 Vasundhara Sharma1
Vasundhara Sharma1 Gary W. Reuther
Gary W. Reuther
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 30 November 2020
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.604142
This article is part of the Research Topic Genetic and Epigenetic Control on Immune Responses Regulating Molecules in Cancer Development, Progression, and Treatment View all 23 articles
The Janus kinase 2 (JAK2)-driven myeloproliferative neoplasms (MPNs) are associated with clonal myelopoiesis, elevated risk of death due to thrombotic complications, and transformation to acute myeloid leukemia (AML). JAK2 inhibitors improve the quality of life for MPN patients, but these approved therapeutics do not readily reduce the natural course of disease or antagonize the neoplastic clone. An understanding of the molecular and cellular changes requisite for MPN development and progression are needed to develop improved therapies. Recently, murine MPN models were demonstrated to exhibit metabolic vulnerabilities due to a high dependence on glucose. Neoplastic hematopoietic progenitor cells in these mice express elevated levels of glycolytic enzymes and exhibit enhanced levels of glycolysis and oxidative phosphorylation, and the disease phenotype of these MPN model mice is antagonized by glycolytic inhibition. While all MPN-driving mutations lead to aberrant JAK2 activation, these mutations often co-exist with mutations in genes that encode epigenetic regulators, including loss of function mutations known to enhance MPN progression. In this perspective we discuss how altered activity of epigenetic regulators (e.g., methylation and acetylation) in MPN-driving stem and progenitor cells may alter cellular metabolism and contribute to the MPN phenotype and progression of disease. Specific metabolic changes associated with epigenetic deregulation may identify patient populations that exhibit specific metabolic vulnerabilities that are absent in normal hematopoietic cells, and thus provide a potential basis for the development of more effective personalized therapeutic approaches.
Myeloproliferative neoplasms (MPN) are BCR-ABL-negative hematological malignancies where mutations in hematopoietic stem cells (HSCs) give rise to aberrant production of myeloid clones leading to three distinct clinical phenotypes. Polycythemia vera (PV) is characterized by trilineage myeloproliferation and extramedullary hematopoiesis, notably leading to elevated red blood cells and hematocrit, while essential thrombocythemia (ET) is characterized in part by dysregulated megakaryopoiesis. In addition to aberrant myeloid cell production, myelofibrosis (MF) is characterized by reactive fibrosis in the bone marrow (1, 2). MPN patients show various symptoms including spleen and liver enlargement, fatigue, pruritus, fever, night sweats, and bone pain. PV and ET can progress to MF and MF patients are at an increased risk of developing acute myeloid leukemia (AML) and have the poorest survival (2, 3). For this reason, clinical assessment of treatment paradigms in MPN have largely been focused on MF patients (4, 5). Advances in our understanding of MPNs from both clinical and pre-clinical studies have defined cell signaling driving mutations which can co-occur with mutations in other genes, including genes that encode epigenetic regulators, that correlate with differential severity of disease (6). Diverse outcomes and responses to current targeted therapies suggest that multiple factors play prominent, yet undefined, roles in MPN pathology, and thus a better understanding of aberrant cellular processes that contribute to MPN remains needed in order to develop effective therapies. Recent preclinical work has suggested significant alterations in metabolic processes may contribute to the development and progression of MPN (7). Herein we highlight and speculate how MPN driving mutations and epigenetic regulators may contribute to reprogrammed metabolism that may provide potential liabilities that can be exploited by future therapeutic intervention for MPN.
Somatic driver mutations in three genes account for about 90% of all MPN cases. These include mutually exclusive activating mutations in the genes that encode Janus Kinase 2 (JAK2) and the thrombopoietin receptor (encoded by the myeloproliferative leukemia virus gene, MPL), as well as inactivating/neo-functional mutations in the gene that encodes the ER chaperone protein calreticulin (CALR) (6, 8). A gain of function mutation encoding JAK2-V617F is the most commonly occurring single mutation, present in more than 95% of patients with PV and 50% to 60% of patients with ET and PMF. The frequency of frameshift mutations in CALR is about 30% in ET and 25% in PMF. Activating mutations in MPL are present in ∼ 2% to 4% of ET patients and ~ 3% to 5% of PMF patients (6, 8). These mutations are designated drivers of human MPN in part because they induce MPN phenotypes in mouse models (9–11). As a common signaling event induced by MPN driving mutations, JAK2 activation is the critical signaling node in MPN, with MPL playing a requisite role in driving myeloproliferation driven by mutant JAK2 and CALR (12–18).
Deregulated JAK2 signaling in MPNs leads to cytokine hyper-sensitivity and enhanced activity of downstream signaling effectors such as the STAT transcription factors, as well as the ERK/MAPK and the PI3K/AKT pathways which play roles in regulating cell survival, proliferation, and apoptosis (1). Genetic removal of STAT5 impedes the MPN phenotype in MPN mouse models, and small molecule inhibitors of the ERK/MAPK pathway and the PI3K/AKT/mTOR pathway antagonize disease, further defining JAK2-mediated signals as playing important roles in MPN (19–27).
Among the current therapies, only allogenic stem cell transplantation is curative, capable of resolving bone marrow fibrosis and the malignant clone (28). Interferon-alpha is similarly promising, and while initially associated with treatment-limiting toxicities, the development of pegylated-interferon has made cytogenetic remission with this agent a real possibility (29, 30). The JAK1/2 kinase inhibitor ruxolitinib was approved by the United States Food and Drug Administration in 2011 for high risk MF patients and in 2014 for certain PV patients (31–33). Ruxolitinib provides symptomatic relief and can improve survival but generally fails to resolve the malignant clone (33–42). Fedratinib, a JAK2 and FLT3 inhibitor approved in 2019 for higher risk MF patients has similar quality of life improvement benefits as seen with other JAK2 inhibitors, but importantly can be effective in some patients who failed ruxolitinib (43–46). Time will determine the extent to which fedratinib improves critical parameters of MPN and survival. However, results from clinical experience suggest targeting JAK2 may not be sufficient or even the best option to reverse the course of disease in MPN patients, and a plethora of pre-clinical studies have suggested a potential benefit of therapeutic combinations. Many of these studies have focused on combining JAK2 inhibitors with inhibitors of signaling proteins associated with JAK2 activation with the hope of enhancing the efficacy of JAK2 inhibitor mono-therapy, which could provide therapeutic opportunities for additional patients, as well as potentially overcome JAK2 inhibitor resistance. Several of these combinations are being assessed in clinical trials (5).
Commonly co-occurring mutations with MPN drivers are in genes that encode epigenetic regulators (6, 8). While such mutations have contributed to refining MPN patient prognostication, studies have also highlighted the potential of epigenetic regulators, or the pathways they affect, as potential therapeutic targets in MPN (6, 8, 47–51).
Some of these epigenetic regulators include DNMT3A, TET2, EZH2, ASXL1, and IDH1/2 (via effects on TET2-mediated methylation). In some cases, concomitant mutation of these genes with MPN driving mutations can enhance disease phenotypes in MPN mouse models (47, 48, 52–57). These studies suggest mutations, most often loss of function, of these epigenetic regulators may contribute to disease progression in MPN patients. In fact, the prognosis and patient response to ruxolitinib is negatively impacted by the presence of many of these mutations, with worse prognosis associated with a greater numbers of mutations present (58–62).
However, other epigenetic regulators have emerged as possible therapeutic targets in MPN. For example, targeting the activity of LSD1 in mouse models of MPN antagonizes disease development (49). The LSD1 inhibitor IMG-7289 is currently being assessed in myelofibrosis patients. Co-expression of mutant IDH1 or IDH2 with JAK2-V617F enhances MPN progression in mice, and a small molecule IDH inhibitor along with ruxolitinib provides enhanced antagonism of disease (48). Inhibitors of BET proteins, which bind acetylated histones to promote gene expression, are effective in MPN models and cooperate with ruxolitinib by antagonizing pro-inflammatory gene signatures, and such inhibitors are under clinical assessment in MF patients (63–66). The loss of EZH2 promotes the progression of MPN in mouse models and confers enhanced sensitivity of disease to BET inhibition (47). Finally, we have recently shown a role for HDAC11 in MPN, but not normal, hematopoiesis, in one of the first reports that shows isoform selective HDAC targeting specifically impedes malignant hematopoiesis without affecting the steady state and transplantation reconstitution of normal bone marrow cells. This suggests HDAC11 may be a therapeutic target to antagonize MPN with minimal adverse effect on normal hematopoiesis (50).
Understanding specific disease-driving effects of aberrant JAK2 signaling in hematopoietic stem and progenitor cells (HSPCs) may provide critical information regarding targets for novel therapies. Inhibition of JAK2 signaling in MPN mouse models elicits a reduced phenotypic disease burden, but it does not readily eradicate the MPN-driving and MPN-initiating cells, reminiscent of JAK2 inhibitor effects in human MPN (67–69). This suggests the HSPC signaling that drives MPN pathogenesis is resistant to the effects of direct JAK2 inhibition and that further understanding the molecular and cellular forces that drive disease are needed to identify potential vulnerabilities that can be targeted to effectively antagonize disease-driving cells. However, in addition to intrinsic abnormalities in MPN-driving cells, microenvironmental contributions play a significant role to malignant hematopoiesis in MPN, including critical roles of megakaryocytes in shaping a disease-driving hematopoietic compartment (16, 63, 70–76). Thus, understanding both intrinsic and extrinsic contributions to MPN-driving HSPCs may be critical for the development of a therapy that effectively induces remission from the neoplastic MPN clone.
Disease-driving HSPCs reside in bone marrow niche microenvironments that become remodeled during malignancy and aging (77). Characteristic of leukemic stem cell survival and expansion are metabolic changes that lead to altered sources of energy production (77, 78). For example, AML leukemia stem cells, unlike leukemic blast cells, depend on oxidative phosphorylation driven by amino acid metabolism, creating a significant therapeutic vulnerability for this disease-driving cell populations (79, 80). Interestingly, this dependency was lost in leukemia stem cells from relapsed patients, where fatty acid metabolism provided the requisite energy source, demonstrating that metabolic changes may drive malignancy and relapse (80). Perhaps more relevant to chronic phase MPN is the report of HSPC subsets that exhibit increased glycolysis associated with myeloid skewing in older people, and the possibility that this may correlate with the natural clonal hematopoiesis associated with aging (81, 82). In addition to JAK2 mutations in DNMT3A, TET2, and ASXL1 are amongst the most commonly detected mutations associated with aging-dependent clonal hematopoiesis in populations not exhibiting hematologic malignancy (83–86). The role of metabolic reprogramming in MPN stem and progenitor cells is not well understood, although several studies have provided important observations.
The JAK2-V617F MPN-driving kinase was shown to enhance the expression the glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) (87). PFKFB3 regulates 6-phosphofructo-1-kinase activity which is a rate limiting enzyme of glycolysis (88). JAK2-V617F-expressing cells exhibited enhanced glucose uptake which correlated with elevated levels of the glucose transporter Glut1, both of which were shown to be under the control of the activity of JAK2-V617F (87). This study demonstrated that JAK2-V617F-dependent regulation of glucose uptake and PFKFB3 have the potential to contribute to enhanced lactate production and metabolic activity. Importantly, significantly elevated PFKFB3 mRNA levels were detected in PV patients compared to healthy controls. The authors demonstrated that PFKB3 expression and activity were required for optimal growth of JAK2-V617F-expressing cells in vitro and as tumors in mice (87). Another study evaluated the previously known increased dependence of cancer cells on free amino acids such as glutamine (89, 90). This study indicated glutaminase (GLS), an enzyme that converts glutamine into glutamate, was not found to be upregulated in primary MPN cells but provides some evidence that targeting its activity may enhance the effect of JAK2 inhibition (91). While these studies provide initial insight into the potential metabolic enzymes may provide a target to improve MPN therapies, they were mostly limited to cellular models.
More recently, Rao et al. elegantly described a reprogramming of metabolic activity in mouse models of JAK2 mutant MPN (7). This report demonstrated that elevated expression of glycolytic enzymes in stem and progenitor cells of JAK2-mutant MPN mice correlated with enhanced glucose uptake, glycolysis and oxidative phosphorylation as well as use of the pentose phosphate pathway (7) (Figure 1A). In fact, enhanced erythropoiesis was so highly dependent on glucose that JAK2-mutant MPN mice became hypoglycemic with disease progression, leading to a state of “energy crisis” that likely contributed to the observed elevation of lipid catabolism. Interestingly, the survival of JAK2-exon 12 mutant MPN model mice was significantly lengthened by a high fat diet, suggesting disease could be impacted by the nutritional energy source. Rao et al. also demonstrated that PFKFB3 was elevated in cells from JAK2-mutant mice. The PFKFB3 inhibitor 3-PO induced apoptosis in MPN model cell lines and primary cells from patients, but only showed additive effects with ruxolitinib in cell lines. Similarly, therapeutic treatment of JAK2-mutant MPN mice with 3-PO modestly antagonized the MPN phenotype, and only showed modest effects in combination with ruxolitinib. Upregulation of proteins that promote enhanced glycolysis may be regulated by HIF1-α, which could provide a therapeutic target to antagonize metabolic dependencies in MPN-driving cells (93). MPN patients often gain weight during ruxolitinib treatment and Rao et al. also suggests PV patients undergoing cytoreductive therapy display increased blood sugar levels (7). Although many factors many contribute to such observations, such as effects on leptin signaling due to JAK2 inhibition or perhaps increased appetite due to reduced splenomegaly, these observations suggest a link between altered metabolic stasis and MPN (94). Ruxolitinib treatment may antagonize enhanced metabolism in MPN progenitor cells, leading to an imbalance of caloric intake and utilization, leading to weight gain (95). Targeting metabolic processes to selectively antagonize malignant progenitor cells thus could have other health concerns for patients. Nonetheless the study by Rao et al. suggests that aberrant metabolic activity of disease-driving MPN progenitor cells may provide a therapeutically targetable liability that may spare cells that aren’t dependent on aberrant metabolic regulation (7).
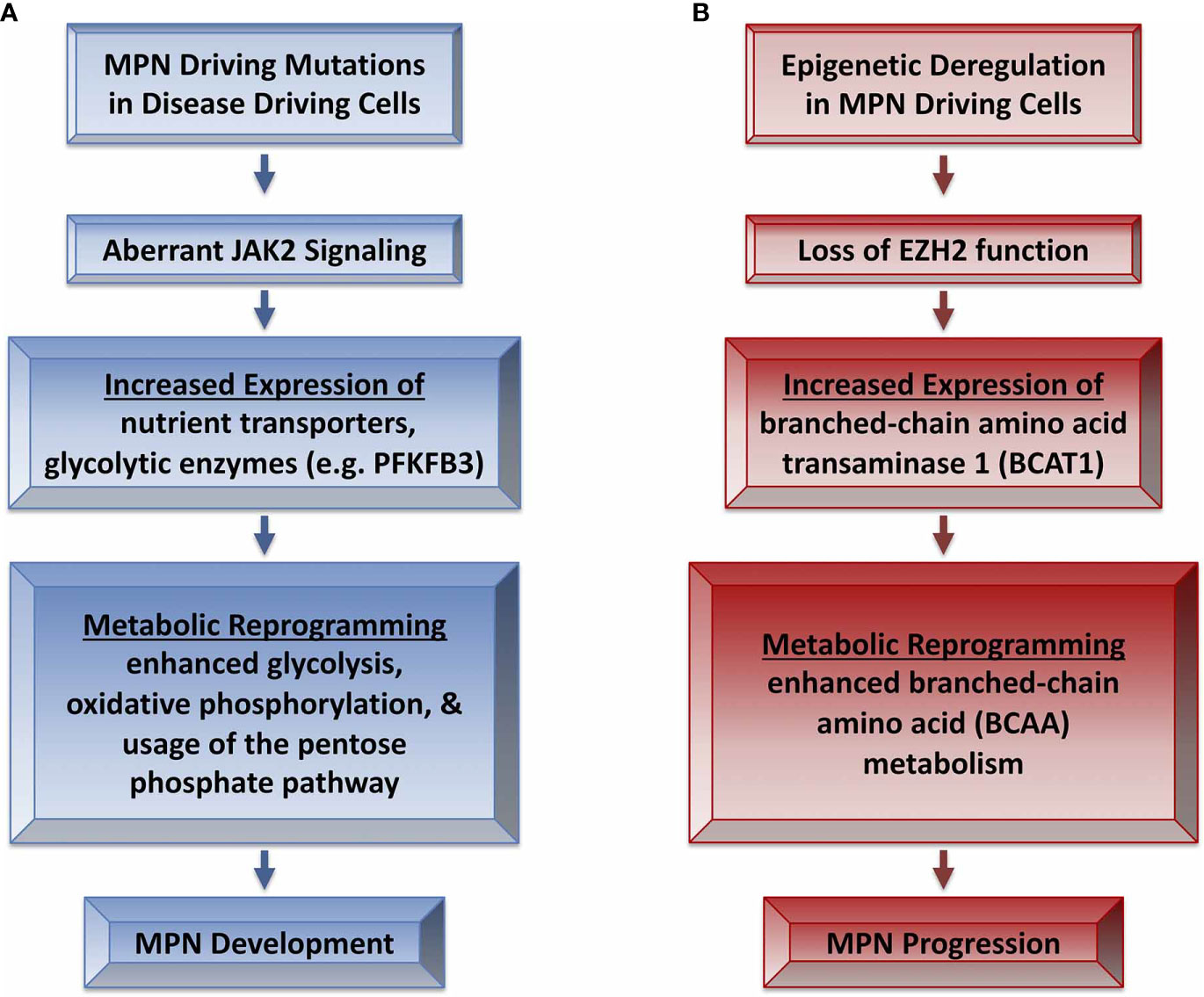
Figure 1 Metabolic reprogramming in MPN development and progression. (A) MPN driving mutations lead to aberrant JAK2 signaling that leads to metabolic reprogramming and MPN development (7). (B) Epigenetic alterations such as loss of EZH2 function in MPN may contribute to progression of disease severity via reprogramming of BCAA metabolism (92).
Numerous signaling pathways activated by JAK2 could impinge on metabolic control in MPN. Induction of PFKFB3 and glycolysis by JAK2-V617F is mediated by STAT5 activation, but other pathways likely contribute to metabolic changes induced by aberrant JAK2 signaling (87). Notably, the mTOR pathway, a well-known regulator of metabolic processes, is regulated by PI3K/AKT signaling, a downstream effector pathway of JAK2 (1, 96). mTOR inhibitors are effective in MPN models, but clinical assessment has not gained much traction, with a more recent focus on PI3K inhibitors, which target mTOR activating signals (5, 19, 23–26, 96, 97). Such signaling pathway inhibitors of course would not affect metabolism specifically. Further understanding of the deregulated metabolic processes that contribute to driving MPN is needed to determine the extent to which these could be targeted more directly, for example, with specific metabolic enzyme inhibitors.
Evidence suggests that deregulated metabolic programs are manifested by altered regulation of gene expression of key proteins in glycolytic processes as well changes of lipid and amino acid metabolism (98–100). Metabolic changes induced in MPN model mice suggest aberrant JAK2 signaling may drive these effects (7). However, other mutations or deregulated signaling associated with MPN may contribute to altered metabolic pathways in MPN driving cells. For example, recent studies have suggested altered epigenetic control of methylation and acetylation have the potential to contribute to the control of metabolic processes, such as amino acid metabolism and glycolysis in MPN.
EZH2, as the enzymatic component of the polycomb repressive complex-2 (PRC2), catalyzes methylation of H3K27 which leads to suppression of gene expression (101). The frequency (~5–10%) of inactivating EZH2 mutations found in MF suggests loss of EZH2-mediated methylation contributes to neoplastic disease (6, 60, 102, 103). Genetic loss of EZH2 leads to a more advanced MPN phenotype and decreased survival in MPN mouse models, suggesting EZH2 plays a tumor suppressive role in MPNs (47, 55, 56, 92).
EZH2 loss/inactivation in an N-RAS-driven mouse model of MPN led to enhanced branched-chain amino acid metabolism (BCAA) during the progression of disease (92). This is consistent with a known role of EZH2 in the regulation of metabolic processes such as glucose, fatty acid, and amino acid metabolism in cancer cells (104). BCAA metabolism plays roles in a variety of cancer types and has been identified as a metabolic vulnerability in myeloid leukemia (105, 106). BCAA metabolism is regulated by branched chain amino acid transaminase 1 (BCAT1) in reversible reactions generating branch-chain alpha-keto acids and glutamate from BCAAs and alpha-ketoglutarate (α-KG) (105). The loss of EZH2 led to the loss of repressive methylation of the BCAT1 promoter, leading to enhanced BCAT1 expression, sustained BCAA levels, progression of MPN (e.g., enhanced myelofibrosis) and transformation to AML (92) (Figure 1B). BCAT1 is generally low or not expressed in hematopoietic cells and exogenous expression of BCAT1 mimics the effects of the loss of EZH2 on leukemia initiating cells (92, 106). Importantly, leukemia initiating cells that lacked EZH2 were more sensitive to BCAT1 inhibition than normal HSPCs, which are unaffected by loss of BCAT1 (92). Thus, the loss of EZH2 can drive MPN severity and the aberrant control of post-MPN leukemic-initiating cells via BCAT1-mediated alteration of metabolic processes. Loss of function EZH2 mutations in human MPN are associated with higher levels of BCAT1 mRNA compared to patients with wildtype EZH2 (92). Therefore, the loss of EZH2-mediated inhibition of BCAT1 expression may contribute to altered metabolic profiles in MPN-driving cells, which may explain the poor prognosis of MPN patients who harbor EZH2 mutations (60).
The increase in BCAT1 levels associated with inactivating mutations of EZH2 could contribute to the available pool of α-KG, an important component of the TCA cycle that also functions as a co-factor for the TET2 methylcytosine dioxygenase that promotes DNA demethylation (107, 108). Loss of function mutations in TET2 are present in 10% to 20% of MPN patients where they contribute to clonal dominance and disease initiation and associate with poor outcomes (6, 61, 109, 110). Loss of TET2 leads to distinct gene expression profiles and enhances disease in an MPN mouse model (53). Likewise, IDH1/2 mutations lead to the production of 2-hydroxyglutarate instead of α-KG, which effectively inhibits the function of TET2, and thus such mutations mimic loss of function of TET2 (111, 112). This is a clear example of the interconnectivity between alterations in metabolic pathways leading to deregulation of epigenetic regulatory mechanisms in cancer. Mutations of IDH1/2 are present in 1% to 3% of MPN patients, are enriched in post-MPN leukemia (~20%) where they associate with poor survival, and enhance the progression of disease in MPN model mice (48, 62). In the MPN model driven by activated N-RAS and loss of EZH2, with subsequent elevation of BCAT1, changes in cellular α-KG levels were not detected (92). However, BCAT1 expression in non IDH or TET2 mutant AML stem cells correlates with decreased α-KG levels and gene expression profiles similar to IDH mutant cells, suggesting deregulation of methylation regulated by TET2 (113). Whether BCAT1 levels affect the activity of TET2-mediated regulation of DNA methylation and gene expression in MPN patients is unknown.
Loss of EZH2 function has been proposed to play a role in the effects elicited by other mutations found in MPN, including ASXL1 and SRSF2, suggesting EZH2 loss of function may contribute to the biology of disease in patients beyond those that have EZH2 mutations (57, 114). As normal HSPCs do not require BCAT1 and these cells have low BCAT1 expression, BCAT1 levels may define subsets of MPN patients that could exhibit BCAT1-driven therapeutic liabilities. BCAT1 inhibition could provide a therapeutic strategy for MPN patients who have elevated BCAT1 (e.g., patients with EZH2, and possibly ASXL1 and SRSF2 mutations) to shift malignant myeloproliferative hematopoiesis back to normal hematopoiesis.
Moreover, the associated decrease in H3K27 methylation with EZH2 loss of function leads to an increase in H3K27 acetylation, providing binding sites for BRD4 and enhancement of gene expression. This epigenetic switch reverts a transcriptional inhibitory mark (methylation) to an activation mark (acetylation), effectively creating a synthetic lethal interaction between loss of function EZH2 mutations and BET inhibition in mouse models (47). As such, loss of EZH2 function in MPN patients may provide enhanced sensitivity to BET inhibitors, which are currently in clinical testing for myelofibrosis patients. It will be interesting to see the relative bromodomain inhibitor responses of MPN or post-MPN AML patients who have inactivating EZH2 mutations, or ASXL1 or SRSF2 mutations which antagonize PRC2/EZH2 function (57, 114).
Finally, BCAA metabolism has recently been associated with tyrosine kinase inhibitor resistance in lung cancer, suggesting such metabolic reprogramming may also contribute to the inefficacy or resistance to JAK2 inhibition (115). In fact, MPN patients with multiple mutations, including in EZH2 and ASXL1, in addition to a JAK2 activating mutation tend to respond poorly to JAK2 inhibitor therapy (59). The possibility exists that BCAA metabolism may affect response to JAK2 inhibition or may contribute to an adaptive response in cells exposed to chronic JAK2 inhibition. If so, inhibition of BCAA metabolism may enhance the efficacy of current targeted therapies in subsets of MPN patients.
Protein lysine acetylation is not limited to histones as it is a key modification found throughout the human proteome, affecting most major cellular processes including metabolism (116–118). One study indicates that almost every enzyme within major metabolic pathways is acetylated, clearly suggesting a likely role of acetylation in metabolic control (118). For example, phosphoenolpyruvate carboxykinase is a gluconeogenic enzyme whose protein stability and levels are antagonized by acetylation (118). Given the breadth of the acetylated proteome and the non-selectivity of clinically assessed HDAC inhibitors, it is likely specific functions of individual HDAC family members need to be identified, along with the development of HDAC-isoform specific inhibitors, in order to selectively target HDACs to impede specific regulatory mechanisms of acetylation. While pan-HDAC inhibitors (e.g., vorinostat, givinostat, pracinostat, panobinostat) have been assessed in clinical trials and some have displayed efficacy in MPN patients, toxicity concerns may limit their potential (119–126). This suggests the development of more specific HDAC inhibitors may minimize adverse effects and allow this class of epigenetic regulators to be therapeutically targeted.
Recent reports have also highlighted a significant role for HDAC11, which has roles in immune tolerance, in regulating metabolic processes (127–130). We, along with our collaborators, demonstrated a role for HDAC11 in HSPCs in a transplantation mouse model of MPN driven by MPL-W515L. Data obtained using HDAC11-null mice and MPN patient samples treated with selective HDAC11 inhibitors suggest that HDAC11 contributes to the neoplastic nature of MPN cells but not normal hematopoiesis (50). Subsequent studies using global acetylomic profiling following HDAC11 inhibition identified glycolytic enzymes (e.g., enolase-1 (ENO-1)) as potential substrates of HDAC11. Pharmacological inhibition as well as knockdown of HDAC11 increased the acetylation of ENO-1, decreased the activity of ENO-1, and reduced the rate of glycolysis as well as oxidative phosphorylation in MPN model cell lines and primary cells from MPN patients but not healthy controls (131). These observations are supported by previous studies linking acetylation to metabolic processes and potentially provide HDAC11-specific functions in regulating metabolic pathways (118).
Therefore, HDAC11 may contribute to the MPN-driver associated metabolic changes that contribute to MPN pathogenesis (7). However, specific metabolic effects of HDAC11 depletion in MPN mouse models have yet to be determined. Similarly, the efficacy of a selective anti-HDAC11 therapeutic in such models and the effects of such a therapeutic on the metabolic profiles of MPN-driving cells are unknown. Continued development of such inhibitors are required to ascertain the extent to which HDAC11 inhibition may provide a therapeutic option to disrupt control of cellular processes requisite for MPN-driving HSPCs, and to determine if the observed metabolic vulnerabilities that have been identified in MPN-driving HSPCs are regulated by HDAC11 (7). HDAC11 also displays a recently identified deacylase activity and the role of this activity in MPN formation is unknown (132–134). Interestingly, HDAC11 was the most highly induced HDAC in response to a variety of HDAC inhibitors in AML cells, suggesting it indeed may have unique and critical properties compared to other HDAC family members (135). HDAC11 inhibition has been suggested to overcome therapy resistance in lung cancer models, and while its role in resistance to targeted therapies such as approved JAK2 inhibitors in MPN is unknown, current data suggest potential for the combination of JAK2 inhibition and HDAC11 inhibition as a possible future therapeutic strategy (136).
MPN-driver mutations enhance JAK2 signaling which promotes neoplastic HSPC expansion, and altered epigenetic control mechanisms play an etiologic role in the development and progression of MPN. Recent studies provide evidence that altered metabolic control may play an important role in MPN and that altered epigenetic regulation may contribute to neoplastic metabolic profiles. Deregulated metabolic processes that support and drive MPN phenotypes may reveal novel vulnerabilities that can be targeted to suppress the malignant clone while sparing healthy HSPCs. The role of disease-associated metabolic states in the upfront inefficacy of JAK2 inhibitors, as well as whether or not JAK2 inhibitor therapy induces changes in metabolic states that contribute to JAK2 inhibitor failure, are questions that remain important to address. Further understanding the metabolic profiles of MPN HSPC clones in MPN subtypes, genotypes, responses to JAK2 inhibitor therapy, and disease progression are required in order to understand the potential relationship between deregulated epigenetics and metabolism in MPN, which could lead to the development of much needed remission-inducing personalized therapies for patients.
All authors contributed to the article and approved the submitted version.
GR receives research funding from Incyte Corporation and Revolution Medicines, Inc. for projects not related to this manuscript. PE-B received research funding from Incyte Corporation and Forma Therapeutics. PE-B is the Chief Scientific Officer of Ibis Therapeutics and works on drug development unrelated to this manuscript. KW received research funding from Forma Therapeutics and Tolero Pharmaceuticals.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer (2007) 7(9):673–83. doi: 10.1038/nrc2210
2. Tefferi A. Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am J Hematol (2018) 93(12):1551–60. doi: 10.1002/ajh.25230
3. Tefferi A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood (2014) 124(16):2507–13; quiz 2615. doi: 10.1182/blood-2014-05-579136
4. Asher S, McLornan DP, Harrison CN. Current and future therapies for myelofibrosis. Blood Rev (2020) 42:100715. doi: 10.1016/j.blre.2020.100715
5. Kuykendall AT, Horvat NP, Pandey G, Komrokji R, Reuther GW. Finding a Jill for JAK: Assessing Past, Present, and Future JAK Inhibitor Combination Approaches in Myelofibrosis. Cancers (Basel) (2020) 12(8):2278. doi: 10.3390/cancers12082278
6. Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood (2017) 129(6):667–79. doi: 10.1182/blood-2016-10-695940
7. Rao TN, Hansen N, Hilfiker J, Rai S, Majewska JM, Leković D, et al. JAK2 mutant hematopoietic cells display metabolic alterations that can be targeted to treat myeloproliferative neoplasms. Blood (2019) 134(21):1832–46. doi: 10.1182/blood.2019000162
8. Grabek J, Straube J, Bywater M, Lane SW. MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and their Effect on Treatment. Cells (2020) 9(8):1901. doi: 10.3390/cells9081901
9. Dunbar A, Nazir A, Levine R. Overview of Transgenic Mouse Models of Myeloproliferative Neoplasms (MPNs). Curr Protoc Pharmacol (2017) 77:14.40.11–14.40.19. doi: 10.1002/cpph.23
10. Jacquelin S, Kramer F, Mullally A, Lane SW. Murine Models of Myelofibrosis. Cancers (Basel) (2020) 12(9):2381. doi: 10.3390/cancers12092381
11. Mullally A, Lane SW, Brumme K, Ebert BL. Myeloproliferative neoplasm animal models. Hematol Oncol Clin North Am (2012) 26(5):1065–81. doi: 10.1016/j.hoc.2012.07.007
12. Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood (2014) 123(22):e123–33. doi: 10.1182/blood-2014-02-55463
13. Marty C, Pecquet C, Nivarthi H, El-Khoury M, Chachoua I, Tulliez M, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood (2016) 127(10):1317–24. doi: 10.1182/blood-2015-11-679571
14. Chachoua I, Pecquet C, El-Khoury M, Nivarthi H, Albu RI, Marty C, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood (2016) 127(10):1325–35. doi: 10.1182/blood-2015-11-681932
15. Elf S, Abdelfattah NS, Chen E, Perales-Paton J, Rosen EA, Ko A, et al. Mutant Calreticulin Requires Both Its Mutant C-terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov (2016) 6(4):368–81. doi: 10.1158/2159-8290.CD-15-1434
16. Zhang Y, Lin CHS, Kaushansky K, Zhan H. JAK2V617F Megakaryocytes Promote Hematopoietic Stem/Progenitor Cell Expansion in Mice Through Thrombopoietin/MPL Signaling. Stem Cells (2018) 36(11):1676–84. doi: 10.1002/stem.2888
17. Vainchenker W, Plo I, Marty C, Varghese LN, Constantinescu SN. The role of the thrombopoietin receptor MPL in myeloproliferative neoplasms: recent findings and potential therapeutic applications. Expert Rev Hematol (2019) 12(6):437–48. doi: 10.1080/17474086.2019.1617129
18. Spivak JL, Merchant A, Williams DM, Rogers O, Zhao W, Duffield A, et al. Thrombopoietin is required for full phenotype expression in a JAK2V617F transgenic mouse model of polycythemia vera. PLoS One (2020) 15(6):e0232801. doi: 10.1371/journal.pone.0232801
19. Bartalucci N, Guglielmelli P, Vannucchi AM. Rationale for targeting the PI3K/Akt/mTOR pathway in myeloproliferative neoplasms. Clin Lymphoma Myeloma Leuk (2013) 13(Suppl 2):S307–309. doi: 10.1016/j.clml.2013.07.011
20. Stivala S, Codilupi T, Brkic S, Baerenwaldt A, Ghosh N, Hao-Shen H, et al. Targeting compensatory MEK/ERK activation increases JAK inhibitor efficacy in myeloproliferative neoplasms. J Clin Invest (2019) 129(4):1596–611. doi: 10.1172/JCI98785
21. Walz C, Ahmed W, Lazarides K, Betancur M, Patel N, Hennighausen L, et al. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood (2012) 119(15):3550–60. doi: 10.1182/blood-2011-12-397554
22. Yan D, Hutchison RE, Mohi G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood (2012) 119(15):3539–49. doi: 10.1182/blood-2011-03-345215
23. Bartalucci N, Calabresi L, Balliu M, Martinelli S, Rossi MC, Villeval JL, et al. Inhibitors of the PI3K/mTOR pathway prevent STAT5 phosphorylation in JAK2V617F mutated cells through PP2A/CIP2A axis. Oncotarget (2017) 8(57):96710–24. doi: 10.18632/oncotarget.18073
24. Bartalucci N, Tozzi L, Bogani C, Martinelli S, Rotunno G, Villeval JL, et al. Co-targeting the PI3K/mTOR and JAK2 signalling pathways produces synergistic activity against myeloproliferative neoplasms. J Cell Mol Med (2013) 17(11):1385–96. doi: 10.1111/jcmm.12162
25. Bogani C, Bartalucci N, Martinelli S, Tozzi L, Guglielmelli P, Bosi A, et al. mTOR inhibitors alone and in combination with JAK2 inhibitors effectively inhibit cells of myeloproliferative neoplasms. PLoS One (2013) 8(1):e54826. doi: 10.1371/journal.pone.0054826
26. Fiskus W, Verstovsek S, Manshouri T, Smith JE, Peth K, Abhyankar S, et al. Dual PI3K/AKT/mTOR inhibitor BEZ235 synergistically enhances the activity of JAK2 inhibitor against cultured and primary human myeloproliferative neoplasm cells. Mol Cancer Ther (2013) 12(5):577–88. doi: 10.1158/1535-7163.Mct-12-0862
27. Khan I, Huang Z, Wen Q, Stankiewicz MJ, Gilles L, Goldenson B, et al. AKT is a therapeutic target in myeloproliferative neoplasms. Leukemia (2013) 27(9):1882–90. doi: 10.1038/leu.2013.167
28. McLornan DP, Yakoub-Agha I, Robin M, Chalandon Y, Harrison CN, Kroger N. State-of-the-art review: allogeneic stem cell transplantation for myelofibrosis in 2019. Haematologica (2019) 104(4):659–68. doi: 10.3324/haematol.2018.206151
29. Hasselbalch HC, Holmström MO. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: minimal residual disease and cure? Semin Immunopathol (2019) 41(1):5–19. doi: 10.1007/s00281-018-0700-2
30. How J, Hobbs G. Use of Interferon Alfa in the Treatment of Myeloproliferative Neoplasms: Perspectives and Review of the Literature. Cancers (Basel) (2020) 12(7):1954. doi: 10.3390/cancers12071954
31. Mascarenhas J, Hoffman R. Ruxolitinib: the first FDA approved therapy for the treatment of myelofibrosis. Clin Cancer Res (2012) 18(11):3008–14. doi: 10.1158/1078-0432.CCR-11-3145
32. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med (2012) 366(9):799–807. doi: 10.1056/NEJMoa1110557
33. Vainchenker W, Leroy E, Gilles L, Marty C, Plo I, Constantinescu SN. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Res (2018) 7:82. doi: 10.12688/f1000research.13167.1
34. Deininger M, Radich J, Burn TC, Huber R, Paranagama D, Verstovsek S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood (2015) 126(13):1551–4. doi: 10.1182/blood-2015-03-635235
35. Verstovsek S, Mesa RA, Gotlib J, Gupta V, DiPersio JF, Catalano JV, et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol (2017) 10(1):55. doi: 10.1186/s13045-017-0417-z
36. Guglielmelli P, Biamonte F, Rotunno G, Artusi V, Artuso L, Bernardis I, et al. Impact of mutational status on outcomes in myelofibrosis patients treated with ruxolitinib in the COMFORT-II study. Blood (2014) 123(14):2157–60. doi: 10.1182/blood-2013-11-536557
37. Pardanani A, Tefferi A. Targeting myeloproliferative neoplasms with JAK inhibitors. Curr Opin Hematol (2011) 18(2):105–10. doi: 10.1097/MOH.0b013e3283439964
38. Tefferi A, Pardanani A. Myeloproliferative Neoplasms: A Contemporary Review. JAMA Oncol (2015) 1(1):97–105. doi: 10.1001/jamaoncol.2015.89
39. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. Efficacy, safety, and survival with ruxolitinib in patients with myelofibrosis: results of a median 3-year follow-up of COMFORT-I. Haematologica (2015) 100(4):479–88. doi: 10.3324/haematol.2014.115840
40. Vannucchi AM, Kantarjian HM, Kiladjian JJ, Gotlib J, Cervantes F, Mesa RA, et al. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica (2015) 100(9):1139–45. doi: 10.3324/haematol.2014.119545
41. Harrison CN, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Gisslinger H, Knoops L, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia (2016) 30(8):1701–7. doi: 10.1038/leu.2016.148
42. Verstovsek S, Gotlib J, Mesa RA, Vannucchi AM, Kiladjian JJ, Cervantes F, et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J Hematol Oncol (2017) 10(1):156. doi: 10.1186/s13045-017-0527-7
43. Bose P, Verstovsek S. Management of myelofibrosis after ruxolitinib failure. Leuk Lymphoma (2020) 61(8):1797–809. doi: 10.1080/10428194.2020.1749606
44. Harrison CN, Schaap N, Mesa RA. Management of myelofibrosis after ruxolitinib failure. Ann Hematol (2020) 99(6):1177–91. doi: 10.1007/s00277-020-04002-9
45. Blair HA. Fedratinib: First Approval. Drugs (2019) 79(15):1719–25. doi: 10.1007/s40265-019-01205-x
46. Harrison CN, Schaap N, Vannucchi AM, Kiladjian JJ, Tiu RV, Zachee P, et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol (2017) 4(7):e317–24. doi: 10.1016/S2352-3026(17)30088-1
47. Sashida G, Wang C, Tomioka T, Oshima M, Aoyama K, Kanai A, et al. The loss of Ezh2 drives the pathogenesis of myelofibrosis and sensitizes tumor-initiating cells to bromodomain inhibition. J Exp Med (2016) 213(8):1459–77. doi: 10.1084/jem.20151121
48. McKenney AS, Lau AN, Somasundara AVH, Spitzer B, Intlekofer AM, Ahn J, et al. JAK2/IDH-mutant-driven myeloproliferative neoplasm is sensitive to combined targeted inhibition. J Clin Invest (2018) 128(2):789–804. doi: 10.1172/JCI94516
49. Jutzi JS, Kleppe M, Dias J, Staehle HF, Shank K, Teruya-Feldstein J, et al. LSD1 Inhibition Prolongs Survival in Mouse Models of MPN by Selectively Targeting the Disease Clone. Hemasphere (2018) 2(3):e54. doi: 10.1097/HS9.0000000000000054
50. Yue L, Sharma V, Horvat NP, Akuffo AA, Beatty MS, Murdun C, et al. HDAC11 deficiency disrupts oncogene-induced hematopoiesis in myeloproliferative neoplasms. Blood (2020) 135(3):191–207. doi: 10.1182/blood.2019895326
51. Pastore F, Bhagwat N, Pastore A, Radzisheuskaya A, Karzai A, Krishnan A, et al. PRMT5 inhibition modulates E2F1 methylation and gene regulatory networks leading to therapeutic efficacy in JAK2V617F mutant MPN. Cancer Discov (2020) 10(11):1742–57. doi: 10.1158/2159-8290.CD-20-0026
52. Jacquelin S, Straube J, Cooper L, Vu T, Song A, Bywater M, et al. Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood (2018) 132(26):2707–21. doi: 10.1182/blood-2018-04-846220
53. Chen E, Schneider RK, Breyfogle LJ, Rosen EA, Poveromo L, Elf S, et al. Distinct effects of concomitant Jak2V617F expression and Tet2 loss in mice promote disease progression in myeloproliferative neoplasms. Blood (2015) 125(2):327–35. doi: 10.1182/blood-2014-04-567024
54. Kameda T, Shide K, Yamaji T, Kamiunten A, Sekine M, Taniguchi Y, et al. Loss of TET2 has dual roles in murine myeloproliferative neoplasms: disease sustainer and disease accelerator. Blood (2015) 125(2):304–15. doi: 10.1182/blood-2014-04-555508
55. Yang Y, Akada H, Nath D, Hutchison RE, Mohi G. Loss of Ezh2 cooperates with Jak2V617F in the development of myelofibrosis in a mouse model of myeloproliferative neoplasm. Blood (2016) 127(26):3410–23. doi: 10.1182/blood-2015-11-679431
56. Shimizu T, Kubovcakova L, Nienhold R, Zmajkovic J, Meyer SC, Hao-Shen H, et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J Exp Med (2016) 213(8):1479–96. doi: 10.1084/jem.20151136
57. Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell (2012) 22(2):180–93. doi: 10.1016/j.ccr.2012.06.032
58. Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia (2013) 27(9):1861–9. doi: 10.1038/leu.2013.119
59. Patel KP, Newberry KJ, Luthra R, Jabbour E, Pierce S, Cortes J, et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood (2015) 126(6):790–7. doi: 10.1182/blood-2015-03-633404
60. Guglielmelli P, Biamonte F, Score J, Hidalgo-Curtis C, Cervantes F, Maffioli M, et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood (2011) 118(19):5227–34. doi: 10.1182/blood-2011-06-363424
61. Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood (2014) 123(14):2220–8. doi: 10.1182/blood-2013-11-537167
62. Tefferi A, Lasho TL, Abdel-Wahab O, Guglielmelli P, Patel J, Caramazza D, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia (2010) 24(7):1302–9. doi: 10.1038/leu.2010.113
63. Kleppe M, Koche R, Zou L, van Galen P, Hill CE, Dong L, et al. Dual Targeting of Oncogenic Activation and Inflammatory Signaling Increases Therapeutic Efficacy in Myeloproliferative Neoplasms. Cancer Cell (2018) 33(1):29–43.e27. doi: 10.1016/j.ccell.2017.11.009
64. Saenz DT, Fiskus W, Manshouri T, Rajapakshe K, Krieger S, Sun B, et al. BET protein bromodomain inhibitor-based combinations are highly active against post-myeloproliferative neoplasm secondary AML cells. Leukemia (2017) 31(3):678–87. doi: 10.1038/leu.2016.260
65. Saenz DT, Fiskus W, Qian Y, Manshouri T, Rajapakshe K, Raina K, et al. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia (2017) 31(9):1951–61. doi: 10.1038/leu.2016.393
66. Wyspiańska BS, Bannister AJ, Barbieri I, Nangalia J, Godfrey A, Calero-Nieto FJ, et al. BET protein inhibition shows efficacy against JAK2V617F-driven neoplasms. Leukemia (2014) 28(1):88–97. doi: 10.1038/leu.2013.234
67. Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell (2010) 17(6):584–96. doi: 10.1016/j.ccr.2010.05.015
68. Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell (2008) 13(4):311–20. doi: 10.1016/j.ccr.2008.02.009
69. Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood (2010) 115(25):5232–40. doi: 10.1182/blood-2009-05-223727
70. Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell (2013) 13(3):285–99. doi: 10.1016/j.stem.2013.06.009
71. Arranz L, Sánchez-Aguilera A, Martín-Pérez D, Isern J, Langa X, Tzankov A, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature (2014) 512(7512):78–81. doi: 10.1038/nature13383
72. Mager LF, Riether C, Schürch CM, Banz Y, Wasmer MH, Stuber R, et al. IL-33 signaling contributes to the pathogenesis of myeloproliferative neoplasms. J Clin Invest (2015) 125(7):2579–91. doi: 10.1172/JCI77347
73. Kleppe M, Kwak M, Koppikar P, Riester M, Keller M, Bastian L, et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov (2015) 5(3):316–31. doi: 10.1158/2159-8290.CD-14-0736
74. Koschmieder S, Mughal TI, Hasselbalch HC, Barosi G, Valent P, Kiladjian JJ, et al. Myeloproliferative neoplasms and inflammation: whether to target the malignant clone or the inflammatory process or both. Leukemia (2016) 30(5):1018–24. doi: 10.1038/leu.2016.12
75. Woods B, Chen W, Chiu S, Marinaccio C, Fu C, Gu L, et al. Activation of JAK/STAT Signaling in Megakaryocytes Sustains Myeloproliferation In Vivo. Clin Cancer Res (2019) 25(19):5901–12. doi: 10.1158/1078-0432.CCR-18-4089
76. Zhan H, Ma Y, Lin CH, Kaushansky K. JAK2(V617F)-mutant megakaryocytes contribute to hematopoietic stem/progenitor cell expansion in a model of murine myeloproliferation. Leukemia (2016) 30(12):2332–41. doi: 10.1038/leu.2016.114
77. Mendez-Ferrer S, Bonnet D, Steensma DP, Hasserjian RP, Ghobrial IM, Gribben JG, et al. Bone marrow niches in haematological malignancies. Nat Rev Cancer (2020) 20(5):285–98. doi: 10.1038/s41568-020-0245-2
78. Rashkovan M, Ferrando A. Metabolic dependencies and vulnerabilities in leukemia. Genes Dev (2019) 33(21-22):1460–74. doi: 10.1101/gad.326470.119
79. Jones CL, Stevens BM, D’Alessandro A, Culp-Hill R, Reisz JA, Pei S, et al. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood (2019) 134(4):389–94. doi: 10.1182/blood.2019898114
80. Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell (2018) 34(5):724–40.e724. doi: 10.1016/j.ccell.2018.10.005
81. Hennrich ML, Romanov N, Horn P, Jaeger S, Eckstein V, Steeples V, et al. Cell-specific proteome analyses of human bone marrow reveal molecular features of age-dependent functional decline. Nat Commun (2018) 9(1):4004. doi: 10.1038/s41467-018-06353-4
82. Poisa-Beiro L, Thoma J, Landry J, Sauer S, Yamamoto A, Eckstein V, et al. Glycogen accumulation, central carbon metabolism, and aging of hematopoietic stem and progenitor cells. Sci Rep (2020) 10(1):11597. doi: 10.1038/s41598-020-68396-2
83. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med (2014) 20(12):1472–8. doi: 10.1038/nm.3733
84. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med (2014) 371(26):2488–98. doi: 10.1056/NEJMoa1408617
85. Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med (2014) 371(26):2477–87. doi: 10.1056/NEJMoa1409405
86. McKerrell T, Park N, Moreno T, Grove CS, Ponstingl H, Stephens J, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep (2015) 10(8):1239–45. doi: 10.1016/j.celrep.2015.02.005
87. Reddy MM, Fernandes MS, Deshpande A, Weisberg E, Inguilizian HV, Abdel-Wahab O, et al. The JAK2V617F oncogene requires expression of inducible phosphofructokinase/fructose-bisphosphatase 3 for cell growth and increased metabolic activity. Leukemia (2012) 26(3):481–9. doi: 10.1038/leu.2011.225
88. Yi M, Ban Y, Tan Y, Xiong W, Li G, Xiang B. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 and 4: A pair of valves for fine-tuning of glucose metabolism in human cancer. Mol Metab (2019) 20:1–13. doi: 10.1016/j.molmet.2018.11.013
89. DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene (2010) 29(3):313–24. doi: 10.1038/onc.2009.358
90. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A (2008) 105(48):18782–7. doi: 10.1073/pnas.0810199105
91. Zhan H, Ciano K, Dong K, Zucker S. Targeting glutamine metabolism in myeloproliferative neoplasms. Blood Cells Mol Dis (2015) 55(3):241–7. doi: 10.1016/j.bcmd.2015.07.007
92. Gu Z, Liu Y, Cai F, Patrick M, Zmajkovic J, Cao H, et al. Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discov (2019) 9(9):1228–47. doi: 10.1158/2159-8290.CD-19-0152
93. Baumeister J, Chatain N, Hubrich A, Maie T, Costa IG, Denecke B, et al. Hypoxia-inducible factor 1 (HIF-1) is a new therapeutic target in JAK2V617F-positive myeloproliferative neoplasms. Leukemia (2020) 34(4):1062–74. doi: 10.1038/s41375-019-0629-z
94. Molle N, Krichevsky S, Kermani P, Silver RT, Ritchie E, Scandura JM. Ruxolitinib can cause weight gain by blocking leptin signaling in the brain via JAK2/STAT3. Blood (2020) 135(13):1062–6. doi: 10.1182/blood.2019003050
95. Mesa RA, Verstovsek S, Gupta V, Mascarenhas JO, Atallah E, Burn T, et al. Effects of ruxolitinib treatment on metabolic and nutritional parameters in patients with myelofibrosis from COMFORT-I. Clin Lymphoma Myeloma Leuk (2015) 15(4):214–21.e211. doi: 10.1016/j.clml.2014.12.008
96. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.02.004
97. Guglielmelli P, Barosi G, Rambaldi A, Marchioli R, Masciulli A, Tozzi L, et al. Safety and efficacy of everolimus, a mTOR inhibitor, as single agent in a phase 1/2 study in patients with myelofibrosis. Blood (2011) 118(8):2069–76. doi: 10.1182/blood-2011-01-330563
98. Kreitz J, Schönfeld C, Seibert M, Stolp V, Alshamleh I, Oellerich T, et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells (2019) 8(8):805. doi: 10.3390/cells8080805
99. Dhall A, Zee BM, Yan F, Blanco MA. Intersection of Epigenetic and Metabolic Regulation of Histone Modifications in Acute Myeloid Leukemia. Front Oncol (2019) 9:432. doi: 10.3389/fonc.2019.00432
100. Maher M, Diesch J, Casquero R, Buschbeck M. Epigenetic-Transcriptional Regulation of Fatty Acid Metabolism and Its Alterations in Leukaemia. Front Genet (2018) 9:405. doi: 10.3389/fgene.2018.00405
101. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature (2011) 469(7330):343–9. doi: 10.1038/nature09784
102. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet (2010) 42(8):722–6. doi: 10.1038/ng.621
103. Abdel-Wahab O, Pardanani A, Patel J, Wadleigh M, Lasho T, Heguy A, et al. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia (2011) 25(7):1200–2. doi: 10.1038/leu.2011.58
104. Zhang T, Gong Y, Meng H, Li C, Xue L. Symphony of epigenetic and metabolic regulation-interaction between the histone methyltransferase EZH2 and metabolism of tumor. Clin Epigenet (2020) 12(1):72. doi: 10.1186/s13148-020-00862-0
105. Ananieva EA, Wilkinson AC. Branched-chain amino acid metabolism in cancer. Curr Opin Clin Nutr Metab Care (2018) 21(1):64–70. doi: 10.1097/MCO.0000000000000430
106. Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J, et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature (2017) 545(7655):500–4. doi: 10.1038/nature22314
107. Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer (2012) 12(9):599–612. doi: 10.1038/nrc3343
108. Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W. The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia (2014) 28(3):485–96. doi: 10.1038/leu.2013.337
109. Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia (2009) 23(5):905–11. doi: 10.1038/leu.2009.47
110. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med (2009) 360(22):2289–301. doi: 10.1056/NEJMoa0810069
111. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell (2010) 18(6):553–67. doi: 10.1016/j.ccr.2010.11.015
112. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19(1):17–30. doi: 10.1016/j.ccr.2010.12.014
113. Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature (2017) 551(7680):384–8. doi: 10.1038/nature24294
114. Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell (2015) 27(5):617–30. doi: 10.1016/j.ccell.2015.04.006
115. Wang Y, Zhang J, Ren S, Sun D, Huang HY, Wang H, et al. Branched-Chain Amino Acid Metabolic Reprogramming Orchestrates Drug Resistance to EGFR Tyrosine Kinase Inhibitors. Cell Rep (2019) 28(2):512–25.e516. doi: 10.1016/j.celrep.2019.06.026
116. Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell (2006) 23(4):607–18. doi: 10.1016/j.molcel.2006.06.026
117. Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep (2012) 2(2):419–31. doi: 10.1016/j.celrep.2012.07.006
118. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science (2010) 327(5968):1000–4. doi: 10.1126/science.1179689
119. Andersen CL, Mortensen NB, Klausen TW, Vestergaard H, Bjerrum OW, Hasselbalch HC. A phase II study of vorinostat (MK-0683) in patients with primary myelofibrosis and post-polycythemia vera myelofibrosis. Haematologica (2014) 99(1):e5–7. doi: 10.3324/haematol.2013.096669
120. Bose P, Verstovsek S. Developmental Therapeutics in Myeloproliferative Neoplasms. Clin Lymphoma Myeloma Leuk (2017) 17S:S43–52. doi: 10.1016/j.clml.2017.02.014
121. Finazzi G, Vannucchi AM, Martinelli V, Ruggeri M, Nobile F, Specchia G, et al. A phase II study of Givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br J Haematol (2013) 161(5):688–94. doi: 10.1111/bjh.12332
122. Rambaldi A, Dellacasa CM, Finazzi G, Carobbio A, Ferrari ML, Guglielmelli P, et al. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol (2010) 150(4):446–55. doi: 10.1111/j.1365-2141.2010.08266.x
123. Quintas-Cardama A, Kantarjian H, Estrov Z, Borthakur G, Cortes J, Verstovsek S. Therapy with the histone deacetylase inhibitor pracinostat for patients with myelofibrosis. Leuk Res (2012) 36(9):1124–7. doi: 10.1016/j.leukres.2012.03.003
124. Mascarenhas J, Lu M, Li T, Petersen B, Hochman T, Najfeld V, et al. A phase I study of panobinostat (LBH589) in patients with primary myelofibrosis (PMF) and post-polycythaemia vera/essential thrombocythaemia myelofibrosis (post-PV/ET MF). Br J Haematol (2013) 161(1):68–75. doi: 10.1111/bjh.12220
125. DeAngelo DJ, Mesa RA, Fiskus W, Tefferi A, Paley C, Wadleigh M, et al. Phase II trial of panobinostat, an oral pan-deacetylase inhibitor in patients with primary myelofibrosis, post-essential thrombocythaemia, and post-polycythaemia vera myelofibrosis. Br J Haematol (2013) 162(3):326–35. doi: 10.1111/bjh.12384
126. Mascarenhas J, Sandy L, Lu M, Yoon J, Petersen B, Zhang D, et al. A phase II study of panobinostat in patients with primary myelofibrosis (PMF) and post-polycythemia vera/essential thrombocythemia myelofibrosis (post-PV/ET MF). Leuk Res (2017) 53:13–9. doi: 10.1016/j.leukres.2016.11.015
127. Bagchi RA, Ferguson BS, Stratton MS, Hu T, Cavasin MA, Sun L, et al. HDAC11 suppresses the thermogenic program of adipose tissue via BRD2. JCI Insight (2018) 3(15):e120159. doi: 10.1172/jci.insight.120159
128. Sun L, Marin de Evsikova C, Bian K, Achille A, Telles E, Pei H, et al. Programming and Regulation of Metabolic Homeostasis by HDAC11. EBioMedicine (2018) 33:157–68. doi: 10.1016/j.ebiom.2018.06.025
129. Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol (2009) 10(1):92–100. doi: 10.1038/ni.1673
130. Yanginlar C, Logie C. HDAC11 is a regulator of diverse immune functions. Biochim Biophys Acta Gene Regul Mech (2018) 1861(1):54–9. doi: 10.1016/j.bbagrm.2017.12.002
131. Sharma V, Yue L, Horvat NP, Christodoulidou A, Akuffo AA, Beatty M, et al. Selective Targeting of Histone Deacetylase 11 Disables Metabolism of Myeloproliferative Neoplasms. Blood (2019) 134(Supplement_1):474–4. doi: 10.1182/blood-2019-127235
132. Moreno-Yruela C, Galleano I, Madsen AS, Olsen CA. Histone Deacetylase 11 Is an ϵ-N-Myristoyllysine Hydrolase. Cell Chem Biol (2018) 25(7):849–56.e848. doi: 10.1016/j.chembiol.2018.04.007
133. Cao J, Sun L, Aramsangtienchai P, Spiegelman NA, Zhang X, Huang W, et al. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc Natl Acad Sci U S A (2019) 116(12):5487–92. doi: 10.1073/pnas.1815365116
134. Kutil Z, Novakova Z, Meleshin M, Mikesova J, Schutkowski M, Barinka C. Histone Deacetylase 11 Is a Fatty-Acid Deacylase. ACS Chem Biol (2018) 13(3):685–93. doi: 10.1021/acschembio.7b00942
135. Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia (2005) 19(10):1751–9. doi: 10.1038/sj.leu.2403910
Keywords: myeloproliferative neoplasm, metabolism, epigenetic, EZHZ2, HDAC11, JAK2, therapy, stem cell
Citation: Sharma V, Wright KL, Epling-Burnette PK and Reuther GW (2020) Metabolic Vulnerabilities and Epigenetic Dysregulation in Myeloproliferative Neoplasms. Front. Immunol. 11:604142. doi: 10.3389/fimmu.2020.604142
Received: 08 September 2020; Accepted: 02 November 2020;
Published: 30 November 2020.
Edited by:
Alejandro Villagra, George Washington University, United StatesReviewed by:
Edwin Chen, University of Westminster, United KingdomCopyright © 2020 Sharma, Wright, Epling-Burnette and Reuther. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gary W. Reuther, Z2FyeS5yZXV0aGVyQG1vZmZpdHQub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.