 Li Li1,2,3
Li Li1,2,3 Yuan-Qiang Lu1,2,3*
Yuan-Qiang Lu1,2,3*- 1Department of Emergency Medicine, School of Medicine, The First Affiliated Hospital, Zhejiang University, Hangzhou, China
- 2Department of Geriatrics, School of Medicine, The First Affiliated Hospital, Zhejiang University, Hangzhou, China
- 3Zhejiang Provincial Key Laboratory for Diagnosis and Treatment of Aging and Physic-Chemical Injury Diseases, Hangzhou, China
High-mobility group box 1 (HMGB1), a prototypical damage-associated molecular pattern (DAMP) molecule, participates in multiple processes of various inflammatory diseases through binding to its corresponding receptors. In the early phase, sepsis is mainly characterized as a multi-bacterial-induced complex, excessive inflammatory response accompanied by the release of pro-inflammatory mediators, which subsequently develops into immune paralysis. A growing number of in vivo and in vitro investigations reveal that HMGB1 plays a pivotal role in the processes of inflammatory response and immunosuppression of sepsis. Therefore, HMGB1 exerts an indispensable role in the immune disorder and life-threatening inflammatory syndrome of sepsis. HMGB1 mainly mediate the release of inflammatory factors via acting on immune cells, pyroptosis pathways and phosphorylating nuclear factor-κB. Moreover HMGB1 is also associated with the process of sepsis-related immunosuppression. Neutrophil dysfunction mediated by HMGB1 is also an aspect of the immunosuppressive mechanism of sepsis. Myeloid-derived suppressor cells (MDSCs), which are also one of the important cells that play an immunosuppressive effect in sepsis, may connect with HMGB1. Thence, further understanding of HMGB1-associated pathogenesis of sepsis may assist in development of promising treatment strategies. This review mainly discusses current perspectives on the roles of HMGB1 in sepsis-related inflammation and immunosuppressive process and its related internal regulatory mechanisms.
Introduction
Sepsis is the body’s maladjusted response to an infection. It is manifest as an excessive systemic, inflammatory immune response in its early stage and as continuous immunosuppression in its later phase. These processes may result in cell dysfunction and ultimately in organ failure (1). Effective sepsis-specific therapies are still lacking. Most current therapeutics focus mainly on supporting dysfunctional organs rather than on cures. Existing therapies include administration of antibiotics to fight against various infections; restoration of fluid balance and use of a vasopressor to maintain perfusion of vital organs; and utilization of mechanical support of failed organs (such as mechanical ventilation in failed lungs, dialysis-like techniques for renal failure, etc.).
Progress has been made on not only in development of technologies but also in comprehensive investigation of the pathogenesis of sepsis and of the diverse interactions between pathogens and hosts. However, sepsis-induced mortality is still unacceptably high typically about 30% and even as much as 40–50% when shock develops (2). In additions, survivors of severe sepsis also show an increased risk of post-sepsis syndrome, such as cognitive impairment and functional disability, which in turn contributes to an elevated risk of various mental and psychological diseases (3).
HMGB1 has been implicated in the pathogenesis of multiple diseases, including cancer, traumatic shock, and autoimmune diseases (4, 5). It has been well proven that systemic HMGB1 levels are markedly increased in murine sepsis models and in patients with sepsis (6). After treatment with lipopolysaccharide (LPS), serum HMGB1 in mice increases from 8 h and peaks in 16 to 32 h (7). The delayed secretion of HMGB1 broadens the time window of treatment opportunities for patients with sepsis using HMGB1 antagonists. Besides, study has shown that reducing the release of HMGB1 can effectively improve the survival of mice suffering from sepsis (8).
In this review, we review some new insights into the roles of HMGB1 associated with inflammation, immunosuppression in sepsis. Understanding the contributions of HMGB1 to the pathogenesis of sepsis may facilitate development of potentially effective therapeutic strategies to shorten the course of the disease as much as possible, and to improve the prognosis and survival rate of sepsis patients.
Insights on HMGB1
HMGB1, a multi-functional and highly conserved nucleoprotein with 215 amino acid residues, includes two tandems, positively charged DNA-binding regions, A box and B box, together with a negatively charged tail consisting of a string of amino acid residues. The A box has no innate pro-inflammatory activity, but can specifically antagonize the cytokine and pro-inflammatory activities of HMGB1 (9). The B box comprises most of the pro-inflammatory properties of HMGB1. Amino acid residues 89-108 of HMGB1, located in the top 20 of B box, are the main region for stimulating cytokine release (10). Two nuclear localization sites (NLS) in the A and B boxes, respectively, regulate the capacity of HMGB1 to be transported into the cytoplasm. HMGB1 consists of three isoforms: disulfide HMGB1, fully reduced HMGB1 and sulfonyl HMGB1, among these disulfide HMGB1 is the only isoform with the pro-inflammatory activity (11).
As one of the earliest confirmed members of the damage-associated molecular pattern family and a prototypical alarm molecule, HMGB1 initiates and maintains the immune response during inflammation (7). The immune function of extracellular HMGB1 is completely dependent on the redox status of its three conserved cysteine residues, at locations 23 and 45 in box A and at location 106 in box B, that regulate receptor binding ability (12). HMGB1 is translocating outside the cell mainly through two pathways: passive release and active secretion. Passive HMGB1 release mainly occurs in different forms of cell death, for instance, pyroptosis, apoptosis, and necrosis (13). HMGB1 released by different forms of dead cells has different redox states. When emancipated after pyroptosis, it is usually in the disulfide form, after apoptosis in the fully oxidized form and after necrosis in fully reduced or disulfide forms (7). Disulfide HMGB1 and fully reduced HMGB1 can be converted to each other according to the redox state of the internal and external environment of the cell, while fully oxidized HMGB1 is an irreversible form (14).
HMGB1 can be actively released by a variety of cells, including endothelial cells, hepatocytes, macrophages, and monocytes (6). Active HMGB1 release is a complex process. First, transport from the nucleus into the cytoplasm depends on posttranslational modifications of NLS such as acetylation, methylation, and phosphorylation, which may in turn be regulated by the JAK/STAT1 signaling pathway and calcium/calmodulin-dependent protein kinase (CaMK) IV (6). Secondly, cytoplasmic HMGB1 is released to the extracellular environment by induction of programmed pro-inflammatory cell pyroptosis or by exocytosis of secretory lysosome the same pathway as IL-1β secretion (15). HMGB1 is commonly detected in nucleated mammalian cells, and its activity is closely related to its cellular location. During homeostasis, nuclear HMGB1 performs an essential role in maintaining the stability of cell functions through its involvement in multiple processes such as DNA replication, transcription, stabilizing nucleosome formation, and regulating gene expression (16). In response to various injuries and inflammatory stimuli, HMGB1 is transported from the nucleus to the cytoplasm and participates in important pathways, for instance, in promotion of inflammasome activation, initiation of autophagy, and regulation of apoptosis (17–19). Once released to the extracellular environment, HMGB1 attains the vital ability to induce chemokine/cytokine production, stimulate neutrophil extracellular trap formation, involvement in neuroimmune or metabolic activities, and other activities (6). Translocation of HMGB1 from the nucleus to the cytoplasm and ultimate extracellular secretion are also key processes for HMGB1-mediated inflammation. Advanced glycation end products (RAGE) and toll-like receptor 4 (TLR4) are important members of the innate immune system (20). HMGB1 exerts its biological function mainly through binding to any of these two receptors (21).
The Role of HMGB1 in Inflammation
Pro-inflammatory cytokines, released from various immune cells, are considered to be essential mediators in the lethal effects of endotoxins. Excessive release of cytokines is closely associated with tissue damage, multiple-organ dysfunction or failure, and even death (22). As an effective pro-inflammatory cytokine, HMGB1 plays a role in various inflammatory diseases, especially in sepsis. In both experimental sepsis models and in sepsis patients (7, 11, 12), the levels of HMGB1 in circulation are remarkably increased, and the concentration of circulating HMGB1 is positively correlated with the severity of inflammation and disease. HMGB1 can also induce inflammatory anemia and cognitive impairment, which is considered to be related to neuroinflammation, among sepsis survivors (15, 23). Compared with other early -phase pro-inflammatory contents, for instance tumor necrosis factor (TNF) and IL-1β, HMGB1 began to appear 8 h and significantly increased after initiation of sepsis (9). Thus, HMGB1 offers a relatively wider time window for clinical treatment of sepsis patients with progressive inflammation.
The early stage of sepsis is accompanied by excessive inflammation, which is the main initial actor of organ dysfunction. Because continued inflammation can have serious consequences, including organ failure, it seems a reasonable therapeutic goal to the auto-inflammatory response to avoid excessive activation of the immune response and a cascade of effects that induce cell and tissue damage. Earlier studies have demonstrated that blocking strong expression of HMGB1 in inflammatory diseases, such as rheumatoid arthritis, myositis, and systemic lupus erythematosus can attenuate inflammation (24). Recent studies have found that targeting HMGB1 can reduce inflammatory response which in turn reduces sepsis associated organ damage (25–28). Therefore, antagonizing HMGB1 in serum or intervening in the pathway of HMGB1 mediated pro-inflammatory pathways may facilitate development of potential therapies in sepsis.
HMGB1 and Immune Cells
HMGB1 promotes sepsis-induced organ dysfunction through suppressing neutrophil ability to clear bacteria, and so enhancing persistent inflammation. In both mice subjected to sepsis and patients surviving septic shock, HMGB1 decreased the capacity of neutrophil to kill bacteria through mediating neutrophil nicotinamide adenine dinucleotide phosphate (NADPH) oxidase dysfunction (21). Interestingly, in an abdominal sepsis mice model, Zhou et al. manifested that platelet-derived HMGB1 facilitated neutrophil activation and reactive oxygen species (ROS) generation, which were critical for the ability of neutrophil to promote bacterial clearance (29). These contradictory results indicate that the functions of HMGB1 in sepsis are complex and different. Perhaps the final balance of these effects determines whether the role of HMGB1 in sepsis is beneficial or harmful. Additionally, investigation showed that platelet-derived HMGB1 stimulated neutrophil extracellular traps (NETs) formation in septic shock patients and sepsis mouse model induced by cecal ligation and puncture (CLP) (30). Research on an abdominal septic animal model also showed that levels of NETs were significantly increased. These increased NETs could recruit neutrophils to the lung and promote the formation of pro-inflammatory compounds in pulmonary (31). Apart from this effect, NETs could also stimulate macrophages to secrete massive inflammatory mediators, tumor necrosis factor-α (TNF-α), HMGB1, which boost systemic inflammation (31). In an animal model of pulmonary fibrosis, interaction of HMGB1 with phosphatidylserine on the surfaces of apoptotic neutrophils inhibited the engulf of apoptotic neutrophils by macrophages (32). Therefore, apoptotic neutrophils remained to generate pro-inflammatory contents of reactive oxygen species (ROS), which aggravated the pulmonary pro-inflammatory environment and enhanced the lung tissue injury. Whether HMGB1 plays the same role in sepsis is question that requires additional investigation. As a member of innate immune cells, macrophages act a vital role in initiating body’s immune response. Studies on both patients with sepsis and sepsis animal model indicated that HMGB1 could facilitate the release of inflammatory cytokines from macrophages, such as: TNF-α, interleukin-6 (IL-6) (33). However, the specific internal regulation mechanism is still unclear. HMGB1 can not only promote the release of inflammatory factors from macrophages, but also facilitate the conversion of macrophages to an inflammatory phenotype. In experimental mouse model of autoimmune myocarditis, HMGB1 promoted macrophage transforming into M1-like Phenotype owning pro-inflammatory activity in vitro (34). Loss of endothelial cells (ECs) integrity can cause inflammatory substances to leak into surrounding tissues, and so facilitate tissue damage and inflammation (35). HMGB1 in circulation from patients subjected to sepsis promoted ECs apoptosis, which in turn increased endothelial (36). The Internal mechanism may be related to the expression levels of some proteins mediated by HMGB1 (Figure 1A).
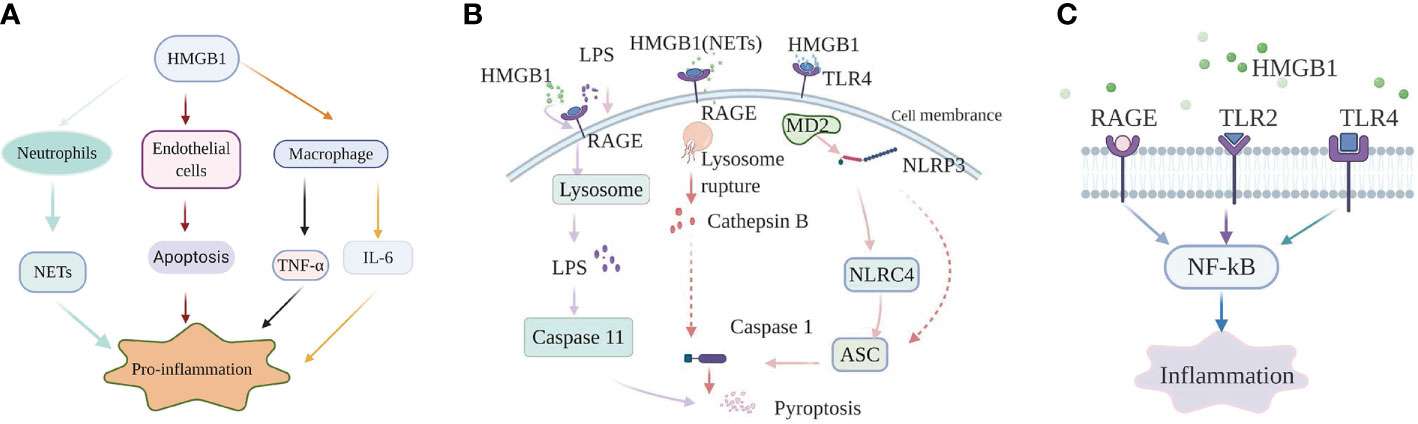
Figure 1 High-mobility group box-1 (HMGB1)-associated regulatory pathways of inflammation in sepsis. (A) HMGB1 and immune cells. HMGB1 promote NETs formation of neutrophil, ECs apoptosis to mediate inflammation; HMGB1 can also directly promote macrophages to release inflammation factors, such as: TNF-α, IL-6. (B) The role of HMGB1 in pyroptosis. Extracellular HMGB1 induces both canonical and noncanonical pyroptosis pathways by binding to their corresponding receptors. (C) HMGB1-RAGE/TLRs-NF-κB signaling pathways. HMGB1 binds to receptors, containing receptor for advanced glycation end products (RAGE), Toll-like receptor 2 (TLR2), and Toll-like receptor 4 (TLR4) on the cell membrane, to phosphorylate nuclear factor-κB, which regulates the generation of inflammatory mediators.
HMGB1 and Pyroptosis
In sepsis, pyroptosis is a special form of programmed cell death, which is accompanied by release of multiple inflammatory compounds, including IL-18 and IL-1β. Normal pyroptosis defends the host from bacterial infection and reduces organ damage. However, excessive pyroptosis can lead to multiple organ dysfunction or failure and septic shock, or to increased chances of secondary infection (37, 38). Pyroptosis contains two chief pathways: one canonical, caspase 1-dependent pathway, and a noncanonical pathway associated with caspase4/5 and caspase11 (39). Caspase4/5 mainly regulates human-related pyroptosis, while caspase11 mainly participates in animal-related pyroptosis.
The nucleotide binding and oligomerization domain-like receptor family pyrin domain-containing protein 3 (NLRP3) inflammasome, a molecule is highly sensitive to both DAMPs and pathogen-associated molecular patterns (PAMPs). When oxidized to form cysteine 23-cysteine 45 bonds, HMGB1 can signal through TLR4-MD2 and act as a priming factor for NLRP3 inflammasome activation (40). When activated by HMGB1, NLRP3 can activate caspase-1 through recruiting nucleotide-binding domain and leucine-rich repeat (NLR) family, caspase recruitment domain (CARD) containing 4 (NLRC4), apoptosis associated speck like protein (ASC), and finally initiating a canonical pathway of pyroptosis and prompting the production of inflammatory factors in sepsis (41). Other research on sepsis model also indicated that hepatocyte-derived HMGB1 transported LPS into the cytosol of macrophages and endothelial cells, via RAGE-mediated internalization (23). Subsequently, LPS activated a noncanonical pyroptosis pathway induced by caspase-11, which in turn increased inflammation and the lethality of sepsis (23). Furthermore, neutralizing extracellular HMGB1 or inhibition of HMGB1-LPS binding could prevent caspase-11-dependent pyroptosis and death in endotoxemia (23). Caspase-11-mediated pyroptosis of renal tubular epithelial cells is also a pivotal point during septic acute kidney injury (42). However, caspase-11 knockout can attenuate pathological kidney damage and improve survival, in both in vitro and in vivo experiments in mice (42). In addition, investigation also revealed that HMGB1 alone initiated ASC-dependent and caspase-11-independent pyroptosis (43). Yang et al. (44) indicated that in hemorrhagic shock, HMGB1 stimulates lung endothelial cell (EC) endocytosis through the RAGE receptor, which in turn facilitates the activation and release of cathepsin B from ruptured lysosomes. This process is then followed by the formation of pyroptosomes and activation of caspase-1, leading to EC pyroptosis and increased lung damage (44). The lung is the most readily targeted organ during the process of sepsis. Thus, it would be worthwhile investigating whether the pathway plays the same role in sepsis and whether manipulation of that pathway could provide an effective intervention. Similarly, in a septic mouse model, NETs-derived HMGB1 induced macrophage pyroptosis through the same signaling pathway demonstrated by Yang et al. (45). Recent investigation performed by Wang et al. (46) manifested that HMGB1 acted as an vital intermediary molecule in the process of TNF-α induce pyroptosis in M1 macrophages in the animal model of acute liver failure induced by LPS (Figure 1B).
HMGB-RAGE/TLRs-NF-κB Signaling Pathways
HMGB1 exerts its pro-inflammatory effects mainly by binding to multiple membrane receptor proteins, including TLR2, RAGE, and TLR4 (16). The important role of the HMGB1-RAGE/TLRs axis is to phosphorylate nuclear factor-κB (NF-κB), facilitating secretion of various proinflammatory cytokines (47).
In sepsis-induced animal model of lung damage, study show that the protective role of Xuebijing (XBJ) may be via inhibiting HMGB1/RAGE axis to decrease pro-inflammatory cytokines releases (48). Additionally, literature documented the combination of HMGB1 and RAGE can trigger a series of signal transduction pathways, which can directly activate NF-κB or indirectly through the mitogen-activated protein kinase (MAPKs) pathway or other ways, thereby promoting the release of inflammatory factors (49). However, the specific details of the activation of the NF-κB pathway by the HMGB1-RAGE axis are not yet clear in sepsis. Moreover, multiple studies on animal models of sepsis found that suppression HMGB1/TLR4/NF-κB signaling pathway could attenuate inflammation response and ultimately reduce the damage of vital organs as well (50, 51). It is worth noting that in a sepsis rat model, serum-derived HMGB1 accumulates in the renal tissue and urine and turn renal tubular epithelial cells (TECs) into inflammatory promoter mediators, which facilitate the release of pro-inflammatory cytokines through binding to TLR4. Moreover, The process accompanied by the activation of MAPK and NF-κB as well (52). Apart from this, study also show that TLR4 and TLR2 receptors convey signals through a MyD88-dependent/independent pathway, which can activate NF-κB (53, 54).
The studies listed above all show that HMGB1 can mediate inflammation response in sepsis by binding to RAGE or TLRs. However, literature also report if TLR4 is functionally inactivated or absent, macrophages containing both RAGE and TLR4 receptors, cannot generate cytokines when initiated by any isoforms of HMGB1 (55). Besides, in LPS-induced mouse model of acute lung injury (ALI), HMGB1 can simultaneously activate the TLR4, TLR2, and RAGE/NF-κB signaling pathways to promote the activation of absent in melanoma 2 (AIM2) inflammasome in macrophage and the polarization of M1 macrophages, followed by production of interleukin−1β (IL-1β) and IL-6 (56). Therefore, the specific mechanism of HMGB1/TLRs/RAGE/NF-κB pathways that mediate inflammation in sepsis need further exploration (Figure 1C).
Therefore, HMGB1 participates in the release of pro-inflammatory factors in sepsis in a variety of ways. Whether these pro-inflammatory pathways affect each other or which is the dominant pathway and the specific regulation process of these pathways need further study.
The Role of HMGB1 in Immunosuppression
It is well known that following an early cytokine storm, late phase sepsis is always accompanied by long-term chronic inflammation and sustained immunosuppression (57, 58). Patients with severe sepsis have a long course of disease and usually exhibit persistent inflammation-immunosuppression and catabolic syndromes, leading to many adverse clinical consequences. The specific mechanisms of the syndrome are currently unknown. However, increasing evidence indicates that alteration of myelopoiesis, expansion of immature myeloid-derived suppressor cells (MDSCs), and reduction of effector T-cell function, are all contributors to the immunosuppressive pathology of sepsis. HMGB1 plays a unique role in the development of post-sepsis immunosuppression. Dynamic monitoring indexes of HMGB1 and cellular immunity are vital for assessing chronic inflammation processes, immune status, and prognoses of patients with severe sepsis (59). However, a consensus on how HMGB1 applies its immunomodulatory function in sepsis remains to be further demonstrated.
HMGB1 and Neutrophils
High level of HMGB1 acts as a specific role in the development of immunosuppression during late phase of sepsis. Research revealed that HMGB1 could inhibit NADPH oxidase activation, leading to a deficiency in oxidative burst and dysfunction of neutrophil-dependent bacterial killing mechanisms in an experimental sepsis model and in patients surviving septic shock (21). Additionally, these neutrophils with immunosuppressive phenotypes not only participate in the development of late-phase immunosuppression, but also affect newly generated neutrophils.
Early research also demonstrated that HMGB1 binding to RAGE receptor could effectively inhibit NADPH oxidase activation in neutrophils (60). However, the mechanism needs to be further confirmed in sepsis.
HMGB1 and MDSCs
Decreasing the numbers of immune cells caused by apoptosis of lymphocytes and imbalance of immune function is one of the main mechanisms of immunosuppression in sepsis (61). Myeloid-derived suppressor cells (MDSCs) have recently been correlated with the profound immunosuppression in sepsis and can suppress T-cell proliferation via a variety of mechanisms (62). MDSCs not only inhibit effector T cells, but they also mediate the expansion of regulatory T cells (Tregs) a recognized type of immunosuppressive cell under inflammatory conditions (63). One important factor is that release of ROS molecules is also one of the main mechanisms by which MDSCs inhibit T cells (64, 65).
Generation of ROS is essential not only for the immunosuppressive function of MDSCs, but also seems to keep these cells in an undifferentiated state (66). ROS is one of the critical regulators in the control of HMGB1 release (5). However, whether ROS derived from MDSCs promotes the production of HMGB1 needs further verification. In the tumor micro-environment, HMGB1 maintains the survival of MDSCs through facilitating autophagy, which promotes the potency of MDSCs to restrain anti-tumor immunity and accelerate tumor progression (67). Thus, it is meaningful to speculate whether HMGBI promotes the survival of MDSCs and maintains the immunosuppressive function of MDSCs via inducing autophagy of MDSCs in sepsis. Li et al. suggested that in tumors, the release of HMGB1 mediated by inflammasomes further induces the expression of CD274/PD-L1, which results in immunosuppression (68). T-cell depletion in sepsis is caused mainly by programmed cell death-1 (PD-1) interaction with its ligand (PD-L1), and the use of antibodies to PD-L1 can improve the survival rate in sepsis (69). Moreover, investigation recently show that MDSCs might act a immunosuppressive effect via PD-L1/PD-1 axis in sepsis mouse model (70). Whether any of these molecules are mediated by HMGB1 or whether HMGB1 promotes the survival of MDSCs through other specific pathways or enhances its immunosuppressive capacity in sepsis is still unknown and needs further research. There have been many reports about MDSCS inhibiting T cell proliferation and function. However, few investigations have focused on the role of MDSCs on B-cells. Recent research shows that human M-MDSCs, significantly inhibits function of human B cells and alters the subsets of B-cell in vitro (71). Therefore, MDSCs could play an important role in immunosuppression, which might be caused by simultaneously inhibiting the functions of T cells and B cells. Thence, exploring the role between HMGB1 and MDSCs is of great significance in discovering new sepsis-related immunotherapy strategies.
In contrast, Liu et al. have concluded that HMGB1 could reverse immunosuppression in sepsis, when released at moderate levels (72). Only when HMGB1 is over-released can it facilitate immune paralysis. Furthermore, once activated by caspase-1, HMGB1 can antagonize apoptosis-induced tolerance through the RAGE signaling pathway in dendritic cells (61). Thence, Whether HMGB1 exerts immunosuppressive effects may also be related to the concentration and activation mode of HMGB1.
Conclusion
HMGB1 is a contradictory molecule in sepsis. It not only mediates inflammation response, but also plays an important role in the immunosuppression of sepsis. In this review, we mainly introduce the relevant pathways of HMGB1 in mediating inflammation in sepsis. At present, there are relatively few studies on the immunosuppressive effect of HMGB1 in sepsis. However, the immunosuppressive effect of MDSCs in sepsis and the related mechanism have been preliminary studied. Exploring the possible regulatory mechanism between HMGB1 and MDSCs in sepsis may provide new ideas for the immunotherapy of sepsis. It is regrettable that there are relatively few investigations on when HMGB1 acts a pro-inflammatory or immunosuppressive effect in sepsis. Therefore, in order to provide more accurate targeted therapy for sepsis, more detailed studies on HMGB1 are needed.
Author Contributions
LL conceived and completed the manuscript. Y-QL supervised and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This review was supported by the grants from the Opening Foundation of State Key Laboratory for the Diagnosis and Treatment of Infectious Diseases (Nos. 2018KF02, and SKLID2019KF06), the National Natural Science Foundation of China (Nos. 81272075 and 81801572), and the Foundation of Key Discipline Construction of Zhejiang Province for Traditional Chinese Medicine (No. 2017-XK-A36).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. van der Poll T, van de Veerdonk F, Scicluna B, Netea M. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol (2017) 17:407–20. doi: 10.1038/nri.2017.36
2. Cohen J, Vincent JL, Adhikari NK, Machado FR, Angus DC, Calandra T, et al. Sepsis: a roadmap for future research. Lancet Infect Dis (2015) 15:581–614. doi: 10.1016/S1473-3099(15)70112-X
3. Huang CY, Daniels R, Lembo A, Hartog C, O’Brien J, Heymann T, et al. Life after sepsis: an international survey of survivors to understand the post-sepsis syndrome. Int J Qual Health Care (2019) 31:191–98. doi: 10.1093/intqhc/mzy137
4. Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME. HMGB1 as biomarker and drug target. Pharmacol Res (2016) 111:534–44. doi: 10.1016/j.phrs.2016.06.031
5. Yu Y, Tang D, Kang R. Oxidative stress-mediated HMGB1 biology. Front Physiol (2015) 6:93. doi: 10.3389/fphys.2015.00093
6. Deng M, Scott MJ, Fan J, Billiar TR. Location is the key to function: HMGB1 in sepsis and trauma-induced inflammation. J Leukoc Biol (2019) 106:161–69. doi: 10.1002/JLB.3MIR1218-497R
7. Yang H, Wang H, Chavan SS, Andersson U. High Mobility Group Box Protein 1 (HMGB1): The Prototypical Endogenous Danger Molecule. Mol Med (2015) 21 Suppl 1:S6–S12. doi: 10.2119/molmed.2015.00087
8. Yang S, Lee W, Lee BS, Lee C, Park EK, Ku SK, et al. Aloin Reduces HMGB1-Mediated Septic Responses and Improves Survival in Septic Mice by Activation of the SIRT1 and PI3K/Nrf2/HO-1 Signaling Axis. Am J Chin Med (2019) 47:613–33. doi: 10.1142/S0192415X19500320
9. Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A (2004) 101:296–301. doi: 10.1073/pnas.2434651100
10. Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, et al. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med (2003) 9:37–45. doi: 10.1007/BF03402105
11. Andersson U, Yang H, Harris H. Extracellular HMGB1 as a therapeutic target in inflammatory diseases. Expert Opin Ther Targets (2018) 22:263–77. doi: 10.1080/14728222.2018.1439924
12. Magna M, Pisetsky DS. The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol Med (2014) 20:138–46. doi: 10.2119/molmed.2013.00164
13. Andersson U, Yang H, Harris H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin Immunol (2018) 38:40–8. doi: 10.1016/j.smim.2018.02.011
14. Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol (2013) 93:865–73. doi: 10.1189/jlb.1212662
15. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature (2012) 488:670–4. doi: 10.1038/nature11290
16. Pellegrini L, Foglio E, Pontemezzo E, Germani A, Russo MA, Limana F. HMGB1 and repair: focus on the heart. Pharmacol Ther (2019) 196:160–82. doi: 10.1016/j.pharmthera.2018.12.005
17. Zhu X, Messer JS, Wang Y, Lin F, Cham CM, Chang J, et al. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J Clin Invest (2015) 125:1098–110. doi: 10.1172/JCI76344
18. Kang R, Livesey KM, Zeh HJ, Loze MT, Tang D. HMGB1: a novel Beclin 1-binding protein active in autophagy. Autophagy (2010) 6:1209–11. doi: 10.4161/auto.6.8.13651
19. Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol (2010) 185:4385–92. doi: 10.4049/jimmunol.1000803
20. Paudel YN, Angelopoulou E, Piperi C, Othman I, Aamir K, Shaikh MF. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells (2020) 9. doi: 10.3390/cells9020383
21. Gregoire M, Tadie JM, Uhel F, Gacouin A, Piau C, Bone N, et al. Frontline Science: HMGB1 induces neutrophil dysfunction in experimental sepsis and in patients who survive septic shock. J Leukoc Biol (2017) 101:1281–87. doi: 10.1189/jlb.5HI0316-128RR
22. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol (2017) 39:517–28. doi: 10.1007/s00281-017-0639-8
23. Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity (2018) 49:740–53.e7. doi: 10.1016/j.immuni.2018.08.016
24. Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol (2012) 8:195–202. doi: 10.1038/nrrheum.2011.222
25. Chen L, Lu Q, Deng F, Peng S, Yuan J, Liu C, et al. miR-103a-3p Could Attenuate Sepsis-Induced Liver Injury by Targeting HMGB1. Inflammation (2020) 43:2075–86. doi: 10.1007/s10753-020-01275-0
26. Xu L, Hu G, Xing P, Zhou M, Wang D. Paclitaxel alleviates the sepsis-induced acute kidney injury via lnc-MALAT1/miR-370-3p/HMGB1 axis. Life Sci (2020) 262:118505. doi: 10.1016/j.lfs.2020.118505
27. Li ZL, Gao M, Yang MS, Xiao XF, Liu JJ, Yang BC. Sesamin attenuates intestinal injury in sepsis via the HMGB1/TLR4/IL-33 signalling pathway. Pharm Biol (2020) 58:898–904. doi: 10.1080/13880209.2020.1787469
28. Yang P, Xiong W, Chen X, Liu J, Ye Z. Overexpression of miR-129-5p Mitigates Sepsis-Induced Acute Lung Injury by Targeting High Mobility Group Box 1. J Surg Res (2020) 256:23–30. doi: 10.1016/j.jss.2020.05.101
29. Zhou H, Deng M, Liu Y, Yang C, Hoffman R, Zhou J, et al. Platelet HMGB1 is required for efficient bacterial clearance in intra-abdominal bacterial sepsis in mice. Blood Adv (2018) 2:638–48. doi: 10.1182/bloodadvances.2017011817
30. Jiao Y, Li W, Wang W, Tong X, Xia R, Fan J, et al. Platelet-derived exosomes promote neutrophil extracellular trap formation during septic shock. Crit Care (2020) 24:380. doi: 10.1186/s13054-020-03082-3
31. Luo L, Zhang S, Wang Y, Rahman M, Syk I, Zhang E, et al. Proinflammatory role of neutrophil extracellular traps in abdominal sepsis. Am J Physiol Lung Cell Mol Physiol (2014) 307:L586–96. doi: 10.1152/ajplung.00365.2013
32. Liu G, Wang J, Park Y, Tsuruta Y, Lorne E, Zhao X, et al. High mobility group protein-1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J Immunol (2008) 181:4240–6. doi: 10.4049/jimmunol.181.6.4240
33. Zhu C, Chen T, Liu B. Inhibitory effects of miR-25 targeting HMGB1 on macrophage secretion of inflammatory cytokines in sepsis. Oncol Lett (2018) 16:5027–33. doi: 10.3892/ol.2018.9308
34. Su Z, Zhang P, Yu Y, Lu H, Liu Y, Ni P, et al. HMGB1 Facilitated Macrophage Reprogramming towards a Proinflammatory M1-like Phenotype in Experimental Autoimmune Myocarditis Development. Sci Rep (2016) 6:21884. doi: 10.1038/srep21884
35. Kumar P, Shen Q, Pivetti C, Lee E, Wu M, Yuan SJ. Erimm: Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med (2009) 11:e19. doi: 10.1017/s1462399409001112
36. Zheng YJ, Xu WP, Ding G, Gao YH, Wang HR, Pan SM. Expression of HMGB1 in septic serum induces vascular endothelial hyperpermeability. Mol Med Rep (2016) 13:513–21. doi: 10.3892/mmr.2015.4536
37. Aglietti RA, Dueber EC. Recent Insights into the Molecular Mechanisms Underlying Pyroptosis and Gasdermin Family Functions. Trends Immunol (2017) 38:261–71. doi: 10.1016/j.it.2017.01.003
38. Pfalzgraff A, Heinbockel L, Su Q, Brandenburg K, Weindl G. Synthetic anti-endotoxin peptides inhibit cytoplasmic LPS-mediated responses. Biochem Pharmacol (2017) 140:64–72. doi: 10.1016/j.bcp.2017.05.015
39. Ruan J, Wang S, Wang J. Mechanism and regulation of pyroptosis-mediated in cancer cell death. Chem Biol Interact (2020) 323:109052. doi: 10.1016/j.cbi.2020.109052
40. Frank MG, Weber MD, Fonken LK, Hershman SA, Watkins LR, Maier SF. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: A role for the NLRP3 inflammasome. Brain Behav Immun (2016) 55:215–24. doi: 10.1016/j.bbi.2015.10.009
41. Gao YL, Zhai JH, Chai YF. Recent Advances in the Molecular Mechanisms Underlying Pyroptosis in Sepsis. Mediators Inflamm (2018) 2018:5823823. doi: 10.1155/2018/5823823
42. Ye Z, Zhang L, Li R, Dong W, Liu S, Li Z, et al. Caspase-11 Mediates Pyroptosis of Tubular Epithelial Cells and Septic Acute Kidney Injury. Kidney Blood Press Res (2019) 44:465–78. doi: 10.1159/000499685
43. Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ (2014) 21:1229–39. doi: 10.1038/cdd.2014.40
44. Yang J, Zhao Y, Zhang P, Li Y, Yang Y, Yang Y, et al. Hemorrhagic shock primes for lung vascular endothelial cell pyroptosis: role in pulmonary inflammation following LPS. Cell Death Dis (2016) 7:e2363. doi: 10.1038/cddis.2016.274
45. Chen L, Zhao Y, Lai D, Zhang P, Yang Y, Li Y, et al. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis (2018) 9:597. doi: 10.1038/s41419-018-0538-5
46. Wang Y, Zhang H, Chen Q, Jiao F, Shi C, Pei M, et al. TNF-alpha/HMGB1 inflammation signalling pathway regulates pyroptosis during liver failure and acute kidney injury. Cell Prolif (2020) 53:e12829. doi: 10.1111/cpr.12829
47. Pillai SS, Sugathan JK, Indira M, Selenium downregulates RAGE. and NFkappaB expression in diabetic rats. Biol Trace Elem Res (2012) 149:71–7. doi: 10.1007/s12011-012-9401-1
48. Wang Q, Wu X, Tong X, Zhang Z, Xu B, Zhou W. Xuebijing Ameliorates Sepsis-Induced Lung Injury by Downregulating HMGB1 and RAGE Expressions in Mice. Evid Based Complement Alternat Med (2015) 2015:860259. doi: 10.1155/2015/860259
49. Chuah YK, Basir R, Talib H, Tie TH, Nordin N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int J Inflam (2013) 2013:403460. doi: 10.1155/2013/403460
50. Sun J, Shi S, Wang Q, Yu K, Wang R. Continuous hemodiafiltration therapy reduces damage of multi-organs by ameliorating of HMGB1/TLR4/NFkappaB in a dog sepsis model. Int J Clin Exp Pathol (2015) 8:1555–64.
51. Li H, Qiu D, Gao Q, Wang H, Sun M. [Selectively activating melanocortin 4 receptor acts against rat sepsis-induced acute liver injury via HMGB1/TLR4/NF-kappaB signaling pathway]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2016) 32:1055–9.
52. Zheng S, Pan Y, Wang C, Liu Y, Shi M, Ding G, et al. HMGB1 Turns Renal Tubular Epithelial Cells into Inflammatory Promoters by Interacting with TLR4 During Sepsis. J Interferon Cytokine Res (2016) 36:9–19. doi: 10.1089/jir.2015.0067
53. Steinle JJ. Role of HMGB1 signaling in the inflammatory process in diabetic retinopathy. Cell Signal (2020) 73:109687. doi: 10.1016/j.cellsig.2020.109687
54. Feng X, Yang R, Tian Y, Miao X, Guo H, Gao F, et al. HMGB1 protein promotes glomerular mesangial matrix deposition via TLR2 in lupus nephritis. J Cell Physiol (2020) 235:5111–19. doi: 10.1002/jcp.29379
55. Yang H, Wang H, Andersson U. Targeting Inflammation Driven by HMGB1. Front Immunol (2020) 11:484. doi: 10.3389/fimmu.2020.00484
56. Wang J, Li R, Peng Z, Hu B, Rao X, Li J. [Corrigendum] HMGB1 participates in LPSinduced acute lung injury by activating the AIM2 inflammasome in macrophages and inducing polarization of M1 macrophages via TLR2, TLR4, and RAGE/NFkappaB signaling pathways. Int J Mol Med (2020) 45:1628. doi: 10.3892/ijmm.2020.4530
57. Otto GP, Sossdorf M, Claus RA, Rodel J, Menge K, Reinhart K, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care (2011) 15:R183. doi: 10.1186/cc10332
58. Stortz JA, Hollen MK, Nacionales DC, Horiguchi H, Ungaro R, Dirain ML, et al. Old Mice Demonstrate Organ Dysfunction as well as Prolonged Inflammation, Immunosuppression, and Weight Loss in a Modified Surgical Sepsis Model. Crit Care Med (2019) 47:e919–e29. doi: 10.1097/CCM.0000000000003926
59. Yu H, Qi Z, Zhao L, Shao R, Fang Y, laboratory LCJC. Prognostic Value of Dynamic Monitoring of Cellular Immunity and HMGB1 in Severe Sepsis: Delayed Chronic Inflammation may be the Leading Cause of Death in Late Severe Sepsis. Clin Lab (2016) 62:2379–85. doi: 10.7754/Clin.Lab.2016.160530
60. Tadie JM, Bae HB, Banerjee S, Zmijewski JW, Abraham E. Differential activation of RAGE by HMGB1 modulates neutrophil-associated NADPH oxidase activity and bacterial killing. Am J Physiol Cell Physiol (2012) 302:C249–56. doi: 10.1152/ajpcell.00302.2011
61. Kumar V. T cells and their immunometabolism: A novel way to understanding sepsis immunopathogenesis and future therapeutics. Eur J Cell Biol (2018) 97:379–92. doi: 10.1016/j.ejcb.2018.05.001
62. Cheng L, Wang J, Li X, Xing Q, Du P, Su L, et al. Interleukin-6 induces Gr-1+CD11b+ myeloid cells to suppress CD8+ T cell-mediated liver injury in mice. PLoS One (2011) 6:e17631. doi: 10.1371/journal.pone.0017631
63. Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol (2011) 11:802–7. doi: 10.1016/j.intimp.2011.01.003
64. Chen X, Song M, Zhang B, Zhang Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid Med Cell Longev (2016) 2016:1580967. doi: 10.1155/2016/1580967
65. Zhang H, Li ZL, Ye SB, Ouyang LY, Chen YS, He J, et al. Myeloid-derived suppressor cells inhibit T cell proliferation in human extranodal NK/T cell lymphoma: a novel prognostic indicator. Cancer Immunol Immunother (2015) 64:1587–99. doi: 10.1007/s00262-015-1765-6
66. Ohl K, Tenbrock K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front Immunol (2018) 9:2499. doi: 10.3389/fimmu.2018.02499
67. Parker KH, Horn LA, Ostrand-Rosenberg S. High-mobility group box protein 1 promotes the survival of myeloid-derived suppressor cells by inducing autophagy. J Leukoc Biol (2016) 100:463–70. doi: 10.1189/jlb.3HI0715-305R
68. Li C, Zhang Y, Cheng X, Yuan H, Zhu S, Liu J, et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev Cell (2018) 46:441–55.e8. doi: 10.1016/j.devcel.2018.07.012
69. Shindo Y, McDonough JS, Chang KC, Ramachandra M, Sasikumar PG, Hotchkiss RS. Anti-PD-L1 peptide improves survival in sepsis. J Surg Res (2017) 208:33–9. doi: 10.1016/j.jss.2016.08.099
70. Ruan WS, Feng MX, Xu J, Xu YG, Song CY, Lin LY, et al. Early Activation of Myeloid-Derived Suppressor Cells Participate in Sepsis-Induced Immune Suppression via PD-L1/PD-1 Axis. Front Immunol (2020) 11:1299. doi: 10.3389/fimmu.2020.01299
71. Jaufmann J, Lelis FJN, Teschner AC, Fromm K, Rieber N, Hartl D, et al. Human monocytic myeloid-derived suppressor cells impair B-cell phenotype and function in vitro. Eur J Immunol (2020) 50:33–47. doi: 10.1002/eji.201948240
Keywords: sepsis, HMGB1, inflammation, pyroptosis, immunosuppression
Citation: Li L and Lu Y-Q (2021) The Regulatory Role of High-Mobility Group Protein 1 in Sepsis-Related Immunity. Front. Immunol. 11:601815. doi: 10.3389/fimmu.2020.601815
Received: 01 September 2020; Accepted: 07 December 2020;
Published: 22 January 2021.
Edited by:
Myoungsun Son, Feinstein Institute for Medical Research, United StatesReviewed by:
Adam C. Soloff, University of Pittsburgh, United StatesMarco De Andrea, University of Turin, Italy
Copyright © 2021 Li and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan-Qiang Lu, bHV5dWFucWlhbmdAemp1LmVkdS5jbg==