy). Those similarities may be further clues for a delayed activation of STING in severe COVID-19 patients, due to DNA damages following severe acute respiratory syndrome coronaviruses (SARS-CoV-2) infection and unusual role of STING in SARS-CoV-2 control. In early stages, Th2 differentiation are noticed in both severe COVID-19 and SAVI syndromes; then, CD4+ and CD8+ T cells functional exhaustion/senescent patterns due to TCR hyper-responsiveness are observed. T cell delayed over-responses can contribute to pneumonitis and delayed cytokine secretion with over-production of IL-6. Last, STING over-activation induces progressive CD4+ and CD8+ T lymphopenia in SAVI syndromes, which parallels what is observed in severe COVID-19. ACE2, the main receptor of SARS-CoV-2, is rarely expressed in immune cells, and it has not been yet proven that some human lymphocytes could be infected by SARS-CoV-2 through CD147 or CD26. However, STING, expressed in humans T cells, might be triggered following excessive transfer of cGAMP from infected antigen presenting cells into activated CD4+ and CD8+ T cells lymphocytes. Indeed, those lymphocytes highly express the cGAMP importer SLC19A1. Whereas STING is not expressed in human B cells, B cells counts are much less affected, either in COVID-19 or SAVI syndromes. The recognition of delayed STING over-activation in severe COVID-19 patients could prompt to target STING with specific small molecules inhibitors already designed and/or aspirin, which inhibits cGAS.
To account for the quite different outcomes of coronavirus disease 2019 (COVID-19), including in young people, as well as the lower prognosis of male, obese, and aged patients, we previously put forward the hypothesis that a delayed over-activation of the stimulator of interferon (IFN) genes (STING) pathway, could be central to the pathogenesis of severe COVID-19 (1, 2). This delayed over-activation could be the consequence of gain of function in the cGAS-STING axis, and/or cytosolic damages induced by severe acute respiratory syndrome coronaviruses (SARS-CoV-2) in epithelial, endothelial, or innate cells (1, 2). This hypothesis was partly deduced from the observation that bats withstand SARS-CoV viruses, thanks to a loss of function mutation of STING, associated with higher synthesis of IFN-α and much lower synthesis of IFN-β (3).
This hypothesis would fit with the Kawasaki-like features and high thrombosis rate observed in severe COVID-19 (2). Indeed, over-activation of the STING pathway can lead to the release of IFNβ and tissue factor (through induction of pyroptosis by the STING-gasdermin pathway) in infected epithelial cells and/or endothelium (2).
As functional consequences of DNA sensing by the cGAS-STING pathway differ according to antigen presenting cells or lymphocytes (4), which had not been the focus of previous articles (1, 2), the present review aims: i) to study arguments for a possible contribution of over-activation of the cGAS-STING pathways to the disturbances of T and B cell responses observed in previous SARS-CoV, and in severe COVID-19 (Table 1); ii) to raise further hypotheses to test in COVID-19 (Table 2).
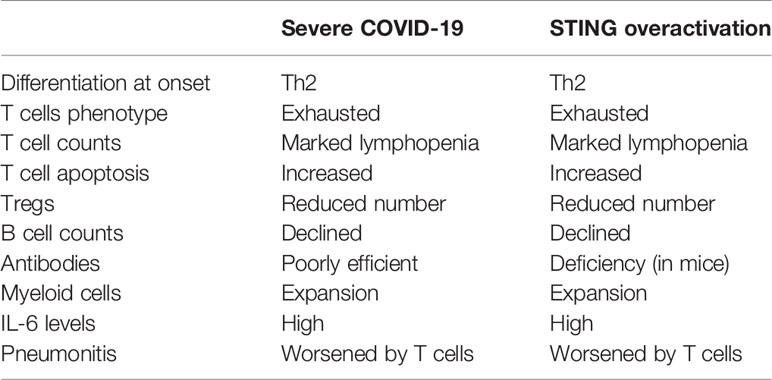
Table 1 Similarities in T and B cells features in severe coronavirus disease 2019 (COVID-19), and STING over-activation, including SAVI (STING-associated vasculopathy with onset in infancy) syndromes.

Table 2 Questions and hypotheses to address.
SARS-CoV-2 invades most host cells via binding of its structural spike glycoprotein to angiotensin-converting enzyme 2 (ACE2) (5, 6). Although ACE2 is upregulated by type I IFN and IFN-γ, and to a lesser extent type II IFNs (7), but not type III IFN (8), it is usually not expressed in immune cells (5, 6), especially in T and B cells.
Nevertheless, it was shown that some immune cells, including T cells, can be infected by the SARS-CoVs and middle-east respiratory syndrome coronavirus (MERS-CoVs) (9, 10) [although they poorly replicate in lymphocytes (9)]. This suggests that other receptors can contribute to entry of those SARS-CoVs in some lymphocytes. A first possibility could be CD147 [also known as basigin (5, 11)]. CD147 is strongly expressed in whole blood, neutrophils, classical monocytes, macrophages, plasmacytoid dendritic cells, NK cells, naïve CD4+ T cells, terminal effector CD4+ T cells, naïve CD8+ T cells, effector memory CD8+ T cells, naïve B cells, and plasmablasts (5, 12). It has also been suggested that CD147 could act as a secondary receptor for SARS-CoV-2 in T cell lines (10) (Table 2).
CD26 (DPP4) is another receptor important in SARS-CoV infections, described in MERS-CoV, and potentially recognizing SARS-CoV-2 (13). Similar to CD147, CD26 is expressed in nearly all immune cells, but, contrary to CD147, not in B cells (5).
However, contributions of CD147 and CD26 to COVID-19 still remain unproven, and ACE2 should be still considered as the only receptor for SARS-CoV-2 (14) (Figure 1).
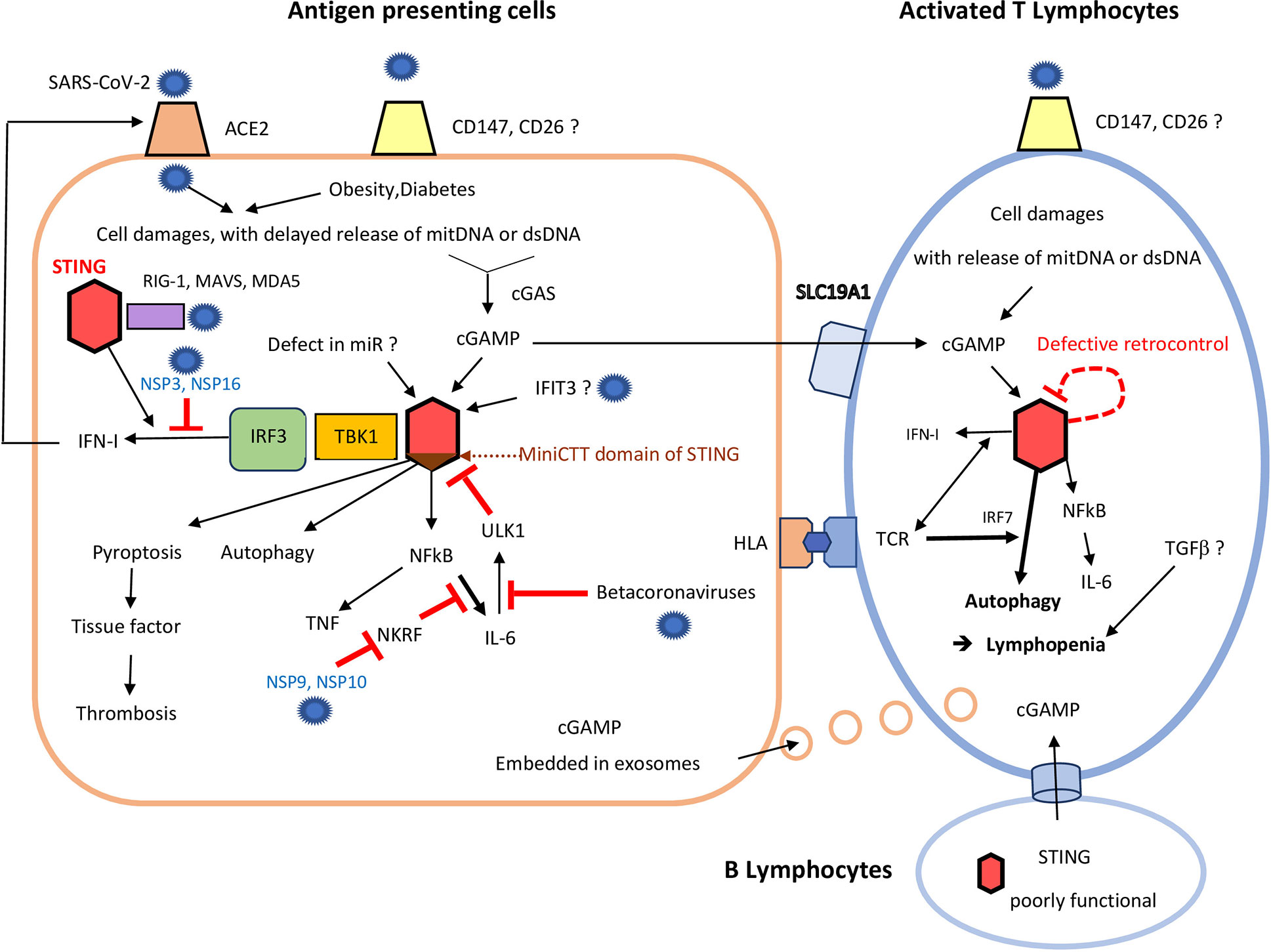
Figure 1 Consequences of STING over-activation on antigen presenting cells and T cells following severe acute respiratory syndrome coronaviruses (SARS-CoV-2) infection. In antigen presenting cells (left), due to poor virus control by RNA sensors (including RIG-1, and MDA5) and despite the help of STING (red hexagon), SARS-CoV-2 induces delayed cell damages, with mitochondrial DNA and dsDNA release. It can add to damaged self-DNA secondary to ageing and/or obesity/diabetes. cGAS catalyzes those self-DNA in cyclic nucleotides, mainly cGAMP, which in turn activates STING (red hexagon). This activation of STING may be enhanced by: i) a lack of miRNAs, (like miR-576-3p, which normally suppress STING translation); ii) excess of IFIT3, which interacts with STING gene to promote its expression. Binding of cGAMP to STING first induces activation of the STING-TBK1-IRF3 pathway, leading to IFN-I synthesis. In COVID-19, the SARS-CoV-2 NSP 3 and 16 lower this IFN secretion, but IFN enhances expression of ACE2 on cell membrane. cGAMP, which can be released to by-stander cells through viral exosomes, can also be transmitted by B cells (low), to activated T cells (right), through specific channels. Independently of the STING-TBK1-IRF3 pathway, binding of cGAMP to STING can also: i) induce pyroptosis; ii) promote autophagy and lymphopenia; iii) activate NF-κB and both TNF and IL-6 synthesis. SARS-CoV-2 NSP 9 and 10 still enhances IL-6 release through inhibition of NKRF. Some beta-coronaviruses can also impede ULK1 and its negative feed-back loop, further enhancing IL-6 secretion. Excess of cGAMP can be internalized by by-stander activated CD4+ and CD8+T cells, which highly express the cGAMP importer SLC19A1. Therefore, even without infection of T lymphocytes, STING activation could also occur in activated T lymphocytes, especially those already stressed by a continuous activation by their hyper-responsive TCR. Defective retro-control of STING (dashed red loop) by IL-6 (see above) could still enhance STING activation in severe COVID-19. In activated T cells, STING overactivation leads to autophagy, up to lymphocyte death. This contributes much to the lymphopenia observed in rodent and human gain of function of STING models, and perhaps also in COVID-19, with subsequent poor virus control.
STING is a pattern recognition receptor localized in the endoplasmic reticulum (ER) membrane. The cGAS-STING pathway plays a central role in sensing cytosolic DNA upon infection with DNA from bacteria and DNA-viruses (including endogenous retroviruses). Lack of expression of STING by some cell types contribute to preferential homing of DNA viruses. For instance, hepatitis B virus has a tropism for human hepatocytes, which have undetectable levels of STING protein (15).
Those DNA viruses can reduce STING signaling by increasing autophagy-mediated turnover of STING or interfering with STING trafficking. They can also interfere with IRF signaling and antiviral IFN I responses, rather than with NF-κB responses. For instance, herpes-virus 1 (HSV-1), reduce the ratio of full-length human STING/truncated STING isoforms, induced by alternative splicing of STING RNAs. The three STING truncated isoforms fail to induce IFN-β, and they act as selective pathway inhibitors of full-length STING, even in combination with upstream inducer cyclic-di-GMP-AMP synthase (16).
Importantly, although mainly a DNA sensor, STING is also necessary for full control of enveloped RNA viruses, like influenza virus and coronaviruses, independently of cGAS. Indeed, sensing of lipid membrane fusion through STING also contributes to the antiviral response against enveloped RNA viruses, so that complete protection against RNA viruses also relies on STING (17). STING can also participate in viral RNA sensing through its interaction with the mitochondrial antiviral signaling protein (MAVS) (17). STING further transduces the signaling induced by RNA-derived PAMPs through retinoic acid-inducible gene (RIG-I)-like sensors (RIG-I and melanoma differentiation-associated protein 5 (MDA-5) (17) (Figure 1).
Multiple Flaviviridae (including zikaviruses, dengue virus, West Nile virus, Japanese encephalitis virus) and hepatitis C virus disrupt STING-mediated signaling (18). Some do so by cleaving STING or cGAS, but others only interfere with the STING-TBK1 interaction (18), explaining why the IFN axis can be affected but not the NF-κB axis (19).
So far, there is no evidence that betacoronaviruses increase autophagy-mediated turnover of STING, interfere with STING trafficking, or cleave STING or cGAS. However, it has been shown that various SARS-CoV-2 protases [non structural protein 3 (NSP3) and NSP16] strongly inhibit the downstream STING-TBK1-IRF3 pathway and IFN synthesis (20). Conversely, SARS-CoV-2 NSP9 and NSP10 protases still increase signaling and IL-6 production by inhibition of NKRF, an endogenous NF-κB repressor (21) (Figure 1).
Importantly, STING also senses intra-cellular damaged self-DNA [mitochondrial DNA, and self-dsDNA released in extra-cellular space, including dsDNA from neutrophil extracellular traps (NETs) (22)] secondary to bacterial or viral infections. Therefore, at late stages of RNA viruses infection, the evasion of STING by RNA viruses can be counterbalanced by its activation following released of damaged self-DNA (Figure 1).
Upon recognition of non-self (microbial) DNA or self-DNA, the cyclic-GMP-AMP synthase (cGAS) of the host catalyzes the production of cyclic dinucleotides, like cGAMP (from GMPs and AMPs) (Figure 1). The length of DNA, rather than its sequence, determines its ability to generate cGAMP following recognition by cGAS (23). Since the endogenous DNA fragments are often short in size, whereas viral and bacterial DNA fragments are generally much longer, this can partly permit discrimination between self and non-self DNA. However, a large amount of self-DNA in cytoplasm outside of the nucleus or mitochondria can also activate the cGAS-STING pathway.
Once activated, STING recruits TANK-binding-kinase (TBK1), which can activate and phosphorylate IFN-regulatory factor 3 (IRF3), to induce type-I IFNs. Those type I IFN then engage the IFN I receptor, thereby activating the JAK/STAT pathways and inducing the transcription of IFN-stimulated genes (ISGs) (23). As the STING gene is itself an ISG, a positive feedback loop can ensue (24). This STING-TBK1-IRF3 axis can be selectively inhibited by viral protease in COVID-19 (20).
However, STING is also a cytosolic hub for other pathways, which can be activated independently of the IFN-TBK1-IRF3 axis. For instance, downstream signaling of STING also lead to NF-κB and/or inflammasomes activation, and secretion of various cytokines, like IL-6 and TNF (1, 16), as also observed in COVID-19 (21) (Figure 1).
STING is highly conserved throughout evolution, being observed >500 million years ago, and has also other primitive functions than secretion of IFN-I and cytokines to fight viruses or intra-cellular bacteria (23, 25, 26).
First, it activates the process of autophagy (critical for the elimination of DNA and viruses in the cytosol of innate immune cells), independently of TBK1 activation and IFN-I signals (25) (Figure 1).
Second, if foreign nucleic acids accumulate despite autophagy, further activation of the STING pathway can lead to growth arrest, up to apoptosis or pyroptosis (23), which can contribute to increase the risk of thrombosis (Figure) (2). Those STING-mediated cell deaths might be critical for preventing pathogen dissemination (23, 26).
Reciprocally, some bacterial pathogens (and possibly some DNA viruses) have learned to exploit those STING-induced deaths to impair T cell response (27). They sort their DNAs into extracellular vesicles and deliver them to T cells, to excessively stimulate the cGAS-STING pathway, after ingestion, which primes those T cells for apoptosis (27, 28). In HSV-1 infections, STING can also be directly transferred to previously uninfected cells through exosomes (29) (Figure 1) in order to force those cells to detect earlier their invasion by HSV-1.
This strategy should less apply to the SARS-CoVs, since they are RNA viruses, less directly recognized by cGAS-STING. However, SARS-CoVs could indirectly tease T cell response through the cGAS-STING pathway. Indeed, viral-induced DNA damage in by-stander cells of T cells (including B cells) can lead to cGAMP synthesis in those neighbor cells (Figure 1). Transfer of this cGAMP to T cells can also activate the cGAS-STING pathway in activated CD4+ and CD8+ T cells. Indeed, those activated T cells highly express the cGAMP importer SLC19A1 (whereas naïve T cells do not) (Figure 1) (28).
SLC19A1 is a folate-organic phosphate antiporter, and the major transporter of CDNs like cGAMP through the cell membranes, of activated T cells (30). Interestingly, in human cell lines and primary cells ex vivo, CDN uptake through SLC19A1 is inhibited by both folates and two anti-folates medications of rheumatoid arthritis and other inflammatory rheumatisms: sulfasalazine and methotrexate (30). Accordingly, the outcome of rheumatoid arthritis with severe COVID-19 treated or not by methotrexate or sulfasalazine could be worth to study, although methotrexate as only limited efficacy against SAVI (STING-associated vasculopathy with onset in infancy) syndromes.
STING is strongly expressed in most cells, except human B cells, and recent studies showed that mouse CD4+ T cells express even higher amounts of STING protein than macrophages and dendritic cells (28). The activation of mouse T cells upon T cell receptor (TCR) recognition of antigen peptide-MHC is regulated by STING (23, 31), which can modulate various T cell functions, such as cytokine production, cell growth, and differentiation (23). STING can make the mouse T lymphocytes more responsive to ER stress induced by T cell receptor (TCR) signaling. It also leads to lymphocytes death following excessive triggering of TCR, in cells with other sources of ER stress (32, 33).
In humans, key components of the DNA sensing machinery are expressed in activated T cells, including STING itself (34). However, as compared with other cells (including myeloid immune cell types and non-immune cells), STING seems much less functional in human T cells, at least regarding IFN secretion (Figure 1) (18). It has been highlighted in the most recent review on STING that lack of an IFN I response downstream of STING in human T and B lymphocytes is in line with the observation that functional DNA sensing pathway in T and B lymphocytes would lead to continuous IFN I production in those highly proliferative cells and detrimental auto-inflammatory diseases (18).
A compromised expression of the cGAS-STING cascade has also been confirmed in CD8+T cells from cancer patients, with reduced stem-like central memory CD8+T cells subsets (35). This is a selective advantage for survival and growth for cancers (36). Several studies showed that STING re-activation can stimulate antitumor immune responses in numerous cancers, including leukemia, melanoma, glioma, and hepatocellular carcinoma (37).
In HIV patients, resting CD4+ T cells, some of which harbor HIV, are also defective in STING-dependent IFN I production (34). A defective STING pathway may render human T cells vulnerable to other pathogens, due to low production of IFN, including other retroviruses (e.g., human T-lymphotropic virus type 1), or viruses (e.g., VZV and HHV 6) that replicate through a DNA (18). Conversely, other downstream signaling of STING than IFN, like NFκB, are less affected in human T cells, as shown in HIV-1 lymphocytes (38). This is also observed in SAVI syndromes (induced by gain of function of STING), where over-activation of STING by viruses can lead to T cell hyper-responsiveness with IL-6 secretion (as well as T cell exhaustion, and autophagy-induced lymphopenia).
The final sources of cGAMP for the activation of STING in T cells should differ according to the possibility (or not) of a direct infection of T cells by SARS-CoV-2 through CD147 or CD26. Such infection has not yet been demonstrated, but might be searched in subsets of activated T cells of severe COVID-19) (Table 2), including memory T cells which upon activation exhibit much greater up-regulation of CD147 than naïve T cells (39) (Figure 1).
In such event, STING might be activated by: i) cGAMP incorporated into viral particles during encapsidation in antigen presenting cells, to accelerate STING activation in neighboring T cells and their apoptosis; ii) endogenous cGAMP synthetized from damaged cytosolic self-DNA within T cells, including virally damaged mitochondrial self-DNA, as shown for dengue virus infected cells (40) (T cells are permissive for dengue virus); iii) a non-canonical mechanism involving lipid membrane alterations, as observed following influenza virus infection in mouse embryonic fibroblast cells (41).
However, even if T cells are not infected by SARS-CoV-2, STING could still be activated within T cells by: iv) exogenous cGAMP released in extra-cellular spaces by dying cells, and internalized by activated CD4+ and CD8+T cells (15) (which highly express the cGAMP importer SLC19A1); v) cGAMP transmitted by B cells to T cells via other cellular channels; vi) DNA damages occurring in T cells themselves, including oxidized mitochondrial-DNA (Figure 1).
At 24h after the first clinical signs of COVID-19, interleukins (IL-1α, IL-1β, IL-6, IL-10), and IFNs (IFN-α2, IFN-β1, IFN-II) are significantly elevated, but the cellular sources of those cytokines are probably more epithelial and innate cells than T lymphocytes (42). After several days, naïve lymphocytes differentiate in Th2, in line with the role of eosinophils in fighting RNA viruses [ascribed to the RNAses inside their granules (43)]. Such Th2 profile had previously been described in SARS patients, and lethal outcomes correlated with elevated Th2 cell serum cytokines (44).
The main ligand for STING, cGAMP, can also preferentially induce a Th2 differentiation (23, 45). Co-stimulation of antigen presenting cells and T cells by foreign or self-DNA similarly suppresses T-bet expression, followed by the induction of GATA-3 and Th2 cytokines (23, 45). The extra-cellular self dsDNA of neutrophil extracellular traps (NETs) induced by rhinovirus infection of upper airways is associated with a preferential Th2 response (46). Accordingly, the shift towards a Th2 response in early stages of COVID-19 would fit with STING activation (Table 1).
However, the expression of the aryl hydrocarbon receptor (AhR) on T cells following CD28 activation can later shift Th1/Th2 balance towards a Th1 response (45). This could explain why, in lungs of mice models of SAVI syndromes, Th1, rather than Th2, seem to play a critical role in tissue damage and the persistence of inflammation (47). Whether SARS-CoV-2 could activate AhR has not been addressed so far (Table 2), but a murine coronavirus does it, contributing to cytokine modulation and viral infection (48).
In most patients, COVID-19 is a benign and even asymptomatic infection, and recognition of some viral peptides by T cells (including some memory T cells already primed by previous encounter with human or animal coronaviruses) could help innate cells to clear SARS-CoV2. Recent findings using immunospot essay assessing IFNγ production, showed that 6/8 contacts of index patients developed a SARS-Cov-2 specific T cell response lasting up to 80 days against structural and/or accessory SARS-CoV-2 proteins, although none developed antibodies, and all were tested negative by PCR (49).
Conversely, due to genetically encoded differences in innate and/or T cells, or epigenetic changes acquired during life, innate and/or adaptive cells could remain inefficient to clear SARS-CoV-2 in the minor subset of patients with severe COVID-19. Those genetically encoded differences might be loss of function mutations polymorphisms: inborn errors of TLR3- and IRF7-dependent type I IFN immunity have been found in 3.5% of life-threatening COVID-19 pneumonia in patients with no prior severe infections (50). However, gain of function mutations in other genes, including genes of the STING pathways, should also be searched, since delayed over-reaction to virus damage might be as important as poor initial control of SARS-CoV-2.
Increased presence of strongly activated T cells, characterized by expression of HLA-DR, CD38, CD69, CD25, CD44, and Ki-67 has been reported in several studies on COVID-19 (51). In intensive care (ICU) units, both virus-specific CD4 and CD8 T cells were detected in all COVID-19 patients (at average frequencies of 1.4 and 1.3%, respectively), with phenotypes suggestive of either CD4 central memory, or CD8 effector memory T cells (52).
Although SARS-CoV-2 restrains antigen presentation by down-regulating MHC class I and II molecules, CD4+ T cells, and even more CD8+ T cells (20, 53), exhibit functional exhaustion/senescent patterns (51). The expression of NK group 2 member A receptor (NKG2A), programmed cell death 1 (PD-1), and T-cell immunoglobulin and mucin containing protein-3 (TIM-3) (51) increased as patients progressed from prodromal to overtly symptomatic stages (54). In critically ill patients, cellular functionality was also shown to be impaired in CD4 and CD8 T cells (20). This could explain why, despite initial over-activation of lymphocytes, at autopsy, patients who succumbed to infection all had high virus levels in the respiratory tract and other tissues (55).
Elevations in CRP (C-reactive protein), strongly associated to interleukin (IL)-6 levels, appear to be unique to COVID-19 patients when compared to other viral infections (20). Those elevated IL-6 levels are associated with ICU admission, respiratory failure, and poor prognosis (20). Various activated and expanded antigen presenting cells, including macrophages and dendritic cells, seem usually responsible for the cytokine storm with high IL-6 secretion, and for the high serum levels of CRP (54). This is in line with previous studies on other betacoronaviruses human infections (SARS-CoV and MERS-CoV). Indeed, most of the exhausted T cells express less cytokines than innate cells, and T cell numbers are negatively correlated to serum IL-6, IL-10, and TNF-α concentration. However, IL-6 increases fairly late during the disease’s course |20] [which much reduces its prognostic value at earlier stages (56)].
A first hypothesis for this delayed raise of IL-6 could be the deferred re-expression of ACE2 receptor (57) on previously infected epithelial cells, following IFN secretion, leading to re-activation of macrophages and dendritic cells (Figure 1). A second hypothesis is an increased peripheral blood frequency of a subset of polyclonal GM-CSF+ CD4 T cells capable of prodigious ex-vivo IL-6 and IFN-γ production, which has been described in COVID-19, although only in critically ill patients (58).
Unlike in innate immune cells, cGAMP alone does not induce IFN-I production by mouse naïve CD4+ and CD8+ T cells, since both TCR stimulation and STING activation are required (23). Conversely, in effector CD4+ and CD8+ T cells, the STING activation by cGAMP can lead to IFN-I and probably various cytokines production, even without TCR co-stimulation (although cGAMP-induced IFN-I production is further increased by IL-2 and TCR binding) (23). The amount of IFN-I produced by mouse effector Th1 and activated CD8+ T cells in response to co-stimulation with TCR and cGAMP is even higher than in dendritic cells (23). This is in line with the much greater ability of previously activated T cells to internalize cGAMP from neighbor cells, as compared to naïve T cells (22).
This continuous secretion of IFNs by effector T cells help to clear intra-cellular pathogens at the early stages of viral infections. Early or preventive treatment by IFN-α or IFN-β improved the outcomes of SARS-CoV and MERS-CoV infection in mice and in non-human primates. However, some viruses, including SARS-CoVs, not only can lower IFN production, but can also take advantage of some subtypes of IFN, like IFN-β, to persist in the host at later stages (7). This could explain why in humans, survival rates in MERS-CoV infection were not increased, possibly because those drugs were given too late (59). At this stage, the delayed IFN-β response in some murine models of severe acute respiratory syndrome (SARS) is, on the opposite, associated with excessive influx of pathogenic inflammatory monocytes-macrophages and a much worse prognosis (60) (Table 2).
STING over-activation might similarly paradoxically enhance SARS-CoV-2 replication in lung monocytes-macrophages through transient increase secretion of IFN-β. This contributes to T lymphocyte exhaustion up to final fall of IFN-I secretion. Indeed, following infection by gamma-herpesvirus 68 (γHV68), the increased secretion of IFN-I also promoted the replication of this virus in cultured macrophages of a mouse model of gain of function of STING (N153S) (61). Furthermore, IFN-β can also induce up-regulation of ACE2 in airway epithelia (7) (Figure 1). This might fuel new infections by SARS-CoV2, and impair antigen-specific T cell responses to SARS-CoV antigens (60).
The contribution of IFN-β to COVID-19 remains quite uncertain however in humans, since: i) IFN-I over-secretion induced early by STING activation in infected cells is quickly counterbalanced by a strong inhibition of the downstream STING-TBK1-IRF3 pathway by various SARS-CoV-2 protases (Figure 1) (20); ii) IFN-I, II and III expression was seemingly not significantly increased in lung tissues infected with SARS-CoV-2 (62); iii) although an increased expression of IFN-I, and IFN-I related genes, were observed in human blood during the first stages of COVID-19, they declined when the patient got worse (63).
Nevertheless, STING activation can lead to nuclear-factor kappa B (NFκB) and cytokine secretion, including IL-6, independently of IFN secretion. First, SARS-CoV-2 non-structural protein (NSP)9 and NSP10 still increase IL-6 production, by inhibition of NF-κB-repressing factor (NKRF), an endogenous NF-κB repressor (21) (Figure 1). Second, whereas the induction of IFN-response genes are maximally up-regulated in PBMCs from SAVI patients (exposure to cGAMP brought about no change), conversely, transcription of the genes encoding tumor necrosis factor (TNF) and IL-6 was elevated in unstimulated peripheral blood mononuclear cells (PBMCs) from SAVI patients, and was still augmented on exposure to cGAMP (64). This is reminiscent of what is observed in severe COVID-19 patients (Table 1) (65).
A subdomain within the C terminus domain (CTT) of STING (mini_CTT), appears to be required for STING-mediated, TBK1/IRF3-independent, NF-κB activation in a human system (31). This mini_CTT domain might also be required for cGAMP-induced inhibition of mammalian target of rapamycin complex 1 (mTORC1) signals in T cells (Figure 1). Accordingly, gain of function of this subdomain of STING could be preferentially studied in young patients with severe COVID-19 (Table 2).
In COVID-19, a sustained decrease in CD4+ and CD8+ T cells, especially CD8+ T cells, is observed, despite an increase in IL-6, IL-10, IL-2, IFN-γ levels, and neutrophil counts. This fall is even more pronounced in patients requiring ICU (54), whatever the age (42). Decreased counts of NK cells, eosinophils (53), and γδ-T cells (66), are also observed in severe cases (53). In most severe COVID-19, this lymphopenia is associated with atrophy of lymphoid organs (67). Regulatory T cells are moderately increased at the onset of mild COVID-19, but similarly later decline in severe cases (68).
In mild cases, this lymphopenia might partly reflect redistribution of lymphocytes to lymphoid organs and tissues, since IFN-I, IL-6, and TNF-α can promote retention of activated lymphocyte in lymphoid organs (54). However, although some autopsy study showed extensive lymphocyte infiltration in the lungs (55), others observed only found neutrophilic infiltration in the lung (69), or death of lymphocytes in spleen and lymph nodes (70).
This T cell lymphopenia appears to predict morbidity and mortality, even at early stages, while elevated levels of CRP, LDH, D-dimer, as well as decreased blood platelets are only late prognosis factors (20).Therefore, rather than a cytokine storm induced by NF-κB stimulation, a marked and widespread virus-induced lymphopenia may be the real cause of death in many patients (71).
Among the numerous mechanisms for the functional exhaustion/senescent patterns of T lymphocytes in severe COVID-19, the release of TGF-β1 could contribute to impair T cell function (and may also be responsible for switch in IgG to IgA production observed in COVID-19 patients). TGF-β1 levels are indeed associated with more severe COVID-19 (72). Similar observations were previously made in patients with severe acute respiratory syndrome (SARS): TGF-β1 was continuously up-regulated during the entirety of SARS, and its prolonged over-production was associated with severity of SARS and memory TCD8 depletion (73). Whether STING also increase TGF-β1 signaling has not been directly addressed in humans, but in rodents STING deficiency in liver effectively reduced the severity of hepatic fibrosis, which was closely associated with the inhibition of TGF-β1 signaling (74). Therefore, STING activation could also reinforce some TGF-β functions in humans (75) and contribute to lymphopenia (Figure 1).
In humans, a marked lymphopenia with reduced memory CD4+ and CD8+ T cells in the periphery (23) is a major feature of SAVI syndrome (76). In a knock-in model carrying an amino acid substitution (V154M) in mouse STING (corresponding to a mutation seen in human patients), the mice also developed a severe combined immunodeficiency disease affecting B, T, and NK cells, with a significant compensatory expansion of monocytes and granulocytes (77). This phenotype is reminiscent of what is observed in severe COVID-19 (Table 1).
B- and T-cell developments were blocked since early immature stages, either in bone marrow or thymus (77). Excess of IFN-I (31) does not contribute to this lymphopenia. Signs of inflammation in lungs and kidneys were also IFN-independent (77), and probably driven by over-reactivity of the TCR (32, 78). This is in line with the observation that in mice STING-associated vasculopathy, STING regulates T cell proliferation in cell culture, independently of IRF3 (32).
Radiations chimeras confirmed that T cell lymphopenia depends on T cell intrinsic expression of the SAVI mutation (79). Co-stimulation of CD4+ T cells by STING and TCR activation can induce growth arrest and lymphopenia by inhibiting mammalian target of rapamycin complex 1 (mTORC1) activation (Table 1). This STING-mediated inhibition of mTORC1 signals following TCR activation is partly dependent on both IFN regulatory factor 3 (IRF3) and IFN regulatory factor 7 (IRF7) (Figure 1), but not on TBK1 and IKKϵ (23). The identification of this unique pathway in T cells is critical for the development of new therapeutic strategies for targeting STING in T cells, and to prevent lymphopenia (23).
STING mutants also induce chronic activation of ER stress and unfolded protein response (UPR) within T lymphocytes. STING-N154S disrupts calcium homeostasis in T cells, and primes them to become hyper-responsive to T cell receptor signaling induced ER stress, leading to cell death, both in CD4+ and CD8+ T cells (32, 78). The mouse CD4+ and CD8+ T cell death through ER stress can be restored (as well as lung disease) by changing TCR specificity (36).
cGAMP-induced STING activation can also lead to the inhibition of IL-2 signaling pathways, which decreases the synthesis of regulatory T cells (Tregs) (23) (Table 1).
Interleukin 7 (IL-7) is essential for lymphocyte survival and expansion (80). IL-7 therapy has been shown to restore lymphocyte counts and functional activity, leading to decreased viral load and clinical improvement in several life-threatening viral infections (71). The effect of compassionate use of IL-7 in 12 critically ill patients with COVID-19 and severe lymphopenia was compared to the outcome of 13 matched controls who did not benefited from IL-7. IL-7 was associated with a restored lymphocyte count, with the IL-7 group having levels more than two-fold greater than the control group (71). However, functional defects of the exhausted lymphocytes could persist, since at day 30, mortality was 42 and 46%, respectively.
SAVI syndromes share other striking similarities with COVID-19: a variable combination of fever, rashes, an inflammatory vasculopathy mimicking lupus chilblains, up to vaso-occlusive process and acral necrosis, and pulmonary inflammation leading to interstitial lung disease (64).
In STING N153S SAVI-like mice model, mice lacking adaptive immunity had no lung disease, and T-cell receptor β chain (Tcrb)−/− STING N153S animals only had mild disease (79). Therefore, T cell over-response seems important for lung disease induction, despite the concurrent lymphopenia induced by STING over-activation (79). Crossing those mice to animals lacking cGAS, IRF3/IRF7, IFN-alpha/beta receptor alpha chain (IFNAR1), adaptive immunity, αβ T cells, and mature B cells, showed that lung disease developed independently of cGAS, IRF3/IRF7, and IFNAR1, suggesting that other triggers than cyclic dinucleotides (the ligands of cGAS) and/or IFN, contribute to STING over-activation within T cells (79). A defective retro-control of STING (Figure 1) might be one explanation for those findings, either in patients with gain of function of STING, and/or severe COVID-19.
In normal cells, IL-6 is a negative feed-back regulator of STING induced by double-stranded DNA, since IL-6 promotes STING degradation by activating/dephosphorylating UNC-51-like kinase (ULK1) (65) (Figure 1). It would be worth testing the hypothesis that this feedback is mitigated in severe COVID-19 by modulation of autophagy (81), and inhibition of molecules like ULK1 by SARS-CoVs (Table 2), in as much they can infect activated/memory T cells expressing CD147. Indeed, HSV-1 can inhibit ULK1 to escape the autophagy process (82), and previous studies on other betacoronaviruses showed that, upon cell infection, these viruses inhibit macro-autophagy (83). For instance, the porcine hemagglutinating encephalomyelitis (PHEV) betacoronavirus inhibits the expression of the ULK1 protein (84). Such virally induced lack of negative feedback on STING expression might indeed contribute, together with gain of function variants of the cGAS-STING pathway variants, to a delayed increase of STING activation in activated CD4+ or CD8+ memory T cells following infection by betacoronaviruses, and worse pneumonitis.
The paradoxical poor control of virus in lung macrophages of SAVI models is partly due to excess of IFN-I and II, as deduced by a strong increase of expression of IFN-stimulated genes, which leads to ACE2 over-expression (Figure 1). The over-response of some T cell subsets to viral antigens in the lung of SAVI and COVID-19, with subsequent fibrosis (61), could also be the consequence of an imbalance between effector T cells and Tregs. Normally, cytosolic DNA sensing via the STING/IFN-β pathway also induces indoleamine 2,3 dioxygenase (IDO). IDO then catabolizes tryptophan to suppress effector and helper T-cell responses and activate Foxp3-lineage CD4(+) Tregs (85). However, Tregs might be even more prone to exhaustion and/or lymphopenia than effector T cells in mouse or humans with over-activation of cGAS-STING, since marked decreases of Tregs have been reported in severe COVID-19 (51) (Table 2).
Like T cells counts, blood B cells count decline with COVID-19 severity [lower counts being associated with increased length of virus shedding (86)], albeit B cell lymphopenia is much less pronounced than T cell lymphopenia (25, 87).
Like T cell, B cells are markedly activated, with highly oligoclonal B cell populations, and profound CD27+CD38+ plasmablasts expansion in some patients. This plasmablast expansion is uncorrelated with decreases in memory B cell subsets, or with the limited changes observed in the circulating T follicular helper cells (cTfh) compartment (87). Long-term studies on serology kinetics in SARS-CoV and MERS-CoV infections, showed that most IgG were neutralizing but waned over time, and usually in less than 3 years, with longer durations of detectable antibody associated with more severe symptoms (88). Whether gain of function and/or activation of STING in subsets of cTfh cells leading to their premature apoptosis due to hyperresponsiveness to TCR signals, contribute to this rather short duration of antibodies towards SARS-CoV infections has not been addressed so far (Table 2).
Sero-conversion occurs between 7 and 14 days after the onset, but this robust antibody response alone is insufficient to avoid severe COVID-19 (20). The high levels of anti-SARS-CoV-2 antibodies in aged COVID-19 do not prevent from severe clinical outcomes, and COVID-19 ICU patients often have high titers SARS-CoV-2 specific antibodies (89).
Another argument suggesting that antibodies might be less important than T cells to control SARS-CoV-2 is the possibility of only mild COVID-19 in patients with severe hypogammaglobulinemia. In a report on seven patients (32 to 79 years-old) with primary antibody deficiencies and COVID-19, five had common variable immune deficiencies (CVIDs) (dysfunctional B lymphocytes), and two had agammaglobulinemia (lacking B lymphocytes). All were substituted and had similar immunoglobulins levels. Whereas the two patients with agammaglobulinemia had a benign COVID-19 course, the five patients with CVIDs presented with a severe form of COVID-19, requiring treatment with multiple drugs, including IL-6–blocking drugs and mechanical ventilation (90). This suggests an active contribution of over-activated but pathogenic subsets of T-bet+ B cells in severe COVID-19 (87), in line with the observation that the granulomatous-lymphocytic interstitial lung diseases occurring in 10% of patients with CVID are partially reversed by B-cell–depleting drugs (90).
B cell receptor (BCR) and STING signaling pathways act synergistically to promote antibodies responses independent of type I IFN (91). However, STING functions autonomously in B cells responding to CDNs, and can be activated by cGAMP without the need of previous BCR ligation (91). This can lead to IFN-β production (92), while IFN-I expression by B cells induces an altered polarization of macrophages toward a regulatory/anti-inflammatory profile, at least in vitro, that might benefit to some pathogens (93).
Mitochondria-mediated apoptosis induced by STING is also more pronounced in normal mouse B cells than in other cells, since upon stimulation, STING is degraded less efficiently in B cells (94).
Activation of MHC class II in human B cells is associated with enhancement of STING signaling (95). However, whereas STING is strongly expressed in mice B cells, in resting humans B cells, STING is poorly expressed and dysfunctional (Figure 1), despite the detection of DNA sensing and signaling proteins of the STING pathway [cGAS, gamma-IFN-inducible protein (IFI16), TBK1 and IRF3] (96). The very poor expression of STING in human SAVI B cells could explain why in humans SAVI severe B lymphopenia and hypo-gammaglobulinemia are not observed [whereas in knock-in mice poor B cell development and an almost complete lack of antibodies are found (77)].
This lack of STING signaling in human B cells might be an explanation for the use of B cells as a reservoir for persistent infection by Epstein-Barr virus (EBV), and other human gamma-herpesviruses (96). Consequently, although human B cells appear equipped to sense invading DNA viruses by other sensors than cGAS-STING, yet they fail to induce an IFN-I response upon cytoplasmic DNA exposure in EBV-negative B cells. EBV-transformed B cell lines do express STING, but these lines, as well as STING-reconstituted EBV-negative B cells, do not produce more IFN-I upon dsDNA or cGAMP stimulation than EBV-negative lines, showing that the cytoplasmic cGAS-STING pathway remains dysfunctional, even in EBV-positive human B cells (96).
Of note, a similar abrogation of signal transduction downstream of STING phenomenon has been reported for some subsets of activated human T lymphocytes that produce substantial levels of STING protein (34), and it might also be worth to study the functionality of the STING pathway in all subsets of memory B and T cells in COVID-19 (Table 2).
Although they poorly express STING, B lymphocytes could indirectly contribute to activate STING in other T cells in COVID-19. Indeed, to alert other cells of viral infections, triggering of cGAS in infected B cells can result in cGAMP production and packaging, and subsequent transfer of this danger message to other cells, including activated T cells (97) (Figure 1).
Consequently, B cells infected by SARS-CoV might contribute to T cell activation and exhaustion in COVID-19, and possibly more frequently in EBV positive patients, which could also contribute to explain why young children have usually much less severe COVID-19 than adults (Table 2).
The striking similarities between clinical and biological features of SAVI syndromes and severe COVID-19, as well as other studies on the role of STING on T and B lymphocytes (Table 1), support the hypothesis of some delayed over-signaling downstream of cGAS-STING in severe COVID-19, despite initial inhibition of the STING-TBK1-IRF3 axis (and IFN secretion) by papain-like-proteases contained within the NSP3 and NSP16 proteins (98) (Figure 1).
A lower IFNα/IFN-β ratio downstream of STING could also promote the replication of SARS-CoV, including SARS-CoV-2, as observed in mice models of SARS-CoV and MERS-CoV (60, 99). Parallel activation of macrophages, dendritic cells, and B cell up to exhaustion, might also lead to excessive triggering of TCR and STING from CD4+ and CD8+ and NK cells, leading to their exhaustion and deaths, and further spreading of SARS-CoV-2.
Cases of severe COVID-19 in very young children suggest that some genetically encoded, and perhaps not yet described, gain of function mutations of the cGAS-STING pathway could be a first explanation for severe SARS-CoV-2 infections in the young. However, most old patients with severe COVID-19 never exhibited previous features of SAVI syndromes, so that other explanations than mutations of cGAS-STING must be discussed, which might also be more specific for SARS-CoV infections.
The first one is mutants in various other molecules controlling the STING pathways. For instance, the Ca2+ sensor stromal interaction molecule 1 (STIM1) ensures correct localization of STING at the ER, and its down-regulation should enhance STING activation (100). A more attractive candidate is IFN induced protein with tetratricopeptide repeats 3 (IFIT3) (Figure 1), which is highly up-regulated in SARS-CoV-2 infected bronchial epithelial cells (101): IFIT3 interacts with both STING and TANK-binding kinase 1 genes, and activate them in some disorders like lupus (102).
The second explanation could be poor control of STING expression levels by microRNAs (miRNAs) (Figure 1). Induction of miR-576-3p (only present in higher primates: humans, chimpanzee, bonobos, gorilla, and orangutan) by IRF3 and IFN-β triggers a feedback mechanism to suppress STING translation and reduce IFN expression (103). During RNA and DNA virus infections, miR-576-3p sets an antiviral response threshold to likely avoid excessive inflammation (103). Deficient production of miR-576-3p has not yet been searched in severe COVID-19 (Table 2).
Therefore, the third and most attractive hypothesis so far is a delayed over-activation of the cGAS-STING pathway, due to a rebound effect following initial STING inhibition by viral proteases. This could occur when damaged self-DNA and mitochondrial DNA [especially in elderly patients, or patients with metabolic disorders (1, 104)], combine with STING over-activation induced by transfer of cGAMP and/or STING from infected antigen presenting cells to T cells (Table 2) (Figure 1).
This rebound effect might be even more severe in patients with mitigated negative feed-back of STING activation by IL-6, induced by disturbances of the autophagy processes, secondary to ULK1 inhibition by beta-coronaviruses, especially in T cells (65, 81) (Figure 1). Lymphopenia, a predictor of poor prognostic, is reversed in 1 week by tocilizumab injection (105). The T CD4+, T CD8+, and NK cells reduced anti-viral cytokine production capabilities and cytotoxic potential, are also partially restored by inhibition of IL-6 by tocilizumab (53). However, inhibition of IL-6 has not yet demonstrated its ability to reduce the death rate in severe COVID-19, although it was associated with seemingly better outcomes of COVID-19 in patients who survived (106). A defect of the negative feed-back of STING activation by IL-6 in most severe cases might contribute to explain those observations.
Confirmation of a delayed over-activation of the STING pathways in severe COVID-19 would prompt to test drugs already designed to specifically and timely control STING activation (107), like endogenous nitro-conjugated linoleic acid (NO2-cLA) (108). Although a reduced release of IFN-I has already been confirmed, those drugs should be tested first in animal models of STING gain of function, to make sure that they also correct STING-induced lymphopenia and NFκB over-activation (108). They may be added to vitamin-D and aspirin, which probably also prevent STING over-activation (2).
STING is indeed a “hub” of the immune response, not restricted to the TBK1-IRF3 pathway, and a driver of cell death, including T lymphocytes death. Directly targeting STING might be more efficient to restrain the delayed cytokine storm and prevent lymphopenia, pneumonitis, and vasculopathy, than blocking only a single cytokine like IL-6. Direct inhibition of STING delayed over-activation in severe COVID-19 might also better protect from premature apoptosis of memory central T cells, and early (109), or late recurrences of SARS-CoV-2 infections, even in young patients.
The striking similarities noticed between SAVI syndromes and signs of severe COVID-19 including pneumonitis (1), inflammatory vasculopathies with acral thrombosis or Kawasaki-like features (2), and the lymphocyte changes described above, are not evidences that the cGAS-STING axis plays a central role in COVID-19 pathogenesis. However, those numerous analogies could prompt to study the contribution of STING activation in T and B cells changes observed during severe COVID-19, and to address some of the questions listed in Table 2.
This could reinforce the rationale of using drugs preventing from over-activation of STING to treat COVID-19, including as cheap and well tolerated drugs as vitamin-D and aspirin (2). Interestingly, in a large retrospective study performed in American ICUs, even low-dose aspirin seemed to decrease by half mechanical ventilation, ICU admission, and in-hospital mortality in hospitalized patients with COVID-19 (110). If confirmed by prospective studies, this finding would have major consequences on the future of the COVID-19 pandemic.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
J-MB and JS had the original idea, and J-MB wrote the first draft of the article. JS, YM, and FL extensively corrected it and suggested new references. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
To Prof. Pierre Youinou for further incentive.
ACE2, angiotensin-converting enzyme 2; AhR, aryl hydrocarbon receptor; BCR, B cell receptor; cGAS, protein cyclic GMP-AMP synthase; CIVD, common variable immune deficiencies; cTfh, circulating T follicular helper cells; COVID-19, coronavirus disease 2019; EBV, Epstein-Barr virus; ER, endoplasmic reticulum; GATA-3, GATA3 binding protein; ICU, intensive care units; IDO, indoleamine 2,3 dioxygenase; IFIT3, IFN induced protein with tetratricopeptide repeats 3; IFN, interferon; ISG, IFN-stimulated genes; MAVS, mitochondrial antiviral signaling protein; MDA-5, melanoma differentiation-associated protein 5; MERS-CoV, middle-east respiratory syndrome coronavirus; mTORC1, mammalian target of rapamycin complex 1; NETs, neutrophil extracellular traps; NFκB, nuclear factor-kappa B; NKG2A, NK group 2 member A receptor; NKRF, NF-κB-repressing factor; nsp, non-structural proteins; PAMPs, pathogens-associated molecular patterns; PD-1, programmed cell death 1; PHEV, porcine hemagglutinating encephamomyelitis virus; RIG-1, retinoic acid-inducible gene; SARS-CoV, severe acute respiratory syndrome coronaviruses; SAVI syndrome, STING-associated vasculopathy with onset in infancy; STIM1, Ca2+ sensor stromal interaction molecule 1; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; TCR, T cell receptor; TIM-3, T-cell immunoglobulin and mucin containing protein-3; ULK1, Unc-51 like autophagy activating kinase 1; UPR, unfolded protein response.
1. Berthelot JM, Lioté F. COVID-19 as a STING Disorder With Delayed Over-Secretion of Interferon-Beta. EBioMedicine (2020) 23(56):102801. doi: 10.1016/j.ebiom.2020.102801
2. Berthelot JM, Drouet L, Lioté F. Kawasaki-like Diseases and Thrombotic Coagulopathy in COVID-19: Delayed Over-Activation of the STING Pathway? Emerg Microbes Infect (2020) 23:1–26. doi: 10.1080/22221751.2020.1785336
3. Xie J, Li Y, Shen X, Goh G, Zhu Y, Cui J, et al. Dampened STING-Dependent Interferon Activation in Bats. Cell Host Microbe (2018) 23:297–301.e4. doi: 10.1016/j.chom.2018.01.006
4. Huang L, Li L, Lemos H, Chandler PR, Pacholczyk G, Baban B, et al. Cutting Edge: DNA Sensing via the STING Adaptor in Myeloid Dendritic Cells Induces Potent Tolerogenic Responses. J Immunol (2013) 191(7):3509–13. doi: 10.4049/jimmunol.1301419
5. Radzikowska U, Ding M, Tan G, Zhakparov D, Peng Y, Wawrzyniak P, et al. Distribution of ACE2, CD147, CD26 and Other SARS-CoV-2 Associated Molecules in Tissues and Immune Cells in Health and in Asthma, COPD, Obesity, Hypertension, and COVID-19 Risk Factors. Allergy (2020) 75:2829–45. doi: 10.1111/all.14429
6. Sun SH, Chen Q, Gu HJ, Yang G, Wang YX, Huang XY, et al. A Mouse Model of SARS-CoV-2 Infection and Pathogenesis. Cell Host Microbes (2020) 2020:S1931–3128(20)30302-4. doi: 10.1016/j.chom.2020.05.020
7. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell (2020) 181:1016–35.e19. doi: 10.1016/j.cell.2020.04.035
8. Busnadiego I, Fernbach S, Pohl MO, Karakus U, Huber M, Trkola A, et al. Antiviral Activity of Type I, II, and III Interferons Counterbalances ACE2 Inducibility and Restricts SARS-CoV-2. mBio (2020) 11(5):e01928–20. doi: 10.1128/mBio.01928-20
9. Chu H, Zhou J, Wong BHY, Li C, Chan JFW, Cheng ZS, et al. Middle East Respiratory Syndrome Coronavirus Efficiently Infects Human Primary T Lymphocytes and Activates the Extrinsic and Intrinsic Apoptosis Pathways. J Infect Dis (2016) 213(6):904–14. doi: 10.1093/infdis/jiv380
10. Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong H, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med (2005) 202(3):415–24. doi: 10.1084/jem.20050828
11. Wang K, Chen W, Zhou YS, Lian JQ, Zhang Z, Du P, et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. BioRxiv Preprint (2020). doi: 10.1101/2020.03.14.988345
12. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science (2015) 347(6220):1260419. doi: 10.1126/science.1260419
13. Huang W, Luo WJ, Zhu P, Tang J, Yu XL, Cui YC, et al. Modulation of CD147-induced matrix metalloproteinase activity: role of CD147 N-glycosylation. Biochem J (2013) 449(2):437–48. doi: 10.1042/BJ20120343
14. Letko M, Marzi A, Munster V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat Microbiol (2020) 5(4):562–9. doi: 10.1038/s41564-020-0688-y
15. Thomsen MK, Nandakumar R, Stadler D, Malo A, Valls RM, Wang F, et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology (2016) 64:746–59. doi: 10.1002/hep.28685
16. Rodríguez-García E, Olagüe C, Ríus-Rocabert S, Ferrero R, Llorens C, Larrea E, et al. TMEM173 Alternative Spliced Isoforms Modulate Viral Replication Through the STING Pathway. Immunohorizons (2018) 11:363–76. doi: 10.4049/immunohorizons.1800068
17. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455:674–8. doi: 10.1038/nature07317
18. Landman SL, Ressing ME, van der Veen AG. Balancing STING in antimicrobial defense and autoinflammation. Cytokine Growth Factor Rev (2020) 55:1–14. doi: 10.1016/j.cytogfr.2020.06.004.doi.org/10.1016/j.cytogfr.2020.06.004
19. Sun L, Xing Y, Chen X, Zheng Y, Yang Y, Nichols DB, et al. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One (2012) 7:e30802 (2012). doi: 10.1371/journal.pone.0030802
20. Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, et al. Immunology of COVID-19: Current State of the Science. Immunity (2020) 52:910–41. doi: 10.1016/j.immuni.2020.05.002
21. Li J, Guo M, Tian X, Liu C, Wang X, Yang X, et al. Virus-host interactome and proteomic survey of PMBCs from COVID-19 patients reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. bioRxiv (2020). doi: 10.1101/2020.03.31.019216
22. Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A (2017) 114:1637–42. doi: 10.1073/pnas.1621363114
23. Imanishi T, Saito T. T Cell Co-stimulation and Functional Modulation by Innate Signals. Trends Immunol (2020) 41:200–12. doi: 10.1016/j.it.2020.01.003
24. Ma F, Li B, Yu Y, Iver SS, Sun M, Cheng G. Positive Feedback Regulation of Type I Interferon by the Interferon-Stimulated Gene STING. EMBO Rep (2015) 16:202–12. doi: 10.15252/embr.201439366
25. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature (2019) 567:262–6. doi: 10.1038/s41586-019-1006-9
26. Yap JKY, Moriyama M, Iwasaki A. Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19. J Immunol (2020) 205:307–12. doi: 10.4049/jimmunol.2000513
27. Nandakumar R, Tschismarov R, Meissner F, Prabakaran T, Krissanaprasit A, Farahani E, et al. Intracellular Bacteria Engage a STING-TBK1-MVB12b Pathway to Enable Paracrine cGAS-STING Signalling. Nat Microbiol (2019) 4:701–13. doi: 10.1038/s41564-019-0367-z
28. Imanishi T, Unno M, Kobayashi W, Yoneda N, Matsuda S, Ikeda K, et al. Reciprocal Regulation of STING and TCR Signaling by mTORC1 for T-cell Activation and Function. Life Sci Alliance (2019), e201800282. doi: 10.26508/lsa.201800282
29. Kalamvoki M, Du T, Roizman B. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc Natl Acad Sci U S A (2014) 111:E4991–6. doi: 10.1073/pnas.1419338111
30. Luteijn RD, Zaver SA, Gowen BG, Wyman SK, Garelis NE, Onia L, et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature (2019) 573(7774):434–8. doi: 10.1038/s41586-019-1553-0
31. Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg MC, et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J Exp Med (2017) 214:1769–85. doi: 10.1084/jem.20161674
32. Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, et al. STING-mediated Disruption of Calcium Homeostasis Chronically Activates ER Stress and Primes T Cell Death. J Exp Med (2019) 216:867–83. doi: 10.1084/jem.20182192
33. Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, et al. Signalling strength determines proapoptotic functions of STING. Nat Commun (2017) 8:1–10. doi: 10.1038/s41467-017-00573-w
34. Berg RK, Rahbek SH, Kofod-Olsen E, Holm CK, Melchjorsen J, Jensen DG, et al. T cells detect intracellular DNA but fail to induce type I IFN responses: implications for restriction of HIV replication. PLoS One (2014) 9:e84513. doi: 10.1371/journal.pone.0084513.10.1371/journal.pone.0084513
35. Li W, Lu L, Lu J, Wang X, Yang C, Jin J, et al. cGAS-STING–mediated DNA sensing maintains CD8+ T cell stemness and promotes antitumor T cell therapy. Sci Transl Med (2020) 12:eaay9013. doi: 10.1126/scitranslmed.aay9013
36. Konno H, Yamauchi S, Berglund A, Putney RM, Mule JJ, Barber GN, et al. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene (2018) 37:2037–51. doi: 10.1038/s41388-017-0120-0
37. Zheng J, Mo J, Zhu T, Zhuo W, Yi Y, Hu S, et al. Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol Cancer (2020) 19(1):133. doi: 10.1186/s12943-020-01250-1
38. Palermo E, Acchioni C, Di Carlo D, Zevini A, Muscolini M, Ferrari M, et al. Activation of latent HIV-1 T cell reservoirs with a combination of innate immune and epigenetic regulators. J Virol (2019) 93:e01194–19. doi: 10.1128/jvi.01194-19
39. Guo N, Ye S, Zhang K, Yu X, Cui H, Yang X, et al. Critical epitope in CD147 facilitates memory CD4+ T-cell hyper-activation in rheumatoid arthritis. Cell Mol Immunol (2019) 16:568–79. doi: 10.1038/s41423-018-0012-4
40. Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection, Nat. Microbiol (2017) 2:1–11. doi: 10.1038/nmicrobiol.2017.37
41. Holm CK, Rahbek SH, Gad HH, Bak RO, Jakobsen MR, Jiang Z, et al. Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat Commun (2016) 7:1–9. doi: 10.1038/ncomms10680
42. Yao Z, Zheng Z, Wu K, Junhua Z. Immune Environment Modulation in Pneumonia Patients Caused by Coronavirus: SARS-CoV, MERS-CoV and SARS-CoV-2. Aging (Albany NY) (2020) 12:7639–51. doi: 10.18632/aging.103101
43. Roncati L, Nasillo V, Lusenti B, Riva G. Signals of Th2 immune response from COVID-19 patients requiring intensive care. Hematol (2020) 99:1419– 20. doi: 10.1007/s00277-020-04066-7
44. Li CKF, Wu H, Yan H, Ma S, Wang L, Zhang M, et al. T cell responses to whole SARS coronavirus in humans. J Immunol (2008) 181:5490–500. doi: 10.4049/jimmunol.181.8.5490
45. Negishi T, Kato Y, Ooneda O, Mimura J, Takada T, Mochizuki H. Effects of Aryl Hydrocarbon Receptor Signaling on the Modulation of TH1/TH2 Balance. J Immunol (2005) 175:7348–56. doi: 10.4049/jimmunol.175.11.7348
46. Toussaint M, Jackson DJ, Swieboda D, Guedán A, Tsourouktsoglou TD, Ching YM, et al. Host DNA Released by NETosis Promotes Rhinovirus-Induced type-2 Allergic Asthma Exacerbation. Nat Med (2017) 23:681–91. doi: 10.1038/nm.4332
47. Nündel K, Marshak-Rothstein A. The Role of Nucleic Acid Sensors and Type I IFNs in Patient Populations and Animal Models of Autoinflammation. Curr Opin Immunol (2019) 61:74–9. doi: 10.1016/j.coi.2019.08.003
48. Grunewald ME, Shaban MG, Mackin SR, Fehr AR, Perlman S. Murine Coronavirus Infection Activates the Aryl Hydrocarbon Receptor in an Indoleamine 2,3-Dioxygenase-Independent Manner, Contributing to Cytokine Modulation and Proviral TCDD-Inducible-PARP Expression. J Virol (2020) 94:e01743–19. doi: 10.1128/JVI.01743-19
49. Gallais F, Velay A, Wendling MJ, Nazon C, Partisani M, Sibilia J, et al. Intrafamilial exposure to SARS-CoV-2 induced cellular immune response without seroconversion. medRxiv (2020). doi: 10.1101/2020.06.21.20132449
50. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science (2020) 370:eabd4570. doi: 10.1126/science.abd4570
51. Paces J, Strizova Z, Smrz D, Cerny J. COVID-19 and the Immune System. Physiol Res (2020) 69:379–88. doi: 10.33549/physiolres.934492
52. Weiskopf D, Schmitz KS, Raadsen MP, Grifoni A, Okba NMA, Endeman H, et al. Phenotype of SARS-CoV-2-specific T-cells in COVID-19 patients with acute respiratory distress syndrome. Sci Immunol (2020) 5(48):eabd2071. doi: 10.1126/sciimmunol.abd2071
53. Mazzoni A, Salvati L, Maggi L, Capone M, Vanni A, Spinicci M, et al. Impaired Immune Cell Cytotoxicity in Severe COVID-19 Is IL-6 Dependent. J Clin Invest (2020) 28:138554. doi: 10.1172/JCI138554
54. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T Cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
55. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
56. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
57. Hirano T, Murakami M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release System. Immunity (2020) 52:731–3. doi: 10.1016/j.immuni.2020.04.003
58. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y, et al. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients. Natl Sci Rev (2020), 2020. doi: 10.1093/nsr/nwaa041
59. Shalhoub S, Farahat F, Al-Jiffri A, Simhairi R, Shamma O, Siddiqi N, et al. IFN-alpha 2a or IFN-beta1a in combination with ribavirin to treat Middle East respiratory syndrome coronavirus pneumonia: A retrospective study. J Antimicrob Chemother (2015) 70:2129–32. doi: 10.1093/jac/dkv085
60. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe (2016) 19:181–93. doi: 10.1016/j.chom.2016.01.007
61. Bennion BG, Ingle H, Ai TL, Miner CA, Platt DJ, Smith AM, et al. A Human Gain-of-Function STING Mutation Causes Immunodeficiency and Gamma-herpesvirus-Induced Pulmonary Fibrosis in Mice. J Virol (2019) 93:e01806–18. doi: 10.1128/JVI.01806-18
62. Chu H, Chan JFW, Wang Y, Yuen TTT, Chai Y, Hou Y, et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: An ex vivo study with implications for the pathogenesis of COVID-19. Clin Infect Dis (2020) 71:1400–9. doi: 10.1093/cid/ciaa410
63. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Pere H, et al. Impaired type I interferon activity and exacerbated inflammatory responses in severe Covid-19 patients. Science (2020) 369:718–24. doi: 10.1126/science.abc6027
64. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
65. Wu X, Yang J, Na T, Zhang K, Davidoff AM, Yuan BZ, et al. RIG-I and IL-6 Are Negative-Feedback Regulators of STING Induced by Double-Stranded DNA. PLoS One (2017) 12:e0182961. doi: 10.1371/journal.pone.0182961
66. Lei L, Qian H, Yang X, Zhou X, Zhang X, Zhang D, et al. The phenotypic changes of gamma-delta T cells in COVID-19 patients. J Cell Mol Med (2020) 24:11603–6. doi: 10.1111/jcmm.15620
67. Terpos E, Ntanasis-Stathopoulos I, Elalamy I, Kastritis E, Sergentanis TN, Politou M, et al. Hematological findings and complications of COVID-19. Am J Hematol (2020). doi: 10.1002/ajh.25829
68. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis (2020) 71:762–8. doi: 10.1093/cid/ciaa248
69. Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, et al. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol (2020) 33:1007–14. doi: 10.1038/s41379-020-0536-x
70. Chen Y, Feng Z, Diao B, Wang R, Wang G, Wang C, et al. The Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Directly Decimates Human Spleens and Lymph Nodes. medRxiv (2020). doi: 10.1101/2020.03.27.20045427
71. Laterre PF, François B, Collienne C, Hantson P, Jeannet R, Remy KE, et al. Association of Interleukin 7 Immunotherapy With Lymphocyte Counts Among Patients With Severe Coronavirus Disease 2019 (COVID-19). JAMA Netw Open (2020) 3(7):e2016485. doi: 10.1001/jamanetworkopen.2020.16485
72. Ghazavi A, Ganji A, Keshavarzian N, Rabiemajd S, Mosayebi G. Cytokine profile and disease severity in patients with COVID-19. Cytokine (2020) 137:155323. doi: 10.1016/j.cyto.2020.155323
73. Huang JL, Huang J, Duan ZH, Wei J, Min J, Luo XH, et al. Th2 predominance and CD8+ memory T cell depletion in patients with severe acute respiratory syndrome. Microbes Infect (2005) 7:427–36. doi: 10.1016/j.micinf.2004.11.017
74. Luo X, Li H, Ma L, Zhou J, Guo X, Woo SL, et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology (2018) 155:1971–1984.e4. doi: 10.1053/j.gastro.2018.09.010
75. Zhang Y, Chen W, Wang Y. STING is an essential regulator of heart inflammation and fibrosis in mice with pathological cardiac hypertrophy via endoplasmic reticulum (ER) stress. Biomed Pharmacother (2020) 125:110022. doi: 10.1016/j.biopha.2020.110022
76. Chia J, Eroglu FK, Özen S, Orhan D, Montealegre-Sanchez G, de Jesus AA, et al. Failure to thrive, interstitial lung disease, and progressive digital necrosis with onset in infancy. J Am Acad Dermatol (2016) 74:186–9. doi: 10.1016/j.jaad.2015.10.007
77. Bouis D, Kirstetter P, Arbogast F, Lamon D, Delgado V, Jung S, et al. Severe Combined Immunodeficiency in Stimulator of Interferon Genes (STING) V154M/wild-type Mice. J Allergy Clin Immunol (2019) 143:712–725.e5. doi: 10.1016/j.jaci.2018.04.034
78. Luksch H, Stinson WA, Platt DJ, Qian W, Kalugotla G, Miner CA, et al. STING-associated Lung Disease in Mice Relies on T Cells but Not Type I Interferon. J Allergy Clin Immunol (2019) 144:254–66.e8. doi: 10.1016/j.jaci.2019.01.044
79. Motwani M, Pawaria S, Bernier J, Moses S, Henry K, Fang T, et al. Hierarchy of Clinical Manifestations in SAVI N153S and V154M Mouse Models. Proc Natl Acad Sci U S A (2019) 116:7941–50. doi: 10.1073/pnas.1818281116
80. Barata JT, Durum SK, Seddon B. Flip the coin: IL-7 and IL-7R in health and disease. Nat Immunol (2020) 20:1584–93. doi: 10.1038/s41590-019-0479-x
81. Carmona-Guttierez D, Bauer MA, Zimmermann A, Kainz K, Hofer SJ, Kroemer G, et al. Digesting the crisis: autophagy and coronaviruses. Microb Cell (2020) 7:119–28. doi: 10.15698/mic2020.05.715
82. Rubio RM, Mohr I. Inhibition of ULK1 and Beclin1 by an α-herpesvirus Akt-like Ser/Thr Kinase Limits Autophagy to Stimulate Virus Replication. Proc Natl Acad Sci U S A (2019) 116:26941–50. doi: 10.1073/pnas.1915139116
83. Shojaei S, Suresh M, Klionsky DJ, Labouta HI, Ghavami S. Autophagy and SARS-CoV-2 Infection: A possible Smart Targeting of the Autophagy Pathway. Virulence (2020) 11:805–10. doi: 10.1080/21505594.2020.1780088
84. Li Z, Zhao K, lv X, Lan Y, Hu S, Shi J, et al. Ulk1 Governs Nerve Growth Factor/TrkA Signaling by Mediating Rab5 GTPase Activation in Porcine Hemagglutinating Encephalomyelitis Virus-Induced Neurodegenerative Disorders. J Virol (2018) 92:e00325–18. doi: 10.1128/JVI.00325-18
85. Lemos H, Huang L, McGaha TL, Mellor AL. Cytosolic DNA Sensing via the Stimulator of Interferon Genes Adaptor: Yin and Yang of Immune Responses to DNA. Eur J Immunol (2014) 44:2847–53. doi: 10.1002/eji.201344407
86. Hao S, Lian J, Lu Y, Jia H, Hu J, Yu G, et al. Decreased B cells on admission was associated with prolonged viral RNA shedding from respiratory tract in Coronavirus Disease 2019: a case control study. J Infect Dis (2020) 31:jiaa311. doi: 10.1093/infdis/jiaa311
87. Mathew D, Giles JR, Baxter AE, Greenplate AR, Wu JE, Alanio C, et al. Deep Immune Profiling of COVID-19 Patients Reveals Patient Heterogeneity and Distinct Immunotypes With Implications for Therapeutic Interventions. Science (2020) 369:eabc8511. doi: 10.1126/science.abc8511
88. Huang AT, Garcia-Carreras B, Hitchings MDT, Yang B, Katznelnik LC, Rattigan SM, et al. A systematic review of antibody mediated immunity to coronaviruses: antibody kinetics, correlates of protection, and association of antibody responses with severity of disease. Nat Commun (2020) 11(1):4704. doi: 10.1038/s41467-020-18450-4
89. Shen C, Wang Z, Zhao F, Yang Y, Li J, Yuan J, et al. Treatment of 5 Critically Ill Patients With COVID-19 With Convalescent Plasma. JAMA (2020) 323:1582–9. doi: 10.1001/jama.2020.4783
90. Quinti I, Lougaris V, Milito C, Cinetto F, Pecoraro A, Mezzaroma I, et al. A Possible Role for B Cells in COVID-19? Lesson From Patients With Agammaglobulinemia. J Allergy Clin Immunol (2020) 146:211–13e4. doi: 10.1016/j.jaci.2020.04.013
91. Walker MM, Crute BW, Cambier JC, Getahun A. B Cell-Intrinsic STING Signaling Triggers Cell Activation, Synergizes With B Cell Receptor Signals, and Promotes Antibody Responses. J Immunol (2018) 201:2641–53. doi: 10.4049/jimmunol.1701405
92. Marcus A, Mao AJ, Lensink-Vasan M, Wang LA, Vance RE, Raulet DH. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity (2018) 49:754–63.e4. doi: 10.1016/j.immuni.2018.09.016
93. Bénard A, Sakwa I, Schierloh P, Colom A, Mercier I, Tailleux L, et al. B Cells Producing Type I IFN Modulate Macrophage Polarization in Tuberculosis. Am J Respir Crit Care Med (2018) 197:801–13. doi: 10.1164/rccm.201707-1475OC
94. Tang CHA, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res (2016) 76:2137–52. doi: 10.1158/0008-5472.CAN-15-1885
95. Katikaneni DS, Jin L. B Cell MHC Class II Signaling: A Story of Life and Death. Hum Immunol (2019) 80:37–43. doi: 10.1016/j.humimm.2018.04.013
96. Gram AM, Sun C, Landman SL, Oosenbrug T, Koppejan HJ, Kwakkenbos MJ. Human B Cells Fail to Secrete Type I Interferons Upon Cytoplasmic DNA Exposure. Mol Immunol (2017) 91:225–37. doi: 10.1016/j.molimm.2017.08.025
97. Bridgeman A, Maelfait J, Davenne T, Partridge T, Peng Y, Mayer A, et al. Viruses Transfer the Antiviral Second Messenger cGAMP Between Cells. Science (2015) 349:1228–32. doi: 10.1126/science.aab3632
98. Nelemans T, Kikkert M. Viral Innate Immune Evasion and the Pathogenesis of Emerging RNA Virus Infections. Viruses (2019) 11:E961. doi: 10.3390/v11100961
99. Oh HLJ, Gan SKE, Bertoletti A, Tan YJ. Understanding the T Cell Immune Response in SARS Coronavirus Infection. Emerg Microbes Infect (2012) 1:e23. doi: 10.1038/emi.2012.26
100. Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, et al. The Ca 2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol (2019) 20:152–62. doi: 10.1038/s41590-018-0287-8
101. Vishnubalaii R, Shaath H, Alaiez NM. Protein Coding and Long Noncoding RNA (lncRNA) Transcriptional Landscape in SARS-CoV-2 Infected Bronchial Epithelial Cells Highlight a Role for Interferon and Inflammatory Response. Genes (Basel) (2020) 7(11):E760. doi: 10.3390/genes11070760
102. Wang J, Dai M, Cui Y, Hou G, Deng J, Gao X, et al. Association of Abnormal Elevations in IFIT3 With Overactive Cyclic GMP-AMP Synthase/Stimulator of Interferon Genes Signaling in Human Systemic Lupus Erythematosus Monocytes. Arthritis Rheumatol (2018) 70:2036–45. doi: 10.1002/art.40576
103. Yarbrough ML, Zhang K, Sakthivel R, Forst CV, Posner BA, Barber GN, et al. Primate-specific miR-576-3p sets host defense signalling threshold. Nat Commun (2014) 5:1–10. doi: 10.1038/ncomms5963
104. Hamann L, Szwed M, Mossakowska M, Chudek J, Puzianowska-Kuznicka M. First evidence for STING SNP R293Q being protective regarding obesity-associated cardiovascular disease in age-advanced subjects – a cohort study. Immun Ageing (2020) 17:7. doi: 10.1186/s12979-020-00176-y
105. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos ND, et al. Complex Immune Dysregulation in COVID-19 Patients With Severe Respiratory Failure. Cell Host Microbe (2020) 27(6):992–1000.e3. doi: 10.1016/j.chom.2020.04.009
106. Xu X, Han M, Li T, Sun W, Wang D, Fu B, et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci USA (2020) 117:10970–5. doi: 10.1073/pnas.2005615117
107. Cui X, Zhang R, Cen S, Zhou J. STING modulators: Predictive significance in drug discovery. Eur J Med Chem (2019) 182:111591. doi: 10.1016/j.ejmech.2019.111591
108. Hansen AL, Buchan GJ, Ruhl M, Mukai K, Salvatore SR, Ogawa E, et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc Natl Acad Sci U S A (2018) 115:E7768–75. doi: 10.1073/pnas.1806239115
109. Lafaie L, Célarier T, Goethals L, Pozzetto B, Grange S, Ojardias E, et al. Recurrence or Relapse of COVID-19 in Older Patients: A Description of Three Cases. J Am Geriatr Soc (2020). doi: 10.1111/jgs.16728
Keywords: SARS-CoV-2, cGAMP, lymphopenia, interferon, STING, COVID-19, lymphocyte, aspirin
Citation: Berthelot J-M, Lioté F, Maugars Y and Sibilia J (2020) Lymphocyte Changes in Severe COVID-19: Delayed Over-Activation of STING? Front. Immunol. 11:607069. doi: 10.3389/fimmu.2020.607069
Received: 16 September 2020; Accepted: 03 November 2020;
Published: 01 December 2020.
Edited by:
Chunfu Zheng, Fujian Medical University, ChinaReviewed by:
Jianzhong Zhu, Yangzhou University, ChinaCopyright © 2020 Berthelot, Lioté, Maugars and Sibilia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Marie Berthelot, amVhbm1hcmllLmJlcnRoZWxvdEBjaHUtbmFudGVzLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.