 Lindsey E. Fox
Lindsey E. Fox Marissa C. Locke
Marissa C. Locke Deborah J. Lenschow
Deborah J. Lenschow- 1Department of Pathology and Immunology, Washington University School of Medicine, Saint Louis, MO, United States
- 2Department of Medicine, Washington University School of Medicine, Saint Louis, MO, United States
Type I interferons (IFNs) are critical effector cytokines of the immune system and were originally known for their important role in protecting against viral infections; however, they have more recently been shown to play protective or detrimental roles in many disease states. Type I IFNs consist of IFNα, IFNβ, IFNϵ, IFNκ, IFNω, and a few others, and they all signal through a shared receptor to exert a wide range of biological activities, including antiviral, antiproliferative, proapoptotic, and immunomodulatory effects. Though the individual type I IFN subtypes possess overlapping functions, there is growing appreciation that they also have unique properties. In this review, we summarize some of the mechanisms underlying differential expression of and signaling by type I IFNs, and we discuss examples of differential functions of IFNα and IFNβ in models of infectious disease, cancer, and autoimmunity.
Introduction
Interferons (IFNs) are cytokines that were originally discovered and named for their ability to interfere with viral replication (1). IFNs are grouped into three classes according to the receptor that mediates their effects: type I IFNs (the focus of this review), type II IFN (IFNγ), and type III IFNs (IFNλs) (2, 3). Broadly speaking, each IFN class signals through receptor-associated Janus kinases (JAKs), which activate various Signal Transducer and Activator of Transcription (STAT)-signaling pathways. Type I IFNs signal through the heterodimeric IFN-α/β receptor 1 (IFNAR1) and IFNAR2, which are associated with the JAKs tyrosine kinase 2 (TYK2) and JAK1, respectively (4). Canonically, activation of TYK2 and JAK1 leads to the formation of the IFN-stimulated gene (ISG) factor 3 (ISGF3) complex, composed of STAT1, STAT2, and interferon regulatory factor 9 (IRF9). The ISGF3 complex then translocates to the nucleus to regulate the expression of hundreds of IFN-stimulated genes. Type I IFN signaling can activate other STAT complexes, often in a cell-type dependent manner. Additionally, alternative signaling cascades, including the mitogen-activated protein kinase p38 pathway and the phosphatidylinositol 3-kinase pathway, are also required for optimal generation of type I IFN responses (4).
Type I IFNs have broad, pleiotropic effects that include antiviral activity, antiproliferative effects, and immunomodulatory properties. There is growing evidence that the overall outcome of type I IFN responses can be beneficial or detrimental for the host depending on the timing, magnitude, and source of IFN production, as well as the specific biological context (5). Moreover, despite signaling through a shared receptor, type I IFN subtypes possess important functional differences, both in vitro and in vivo. The purpose of this review is to summarize the current understanding of differential type I IFN properties, focusing on the role of human and mouse IFNα and IFNβ in infectious disease, cancer, and autoimmunity. In particular, we seek to highlight the few examples that demonstrate or suggest differential activities for type I IFN subtypes in vivo.
Type I IFNS: A Multigene Family
Type I IFNs exist as a multigene family across many species (Figure 1) (6). IFNαs, IFNβ, IFNϵ, IFNκ, and IFNω are found in many species, whereas IFNδ and IFNτ are only found in pigs and cattle (7). In humans (HuIFN), the type I IFN genes are located on chromosome 9 and encode 13 IFNα subtypes and single forms of IFNβ, IFNϵ, IFNκ, and IFNω (7). Type I IFNs in mice (MuIFN) are located on chromosome 4, and likewise, consist of multiple genes with some differences compared to human. MuIFNs include 14 IFNα subtypes, IFNβ, IFNϵ, IFNκ, an IFN-like cytokine IFNζ (also known as limitin), but lack a functional IFNω, which is present as a pseudogene (8).
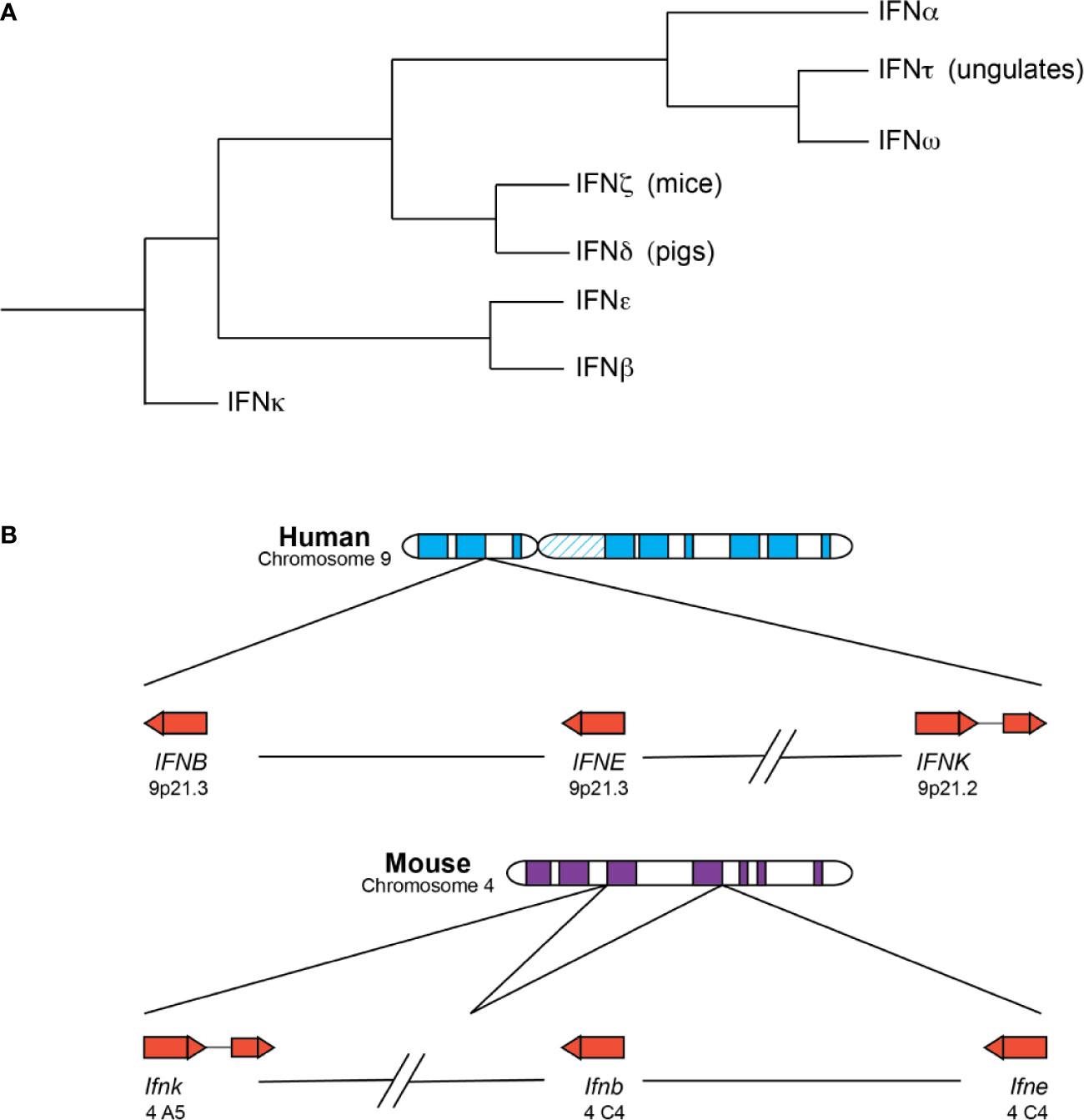
Figure 1 Type I IFNs are a closely related family of related cytokines. (A) Depicted is a summary of existing phylogenetic analyses of the type I IFNs. The branches are not drawn to scale. IFNκ, IFNβ, and IFNϵ are mostly present in placental mammals as single copies and the first subtypes to diverge from the other type I IFNs. IFNβ and IFNϵ are especially similar and can be found within the same clade in some analyses. IFNδ and IFNζ are the next subtypes to diverge and are only found in pigs and mice, respectively. IFNτ and IFNω are closely related, despite their differences in function and distribution—IFNτ is only expressed in placental tissues of ungulate species and involved in pregnancy, whereas IFNω is found in many species and possesses the more canonical antiviral and immunomodulatory functions. IFNω and IFNα loci are expanded to include many subtypes in a number of species. (B) The chromosomal locations of human (top) and murine (bottom) IFNκ, IFNβ, and IFNϵ genes are depicted. The arrow direction indicates on which strand the gene is encoded: a left-to-right arrow depicts the forward or positive strand and a right-to-left arrow indicates the reverse or negative strand. IFNκ is the only subtype to contain an intron and is situated further away from the other type I IFNs, though its positioning relative to the other IFNs is different in mice and humans. IFNβ and IFNϵ roughly form the boundaries of the type I IFN locus, with the other type I IFNs falling between the two genes.
Phylogenetic analyses reveal that the type I IFN subtypes form clades consistent with mammalian speciation (7, 9, 10). For the most part, placental mammals possess single copies of the genes encoding IFNκ, IFNβ, and IFNϵ, and these unduplicated subtypes represent the first major clade within mammalian IFNs (11). IFNκ is the first subtype to diverge within mammalian type I IFNs and forms an outgroup, possibly the result of a unique evolutionary route for IFNκ relative to IFNβ and IFNϵ (11). IFNκ is additionally distinctive as the only mammalian type I IFN that contains an intron, and for many species, the gene encoding IFNκ is situated further away from the IFN locus (7, 9, 11). Depending on the analysis, IFNβ or IFNϵ is the next subtype to diverge from mammalian type I IFNs, and in some analyses IFNβ and IFNϵ fall within the same clade, suggesting that these subtypes might be more closely related to each other than the other type I IFN subtypes (7, 9, 11, 12). The genes encoding IFNβ and IFNϵ are situated at the “beginning” and “end” of the type I IFN locus across many species, which is relatively conserved across mammalian species. IFNδ and IFNζ (limitin) are the next type I IFNs to diverge within mammalian IFNs and are only found in pigs and mice, respectively (7). However, recent identification of a putative HuIFNδ gene calls this into question (11).
The last subtypes to diverge are the IFNαs, IFNωs, and IFNτs. These subtypes are thought to be exclusively found in placental mammals and are usually situated between the IFNϵ and IFNβ genes within the type I IFN locus. IFNω and IFNτ are closely related, even though they possess different functions (7, 11). IFNτ is only found in placental tissues of ungulate species, is involved in pregnancy, and may have arisen from an IFNω subtype (10, 13). In contrast, IFNω is an antiviral and immunomodulatory molecule, like IFNα, and functional copies have been identified in humans and other animal groups including felines, pigs, cattle, serotine bats, and others but are not present in canines or mice (14). Notably, humans have only one IFNω, but there is evidence that IFNω is still expanding and diversifying in many species, including bats and pigs (15–17). Lastly, the genes encoding IFNα are found in all placental mammals and form species-specific clades, with some exceptions for closely related organisms (e.g. chimpanzees, humans, and gorillas); a combination of gene duplication and gene conversion events likely gave rise to the expanded IFNα genes present in many mammals (6). Of note, a recent study found that for some IFNα subtypes, such as HuIFNα6, α8, α13, and α14, amino acid-altering variation was more constrained in the human population, suggesting that they might perform non-redundant functions in host responses (18).
As sequenced genomes of other species become available, the phylogenetic clustering of some type I IFNs may change. However, the key point is that the multigene nature of type I IFNs is conserved across many species. Both IFNα and IFNω subtypes expanded independently and multiple times, suggesting that it is advantageous for the host to possess a large repertoire of at least several type I IFN subtypes. Unfortunately, the fact that type I IFNs expanded multiple times complicates directly applying results of IFN studies from animal models to clinical settings, and caution is warranted in drawing conclusions about specific human IFNα subtypes from studies of murine IFNα subtypes.
Molecular Mechanisms Underlying Distinct Functions of Type I IFNs
Though type I IFNs possess many overlapping functions, it is now appreciated that the individual subtypes have different potencies of their shared functions and some unique functions in vitro. An important early example demonstrating this was the finding that HuIFNβ was 100-fold more potent than HuIFNα2 in inhibiting osteoclastogenesis through its ability to preferentially induce the chemokine CXCL11 (19). Since this observation, it is now appreciated that the pleiotropic activities ascribed to different type I IFN subtypes are the product of distinct patterns and kinetics of expression, as well as signaling differences that arise from differential binding affinities and susceptibility to negative feedback loops (20, 21). The ability of the type I IFN receptor to have fine-tuned responses to many ligands is likely advantageous considering the array of pathogens that have co-evolved alongside humans, mice, and other animals.
Differential Dependence on IRF3 and IRF7 for Transcription
Before examining the signaling and functional properties of IFN subtypes, it should be noted that type I IFNs are differentially induced downstream of pattern recognition receptor (PRR) signaling, except for IFNϵ, which is hormonally regulated (see below). PRR signaling converges on the phosphorylation and activation of the transcription factors IRF3 and IRF7, though other IRFs can be involved in IFN-dependent antiviral responses (22, 23). For most cell types IRF3 is constitutively expressed, whereas IRF7 is induced downstream of type I IFN signaling to then amplify and diversify the type I IFN response (22). The exception to this rule is plasmacytoid dendritic cells (pDCs), which constitutively express IRF7 and are thus poised to rapidly secrete large amounts of type I IFN (24). The promoters of specific type I IFN genes differ in their requirement of IRF3 or IRF7 binding for maximal transcription. Thus, the temporal regulation of IRFs dictates the expression of IFN subtypes.
Early in a response, IRF3 activation first induces transcription of MuIFNβ and MuIFNα4 via unique IRF3 binding sites within their promoters (25–31). For the most part, the other MuIFNα subtypes require both IRF3 and IRF7 for maximal transcription, and so they depend on type I IFN-mediated upregulation of IRF7 (32–34). Similar to mice, IRF3 also initiates human type I IFN responses by upregulating transcription of HuIFNβ and HuIFNα1, while the other HuIFNA genes require both IRF3 and IRF7 (35, 36). Altogether, these findings demonstrate that for most cell types, activation of constitutive IRF3 by PRR signaling initiates a first wave of HuIFNβ and HuIFNα1 (or MuIFNβ and MuIFNα4 for mice). Subsequently, a second, amplified wave of diverse IFNα subtypes follows that is IRF7-dependent. As the ratio of IRF3 to IRF7 or other IRFs changes over time, the repertoire of IFN subtypes expressed changes as well.
There are several intriguing deviations from this paradigm. First, the IFNβ promoter has additional response elements that make it responsive to NF-κB signaling through activating transcription factor 2 (ATF-2) and c-Jun, which allows other signaling pathways to augment IFNβ production (29, 37, 38). This unique promoter feature also permits IRF3-independent basal expression of low amounts of IFNβ in the absence of infection, which can have significant impact on mounting successful innate immune responses against a variety of infections (39–47). IFNκ may have somewhat restricted expression, as it was named for its high expression in keratinocytes; however, other cell types, including immune cells and lung epithelial cells, can upregulate IFNκ expression (48–50). Further characterization is needed to determine which cells are capable of expressing IFNκ in different contexts. Lastly, IFNϵ is the most notable exception to the IRF-mediated IFN induction paradigm, as it is not regulated at all by PRR signaling and IRF3/7. Instead, it is constitutively expressed in the epithelium of reproductive organs and hormonally regulated, and this is reflected in its unique promoter (51–53).
Differential Binding Affinity Determines Signaling and Function
All type I IFNs bind to and signal through the heterodimeric receptor IFNAR1 and IFNAR2 to activate canonical JAK/STAT signaling pathways (4). A unique feature of type I IFN signaling is that the signaling outcome can vary depending on the cell type, specific ligand, and concentration of the type I IFN subtype. The molecular mechanisms that underlie the plasticity of type I IFN signaling have been extensively reviewed elsewhere, so only key features will be outlined in this review (20, 54, 55).
In general, IFNAR2 is the primary ligand binding receptor subunit and binds type I IFNs with high affinity (typically nanomolar affinity); IFNAR1 is subsequently recruited to the receptor-ligand complex and binds with relatively lower affinity (approximately micromolar affinity) (54). HuIFNβ has the highest natural binding affinity to the type I IFN receptors with picomolar affinity for IFNAR2 and nanomolar affinity for IFNAR1, whereas HuIFNα2 possesses nanomolar affinity for IFNAR2 and micromolar affinity for IFNAR1 (56–58). This higher affinity interaction may enable IFNβ to uniquely signal through IFNAR1 in an IFNAR2-independent manner, but further work is needed to corroborate this finding and to determine if other receptors are involved in this phenomenon (59, 60). Engineered IFNα2 and IFNω mutants that mimic the range of affinities for the receptor complex have demonstrated that type I IFN signaling outcomes can be directly linked to IFN affinity to the receptor complex. Hence, type I IFN mutants that acquire IFNβ-like affinity acquire IFNβ-like potency (61, 62).
In line with these findings for IFNα, IFNβ, and IFNω, recent work showed that HuIFNϵ and HuIFNκ bound IFNAR2 with particularly weak affinity and demonstrated approximately 1000-fold decreased potency in ISGF3-mediated gene expression compared to HuIFNα2, whereas their affinity for IFNAR1 was comparable to other type I IFN subtypes (63). HuIFNϵ and HuIFNκ also bound the poxvirus antagonist B18R with weaker affinity relative to the other IFN subtypes, perhaps suggesting a fitness advantage for the host to have some weaker binding IFN subtypes in order to avoid virus inhibition (63). In influenza A virus (IAV) infection, HuIFNκ, but not IFNα or IFNβ, relied on chromodomain helicase DNA binding protein 6 (CHD6) to efficiently suppress viral replication (50). Moreover, induction of CHD6 was not dependent on STAT1, but rather, IFNκ signaled through the mitogen-activated protein kinase (MAPK) p38 and the transcription factor c-Fos to mediate its antiviral effects. Altogether, these findings suggest that in addition to having unique expression patterns, IFNϵ and IFNκ may possess additional biochemical and signaling features that grant unique properties in vivo.
Differential Sensitivity to Feedback Loops
The affinity of individual subtypes, as outlined above, is a key component in determining the signaling outcome from IFNAR1/2 engagement, but negative feedback loops are an additional level of regulation and fine-tuning. IFNAR1/2 surface abundance is typically quite low, and modulating the surface receptor expression is one means of regulating type I IFN signaling after type I IFN induction (64). Manipulation of a cell line’s IFNAR expression demonstrated that the antiproliferative and proapoptotic activities induced by HuIFNβ are less sensitive to decreased receptor levels than those induced by HuIFNα2 (65, 66). The physiological relevance of receptor expression influencing type I IFN signaling is demonstrated in the number of IFN-dependent mechanisms that downregulate IFNAR1 and IFNAR2 levels. We will outline a few examples.
First, protein kinase D2 (PKD2) is a negative regulator activated downstream of IFN signaling. It phosphorylates IFNAR1, enabling interaction with a ubiquitin E3 ligase, and subsequent ubiquitination leads to endocytosis of the IFN signaling complex (67, 68). Endosomes with short-lived receptor-ligand complexes formed by lower affinity IFNαs are more likely to be recycled to the cell surface; endosomes with longer-lived complexes formed by higher affinity IFNβ ultimately fuse with the lysosome, but signaling can continue to take place as trafficking progresses through the endosomal compartment (69–72). Second, Suppressor of Cytokine Signaling 1 (SOCS1) can directly dampen the type I IFN response by interacting with TYK2 to disrupt TYK2-STAT signaling, but it also decreases surface levels of IFNAR1, which requires TYK2 for stability at the cell surface (73). Lastly, ubiquitin-specific peptidase 18 (USP18) can bind the cytoplasmic domain of IFNAR2 and interfere with IFNAR1 recruitment and ternary receptor complex formation without decreasing surface IFNAR2 levels (74, 75). The USP18-IFNAR2 interaction makes it so that only higher affinity ligands such as IFNβ are able to recruit IFNAR1 into the receptor complex, making the cell less responsive to weaker affinity type I IFNs (76, 77).
Key Principles for Differential Activities
Altogether, differential expression, binding affinity to the receptor, and downstream feedback loops enable IFNAR1/2 to have graded responses to multiple ligands. Redundancy and pleiotropy are key features of type I IFN responses. Essentially, any type I IFN subtype can induce robust (or redundant) properties, such as antiviral activity, even at low surface receptor density. In contrast, tunable (or pleiotropic) functions, like antiproliferative activity, are more heavily influenced by affinity of the ligand, receptor density, and intracellular negative regulators, and so higher affinity ligands, like IFNβ, tend to be more potent (21). However, as noted above, some type I IFN subtypes may be able to signal through alternative pathways, in spite of or, more likely, because of possessing lower binding affinity. Understanding the molecular mechanisms underlying differential signaling by IFNs is an active area of research and how the differential activities of IFNα and IFNβ impact disease will be explored in the remaining sections.
Infectious Diseases
Type I IFNs have been extensively studied in the context of infectious diseases, and this body of work includes most of the studies that have directly compared the functions of IFNα and IFNβ in vivo. In the following subsections we highlight key findings from animal models and human studies that have contributed to understanding the mechanisms of differential properties of IFNα and IFNβ in viral, bacterial, and parasitic infections.
Viral Infections
The important role that viral infections have served in helping us understand type I IFN biology cannot be understated. Viral infections were key instruments in the discovery of the antiviral properties of type I IFNs (1). It is now widely appreciated that type I IFNs play a much larger role in coordinating protective immunity beyond directly eliciting an antiviral state, including their role in DC maturation, augmenting antibody production by B cells, and improving cytolytic T cell effector functions (5). Intriguingly, type I IFNs can also play a detrimental role in certain contexts, such as persistent viral infections. Given their key roles in disease outcome, viral systems also include some of the clearest examples of differential functions of IFNα and IFNβ in vivo (Table 1). The following viral models collectively highlight that differential functions of IFNαs and IFNβ can profoundly influence disease pathogenesis and that the mechanisms underlying differential functions vary depending on the biological context.

Table 1 Summary of IFNα and IFNβ functions in mouse models of viral infections.
Lymphocytic Choriomeningitis Virus
Lymphocytic choriomeningitis virus (LCMV) is a nonlytic, negative-strand RNA virus and a prototypic member of the Arenaviridae family, which are causative agents of hemorrhagic fevers in humans (100). The host genetics, viral strain, dose, and inoculation route all have profound impacts on host responses and disease outcome, and this remains true for the role of type I IFN responses during LCMV pathogenesis (101). LCMV infection serves as an excellent example of the pathogenic potential of type I IFNs.
LCMV-Clone-13 (Cl-13), which differs from its parent strain LCMV-Armstrong (Arm) by just three amino acids, causes a persistent infection, whereas LCMV-Arm is acutely and effectively cleared by immunocompetent mice (102). A clear pathogenic role for type I IFNs during persistent LCMV-Cl-13 infection has been established (78, 79, 103–105). Loss of IFNAR1 caused increased viral loads early during infection but ultimately restored splenic organization, decreased expression of the negative immune regulators IL-10 and programmed death-ligand 1 (PD-L1), increased protective adaptive immune responses, and accelerated clearance of persistent virus (78, 79, 105). While both LCMV-Arm and LCMV-Cl-13 infection led to high IFNα levels in the serum, only LCMV-Cl-13 induced significant serum IFNβ (79). In a seminal study, Ng and colleagues showed that the pathogenic activity of type I IFNs in persistent LCMV infection could be ascribed to just one subtype—IFNβ. Using monoclonal antibody (mAb) blockade and genetic deletion, they showed that IFNβ was dispensable for controlling early LCMV-Cl-13 viral loads, suggesting that IFNα or other subtypes mediate these antiviral responses (80). Instead, blockade of IFNβ but not IFNα improved splenic architecture, decreased infection of CD8α− DC, and enhanced antiviral T cell responses that led to clearance of persistent virus, mimicking many of the effects seen with IFNAR1 blockade. Altogether, persistent LCMV-Cl-13 infection serves as an important example that the type I IFN subtypes can have distinct properties in vivo that have profound impacts on viral pathogenesis.
As discussed above, LCMV-Cl-13 infection causes persistent infection in certain mouse strains (C57BL/6, BALB/C, C3H, or SWR/J); however, LCMV-Cl-13 infection of other strains (NZB, SJL/J, PL/J, NZO, or FVB/N mice) causes type I IFN- and CD8 T cell-dependent severe vascular leakage and death by about 6–8 days post infection (dpi) (81, 82, 106, 107). NZB.Ifnar1−/− but not NZB.Ifnb−/− mice were protected from LCMV-Cl-13 induced lethal vascular leakage, suggesting that IFNβ is dispensable for the detrimental effects of type I IFN in this model and that other subtypes like IFNα may drive this phenotype (81). However, this is challenged by the fact that blockade of IFNβ alone, pan-IFNα (α1, α4, α5, α11, and α13) alone, or combined pan-IFNα/β did not replicate the protection provided by anti-IFNAR1 treatment in FVB/N mice (82). The inability of IFNβ or IFNα blockade to phenocopy IFNAR1 blockade could be due to dosing issues, as the serum levels of IFNα were severely elevated (roughly 18-fold over IFNβ levels), involvement of IFNα subtypes not blocked by the mAb, or involvement other type I IFN subtypes altogether could be responsible for the lethal phenotype. Nevertheless, type I IFNs are clearly important host determinants of lethal LCMV infection, and the individual IFN subtype(s) responsible remains an open question.
Chikungunya and West Nile Viruses
Chikungunya virus (CHIKV) is a mosquito-transmitted, reemerging alphavirus that causes outbreaks of acute fever, rash, polyarthritis, arthralgia, and myositis (108). West Nile virus (WNV) is a mosquito-transmitted flavivirus that can cause encephalitis in severe cases (109). It is helpful to consider these models together because both models utilize a peripheral route of infection by inoculating the footpad subcutaneously (s.c.), and type I IFNs are essential for controlling both CHIKV and WNV, as Ifnar1−/− mice rapidly succumb to a severe, disseminated infection with either virus (83, 84, 88, 89). The collective evidence from these models suggest that IFNα and IFNβ play nonredundant protective roles.
Loss of IRF7, the master transcriptional regulator of IFNα subtypes, in acute WNV infection increased lethality and viral loads in both peripheral and central nervous system (CNS) tissues compared to WT animals (34, 85). Similarly, Irf7−/− mice infected with CHIKV developed worse clinical disease (foot swelling) and sustained high viral loads at the site of infection and sites of dissemination (90–92). The poor clinical outcome of Irf7−/− mice during WNV and CHIKV infection may be the result of decreased IFNα activity in the serum (85, 86, 91, 92). This postulation is supported by the observation that Irf7−/− mice produce little to no systemic IFNα activity when infected with a number of viruses, including Dengue virus (DENV), herpes simplex virus 1 (HSV-1), and encephalomyocarditis virus (EMCV), and this loss of systemic IFNα activity correlated with increased susceptibility to those infections (34, 110, 111). Pan-IFNα mAb blockade closely mimicked the clinical and virologic phenotype of Irf7−/− mice in CHIKV infection and phenocopied the lethality observed in WNV infection (86, 90). Altogether, these findings suggest that an important protective function of IRF7 is the production and amplification of IFNα responses and that IFNαs are important for controlling viral replication and dissemination.
In contrast with IFNα, the role of IFNβ in vivo is more varied and dependent on the biological context. Ifnb−/− mice are more susceptible than WT mice to WNV infection, and this increased lethality was accompanied with elevated viral burden in some but not all tissues (87). Specifically, WT and Ifnb−/− mice similarly controlled WNV replication in the spleen and serum, consistent with IFNα subtypes dominating serum IFN activity. WNV did replicate to a larger extent in the brain, spinal cord, and the draining lymph in Ifnb−/− mice compared to WT mice (87). An antiviral role for IFNβ has also been described for vaccinia virus and IAV infections (93, 94). In contrast to WNV infection, loss of IFNβ exacerbated CHIKV-induced disease but with minimal impact on viral burden at the inoculation site or distant tissues, suggesting that IFNβ may be important in restricting viral replication within certain but not all tissues (90). Rather, the increased disease severity of CHIKV-infected Ifnb−/− mice correlated with increased neutrophil accumulation at the site of infection, and depletion of neutrophils in Ifnb−/− mice reversed the disease exacerbation to WT levels. Altogether, these data from CHIKV and WNV infections point to the particular importance of IFNα subtypes in restricting viral replication and spread and highlight that the primary role of IFNβ varies depending on the specific context.
Human Immunodeficiency Virus 1 and Friend Retrovirus
Human immunodeficiency virus 1 (HIV-1) is a highly pathogenic retrovirus that leads to acquired immunodeficiency syndrome (AIDS). The relationship between type I IFNs and HIV-1 pathogenesis is complex, and it is outside the scope of the this review to cover all the protective and pathogenic functions, which have been extensively reviewed elsewhere (112–114). The purpose of reviewing HIV and Friend retrovirus (FV) infection is not to delve into whether type I IFNs have a net protective or pathogenic role, but rather, we seek to underscore that the IFNα subtypes are not equivalent in their antiviral or immunomodulatory properties in vivo.
Harper and colleagues evaluated the mRNA expression of specific IFNα subtypes in human pDCs following HIV-1 exposure (115). Intriguingly, they found an inverse relationship between the subtypes induced and their antiviral potency. HuIFNα1/13 and HuIFNα2 were highly expressed, but they demonstrated weaker antiviral activity in vitro, whereas HuIFNα6, α8, and α14 represented a smaller fraction of the IFNα subtypes induced but demonstrated the highest antiviral activity against HIV-1. Likewise, a study from Lavender and colleagues showed that therapeutic administration of HuIFNα14 was more beneficial than administration of HuIFNα2 in controlling HIV-1 replication in a humanized mouse model (116). The efficacy of IFNα14 was associated with increased ability to stimulate intrinsic immune responses including expression of tetherin and Mx2 as well as a greater frequency of TRAIL+ natural killer (NK) cells. Conversely, IFNα2 was superior in increasing the frequency of CD8+ T cells. An additional study used humanized mice that lack pDCs (Hu-PBL mice) and do not express much endogenous type I IFN during acute HIV-1 infection to study the impact of IFNα subtypes. They performed a single hydrodynamic injection of plasmid encoding different type I IFN subtypes (HuIFNα2, α6, α8, α14, or β) into Hu-PBL mice prior to HIV-1 infection (117). The authors found that all subtypes tested limited HIV-1 replication and prevented HIV-induced CD4+ T cell depletion by 10 dpi, but only HuIFNα14- and HuIFNβ-expressing mice demonstrated this protective effect out to 40 dpi. Altogether these findings demonstrate nonredundant functions of IFNα subtypes, with HuIFNα14 emerging as an intriguing subtype for further studies during HIV-1 infection.
Distinct properties of murine IFNα subtypes have also been observed in FV infection, a commonly used murine retrovirus model. A protective role for type I IFNs in controlling FV infection in vivo was demonstrated with Ifnar1−/− and Ifnb−/− mice both having increased viral loads in the spleen. However, only Ifnar1−/− mice showed a significant increase in viremia (95). These findings suggest that both IFNα and IFNβ protect against FV infection, but IFNα may be more important for controlling systemic infection and dissemination. Different potencies among IFNα subtypes have also been revealed. Ex vivo stimulation of FV-specific CD8+ T cells demonstrated differential activities among the IFNα subtypes. IFNα4, α6, and α9 had the strongest effects on CD8+ T cells, including inhibiting proliferation, stimulating cytokine production, and enhancing cytotoxicity (118). Treatment of FV-infected mice with MuIFNα1, α4, or α9, but not α6, significantly decreased viral loads, and subtype effectiveness was associated with different mechanisms (96). Only IFNα1 treatment correlated with activated FV-specific CD8+ T cells in the spleen, whereas NK cell activation was observed after treatment with all examined IFNα subtypes. Another study demonstrated that prophylactic administration of MuIFNα11, but not α2 or α5, significantly reduced viral loads by activating NK cells and ultimately provided long-term protection (6 weeks) (97). Together with the HIV-1 studies, retroviruses have proven to be effective tools for probing the diverse functions IFNα subtypes.
Hepatitis B and Hepatitis C Viruses
Hepatitis B (HBV) and hepatitis C viruses (HCV) are drastically distinct pathogens from a virological perspective—HBV is a double-stranded DNA virus belonging to the Hepadnaviridae family, whereas HCV is a positive-strand RNA virus and a member of Flaviviridae. However, both viruses display tropism for hepatocytes, and chronic infection with either virus can lead to liver failure, cirrhosis, and hepatocellular carcinoma (119). Beginning in the 1980s, derivatives of recombinant HuIFNα2 were used to treat chronic HBV and HCV, but treatment was successful in a limited subset of patients and severe side effects were common [reviewed in reference (120)]. These issues have led to the phasing out of type I IFN-based therapeutics in favor of direct-acting antiviral drugs (120). Though HuIFNα2-based therapeutics are the only approved type I IFN therapies for HCV or HBV treatment, pilot studies of IFNβ therapy in IFNα-nonresponding HBV or HCV patients suggest some beneficial effects of IFNβ as well (121–123). These findings suggest that other IFN subtypes in addition to IFNα2 may offer protective effects against hepatitis viruses.
Indeed, one study with the HBV hydrodynamic injection model demonstrated that prophylactic treatment with MuIFNα4 or α5 was more effective than other IFNα subtypes in decreasing HBV replication in vivo, and both α4 or α5 also increased effector NK and CD8+ T cell frequencies in the liver and spleen (98). Hydrodynamic injection of plasmids expressing MuIFNα4, α5, or combined α4 and α5 was more effective than treatment with the respective recombinant proteins, highlighting the importance of long-lasting endogenous IFNα expression in the liver during HBV infection. Another study directly showed differential effects of IFNα4 and IFNβ in the hydrodynamic injection HBV model (99). Co-injection of a plasmid encoding MuIFNα4 with HBV DNA decreased HBV serum markers, elevated liver ISG expression, and reduced HBV+ cells in the liver, whereas co-injection of an IFNβ-expressing plasmid demonstrated weaker inhibition of HBV and surprisingly led to a transient increase in HBV+ hepatocytes. This increase in HBV+ hepatocytes was not observed if the IFNβ plasmid was injected 14 dpi instead of co-injected with HBV (99). Even as the currently approved type I IFN therapies are being phased out of clinical use, these findings add to the accumulating evidence of distinct potencies and functions of IFNα and IFNβ subtypes in mouse models of relevant human pathogens.
IFNω Subtype Differences
IFNω is understudied compared to IFNα/β subtypes likely because mice lack a functional IFNω, but there is much in vitro evidence that it signals and functions similarly to IFNα/β (61, 124). Humans have only one IFNω subtype, but several species possess an expansion of IFNω genes (15–17, 125, 126). A number of these IFNω subtypes have been cloned from several species and have been demonstrated to be functional type I IFNs (127–129). Just as there is growing appreciation that expanded IFNα subtypes provide an evolutionary advantage beyond redundancy, it stands to reason that the expansion of IFNω genes likewise imparts a fitness advantage for those species. Indeed, a recent study compared two different IFNω subtypes from Rousettus aegyptiacus bats and found that IFNω9 displayed more effective antiviral activity against several RNA viruses in vitro compared to IFNω4 (130). Additionally, differences in expression and activity of porcine IFNω subtypes have also been demonstrated, with IFNω7 demonstrating the best antiviral activity in vitro (131). Several of these animals with expanded IFNω subtypes represent important reservoirs and transmitters of relevant human pathogens, so IFNω functional studies may provide valuable information on understanding the interactions between pathogens and their natural hosts.
Remarks on Viral Infections
When type I IFNs act on the proper cell type at the opportune time, they can induce an antiviral state, promote apoptosis of virally infected cells, coordinate recruitment of immune cells, enhance activation of antigen-presenting cells, and augment protective B and T cell responses. Not all IFNs are equal in their ability to induce these protective effects, and exploring this idea in vivo is an active area of research. Studies from infection with LCMV, WNV, and CHIKV have made it evident that endogenous IFNα subtypes are particularly important for limiting viremia and viral spread, likely due to their abundant activity in the serum in a number of viral infections. In peripheral tissues, IFNαs and IFNβ can exert important antiviral or immunomodulatory activity. Whether a particular subtype emerges as more important than others is likely going to depend on its biochemical properties, the cellular tropism of the virus, the source and magnitude of its induction, how long its expression is sustained, and the specific cell types responding to IFN.
If type I IFN signaling is sustained too long, immunosuppression and viral persistence can occur through the upregulation of negative immune regulators, like IL-10 and PD-L1. LCMV infection is a good example of this scenario, and strikingly, IFNβ was critical in promoting many detrimental features of type I IFN signaling in this model. We did not have space to discuss the growing evidence that type I IFNs can promote tissue damage during acute viral infections by promoting excessive inflammation and cell death [discussed in references (132, 133)]. This has been observed for mouse strains highly susceptible to influenza or coronavirus infection (134–136). The mechanisms responsible for these detrimental effects of type I IFN are an active area of research, but initial observations suggest that excessive or delayed IFN induction may play a role. It is also unknown whether specific IFN subtypes are responsible for these effects. Future studies exploring this possibility could have an important impact on human disease.
Bacterial Infections
Type I IFNs can play a pathogenic or protective role during bacterial infection depending on the pathogen. The mechanisms underlying the beneficial or detrimental roles during bacterial infection remain poorly understood and warrant further study. Below we explore some of the properties of type I IFNs during models of bacterial infections (Table 2). However, compared to the examples from viral infections, few of these studies directly compare the functions of IFNα and IFNβ. We draw attention to a few instances in which specific subtypes have been examined and highlight areas where this may be an interesting avenue to explore.
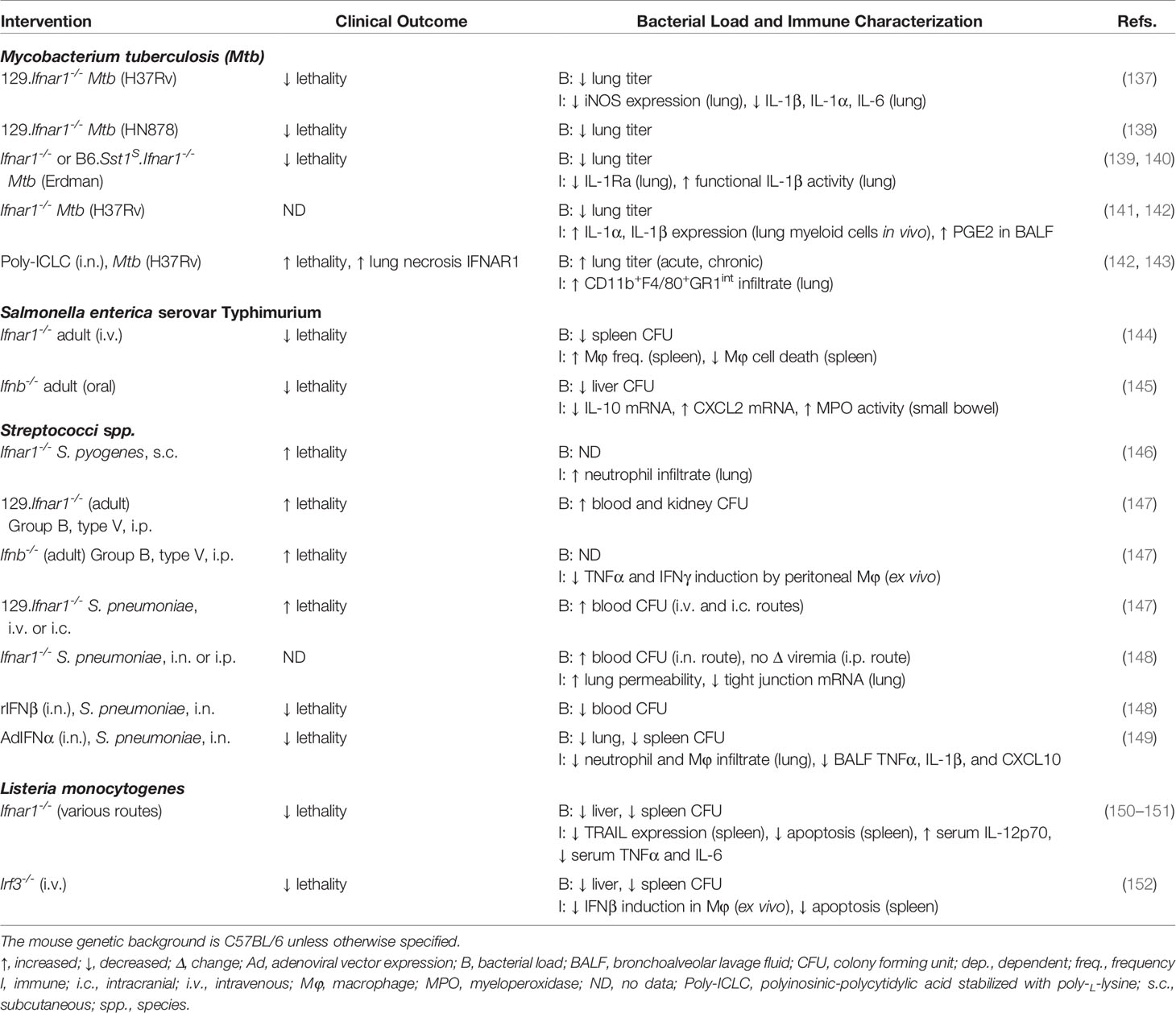
Table 2 Summary of IFNα and IFNβ functions in mouse models of bacterial infections.
Mycobacterium Tuberculosis
Mycobacterium tuberculosis (Mtb) causes the disease tuberculosis and represents a global health burden. This intracellular pathogen primarily infects the lungs, and it can enter latency if it is not eliminated, persisting in granulomas (154). The actions of type I IFNs during Mtb infections are complex, and there are numerous examples of contradictory findings. Overall, there is strong evidence that type I IFNs are detrimental to the host, but depending on the timing of IFN induction, the bacterial strain, and host genetics, IFNs may occasionally benefit the host during infection [reviewed in reference (155)].
Numerous studies have shown a type I IFN-inducible transcriptional profile in blood isolated from patients with active tuberculosis, but this signature is typically absent in patients with latent infection or patients who have undergone successful treatment (156–158). Concordantly, infection with hypervirulent Mtb laboratory strains showed increased recruitment of type I IFN-producing pDCs and classical DCs and elevated expression of IFNα or IFNβ in the lung, depending on the study (138, 139, 159–162). Multiple studies with human and mouse models have shown that type I IFNs are associated with impaired IFNγ-mediated antibacterial effects, decreased expression of IL-1α and IL-1β, decreased production of prostaglandin E2 (PGE2), and upregulation of IL-10 (138–142, 159, 162–165). Type I IFNs are also associated with increased cell death of macrophages and increased recruitment of myeloid cells permissive to Mtb infection (137, 143). Limited work has addressed the pathogenic potential of individual type I IFNs, but one recent study found that in vitro blockade of IFNα (subtypes unspecified), but not IFNβ blockade, significantly decreased intracellular Mtb bacterial load in a macrophage cell line (166). It remains to be determined if a similar effect could be observed in vivo.
Despite all of the evidence pointing to detrimental effects of type I IFNs in Mtb infection, type I IFNs may play a beneficial role in particular circumstances. First, several case reports have suggested that coadministration of IFNα with antimycobacterial therapy decreased bacterial burden in individuals who failed to respond to antimycobacterial therapy alone (167–170). However, these studies were employed before the pathogenic effects of type I IFNs were appreciated, and the mechanisms driving the apparent protection remain elusive. Second, in agreement with the findings that the detrimental effects of type I IFNs are largely due to inhibition of IFNγ, type I IFNs appear to be protective in contexts of IFNγ deficiency. Mice lacking both type I and type II IFN receptors displayed increased mortality and pathology compared to mice lacking only the type II IFN receptor in Mtb infection (171, 172). Mechanistically, type I IFNs may dampen recruitment of Mtb-permissible macrophages and suppress macrophages from entering an alternative activation state. In accord with these mice studies, administration of IFNα2b combined with antimycobacterial chemotherapy had beneficial effects in Mtb-infected children with underlying IFNγ signaling deficiencies (173, 174). It is unclear whether IFNβ can induce these effects as well. Further head-to-head comparison studies of IFNα and IFNβ are needed to determine if this protective effect of type I IFNs is unique to IFNα.
Type I IFNs may also benefit the host in infection with less virulent Mycobacterium strains, such as the bacille Calmette-Guérin (BCG) vaccine derived from M. bovis (175, 176). Administration of IFNα at the time of BCG vaccination (s.c.) in mice followed by intramuscular IFNα boosts (subtype not disclosed) promoted production of IFNγ, tumor necrosis factor (TNF), and IL-12, thus slightly increasing the protection seen upon re-challenge with Mtb intranasal (i.n.) compared to immunization with BCG alone (175). Moreover, the bacterial ESX-1 secretion system promotes type I IFN induction, and its recombinant expression in the BCG vaccine better protected against Mtb infection than other versions of the vaccine (176–179). In vitro data also highlight the complexity of type I IFN functions, as pretreatment of permissible cells with IFN before Mycobacterium infection can promote bacterial growth or increase immune activation, depending on the cell type and bacterial strain (180, 181). Thus, type I IFNs may play a protective role in vaccination with weaker Mycobacterium strains.
Salmonella enterica Serovar Typhimurium
Salmonella is a common, pathogenic genus of bacteria that causes acute gastroenteritis. Type I IFNs largely play a pathogenic role in Salmonella infection by promoting necroptosis and suppressing protective innate cell recruitment and proinflammatory responses. Deletion of IFNAR1 increased survival of adult mice infected (i.v.) with S. enterica serovar Typhimurium (S. Typhimurium) and decreased splenic bacterial loads (144). Additionally, splenic macrophages in Ifnar1−/− mice were resistant to S. Typhimurium-induced necroptosis ex vivo, and a follow-up mechanistic study further determined that type I IFN signaling impaired antioxidative stress responses to S. Typhimurium infection of bone marrow-derived macrophages (144, 182). IFNβ may be the dominant type I IFN subtype driving this necroptosis phenotype, as blockade of IFNβ, but not IFNα, prevented necroptosis and enhanced survival of bone marrow-derived macrophages during S. Typhimurium infection in vitro (144). It is unclear how many IFNα subtypes the antibody used blocks (clone: RMMA-1), so it is premature to rule out a contribution of IFNα. A role for IFNβ was further demonstrated in a separate study which showed that Ifnb−/− mice were more resistant to oral infection of S. Typhimurium, which was characterized by decreased bacterial burden, dampened expression of IL-10, and increased levels of CXCL2 and myeloperoxidase activity in the liver (145). Altogether, these findings suggest that IFNβ may play a detrimental role in S. Typhimurium infection by negatively regulating protective immune responses, but further studies are needed to rule out the involvement of other type I IFN subtypes.
Listeria monocytogenes
Listeria monocytogenes is an intracellular, pathogenic bacteria that causes sepsis and meningitis in immunocompromised and pregnant individuals (183). Many groups have shown that type I IFN signaling is detrimental to the host in systemic L. monocytogenes infection, but not in all routes of infection (150–153, 184, 185). Despite the important role that type I IFNs play in L. monocytogenes pathogenesis, the contribution of individual subtypes remains unknown. Irf3−/− mice displayed increased resistance to L. monocytogenes infection (60% survival), which almost phenocopied the resistance seen in Ifnar1−/− mice (80% survival) (152). Additionally, C57BL/6ByJ mice, which have a polymorphism in Irf3 causing inefficient splicing of its mRNA, demonstrated reduced IFNβ induction and increased resistance to Mtb infection (186). These observations may suggest an important role for IFNβ in susceptibility to L. monocytogenes infection. However, these studies did not assess IFNα induction, and characterization of Ifnb−/− mice is needed to confirm this hypothesis. Mechanistically, loss of type I IFN attenuated Listeria-induced cell death in myeloid cells and lymphocytes in vivo and ex vivo (150, 152, 187, 188). Antigen-stimulated T cells were more sensitive to lysteriolysin O (LLO)-induced apoptosis after exposure to IFNα compared to cells only treated with LLO (150). Thus, a role for IFNα subtypes should not be discounted. Altogether, it is impossible to draw firm conclusions about the roles of individual type I IFNs in L. monocytogenes infection with the currently available information. Studies that specifically block IFNα or IFNβ in Listeria infection might yield important insight into the functions of type I IFN subtypes.
Streptococci Species
Streptococci species often colonize mucosal surfaces and skin of healthy individuals without causing disease, but they can cause a variety of serious diseases in immunocompromised individuals or newborns (189). Type I IFNs appear to play a protective role during infection with a variety of Streptococci species (146–149).
S. pneumoniae, an alpha-hemolytic species commonly known as pneumococcus, is an opportunistic pathogen that colonizes the mucosal surfaces of the upper respiratory tract and is a leading bacterial cause of otitis media, pneumonia, sepsis, and meningitis (190). Type I IFNs play a beneficial role during pneumococcal infection, though the route of infection matters (147, 148). Loss of IFNAR1 increased lung permeability by decreasing tight junction protein expression, which is consistent with increased bacterial titer in the blood if S. pneumoniae was inoculated via an i.n. route but not via an intraperitoneal (i.p.) route (148). IFNβ played a role in mediating these protective effects because pre-treatment of mice with recombinant IFNβ i.n. significantly increased survival following S. pneumoniae challenge and decreased blood bacterial titer. However, IFNα subtypes likely provide beneficial effects as well since a separate study showed that prophylactic or therapeutic administration (i.n.) of an adenoviral vector expressing IFNα enhanced survival after pneumococcal infection and decreased lung and spleen bacterial burden (149). It is unclear which IFNα subtype was used in this study, so more work is needed to determine if some IFNα subtypes are more potent than others.
A protective role of type I IFNs was also demonstrated in infection with the beta-hemolytic species S. pyogenes (group A streptococcus, GAS) and S. agalactiae (group B streptococcus, GBS) (146, 147). In GBS i.v. challenge, IFNβ transcript was more robustly induced in the spleen compared to IFNα4, and Ifnb−/− mice demonstrated increased lethality compared to WT mice (147). Additionally, in vitro GBS infection poorly activated peritoneal macrophages from Ifnar1−/− or Ifnb−/− mice compared to WT controls, suggesting that IFNβ may function to augment macrophage antibacterial properties. However, carefully controlled experiments need to be performed in order to determine if IFNβ is directly modulating macrophage activation or if IFNβ acts indirectly by influencing bacterial loads. The role of specific subtypes was not evaluated in GAS infection; however, macrophages and DCs were found to induce IFNβ downstream of unique pathways. Macrophages required IRF3, STING, TBK1, MyD88, and stimulation with streptococcal DNA, whereas DCs depended on MyD88, IRF5, and streptococcal RNA (146). It might be interesting to evaluate Irf3−/−, Irf5−/−, and Ifnb−/− mice in S. pyogenes infection to determine if the cellular source of IFN affects pathogenesis. Additionally, better characterization of the IFNα subtypes induced and their role in GAS and GBS is needed.
Remarks on Bacterial Infections
Similar to viral infections, type I IFNs can be either detrimental or beneficial to the host during bacterial infections, depending on the specific pathogen. The mechanisms underlying these divergent outcomes share many features with viral infections. The ability of type I IFNs to regulate cell death, suppress protective IFNγ responses, and/or upregulate IL-10 can account for the detrimental functions of type I IFNs during Mtb, Salmonella, and L. monocytogenes infection. These activities are reminiscent of the type I IFN-driven increases in IL-10 and PD-L1 observed in LCMV, as well as the increased cell death observed in acute influenza infection (132, 135). Even though a detrimental role for type I IFNs is well documented in Mtb infection, in special contexts type I IFNs may be able to serve a protective function. Of particular interest is the possibility of type I IFN serving as an adjuvant with certain, less virulent Mycobacterium vaccination strains. As is the case with some viral infections, the timing, magnitude, and cellular source of type I IFNs underlie these distinct outcomes. In the future it will be interesting to explore if these divergent phenomena are also due to differential induction or functions of type I IFN subtypes.
There are also examples of type I IFNs having a protective role in bacterial infections, such as with several Streptococcus species. This net beneficial effect may reflect many of the functions commonly observed in viral infections, such as coordinating protective immune cell recruitment and activation and promoting the right level of inflammation needed to clear the bacterial infection. The exact mechanisms underlying these protective effects are understood at a very general level and questions remain. Which cells do IFNs signal on to mediate these protective effects? What ISGs are responsible for mediating protection, and are they different from those acting in viral infections? Importantly, do specific type I IFN subtypes drive particular protective functions? We are only beginning to grasp how type I IFNs contribute to protective antibacterial immune responses, and there are many interesting avenues to explore relevant to human health.
Parasitic Infections
Parasites include single-cellular protozoa (e.g. Plasmodium and Leishmania species) and multicellular helminths, which include flatworms (e.g. Schistosoma species) and roundworms (e.g. Ascaris species) (191–194). Previously, parasite-host interaction studies have not investigated the functions of type I IFNs, but recent studies in malaria have identified both protective and pathogenic properties of IFNα/β [reviewed in references (195, 196)]. Below we explore the roles of IFNα and IFNβ during Plasmodium infection, the causative agent of malaria (Table 3).

Table 3 Summary of IFNα and IFNβ functions in mouse models of malaria infection.
Plasmodium Overview
Malaria initially presents as a wide variety of symptoms, including periodic fever, chills, headache, malaise, and muscle and joint aches, but as disease progresses severe anemia, blood acidosis, splenomegaly, acute respiratory distress syndrome, and spread to the brain are possible, which can be fatal (210). Infected mosquitoes transmit Plasmodium sporozoites to humans during a blood meal. The sporozoites initially infect hepatocytes, where they replicate as merozoites (liver stage), and eventually, merozoites enter the blood stream to infect red blood cells, where they begin asexual reproduction (blood stage) (191). Symptoms in humans usually begin developing several days after release of parasites into the blood. P. falciparum and P. vivax are the most common species responsible for malaria disease in humans, and several Plasmodium species (P. berghei, P. yoelii, P. chabaudi, and P. vinckei) infect rodents and recapitulate various stages of human disease (210).
Liver-Stage Malaria
Two important studies recently revealed a protective role for type I IFNs in controlling liver-stage Plasmodium infection. First, Liehl and colleagues showed that all of the early upregulated genes in the liver from mice infected with P. berghei (ANKA) were classified as IFN-stimulated genes or linked to the type I IFN signaling pathway (197). Similarly, Miller et al. also uncovered an early type I IFN signature in the liver of mice infected with P. yoelii (Py17XNL) (198). Upon global IFNAR1 deficiency or conditional deletion of IFNAR1 on hepatocytes (Albumin-Cre), mice failed to control parasite replication in the liver (197, 198). These studies suggest that type I IFN signaling protects against malaria infection by controlling early parasite replication in the liver. Further characterization revealed that Irf3−/− mice, but not Irf7−/− mice, showed a similar early increase in liver parasite burden as Ifnar1−/− mice following P. yoelii (Py17XNL) infection (198). This is consistent with the observation that Irf3−/− mice demonstrated a more severe decrease in early liver ISG induction compared to Irf7−/− mice following P. berghei (ANKA) infection (197). Given that IRF3 is a key regulator of IFNβ induction, these findings could suggest that endogenous IFNβ is more important than IFNα subtypes for controlling parasite burden in liver stage malaria. Additional studies are needed to confirm this hypothesis.
Blood-Stage Malaria
There is conflicting evidence for whether type I IFNs have a net beneficial or detrimental effect during the blood stage of malaria. Evidence for a protective role is as follows. First, treatment of mice with recombinant hybrid HuIFNα1/α8, which has activity on murine cells, concurrent with P. yoelii (265 BY) infection decreased early parasitemia, and the authors proposed that this was due to IFNα-dependent inhibition of reticulocyte (immature red blood cell) development, as opposed to direct anti-plasmodium effects (202, 211). Moreover, deletion of inflammasome components or some intracellular PRR sensing components decreased parasitemia and increased resistance to lethal P. yoelii infection through alleviation of SOCS1-mediated suppression of type I IFN responses (201, 212).
Other studies have demonstrated that type I IFNs might play a detrimental role during blood-stage malaria. First, a group showed that Ifnar1−/− and Irf7−/− mice better controlled parasitemia in non-lethal P. chabaudi infection compared to WT controls (199). Additionally, Sebina and colleagues showed that IFNAR1 deletion in P. yoelii (Py17XNL) infection increased pathogen-specific antibody titers and decreased parasitemia late in infection (17–21 dpi) (200). Mechanistically, type I IFN signaled on DCs to limit their activation of T follicular helper cells in an inducible T cell co-stimulator (ICOS) signaling-dependent manner, and this interaction ultimately influenced downstream germinal center B cell responses (200). However, it should be noted that IFNAR1 deletion in the Sebina et al. study also trended toward increased parasitemia early in infection (6–11 dpi), suggesting that these findings are not completely incongruous with the studies that found a protective role for type I IFNs. Altogether, type I IFNs might be detrimental in the blood stage malaria by impeding humoral immunity later in infection, but the Plasmodium strain and timing of IFN action may influence the overall effect of type I IFNs on disease outcome. It would be interesting to determine if this effect is dependent on certain type I IFN subtypes.
Cerebral Malaria
Similar to the blood stage, the role of type I IFNs during cerebral malaria remains controversial. Several independent groups have demonstrated that Ifnar1−/− mice are either completely or partially protected from lethal experimental cerebral malaria (P. berghei ANKA sporozoite infection), demonstrating a net pathogenic effect for type I IFNs in this context (199, 203–207). Loss of type I IFN signaling may increase IFNγ-producing CD4+ T cells, reduce pathogenic CD8+ T cell recruitment and/or activation in the brain, improve DC priming of CD4+ T cell responses, or some combination thereof (199, 204–207). Irf7−/− mice only partially recapitulated the decreased brain pathology and protection from P. berghei (ANKA) lethality observed in Ifnar1−/− mice, but loss of IRF7 perfectly phenocopied the decreased parasitemia observed in Ifnar1−/− mice (199). These findings may suggest IFNαs are more important in promoting parasitemia, whereas IFNβ and IFNα might both contribute to brain pathology, but specific antibody blockade of type I IFN subtypes would confirm this hypothesis.
Paradoxically, a few groups have shown that systemically administering either recombinant IFNβ or hybrid IFNα1/α8 concurrently with infection alleviated cerebral malaria (P. berghei ANKA) (208, 209). Both IFN treatments reduced parasite burden in the brain and decreased infiltrating CD8+ T cells in the brain compared to control mice, but only IFNα1/α8 treatment decreased blood parasitemia (208, 209). A more recent study identified receptor transporter protein 4 (RTP4) as a positive regulator of type I IFN responses, and Rtp4−/− mice were completely protected from P. berghei (ANKA) lethality and brain pathology (213). This protection in Rtp4−/− mice correlated with increased type I IFN responses in microglia isolated from the brain, suggesting a protective role for IFNs, but blockade of type I IFN signaling in Rtp4−/− mice is needed to confirm a causal link (213). Overall, an issue of magnitude and timing of IFN response might underlie these apparent discrepancies with the protective phenotypes of Ifnar1−/− mice (discussed below). Indeed, antibody blockade of IFNAR1 as late as 5 dpi was almost as protective as Ifnar1−/− mice, suggesting that the detrimental effects of type I IFNs occurred during priming of adaptive immune responses (199).
Remarks on Parasitic Infections
It is clear that the role of type I IFNs in malaria is complex and depends on the stage of Plasmodium life cycle. Type I IFNs seem to play a protective role during the liver stage, but there are contradictory findings from various models of blood-stage and cerebral malaria. Perhaps infection with some strains of Plasmodium yields suboptimal type I IFN production very early in infection, ultimately leading to delayed and higher levels later in infection when parasite burden is not effectively controlled. Proper intervention at either step would benefit the host, and this could explain why loss of IFN signaling or exogenous IFN treatment can both be protective. The contribution of individual IFN subtypes remains unclear, though divergent phenotypes in Irf3−/− and Irf7−/− mice suggest this could be an interesting question to explore. Importantly, genetic variants in IFNAR1 have been associated with either greater or lower risk of severe malaria disease (205, 214–217). The impact of each genetic variant on IFNAR1 expression and function still need to be determined, but these findings suggest that type I IFNs are important regulators of malaria disease in humans.
Overall, parasitic pathogens are biologically very diverse, so data from other parasitic infection models are needed to begin drawing broad conclusions. A recent study demonstrated that the TLR4-IRF1-IFNβ axis played a protective role in mice infected with Leishmania infantum by dampening proinflammatory pathways and IFNγ production by CD4+ T cells (218). RNA sequencing analysis of human samples revealed that upregulation of TLR4 and type I IFN pathways was associated with asymptomatic individuals compared to patients with visceral leishmaniasis (218). Another group found that Ifnar1−/− mice were more susceptible to Toxoplasma gondii infection (219). It would be interesting to know if IFNs are generally more important in single-cellular parasitic infections. That said, the multicellular helminth Schistosoma mansoni can induce a systemic type I IFN signature in mice and activate TLR3 in DCs in vitro, suggesting that a role for type I IFNs in parasitic worm infections is certainly possible (220, 221). Continued work to delineate the cellular sources and functions of type I IFNs in malaria and other parasitic diseases may reveal novel opportunities for therapeutic intervention and help uncover novel functions of type I IFNs.
Cancer
The majority of reports from animal models and the clinic demonstrate that type I IFNs play an important protective role in enhancing anti-tumor immune responses and restricting tumor growth [reviewed in (222, 223)]. However, similar to persistent viral infections, the functions of type I IFNs in cancer can change throughout disease course, and there is evidence that, in certain contexts, IFN might act as a barrier to efficacious checkpoint-blockade therapy [reviewed in (224)]. Below we discuss the actions of endogenous IFNα/β and IFN-based therapies in animal models and clinical studies (Figure 2).
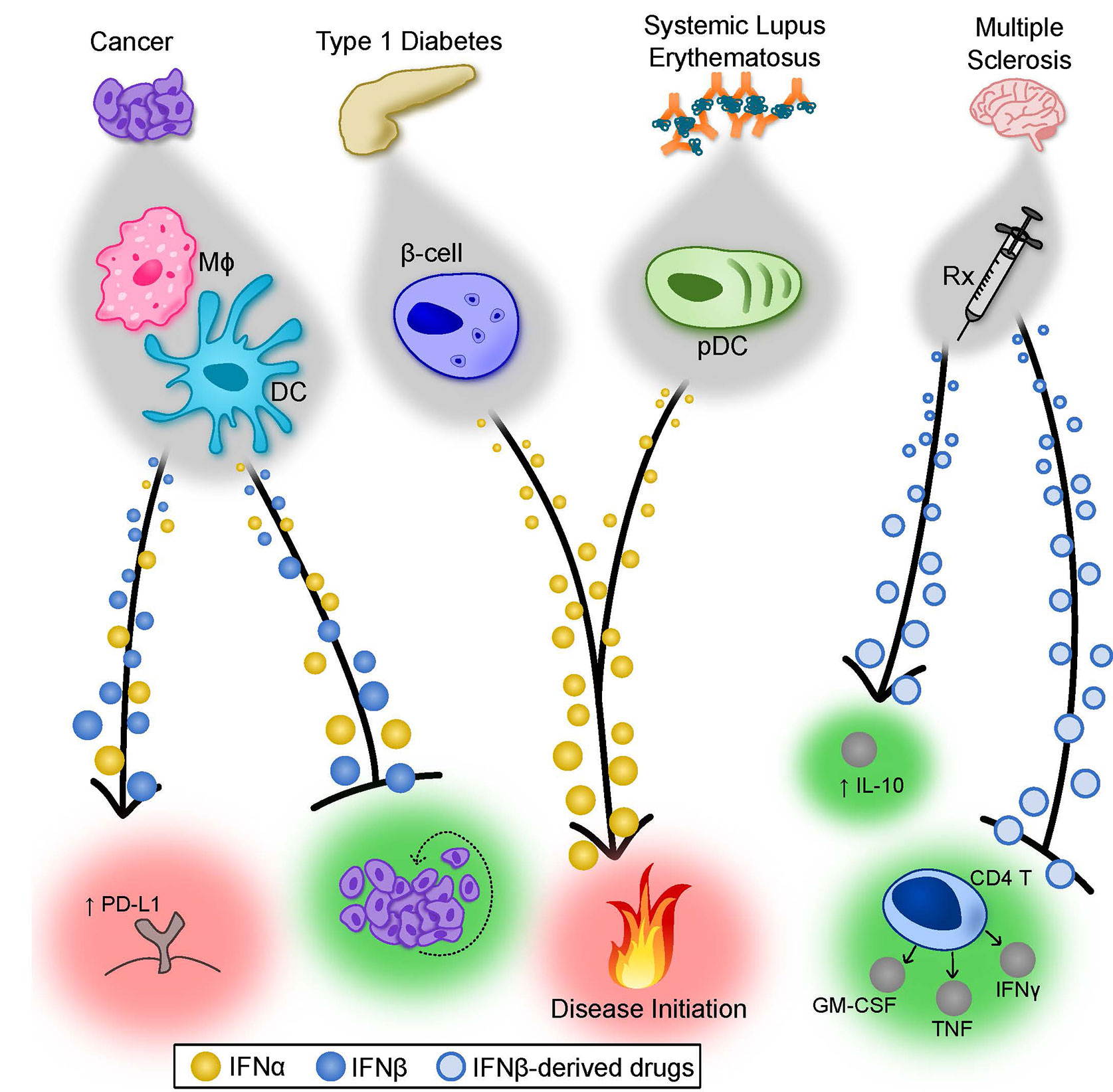
Figure 2 Summary of the Properties of IFNα and IFNβ in cancer and autoimmunity. Type I IFNs display both unique and overlapping properties in various disease states. In cancer, depending on the tumor and degree of metastases, both IFNα and IFNβ can contribute to tumor rejection by directly limiting tumor cell proliferation (depicted) but also through modulation of antitumor immune responses (not depicted). In certain cases, type I IFNs can induce PD-L1 expression on tumor cells, suppressing immune-mediated killing of the tumor. The factors that cause type I IFNs to exert detrimental effects remain poorly understood. In T1D, there is evidence that IFNα subtypes play an important role in pathogenesis. Forced expression of IFNα by pancreatic β-cells accelerated the onset and severity of T1D in a mouse model, and patients receiving IFNα therapy for treatment of other diseases have a higher incidence of T1D. Similarly, immune complex-driven activation of pDCs induces robust IFNα production, which may participate in initiation of SLE. Finally, IFNβ-derived therapeutics have well-established efficacy for treating MS patients. Though still largely debated, the mechanism of protection mediated by IFNβ is complex and possibly includes limiting cytokine production from pathogenic CD4+ T cells and augmenting IL-10 production in a number of cell types. β-cell, pancreatic β-cell; DC, dendritic cell; IL, interleukin; Mφ, macrophage; MS, multiple sclerosis; pDC, plasmacytoid dendritic cell; Rx, prescription drug; SLE, systemic lupus erythematosus; T1D, type I diabetes.
Animal Studies: Endogenous Type I IFN Activity
An early study showed that mice transplanted with human tumors and treated with neutralizing antibodies to type I IFNs demonstrated exacerbated tumor growth and metastasis compared to controls, suggesting a protective role for endogenous type I IFN activity (225). Since this finding, we now know that endogenous type I IFN can mediate tumor rejection through signaling on immune cells or tumor cells.
A seminal paper showed that type I IFN signaling on host hematopoietic cells was crucial for development of anti-tumor immune responses (226). Using conditional IFNAR1 deletion, bone marrow chimeras, and adoptive transfer experiments, a number of studies have shown that type I IFN signaling on several types of immune cells is important for immunity in cancer. For instance, type I IFN signaling on DCs, but not granulocytes or macrophages, was required for rejection of highly immunogenic tumors (227). Additionally, Itgax-Cre+ Ifnar1fl/fl (CD11c-Cre) mice showed diminished cross presentation by DCs to CD8+ T cells, which likely contributed to their failed tumor rejection (227, 228). In an NK cell sensitive tumor model, endogenous type I IFN was required for NK cell-mediated tumor rejection and homeostasis (229).
Other studies have shown that type I IFN signaling on tumor stromal cells may be important for controlling tumor burden. In vivo, both IFNα and IFNβ have antiangiogenic activity via signaling on vascular endothelial cells to downregulate growth factors such as vascular endothelial growth factor (230, 231). Stromal cells such as mesenchymal stem cells may play a role in controlling tumor growth by producing IFNα in order to enhance NK and CD8+ T cell responses (232). However, extended low level IFN signaling on tumor cells may render them resistant to apoptosis and immune-mediated killing (233, 234). These differences highlight the complexities of type I IFN actions and the need to delineate cell-type specific IFN signaling and consequent gene regulation.
Limited studies have directly compared the endogenous functions of individual IFNα/β subtypes in cancer models, but there have been a few studies conducted with IFNβ-deficient mice. Ifnb−/− mice showed expedited tumor growth, enhanced angiogenesis, and increased neutrophil infiltration to the tumor compared to WT mice (235–238). These findings demonstrate that endogenous IFNβ is important for the host anti-tumor response, but the specific signaling pathways downstream of IFNβ and cell types mediating these effects remain unclear. The direct contributions of endogenous IFNα remain uninvestigated, so much work is needed to fully characterize the contribution of endogenous IFN in tumor rejection.
Animal Studies: Type I IFN-Based Therapies
The possibility that IFNs might be therapeutically useful in cancer was first shown in the early 1970s, when crude preparations of were administered to mice with syngeneic tumors increased their survival compared to untreated mice (239, 240). IFN therapies have been quite effective against hematological cancers, including hairy cell leukemia and chronic myelogenous leukemia but vary in efficacy against solid tumors, such as melanoma [reviewed in (222, 223, 241, 242)]. Below we discuss various therapeutic strategies derived from either IFNα or IFNβ subtypes. Collectively, these studies show that IFNα and IFNβ are able to promote a similar range of immunomodulatory and antitumor effects, so studies that directly compare the activities of IFNαs and IFNβ are needed to discern if there are bona fide differential properties.
IFNα-Based Therapies
Derivatives of IFNα2b have long been used in the clinic, but toxicity issues are associated with systemic administration and persistent use. Consequently, many groups have sought ways to increase IFNα expression with more precision. An influential study developed RNA-lipoplexes encoding neoantigens or endogenous self-antigens, which yielded rapid and robust IFNα production by macrophages and DCs (IFNβ induction was not determined) (243). Importantly, these RNA-lipoplex vaccines were able to mediate rejection of several different types of aggressive tumors in mice (243). Another group developed a method to genetically modify human hematopoietic stem cells (HSCs) to express HuIFNα2b, but only in differentiated monocytes (244). The engineered HSCs were able to repopulate immunodeficient mice and effectively inhibit tumor progression in a murine breast cancer model (244). AcTakines (Activity-on-Target), which are optimized cytokines that only act on cells for which they are targeted, represent another interesting alternative to traditional IFN therapies. Indeed, CD20-targeted IFNα2b-derived AcTaferon reduced lymphoma and melanoma tumors engineered to express CD20 (245, 246). Increasing tumor cell production of IFNα is another approach, and a very recent study demonstrated that IFNα subtypes are not all equal in their antitumor properties. B16 melanoma cells were engineered to overexpress IFNα2, α4, α5, α6, or α9, but only IFNα2- and α9-expressing tumors were effectively controlled in an adaptive-immunity dependent manner (247). Other studies have used a variety of genetic engineering methods to augment IFNα production in the tumor microenvironment and improve antitumor immunity (248–251).
IFNβ-Based Therapies
Derivatives of IFNα2 have been the focus of most IFN-based therapies, but several studies have explored the effect of IFNβ during various models of cancer. IFNβ treatment of transformed human mammary epithelial cells in vitro led to a less aggressive state (252). Another group showed that treating mice with an anti-tumor antibody fused to IFNβ increased clearance of antibody-resistant tumor cells by increasing cross presentation by tumor-infiltrating DCs and activation of CD8+ T cells (253). Unfortunately, this treatment also upregulated the inhibitory molecule PD-L1 on tumor cells, but this negative effect was overcome with co-administration of anti-PD-L1 antibody (253). Another group transduced induced pluripotent stem cell (iPSC)-derived myeloid cells with an IFNβ-encoding lentivirus to treat disseminated gastric cancer (254). When injected into immunocompromised mice, the modified myeloid cells accumulated in the tumors and inhibited growth of the peritoneally disseminated cancer (254). Lastly, intratumoral injection of an mRNA encoding a fusion protein consisting of IFNβ and the ectodomain of transforming growth factor-β receptor II enhanced DC activation of CD8+ T cells in vitro and promoted rejection of the TC-1 tumor cell line in vivo (255).
Human Studies
The antitumor and immunomodulatory effects of IFNα therapy have been demonstrated in the treatment of a variety of cancers, and here we present a few representatives. IFNα-derived therapies are the only approved adjuvant therapies in melanoma patients after surgical resection, and immunomodulatory actions, such as increased tumor-infiltrating cells and decreased circulating T-regulatory cells, are key mechanisms of action [reviewed in reference (242)]. After being replaced with tyrosine kinase inhibitors like imatinib, interest in IFNα-based therapy has recently reemerged for treatment of chronic myeloid leukemia (CML) [reviewed in reference (241)]. This is because there is evidence that IFNα therapy is able to target and sensitize the rare CML stem cell population to subsequent killing by chemotherapy, whereas imatinib is more effective against more differentiated CML progenitors (256, 257). Lastly, an analysis of matched primary breast cancer tumors and bone metastases revealed that primary tumor cells expressed IRF7, whereas metastases consistently demonstrated downregulation of IRF7 expression (258). This may suggest that IRF7-mediated IFNα production in primary tumors is an important factor for limiting metastases, but further studies are needed to determine if this is an IFNα-specific effect or if there is also a role for IFNβ. Fewer clinical studies have been conducted with IFNβ-derived therapies, but there is evidence that IFNβ also plays a protective role in tumor rejection. Increased IFNβ mRNA expression significantly correlated with improved survival in patients with triple-negative breast cancer, though the mechanism is undetermined (252). In vitro studies have shown that IFNβ is more potent in inducing apoptosis in melanoma cells compared to IFNα (259). The relevance of this differential potency has yet to be extensively explored in vivo.
Detrimental Effects of Type I IFNs in Cancer
Despite all the evidence that type I IFNs can facilitate protective antitumor immune responses, IFNs can also impede cancer therapies. We provide just a few mechanistic examples. Persistent type II IFN signaling on tumors can result in PD-L1-dependent and PD-L1-independent resistance to immune checkpoint blockade, and the authors identified a role for type I IFNs in maintaining PD-L1-independent resistance (233). Radiation and chemotherapy stimulate immune-mediated destruction of tumor cells partly through induction of type I IFNs (260–264). However, recent work showed that conditional deletion of IFNAR1 on tumor cells enhanced responsiveness to radiation therapy through increased susceptibility to CD8+ T cell-mediated killing (265). Lastly, oncolytic viruses can preferentially kill cancer cells, but tumor responsiveness to type I IFN activity confers resistance to this therapeutic method. One study showed that IFNα and IFNβ differ in their ability to confer resistance to oncolytic virus treatment in vitro. Exogenous IFNβ more effectively prevented oncolysis of human head and neck squamous cell carcinoma cells by vesicular stomatitis virus compared to IFNα, but differential effects were not observed for normal keratinocytes or endothelial cells (266).
Remarks on Cancer Studies
Collectively, this large body of cancer studies has shown that the roles of type I IFNs are complex and likely context specific. The extensive use of IFNα-derived therapies to treat a number of cancers in the clinic has greatly increased our understanding of the range of IFNα properties in vivo. Cancer models are uniquely advantageous for studying protective immunomodulatory effects of IFNs compared to infection models because pathogen load is not a confounding factor. Despite the large body of work suggesting the benefits of type I IFN signaling in cancer, the actions of specific IFN subtypes, for the most part, remain undefined. The beneficial effects of indirect activators of type I IFNs, such as the RNA-lipoplexes (discussed above) or STING agonists, may be due to their ability to induce multiple IFN subtypes with either overlapping or unique functions (222, 244). The heterogeneity of cancer makes it all the more important to appropriately stratify patients to ensure a beneficial effect of treatment.
Autoimmunity
Type I IFNs have emerged as critical mediators of autoimmunity. Patients with a variety of autoimmune diseases display serum type I IFN signatures, and IFN treatments for other diseases have correlated with the development of autoimmunity. These observations have led to the assumption that type I IFNs may contribute to autoimmunity pathogenesis. However, IFNβ-derived therapeutics have been used to treat multiple sclerosis, highlighting that caution is warranted in attempting to summarize the mechanisms of autoimmune disorders. Below we outline the current understanding of the roles of IFNα and IFNβ during systemic lupus erythematosus, type 1 diabetes, and multiple sclerosis (Figure 2). This is not an exhaustive analysis of autoimmune disorders, and active research is exploring the function of type I IFNs in other disorders, such as rheumatoid arthritis and Sjögren’s syndrome (267, 268).
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease that affects organs such as the skin, joints, kidneys, and CNS (269). A type I IFN gene signature in the blood of SLE patients is well established (270–272). Additionally, a number of genetic risk factors for SLE are associated with type I IFN production or signaling, including IRF5, IRF7, IRAK1, and TYK2 [reviewed in reference (273)]. The majority of patients (70–80%) develop anti-nuclear autoantibodies (ANA), which form immune complexes with extracellular nucleic acids and induce production of type I IFN, especially IFNα, by pDCs (274). Type I IFNs promote disease by signaling on a variety of immune cells, including DCs, B cells, and T cells (275–277). It has been shown that IFNα or IFNβ treatment in vitro induced different transcriptional programs in DCs, with IFNα-primed DCs demonstrating increased phagocytic uptake of apoptotic cells and nucleic acids (278). Given the prevalence of IFNα in the serum of SLE patients and role of pathogenic responses to nucleic acids, the impact of IFNα versus IFNβ on DC activation in the context of SLE might be an interesting topic to interrogate.
A recent study from Klarquist et al. sought to dissect the effect of type I IFN signaling on CD4+ T cells and B cells on the development of T follicular helper cells, germinal center B cells, and plasmablasts. They found that IFN signaling decreased the threshold for B cell receptor signaling, increased MHC-II expression, and promoted germinal center B cell function, thus lowering the threshold for autoreactive B cell activation (276). They also found that type I IFN protected T follicular helper cells from NK cell-mediated death, thus further promoting B cell responses (276). Other studies suggest that IFNα may further drive SLE by increasing production of multiple TNF family members, such as BAFF and APRIL, which promote B cell survival and can drive SLE pathogenesis (279–281). Due to the apparent pathogenic role of IFNα during SLE, attempts have been made to neutralize type I IFNs in SLE patients (282–287). Both anti-IFNα and, more recently, anti-IFNAR1 therapies have been tested (282–287). Both treatment strategies showed disparate efficacy in patients, so further work is needed to clarify if this type of therapeutic intervention would be beneficial for patients. It might be that IFNα only plays a key role in the initiation and early stages of disease, so the disease stage may be important in stratifying patients [reviewed in reference (288)].
Type 1 Diabetes
Type 1 diabetes (T1D) is a chronic, autoimmune disease caused by the immune-mediated destruction of pancreatic β-cells that leads to insulin deficiency and hyperglycemia (289). A blood type I IFN signature in T1D patients precedes the development of autoantibodies and disease (290–293). One study detected a significant increase in expression of IFNα subtypes, but not IFNβ, in postmortem pancreas specimens from T1D patients compared to control subjects (290). Moreover, many genetic polymorphisms associated with T1D are involved in the type I IFN response such as MDA5 and TYK2 (294–296). Altogether, these findings suggest a detrimental role for type I IFNs in T1D. A role for type I IFNs in the development of T1D is supported in animal models. An early study showed that forced constitutive IFNα expression by pancreatic β-cells in mice resulted in hypoinsulinemic diabetes and pancreatic inflammation (297). Additionally, non-obese diabetic (NOD) mice, a common model for T1D, showed elevated IFN-inducible transcripts in the pancreatic islets prior to disease onset, and treatment of young NOD mice with anti-IFNAR1 mAb delayed the onset and decreased the occurrence of T1D (298, 299). Collectively, these findings suggest that type I IFN signaling, especially in the pancreas, may play a key role in initiating T1D.
LCMV can be employed as a viral model of T1D, in which mice transgenically express LCMV glycoprotein (GP) under the control of the rat insulin promoter (Rip-LCMV) (300). Development of Rip-LCMV T1D is dependent on type I IFN (301, 302). Recent work showed that anti-IFNAR1 mAb treatment reduced blood glucose to normal levels and prevented destruction of pancreatic islets (302). Importantly, they also showed that pan-IFNα (α1, α4, α5, α11, and α13) mAb blockade, but not IFNβ blockade, was able to recapitulate the anti-IFNAR1 phenotype, demonstrating a distinct role for IFNα subtypes in promoting pathogenesis in the Rip-LCMV T1D model. A similar detrimental role for IFNα is suggested in human disease. IFNα therapy for HCV in individuals genetically predisposed to T1D induced or exacerbated the development of T1D (303). Moreover, a recent study showed that a subset of AIRE-deficient patients who developed autoantibodies specific for IFNα, especially IFN-α1/13, IFN-α5, and IFN-α14, were less likely to develop T1D, whereas patients who failed to generate these antibodies developed T1D (304). Altogether, animal and human studies suggest a detrimental role of type I IFNs in T1D, and IFNα subtypes appear to play a dominant role in disease development and pathogenesis.
Multiple Sclerosis
Multiple sclerosis (MS) is a chronic, autoimmune disease of the CNS in which immune cells target and destroy the myelin sheath surrounding neurons, leading to neurodegeneration (305). Similar to other autoimmune conditions, MS patients can show a serum type I IFN signature, but this signature is relatively low when compared to SLE patients (306, 307). However, in strong contrast to SLE and T1D, type I IFNs, do not appear to play a detrimental role. In fact, IFNβ was the first FDA-approved therapy for MS (308–311). However, due to its flu-like side effects and the availability of more effective treatments, it is no longer the preferred therapy for MS patients (312). Even though IFNβ treatment is currently less preferred in clinical use, animal models and clinical studies (discussed below) have revealed important insight into the properties of IFNβ in vivo.
In Vitro and Animal Studies
Experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, has provided mechanistic insight into the protective actions of IFNβ (313). Mice lacking IFNβ, IFNAR1, or IRF7 showed exacerbated clinical EAE compared to WT mice, perhaps due to greater T cell infiltration and increased proinflammatory cytokine production in the CNS (314–316). Unexpectedly, mice that lack IRF3 showed significantly lessened clinical disease compared to WT mice, and this seemed to be due to a cell-intrinsic defect in the development of T helper type 17 (TH17) cells (317). Indeed, TH17 versus TH1 skewing can drastically influence the impact of IFNβ treatment in EAE. IFNβ treatment was effective in reducing EAE severity in TH1-induced EAE but worsened disease in TH17-induced EAE (318). Thus, depending on the skewing of the T helper responses and method of induction of EAE, IFNβ may be protective or pathogenic.
Many cell types respond to IFNβ therapy in EAE. Deletion of Ifnar1 on myeloid cells including macrophages, monocytes, granulocytes, and microglia, but not neuroectodermal cells, resulted in increased severity of EAE symptoms, suggesting that IFNβ mediates its protective effects, in part, by acting on these cells (315). Mice treated with TLR3 or TLR7 agonists display reduced disease severity associated with increased type I IFN production by pDCs and other antigen presenting cells (319, 320). Other reports have also suggested that IFNβ signaling on T cells curbs their pathogenicity (321, 322). Furthermore, type I IFN signaling on conventional DCs limited their migration to the CNS and prevented their activation of TH17 cells during EAE (323, 324). The tissue resident antigen presenting cells in the CNS, microglia, may also play a role in the type I IFN response during EAE. Type I IFN signaling on microglia promoted clearance of myelin debris by increasing their phagocytic activity (325, 326). Finally, a study identified a role for type I IFN signaling on astrocytes to suppress CNS inflammation during EAE (327).
Clearly IFNβ is able to induce protective effects during EAE, and a recent report demonstrated that sustained low-dose IFNα1 delivery via an adeno-associated viral system prevented the onset of disease in EAE (328). This therapeutic effect was associated with regulatory T cell expansion, and myelin-specific effector T cells displayed reduced proliferative capacity, decreased proinflammatory cytokine production, and increased expression of IL-10 and PD-1 (programmed cell death protein 1) (328). Another study showed that a systemic high dose of MuIFNα11 was able to initially delay EAE in mice but ultimately caused significant toxicity and mortality; however, when IFNα activity was targeted to DCs (Clec9A-targeted AcTaferon), they found efficient protection from EAE (329). These findings suggest that IFNβ might not be unique in its ability to confer protection in EAE, but more work is needed to determine what factors cause IFNα treatments to yield detrimental effects or protective effects.
Human Studies
IFNβ was the first FDA-approved therapy for MS (308–311). However, due to its flu-like side effects and the availability of more effective treatments, it is no longer the preferred therapy for MS patients (312). Observations from patients suggest that IFNβ therapy likely acts through multiple mechanisms, such as influencing immune cell recruitment and activation. First, IFNβ treatment correlated with decreased new brain lesions and increased soluble VCAM-1 in patient serum, suggesting that modulating immune cell entry to the CNS is one potential mechanism of IFNβ therapy (330). In addition to impacting cell recruitment, IFNβ treatment may also regulate survival of immune cells since an increase in proapoptotic genes was observed in peripheral immune cells isolated from IFNβ-treated patients (331, 332).
Pathogenic TH1 and/or TH17 cells likely play an important role in MS, and IFNβ therapy may limit the proliferation of pathogenic T cells and modulate their cytokine production (332, 333). IFNβ therapy is likely more effective in individuals with a TH1 driven disease, since high serum IL-17F levels correlated with a poor response to IFNβ therapy (318). A number of cell types are likely involved in protective IFNβ treatment. For example, IFNβ treatment of MS patients can induce IL-10 production by myeloid cells, but treatment can also suppress production of granulocyte-macrophage colony-stimulating factor (GM-CSF), IFNγ, and TNF by effector T cells (334–340). Additionally, in patients that responded to IFNβ therapy, treatment induced T regulatory cells that produced IL-10 and expressed PD-L1 (341, 342). Altogether, the protective mechanisms that underlie IFNβ therapeutic effects likely involve direct or indirect actions on effecter T cells. A better understanding of these mechanisms would likely reveal important information about the functional capacity of IFNβ in vivo.
Remarks on Autoimmune Studies
A large proportion of patients with SLE or T1D show a type I IFN signature in their blood, and many studies have shown that type I IFNs promote pathogenesis in these autoimmune disorders. There is strong evidence implicating the IFNα subtypes in initiation and progression of SLE and T1D, but at this time, a role for IFNβ cannot be entirely ruled out—direct functional comparisons of IFNα versus IFNβ would be needed to draw that conclusion. Altogether, the specific pathogenic functions of type I IFNs during autoimmune disorders are likely tissue specific. A recent study performed gene-expression profiling of structural cells from 12 different tissues and found that the responses of the cells to stimuli were tissue-specific, thus identifying the stroma as an important regulator of tissue-specific immune responses (343). While there is clear evidence that type I IFNs can modulate pathogenic autoimmune responses, it is important to know how systemic IFNα activity might promote cell-type specific effects in diseased versus nondiseased tissues in disorders like T1D that target a particular tissue, but also in diseases like SLE that have multi-organ effects.
In contrast, blood from MS patients do not display as robust a type I IFN signature as SLE or T1D patients, and many studies have demonstrated that IFNβ treatment has therapeutic properties in animal models of MS and in affected individuals. The protective functions of IFNβ are complex and likely include modulating immune cell recruitment and activation directly through action on immune cells and indirectly through action on brain resident cells. The functions of IFNαs in MS are less clear. There might be conditions, such as very low doses or when targeted to a specific cell type, in which IFNα subtypes are also protective. Careful comparison of IFNβ versus IFNα dose responses in EAE might uncover novel mechanisms for differential functions among type I IFNs in vivo.
Concluding Remarks
Whether type I IFNs have a net beneficial or detrimental effect on disease outcome depends on a variety of factors including the timing and magnitude of induction relative to disease onset, the duration of expression, the specific subtypes induced, the cell types responding, and likely other factors. Progress is needed in understanding the spatiotemporal induction of the various type I IFN subtypes in vivo, as well as the cell types responsible for type I IFN production. A lack of tools to differentiate between different subtypes has hindered progress in this area. Quantitative reverse transcription polymerase chain reaction has been a useful technique for quantifying specific IFN subtypes, and single-molecule array (Simoa) digital ELISA technology was demonstrated to detect IFN in blood with high sensitivity (344). However, there is a need for licensed antibodies against individual subtypes that are able to neutralize in animal models and reliably stain tissue sections to more accurately determine the timing of expression at the tissue level.
Transcriptomic approaches have successfully differentiated type I and type III ISG signatures in organoid cultures (345). Because the effects of type I IFN are pleiotropic, there is a need to delineate the ISGs responsible for the protective and pathogenic functions of type I IFN subtypes in a given context and to understand how cell-type specificity might affect expression of those genes. A recent report profiled gene-expression networks of fibroblasts, endothelial, and epithelial cells isolated from multiple tissues and revealed tissue-specific signaling networks (343). A similar approach or spatial transcriptomics, which yields gene expression profiles in intact tissue sections, would be powerful tools to unravel the cell type-specific responses to different type I IFN subtypes in vivo (346).
Lastly, given that many type I IFN subtypes have expanded independently after mammalian speciation, there is a great need for tools to allow the study of human type I IFN subtypes in animal models. Immune-humanized mice and hybrid IFNAR (HyBNAR) mice, which transgenically encode variants of IFNAR1/2 that contain the human extracellular domains fused to the transmembrane and cytoplasmic segments of murine IFNAR, have both been used to study HuIFN in mice (347). These two systems are helpful in contexts where immune cells are the predominant sources of and responders to type I IFN or in studies administering exogenous HuIFN, but they do not permit loss-of-function studies, exclude the impact of endogenous IFN expression by stromal cells, and IFNAR1/2 transgenes are likely more highly expressed than endogenous IFNAR1/2. Overall, a concerted effort to address this lack of tools will go a long way toward increasing our ability to directly compare the expression and functions of distinct type I IFN subtypes, which will undoubtedly generate new strategies to augment or dampen the type I IFN pathway for biomedical purposes.
Author Contributions
LEF and MCL conceptualized and drafted the manuscript, as well as created the figures and tables. LEF, MCL, and DJL all reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
LEF was supported by a National Institutes of Health (NIH) postdoctoral research training grant (T32 CA009547) (https://www.nih.gov/). MCL was supported by a predoctoral research training grant (T32 AI007163) and by an F31 fellowship (AI149999-01A1) (Ruth L. Kirschstein Predoctoral Individual National Research Service Award) from the NIH (https://www.nih.gov/). DJL was supported by the NIH (R01 AI127513 and R21 AI135490) (https://www.nih.gov/).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
Acknowledgments
We thank Anne Robinson and Amanda Dicks for designing and editing Figure 2 in association with InPrint at Washington University in St. Louis.
References
1. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc London Ser B - Biol Sci (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
2. Lazear HM, Schoggins JW, Diamond MS. Shared and Distinct Functions of Type I and Type III Interferons. Immunity (2019) 50:907–23. doi: 10.1016/j.immuni.2019.03.025
3. Schoenborn JR, Wilson CB. Regulation of Interferon-γ During Innate and Adaptive Immune Responses. Adv Immunol (2007) 96:41–101. doi: 10.1016/S0065-2776(07)96002-2
4. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5:375–86. doi: 10.1038/nri1604
5. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15:87–103. doi: 10.1038/nri3787
6. Woelk CH, Frost SDW, Richman DD, Higley PE, Kosakovsky Pond SL. Evolution of the interferon alpha gene family in eutherian mammals. Gene (2007) 397:38–50. doi: 10.1016/j.gene.2007.03.018
7. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev (2004) 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x
8. van Pesch V, Lanaya H, Renauld J-C, Michiels T. Characterization of the Murine Alpha Interferon Gene Family. J Virol (2004) 78:8219–28. doi: 10.1128/JVI.78.15.8219-8228.2004
9. Hardy MP, Owczarek CM, Jermiin LS, Ejdebäck M, Hertzog PJ. Characterization of the type I interferon locus and identification of novel genes. Genomics (2004) 84:331–45. doi: 10.1016/j.ygeno.2004.03.003
10. Chen J, Baig E, Fish EN. Diversity and relatedness among the type I interferons. J Interferon Cytokine Res (2004) 24:687–98. doi: 10.1089/jir.2004.24.687
11. Xu L, Yang L, Liu W. Distinct evolution process among type I interferon in mammals. Protein Cell (2013) 4:383–92. doi: 10.1007/s13238-013-3021-1
12. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol (2003) 4:69–77. doi: 10.1038/ni875
13. Roberts RM, Ezashi T, Rosenfeld CS, Ealy AD, Kubisch HM. Evolution of the interferon tau genes and their promoters, and maternal-trophoblast interactions in control of their expression. Reprod Suppl (2003) 61:239–51. doi: 10.1530/biosciprocs.5.018
14. Li S, Gong M, Zhao F, Shao J, Xie Y, Zhang Y, et al. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell Physiol Biochem (2018) 51:2377–96. doi: 10.1159/000495897
15. Walker AM, Roberts RM. Characterization of the bovine type I IFN locus: Rearrangements, expansions, and novel subfamilies. BMC Genomics (2009) 10:187. doi: 10.1186/1471-2164-10-187
16. Sang Y, Bergkamp J, Blecha F. Molecular evolution of the porcine type I interferon family: Subtype-specific expression and antiviral activity. PloS One (2014) 9:e112378. doi: 10.1371/journal.pone.0112378
17. Shields LE, Jennings J, Liu Q, Lee J, Ma W, Blecha F, et al. Cross-Species Genome-Wide Analysis Reveals Molecular and Functional Diversity of the Unconventional Interferon-ω Subtype. Front Immunol (2019) 10:1431. doi: 10.3389/fimmu.2019.01431
18. Manry J, Laval G, Patin E, Fornarino S, Itan Y, Fumagalli M, et al. Evolutionary genetic dissection of human interferons. J Exp Med (2011) 208:2747–59. doi: 10.1084/jem.20111680
19. Coelho LFL, Magno de Freitas Almeida G, Mennechet FJD, Blangy A, Uzé G. Interferon-alpha and -beta differentially regulate osteoclastogenesis: role of differential induction of chemokine CXCL11 expression. Proc Natl Acad Sci USA (2005) 102:11917–22. doi: 10.1073/pnas.0502188102
20. Schreiber G. The molecular basis for differential type I interferon signaling. J Biol Chem (2017) 292:7285–94. doi: 10.1074/jbc.R116.774562
21. Hertzog PJ, Williams BRG. Fine tuning type I interferon responses. Cytokine Growth Factor Rev (2013) 24:217–25. doi: 10.1016/j.cytogfr.2013.04.002
22. Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol (2006) 6:644–58. doi: 10.1038/nri1900
23. Ryzhakov G, Eames HL, Udalova IA. Activation and function of interferon regulatory factor 5. J Interferon Cytokine Res (2015) 35:71–8. doi: 10.1089/jir.2014.0023
24. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol (2015) 15:471–85. doi: 10.1038/nri3865
25. Au WC, Moore PA, Lowther W, Juang YT, Pitha PM. Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc Natl Acad Sci USA (1995) 92:11657–61. doi: 10.1073/pnas.92.25.11657
26. Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol (1998) 18:2986–96. doi: 10.1128/MCB.18.5.2986
27. Sato M, Tanaka N, Hata N, Oda E, Taniguchi T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-β gene. FEBS Lett (1998) 425:112–6. doi: 10.1016/S0014-5793(98)00210-5
28. Schafer SL, Lin R, Moore PA, Hiscott J, Pitha PM. Regulation of Type I Interferon Gene Expression by Interferon Regulatory Factor-3. J Biol Chem (1998) 273:2714–20. doi: 10.1074/jbc.273.5.2714
29. Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus Infection Induces the Assembly of Coordinately Activated Transcription Factors on the IFN-β Enhancer In Vivo. Mol Cell (1998) 1:507–18. doi: 10.1016/S1097-2765(00)80051-9
30. Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J (1998) 17:1087–95. doi: 10.1093/emboj/17.4.1087
31. Juang YT, Lowther W, Kellum M, Au WC, Lin R, Hiscott J, et al. Primary activation of interferon A and interferon B gene transcription by interferon regulatory factor 3. Proc Natl Acad Sci U S A (1998) 95:9837–42. doi: 10.1073/pnas.95.17.9837
32. Marié I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J (1998) 17:6660–9. doi: 10.1093/emboj/17.22.6660
33. Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett (1998) 441:106–10. doi: 10.1016/S0014-5793(98)01514-2
34. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434:772–7. doi: 10.1038/nature03464
35. MacDonald NJ, Kuhl D, Maguire D, Näf D, Gallant P, Goswamy A, et al. Different pathways mediate virus inducibility of the human IFN-alpha 1 and IFN-beta genes. Cell (1990) 60:767–79. doi: 10.1016/0092-8674(90)90091-R
36. Génin P, Lin R, Hiscott J, Civas A. Differential Regulation of Human Interferon A Gene Expression by Interferon Regulatory Factors 3 and 7. Mol Cell Biol (2009) 29:3435–50. doi: 10.1128/MCB.01805-08
37. Gessani S, Belardelli F, Pecorelli A, Puddu P, Baglioni C. Bacterial lipopolysaccharide and gamma interferon induce transcription of beta interferon mRNA and interferon secretion in murine macrophages. J Virol (1989) 63:2785–9. doi: 10.1128/JVI.63.6.2785-2789.1989
38. Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-beta enhanceosome. Cell (2007) 129:1111–23. doi: 10.1016/j.cell.2007.05.019
39. Hata N, Sato M, Takaoka A, Asagiri M, Tanaka N, Taniguchi T. Constitutive IFN-alpha/beta signal for efficient IFN-alpha/beta gene induction by virus. Biochem Biophys Res Commun (2001) 285:518–25. doi: 10.1006/bbrc.2001.5159
40. Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity (2012) 37:158–70. doi: 10.1016/j.immuni.2012.04.011
41. Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity (2012) 37:171–86. doi: 10.1016/j.immuni.2012.05.020
42. Kawashima T, Kosaka A, Yan H, Guo Z, Uchiyama R, Fukui R, et al. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-β. Immunity (2013) 38:1187–97. doi: 10.1016/j.immuni.2013.02.024
43. Winkler ES, Shrihari S, Hykes BL, Handley SA, Andhey PS, Huang Y-JS, et al. The Intestinal Microbiome Restricts Alphavirus Infection and Dissemination through a Bile Acid-Type I IFN Signaling Axis. Cell (2020) 182:901–18.e18. doi: 10.1016/j.cell.2020.06.029
44. Bradley KC, Finsterbusch K, Schnepf D, Crotta S, Llorian M, Davidson S, et al. Microbiota-Driven Tonic Interferon Signals in Lung Stromal Cells Protect from Influenza Virus Infection. Cell Rep (2019) 28:245–56.e4. doi: 10.1016/j.celrep.2019.05.105
45. Morin P, Bragança J, Bandu MT, Lin R, Hiscott J, Doly J, et al. Preferential binding sites for interferon regulatory factors 3 and 7 involved in interferon-A gene transcription. J Mol Biol (2002) 316:1009–22. doi: 10.1006/jmbi.2001.5401
46. Civas A, Génin P, Morin P, Lin R, Hiscott J. Promoter organization of the interferon-A genes differentially affects virus-induced expression and responsiveness to TBK1 and IKKϵ. J Biol Chem (2006) 281:4856–66. doi: 10.1074/jbc.M506812200
47. Gough DJ, Messina NL, Clarke CJP, Johnstone RW, Levy DE. Constitutive Type I Interferon Modulates Homeostatic Balance through Tonic Signaling. Immunity (2012) 36:166–74. doi: 10.1016/j.immuni.2012.01.011
48. LaFleur DW, Nardelli B, Tsareva T, Mather D, Feng P, Semenuk M, et al. Interferon-κ, a Novel Type I Interferon Expressed in Human Keratinocytes. J Biol Chem (2001) 276:39765–71. doi: 10.1074/jbc.M102502200
49. Nardelli B, Zaritskaya L, Semenuk M, Cho YH, LaFleur DW, Shah D, et al. Regulatory effect of IFN-kappa, a novel type I IFN, on cytokine production by cells of the innate immune system. J Immunol (2002) 169:4822–30. doi: 10.4049/jimmunol.169.9.4822
50. He Y, Fu W, Cao K, He Q, Ding X, Chen J, et al. IFN-κ suppresses the replication of influenza A virusesthrough the IFNAR-MAPK-Fos-CHD6 axis. Sci Signal (2020) 13:eaaz3381. doi: 10.1126/scisignal.aaz3381
51. Fung KY, Mangan NE, Cumming H, Horvat JC, Mayall JR, Stifter SA, et al. Interferon-ϵ protects the female reproductive tract from viral and bacterial infection. Science (2013) 339:1088–92. doi: 10.1126/science.1233321
52. Hermant P, Francius C, Clotman F, Michiels T. IFN-ϵ Is Constitutively Expressed by Cells of the Reproductive Tract and Is Inefficiently Secreted by Fibroblasts and Cell Lines. PloS One (2013) 8:1–9. doi: 10.1371/journal.pone.0071320
53. Demers A, Kang G, Ma F, Lu W, Yuan Z, Li Y, et al. The mucosal expression pattern of interferon-ε in rhesus macaques. J Leukoc Biol (2014) 96:1101–7. doi: 10.1189/jlb.3A0214-088RRR
54. Schreiber G, Piehler J. The molecular basis for functional plasticity in type I interferon signaling. Trends Immunol (2015) 36:139–49. doi: 10.1016/j.it.2015.01.002
55. Ng CT, Mendoza JL, Garcia KC, Oldstone MBA. Alpha and Beta Type 1 Interferon Signaling: Passage for Diverse Biologic Outcomes. Cell (2016) 164:349–52. doi: 10.1016/j.cell.2015.12.027
56. Piehler J, Roisman LC, Schreiber G. New Structural and Functional Aspects of the Type I Interferon-Receptor Interaction Revealed by Comprehensive Mutational Analysis of the Binding Interface. J Biol Chem (2000) 275:40425–33. doi: 10.1074/jbc.M006854200
57. Roisman LC, Jaitin DA, Baker DP, Schreiber G. Mutational Analysis of the IFNAR1 Binding Site on IFNα2 Reveals the Architecture of a Weak Ligand–Receptor Binding-site. J Mol Biol (2005) 353:271–81. doi: 10.1016/j.jmb.2005.08.042
58. Lavoie TB, Kalie E, Crisafulli-Cabatu S, Abramovich R, DiGioia G, Moolchan K, et al. Binding and activity of all human alpha interferon subtypes. Cytokine (2011) 56:282–9. doi: 10.1016/j.cyto.2011.07.019
59. de Weerd NA, Matthews AY, Pattie PR, Bourke NM, Lim SS, Vivian JP, et al. A hot spot on interferon α/β receptor subunit 1 (IFNAR1) underpins its interaction with interferon-β and dictates signaling. J Biol Chem (2017) 292:7554–65. doi: 10.1074/jbc.M116.773788
60. de Weerd NA, Vivian JP, Nguyen TK, Mangan NE, Gould JA, Braniff S-J, et al. Structural basis of a unique interferon-β signaling axis mediated via the receptor IFNAR1. Nat Immunol (2013) 14:901–7. doi: 10.1038/ni.2667
61. Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell (2011) 146:621–32. doi: 10.1016/j.cell.2011.06.048
62. Jaitin DA, Roisman LC, Jaks E, Gavutis M, Piehler J, Van der Heyden J, et al. Inquiring into the differential action of interferons (IFNs): an IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol Cell Biol (2006) 26:1888–97. doi: 10.1128/MCB.26.5.1888-1897.2006
63. Harris BD, Schreiter J, Chevrier M, Jordan JL, Walter MR. Human interferon-ϵ and interferon-κ exhibit low potency and low affinity for cell-surface IFNAR and the poxvirus antagonist B18R. J Biol Chem (2018) 293:16057–68. doi: 10.1074/jbc.RA118.003617
64. Moraga I, Spangler J, Mendoza JL, Garcia KC. Multifarious Determinants of Cytokine Receptor Signaling Specificity. Adv Immunol (2014) 121:1–39. doi: 10.1016/B978-0-12-800100-4.00001-5
65. Moraga I, Harari D, Schreiber G, Uze G, Pellegrini S. Receptor Density Is Key to the Alpha2/Beta Interferon Differential Activities. Mol Cell Biol (2009) 29:4778–87. doi: 10.1128/MCB.01808-08
66. Levin D, Harari D, Schreiber G. Stochastic Receptor Expression Determines Cell Fate upon Interferon Treatment. Mol Cell Biol (2011) 31:3252–66. doi: 10.1128/MCB.05251-11
67. Zheng H, Qian J, Varghese B, Baker DP, Fuchs S. Ligand-Stimulated Downregulation of the Alpha Interferon Receptor: Role of Protein Kinase D2. Mol Cell Biol (2011) 31:710–20. doi: 10.1128/MCB.01154-10
68. Kumar KGS, Tang W, Ravindranath AK, Clark WA, Croze E, Fuchs SY. SCF(HOS) ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-alpha receptor. EMBO J (2003) 22:5480–90. doi: 10.1093/emboj/cdg524
69. Marchetti M, Monier MN, Fradagrada A, Mitchell K, Baychelier F, Eid P, et al. Stat-mediated signaling induced by type I and type II interferons (IFNs) is differentially controlled through lipid microdomain association and clathrin-dependent endocytosis of IFN receptors. Mol Biol Cell (2006) 17:2896–909. doi: 10.1091/mbc.e06-01-0076
70. Marijanovic Z, Ragimbeau J, van der Heyden J, Uzé G, Pellegrini S. Comparable potency of IFNα2 and IFNβ on immediate JAK/STAT activation but differential down-regulation of IFNAR2. Biochem J (2007) 407:141–51. doi: 10.1042/BJ20070605
71. Payelle-Brogard B, Pellegrini S. Biochemical monitoring of the early endocytic traffic of the type I interferon receptor. J Interf Cytokine Res (2010) 30:89–98. doi: 10.1089/jir.2009.0044
72. Chmiest D, Sharma N, Zanin N, Viaris de Lesegno C, Shafaq-Zadah M, Sibut V, et al. Spatiotemporal control of interferon-induced JAK/STAT signalling and gene transcription by the retromer complex. Nat Commun (2016) 7:13476. doi: 10.1038/ncomms13476
73. Piganis RAR, De Weerd NA, Gould JA, Schindler CW, Mansell A, Nicholson SE, et al. Suppressor of Cytokine Signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferon α receptor (IFNAR1)-associated tyrosine kinase tyk2. J Biol Chem (2011) 286:33811–8. doi: 10.1074/jbc.M111.270207
74. Morales DJ, Lenschow DJ. The antiviral activities of ISG15. J MolBiol (2013)425:4995–5008. doi: 10.1016/j.jmb.2013.09.041
75. Malakhova OA, Kim K IL, Luo JK, Zou W, Kumar KGS, Fuchs SY, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J (2006) 25:2358–67. doi: 10.1038/sj.emboj.7601149
76. Wilmes S, Beutel O, Li Z, Francois-Newton V, Richter CP, Janning D, et al. Receptor dimerization dynamics as a regulatory valve for plasticity of type I interferon signaling. J Cell Biol (2015) 209:579–93. doi: 10.1083/jcb.201412049
77. François-Newton V, Magno de Freitas Almeida G, Payelle-Brogard B, Monneron D, Pichard-Garcia L, Piehler J, et al. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PloS One (2011) 6:e22200. doi: 10.1371/journal.pone.0022200
78. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science (2013) 340:202–7. doi: 10.1126/science.1235208
79. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KCF, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science (2013) 340:207–11. doi: 10.1126/science.1235214
80. Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, Rice S, et al. Blockade of Interferon Beta, but Not Interferon Alpha, SignalingControls Persistent Viral Infection. Cell Host Microbe(2015) 17:653–61. doi: 10.1016/j.chom.2015.04.005
81. Baccala R, Welch MJ, Gonzalez-Quintial R, Walsh KB, Teijaro JR, Nguyen A, et al. Type I interferon is a therapeutic target for virus-induced lethal vascular damage. Proc Natl Acad Sci USA (2014) 111:8925–30. doi: 10.1073/pnas.1408148111
82. Oldstone MBA, Ware BC, Horton LE, Welch MJ, Aiolfi R, Zarpellon A, et al. Lymphocytic choriomeningitis virus Clone 13 infection causes either persistence or acute death dependent on IFN-1, cytotoxic T lymphocytes (CTLs), and host genetics. Proc Natl Acad Sci (2018) 115:E7814–23. doi: 10.1073/pnas.1804674115
83. Samuel MA, Diamond MS. Alpha/Beta Interferon Protects against Lethal West Nile Virus Infection by Restricting Cellular Tropism and Enhancing Neuronal Survival. J Virol (2005) 79:13350–61. doi: 10.1128/JVI.79.21.13350-13361.2005
84. Pinto AK, Ramos HJ, Wu X, Aggarwal S, Shrestha B, Gorman M, et al. Deficient IFN Signaling by Myeloid Cells Leads to MAVS-Dependent Virus-Induced Sepsis. PloS Pathog (2014) 10:e1004086. doi: 10.1371/journal.ppat.1004086
85. Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M, Diamond MS. Interferon Regulatory Factor IRF-7 Induces the Antiviral Alpha Interferon Response and Protects against Lethal West Nile Virus Infection. J Virol (2008) 82:8465–75. doi: 10.1128/JVI.00918-08
86. Sheehan KCF, Lazear HM, Diamond MS, Schreiber RD. Selective Blockade of Interferon-α and -β Reveals Their Non-Redundant Functions in a Mouse Model of West Nile Virus Infection. PloS One (2015) 10:e0128636. doi: 10.1371/journal.pone.0128636
87. Lazear HM, Pinto AK, Vogt MR, Gale M, Diamond MS. Beta Interferon Controls West Nile Virus Infection and Pathogenesis in Mice. J Virol (2011) 85:7186–94. doi: 10.1128/JVI.00396-11
88. Couderc T, Chrétien F, Schilte C, Disson O, Brigitte M, Guivel-Benhassine F, et al. A mouse model for Chikungunya: Young age and inefficient type-I interferon signaling are risk factors for severe disease. PloS Pathog (2008) 4:e29. doi: 10.1371/journal.ppat.0040029
89. Schilte C, Couderc T, Chretien F, Sourisseau M, Gangneux N, Guivel-Benhassine F, et al. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J Exp Med (2010) 207:429–42. doi: 10.1084/jem.20090851
90. Cook LE, Locke MC, Young AR, Monte K, Hedberg ML, Shimak RM, et al. Distinct Roles of Interferon Alpha and Beta in Controlling Chikungunya Virus Replication and Modulating Neutrophil-Mediated Inflammation. J Virol (2019) 94:e00841–19. doi: 10.1128/JVI.00841-19
91. Rudd PA, Wilson J, Gardner J, Larcher T, Babarit C, Le TT, et al. Interferon Response Factors 3 and 7 Protect against Chikungunya Virus Hemorrhagic Fever and Shock. J Virol (2012) 86:9888–98. doi: 10.1128/JVI.00956-12
92. Schilte C, Buckwalter MR, Laird ME, Diamond MS, Schwartz O, Albert ML. Cutting Edge: Independent Roles for IRF-3 and IRF-7 in Hematopoietic and Nonhematopoietic Cells during Host Response to Chikungunya Infection. J Immunol (2012) 188:2967–71. doi: 10.4049/jimmunol.1103185
93. Koerner I, Kochs G, Kalinke U, Weiss S, Staeheli P. Protective Role of Beta Interferon in Host Defense against Influenza A Virus. J Virol (2007) 81:2025–30. doi: 10.1128/JVI.01718-06
94. Deonarain R, Alcamí A, Alexiou M, Dallman MJ, Gewert DR, Porter AC. Impaired antiviral response and alpha/beta interferon induction in mice lacking beta interferon. J Virol (2000) 74:3404–9. doi: 10.1128/JVI.74.7.3404-3409.2000
95. Gerlach N, Schimmer S, Weiss S, Kalinke U, Dittmer U. Effects of Type I Interferons on Friend Retrovirus Infection. J Virol (2006) 80:3438–44. doi: 10.1128/JVI.80.7.3438-3444.2006
96. Gerlach N, Gibbert K, Alter C, Nair S, Zelinskyy G, James CM, et al. Anti-retroviral effects of type I IFN subtypes in vivo. Eur J Immunol (2009) 39:136–46. doi: 10.1002/eji.200838311
97. Gibbert K, Joedicke JJ, Meryk A, Trilling M, Francois S, Duppach J, et al. Interferon-alpha subtype 11 activates NK cells and enables control of retroviral infection. PloS Pathog (2012) 8:e1002868. doi: 10.1371/journal.ppat.1002868
98. Song J, Li S, Zhou Y, Liu J, Francois S, Lu M, et al. Different antiviral effects of IFNα subtypes in a mouse model of HBV infection. Sci Rep (2017) 7:334. doi: 10.1038/s41598-017-00469-1
99. Zhou Y, Li S, Tang Z, Xu C, Huang S, Wu J, et al. Different antiviral effects of IFNα and IFNβ in an HBV mouse model. Immunobiology (2017) 222:562–70. doi: 10.1016/j.imbio.2016.11.003
100. Oldstone MBA. Biology and pathogenesis of lymphocytic choriomeningitis virus infection. Curr Top Microbiol Immunol (2002) 263:83–117. doi: 10.1007/978-3-642-56055-2_6
101. Suprunenko T, Hofer MJ. Complexities of Type I Interferon Biology: Lessons from LCMV. Viruses (2019) 11:172. doi: 10.3390/v11020172
102. Sullivan BM, Emonet SF, Welch MJ, Lee AM, Campbell KP, De La Torre JC, et al. Point mutation in the glycoprotein of lymphocytic choriomeningitis virus is necessary for receptor binding, dendritic cell infection, and long-term persistence. Proc Natl Acad Sci USA (2011) 108:2969–74. doi: 10.1073/pnas.1019304108
103. Osokine I, Snell LM, Cunningham CR, Yamada DH, Wilson EB, Elsaesser HJ, et al. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc Natl Acad Sci (2014) 111:7409–14. doi: 10.1073/pnas.1401662111
104. Raju S, Verbaro DJ, Egawa T. PD-1 Signaling Promotes Control of Chronic Viral Infection by Restricting Type-I-Interferon-Mediated Tissue Damage. Cell Rep (2019) 29:2556–64.e3. doi: 10.1016/j.celrep.2019.10.092
105. Moseman EA, Wu T, de la Torre JC, Schwartzberg PL, McGavern DB. Type I interferon suppresses virus-specific B cell responses by modulating CD8+ T cell differentiation. Sci Immunol (2016) 1:eaah3565. doi: 10.1126/sciimmunol.aah3565
106. Iannacone M, Sitia G, Isogawa M, Whitmire JK, Marchese P, Chisari FV, et al. Platelets prevent IFN-α/β-induced lethal hemorrhagepromoting CTL-dependent clearance of lymphocytic choriomeningitis virus.Proc Natl Acad Sci USA (2008) 105:629–34. doi: 10.1073/pnas.0711200105
107. Schnell FJ, Sundholm S, Crumley S, Iversen PL, Mourich DV. Lymphocytic Choriomeningitis Virus Infection in FVB Mouse Produces Hemorrhagic Disease. PloS Pathog (2012) 8:e1003073. doi: 10.1371/journal.ppat.1003073
108. Silva LA, Dermody TS. Chikungunya virus: epidemiology, replication, disease mechanisms, and prospective intervention strategies. J Clin Invest (2017) 127:737–49. doi: 10.1172/JCI84417
109. Suthar MS, Diamond MS, Gale M. West Nile virus infection and immunity. Nat Rev Microbiol (2013) 11:115–28. doi: 10.1038/nrmicro2950
110. Chen H-W, King K, Tu J, Sanchez M, Luster AD, Shresta S. The roles of IRF-3 and IRF-7 in innate antiviral immunity against dengue virus. J Immunol (2013) 191:4194–201. doi: 10.4049/jimmunol.1300799
111. Lang PA, Cervantes-Barragan L, Verschoor A, Navarini AA, Recher M, Pellegrini M, et al. Hematopoietic cell-derived interferon controls viral replication and virus-induced disease. Blood (2008) 113:1045–52. doi: 10.1182/blood-2007-10-117861
112. Soper A, Kimura I, Nagaoka S, Konno Y, Yamamoto K, Koyanagi Y, et al. Type I interferon responses by HIV-1 infection: Association with disease progression and control. Front Immunol (2018) 8:1823. doi: 10.3389/fimmu.2017.01823
113. Acchioni C, Marsili G, Perrotti E, Remoli AL, Sgarbanti M, Battistini A. Type I IFN – A blunt spear in fighting HIV-1 infection. Cytokine Growth Factor Rev (2015) 26:143–58. doi: 10.1016/j.cytogfr.2014.10.004
114. Doyle T, Goujon C, Malim MH. HIV-1 and interferons: who’s interfering with whom? Nat Rev Microbiol (2015) 13:403–13. doi: 10.1038/nrmicro3449
115. Harper MS, Guo K, Gibbert K, Lee EJ, Dillon SM, Barrett BS, et al. Interferon-α Subtypes in an Ex Vivo Model of Acute HIV-1 Infection: Expression, Potency and Effector Mechanisms. PloS Pathog (2015) 11:e1005254. doi: 10.1371/journal.ppat.1005254
116. Lavender KJ, Gibbert K, Peterson KE, Van Dis E, Francois S, Woods T, et al. Interferon Alpha Subtype-Specific Suppression of HIV-1 Infection In Vivo. J Virol (2016) 90:6001–13. doi: 10.1128/JVI.00451-16
117. Abraham S, Choi JG, Ortega NM, Zhang J, Shankar P, Swamy NM. Gene therapy with plasmids encoding IFN-β or IFN-α14 confers long-term resistance to HIV-1 in humanized mice. Oncotarget (2016) 7:78412–20. doi: 10.18632/oncotarget.12512
118. Dickow J, Francois S, Kaiserling R-L, Malyshkina A, Drexler I, Westendorf AM, et al. Diverse Immunomodulatory Effects of Individual IFNα Subtypes on Virus-Specific CD8+ T Cell Responses. Front Immunol (2019) 10:2255. doi: 10.3389/fimmu.2019.02255
119. Rehermann B, Bertoletti A. Immunological aspects of antiviral therapy of chronic hepatitis B virus and hepatitis C virus infections. Hepatology (2015) 61:712–21. doi: 10.1002/hep.27323
120. Heim MH. 25 years of interferon-based treatment of chronic hepatitis C: An epoch coming to an end. Nat Rev Immunol (2013) 13:535–42. doi: 10.1038/nri3463
121. Muoz R, Castellano G, Fernández I, Álvarez MV, Manzano ML, Marcos MS, et al. A pilot study of β-interferon for treatment of patients with chronic hepatitis B who failed to respond to α-interferon. J Hepatol (2002) 37:655–9. doi: 10.1016/S0168-8278(02)00261-1
122. Ruíz-Moreno M, Fernández P, Leal A, Bartolomé J, Castillo I, Oliva H, et al. Pilot interferon-β trial in children with chronic hepatitis B who had previously not responded to interferon-α therapy. Pediatrics (1997) 99:222–5. doi: 10.1542/peds.99.2.222
123. Arase Y, Suzuki Y, Suzuki F, Matsumoto N, Akuta N, Imai N, et al. Efficacy and safety of combination therapy of natural human interferon beta and ribavirin in chronic hepatitis C patients. Intern Med (2011) 50:2083–8. doi: 10.2169/internalmedicine.50.5767
124. Flores I, Mariano TM, Pestka S. Human interferon omega (ω) binds to the α/β receptor. J Biol Chem (1991) 266:19875–7.
125. Yang LM, Xue QH, Sun L, Zhu YP, Liu WJ. Cloning and characterization of a novel feline IFN-ω. J Interf Cytokine Res (2007) 27:119–27. doi: 10.1089/jir.2006.0094
126. Pavlovich SS, Lovett SP, Koroleva G, Guito JC, Arnold CE, Nagle ER, et al. The Egyptian Rousette Genome Reveals Unexpected Features of Bat Antiviral Immunity. Cell (2018) 173:1098–110.e18. doi: 10.1016/j.cell.2018.03.070
127. He X, Koryta T, Schatz J, Freuling CM, Muller T, Kollner B. Anti-Lyssaviral Activity of Interferons κ and ω from the Serotine Bat, Eptesicus serotinus. J Virol (2014) 88:5444–54. doi: 10.1128/JVI.03403-13
128. Li SF, Zhao FR, Gong MJ, Shao JJ, Xie YL, Chang HY, et al. Antiviral activity of porcine interferon omega 7 against foot-and-mouth disease virus in vitro. J Med Virol (2019) 91:208–14. doi: 10.1002/jmv.25272
129. Wang X, Li F, Han M, Jia S, Wang L, Qiao X, et al. Cloning, prokaryotic soluble expression, and analysis of antiviral activity of two novel feline IFN-ω proteins. Viruses (2020) 12:335. doi: 10.3390/v12030335
130. Pavlovich SS, Darling T, Hume AJ, Davey RA, Feng F, Mühlberger E, et al. Egyptian Rousette IFN-ω Subtypes Elicit Distinct Antiviral Effects and Transcriptional Responses in Conspecific Cells. Front Immunol (2020) 11:435. doi: 10.3389/fimmu.2020.00435
131. Zhao X, Cheng G, Yan W, Liu M, He Y, Zheng Z. Characterization and virus-induced expression profiles of the porcine interferon-ω multigene family. J Interf Cytokine Res (2009) 29:687–93. doi: 10.1089/jir.2008.0060
132. Davidson S, Maini MK, Wack A. Disease-Promoting Effects of Type I Interferons in Viral, Bacterial, and Coinfections. J Interf Cytokine Res (2015) 35:252–64. doi: 10.1089/jir.2014.0227
133. Park A, Iwasaki A, Type I. and Type III Interferons – Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe (2020) 27:870–8. doi: 10.1016/j.chom.2020.05.008
134. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe (2016) 19:181–93. doi: 10.1016/j.chom.2016.01.007
135. Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon αβ in acute influenza infection. Nat Commun (2014) 5:3864. doi: 10.1038/ncomms4864
136. Major J, Crotta S, Llorian M, McCabe TM, Gad HH, Priestnall SL, et al. Type Iand III interferons disrupt lung epithelial repair during recovery from viral infection. Science (2020) 369:712–7. doi: 10.1126/science.abc2061
137. Dorhoi A, Yeremeev V, Nouailles G, Weiner J, Jörg S, Heinemann E, et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur J Immunol (2014) 44:2380–93. doi: 10.1002/eji.201344219
138. Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res (2005) 25:694–701. doi: 10.1089/jir.2005.25.694
139. Ji DX, Yamashiro LH, Chen KJ, Mukaida N, Kramnik I, Darwin KH, et al. Type I interferon-driven susceptibility to Mycobacterium tuberculosis is mediated by IL-1Ra. Nat Microbiol (2019) 4:2128–35. doi: 10.1038/s41564-019-0578-3
140. Kimmey JM, Campbell JA, Weiss LA, Monte KJ, Lenschow DJ, Stallings CL. The impact of ISGylation during Mycobacterium tuberculosis infection in mice. Microbes Infect (2017) 19:249–58. doi: 10.1016/j.micinf.2016.12.006
141. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity (2011) 35:1023–34. doi: 10.1016/j.immuni.2011.12.002
142. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature (2014) 511:99–103. doi: 10.1038/nature13489
143. Antonelli LRV, Gigliotti Rothfuchs A, Gonçalves R, Roffê E, Cheever AW, Bafica A, et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest (2010) 120:1674–82. doi: 10.1172/JCI40817
144. Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol (2012) 13:954–62. doi: 10.1038/ni.2397
145. Perkins DJ, Rajaiah R, Tennant SM, Ramachandran G, Higginson EE, Dyson TN, et al. Salmonella Typhimurium Co-Opts the Host Type I IFN System To Restrict Macrophage Innate Immune Transcriptional Responses Selectively. J Immunol (2015) 195:2461–71. doi: 10.4049/jimmunol.1500105
146. Gratz N, Hartweger H, Matt U, Kratochvill F, Janos M, Sigel S, et al. Type I interferon production induced by Streptococcus pyogenes-derived nucleic acids is required for host protection. PloS Pathog (2011) 7:e1001345. doi: 10.1371/journal.ppat.1001345
147. Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol (2007) 178:3126–33. doi: 10.4049/jimmunol.178.5.3126
148. LeMessurier KS, Häcker H, Chi L, Tuomanen E, Redecke V. Type I interferon protects against pneumococcal invasive disease by inhibiting bacterial transmigration across the lung. PloS Pathog (2013) 9:e1003727. doi: 10.1371/journal.ppat.1003727
149. Damjanovic D, Khera A, Medina MF, Ennis J, Turner JD, Gauldie J, et al. Type 1 interferon gene transfer enhances host defense against pulmonary Streptococcus pneumoniae infection via activating innate leukocytes. Mol Ther Methods Clin Dev (2014) 1:14005. doi: 10.1038/mtm.2014.5
150. Carrero JA, Calderon B, Unanue ER. Type I Interferon Sensitizes Lymphocytes to Apoptosis and Reduces Resistance to Listeria Infection. J Exp Med (2004) 200:535–40. doi: 10.1084/jem.20040769
151. Osborne SE, Sit B, Shaker A, Currie E, Tan JMJ, van Rijn J, et al. Type I interferon promotes cell-to-cell spread of Listeria monocytogenes. Cell Microbiol (2017) 19:e12660. doi: 10.1111/cmi.12660
152. O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, et al. Type I Interferon Production Enhances Susceptibility to Listeria monocytogenes Infection. J Exp Med (2004) 200:437–45. doi: 10.1084/jem.20040712
153. Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA. Mice Lacking the Type I Interferon Receptor Are Resistant to Listeria monocytogenes. J Exp Med (2004) 200:527–33. doi: 10.1084/jem.20040976
154. Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, et al. Tuberculosis. Nat Rev Dis Prim (2016) 2:16076. doi: 10.1038/nrdp.2016.77
155. Moreira-Teixeira L, Mayer-Barber K, Sher A, O’Garra A. Type I interferons in tuberculosis: Foe and occasionally friend. J Exp Med (2018) 215:1273–85. doi: 10.1084/jem.20180325
156. Berry MPR, Graham CM, McNab FW, Xu Z, Bloch SAA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature (2010) 466:973–7. doi: 10.1038/nature09247
157. Ottenhoff THM, Dass RH, Yang N, Zhang MM, Wong HEE, Sahiratmadja E, et al. Genome-Wide Expression Profiling Identifies Type 1 Interferon Response Pathways in Active Tuberculosis. PloS One (2012) 7:e45839. doi: 10.1371/journal.pone.0045839
158. Maertzdorf J, Repsilber D, Parida SK, Stanley K, Roberts T, Black G, et al. Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes Immun (2011) 12:15–22. doi: 10.1038/gene.2010.51
159. Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-α/β. Proc Natl Acad Sci USA (2001) 98:5752–7. doi: 10.1073/pnas.091096998
160. Ordway D, Henao-Tamayo M, Harton M, Palanisamy G, Troudt J, Shanley C, et al. The Hypervirulent Mycobacterium tuberculosis Strain HN878 Induces a Potent TH1 Response followed by Rapid Down-Regulation. J Immunol (2007) 179:522–31. doi: 10.4049/jimmunol.179.1.522
161. Carmona J, Cruz A, Moreira-Teixeira L, Sousa C, Sousa J, Osorio NS, et al. Mycobacterium tuberculosis Strains Are Differentially Recognized by TLRs with an Impact on the Immune Response. PloS One (2013) 8:e67277. doi: 10.1371/journal.pone.0067277
162. Moreira-Teixeira L, Tabone O, Graham CM, Singhania A, Stavropoulos E, Redford PS, et al. Mouse transcriptome reveals potential signatures of protection and pathogenesis in human tuberculosis. Nat Immunol (2020) 21:464–76. doi: 10.1038/s41590-020-0610-z
163. de Paus RA, van Wengen A, Schmidt I, Visser M, Verdegaal EME, van Dissel JT, et al. Inhibition of the type I immune responses of human monocytes by IFN-α and IFN-β. Cytokine (2013) 61:645–55. doi: 10.1016/j.cyto.2012.12.005
164. Teles RMB, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science (2013) 339:1448–53. doi: 10.1126/science.1233665
165. McNab FW, Ewbank J, Howes A, Moreira-Teixeira L, Martirosyan A, Ghilardi N, et al. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-γ for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J Immunol (2014) 193:3600–12. doi: 10.4049/jimmunol.1401088
166. Taneja V, Kalra P, Goel M, Khilnani GC, Saini V, Prasad GBKS, et al. Impact and prognosis of the expression of IFN-α among tuberculosis patients. PloS One (2020) 15:e0235488. doi: 10.1371/journal.pone.0235488
167. Giosué S, Casarini M, Alemanno L, Galluccio G, Mattia P, Pedicelli G, et al. Effects of aerosolized interferon-alpha in patients with pulmonary tuberculosis. Am J Respir Crit Care Med (1998) 158:1156–62. doi: 10.1164/ajrccm.158.4.9803065
168. Palmero D, Eiguchi K, Rendo P, Castro Zorrilla L, Abbate E, González Montaner LJ. Phase II trial of recombinant interferon-alpha2b in patients with advanced intractable multidrug-resistant pulmonary tuberculosis: long-term follow-up. Int J Tuberc Lung Dis (1999) 3:214–8.
169. Zarogoulidis P, Kioumis I, Papanas N, Manika K, Kontakiotis T, Papagianis A, et al. The effect of combination IFN-alpha-2a with usual antituberculosis chemotherapy in non-responding tuberculosis and diabetes mellitus: a case report and review of the literature. J Chemother (2012) 24:173–7. doi: 10.1179/1973947812Y.0000000005
170. Giosuè S, Casarini M, Ameglio F, Zangrilli P, Palla M, Altieri AM, et al. Aerosolized interferon-alpha treatment in patients with multi-drug-resistant pulmonary tuberculosis. Eur Cytokine Netw (2000) 11:99–104.
171. Moreira-Teixeira L, Sousa J, McNab FW, Torrado E, Cardoso F, Machado H, et al. Type I IFN Inhibits Alternative Macrophage Activation during Mycobacterium tuberculosis Infection and Leads to Enhanced Protection in the Absence of IFN-γ Signaling. J Immunol (2016) 197:4714–26. doi: 10.4049/jimmunol.1600584
172. Desvignes L, Wolf AJ, Ernst JD. Dynamic Roles of Type I and Type II IFNs in Early Infection with Mycobacterium tuberculosis. J Immunol (2012) 188:6205–15. doi: 10.4049/jimmunol.1200255
173. Ward CM, Jyonouchi H, Kotenko SV, Smirnov SV, Patel R, Aguila H, et al. Adjunctive treatment of disseminated Mycobacterium avium complex infection with interferon alpha-2b in a patient with complete interferon-gamma receptor R1 deficiency. Eur J Pediatr (2007) 166:981–5. doi: 10.1007/s00431-006-0339-1
174. Bax HI, Freeman AF, Ding L, Hsu AP, Marciano B, Kristosturyan E, et al. Interferon Alpha Treatment of Patients with Impaired Interferon Gamma Signaling. J Clin Immunol (2013) 33:991–1001. doi: 10.1007/s10875-013-9882-5
175. Rivas-Santiago CE, Guerrero GG. IFN-α Boosting of Mycobacterium bovis Bacillus Calmette Güerin-Vaccine Promoted Th1 Type Cellular Response and Protection against M. tuberculosis Infection. BioMed Res Int (2017) 2017:8796760. doi: 10.1155/2017/8796760
176. Gröschel M I, Sayes F, Shin SJ, Frigui W, Pawlik A, Orgeur M, et al. Recombinant BCG Expressing ESX-1 of Mycobacterium marinum Combines Low Virulence with Cytosolic Immune Signaling and Improved TB Protection. Cell Rep (2017) 18:2752–65. doi: 10.1016/j.celrep.2017.02.057
177. Bottai D, Frigui W, Clark S, Rayner E, Zelmer A, Andreu N, et al. Increased protective efficacy of recombinant BCG strains expressing virulence-neutral proteins of the ESX-1 secretion system. Vaccine (2015) 33:2710–8. doi: 10.1016/j.vaccine.2015.03.083
178. Lienard J, Nobs E, Lovins V, Movert E, Valfridsson C, Carlsson F. The Mycobacterium marinum ESX-1 system mediates phagosomal permeabilization and type I interferon production via separable mechanisms. Proc Natl Acad Sci (2020) 117:1160–6. doi: 10.1073/pnas.1911646117
179. Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The Type I IFN Response to Infection with Mycobacterium tuberculosis Requires ESX-1-Mediated Secretion and Contributes to Pathogenesis. J Immunol (2007) 178:3143–52. doi: 10.4049/jimmunol.178.5.3143
180. Giacomini E, Remoli ME, Gafa V, Pardini M, Fattorini L, Coccia EM. IFN-β improves BCG immunogenicity by acting on DC maturation. J Leukoc Biol (2009) 85:462–8. doi: 10.1189/jlb.0908583
181. Bouchonnet F, Boechat N, Bonay M, Hance AJ. Alpha/Beta Interferon Impairs the Ability of Human Macrophages To Control Growth of Mycobacterium bovis BCG. Infect Immun (2002) 70:3020–5. doi: 10.1128/IAI.70.6.3020-3025.2002
182. Hos NJ, Ganesan R, Gutiérrez S, Hos D, Klimek J, Abdullah Z, et al. Type I interferon enhances necroptosis of Salmonella Typhimurium–infected macrophages by impairing antioxidative stress responses. J Cell Biol (2017) 216:4107–21. doi: 10.1083/jcb.201701107
183. Radoshevich L, Cossart P. Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nat Rev Microbiol (2018) 16:32–46. doi: 10.1038/nrmicro.2017.126
184. Pitts MG, Myers-Morales T, D’Orazio SEF. Type I IFN Does Not Promote Susceptibility to Foodborne Listeria monocytogenes. J Immunol (2016) 196:3109–16. doi: 10.4049/jimmunol.1502192
185. Kernbauer E, Maier V, Rauch I, Müller M, Decker T. Route of Infection Determines the Impact of Type I Interferons on Innate Immunity to Listeria monocytogenes. PloS One (2013) 8:e65007. doi: 10.1371/journal.pone.0065007
186. Garifulin O, Qi Z, Shen H, Patnala S, Green MR, Boyartchuk V. Irf3 Polymorphism Alters Induction of Interferon Beta in Response to Listeria monocytogenes Infection. PloS Genet (2007) 3:e152. doi: 10.1371/journal.pgen.0030152
187. Stockinger S, Materna T, Stoiber D, Bayr L, Steinborn R, Kolbe T, et al. Production of Type I IFN Sensitizes Macrophages to Cell Death Induced by Listeria monocytogenes. J Immunol (2002) 169:6522–9. doi: 10.4049/jimmunol.169.11.6522
188. Merrick JC, Edelson BT, Bhardwaj V, Swanson PE, Unanue ER. Lymphocyte apoptosis during early phase of Listeria infection in mice. Am J Pathol (1997) 151:785–92.
189. Mitchell TJ. The pathogenesis of streptococcal infections: from tooth decay to meningitis. Nat Rev Microbiol (2003) 1:219–30. doi: 10.1038/nrmicro771
190. Weiser JN, Ferreira DM, Paton JC. Streptococcus pneumoniae: transmission, colonization and invasion. Nat Rev Microbiol (2018) 16:355–67. doi: 10.1038/s41579-018-0001-8
191. Phillips MA, Burrows JN, Manyando C, van Huijsduijnen RH, Van Voorhis WC, Wells TNC. Malaria. Nat Rev Dis Prim (2017) 3:17050. doi: 10.1038/nrdp.2017.50
192. Kaye P, Scott P. Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol (2011) 9:604–15. doi: 10.1038/nrmicro2608
193. Hotez PJ, Bethony JM, Diemert DJ, Pearson M, Loukas A. Developing vaccines to combat hookworm infection and intestinal schistosomiasis. Nat Rev Microbiol (2010) 8:814–26. doi: 10.1038/nrmicro2438
194. Savioli L, Albonico M. Focus: Soil-transmitted helminthiasis. Nat Rev Microbiol (2004) 2:618–9. doi: 10.1038/nrmicro962
195. Sebina I, Haque A. Effects of type I interferons in malaria. Immunology (2018) 155:176–85. doi: 10.1111/imm.12971
196. Silva-Barrios S, Stäger S. Protozoan Parasites and Type I IFNs. Front Immunol (2017) 8:14. doi: 10.3389/fimmu.2017.00014
197. Liehl P, Zuzarte-Luís V, Chan J, Zillinger T, Baptista F, Carapau D, et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat Med (2014) 20:47–53. doi: 10.1038/nm.3424
198. Miller JL, Sack BK, Baldwin M, Vaughan AM, Kappe SH II. Interferon-Mediated Innate Immune Responses against Malaria Parasite Liver Stages. Cell Rep (2014) 7:436–47. doi: 10.1016/j.celrep.2014.03.018
199. Edwards CL, Best SE, Gun SY, Claser C, James KR, de Oca MM, et al. Spatiotemporal requirements for IRF7 in mediating type I IFN-dependent susceptibility to blood-stage Plasmodium infection. Eur J Immunol (2015) 45:130–41. doi: 10.1002/eji.201444824
200. Sebina I, James KR, Soon MSF, Fogg LG, Best SE, de Labastida Rivera F, et al. IFNAR1-Signalling Obstructs ICOS-mediated Humoral Immunity during Non-lethal Blood-Stage Plasmodium Infection. PloS Pathog (2016) 12:e1005999. doi: 10.1371/journal.ppat.1005999
201. Yu X, Cai B, Wang M, Tan P, Ding X, Wu J, et al. Cross-Regulation of Two Type I Interferon Signaling Pathways in Plasmacytoid Dendritic Cells Controls Anti-malaria Immunity and Host Mortality. Immunity (2016) 45:1093–107. doi: 10.1016/j.immuni.2016.10.001
202. Vigário AM, Belnoue E, Cumano A, Marussig M, Miltgen F, Landau I, et al. Inhibition of Plasmodium yoelii blood-stage malaria by interferon α through the inhibition of the production of its target cell, the reticulocyte. Blood (2001) 97:3966–71. doi: 10.1182/blood.V97.12.3966
203. Sharma S, DeOliveira RB, Kalantari P, Parroche P, Goutagny N, Jiang Z, et al. Innate Immune Recognition of an AT-Rich Stem-Loop DNA Motif in the Plasmodium falciparum Genome. Immunity (2011) 35:194–207. doi: 10.1016/j.immuni.2011.05.016
204. Haque A, Best SE, Ammerdorffer A, Desbarrieres L, de Oca MM, Amante FH, et al. Type I interferons suppress CD4+ T-cell-dependent parasite control during blood-stage Plasmodium infection. Eur J Immunol (2011) 41:2688–98. doi: 10.1002/eji.201141539
205. Ball EA, Sambo MR, Martins M, Trovoada MJ, Benchimol C, Costa J, et al. IFNAR1 Controls Progression to Cerebral Malaria in Children and CD8 + T Cell Brain Pathology in Plasmodium berghei –Infected Mice. J Immunol (2013) 190:5118–27. doi: 10.4049/jimmunol.1300114
206. Palomo J, Fauconnier M, Coquard L, Gilles M, Meme S, Szeremeta F, et al. Type I interferons contribute to experimental cerebral malaria development in response to sporozoite or blood-stage Plasmodium berghei ANKA. Eur J Immunol (2013) 43:2683–95. doi: 10.1002/eji.201343327
207. Haque A, Best SE, Montes de Oca M, James KR, Ammerdorffer A, Edwards CL, et al. Type I IFN signaling in CD8- DCs impairs Th1-dependent malaria immunity. J Clin Invest (2014) 124:2483–96. doi: 10.1172/JCI70698
208. Morrell CN, Srivastava K, Swaim A, Lee MT, Chen J, Nagineni C, et al. Beta Interferon Suppresses the Development of Experimental Cerebral Malaria. Infect Immun (2011) 79:1750–8. doi: 10.1128/IAI.00810-10
209. Vigário AM, Belnoue E, Grüner AC, Marussig M, Kayibanda M, Deschemin J-C, et al. Recombinant Human IFN-α Inhibits Cerebral Malaria and Reduces Parasite Burden in Mice. J Immunol (2007) 178:6416–25. doi: 10.4049/jimmunol.178.10.6416
210. Gowda DC, Wu X. Parasite Recognition and Signaling Mechanisms in Innate Immune Responses to Malaria. Front Immunol (2018) 9:3006. doi: 10.3389/fimmu.2018.03006
211. Meister A, Uze G, Mogensen KE, Gresser I, Tovey MG, Grutter M, et al. Biological Activities and Receptor Binding of Two Human Recombinant Interferons and their Hybrids. J Gen Virol (1986) 67:1633–43. doi: 10.1099/0022-1317-67-8-1633
212. Yu X, Du Y, Cai C, Cai B, Zhu M, Xing C, et al. Inflammasome activation negatively regulates MyD88-IRF7 type I IFN signaling and anti-malaria immunity. Nat Commun (2018) 9:4964. doi: 10.1038/s41467-018-07384-7
213. He X, Ashbrook AW, Du Y, Wu J, Hoffmann H-H, Zhang C, et al. RTP4 inhibits IFN-I response and enhances experimental cerebral malaria and neuropathology. Proc Natl Acad Sci USA (2020) 117:19465–74. doi: 10.1073/pnas.2006492117
214. Aucan C, Walley AJ, Hennig BJW, Fitness J, Frodsham A, Zhang L, et al. Interferon-alpha receptor-1 (IFNAR1) variants are associated with protection against cerebral malaria in the Gambia. Genes Immun (2003) 4:275–82. doi: 10.1038/sj.gene.6363962
215. Khor CC, Vannberg FO, Chapman SJ, Walley A, Aucan C, Loke H, et al. Positive replication and linkage disequilibrium mapping of the chromosome 21q22.1 malaria susceptibility locus. Genes Immun (2007) 8:570–6. doi: 10.1038/sj.gene.6364417
216. Zhang G, DeWeerd NA, Stifter SA, Liu L, Zhou B, Wang W, et al. A proline deletion in IFNAR1 impairs IFN-signaling and underlies increased resistance to tuberculosis in humans. Nat Commun (2018) 9:85. doi: 10.1038/s41467-017-02611-z
217. Feintuch CM, Tare A, Cusumano LR, Benayoun J, Ryu S, Sixpence A, et al. Type I Interferon Receptor Variants in Gene Regulatory Regions are Associated with Susceptibility to Cerebral Malaria in Malawi. Am J Trop Med Hyg (2018) 98:1692–8. doi: 10.4269/ajtmh.17-0887
218. Sacramento LA, Benevides L, Maruyama SR, Tavares L, Fukutani KF, Francozo M, et al. TLR4 abrogates the Th1 immune response through IRF1 and IFN-β to prevent immunopathology during L. infantum infection. PloS Pathog (2020) 16:e1008435. doi: 10.1371/journal.ppat.1008435
219. Matta SK, Olias P, Huang Z, Wang Q, Park E, Yokoyama WM, et al. Toxoplasma gondii effector TgIST blocks type I interferon signaling to promote infection. Proc Natl Acad Sci USA (2019) 116:17480–91. doi: 10.1073/pnas.1904637116
220. Obieglo K, Costain A, Webb LM, Ozir-Fazalalikhan A, Brown SL, MacDonald AS, et al. Type I interferons provide additive signals for murine regulatory B cell induction by Schistosoma mansoni eggs. Eur J Immunol (2019) 49:1226–34. doi: 10.1002/eji.201847858
221. Aksoy E, Zouain CS, Vanhoutte F, Fontaine J, Pavelka N, Thieblemont N, et al. Double-stranded RNAs from the helminth parasite Schistosoma activate TLR3 in dendritic cells. J Biol Chem (2005) 280:277–83. doi: 10.1074/jbc.M411223200
222. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer (2016) 16:131–44. doi: 10.1038/nrc.2016.14
223. Budhwani M, Mazzieri R, Dolcetti R. Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-tumor Immunity to Resistance. Front Oncol (2018) 8:322. doi: 10.3389/fonc.2018.00322
224. Snell LM, McGaha TL, Brooks DG. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol (2017) 38:542–57. doi: 10.1016/j.it.2017.05.005
225. Reid LM, Minato N, Gresser I, Holland J, Kadish A, Bloom BR. Influence of anti-mouse interferon serum on the growth and metastasis of tumor cells persistently infected with virus and of human prostatic tumors in athymic nude mice. Proc Natl Acad Sci (1981) 78:1171–5. doi: 10.1073/pnas.78.2.1171
226. Dunn GP, Bruce AT, Sheehan KCF, Shankaran V, Uppaluri R, Bui JD, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol (2005) 6:722–9. doi: 10.1038/ni1213
227. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med (2011) 208:1989–2003. doi: 10.1084/jem.20101158
228. Schiavoni G, Mattei F, Gabriele L. Type I Interferons as Stimulators of DC-Mediated Cross-Priming: Impact on Anti-Tumor Response. Front Immunol (2013) 4:1–7. doi: 10.3389/fimmu.2013.00483
229. Swann JB, Swann JB, Hayakawa Y, Zerafa N, Sheehan KCF, Scott B, Schreiber RD, et al. Type I IFN Contributes to NK Cell Homeostasis, Activation, and Antitumor Function. J Immunol (2007) 178:7540–9. doi: 10.4049/jimmunol.178.12.7540
230. von Marschall Z, Scholz A, Cramer T, Schafer G, Schirner M, Oberg K, et al. Effects of Interferon Alpha on Vascular Endothelial Growth Factor Gene Transcription and Tumor Angiogenesis. JNCI J Natl Cancer Inst (2003) 95:437–48. doi: 10.1093/jnci/95.6.437
231. Spaapen RM, Leung MYK, Fuertes MB, Kline JP, Zhang L, Zheng Y, et al. Therapeutic Activity of High-Dose Intratumoral IFN-β Requires Direct Effect on the Tumor Vasculature. J Immunol (2014) 193:4254–60. doi: 10.4049/jimmunol.1401109
232. Shi Y. Interferon-α-secreting mesenchymal stem cells exert potent antitumor effect in vivo. Oncogene (2014) 33:5047–52. doi: 10.1038/onc.2013.458
233. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell (2016) 167:1540–54.e12. doi: 10.1016/j.cell.2016.11.022
234. Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, et al. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J (2013) 32:2751–63. doi: 10.1038/emboj.2013.203
235. Andzinski L, Kasnitz N, Stahnke S, Wu C-FF, Gereke M, Von Köckritz-Blickwede M, et al. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer (2016) 138:1982–93. doi: 10.1002/ijc.29945
236. Wu C-F, Andzinski L, Kasnitz N, Kröger A, Klawonn F, Lienenklaus S, et al. The lack of type I interferon induces neutrophil-mediated pre-metastatic niche formation in the mouse lung. Int J Cancer (2015) 137:837–47. doi: 10.1002/ijc.29444
237. Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest (2010) 120:1151–64. doi: 10.1172/JCI37223
238. Jablonska J, Wu C-F, Andzinski L, Leschner S, Weiss S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β. Int J Cancer (2014) 134:1346–58. doi: 10.1002/ijc.28551
239. Gresser I, Maury C, Brouty-Boyé D. Mechanism of the antitumour effect of interferon in mice. Nature (1972) 239:167–8. doi: 10.1038/239167a0
240. Gresser I, Bourali C, Lévy JP, Fontaine-Brouty-Boyé D, Thomas MT. Increased survival in mice inoculated with tumor cells and treated with interferon preparations. Proc Natl Acad Sci USA (1969) 63:51–7. doi: 10.1073/pnas.63.1.51
241. Talpaz M, Hehlmann R, Quintás-Cardama A, Mercer J, Cortes J. Re-emergence of interferon-α in the treatment of chronic myeloid leukemia. Leukemia (2013) 27:803–12. doi: 10.1038/leu.2012.313
242. Di Trolio R, Simeone E, Di Lorenzo G, Buonerba C, Ascierto PA. The use of interferon in melanoma patients: a systematic review. Cytokine Growth Factor Rev (2015) 26:203–12. doi: 10.1016/j.cytogfr.2014.11.008
243. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature (2016) 534:396–401. doi: 10.1038/nature18300
244. Escobar G, Moi D, Ranghetti A, Ozkal-Baydin P, Squadrito ML, Kajaste-Rudnitski A, et al. Genetic Engineering of Hematopoiesis for Targeted IFN- Delivery Inhibits Breast Cancer Progression. Sci Transl Med (2014) 6:217ra3–3. doi: 10.1126/scitranslmed.3006353
245. Cauwels A, Van Lint S, Garcin G, Bultinck J, Paul F, Gerlo S, et al. A safe and highly efficient tumor-targeted type I interferon immunotherapy depends on the tumor microenvironment. Oncoimmunology (2018) 7:e1398876. doi: 10.1080/2162402X.2017.1398876
246. Garcin G, Paul F, Staufenbiel M, Bordat Y, Van der Heyden J, Wilmes S, et al. High efficiency cell-specific targeting of cytokine activity. Nat Commun (2014) 5:3016. doi: 10.1038/ncomms4016
247. Buzzai AC, Wagner T, Audsley KM, Newnes HV, Barrett LW, Barnes S, et al. Diverse Anti-Tumor Immune Potential Driven by Individual IFNα Subtypes. Front Immunol (2020) 11:542. doi: 10.3389/fimmu.2020.00542
248. Willemen Y, Van den Bergh JMJ, Lion E, Anguille S, Roelandts VAE, Van Acker HH, et al. Engineering monocyte-derived dendritic cells to secrete interferon-α enhances their ability to promote adaptive and innate anti-tumor immune effector functions. Cancer Immunol Immunother (2015) 64:831–42. doi: 10.1007/s00262-015-1688-2
249. Jiang W, Zhang C, Tian Z, Zhang J. hIFN-α gene modification augments human natural killer cell line anti-human hepatocellular carcinoma function. Gene Ther (2013) 20:1062–9. doi: 10.1038/gt.2013.31
250. Hashimoto H, Ueda R, Narumi K, Heike Y, Yoshida T, Aoki K. Type I IFN gene delivery suppresses regulatory T cells within tumors. Cancer Gene Ther (2014) 21:532–41. doi: 10.1038/cgt.2014.60
251. Escobar G, Barbarossa L, Barbiera G, Norelli M, Genua M, Ranghetti A, et al. Interferon gene therapy reprograms the leukemia microenvironment inducing protective immunity to multiple tumor antigens. Nat Commun (2018) 9:2896. doi: 10.1038/s41467-018-05315-0
252. Doherty MR, Cheon H, Junk DJ, Vinayak S, Varadan V, Telli ML, et al. Interferon-beta represses cancer stem cell properties in triple-negative breast cancer. Proc Natl Acad Sci USA (2017) 114:13792–7. doi: 10.1073/pnas.1713728114
253. Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, et al. Targeting the tumor microenvironment with interferon-β bridges innate and adaptive immune responses. Cancer Cell (2014) 25:37–48. doi: 10.1016/j.ccr.2013.12.004
254. Koba C, Haruta M, Matsunaga Y, Matsumura K, Haga E, Sasaki Y, et al. Therapeutic Effect of Human iPS-Cell–Derived Myeloid Cells Expressing IFN-β against Peritoneally Disseminated Cancer in Xenograft Models. PloS One (2013) 8:e67567. doi: 10.1371/journal.pone.0067567
255. Van der Jeught K, Joe PT, Bialkowski L, Heirman C, Daszkiewicz L, Liechtenstein T, et al. Intratumoral administration of mRNA encoding a fusokine consisting of IFN-β and the ectodomain of the TGF-β receptor ii potentiates antitumor immunity. Oncotarget (2014) 5:10100–13. doi: 10.18632/oncotarget.2463
256. Angstreich GR, Matsui W, Huff CA, Vala MS, Barber J, Hawkins AL, et al. Effects of imatinib and interferon on primitive chronic myeloid leukaemia progenitors. Br J Haematol (2005) 130:373–81. doi: 10.1111/j.1365-2141.2005.05606.x
257. Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNα activates dormant haematopoietic stem cells in vivo. Nature (2009) 458:904–8. doi: 10.1038/nature07815
258. Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med (2012) 18:1224–31. doi: 10.1038/nm.2830
259. Chawla-Sarkar M, Leaman DW, Borden EC. Preferential induction of apoptosis by interferon (IFN)-β compared with IFN-α: Correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin Cancer Res (2001) 7:1821–31.
260. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med (2014) 20:1301–9. doi: 10.1038/nm.3708
261. Lim JYH, Gerber SA, Murphy SP, Lord EM. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8+ T cells. Cancer Immunol Immunother (2014) 63:259–71. doi: 10.1007/s00262-013-1506-7
262. Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, et al. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res (2011) 71:2488–96. doi: 10.1158/0008-5472.CAN-10-2820
263. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity (2014) 41:843–52. doi: 10.1016/j.immuni.2014.10.019
264. Woo S-R, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity (2014) 41:830–42. doi: 10.1016/j.immuni.2014.10.017
265. Chen J, Cao Y, Markelc B, Kaeppler J, Vermeer JAF, Muschel RJ. Type I IFN protects cancer cells from CD8+ T cell-mediated cytotoxicity after radiation. J Clin Invest (2019) 129:4224–38. doi: 10.1172/JCI127458
266. Westcott MM, Liu J, Rajani K, D’Agostino R, Lyles DS, Porosnicu M. Interferon Beta and Interferon Alpha 2a Differentially Protect Head and Neck Cancer Cells from Vesicular Stomatitis Virus-Induced Oncolysis. J Virol (2015) 89:7944–54. doi: 10.1128/JVI.00757-15
267. Muskardin TLW, Niewold TB. Type I interferon in rheumatic diseases. Nat Rev Rheumatol (2018) 14:214–28. doi: 10.1038/nrrheum.2018.31
268. Yao Y, Liu Z, Jallal B, Shen N, Rönnblom L. Type I interferons in Sjögren’s syndrome. Autoimmun Rev (2013) 12:558–66. doi: 10.1016/j.autrev.2012.10.006
269. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Prim (2016) 2:16039. doi: 10.1038/nrdp.2016.39
270. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA (2003) 100:2610–5. doi: 10.1073/pnas.0337679100
271. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med (2003) 197:711–23. doi: 10.1084/jem.20021553
272. Crow MK, Kirou KA, Wohlgemuth J. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity (2003) 36:481–90. doi: 10.1080/08916930310001625952
273. Bronson PG, Chaivorapol C, Ortmann W, Behrens TW, Graham RR. The genetics of type I interferon in systemic lupus erythematosus. Curr Opin Immunol (2012) 24:530–7. doi: 10.1016/j.coi.2012.07.008
274. Mustelin T, Lood C, Giltiay NV. Sources of Pathogenic Nucleic Acids in Systemic Lupus Erythematosus. Front Immunol (2019) 10:1028. doi: 10.3389/fimmu.2019.01028
275. Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of Dendritic Cell Differentiation by IFN-alpha in Systemic Lupus Erythematosus. Science (80- ) (2001) 294:1540–3. doi: 10.1126/science.1064890
276. Klarquist J, Cantrell R, Lehn MA, Lampe K, Hennies CM, Hoebe K, et al. Type I IFN Drives Experimental Systemic Lupus Erythematosus by Distinct Mechanisms in CD4 T Cells and B Cells. ImmunoHorizons (2020) 4:140–52. doi: 10.4049/immunohorizons.2000005
277. Banchereau J, Pascual V, Palucka AK. Autoimmunity through cytokine-induced dendritic cell activation. Immunity (2004) 20:539–50. doi: 10.1016/S1074-7613(04)00108-6
278. Garcin G, Bordat Y, Chuchana P, Monneron D, Law HKW, Piehler J, et al. Differential Activity of Type I Interferon Subtypes for Dendritic Cell Differentiation. PloS One (2013) 8:1–11. doi: 10.1371/journal.pone.0058465
279. Palucka AK, Blanck J-P, Bennett L, Pascual V, Banchereau J. Cross-regulation of TNF and IFN-α in autoimmune diseases. Proc Natl Acad Sci (2005) 102:3372–7. doi: 10.1073/pnas.0408506102
280. Sharif MN, Tassiulas I, Hu Y, Mecklenbräuker I, Tarakhovsky A, Ivashkiv LB. IFN-α Priming Results in a Gain of Proinflammatory Function by IL-10: Implications for Systemic Lupus Erythematosus Pathogenesis. J Immunol (2004) 172:6476–81. doi: 10.4049/jimmunol.172.10.6476
281. MacLennan IC, Vinuesa CG. Dendritic Cells, BAFF, and APRIL. Immunity (2002) 17:235–8. doi: 10.1016/S1074-7613(02)00398-9
282. Petri M, Wallace DJ, Spindler A, Chindalore V, Kalunian K, Mysler E, et al. Sifalimumab, a Human Anti-Interferon-α Monoclonal Antibody, in Systemic Lupus Erythematosus: A Phase I Randomized, Controlled, Dose-Escalation Study. Arthritis Rheumatol (2013) 65:1011–21. doi: 10.1002/art.37824
283. Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, et al. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheumatol Dis (2016) 75:1909–16. doi: 10.1136/annrheumdis-2015-208562
284. Lauwerys BR, Hachulla E, Spertini F, Lazaro E, Jorgensen C, Mariette X, et al. Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon α-kinoid. Arthritis Rheumatol (2013) 65:447–56. doi: 10.1002/art.37785
285. Yao Y, Richman L, Higgs BW, Morehouse CA, De Los Reyes M, Brohawn P, et al. Neutralization of interferon-α/β-inducible genes and downstream effect in a phase I trial of an anti-interferon-α monoclonal antibody in systemic lupus erythematosus. Arthritis Rheumatol (2009) 60:1785–96. doi: 10.1002/art.24557
286. Riggs JM, Hanna RN, Rajan B, Zerrouki K, Karnell JL, Sagar D, et al. Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Sci Med (2018) 5:e000261. doi: 10.1136/lupus-2018-000261
287. Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an Anti–Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol (2017) 69:376–86. doi: 10.1002/art.39962
288. Crow MK. Interferon-alpha: a therapeutic target in SLE. Rheum Dis Clin North Am (2010) 36:1–13. doi: 10.1016/j.rdc.2009.12.008
289. Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Prim (2017) 3:17016. doi: 10.1038/nrdp.2017.16
290. Huang X, Yuang J, Goddard A, Foulis A, James RFL, Lernmark A, et al. Interferon Expression in the Pancreases of Patients With Type I Diabetes. Diabetes (1995) 44:658–64. doi: 10.2337/diabetes.44.6.658
291. Ferreira RC, Guo H, Coulson RMR, Smyth DJ, Pekalski ML, Burren OS, et al. A type I Interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes (2014) 63:2538–50. doi: 10.2337/db13-1777
292. Kallionpaa H, Elo LL, Laajala E, Mykkanen J, Ricano-Ponce I, Vaarma M, et al. Innate Immune Activity Is Detected Prior to Seroconversion in Children With HLA-Conferred Type 1 Diabetes Susceptibility. Diabetes (2014) 63:2402–14. doi: 10.2337/db13-1775
293. Foulis AK, Farquharson MA, Meager A. Immunoreactive α-interferon in insulin-secreting β cells in type 1 diabetes mellitus. Lancet (London England) (1987) 2:1423–7. doi: 10.1016/S0140-6736(87)91128-7
294. Todd JA. Etiology of Type 1 Diabetes. Immunity (2010) 32:457–67. doi: 10.1016/j.immuni.2010.04.001
295. Sepe V, Loviselli A, Bottazzo GF. Genetics of Type 1A Diabetes. N Engl J Med (2009) 361:211. doi: 10.1056/NEJMc091064
296. Marroqui L, Dos Santos RS, Fløyel T, Grieco FA, Santin I, Op de beeck A, et al. TYK2, a Candidate Gene for Type 1 Diabetes, Modulates Apoptosis and the Innate Immune Response in Human Pancreatic β-Cells. Diabetes (2015) 64:3808–17. doi: 10.2337/db15-0362
297. Stewart TA, Hultgren B, Huang X, Pitts-Meek S, Hully J, MacLachlan NJ. Induction of type I diabetes by interferon-alpha in transgenic mice. Science (1993) 260:1942–6. doi: 10.1126/science.8100367
298. Carrero JA, Calderon B, Towfic F, Artyomov MN, Unanue ER. Defining the Transcriptional and Cellular Landscape of Type 1 Diabetes in the NOD Mouse. PloS One (2013) 8:e59701. doi: 10.1371/journal.pone.0059701
299. Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO. Interferon-α initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci USA (2008) 105:12439–44. doi: 10.1073/pnas.0806439105
300. Oldstone MBA, Nerenberg M, Southern P, Price J, Lewicki H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: Role of anti-self (virus) immune response. Cell (1991) 65:319–31. doi: 10.1016/0092-8674(91)90165-U
301. Lang KS, Recher M, Junt T, Navarini AA, Harris NL, Freigang S, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med (2005) 11:138–45. doi: 10.1038/nm1176
302. Marro BS, Ware BC, Zak J, de la Torre JC, Rosen H, Oldstone MBA. Progression of type 1 diabetes from the prediabetic stage is controlled by interferon-α signaling. Proc Natl Acad Sci (2017) 114:3708–13. doi: 10.1073/pnas.1700878114
303. Fabris P, Floreani A, Tositti G, Vergani D, de Lalla F, Betterle C. Type 1 diabetes mellitus in patients with chronic hepatitis C before and after interferon therapy. Aliment Pharmacol Ther (2003) 18:549–58. doi: 10.1046/j.1365-2036.2003.01681.x
304. Meyer S, Woodward M, Hertel C, Vlaicu P, Haque Y, Kärner J, et al. AIRE-Deficient Patients Harbor Unique High-Affinity Disease-Ameliorating Autoantibodies. Cell (2016) 166:582–95. doi: 10.1016/j.cell.2016.06.024
305. Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, et al. Multiple sclerosis. Nat Rev Dis Prim (2018) 4:1–27. doi: 10.1038/s41572-018-0050-3
306. Comabella M, Lünemann JD, Río J, Sánchez A, López C, Julià E, et al. A type I interferon signature in monocytes is associated with poor response to interferon-β in multiple sclerosis. Brain (2009) 132:3353–65. doi: 10.1093/brain/awp228
307. Feng X, Reder NP, Yanamandala M, Hill A, Franek BS, Niewold TB, et al. Type I interferon signature is high in lupus and neuromyelitis optica but low in multiple sclerosis. J Neurol Sci (2012) 313:48–53. doi: 10.1016/j.jns.2011.09.032
308. Jacobs L, Salazar AM, Herndon R, Reese PA, Freeman A, Josefowicz R, et al. Multicentre double-blind study of effect of intrathecally administered natural human fibroblast interferon on exacerbations of multiple sclerosis. Lancet (London England) (1986) 2:1411–3. doi: 10.1016/S0140-6736(86)92730-3
309. Comi G, Filippi M, Barkhof F, Durelli L, Edan G, Fernández O, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: A randomised study. Lancet (2001) 357:1576–82. doi: 10.1016/S0140-6736(00)04725-5
310. Jacobs LD, Cookfair DL, Rudick RA, Herndon RM, Richert JR, Salazar AM, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol (1996) 39:285–94. doi: 10.1002/ana.410390304
311. Calabresi PA, Kieseier BC, Arnold DL, Balcer LJ, Boyko A, Pelletier J, et al. Pegylated interferon β-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. Lancet Neurol (2014) 13:657–65. doi: 10.1016/S1474-4422(14)70068-7
312. Granqvist M, Boremalm M, Poorghobad A, Svenningsson A, Salzer J, Frisell T, et al. Comparative effectiveness of rituximab and other initial treatment choices for multiple sclerosis. JAMA Neurol (2018) 75:320–7. doi: 10.1001/jamaneurol.2017.4011
313. Mendel I, de Rosbo NK, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: Fine specificity and T cell receptor Vβ expression of encephalitogenic T cells. Eur J Immunol (1995) 25:1951–9. doi: 10.1002/eji.1830250723
314. Salem M, Mony JT, Løbner M, Khorooshi R, Owens T. Interferon regulatory factor-7 modulates experimental autoimmune encephalomyelitis in mice. J Neuroinflam (2011) 8:181. doi: 10.1186/1742-2094-8-181
315. Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, et al. Distinct and Nonredundant In Vivo Functions of IFNAR on Myeloid Cells Limit Autoimmunity in the Central Nervous System. Immunity (2008) 28:675–86. doi: 10.1016/j.immuni.2008.03.011
316. Teige I, Treschow A, Teige A, Mattsson R, Navikas V, Leanderson T, et al. IFN-beta gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J Immunol (2003) 170:4776–84. doi: 10.4049/jimmunol.170.9.4776
317. Fitzgerald DC, O’Brien K, Young A, Fonseca-Kelly Z, Rostami A, Gran B. Interferon regulatory factor (IRF) 3 is critical for the development of experimental autoimmune encephalomyelitis. J Neuroinflam (2014) 11:1–7. doi: 10.1186/1742-2094-11-130
318. Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, et al. T helper type 1 and 17 cells determine efficacy of interferon-βin multiple sclerosis and experimental encephalomyelitis. Nat Med(2010) 16:406–12. doi: 10.1038/nm.2110
319. Touil T, Fitzgerald D, Zhang G-X, Rostami A, Gran B. Cutting Edge: TLR3 Stimulation Suppresses Experimental Autoimmune Encephalomyelitis by Inducing Endogenous IFN-β. J Immunol (2006) 177:7505–9. doi: 10.4049/jimmunol.177.11.7505
320. O’Brien K, Fitzgerald D, Rostami A, Gran B. The TLR7 agonist, imiquimod, increases IFN-β production and reduces the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol (2010) 221:107–11. doi: 10.1016/j.jneuroim.2010.01.006
321. Naves R, Singh SP, Cashman KS, Rowse AL, Axtell RC, Steinman L, et al. The Interdependent, Overlapping, and Differential Roles of Type I and II IFNs in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. J Immunol (2013) 191:2967–77. doi: 10.4049/jimmunol.1300419
322. Zhang L, Yuan S, Cheng G, Guo B. Type I IFN Promotes IL-10 Production from T Cells to Suppress Th17 Cells and Th17-Associated Autoimmune Inflammation. PloS One (2011) 6:e28432. doi: 10.1371/journal.pone.0028432
323. Pennell LM, Fish EN. Interferon-β regulates dendritic cell activation and migration in experimental autoimmune encephalomyelitis. Immunology (2017) 152:439–50. doi: 10.1111/imm.12781
324. Shinohara ML, Kim J-H, Garcia VA, Cantor H. Engagement of the Type I Interferon Receptor on Dendritic Cells Inhibits T Helper 17 Cell Development: Role of Intracellular Osteopontin. Immunity (2008) 29:68–78. doi: 10.1016/j.immuni.2008.05.008
325. Scheu S, Ali S, Mann-Nüttel R, Richter L, Arolt V, Dannlowski U, et al. Interferon β-mediated protective functions of microglia in central nervous system autoimmunity. Int J Mol Sci (2019) 20:190. doi: 10.3390/ijms20010190
326. Kocur M, Schneider R, Pulm AK, Bauer J, Kropp S, Gliem M, et al. IFNβ secreted by microglia mediates clearance of myelin debris in CNS autoimmunity. Acta Neuropathol Commun (2015) 3:20. doi: 10.1186/s40478-015-0192-4
327. Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med (2016) 22:586–97. doi: 10.1038/nm.4106
328. Vasquez M, Consuegra-Fernández M, Aranda F, Jimenez A, Tenesaca S, Fernandez-Sendin M, et al. Treatment of Experimental Autoimmune Encephalomyelitis by Sustained Delivery of Low-Dose IFN-α. J Immunol (2019) 203:696–704. doi: 10.4049/jimmunol.1801462
329. Cauwels A, Van Lint S, Catteeuw D, Pang S, Paul F, Rogge E, et al. Targeting interferon activity to dendritic cells enables in vivo tolerization and protection against EAE in mice. J Autoimmun (2019) 97:70–6. doi: 10.1016/j.jaut.2018.10.010
330. Calabresi PA, Tranquill LR, Dambrosia JM, Stone LA, Maloni H, Bash CN, et al. Increases in soluble VCAM-1 correlate with a decrease in MRI lesions in multiple sclerosis treated with interferon β-1b. Ann Neurol (1997) 41:669–74. doi: 10.1002/ana.410410517
331. Yu L, Croze E, Yamaguchi KD, Tran T, Reder AT, Litvak V, et al. Induction of a unique isoform of the NCOA7 oxidation resistance gene by interferon β-1b. J Interf Cytokine Res (2015) 35:186–99. doi: 10.1089/jir.2014.0115
332. Gniadek P, Aktas O, Wandinger K-P, Bellmann-Strobl J, Wengert O, Weber A, et al. Systemic IFN-beta treatment induces apoptosis of peripheral immune cells in MS patients. J Neuroimmunol (2003) 137:187–96. doi: 10.1016/S0165-5728(03)00074-2
333. Pette M, Pette DF, Muraro PA, Farnon E, Martin R, McFarland HF, et al. Interferon-β interferes with the proliferation but not with the cytokine secretion of myelin basic protein-specific, T-helper type 1 lymphocytes. Neurology (1997) 49:385–92. doi: 10.1212/WNL.49.2.385
334. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T Cells Is Increased in Multiple Sclerosis and Suppressed by IFN-β Therapy. J Immunol (2015) 194:5085–93. doi: 10.4049/jimmunol.1403243
335. Börnsen L, Romme Christensen J, Ratzer R, Hedegaard C, Søndergaard HB, Krakauer M, et al. Endogenous Interferon-β-Inducible Gene Expression and Interferon-β-Treatment Are Associated with Reduced T Cell Responses to Myelin Basic Protein in Multiple Sclerosis. PloS One (2015) 10:e0118830. doi: 10.1371/journal.pone.0118830
336. Noronha A, Toscas A, Jensen MA. Interferon β decreases T cell activation and interferon γ production in multiple sclerosis. J Neuroimmunol (1993) 46:145–53. doi: 10.1016/0165-5728(93)90244-S
337. Özenci V, Kouwenhoven M, Huang YM, Kivisäkk P, Link H. Multiple sclerosis is associated with an imbalance between tumour necrosis factor-alpha (TNF-α)- and IL-10-secreting blood cells that is corrected by interferon-beta (IFN-β) treatment. Clin Exp Immunol (2000) 120:147–53. doi: 10.1046/j.1365-2249.2000.01175.x
338. Krakauer M, Sorensen P, Khademi M, Olsson T, Sellebjerg F. Increased IL-10 mRNA and IL-23 mRNA expression in multiple sclerosis: Interferon-β treatment increases IL-10 mRNA expression while reducing IL-23 mRNA expression. Mult Scler (2008) 14:622–30. doi: 10.1177/1352458507087136
339. Kozovska ME, et al. Interferon beta induces T-helper 2 immune deviation in MS. Neurology (1999) 53:1692–7. doi: 10.1212/WNL.53.8.1692
340. Reder AT, Feng X. How Type I Interferons Work in Multiple Sclerosis and Other Diseases: Some Unexpected Mechanisms. J Interf Cytokine Res (2014) 34:589–99. doi: 10.1089/jir.2013.0158
341. Liu Y, Carlsson R, Comabella M, Wang J, Kosicki M, Carrion B, et al. FoxA1 directs the lineage and immunosuppressive properties of a novel regulatory T cell population in EAE and MS. Nat Med (2014) 20:272–82. doi: 10.1038/nm.3485
342. Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-”like, Foxp3 + regulatory T cells in human autoimmune disease. Nat Med (2011) 17:673–5. doi: 10.1038/nm.2389
343. Krausgruber T, Fortelny N, Fife-Gernedl V, Senekowitsch M, Schuster LC, Lercher A, et al. Structural cells are key regulators of organ-specific immune responses. Nature (2020) 583:296–302. doi: 10.1038/s41586-020-2424-4
344. Rodero MP, Decalf J, Bondet V, Hunt D, Rice GI, Werneke S, et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J Exp Med (2017) 214:1547–55. doi: 10.1084/jem.20161451
345. Pervolaraki K, Rastgou Talemi S, Albrecht D, Bormann F, Bamford C, Mendoza JL, et al. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PloS Pathog (2018) 14:e1007420. doi: 10.1371/journal.ppat.1007420
346. Vickovic S, Eraslan G, Salmén F, Klughammer J, Stenbeck L, Schapiro D, et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat Methods (2019) 16:987–90. doi: 10.1038/s41592-019-0548-y
Keywords: type I interferons, infection, autoimmunity, cancer, IFNα subtypes, IFNβ
Citation: Fox LE, Locke MC and Lenschow DJ (2020) Context Is Key: Delineating the Unique Functions of IFNα and IFNβ in Disease. Front. Immunol. 11:606874. doi: 10.3389/fimmu.2020.606874
Received: 15 September 2020; Accepted: 11 November 2020;
Published: 21 December 2020.
Edited by:
Mark R. Walter, University of Alabama at Birmingham, United StatesReviewed by:
Gilles Uzé, Centre National de la Recherche Scientifique (CNRS), FranceMario Santiago, University of Colorado, United States
Copyright © 2020 Fox, Locke and Lenschow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deborah J. Lenschow, ZGxlbnNjaG93QHd1c3RsLmVkdQ==
†These authors have contributed equally to this work