 Caio S. Bonilha
Caio S. Bonilha Robert A. Benson
Robert A. Benson James M. Brewer
James M. Brewer Paul Garside
Paul Garside- 1College of Medical, Veterinary and Life Sciences, Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow, United Kingdom
- 2Research and Development Department, Antibody Analytics Ltd., Newhouse, Lanarkshire, United Kingdom
The junctional adhesion molecule-A (JAM-A) is a cell surface adhesion molecule expressed on platelets, epithelial cells, endothelial cells and leukocytes (e. g. monocytes and dendritic cells). JAM-A plays a relevant role in leukocyte trafficking and its therapeutic potential has been studied in several pathological conditions due to its capacity to induce leukocyte migration out of inflamed sites or infiltration into tumor sites. However, disruption of JAM-A pathways may worsen clinical pathology in some cases. As such, the effects of JAM-A manipulation on modulating immune responses in the context of different diseases must be better understood. In this mini-review, we discuss the potential of JAM-A as a therapeutic target, summarizing findings from studies manipulating JAM-A in the context of inflammatory diseases (e.g. autoimmune diseases) and cancer and highlighting described mechanisms.
Introduction
The junctional adhesion molecule-A (JAM-A), also called junctional adhesion molecule-1 (JAM-1), is a member of the immunoglobulin superfamily and received its first denomination as F11 receptor (F11R), a molecule expressed on the surface of human platelets (1). Only a few years later, this molecule was detected in epithelial and endothelial intercellular tight junctions (2). JAM-A interactions with extracellular ligands assure firm cell-cell adhesion, playing important roles in endothelial cell migration (3–5) and proliferation (6) and epithelial cell barrier functions (7). Changes in barrier integrity caused by disruption of JAM-A pathways can indirectly modulate immune responses by modifying migratory patterns of antigen presenting cells (APC). However, JAM-A is also expressed by immune cells themselves, such as monocytes (2) and dendritic cells (DC) (8, 9). Thus, immune mediated processes are also likely to be directly influenced by JAM-A activity. As our understanding of the contributions of JAM-A to inflammatory process increases so does interest in the therapeutic value in targeting of JAM-A. In this regard, we summarize findings on the disruption of JAM-A pathways in murine models of inflammation and cancer, highlighting possible opposing immunological mechanisms mainly involving leukocyte migration.
JAM-A Biology
JAM-A is a transmembrane glycoprotein composed of a cytoplasmatic tail and an extra-cellular region consisting of a membrane-distal domain (D1) and a membrane-proximal domain (D2) (Figure 1). JAM-A homophilic binding (JAM-A-JAM-A) was first suggested to occur in epithelial-epithelial and endothelial-endothelial cell interactions, due to the presence of JAM-A in tight junctions (2). The detection of JAM-A dimers on the surface of JAM-A-transfected epithelial cells strengthened this hypothesis (11), being confirmed by protein- (11, 12) and cell-based (12, 13) assays. Homophilic ligation is mediated by JAM-A D1 domain and allows the formation of JAM-A dimers (14). Dimerization occurs by interaction of JAM-A monomers within the surface of the same cell (cis interactions). JAM-A dimers can then interact with other JAM-A dimers (trans interactions) to bridge epithelial cells in tight junctions (see Figure 1C).
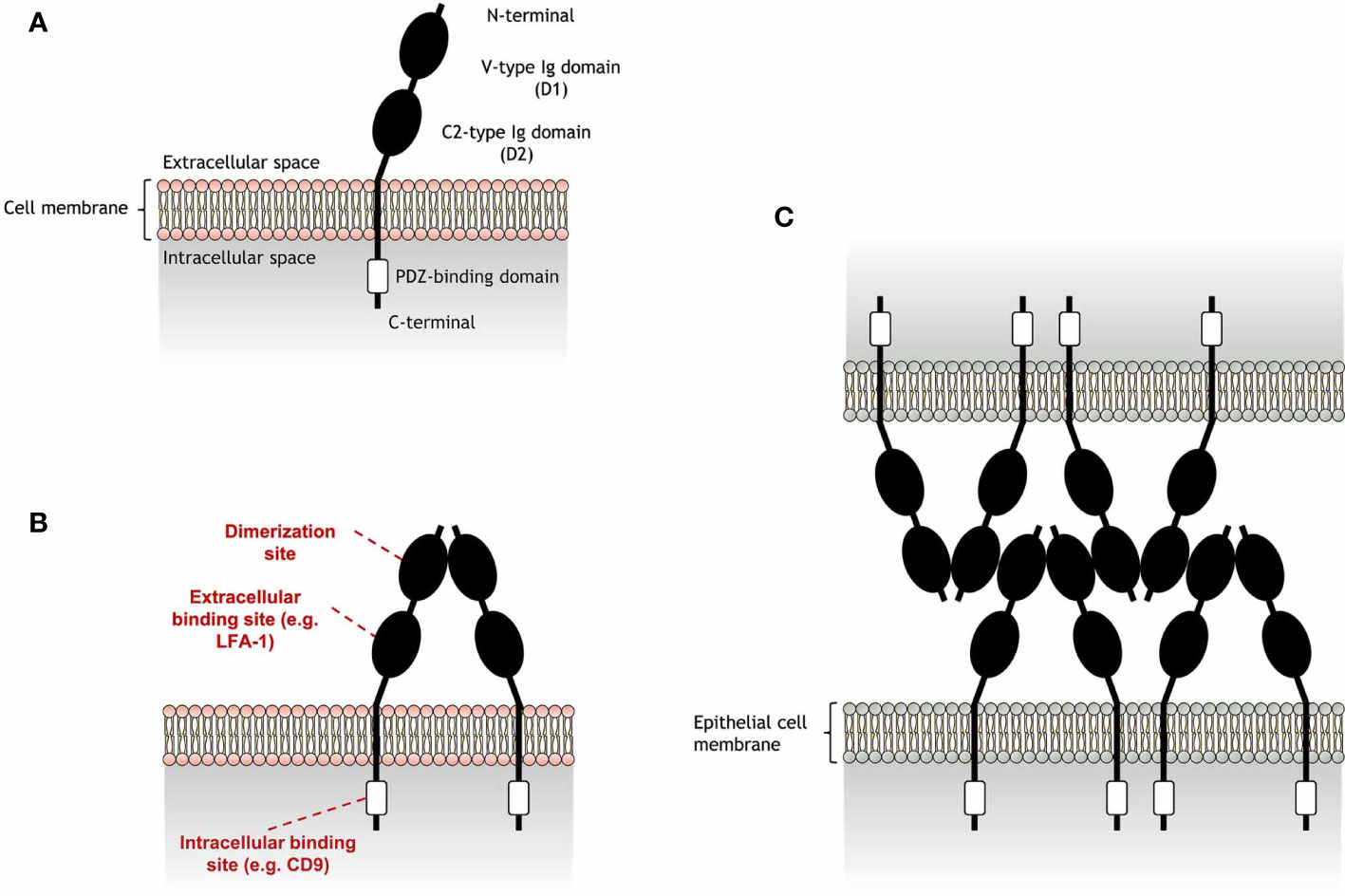
Figure 1 Schematic representation of junctional adhesion molecule-A (JAM-A) structure and homophilic adhesion. (A) JAM-A is composed by an extracellular region and a cytoplasmatic tail, connected by a transmembrane portion. JAM-A extracellular portion is formed by a membrane-distal V-type Ig-like domain (D1), which also includes a N-terminal portion, and a membrane-proximal C2-type Ig domain (D2). These Ig-like domains are linked by a short connector region. JAM-A cytoplasmatic tail contains a PDZ-binding motif that is linked to a C-terminal portion. JAM-A can be expressed on the cell surface as monomers, but can also interact with JAM-A monomers in cis interactions to form (B) homodimers in a process called dimerization. While the D1 domain is the region in which JAM-A monomers interact with each other, the D2 region can bind to other extracellular ligands in trans interactions [e.g. lymphocyte function-associated antigen 1 (LFA-1)]. In addition, the PDZ-binding domain allows JAM-A to bind to scaffold proteins, such as CD9, responsible to link JAM-A to β3 integrin. (C) Endothelial JAM-A homophilic adhesion consists of dimers on opposing cells forming contacts via the D1 domain and allows strong cell-cell adhesion in tight junctions for the formation of a molecular barrier that ensures the homeostasis of epithelial barrier integrity. JAM-A trans cell-cell arrangement was reported by Kostrewa et al. (10).
Besides homophilic binding, JAM-A can also undergo heterophilic cis or trans interactions with other extracellular ligands (Table 1). JAM-A can bind to the β2 chain of the lymphocyte function-associated antigen 1 (LFA-1), but not of the macrophage-1 antigen (MAC-1), via the JAM-A D2 domain (16, 18). The D2 domain was also found to be important in stabilizing homophilic interactions (13). However, the ligation of JAM-A D2 domain to LFA-1 was found to reduce the dynamic strength of JAM-A homophilic interactions in assays with immobilized JAM-A and Jurkat T cells (13), a human cell line expressing considerable levels of JAM-A (16). Immunoprecipitation assays revealed formation of JAM-A and β3 integrin (CD61) aggregates in endothelial cell lysates (3), suggesting a direct interaction between these molecules. However, this interaction was later found to be dependent on CD9, as absence of this tetraspanin inhibited JAM-A coimmunoprecipitation with β3 integrin (3). Other immunoprecipitation assays suggest that JAM-A can also bind to αIIb integrin (CD41) (15), as well as the β2 chain of LFA-1, as previously mentioned. However, CD9 is also known to interact with some of these integrins (19), which may point a dependence on this tetraspanin for the interaction of these integrins with JAM-A. In addition to these ligands, JAM-A can bind to another member of its family, the junctional adhesion molecule-B (JAM-B) (17), and can also work as a receptor for a few strains of murine and human viruses (14, 20–22). While JAM-A cis interactions are mainly responsible for cell signaling processes that may indirectly regulate cell migration (4, 15, 23), trans interactions directly mediate cell-cell adhesion and are essential for JAM-A role in leukocyte migration.

Table 1 Junctional adhesion molecule-A (JAM-A) described extracellular ligands.
JAM-A Role in Leukocyte Trafficking
The first report of a role for JAM-A in leukocyte migration comes from Martìn-Padura et al. (2), in which JAM-A blockade was found to inhibit spontaneous and chemokine-induced monocyte transmigration through endothelial cell monolayers. Tumor necrosis factor alpha (TNF-α) and interferon-gamma (IFN-γ)-stimulated endothelial cells redistribute JAM-A on their surface from intercellular junctions to the apical region of the cells, increasing leukocyte adhesion to the inflamed endothelia (16, 24). As such, several other studies have shown endothelial and epithelial cell expression of JAM-A contributes to leukocyte trafficking.
JAM-A blockade was found to inhibit chemokine stimulated neutrophil transmigration across TNF-α and IFN-γ-inflamed endothelium, but not neutrophil arrest (16). Treatment with anti-JAM-A antibody or JAM-A-Fc fusion protein (human JAM-A extracellular domains fused to the Fc portion of human IgG1) inhibited transendothelial migration of human memory (CD45RO+) CD4+ T cells triggered by para-methoxyamphetamine (PMA) and CXC chemokine ligand 12a (CXCL12a) in a LFA-1-depedent manner (16, 25). Splenic CD11c+CD11b-B220+ plasmacytoid DCs (pDC) treated with a different anti-JAM-A mAb also had diminished transmigration through layers of high endothelial venule (HEV) cells, whereas cell adhesion remained unaffected (26). In vivo, treatment with anti-JAM-A mAb decreased leukocyte transendothelial migration through cremaster venules induced by IL-1β, but not by chemoattractants, leukotriene B4 (LTB4) or platelet-activating factor (PAF) (27). This same effect was found in JAM-A deficient mice in comparison with wild-type (WT) mice. This reduction in leukocyte transendothelial migration through venules was found to be mediated by JAM-A expressed on endothelial cells, but not on leukocytes.
Although studies in monocytes, neutrophils, memory T cells and pDCs suggest that JAM-A blockade could have an inhibitory effect on leukocyte migration, conventional DCs (cDC) show an increased propensity to cross lymphatic endothelial cells when JAM-A activity is lacking (8). While monocytes, neutrophils (2, 8, 9) and pDCs (26) express very low or undetectable levels of JAM-A, cDCs express high levels of this transmembrane protein (8, 9). Bone marrow-derived DCs (BMDC) treated with anti-JAM-A mAb, or originating from JAM-A knockout (KO) mice, showed increased random motility in vitro compared to their respective controls (8). These JAM-A-deficient BMDCs expressed similar levels of maturation markers (CD80 and CD86), surface molecules related to DC migration (CD11a, CD11b, CD11c, CD62L, JAM-B, and JAM-C) and antigen uptake capacity in comparison to WT BMDCs, suggesting that JAM-A may not participate in early stages of DC differentiation and maturation. However, JAM-A deficient BMDCs display increased transmigration across monolayers of lymphatic endothelial cells, but unaffected transmigration across microvascular endothelial cells. In vivo, BMDCs from JAM-A deficient mice had increased migration from FITC-painted skin to the LN. The selective transmigration of JAM-A-deleted DCs suggests that DC JAM-A plays a role in homing steps during DC trafficking to the LN from tissues but may not participate in DC tissue infiltration from the vasculature. When analyzing the role of endothelial JAM-A in DC migration, another study reported higher in vitro transmigration of JAM-A-expressing BMDCs through layers of JAM-A-deficient lung endothelial cells in comparison with endothelial cells from JAM-A-/- mice reconstituted with full-length JAM-A complementary DNA (cDNA) (28). These studies indicate that while DC JAM-A participates in DC trafficking through the lymphatics, endothelial JAM-A play a dominant role in DC arrest and migration functions through the vascular endothelium. The effects of JAM-A manipulation to the migration of DCs and other immune cells have raised interest in potential JAM-A-targeted therapies for inflammatory diseases and cancer.
JAM-A Manipulation in Disease Models of Inflammation
Due to its expression in different cell types (platelets, endothelial cells and leukocytes), its capacity to modulate cell adhesion and migration and its upregulation in inflamed tissues, JAM-A has been studied as a therapeutic target in a number of disease models.
Skin Inflammation
Inflammatory skin sites are characterized by proliferation of dermal cells, dilated blood vessels and accumulation of immune cells (29). In a model of skin inflammation, systemic treatment with an antagonistic anti-JAM-A mAb inhibited leukocyte infiltration upon chemokine administration in subcutaneous air pouches (2). However, in a model of ear skin inflammation driven in an antigen specific manner, JAM-A-deficient mice displayed enhanced contact hypersensitivity (8). The increase in ear swelling in this T-cell mediated model was linked to activity of JAM-A-deficient DCs, most likely arising through their augmented migration to LNs and enhancing activation of antigen specific T cells.
Vascular Disease
Adhesion molecules have been implicated in vascular wall integrity and play an important role in vascular diseases (30). JAM-A ability to control platelet aggregation (12) have triggered particular interest in this molecule in studies of vascular diseases such as atherosclerosis, a cardiovascular disease caused by the development of plaques on artery walls restricting or blocking blood flow to specific organs or regions of the body (31). In humans, elevated JAM-A gene expression was described in atherosclerotic plaques compared with artery segments of normal patients (32, 33) and in unstable carotid plaques in comparison to stable plaques (33). In addition, JAM-A has been found to be required for human platelet adhesion to inflamed endothelial cells (34).
In models of ischemia-reperfusion (I/R) injury, both JAM-A genetic depletion and blockade with anti-JAM-A mAb suppressed leukocyte infiltration in response to cremaster muscle (27) and liver I/R injury (35). However, no protective effects on microvascular and hepatocellular injury were reported, evidenced by unaffected levels of sinusoidal perfusion and liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST). The use of mice with JAM-A selective depletion in endothelial cells identified the requirement of endothelial JAM-A in the reduction of T cell, but not neutrophil, transmigration. This dependence on endothelial JAM-A was also reported in a model of heart I/R injury, in which JAM-A absence reduced leukocyte infiltration in the myocardium by affecting transendothelial migration (36).
In mouse models of atherosclerosis, increased f11r messenger ribonucleic acid (mRNA) expression was found in both early atherosclerotic endothelium of carotid arteries (25) and advanced atherosclerotic plaques (32). A JAM-A-Fc fusion protein was used in an ex vivo perfusion model to demonstrate JAM-A role in early atherosclerosis. Treatment with this fusion protein inhibited the arrest of human monocytes and memory CD4+ T cells in murine atherosclerotic endothelium under blockade of very late antigen 4 (VLA-4), an intercellular adhesion molecule 1 (ICAM-1) ligand (25). These results show the capacity of JAM-A targeting to modulate the recruitment of leukocytes to atherosclerotic endothelium by blocking pathways of competitor ligands.
In vitro, treatment with a JAM-A antagonistic peptide capable of blocking homophilic binding (peptide 4D), decreased platelet adhesion to inflamed endothelial cells, whereas agonistic reagents promoted platelet aggregation (12, 37). This antagonistic peptide also decreased plaque number and size and increased survival of mice in an atherosclerosis model in comparison with mice treated with a scrambled peptide (38), due to a reduction on platelet adhesion to the inflamed endothelium. However, in a murine model of thrombosis, lack of JAM-A resulted in increased thrombus formation due to enhanced aggregation of platelets (39). JAM-A-/- mice also had shorter tail-bleeding time and faster vessel occlusion. Although no studies have evaluated the capacity of the antagonistic peptide to block other JAM-A pathways, these studies suggest that, while JAM-A homophilic binding by platelets may be a useful target for avoiding formation of atheroma, other JAM-A pathways may have platelet stimulatory functions and promote blood coagulation that can obstruct and impair the homeostasis of the circulatory system.
Inflammatory Bowel Disease
The intestinal mucosa forms an important barrier against potential hostile microorganisms and other foreign antigens. The permeability of the intestinal epithelium is mediated by tight junctions, formed of molecular components, such as adhesion molecules, that link intestinal epithelial cells and exert a control on the passage of environmental molecules. Intestinal barrier defects have been associated with chronic mucosal inflammation and inflammatory bowel diseases (IBDs) (40). A few studies have addressed the role of JAM-A in the regulation of intestinal barrier functions during IBDs. Although JAM-A deficient mice displayed higher susceptibility for development of dextran sulfate sodium (DSS)-induced colitis in comparison with WT mice (41), JAM-A depletion increased epithelial proliferation, which resulted in faster repair of epithelial defects, evidenced by a reduction in damaged colonic mucosa. The increase in clinical disease was attributed to a higher permeability in the colonic mucosa of JAM-A-/- mice, supposedly enhancing its vulnerability to acute DSS-induced colitis (42). To address if this increased susceptibility was mediated by the higher number of B and T cells found in the lamina propria of JAM-A-/- mice, JAM-A-deficient mice crossed to recombination activating gene 1 (RAG1) knockout animals lacking B and T cell development were also investigated (43). Lack of adaptive immunity in JAM-A-deficient RAG1-/- mice promoted an even higher susceptibility for development of DSS-induced colitis in comparison with JAM-A-/- animals. However, administration of antibiotics in JAM-A-deficient RAG1-/- mice reduced the susceptibility to disease development. These results suggest an important compensatory role of the adaptive immune system in JAM-A-/- mice that limits bacterial-driven colitis. CD4+ T cells played a relevant role in this compensatory mechanism, as depletion of these cells, but not CD8+ T cells, enhanced susceptibility to colitis. In addition, increased gene and protein expression of transforming growth factor beta 1 (TGF-β1) in colonic tissues pointed to a possible role of this growth factor in the compensatory mechanism. Treatment with anti-TGF-β1 mAb decreased body weight and increased disease activity index scores in JAM-A-deficient mice in comparison to anti-TGF-β1-treated WT mice and to isotype-treated JAM-A-/- mice, demonstrating a protective role of TGF-β1 in JAM-A-deficient mice from developing a more severe acute colonic inflammation.
Mice with selective loss of JAM-A in myelomonocytic cells, progenitors that can differentiate into monocytes, macrophages and subtypes of conventional DCs (44), were used to investigate the role of JAM-A expression on these cells during intestinal inflammation. These mice showed no difference in neutrophil recruitment into the peritoneum and macrophage chemokine production in response to lipopolysaccharides (LPS) or zymosan in comparison with control mice (45). However, these parameters were significantly reduced in global JAM-A-/- mice stimulated by these inflammatory mediators. Mice with selective loss of JAM-A on intestinal epithelial cells demonstrated increased intestinal permeability and reduced peritoneal neutrophil migration and macrophage chemokine production. These findings suggest that JAM-A expression in the epithelium is fundamental for JAM-A -mediated intestinal inflammation.
Exposure of human intestinal epithelial cells to cytokines (TNF-α, IFN-γ, IL-22, or IL-17A) was found to induce JAM-A cytoplasmatic tail tyrosine Y280 phosphorylation (46). Elevated levels of this phosphorylated form were also detected in the colonic mucosa of DSS-induced colitis mice and humans with ulcerative colitis in comparison to healthy mucosa. Further studies will investigate the role of JAM-A cytoplasmatic tail phosphorylation as a regulator of epithelial intestinal barrier functions during bowel inflammation.
Neurological Disorders
Disruption of the blood-brain barrier of the central nervous system (CNS) leads to leukocyte accumulation in the cerebrospinal fluid, a main component of brain disorders such as meningitis (47) and multiple sclerosis (MS) (48). Surface JAM-A is expressed in brain vessels of MS patients (49) and in vessels of the brain parenchyma and choroid plexus of mice subject to cytokine-induced meningitis (50). In this model of cytokine-induced meningitis, intravenous treatment with anti-JAM-A mAb attenuated meningeal inflammation (50). This therapeutic effect was attributed to inhibition of monocyte and neutrophil accumulation in the cerebrospinal fluid and neutrophil infiltration into the brain parenchyma arising from reduced blood–brain barrier permeability. Further pre-clinical models that aim to discover the effects of JAM-A blockade/promotion on other immunological components involved in neurological disorders may identify JAM-A as a potential target for disease treatment.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a chronic systemic inflammatory autoimmune disease that mainly affects the joints, with an essential participation of the adaptive immune system in its induction phase (51). A breach of self-tolerance led by failure in immune regulatory mechanisms results in activation of immune responses (52) and production of several autoantibodies, against host proteins such as cartilage components, nuclear proteins, stress proteins and citrullinated proteins (53). At the center of these immune responses are leukocytes, that accumulate in the joint, contributing to chronic joint pain (54). Evidence for of JAM-A gene upregulation in human hypoxic cells (55, 56) suggest that the hypoxia present in the synovial tissue of RA patients (57–59) could potentially lead to increased JAM-A gene and protein expression on cells from inflamed joints of these patients. Recently, a study described increased expression of f11r mRNA on PMBCs of RA patients (60). Nevertheless, this upregulation could have been driven by the systemic inflammation itself, as healthy individuals were used as control. In addition, JAM-A is expressed in inflamed joints of K/BxN mice (61), animals expressing transgenic T cell receptor (TCR) and major histocompatibility complex class II (MHCII) that develop severe inflammatory arthritis (62).
In a model of RA in which autoantibodies from arthritogenic K/BxN drive inflammation and tissue destruction in serum-recipient mice, treatment with anti-JAM-A mAb delayed the disease onset and partially ameliorated overall disease (61). Similar effects were found on mice treated with anti-ICAM-1 mAb, and a more prominent amelioration was found on treatment with mAb targeting the alpha chain of LFA-1, but not the beta chain. ICAM-1 is another ligand for LFA-1 (63), which may suggest that the therapeutic effects in these arthritic mice treated with both anti-JAM-A and anti-ICAM-1 mAbs could have been caused by disruption of LFA-1 pathways. However, more studies are required to investigate the mechanisms involved in JAM-A blockade in models of autoimmune diseases, which may be related to a modulation on immune cells trafficking, as proposed in Figure 2.
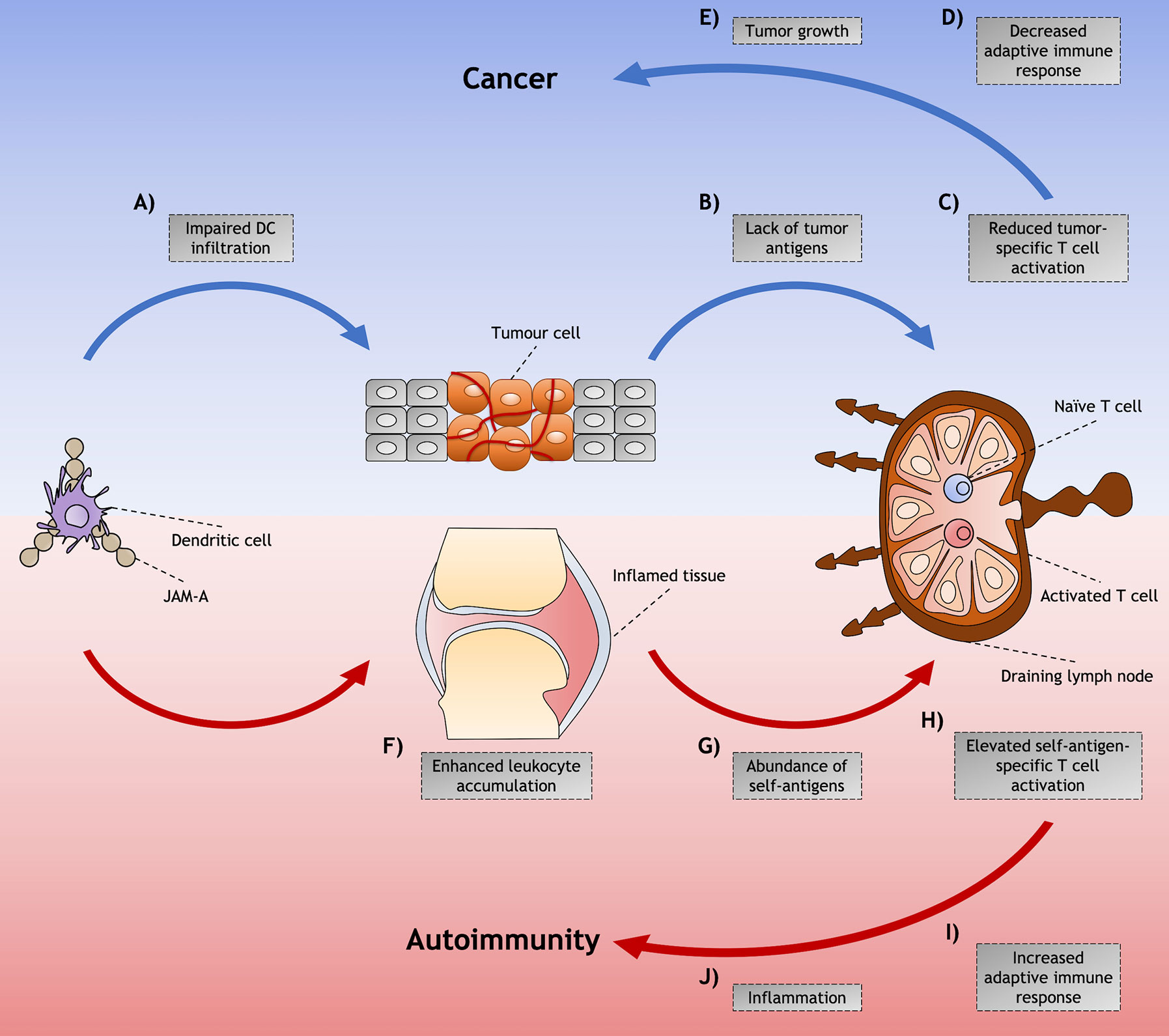
Figure 2 Proposed model for junctional adhesion molecule-A (JAM-A)-mediated dendritic cell (DC) role in cancer and autoimmunity. Although JAM-A may contribute to disease through diverse cell types and signaling processes, mechanisms of DC trafficking mediated by JAM-A and their indirect effects in the immune response might play an important role in the induction and/or promotion of cancer and autoimmunity. Among these is the (A) impaired infiltration of DCs that is found in tumors that have achieved the hallmarks of cancer. (B) This diminished presence of DCs impairs the availability of tumor antigens that would be presented to tumor-specific T cells in the LNs, (C) leading to reduced T cell activation and (D) decreased adaptive immune responses against the abnormal cells. (E) The lack of immunity against these cells allows tumor to grow and cancer to be set. (F) On the other hand, in autoimmunity, JAM-A may assist the accumulation of immune cells in the inflamed tissues by enhancing leukocyte adhesion to the inflamed endothelia. (G) The accumulation of DCs increases the availability of self-antigen not only in peripheral tissues - such as arthritic joints of RA patients, where antigen presentation can occur - but also in the draining LN, by DCs carrying self-antigen captured in the affected tissue. (H) This abundance of self-antigens possibly leads to the activation of a higher proportion of self-antigen specific T cells. (I) The induction of adaptive immune responses against self-antigens leads to (J) inflammation and tissue destruction characteristic of autoimmune diseases.
JAM-A Manipulation in Cancer Models
The six hallmarks of cancer are sustained proliferative signaling, resistance to cell death, replicative immortality, induction of angiogenesis, evasion of growth suppressors and activation of invasion and metastasis mechanisms (64). Interventions that aim to enhance immune responses to tumor cells, such as increasing tumor-derived antigen presentation to T cells are desirable for cancer treatment. Evidence from the literature suggest that circulating, soluble JAM-A could be used as a biomarker for the detection of some types of cancer, such as multiple myeloma (65) and head and neck squamous cell carcinoma (66). Although few studies show that JAM-A can be downregulated in tumor sites of metastatic breast (67) and anaplastic thyroid carcinoma tissues (68), high levels of JAM-A protein expression in tumor tissues have been correlated with poor prognosis in breast cancer (69) and nasopharyngeal carcinoma (70) patients.
In vitro studies using human triple negative breast cancer or thyroid carcinoma cell lines showed increased tumor cell proliferation and migration under JAM-A gene silencing (67, 68). Transfection of a JAM-A plasmid to these cell lines impaired transendothelial migration and colony formation. However, treatment with a JAM-A antagonistic peptide inhibited transmigration of breast cancer cells through inflamed endothelium (71), an important step in the initiation of metastasis formation. Additionally, JAM-A inhibition increased apoptosis and decreased proliferation of multiple myeloma cells in vitro and inhibited the progression of this type of cancer in vivo (65). In RIP1Tag2 mice, which express the SV40 T-antigen under the rat insulin promoter resulting in carcinogenesis of β cells in pancreatic islets, JAM-A absence decreased growth and aggressiveness of tumors in comparison with control mice (28). An increased tumor specific immune response resulted in diminished angiogenesis and increased apoptosis that was attributed to a more efficient infiltration of JAM-A-deficient DCs, but not macrophages, into tumor sites. This mechanism was dependent on both CD4+ and CD8+ T cells. In a model of multifocal mammary adenocarcinoma, mice lacking JAM-A developed smaller mammary tumors than control mice (72). Disruption of JAM-A pathways increased cell apoptosis and decreased proliferation, however, no differences in angiogenesis or infiltration of DCs or macrophages were found. In addition, treatment with anti-JAM-A mAb suppressed progression of malignant tumors by impairing cell proliferation and angiogenesis in malignant myeloma xenograft murine models (73). Altogether, these studies suggest that although JAM-A expressed by tumor cells may have a protective role on progression of cancer, JAM-A antagonism could enhance immune response against these abnormal cells by possibly facilitating leukocyte infiltration into tumors and DC egress to LNs (see Figure 2). Nevertheless, in a recent study, JAM-A-deficient female mice developed a more aggressive phenotype of brain tumor in comparison with WT females and JAM-A–deficient and WT males (74). This study highlights the impact of other factors, such as sex difference, in the therapeutic effects that JAM-A blockade might promote in the development of tumors and demonstrates the complexity and challenges involved in the potential development of a JAM-A-targeted drug for cancer treatment.
Conclusion and Future Perspectives
Upon recruitment of its extracellular ligands, JAM-A ensures firm adhesion of leukocytes and platelets to the endothelia and plays a definitive role in immune cell transmigration. Antagonistic JAM-A targeting in preclinical models of inflammatory diseases show promising results for controlling inflammation caused by leukocyte or platelet accumulation. In addition, in some cancer models, manipulation of JAM-A pathways achieved agonism of immune responses by affecting leukocyte infiltration into tumors, controlling the progression of the disease. However, the possible disruption in multiple pathways (endothelial cell-endothelial cell, leukocyte-endothelial cell, leukocyte-leukocyte, platelet-endothelial cell, platelet-leukocyte and platelet-platelet interactions) caused by JAM-A targeting suggests precaution in the interpretation of results from preclinical model studies. As such, studies with cell-selective JAM-A disruption that aim to distinguish pathway-specific effects in different pathological conditions will further our understanding of JAM-A role in autoimmunity and cancer and may highlight JAM-A as a potential therapeutic target for human disease.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
CB was supported by CAPES Foundation, an agency under the Ministry of Education of Brazil (grant number 88881.129556/2016-01). CB, RB, JB, and PG were supported by the Arthritis Research UK (ARUK) programme grant number 19788 and the Innovative Medicines Initiative EU-funded project Be The Cure (BTCURE) [grant number 115142-2]. JB and PG were also supported by the Research into Inflammatory Arthritis Centre Versus Arthritis (RACE) (grant number 22072).
Conflict of Interest
Author RB was employed by the company Antibody Analytics Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kornecki E, Walkowiak B, Naik U, Ehrlich Y. Activation of human platelets by a stimulatory monoclonal antibody. J Biol Chem (1990) 265:10042–8.
2. Martìn-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol (1998) 142:117–27. doi: 10.1083/jcb.142.1.117
3. Naik MU, Naik UP. Junctional adhesion molecule-A-induced endothelial cell migration on vitronectin is integrin αVβ3 specific. J Cell Sci (2006) 119:490–9. doi: 10.1242/jcs.02771
4. Peddibhotla SSD, Brinkmann BF, Kummer D, Tuncay H. Tetraspanin CD9 links junctional adhesion molecule-A to αvβ3 integrin to mediate basic fibroblast growth factor-specific angiogenic signaling. Mol Biol Cell (2013) 24:933–44. doi: 10.1091/mbc.E12-06-0481
5. Naik MU, Vuppalanchi D, Naik UP. Essential role of Junctional adhesion molecule-1 in basic fibroblast growth factor-induced endothelial cell migration. Arterioscler Thromb Vasc Biol (2003) 23:2165–71. doi: 10.1161/01.ATV.0000093982.84451.87
6. Naik MU, Mousa SA, Parkos CA, Naik UP. Signaling through JAM-1 and αvβ3 is required for the angiogenic action of bFGF: Dissociation of the JAM-1 and αvβ3 complex. Blood (2003) 102:2108–14. doi: 10.1182/blood-2003-04-1114
7. Mandell KJ, McCall IC, Parkos CA. Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J Biol Chem (2004) 279:16254–62. doi: 10.1074/jbc.M309483200
8. Cera MR, Del Prete A, Vecchi A, Corada M, Martin-Padura I, Motoike T, et al. Increased DC trafficking to lymph nodes and contact hypersensitivity in junctional adhesion molecule-A–deficient mice. J Clin Invest (2004) 114:729–38. doi: 10.1172/jci21231
9. Ogasawara N, Kojima T, Go M, Fuchimoto J, Kamekura R, Koizumi J, et al. Induction of JAM-A during differentiation of human THP-1 dendritic cells. Biochem Biophys Res Commun (2009) 389:543–9. doi: 10.1016/j.bbrc.2009.09.024
10. Kostrewa D, Brockhaus M, D’Arcy A, Dale GE, Nelboeck P, Schmid G, et al. X-ray structure of junctional adhesion molecule: Structural basis for homophilic adhesion via a novel dimerization motif. EMBO J (2001) 20:4391–8. doi: 10.1093/emboj/20.16.4391
11. Bazzoni G, Martìnez-Estrada OM, Mueller F, Nelboeck P, Schmid G, Bartfai T, et al. Homophilic interaction of junctional adhesion molecule. J Biol Chem (2000) 275:30970–6. doi: 10.1074/jbc.M003946200
12. Babinska A, Kedees MH, Athar H, Ahmed T, Batuman O, Ehrlich YH, et al. F11-receptor (F11R/JAM) mediates platelet adhesion to endothelial cells: role in inflammatory thrombosis. Thromb Haemost (2002) 88:843–50. doi: 10.1055/s-0037-1613312
13. Wojcikiewicz EP, Koenen RR, Fraemohs L, Minkiewicz J, Azad H, Weber C, et al. LFA-1 binding destabilizes the JAM-A homophilic interaction during leukocyte transmigration. Biophys J (2009) 96:285–93. doi: 10.1529/biophysj.108.135491
14. Prota AE, Campbell JA, Schelling P, Forrest JC, Watson MJ, Peters TR, et al. Crystal structure of human junctional adhesion molecule 1: Implications for reovirus binding. Proc Natl Acad Sci (2003) 100:5366–71. doi: 10.1073/pnas.0937718100
15. Naik MU, Caplan JL, Naik UP. Junctional adhesion molecule-A suppresses platelet integrin αIIbβ3 signaling by recruiting Csk to the integrin-c-Src complex. Blood (2014) 123:1393–402. doi: 10.1182/blood-2013-04-496232
16. Ostermann G, Weber KSC, Zernecke A, Schröder A, Weber C. JAM-I is a ligand of the β2 integrin LFA-I involved in transendothelial migration of leukocytes. Nat Immunol (2002) 3:151–8. doi: 10.1038/ni755
17. Kobayashi I, Kobayashi-Sun J, Kim AD, Pouget C, Fujita N, Suda T, et al. Jam1a-Jam2a interactions regulate haematopoietic stem cell fate through Notch signalling. Nature (2014) 512:319–23. doi: 10.1038/nature13623
18. Fraemohs L, Koenen RR, Ostermann G, Weber C, Fraemohs L, Koenen RR, et al. The functional interaction of the β2 integrin lymphocyte function-associated antigen-1 with junctional adhesion molecule-A is mediated by the I domain. J Immunol (2004) 173:6259–64. doi: 10.4049/jimmunol.173.10.6259
19. Longhurst CM, White MM, Wilkinson DA, Jennings LK. A CD9, α(IIb)β3, integrin-associated protein, and GPlb/V/IX complex on the surface of human platelets is influenced by α(IIb)β3 conformational states. Eur J Biochem (1999) 263:104–10. doi: 10.1046/j.1432-1327.1999.00467.x
20. Campbell JA, Schelling P, Wetzel JD, Johnson EM, Forrest JC, Wilson GAR, et al. Junctional adhesion molecule A serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J Virol (2005) 79:7967–78. doi: 10.1128/jvi.79.13.7967-7978.2005
21. Sosnovtsev SV, Sandoval-Jaime C, Parra GI, Tin CM, Jones RW, Soden J, et al. Identification of human junctional adhesion molecule 1 as a functional receptor for the hom-1 calicivirus on human cells. MBio (2017) 8:1–19. doi: 10.1128/mBio.00031-17
22. Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, et al. Junction adhesion molecule is a receptor for reovirus. Cell (2001) 104:441–51. doi: 10.1016/S0092-8674(01)00231-8
23. Severson EA, Jiang L, Ivanov AI, Mandell KJ, Nusrat A, Parkos CA. Cis-Dimerization Mediates Function of Junctional Adhesion Molecule A. Mol Biol Cell (2008) 19:1862–72. doi: 10.1091/mbc.E07-09-0869
24. Ozaki H, Ishii K, Horiuchi H, Arai H, Kawamoto T, Okawa K, et al. Cutting edge: combined treatment of TNF-alpha and IFN-gamma causes redistribution of junctional adhesion molecule in human endothelial cells. J Immunol (1999) 163:553–7.
25. Ostermann G, Fraemohs L, Baltus T, Schober A, Lietz M, Zernecke A, et al. Involvement of JAM-A in mononuclear cell recruitment on inflamed or atherosclerotic endothelium inhibition by soluble JAM-A. Arterioscler Thromb Vasc Biol (2005) 25:729–35. doi: 10.1161/01.ATV.0000157154.14474.3b
26. Matsutani T, Tanaka T, Tohya K, Otani K, Jang MH, Umemoto E, et al. Plasmacytoid dendritic cells employ multiple cell adhesion molecules sequentially to interact with high endothelial venule cells - molecular basis of their trafficking to lymph nodes. Int Immunol (2007) 19:1031–7. doi: 10.1093/intimm/dxm088
27. Woodfin A, Reichel CA, Khandoga A, Corada M, Voisin M-B, Scheiermann C, et al. JAM-A mediates neutrophil transmigration in a stimulus-specific manner in vivo: evidence for sequential roles for JAM-A and PECAM-1 in neutrophil transmigration. Blood (2007) 110:1848–56. doi: 10.1182/blood-2006-09-047431
28. Murakami M, Francavilla C, Torselli I, Corada M, Maddaluno L, Sica A, et al. Inactivation of junctional adhesion molecule-A enhances antitumoral immune response by promoting dendritic cell and T lymphocyte infiltration. Cancer Res (2010) 70:1759–65. doi: 10.1158/0008-5472.CAN-09-1703
29. Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol (2014) 14:289–301. doi: 10.1038/nri3646
30. Krieglstein CF, Granger DN. Adhesion molecules and their role in vascular disease. Am J Hypertens (2001) 14:44–54. doi: 10.1016/s0895-7061(01)02069-6
31. MacRitchie N, Grassia G, Noonan J, Garside P, Graham D, Maffia P. Molecular imaging of atherosclerosis: spotlight on Raman spectroscopy and surface-enhanced Raman scattering. Heart (2018) 104:460–7. doi: 10.1136/heartjnl-2017-311447
32. Babinska A, Azari BM, Salifu MO, Liu R, Jiang X, Sobocka MB, et al. The F11 receptor (F11R/JAM-A) in atherothrombosis: overexpression of F11R in atherosclerotic plaques. Thromb Haemost (2007) 97:272–81. doi: 10.1160/TH06
33. Slevin M, Elasbali AB, Turu MM, Krupinski J, Badimon L, Gaffney J. Identification of differential protein expression associated with development of unstable human carotid plaques. Am J Pathol (2006) 168:1004–21. doi: 10.2353/ajpath.2006.050471
34. Azari BM, Marmur JD, Salifu MO, Ehrlich YH, Kornecki E, Babinska A. Transcription and translation of human F11R gene are required for an initial step of atherogenesis induced by inflammatory cytokines. J Transl Med (2011) 9:98. doi: 10.1186/1479-5876-9-98
35. Khandoga A, Kessler JS, Meissner H, Hanschen M, Corada M, Motoike T, et al. Junctional adhesion molecule-A deficiency increases hepatic ischemia-reperfusion injury despite reduction of neutrophil transendothelial migration. Blood (2019) 106:725–34. doi: 10.1182/blood-2004-11-4416
36. Corada M, Chimenti S, Cera MR, Vinci M, Salio M, Fiordaliso F, et al. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc Natl Acad Sci U S A (2005) 102:10634–9. doi: 10.1073/pnas.0500147102
37. Babinska A, Clement CC, Swiatkowska M, Szymanski J, Shon A, Ehrlich YH, et al. Development of new antiatherosclerotic and antithrombotic drugs utilizing F11 receptor (F11r/JAM-A) peptides. Biopolymers (2014) 102:322–34. doi: 10.1002/bip.22503
38. Babinska A, Clement CC, Przygodzki T, Talar M, Li Y, Braun M, et al. A peptide antagonist of F11R/JAM-A reduces plaque formation and prolongs survival in an animal model of atherosclerosis. Atherosclerosis (2019) 284:92–101. doi: 10.1016/j.atherosclerosis.2019.02.014
39. Naik MU, Stalker TJ, Brass LF, Naik UP. JAM-A protects from thrombosis by suppressing integrin αIIbβ3-dependent outside-in signaling in platelets. Blood (2012) 119:3352–60. doi: 10.1182/blood-2011-12-397398
40. Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: Lessons learned from animal models and human genetics. Front Immunol (2013) 4:280. doi: 10.3389/fimmu.2013.00280
41. Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med (2007) 204:3067–76. doi: 10.1084/jem.20071416
42. Ahmad R, Sorrell MF, Batra SK, Dhawan P, Singh AB. Gut permeability and mucosal inflammation: bad, good or context dependent. Mucosal Immunol (2017) 10:307–17. doi: 10.1038/mi.2016.128
43. Khounlotham M, Kim W, Peatman E, Nava P, Medina- O, Addis C, et al. Compromised intestinal epithelial barrier induces adaptive immune compensation that protects from colitis. Immunity (2012) 37:563–73. doi: 10.1016/j.immuni.2012.06.017
44. Ardavín C. Origin, precursors and differentiation of mouse dendritic cells. Nat Rev Immunol (2003) 3:582–90. doi: 10.1038/nri1127
45. Luissint AC, Williams HC, Kim W, Flemming S, Azcutia V, Hilgarth RS, et al. Macrophage-dependent neutrophil recruitment is impaired under conditions of increased intestinal permeability in JAM-A-deficient mice. Mucosal Immunol (2019) 12:668–78. doi: 10.1038/s41385-019-0143-7
46. Fan S, Weight CM, Luissint AC, Hilgarth RS, Brazil JC, Ettel M, et al. Role of JAM-A tyrosine phosphorylation in epithelial barrier dysfunction during intestinal inflammation. Mol Biol Cell (2019) 30:566–78. doi: 10.1091/mbc.E18-08-0531
47. Niu Z, Chen Y, Zhang K, De Bernard M. Polymorphonuclear Leukocyte Transendothelial Migration Proceeds at Blood-Brain Barrier in Neonatal Meningitis. Front Microbiol (2020) 11:969. doi: 10.3389/fmicb.2020.00969
48. Ruiz F, Vigne S, Pot C. Resolution of inflammation during multiple sclerosis. Semin Immunopathol (2019) 41:711–26. doi: 10.1007/s00281-019-00765-0
49. Padden M, Leech S, Craig B, Kirk J, Brankin B, McQuaid S. Differences in expression of junctional adhesion molecule-A and β-catenin in multiple sclerosis brain tissue: Increasing evidence for the role of tight junction pathology. Acta Neuropathol (2007) 113:177–86. doi: 10.1007/s00401-006-0145-x
50. Del Maschio A, De Luigi A, Martin-Padura I, Brockhaus M, Bartfai T, Fruscella P, et al. Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM). J Exp Med (1999) 190:1351–6. doi: 10.1084/jem.190.9.1351
51. Scott DL, Wolfe F, Huizinga TWJ. Rheumatoid arthritis. Lancet (2010) 376:1094–108. doi: 10.1016/S0140-6736(10)60826-4
52. Mueller DL, Jenkins MK. Autoimmunity: when self-tolerance breaks down. Curr Biol (1997) 7:255–7. doi: 10.1016/S0960-9822(06)00115-1
53. Song YW, Kang EH. Autoantibodies in rheumatoid arthritis: rheumatoid factors and anticitrullinated protein antibodies. QJM (2010) 103:139–46. doi: 10.1093/qjmed/hcp165
54. Palmer G, Gabay C, Imhof BA. Leukocyte migration to rheumatoid joints: enzymes take over. Arthritis Rheum (2006) 54:2707–10. doi: 10.1002/art.22062
55. Mense SM, Sengupta A, Zhou M, Lan C, Bentsman G, Volsky DJ, et al. Gene expression profiling reveals the profound upregulation of hypoxia-responsive genes in primary human astrocytes. Physiol Genomics (2006) 25:435–49. doi: 10.1152/physiolgenomics.00315.2005
56. Ben-Zvi M, Amariglio N, Paret G, Nevo-Caspi Y. F11R expression upon hypoxia is regulated by RNA editing. PLoS One (2013) 8:1–12. doi: 10.1371/journal.pone.0077702
57. Akhavani MA, Madden L, Buysschaert I, Sivakumar B, Kang N, Paleolog EM. Hypoxia upregulates angiogenesis and synovial cell migration in rheumatoid arthritis. Arthritis Res Ther (2009) 11:1–11. doi: 10.1186/ar2689
58. Sivakumar B, Akhavani MA, Winlove CP, Taylor PC, Paleolog EM, Kang N. Synovial hypoxia as a cause of tendon rupture in rheumatoid arthritis. J Handb Surg Am (2008) 33:49–58. doi: 10.1016/j.jhsa.2007.09.002
59. Lund-Olesen K. Oxygen tension in synovial fluids. Arthritis Rheum (1970) 13:769–76. doi: 10.1002/art.1780130606
60. Fang T-J, Lin C-H, Lin Y-Z, Li R-N, Ou T-T, Wu C-C, et al. F11R mRNA expression and promoter polymorphisms in patients with rheumatoid arthritis. Int J Rheum Dis (2016) 19:127–33. doi: 10.1111/1756-185x.12663
61. Watts GM, Beurskens FJM, Martin-Padura I, Ballantyne CM, Klickstein LB, Brenner MB, et al. Manifestations of inflammatory arthritis are critically dependent on LFA-1. J Immunol (2005) 174:3668–75. doi: 10.4049/jimmunol.174.6.3668
62. Christensen AD, Haase C, Cook AD, Hamilton JA. K/BxN serum-transfer arthritis as a model for human inflammatory arthritis. Front Immunol (2016) 7:213. doi: 10.3389/fimmu.2016.00213
63. Kandula S, Abraham C. LFA-1 on CD4 + T cells is required for optimal antigen-dependent activation in vivo. J Immunol (2004) 173:4443–51. doi: 10.4049/jimmunol.173.7.4443
64. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
65. Solimando AG, Brandl A, Mattenheimer K, Graf C, Ritz M, Ruckdeschel A, et al. JAM-A as a prognostic factor and new therapeutic target in multiple myeloma. Leukemia (2018) 32:736–43. doi: 10.1038/leu.2017.287
66. Kakuki T, Kurose M, Ichi Takano K, Kondoh A, Obata K, Nomura K, et al. Dysregulation of junctional adhesion molecule-A via p63/GATA-3 in head and neck squamous cell carcinoma. Oncotarget (2016) 7:33887–900. doi: 10.18632/oncotarget.8432
67. Naik MU, Naik TU, Suckow AT, Duncan MK, Naik UP. Attenuation of junctional adhesion molecule-A is a contributing factor for breast cancer cell invasion. Cancer Res (2008) 68:2194–203. doi: 10.1158/0008-5472.CAN-07-3057
68. Orlandella FM, Mariniello RM, Iervolino PLC, Auletta L, De Stefano AE, Ugolini C, et al. Junctional adhesion molecule-A is down-regulated in anaplastic thyroid carcinomas and reduces cancer cell aggressiveness by modulating p53 and GSK3 α/β pathways. Mol Carcinog (2019) 58:1181–93. doi: 10.1002/mc.23001
69. McSherry EA, McGee SF, Jirstrom K, Doyle EM, Brennan DJ, Landberg G, et al. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int J Cancer (2009) 125:1343–51. doi: 10.1002/ijc.24498
70. Tian Y, Tian Y, Zhang W, Wei F, Yang J, Luo X, et al. Junctional adhesion molecule-A, an epithelial-mesenchymal transition inducer, correlates with metastasis and poor prognosis in human nasopharyngeal cancer. Carcinogenesis (2015) 36:41–8. doi: 10.1093/carcin/bgu230
71. Bednarek R, Selmi A, Wojkowska D, Karolczak K, Popielarski M, Stasiak M, et al. Functional inhibition of F11 receptor (F11R/junctional adhesion molecule-A/JAM-A) activity by a F11R-derived peptide in breast cancer and its microenvironment. Breast Cancer Res Treat (2020) 179:325–35. doi: 10.1007/s10549-019-05471-x
72. Murakami M, Giampietro C, Giannotta M, Corada M, Torselli I, Orsenigo F, et al. Abrogation of junctional adhesion molecule-A expression induces cell apoptosis and reduces breast cancer progression. PLoS One (2011) 6(6):e21242. doi: 10.1371/journal.pone.0021242
73. Solimando AG, Da Vià MC, Leone P, Borrelli P, Croci GA, Tabares P, et al. Halting the vicious cycle within the multiple myeloma eco-system: blocking JAM-A on bone marrow endothelial cells restores the angiogenic homeostasis and suppresses tumor progression. Haematologica (2020) 105. doi: 10.3324/haematol.2019.239913
Keywords: autoimmune diseases, cell adhesion, epithelial barrier, F11 receptor, inflammation, junctional adhesion molecule-A
Citation: Bonilha CS, Benson RA, Brewer JM and Garside P (2020) Targeting Opposing Immunological Roles of the Junctional Adhesion Molecule-A in Autoimmunity and Cancer. Front. Immunol. 11:602094. doi: 10.3389/fimmu.2020.602094
Received: 02 September 2020; Accepted: 02 November 2020;
Published: 25 November 2020.
Edited by:
Helen Michelle McGettrick, University of Birmingham, United KingdomReviewed by:
Stefano Angiari, Medical University of Graz, AustriaChristopher V. Carman, Harvard University, United States
Mathieu-Benoit Voisin, Queen Mary University of London, United Kingdom
Scott I. Simon, University of California, Davis, United States
Copyright © 2020 Bonilha, Benson, Brewer and Garside. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul Garside, cGF1bC5nYXJzaWRlQGdsYXNnb3cuYWMudWs=