 Thais Fernanda de Campos Fraga-Silva1,2Sandra Regina Maruyama3Carlos Arterio Sorgi4Elisa Maria de Sousa Russo5Ana Paula Morais Fernandes6Cristina Ribeiro de Barros Cardoso1,5Lucia Helena Faccioli1,5Marcelo Dias-Baruffi5†Vânia Luiza Deperon Bonato1,2*†
Thais Fernanda de Campos Fraga-Silva1,2Sandra Regina Maruyama3Carlos Arterio Sorgi4Elisa Maria de Sousa Russo5Ana Paula Morais Fernandes6Cristina Ribeiro de Barros Cardoso1,5Lucia Helena Faccioli1,5Marcelo Dias-Baruffi5†Vânia Luiza Deperon Bonato1,2*†- 1Basic and Applied Immunology Program, Ribeirao Preto Medical School, University of Sao Paulo, Ribeirao Preto, Sao Paulo, Brazil
- 2Department of Biochemistry and Immunology, Ribeirao Preto Medical School, University of Sao Paulo, Ribeirao Preto, Sao Paulo, Brazil
- 3Department of Genetics and Evolution, Federal University of Sao Carlos, Sao Carlos, Brazil
- 4Department of Chemistry, Faculty of Philosophy, Sciences and Letters of Ribeirao Preto, University of Sao Paulo, Ribeirao Preto, Brazil
- 5Department of Clinical Analysis, Toxicological and Food Science Analysis, School of Pharmaceutical Sciences of Ribeirao Preto, University of Sao Paulo, Ribeirao Preto, Brazil
- 6Department of General and Specialized Nursing, School of Nursing of Ribeirao Preto, University of Sao Paulo, Ribeirao Preto, Brazil
In the last few months, the coronavirus disease 2019 (COVID-19) pandemic has affected millions of people worldwide and has provoked an exceptional effort from the scientific community to understand the disease. Clinical evidence suggests that severe COVID-19 is associated with both dysregulation of damage tolerance caused by pulmonary immunopathology and high viral load. In this review article, we describe and discuss clinical studies that show advances in the understanding of mild and severe illness and we highlight major points that are critical for improving the comprehension of different clinical outcomes. The understanding of pulmonary immunopathology will contribute to the identification of biomarkers in an attempt to classify mild, moderate, severe and critical COVID-19 illness. The interface of pulmonary immunopathology and the identification of biomarkers are critical for the development of new therapeutic strategies aimed to reduce the systemic and pulmonary hyperinflammation in severe COVID-19.
Introduction
Between the end of December 2019 and the beginning 2020 a progressive number of deaths due to aggressive pneumonia in the province of Wuhan, China, led to the identification of a new coronavirus, called Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), as the cause of Coronavirus Disease 2019 (COVID-19) (1–3). The infection spread globally becoming a pandemic event in March 2020 (4). COVID-19 can be asymptomatic or symptomatic, which is mainly characterized by dry cough, intense throat pain and fever, which may be accompanied by headache, diarrhea, and mild, moderate, or severe respiratory failure. Around 80% of the people infected with SARS-CoV-2 develop a mild form of COVID-19. The remaining ~20% develop more severe forms of the disease, and require hospitalization, often accompanied by intensive care and mechanical ventilation. Approximately 5% of these patients may progress to sepsis and critical clinical conditions with unfavorable outcomes until death, which could be associated with pulmonary hyperinflammation and respiratory failure (5–7). The severity of COVID-19 is associated with comorbidities, which include senescence, obesity, hypertension, and cardiovascular diseases, and chronic lung diseases (8). The constellation of factors that coordinate the generation of mild, moderate, and severe forms of the disease and how the comorbidities affect the magnitude of inflammation and anti-viral mechanisms that eliminate viral loads are critical aspects for the development of strategies for physicians to face COVID-19.
Many physicians and researchers worldwide have mobilized to study this new disease. After nine months of the pandemic, we see a significant number of scientific studies on different topics, such as those focused on the characterization of hyperinflammation, cytokine storm syndrome, leukocyte subsets, antibody response, viral antigens profiling, laboratorial, and clinical aspects from patients, vaccine development, and drug repurposing. This reflects the efforts of the global scientific community in the search for understanding and solutions for COVID-19.
COVID-19 has fostered a deep reflection, prompting us to share a more comprehensive view about this pandemic. From our perspective, it is possible to develop an efficient pathway to track and control COVID-19. This would involve the articulation of leaders from each country, including leaders from Universities that show a collaborative research tradition, along with the articulation of researchers with common research interests at the national and international levels. Unfortunately, Brazil represents an example that is far from the best practice, where a very feeble interaction has occurred between the government and the scientific community. Brazilian authorities denied or minimized the threat that COVID-19 represents, as recently reported in The Lancet (9, 10). However, Brazilian foundations for research development have encouraged COVID-19 research, and Brazilian scientists have shown important knowledge about the epidemic transmission and evolutionary trajectories of SARS-CoV-2 lineages in Brazil (11) and the epidemiological and clinical characteristics of the COVID-19 epidemic in Brazil (12). Moreover, Brazilian researchers conducted a multicenter, randomized, open-label, three-group, controlled trial involving hospitalized patients at 55 hospitals with suspected or confirmed mild-moderate COVID-19 to evaluate the effect of hydroxychloroquine, alone or with azithromycin, and reported no benefits on the use of both drugs (13). In other investigation from the same group, a randomized, open label, clinical trial at 57 centers was conducted with 397 severe COVID-19 patients. The inclusion of azithromycin to standard of care showed no improvement in clinical outcomes compared to 183 patients to the control group (standard of care, which included hydroxychloroquine) (14).
Considering that we have experienced a pandemic for the first time, the actions of researchers with common investigation objectives are critical worldwide. COVID-19 has affected all countries and ethnicities; therefore we should use it as an example to generate a worldwide research consortium, to encourage scientific cooperation between different research groups with common interests worldwide, promoting internationalization by increasing partnerships, using standardized approaches and methodologies among the different laboratories.
After this brief reflection, in this review, we will address the advances, key points, and critically analyze the comprehension of pulmonary immunopathology in the COVID-19. The understanding of pulmonary immunopathology will contribute to the identification of biomarkers, the other issue that we will address here in an attempt to classify mild, moderate, and severe COVID-19 illness. The broad understanding of immunopathology and the development of biomarkers are both crucial to follow the progression of infection, to predict the disease severity, and choose the best therapy. These topics are critical for the development of new therapeutic strategies aimed to reduce the hyperinflammation in severe COVID-19.
Innate and Adaptive Immune Response and Inflammation During COVID-19
Biobanking Molecular Profiles, Transcriptome, and COVID-19
Collaborative efforts have been launched worldwide (COVID-19 host genetics initiative; COVID Human Genetic Effort). Countries with well-established health care policies for biobanking molecular profiles of the population are now benefiting from retrospective studies using genomic data linked to COVID-19 registries. Moreover, hundreds of prospective studies to pursue genetic variants associated with susceptibility or resistance to SARS-CoV-2 infection and its complications are being carried out. A genome-wide association study (GWAS) with 1980 COVID-19 patients from Italy and Spain has found a 3p21.31 gene cluster linked to susceptibility to severe disease (15). Within this genomic cluster, one of the target candidates to be investigated is the SLC6A20 gene that codes amino acid (proline) transporter (SIT1), which interacts with ACE-2 (16), the main cell entry for SARS-CoV-2.
The analysis of 700 lung transcriptome samples of patients with comorbidities associated with COVID-19, such as hypertension, chronic obstructive pulmonary disease (COPD) and diabetes, showed that the expression of ACE-2 receptor was higher in those patients compared to controls (17). ACE-2 was identified as the cell entry receptor for SARS-CoV as well as SARS-CoV-2 (2). Therefore, one of the reasons that comorbidities contribute to the development of severe COVID-19 might be the virus entry facilitated by higher expression of the receptor. However, the inflammatory process that characterizes the comorbidities associated with COVID-19 might also play a role in the pulmonary immunopathology and dysfunctions that affect other organs. Of note, a GWAS combined with a transcriptome-wide association study with 2244 patients with a critical COVID-19 form treated in 208 United Kingdom intensive care units (ICU) indicated that COVID-19 severity and mortality might be related to two genetic mechanisms. The first one is associated with susceptibility to SARS-CoV-2 infection in the early disease stages due to low expression of interferon receptor (IFNAR2) and interferon-inducible oligoadenylate synthetase (OAS) genes. The second mechanism is related to late disease stages and lung non-homeostatic inflammation associated with upregulation of tyrosine kinase 2 (TYK2), dipeptidyl peptidase 9 (DPP9), and CC-chemokine receptor 2 (CCR2) genes (18).
The comorbidities suggest that COVID-19 is heterogeneous. Besides older age, comorbidities such as hypertension, coronary artery disease, obesity, type II diabetes, chronic kidney disease, and chronic pulmonary disease are risk factors for severe disease (8). In a meta-analysis with seven COVID-19 studies, it was reported that the most prevalent comorbidities were hypertension (21.1%) and diabetes (9.7%), followed by cardiovascular disease (8.4%) and respiratory system disease (1.5%) (19). In a study with 41 patients, 13 (32%) had underlying diseases including diabetes, cardiovascular disease, hypertension or chronic obstructive pulmonary disease (COPD) (20), while the evaluation of 138 patients in another study revealed that 64 (46.4%) had comorbidities (21). Moreover, patients admitted in the ICU had a higher number of comorbidities (72.2%) compared to those with no admission on ICU (37.3%) (21), suggesting that comorbidities represent additional risk factor for the development of severe COVID-19. This highlights that a single type of therapy may not treat the different clinical presentations of COVID-19, and specific treatments might be important for clearance of viral load versus mitigation of hyperinflammation.
Obesity-induced inflammation and metabolic dysregulation could aggravate COVID-19 illness by pathways dependent on the induction of hypercoagulopathy and the infection of adipose tissue (22). The white adipose tissue (WAT) could represent an additional niche for SARS-CoV-2 replication once obesity induces ACE-2 receptor expression on adipocytes. Moreover, obesity generates deposition of WAT on large airway walls inducing an increase of lung wall thickness. Obesity-induced inflammation causes neutrophil influx, pulmonary damage and fibrosis, while lung wall thickness impairs gas exchange (22). Unexpectedly, transcriptional analysis from two COVID-19 patients showed that levels of lung IL-6 mRNA expression, a biomarker associated to COVID-19 hyperinflammation (23), were similar to those in healthy individuals. Despite that, two types of human lung epithelial adenocarcinoma cell (Calu-3 and A549) and normal human bronchial epithelial cell (NHBE) infected with SARS-CoV-2 showed higher levels of IL-6 mRNA in comparison with uninfected cells (24). Certainly, the mechanisms that trigger pulmonary immunopathology during COVID-19 deserve investigation in an attempt to identify different targets to control inflammation.
Given the difficulty of accessing lung samples from individuals with mild disease and considering the limitation of immunological and inflammatory molecules that can be measured in peripheral blood, the evaluation of receptors and mediators associated with the activation of inflammatory response and other biological pathways through transcriptome analysis is an extremely important approach. Transcriptomics is powerful in providing a landscape of molecular mechanisms involved in any condition whether homeostatic or pathological. A valuable number of pioneering studies focused on transcriptomics of systemic and local responses in COVID-19 patients not only has corroborated the immunological findings, but also has shown how complex and diverse the disease might be. Transcriptome data at single-cell resolution of peripheral response by peripheral blood mononuclear cells (PBMC) transcriptional profiling (25, 26) and local response by bronchoalveolar lavage fluid (BALF) transcriptional profiling (27, 28) depicted the role of type I IFN and chemokines signatures, respectively. At the local site, the participation of neutrophils and monocyte-derived macrophages (24, 27–29) has been highlighted in the inflammatory response of severe cases of COVID-19. However, the transcriptional and functional response of peripheral neutrophils in COVID-19 remains to be explored, because the systemic studies so far have employed PBMC rather than total leukocytes.
Pro-inflammatory responses mediated by IL-1β have been featured in severe outcomes of infection, with transcriptional signature observed both at peripheral (26) and lungs (28). Transcriptome analysis of BALF also shed light on physiopathological processes triggered by SARS-CoV-2 infection, unveiling the role of club cells in mucin hypersecretion and ciliary dysfunction in lung epithelial cells can lead to severe COVID-19 (28). Overall, the transcriptome studies have raised convergent observations, but also have shown differences likely due to targeted cell populations and methodological approaches and mainly, because of the small number of analyzed samples. As soon other transcriptome data become available inquiring a plethora of cohorts, more mechanistic insights underlying the disease will be provided, fostering the search for drug repurposing, as well as novel therapies. Importantly, the role of aging and comorbidities should be taken in consideration in such studies, as they can dramatically affect the transcriptional patterns in tissues.
In the last three months, the differences between COVID-19 patients with or without diabetes started to be reported. Eighty COVID-19 patients were classified in diabetes, secondary hyperglycemia or euglycemia groups. The frequency of severe cases was higher in diabetes patients that showed higher concentrations of C-reactive protein (CRP), lactate dehydrogenase (LDH) and IL-6, lower CD4/CD8 cell ratio compared to other two groups. Moreover, diabetes patients presented diverse multifocal features in CT images (30). In other study with 33 diabetes and 37 non-diabetes COVID-19 patients, those diabetes patients showed no differences in IgG, IgM, IgA, C3, and C4 complement proteins, and lymphocytes compared to non-diabetes patients. However, diabetes patients showed higher neutrophil counts, higher CRP, IgE, and IL-6, TNF and IFN-γ levels (31). These studies show that inflammation triggered by diabetes courses with worse prognosis, severe COVID-19, and pulmonary damage.
Peripheral Inflammation and Immune Response
Higher concentrations of IL-2, IL-6, IL-7, IL-10, IFN-γ, G-CSF, TNF, and chemokines such as CCL2/MCP-1, CCL3/MIP-1a, CXCL10/IP-10 were found in the serum of patients compared with healthy donors (20, 32). The same was reported in a study with 187 patients, with 145 survivors and 28 non-survivors: increased concentrations of IL-6 and IL-10 in all patients compared to normal values (33). A comparison performed with 40 patients showed an increase of IL-2, IL-6, IL-10, and IFN-γ in the serum of 13 patients with the severe form of COVID-19 compared to 27 cases of mild disease (34). A study with 102 patients and 45 healthy controls showed differences only in the concentrations of IL-6 and IL-10 in critical patients compared to those with severe and moderate disease (32). This evidence was also reported in the study that described an increase of IL-6 levels in 31 (30.39%) of 102 patients with mild disease, and in 16 (76.19%) of 21 patients with severe disease (35). Both studies suggest that IL-6 and IL-10 are predictive factors of COVID-19 severity and progression. Moreover, the differences among the reported findings might be associated with comorbidities. In addition, the detrimental role of IL-6-IL-6 receptor (IL-6R) axis in the early phase of SARS-CoV-2 infection was not confirmed in a randomized, double-blind, placebo-controlled trial. In this study, the authors reported that the treatment of 161 COVID-19 patients with a single dose (8 mg/kg) of tocilizumab (anti-IL-6R monoclonal antibody) did not prevent death and intubation of patients with moderate disease (36). It would be important to evaluate the tocilizumab treatment in patients with severe and critical COVID-19 illness.
The cytokine storm in the circulation and the diffuse alveolar damage in the pneumonia of severe COVID-19 patients are features shared with Acute Respiratory Distress Syndrome (ARDS) (37). The peripheral hyperinflammation could also be a consequence of secondary haemophagocytic lymphohistiocytosis (sHLH), a rare syndrome that causes immunological hyperactivation, which was previously associated with viral infections, including SARS-CoV-2 (38). sHLH is characterized by high levels of circulating cytokines and chemokines, and by a reduction in CD8+ T lymphocytes and NK cells (38, 39). Similarly, severe cases of COVID-19 are accompanied by a reduction in all lymphocyte populations: B, CD4+ T, CD8+ T, and NK cells, and by neutrophilia (5, 40). Moreover, serum levels of IL-6, IL-10, and TNF were negatively correlated with the number of circulating T cells (41). Considering that circulating human lymphocytes and monocytes are infected by SARS-CoV-2, as reported in two studies that have not been certified by peer review yet (42, 43), the blood circulating leukocytes may have important implications for disease pathogenesis.
An analysis of 452 patients, 286 with severe COVID-19, exhibited a reduction of CD4+, CD8+ and regulatory T lymphocytes, a reduction of monocytes, eosinophils and basophils, and an increase in the neutrophil-to-lymphocyte ratio (NLR) (44). In an evaluation of 210 patients, 87 developed severe illness and exhibited higher NLR compared to the mild illness group (45). Out of 95 patients, 56 were cured and 39 died, a mortality rate predicted by the NLR (46). Thus, NLR appears to be an early risk factor for severe COVID-19 and should be considered as criteria for classification of severe COVID-19, as well as a criteria for monitoring the treatment (44–46). The increase in the NLR was significantly higher for neutrophils and CD8+ T, but not CD4+ T lymphocytes, in patients needing ICU compared to non-ICU, suggesting that neutrophil/CD8+ T lymphocytes ratio could be a more accurate biomarker for severe COVID-19 (47). However, a high neutrophil/CD4+ T lymphocytes ratio was associated with higher incidence of severe disease during admission, and the authors suggest that a reduction in the neutrophil/CD4+ T lymphocytes ratio could be a biomarker for virus negative conversion (48). These results were confirmed in a multicenter retrospective cohort with 548 cases with discharged or deceased outcomes from 575 hospitals throughout China: patients were categorized on admission into mild/moderate (345), severe (155) and critical (48) degree (49).
The dynamic changes in hematologic and inflammatory biomarkers in survivors and non-survivors during progression of COVID-19 on admission, hospitalization, and end-hospitalization were used to classify these biomarkers into 3 distinct patterns. The pattern 1, including eosinophils, lymphocytes, and platelets, showed an upward trend in survivors, but a downward trend or kept low in non-survivors. The pattern 2, including neutrophils, NLR, IL-6, procalcitonin, D-dimer, prothrombin time, amyloid A protein, CRP, and ferritin were maintained at lower levels or downward trend in survivors and upward trend or maintained higher levels in non-survivors. Whereas, in the pattern 3, all biomarkers were in normal range and this pattern was associated with survivors. The authors suggested that restored levels of lymphocytes, eosinophils, and platelets could serve as predictors for recovery, whereas progressive increases in neutrophils, basophils, and IL-6 were associated with fatal outcome. In addition, an increase in naive T lymphocytes, and a reduction of memory T lymphocytes, CD4+ T cells and regulatory T cells was described in the peripheral blood of patients with the severe COVID-19 (44). Significant changes in peripheral blood parameters associated with different outcomes of COVID-19 patients are summarized in Table 1.

Table 1 Significant changes in peripheral blood parameters associated with different outcomes of COVID-19 patients.
A study that evaluated circulating T lymphocytes in 4 elderly patients showed differences in the populations of CD4+ and CD8+ T cells expressing markers associated with activation, immune memory and suppression, such as PD-1 (50). However, this analysis was not accompanied by a description of the outcome of COVID-19 in those individuals. Still, peripheral lymphopenia was followed by markers that characterize lymphocyte senescence/exhaustion, such as an increase of NKG2, CD57, or PD-1 expression on CD8+ T lymphocytes and NK cells (51, 52). Twenty eight studies were reviewed involving 3939 COVID-19 patients in China, and the authors described the reduction of lymphocyte subsets and neutrophilia followed by increased levels of cytokines, specifically IL-6 (40). Antagonists of IL-6 receptor have shown promising results in clinical trials, and antibodies against IL-1, IL-1R, IL-8, TNF, GM-CSF, GM-CSF receptor, IFN-γ, and IL-17 are under evaluation (40). Therefore, several lines of evidence indicate that a cytokine storm and neutrophilia, which are hallmarks of activation of innate response and reveal dysfunction of the immune system, induce down-regulation of the adaptive immune responses that are crucial for infection control. In this scenario, high levels of IL-6 negatively regulate perforin and granzyme B levels, one of the reasons that may affect the activity of NK cells (53). The detrimental role of neutrophils was described in a study with 32 patients showing that the concentrations of neutrophil extracellular traps (NETs) were higher in plasma and tracheal aspirate from severe COVID-19 patients compared to the controls. The authors showed that NETs induced the death of A549 airway epithelial cell line (54). It will be important to assess the effect of NETs on lymphocytes. In this context, COVID-19 patients displaying alterations in the T cell compartment that leans toward the functional phenotype Th17 could favor the occurrence of hyperinflammation by the exacerbated release of IL-17, a potent chemoattract/activator for human neutrophils (51).
The early induction of the adaptive immune response seems to be important for the best prognosis of the disease, since a case-report study described that the recovery of an infected woman in China was associated with an increase in antibody-producing cells and follicular CD4+ T cells, accompanied by increased levels of IgM and of IgG in the peripheral blood on days 7 and 8 of symptoms. Such parameters remained high on day 20, when the patient was already recovered. The concentrations of inflammatory cytokines and chemokines were reduced and the patient did not use any medication until recovery (55). It will be essential to assess whether this type of immune response indeed characterizes individuals with mild disease or whether other biomarkers could more accurately define the stratification between mild and severe forms. The characterization of leukocyte subsets and phenotypes, and functional activity of these cells in the peripheral blood of asymptomatic individuals, as those in close contact with patients, may provide information associated with the protective immune response, while the study of oligo-symptomatic patients in different cohorts will provide the dimensions of a balanced immune response that reflects pathogen resistance and damage tolerance. A Chinese study reported the evaluation of 25 asymptomatic COVID-19 patients, and showed that asymptomatic patients exhibited normal clinical indicators characterized by increased number of T, B, and NK cells, lower LDH compared to 27 symptomatic patients, which exhibited liver damage. Asymptomatic patients showed a faster viral clearance, although they may represent a greater risk of virus transmission (56). The increase of CD8+ T cells might be a biomarker for the efficacy of treatment for COVID-19, as patients who had a positive response for the treatment exhibited increases of those cells (57). Lastly, the evaluation of functional activity of lymphocytes in the peripheral blood of recovered individuals could make a huge contribution in attempts to characterize the protective immune response against SARS-CoV-2 infection.
There are contradictory indications in the literature about the role of lymphocytes in the immunopathology of COVID-19, since it has been reported that SARS-CoV-2 infection could provoke an immunological hyperactivation and accumulation of T lymphocytes in the lungs of patients with severe illness (38, 58) or T cell exhaustion/senescence (41, 51, 52, 59).
In this scenario, two very elegant studies appear to be the first to show the stratification of different COVID-19 clinical forms associated with different immunotypes within the same clinical form and depending on the phase of disease. These two studies used different approaches to perform longitudinal evaluation and analyze immune and clinical features. On study involved a longitudinal evaluation from 1 to 25 days from symptoms onset with 113 COVID-19 patients with moderate (non-ICU) and severe (ICU) disease. This study indicated that moderate cases were grouped in a cluster 1 that characterizes low expression of inflammatory cytokines and an increase in gene expression associated with growth factors or tissue repair genes. By contrast, severe cases were grouped in clusters 2 and 3 that characterize inflammatory cytokines and type 2 cytokines. Cluster 3 represented a more intense pro-inflammatory cytokine signature, worse disease and eventually death. In addition, this study indicated that although COVID-19 patients with severe illness in the first days from symptoms onset exhibited augment of IL-6 and IL-10, at late days from symptoms onset, they showed increased levels of IFN-α and IFN- λ, IL-17, IL-22, eotaxin, IL-13, and a reduction of IL-6. Moreover, severe patients displayed an increase of monocytes with down-regulation of HLA-DR (60). This study reports the novel finding that in the initial or in the later symptoms onset, patients with severe illness show different status of immune activation, defined as immunotypes.
In a study with 125 patients hospitalized at the University of Pennsylvania, Mathew and coworkers confirmed what has been already reported, i.e., patients were older age than recovery patients and healthy controls, and cardiovascular disease was the mostly associated comorbidity to COVID-19. The great majority of patients exhibited increased CRP, high LDH, D-dimer, and ferritin. Lymphopenia was reported in approximately half of the patients and high levels of IL-6 were detected in 31 from 46 subjects evaluated (61). However, an amazing aspect to this study is the elegant way in which it was shown that different immunotypes occur in hospitalized patients and the differences in the time-dependent trajectory of severe disease, using a multidimensional integrated analysis with several immunological and clinical parameters. Immunotype 1, identified in more severe cases, was characterized by intense activation and proliferation of CD4+ T cells, activation of CD8+ T cells, presence of exhausted CD8+ T cells and low frequency of circulating follicular helper cells (cTfh). Immunotype 2 was defined by the presence of T betbright CD8+ cells, reduced activation of CD4+ T cells compared to immunotype 1, and presence of memory B cells and proliferating plasmablasts (PB). Immunotype 3, which was identified in ~20% of patients, showed minimal activation of T cells, suggesting fails in the activation of immune response. The longitudinal analysis of severe cases was associated with immunotype 1 (61), suggesting an immunopathology induced by unbalanced adaptive immune response.
Proteomics and metabolomics analysis revealed with 93.5% accuracy that serum contains 22 proteins and seven metabolites that may help to classify severe COVID-19. Besides CRP, other acute phase proteins including SAA1 and SAA2, SERPINA3, VCAM-1, C6 protein of complement system, and steroid hormones were increased, while sphingolipids, fatty acids, among other lipid metabolites were decreased (62).
In this context, the field needs to further research larger cohorts that compare the mild, moderate, severe and critical forms of the disease. At the moment there are a small amount of evidence in this regard, each one evaluating different parameters of immune response and inflammation.
Lessons From Coronavirus Infections
During epidemics resulting from coronavirus infections in 2002/2003 (Severe Acute Respiratory Syndrome Coronavirus - SARS-CoV) and 2012/2013 (Middle East Respiratory Syndrome - MERS), respectively, researchers suggested, based on lung histopathological analysis from individuals who died of pulmonary disease, that the progressive and late form of SARS-CoV disease was more associated with tissue damage caused by immunopathology than with viral load (63). Although SARS-CoV and MERS were more lethal than COVID-19, the hallmark of this last is its higher transmissibility by airways. The oro-fecal transmission was not confirmed yet despite the detection of viral particles in feces (64). Even though high serum levels of IgM against SARS-CoV-2 and inflammation/tissue injury biomarkers (IL-6, CRP, and LDH) were reported to a neonate born to an infected mother, there is still no robust evidence that confirms potential vertical transmission of this coronavirus (65–67).
As SARS-CoV and MERS, COVID-19 causes a huge inflammation, described as a cytokine storm, and diffuse alveolar damage that characterize the severe form of illness (40). Pulmonary pathology induces respiratory failure, which is followed by damage in other organs, such as kidneys, liver and cardiovascular and nervous systems. The fact that SARS-CoV-2 infection causes intense pathological inflammation shows that surviving the infection is more a matter of tolerating lung damage (damage tolerance) than properly controlling viral load (pathogen tolerance), as already described (68, 69). Therefore, although the development of vaccines against SARS-CoV-2 is crucial for the protection of population worldwide and the development of new drugs or drug repurposing is critical for viral clearance, the understanding of pulmonary immunopathology during COVID-19 is the key for treatment aimed to reduce inflammation in the severe forms of the disease. Additionally, this knowledge is of great value for the development of safe vaccines that avoid clinical severity to subsequent infections and potential side effects of convalescent plasma therapy for COVID-19 (70–72). New perspectives for the treatment of pneumonia involve host-directed therapies and should include therapeutic measures aimed at damage tolerance mechanisms (69). The concept of host-directed therapies is particularly important for COVID-19, which shows heterogeneous clinical forms and requires the study of intrinsic factors as the genetic background, and extrinsic factors, associated with environmental factors and comorbidities.
Pulmonary Inflammation and Immunopathology
Considering that most studies published to date evaluated inflammatory mediators and leukocytes in peripheral blood and that immune response may be under compartmentalization, what is found in the periphery sites does not necessarily occur in the lungs. The understanding of the immunological and inflammatory response at the first and main site of the disease is fundamental for the identification of mediators, receptors and cells that cause dysfunction of immune response and how such dysfunction affects the progression of disease. The focus on pathological immune response that cause hyperinflammation and, as consequence, ARDS, will probably determine the design of therapies that may be efficient to down regulate the pulmonary inflammation.
In a recent review article, 23 studies were reported with post-mortem patients and evaluation of pulmonary pathology (73). The first of them, published in February, described a diffuse alveolar damage in a 50 year-old male, similar to those already described for MERS and SARS-CoV (74). The diffuse alveolar damage and histological findings compatible with ARDS were also reported in subsequent studies with deceased COVID-19 patients (75, 76). However, while one study showed extensive lymphocyte infiltration in the lungs (74), the other study that evaluated 4 COVID-19 patients reported only neutrophilic infiltration (75). The number of articles with post-mortem and pulmonary pathology, increased from April, and describe endotheliitis, coagulopathy and pulmonary emboli associated with influx of neutrophils in the alveolar space and trachea mucosal, presence of NETs and activation of complement system. An example is the post-mortem examination of 21 COVID-19 patients from Switzerland that described a diffuse alveolar damage accompanied by capillary congestion and presence of microthrombi. Bronchopneumonia was reported in 10 patients, 4 with pulmonary embolism, 3 with alveolar haemorrhage and 1 with vasculitis. The disease preferentially affected males with cardiovascular comorbidities and individuals with blood group A (77). Although it was described that co-localization occurred between viral particles and endothelial cells in different organs as lungs, kidneys and heart (78), another study with 11 post-mortem individuals showed that SARS-CoV-2 was detected in different organs such as intestine, liver and kidneys besides lungs, but the inflammation was focused on lungs and secondary lymphoid organs (7). Interestingly, there was no association between viral loads or the presence of viral antigens and inflammation. Although the lung inflammation is a consequence of SARS-CoV-2 infection, the hyperinflammation that induces immunopathology and respiratory failure may be consequence of dysfunction in the activation of immune response caused by alveolar cell death and alveolar congestion that reduces oxygen flow.
A very interesting and elucidating study that has not been certified by peer review yet reported analysis of lung autopsy material from 24 deceased COVID-19 patients (14 men and 10 women; mean age of 62.5 years old) collected in two institutions of the United States of America. First, they confirmed the data from different studies that COVID-19 is more prevalent in men and in older adults. The patients were classified as high or low viral RNA load, with the predominance of intracellular viral RNA in pneumocytes associated with extracellular viral RNA detection in hyaline membranes (79). In contrast to the results reported by Dorward and coworkers (7), a high viral RNA load was associated with high expression of IFN pathway, endothelial and wound healing genes, which corroborate the findings of lung damage characterized by exudative diffuse alveolar damage, fibrosis, infiltration of macrophages, monocytes, T cells and NK cells in areas positive or negative for SARS-CoV-2, and high expression of CTLA-4, PD-L1, and IDO. The low viral RNA load was associated with the presence of M0 and M2 macrophages, wound healing and presence of more intact pneumocytes (79). Dorward and coworkers also reported the decreased counts of resident macrophages in bronchoalveolar lavage fluid and pulmonary vasculitis characterized by the predominance of myeloid cells and monocytes; however, vasculitis was not necessarily associated with viral S protein on endothelial cells. Myeloid cells and CD4+ and CD8+ cells (in a lower extent) accumulated in the pulmonary parenchyma (7). Recently, two immunopathological profiles associated with signature of high or low interferon-stimulated genes (ISGhigh and ISGlow, respectively) were described in the lungs of deceased COVID-19 patients. Patients with ISGhigh signature remained in the hospital for a shorter period than patients with ISGlow signature. Lung samples from ISGhigh patients exhibited higher viral load and higher production of inflammatory cytokines and chemokines compared to lung samples of ISGlow patients, which showed higher number of CD4+, CD8+, CD20+ (B cells), CD163+ (monocytes), and CD64+ (macrophages) cells, complement deposition, diffuse alveolar damage, and coagulopathy, characterizing an immunopathology profile (80). The authors describe that the variability in the profiles is due to initial infectious dose and genetic background to generate an immune response.
To gain insights about the mechanisms that trigger SARS-CoV-2-induced inflammation, Li and coworkers showed that viral infection of Calu-3 cells induced apoptosis dependent on caspase 8 and production of IL-7, IL-8, TNF, CXCL-10, and CCL5. They also showed the induction of apoptosis, necroptosis and inflammation in the lungs using the ACE-2 transgenic mouse model. Furthermore, the analysis of lung sections from deceased COVID-19 patients confirmed the presence of apoptosis, necroptosis, necrotic cell debris and interstitial fibrosis (81). Therefore, two types of cell death and caspase-8-dependent inflammation may induce pulmonary immunopathology. Using a proteomic approach combined with bioinformatics analysis, Leng and coworkers reported the pathogenesis of SARS-CoV-2 at the molecular level using lung tissue samples from four COVID-19 patients. For proteomics analysis, lung samples were obtained from two elderly patients, one male and one female. From 641 proteins differentially expressed, 222 proteins were upregulated and 419 were downregulated compared with control (paracancerous tissue of lung cancer with no COVID-19). Most of proteins, up or downregulated, were localized in the nucleus. RIG-I pathway was activated. Downregulated proteins were found to be associated with endoplasmatic reticulum, lysosomes, ribosomes, cytoskeleton, and cell membrane. TLR4, CD40, IL-6, IL-8, TNF, IFN-α, ICAM1, and CXCL12, which are associated with NF-kB pathways, and TRAF protein family members were upregulated in the lungs of COVID-19 patients. Angiogenesis pathway, adhesion, and interaction with the extracellular matrix receptors were decreased, while activation of coagulation cascade, MMP2, MMP8 and CTSS cathepsin were upregulated (82). Although with a small number of lung samples from patients evaluated, this is a very interesting study because it identifies at the site of inflammation, possible molecular targets to be investigated as therapy to balance immunopathology.
The mechanisms underlying lymphopenia and lymphocyte exhaustion in peripheral blood, as well as lymphocyte accumulation in the lungs remain to be investigated. The findings reported herein clearly depict the heterogeneous immune response in the later phase of critical disease and highlight the efforts to design different immune therapies. Some characteristics mentioned above are summarized in Table 2.

Table 2 Lung immunopathological characteristics of COVID-19 patients.
Although these studies (7, 78, 79) show an evident progress forward to characterization of pulmonary pathology, much remains to be learned about damage tolerance for COVID-19 to translate this knowledge into clinic application. In this sense, the functional activity of T cell subsets in the lungs was not yet reported, but this information will be important to characterize the scenario of inflammation in the lung microenvironment. Other key point to address is the quantification of regulatory T cells, their influx and suppressive activity in the pulmonary parenchyma of severe patients because the balance of effector T cells and regulatory T cells is critical for damage tolerance.
This issue brings us back to the cytokine storm. The overproduction of IL-2, IFN-γ, IL-6, and IL-10 in the peripheral blood would also be the hallmark within lungs? If this clinical evidence in blood finds a confirmation in the lungs, would the increase in IL-2 concentrations in the lungs be associated with the expansion of regulatory T cells? In this context, it would be interesting to assess the ratio of CD4+ T and regulatory T cells, although both are reduced in the circulation of patients with severe forms of the disease. Therefore, could a reduction of T lymphocytes and regulatory T cells in the blood represent their accumulation in the lungs? Considering the lymphopenia and expression of exhaustion markers on lymphocytes in severe illness, could this reflect a lower influx within the lungs, and as consequence, a low strength for an effective adaptive immune response? The case-report with an infected woman in China, previously mentioned here, describes that during the symptomatic phase, lymphocyte, neutrophil and NK cell counts in the serum were comparable to basal levels. However, the patient exhibited a reduction in the number of CD14+CD16+ monocytes (55). A reduction of CD14+CD16+ monocytes could be an indicative for the monocyte migration to the lungs. Considering the participation of reticulo-endothelial system and accumulation of macrophages and monocytes in the pulmonary parenchyma of post-mortem patients, and the congestion of myeloid cells in the pulmonary arteries (7), it is also critical to evaluate the phenotype of monocytes and macrophages as well as their functional activity in the lungs of patients with severe COVID-19. The density of monocytes/macrophages and CD4+ and CD8+ T lymphocytes in the lungs of patients with severe COVID-19 as well as their functional role are key factors to predict the extension of immunopathology. For sure, comorbidities play a role on that because obesity was associated with a high peripheral neutrophil/CD8+ T lymphocyte ratio, an indicative of poor prognosis in patients with COVID-19 (47).
In addition, obesity might also play a role in the coagulopathy described in severe form of COVID-19 because it induces overproduction of procoagulant factors that induce thrombosis (22). Interestingly, IL-6 may participate in the induction of coagulation cascades (83) and levels of IL-6, IL-10 and TNF were negatively correlated with the number of circulating T cells (41). Moreover, severe COVID-19 patients exhibited increased platelet activation and platelet-monocytes aggregates compared with mild COVID-19 patients. Platelet activation and expression of tissue factor by monocytes were associated with increased fibrinogen and D-dimer, markers of coagulation dysfunction, in severe COVID-19 patients (84). It remains to be investigated whether other COVID-19-associated comorbidities have similar effect on pulmonary pathology. This understanding is critical for the development of host-directed therapies considering that infection by SARS-CoV-2 results in different clinical outcomes.
This rationale prompts us to consider the initial steps of SARS-CoV-2 infection. To gain insights into early infectivity and disease pathogenesis to SARS-CoV-2, Hou and coworkers used GFP reporter virus to characterize virus tropism and reported that a higher nasal expression of ACE-2 compared to bronchial airway epithelia, along with high infectivity of nasal epithelium followed by reduction in bronchioles and alveoli (85). The high expression of ACE-2 and transmembrane protease serine 2 (TMPRSS2) in nasal epithelium compared to lower airways suggest that therapeutic interventions must also consider nasal cavity. In lung autopsies, the authors found that ciliated cells were infected, but not mucus secretory club cells, despite ACE-2 and TMPRSS2 expression (85). Therefore, it remains to be investigated other factors that mediate infection in the lower airways. After infection mediated by ACE-2 receptor expressed on type II alveolar epithelial cells, the lysis of epithelial cells generates the release of DAMPs (danger associated molecular patterns), that are recognized by PRRs (pattern recognition receptors) and perpetuates cytokine release (37) and the recruitment of neutrophils and monocytes (7). IL-18 was correlated with poor outcome of disease and NLRP3 appears to be increased in the lung autopsies of deceased patients (86). However, the treatment of SARS-CoV2-infected Calu-3 cells with caspase-1 or NLRP3 inhibitors did not affect the secretion of IL-1β, suggesting that caspase-8-induced inflammation mediates the production of IL-1β (81). The fact is that we have still a very limited comprehension about initial steps that generate the innate response (87). Initial events may involve also the secretion of type I IFN by most of cells and type III IFN predominantly by epithelial cells during viral airway infections (88). It is been established that SARS-CoV-2 impairs the production of type I and type III IFNs by human epithelial cell lineage. Moreover, normal human bronchial epithelial cells produce no detectable level of type I and type III IFNs and induce limited subsets of ISG, and post-mortem lung samples from COVID-19 patients revealed also no detectable type I or type III IFNs (24). However, epithelial cells stimulated with type I IFN showed an increased expression of ACE-2, which could contribute for viral invasion (89). Post-mortem lung samples from COVID-19 patients showed a robust chemokine gene expression that induces the recruitment of inflammatory monocytes, as previously described in a mouse model for SARS-CoV (90). Monocytes and macrophages express also ACE-2 receptor and induce inflammation (91). Although there is a delay and low levels of secreted type I IFN, ISG are induced and the viral replication is moderate in vivo, in a ferret model (24). Although IFN-α has been associated with a protective effect in SARS-CoV infection (92), these mediators may play a pathological role because high levels of them were correlated with sequelae (93). As mentioned above, ISGhigh signatures in the lungs of deceased COVID-19 patients were associated with high viral load and more severe disease, although both ISGhigh and ISGlow lung signatures were fatal in COVID-19 (80). Furthermore, as also pointed out previously here, a genetic mechanism associated with susceptibility to SARS-CoV-2 infection in the early disease stages is the low expression of interferon receptor (IFNAR2) and interferon-inducible oligoadenylate synthetase (OAS) genes (15). Therefore, the role of type I IFN appears to be time-dependent and concentration-dependent: protective at moderate levels in the early disease and detrimental at high levels in the progressive disease. Interestingly, the expression of type I IFN in the early disease could be a factor associated with lower susceptibility of female compared to male to COVID-19, considering that the type I IFN is more efficiently produced by plasmacytoid dendritic cells (pDC) in female. Epigenetic modification of X-linked genes would induce higher type I IFN production by female pDC, IFN-α-dependent B cell activation and antibodies secretion associated with viral clearance (94). In addition, Zhou and coworkers showed that although patients with acute severe illness generated neutralizing antibodies, they exhibited a weak specific CD8+ T cell response, an increase of conventional DC:pDC ratio and an impairment of DC activation (95).
Therefore, the dysfunction in the innate response with improper recruitment of myeloid cells (neutrophils and monocytes) suggests that treatments for COVID-19 need be driven to controlling inflammation (24). Soluble CD14, a classical marker of monocytes, is increased in the plasma of COVID-19 patients and is associated with severity of disease (37). Considering that CD14-deficient mice infected by influenza A virus did not develop inflammatory response, CD14 could be a target focused on blocking steps in innate immunity to mitigate pulmonary immunopathology (37). To understand the protective or pathological role of monocytes and the magnitude of monocyte influx to the lungs, it will be important to evaluate the severe cases of COVID-19 associated with different comorbidities, which contribute for the diverse degree of immunopathology. Moreover, it will be important to understand the participation of epithelial cell in the generation of inflammation, which is the first entrance door for SARS-CoV-2.
Because COVID-19 is heterogeneous and pulmonary immunopathology is multifactorial, it will be important to address also the role of steroid hormones, lipid mediators and HLA-G protein in COVID-19. Figure 1 depicts cells and molecules/mediators that participate of peripheral and pulmonary inflammation and immunopathology during COVID-19.
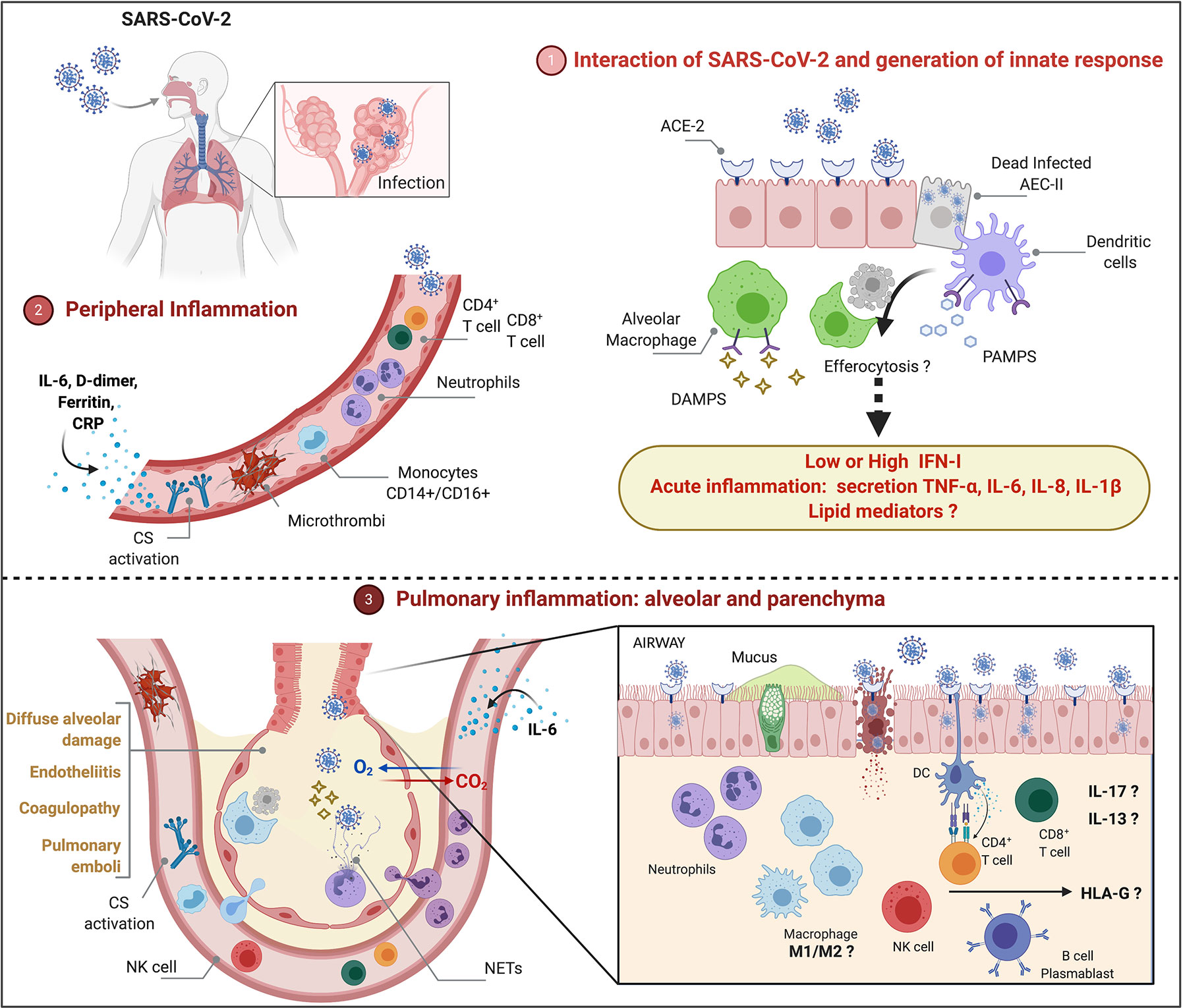
Figure 1 Peripheral and pulmonary inflammation and immunopathology during severe COVID-19 (1). SARS-CoV-2 infects type II alveolar epithelial cells (AEC-II) that express ACE-2 receptor. Cytolysis of infected AEC-II release DAMPs and PAMPs that may contribute for activation of innate response and induction of acute inflammation. Although SARS-CoV-2 inhibits type I-IFN (IFN-I)-induced cell activation pathways, longitudinal evaluation of patients with severe illness show increased IFN-I that may play a pathological role. Functional activation of alveolar macrophages and dendritic cells, efferocytosis and lipid mediators remain to be investigated (2). Peripheral inflammation is characterized by neutrophilia, lymphopenia (CD4+ T, CD8+ T, regulatory T, NK cells), increased monocytes (CD14+CD16+), activation of complement system (CS), increased levels of IL-6, C-reactive protein (CRP), D-dimer and ferritin (3). Severe COVID-19 courses with diffuse alveolar damage, endotheliitis, coagulopathy, and pulmonary emboli associated with neutrophilic/myeloid inflammation and lymphocyte inflammation in lower extent, increased levels of IL-13 and IL-17, accumulation of NETs. The role of M1/M2 macrophages as well as the participation of sex steroid hormones and HLA-G in the immunopathology remains to be investigated.
Sex Steroid Hormones and COVID-19
The communication between the endocrine and immune systems is fundamental for host homeostasis (96). This interaction depends on mediators such as cytokines and lipid-derived steroid hormones, which may be produced by leukocytes or be influenced by them. Otherwise, classical hormones released by endocrine glands may regulate the immune response and the consequent tissue damage in uncontrolled inflammatory processes (97).
The gonads and adrenal glands produce sexual hormones such as androgens that together with estrogens drive the sexual dimorphism in men and women. Androgens and estrogens activate specific receptors in leukocytes, thus influencing immune function and susceptibility to infection or inflammation (98). There are clear differences between the immunological status of males and females, as observed by the incidence, susceptibility and outcome of diseases in which sexual hormones modulate the patient’s immunity (99–101). In accordance, estrogens play a fundamental role in numerous events in the human physiology and diseases, as observed by the higher incidence of autoimmunity in women compared to men (102–104). In contrast, the androgen testosterone, mainly produced by males, negatively regulates the cytokine BAFF that is an essential factor for B lymphocytes activation and antibody production in autoimmune diseases (98).
Earlier studies from the 1960s decade suggested the increased capacity of women to produce antibodies (100), which are findings that could be related to recent data showing augmented immune reactivity of females as well as their resistance to infectious diseases, including those caused by respiratory viruses such as severe acute respiratory syndrome (SARS) (105), Middle East Respiratory Syndrome (MERS) (106) and influenza (107, 108). Interestingly, while this viral infection results in a more severe disease in men, with increased hospitalization occurrences (108, 109), women also produce higher levels of antibodies in response to influenza vaccination (110). Then, the sex steroid hormones may contribute to the disease worsening or protection, depending on the patient’s disorder and immune-endocrine status.
Despite hormones, various hypotheses have been proposed to explain the sex differences in the COVID-19 outcome, including genetics, immune responses, environmental and cultural factors such as smoking, as well as working routine (111). Men are at an increased risk for SARS-CoV2-induced complications (112), with notable sex differences in the incidence, severity and fatality of this viral disease (113, 114). Indeed, women exhibit reduced incidence and morbidity rates of COVID-19 (113).
The clear genetic discrepancy between males and females potentially contribute to the disease outcome (115). The presence of a single X chromosome in men (in contrast to XX in women) could predispose to reduced alternative mechanisms for protection upon exposure to the virus. Notably, ACE-2 gene is located on X chromosomes and is affected by androgens (116–119), thus accounting for a higher susceptibility to infection in men. On the contrary, estrogen can down regulate the expression of ACE-2 in differentiated airway epithelial cells (120). Furthermore, since the androgen receptor (AR) gene is also located on the X chromosome, the sensitivity to hormones such as testosterone could represent an important determinant factor for the severity of COVID-19 in men (121).
SARS-CoV-2 infects the cells by the binding of its spike protein to the cellular ACE-2 receptor. The TMPRSS2, whose activation is dependent on androgens, primes spike protein for virus entry into the host’s cells and interact with ACE-2 receptors, facilitating viral entry (122). It is of note that the transcription of TMPRSS2 gene depends on AR and increases after exposure to androgens (123, 124). In agreement, there is up-regulation of TMPRSS2 mRNA upon activation of AR (125), while androgen deprivation therapy (ADT) suppresses this axis (126, 127). Moreover, the in vitro inhibition of TMPRSS2 can prevent SARS-CoV-2 infection of cultured cells (122). Altogether, this evidence suggest that the elevated expression of TMPRSS2 induced by androgens could be one of the explanations for the increased male susceptibility to COVID-19 and the milder disease in children who still do not have high expression of AR (128).
As discussed above, one of the reasons for such sex dichotomy in COVID-19 could be the difference between the immune response of males and females (129, 130). Higher levels of estrogens are related to stronger reactions that facilitate the pathogen clearance, while testosterone usually inhibits the immune processes, thus supporting the overall assumption that men have reduced autoimmunity as well as attenuated responses against infectious agents. Indeed, the androgen’s effects on innate and adaptive immune responses depends on the hormone signaling through the AR (98, 131–133). A recent study found that critically ill patients with COVID-19 had lower amounts of circulating and free testosterone compared to subjects in a more stable condition, indicating that this androgen could predict the worst disease outcome (134). In line with that, a drug screening study confirmed the capacity of a 5-alpha reductase inhibitor, which prevent the conversion of testosterone into the potent dihydrotestosterone, to reduce ACE-2 levels in cardiac and human alveolar epithelial cells treated in vitro with this drug. Patient data also showed that altered androgen conditions are associated to severe COVID-19 complications as well as cardiac injury, thus clarifying some important mechanisms that could lead to augmented men’s susceptibility to this disease (135). Furthermore, besides ACE-2 and AR, other genes related to the immune response are also located on the X chromosomes (136, 137) and could potentially interfere in the virus elimination or inflammation control. Indeed, males had increased IL-8, IL-18, CCL5, and prominent non-classical monocytes, while female patients presented stronger T cell activation, compared to men (138).
Hence, considering that sex hormones such as estrogens and androgens may differentially modulate the inflammatory responses, it is plausible to assume that these steroid immune-endocrine mediators may interfere in the anti-viral immunity and the outcome of COVID-19 in both men and women.
Lipid Mediators and COVID-19
Lipid mediators, such as leukotrienes (LT) and prostaglandins (PGs) derived from arachidonic acid (AA) metabolism (139, 140), are involved in the induction and in the regulation of immune responses in bacteria, fungi, and parasites infections, as well as in envenomation (141–147). Although LT are considered deleterious, as cysteinyl leukotriene (CysLT) induces muscle contraction when released in upper bronchoalveolar space inducing bronchospasms (148), LTB4 has been considerate beneficial to the control of infections, especially in the lungs. LTB4 increases phagocytosis and microbial killing (149–152), induces the production of antimicrobial peptides (153), decreases IL-1β production (144). Moreover, pharmacological or genetic inhibition of LTB4 depresses immune response against bacteria, fungi, and parasites (142, 149, 152, 154), and LTB4 administration improves survival in fungal and bacterial infection (153, 155).
The participation of LT has been also described in viral infection (156). Bronchial epithelial cells infected with respiratory syncytial virus produce LT (157), suggesting a role for 5-LO-derived compound in viral infections. Moreover, experimental models in mice validated the protective role of LTB4 as antiviral mediator. The investigators showed that LTB4, via nucleotide-binging oligomerization domain 2 (NOD2) pathway enhances the anti-influenza A response, and that the treatment of influenza A-infected mice with LTB4 increased the release of IFN-β, IL-6, and TNF in the lungs, ameliorating the innate response and increasing mice survival (158). We have previously showed that the increase in circulating LTB4 and CysLT is associated with the neuroinflammatory phase of HTLV-1 infection in human (HAM/TSP), although we were enable to demonstrate the it role in the infection (159).
As above mentioned, AA metabolism via cyclooxygenase (COX)-1 and COX2 originate prostanoids, such as PGE2, PGD2, TXA2, PGI2 (160), some of them with opposite functions (161). After binding to the receptors EP1, EP2, EP3, EP4, PGE2 induces inflammation, apoptosis, and cell proliferation (140, 144, 145, 162). In this context, the role of PGE2 in viral infection has been documented (163). In influenza A virus infection, it was described that the augment of PGE2 secretion inhibits type I interferon release, macrophage apoptosis, antigen presentation and T cell mediated immunity, with consequent increase in virus burden (164). Using genetic and pharmacological tools to inhibit PGE2 production, it was shown an increase in the survival of lethally virus-infected mice, and PGE2 inhibition was suggested as a treatment for influenza A infection (164). More recently, in H1N1 infected obese mice, an immune suppressive role of PGE2 was also reported, and the investigators suggested the treatment with paracetamol to restore cytokine production and to rescue lethally H1N1 infected mice from death (165).
Considering the participation of lipid mediators in COVID-19, eicosanoids and docosanoids could be markers of disease severity (166, 167). Overmyer and coworkers quantified and compared metabolites and lipids from COVID-19 and non-COVID-19 patients blood samples and reported that dysregulation of lipid transport and neutrophil degranulation were associated with severe COVID-19 (168). The diminished production of immune regulatory PGs and the increase of AA-derived products, 5-lipoxygenase-derived products (5-LOX) and cytochrome P450 (CYP450) species in the serum were associated with severe disease (166). By contrast, it was suggested that AA and others lipid mediators potentially could inactivate SARS-CoV-2 (169). A lipid storm within the lungs of severe COVID-19 patients was characterized by the oxylipids quantification on BALF of patients, under mechanical ventilation support. The authors showed a predominance of COX metabolites, such as TXB2, PGE2, 12-HHTrE and PGD2; LTs, notably LTB4, 20-COOH-LTB4, LTE4, and eoxin E4; 15-lipoxygenase metabolites derived from linoleic, arachidonic, eicosapentaenoic, and docosahexaenoic; and specialized pro-resolving mediators, such as lipoxin A4 and the D-series resolvins (170). These studies show that the role of lipid mediators in the SARS-CoV-2 infection requires further investigations.
HLA-G and COVID-19
The genetic susceptibility to intracellular pathogens has been demonstrated for a variety of infectious diseases, such as leprosy, tuberculosis, malaria and noteworthy those caused by RNA viruses, such as HIV-1/AIDS, hepatitis B and C and dengue (171), including SARS-CoV (172, 173). In that sense, it has been reported that the host genetic polymorphisms such as ABO, Human Leukocyte Antigen (HLA), IL-6, complement and angiotensin-converting enzyme 2 (ACE-2) genes may be associated with SARS-CoV-2 susceptibility and COVID-19 severity (15, 174–178). Understanding the contribution of the host genomics as an intrinsic factor to the variable outcomes of SARS-CoV-2 infections, from asymptomatic to severe form or death, will enable to improve the medical practice and patient care. For this, it is necessary to translate the host genetic variations into predictive risk factors that will foster the clinical management.
Human leukocyte antigens (HLA) are involved in several important functions of the immune system, starting from antigen presentation to lymphocytes, performed by the classical class I (HLA-A/B/C) and class II (HLA-DR/DQ/DP) histocompatibility molecules), and extending until the control of the immune response, primarily performed by the non-classical class I molecules (HLA-E/F/G) (179). Among these molecules, HLA-G is the most studied, and its major function is the down-regulation of the function of the innate and adaptive immune system cells, by means of the interaction with inhibitory receptors (180). Accordingly, HLA-G may inhibit the proliferation of T and B lymphocytes, the activity of antigen-presenting cells (APC) and the cytotoxicity of CD8+ T cells and NK cells (181, 182). Because of these properties, HLA-G has been recognized as a major immune check point molecule. In certain conditions, like viral infections, the overexpression of HLA-G can create a tolerogenic environment; inhibiting several steps of the immune response, propitiating the spreading of infectious (183).
HLA-G is encoded within the human Major Histocompatibility Complex (NCBI gene ID: 3135), and in contrast to normal conditions in which the expression of HLA-G is very restricted to few tissues, ectopic HLA-G expression may occur in pathological conditions, such as viral infection, favoring the virus escape from the patient’s immune surveillance (184). Several HLA-G isoforms have been described that can be expressed on cell membranes and reach distant immunological sites as soluble forms (sHLA-G). Both membrane-bound and sHLA-G induce regulatory mechanisms, such as apoptosis of CD8+ T and NK cells, inhibition of B-cell proliferation, differentiation and antibody secretion (185).
Overall, all segments of HLA-G may contribute to its immunoregulatory properties. The short cytoplasmic tail retains HLA-G longer in the endoplasmic reticulum and prolongs the half-life of the molecule on the cell surface, because of the lack of an endocytosis motif, permitting multiple interactions with cells of the immune system. The extracellular domains interact with leukocyte receptors, including CD8, ILT-2/LILRB1, and ILT-4/LILRB2, and the NK immunoglobulin-like receptor KIR2DL4 (CD158d) (183).
Although HLA-G may interact with KIR molecules, the most important interaction are with the LILRB1s (expressed on the surface of NK and lymphomononuclear cells) and LILRB2s (expressed on the surface of monocytes and dendritic cells) (186). Both LILRB1 and LILRB2 have several ITIM receptors in their cytoplasmic tail, inhibiting signaling events triggered by stimulatory receptors (187). Both receptors interact with classical HLA class I molecules; however, the binding with HLA-G presents three- to four-fold higher affinity when compared to classical molecules (188). The CD8 molecule also interacts with the α3 domain of classical and non-classical molecules, including HLA-G and HLA-E, although exhibiting different affinities (183).
Besides the inhibitory role of HLA-G in cytotoxic cells exhibiting CD8 on their surface, other interactions of HLA-G molecules with CD8 cells deserve attention. Soluble HLA-A-B-C and -G molecules can induce apoptosis in activated CD8+ T lymphocytes as well as in CD8+ NK cells (lacking the T cell receptor) at similar rates. The binding of soluble HLA molecules to CD8 leads to apoptosis, upregulating Fas production and Fas/FasL interaction (189, 190). Classical and non-classical soluble molecules may produce similar effects on apoptosis of CD8+ cells; however, in conditions in which soluble HLA-G is elevated, including pregnancy, some tumors and allografts with good prognosis, this mechanism may represent an additional immunomodulatory effect of HLA-G. Although the precise mechanism of this interaction has not been elucidated, the interaction of soluble forms of HLA-G containing the α3 domain is plausible (183). Taken together, these observations suggest that, in the context of viral infections, the expression of HLA-G is a complex process modulated by many factors such as HLA-G polymorphism, stage of infection, drug therapy, and cytokine expression patterns, which may contribute to an immunological environment affecting the outcome of infection (191).
Viruses have also evolved several strategies to evade the cytotoxic effect of immune effector cells, including downregulation of HLA classical class I molecules and the upregulation of non-classical molecules, or both. As a result, the increased HLA-G expression, induced by the virus itself or by the presence of an inflammatory milieu containing transcription and post-transcription factors that positively modulate HLA-G expression, may exacerbate virus morbidity and/or patient mortality, the expression of HLA-G in these disorders may predict a worse outcome and greater susceptibility to cell transformation (192). Specifically in the SARS-CoV-2 infection, few studies have been conducted so far. Zhang and coworkers showed that HLA‐G expression in peripheral T cells, B cells and monocytes reflected a high, low, high pattern, respectively, to viral replication and clearance, suggesting that SARS‐CoV‐2 infection might be involved in the regulation of HLA‐G expression, which impairs CD8+ cell‐mediated recognition and supports a mechanism of immune evasion (193). Zidi suggested that the decrease of HLA-G expression on cell surface could be a consequence of cleavage by MMP. The author still suggested that the follow-up of soluble HLA-G could be used to identify a worse prognostic of COVID-19 among patients with high levels of soluble HLA-G, and therefore the blocking of HLA-G interaction with their receptors may enhance immune response (194).
Although many clinical trials have been conducted, presently, no effective drug treatment is available for SARS‐CoV‐2. However, host humoral and cellular immune responses are imperative to control viral infections. In this review article, we discuss the role important soluble mediators, antibodies, and leukocytes that are involved on the pathogenesis of SARS‐CoV‐2-mediated infection and on the outcome of COVID-19 patients. In this scenario, we highlight the role the well-recognized tolerogenic role of the HLA‐G molecule.
Concluding Remarks
From the identification of SARS-CoV-2 in January and February to COVID-19 being declared a pandemic by WHO in March 2020, we have followed the movement of the Chinese scientific community in the dissemination of clinical and laboratory findings of mild and severe clinical forms of this new disease that mobilized the world in a way that we had not yet witnessed. As the SARS-CoV-2 infection spread to different continents and countries, we have followed the rapid mobilization of the other scientific communities worldwide, which seek a better understanding of the generation of the immune response, leukocyte populations and soluble mediators involved in the protective response or unfavorable disease outcome. From April 2020, we observed an increasing number of studies focused on the description of pulmonary immunopathology and the functional characterization of circulating leukocyte populations. Undoubtedly, a huge advance in the characterization of infected cells, with studies that were initiated with cell lines and advanced to the autopsies of lung tissue of deceased individuals, suggesting that ACE-2 receptor may not be the only cell entry for the virus. From now on, it will be essential to investigate how immunopathology is generated and the mechanisms behind the damage tolerance dysregulation. In this sense, the study of the main comorbidities that participate of COVID-19 immunopathology will be essential to understand the different clinical outcomes. For this, experimental models and well-defined experimental approaches in vitro will have an important role for the understanding of pulmonary immunopathology to advance from the descriptive phenomenon to the investigation of the triggering mechanism. In this scenario, researchers should be able to identify targets for therapies aimed at reducing pulmonary immunopathology and biomarkers that make it possible to differentiate mild, moderate, severe, and critical clinical forms, and that make it feasible to monitor the evolution of the disease.
Author Contributions
VB participated in the conceptual design. TF-S, MD-B, and VB drafted, wrote, reviewed, and edited the manuscript. SM reviewed and edited transcriptome section. ER reviewed and edited the manuscript. AF reviewed and edited HLA-G section. CC reviewed and edited sex steroid hormone section. CS and LF reviewed and edited lipid mediator section. All authors contributed to the article and approved the submitted version.
Funding
The authors gratefully acknowledge São Paulo Research Foundation - FAPESP (2020/05270-0, 2020/05207-6, and 2017/21629-5) and National Council for Scientific and Technological Development - CNPq (312606/2019-2, 311639/2017-8, and 309583/2019-5).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are thankful to Professor Richard D. Cummings from Harvard Medical School - Beth Israel Deaconess Medical Center - for carefully revising and making important comments to this review.
References
1. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet (2020) 395:565–74. doi: 10.1016/S0140-6736(20)30251-8
2. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
3. Chavez S, Long B, Koyfman A, Liang SY. Coronavirus Disease (COVID-19): A primer for emergency physicians. Am J Emerg Med (2020). doi: 10.1016/j.ajem.2020.03.036
4. WHO. Coronavirus Disease 2019. In: World Heal Organ (2020). Available at: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200331-sitrep-71-covid-19.pdf?sfvrsn=4360e92b_8.
5. Cao X. COVID-19: immunopathology and its implications for therapy. Nat Rev Immunol (2020) 20:269–70. doi: 10.1038/s41577-020-0308-3
6. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA (2020) 323:1239–42. doi: 10.1001/jama.2020.2648
7. Dorward DA, Russell CD, Um IH, Elshani M, Armstrong SD, Penrice-Randal R, et al. Tissue-specific Immunopathology in Fatal COVID-19. Am J Respir Crit Care Med (2020) rccm.202008–3265OC. doi: 10.1164/rccm.202008-3265OC
8. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med (2020) 180:934–43. doi: 10.1001/jamainternmed.2020.0994
9. The Lancet. COVID-19 in Brazil: “So what?” Lancet (2020) 395:1461. doi: 10.1016/S0140-6736(20)31095-3
10. Pontes M, Lima J. Brazil’s COVID-19 response. Lancet (London England) (2020) 396:e34. doi: 10.1016/S0140-6736(20)31919-X
11. Candido DS, Claro IM, de Jesus JG, Souza WM, Moreira FRR, Dellicour S, et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Sci (80-) (2020) 369:1255–60. doi: 10.1126/science.abd2161
12. de Souza WM, Buss LF, Candido D da S, Carrera J-P, Li S, Zarebski AE, et al. Epidemiological and clinical characteristics of the COVID-19 epidemic in Brazil. Nat Hum Behav (2020) 4:856–65. doi: 10.1038/s41562-020-0928-4
13. Cavalcanti AB, Zampieri FG, Rosa RG, Azevedo LCP, Veiga VC, Avezum A, et al. Hydroxychloroquine with or without Azithromycin in Mild-to-Moderate Covid-19. N Engl J Med (2020) 383:2041–52. doi: 10.1056/NEJMoa2019014
14. Furtado RHM, Berwanger O, Fonseca HA, Corrêa TD, Ferraz LR, Lapa MG, et al. Azithromycin in addition to standard of care versus standard of care alone in the treatment of patients admitted to the hospital with severe COVID-19 in Brazil (COALITION II): a randomised clinical trial. Lancet (2020) 396:959–67. doi: 10.1016/S0140-6736(20)31862-6
15. Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, Invernizzi P, et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N Engl J Med (2020) 383:1522–34. doi: 10.1056/NEJMoa2020283
16. Vuille-Dit-Bille RN, Camargo SM, Emmenegger L, Sasse T, Kummer E, Jando J, et al. Human intestine luminal ACE2 and amino acid transporter expression increased by ACE-inhibitors. Amino Acids (2015) 47:693–705. doi: 10.1007/s00726-014-1889-6
17. Pinto BGG, Oliveira AER, Singh Y, Jimenez L, Gonçalves ANA, Ogava RLT, et al. ACE2 expression is increased in the lungs of patients with comorbidities associated with severe COVID-19. J Infect Dis (2020) 222:556–63. doi: 10.1093/infdis/jiaa332
18. Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, et al. Genetic mechanisms of critical illness in Covid-19. Nature (2020). doi: 10.1038/s41586-020-03065-y
19. Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, et al. Prevalence of comorbidities in the novel Wuhan coronavirus (COVID-19) infection: a systematic review and meta-analysis. Int J Infect Dis (2020) 94:91–5. doi: 10.1016/j.ijid.2020.03.017
20. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
21. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA - J Am Med Assoc (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
22. Pasquarelli-Nascimento G, Braz-de-melo HA, Faria SS, Santos IDO, Kobinger GP. Hypercoagulopathy and Adipose Tissue Exacerbated Inflammation May Explain Higher Mortality in COVID-19 Patients With Obesity. Front Endocrinol (2020) 11:1–16. doi: 10.3389/fendo.2020.00530
23. Kermali M, Khalsa RK, Pillai K, Ismail Z, Harky A. The role of biomarkers in diagnosis of COVID-19 – A systematic review. Life Sci (2020) 254:117788. doi: 10.1016/j.lfs.2020.117788
24. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell (2020) 181:1036–45.e9. doi: 10.1016/j.cell.2020.04.026
25. Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med (2020) 26:1070–6. doi: 10.1038/s41591-020-0944-y
26. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol (2020) 5:eabd1554. doi: 10.1126/sciimmunol.abd1554
27. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
28. He J, Cai S, Feng H, Cai B, Lin L, Mai Y, et al. Single-cell analysis reveals bronchoalveolar epithelial dysfunction in COVID-19 patients. Protein Cell (2020) 11:680–7. doi: 10.1007/s13238-020-00752-4
29. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect (2020) 9:761–70. doi: 10.1080/22221751.2020.1747363
30. Zhou W, Ye S, Wang W, Li S, Hu Q, Masaki T. Clinical Features of COVID-19 Patients with Diabetes and Secondary Hyperglycemia. J Diabetes Res (2020) 2020:1–9. doi: 10.1155/2020/3918723
31. Zhao R, Sun Y, Zhang Y, Wang W, Wang S, Wang C, et al. Distinguishable Immunologic Characteristics of COVID-19 Patients with Comorbid Type 2 Diabetes Compared with Nondiabetic Individuals. Mediators Inflammation (2020) 2020:1–10. doi: 10.1155/2020/6914878
32. Han H, Ma Q, Li C, Liu R, Zhao L, Wang W, et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg Microbes Infect (2020) 9:1123–30. doi: 10.1080/22221751.2020.1770129
33. XU B, Fan C, Wang A, Zou Y, Yu Y, He C, et al. Suppressed T cell-mediated immunity in patients with COVID-19: A clinical retrospective study in Wuhan, China. J Infect (2020) 81:e51–60. doi: 10.1016/j.jinf.2020.04.012
34. Liu J, Li S, Liu J, Liang B, Wang X, Wang H, et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine (2020) 55:102763. doi: 10.1016/j.ebiom.2020.102763
35. Wan S, Yi Q, Fan S, Lv J, Zhang X, Guo L, et al. Relationships among lymphocyte subsets, cytokines, and the pulmonary inflammation index in coronavirus (COVID-19) infected patients. Br J Haematol (2020) 189:428–37. doi: 10.1111/bjh.16659
36. Stone JH, Frigault MJ, Serling-Boyd NJ, Fernandes AD, Harvey L, Foulkes AS, et al. Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. N Engl J Med (2020) 383:2333–44. doi: 10.1056/NEJMoa2028836
37. Martin TR, Wurfel MM, Zanoni I, Ulevitch R. Targeting innate immunity by blocking CD14: Novel approach to control inflammation and organ dysfunction in COVID-19 illness. EBioMedicine (2020) 57:102836. doi: 10.1016/j.ebiom.2020.102836
38. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet (2020) 395:1033–4. doi: 10.1016/S0140-6736(20)30628-0
39. Mahmoudi S, Rezaei M, Mansouri N, Marjani M, Mansouri D. Immunologic Features in Coronavirus Disease 2019: Functional Exhaustion of T Cells and Cytokine Storm. J Clin Immunol (2020) 40:974–6. doi: 10.1007/s10875-020-00824-4
40. Wang J, Jiang M, Chen X, Montaner LJ. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol (2020) 108:17–41. doi: 10.1002/JLB.3COVR0520-272R
41. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
42. Pontelli MC, Castro IA, Martins RB, Veras FP. Infection of human lymphomononuclear cells by SARS-CoV-2. bioRxiv (2020). doi: 10.1101/2020.07.28.225912
43. Davanzo GG, Codo AC, Brunetti NS, Boldrini V, Knittel TL, Monterio LB, et al. SARS-CoV-2 Uses CD4 to Infect T Helper Lymphocytes. medRxiv (2020), 2020.09.25.20200329. doi: 10.1101/2020.09.25.20200329
44. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis (2020) 71:762–8. doi: 10.1093/cid/ciaa248
45. Kong M, Zhang H, Cao X, Mao X, Lu Z. Higher level of Neutrophil-to-Lymphocyte is associated with severe COVID-19. Epidemiol Infect (2020) 148:E139. doi: 10.1017/S0950268820001557
46. Gan J, Li J, Li S, Yang C. Leucocyte Subsets Effectively Predict the Clinical Outcome of Patients With COVID-19 Pneumonia: A Retrospective Case-Control Study. Front Public Heal (2020) 8:299. doi: 10.3389/fpubh.2020.00299
47. Urra JM, Cabrera CM, Porras L, Ródenas I. Selective CD8 cell reduction by SARS-CoV-2 is associated with a worse prognosis and systemic inflammation in COVID-19 patients. Clin Immunol (2020) 217:108486. doi: 10.1016/j.clim.2020.108486
48. Wang H, Zhang Y, Mo P, Liu J, Wang H, Wang F, et al. Neutrophil to CD4+ lymphocyte ratio as a potential biomarker in predicting virus negative conversion time in COVID-19. Int Immunopharmacol (2020) 85:106683. doi: 10.1016/j.intimp.2020.106683
49. Chen R, Sang L, Jiang M, Yang Z, Jia N, Fu W, et al. Longitudinal hematologic and immunologic variations associated with the progression of COVID-19 patients in China. J Allergy Clin Immunol (2020) 146:89–100. doi: 10.1016/j.jaci.2020.05.003
50. Cossarizza A, De Biasi S, Guaraldi G, Girardis M, Mussini C. Modena Covid-19 Working Group (MoCo19). SARS-CoV-2, the Virus that Causes COVID-19: Cytometry and the New Challenge for Global Health. Cytometry A (2020) 97:340–3. doi: 10.1002/cyto.a.24002
51. De Biasi S, Meschiari M, Gibellini L, Bellinazzi C, Borella R, Fidanza L, et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat Commun (2020) 11:3434. doi: 10.1038/s41467-020-17292-4
52. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol (2020) 17:533–5. doi: 10.1038/s41423-020-0402-2
53. Cifaldi L, Prencipe G, Caiello I, Bracaglia C, Locatelli F, De Benedetti F, et al. Inhibition of Natural Killer Cell Cytotoxicity by Interleukin-6: Implications for the Pathogenesis of Macrophage Activation Syndrome. Arthritis Rheumatol (2015) 67:3037–46. doi: 10.1002/art.39295
54. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2–triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med (2020) 217:e20201129. doi: 10.1084/jem.20201129
55. Thevarajan I, Nguyen THO, Koutsakos M, Druce J, Caly L, van de Sandt CE, et al. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat Med (2020) 26:453–5. doi: 10.1038/s41591-020-0819-2
56. Han H, Xu Z, Cheng X, Zhong Y, Yuan L, Wang F, et al. Descriptive, Retrospective Study of the Clinical Characteristics of Asymptomatic COVID-19 Patients. mSphere (2020) 5:e00922-20. doi: 10.1128/mSphere.00922-20
57. Wang F, Nie J, Wang H, Zhao Q, Xiong Y, Deng L, et al. Characteristics of Peripheral Lymphocyte Subset Alteration in COVID-19 Pneumonia. J Infect Dis (2020) 221:1762–9. doi: 10.1093/infdis/jiaa150
58. Zhang B, Zhou X, Qiu Y, Song Y, Feng F, Feng J, et al. Clinical characteristics of 82 cases of death from COVID-19. PloS One (2020) 15:e0235458. doi: 10.1371/journal.pone.0235458
59. Zheng HY, Zhang M, Yang CX, Zhang N, Wang XC, Yang XP, et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell Mol Immunol (2020) 17:541–3. doi: 10.1038/s41423-020-0401-3
60. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature (2020) 584:463–9. doi: 10.1038/s41586-020-2588-y
61. Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Sci (80-) (2020) 369:eabc8511. doi: 10.1126/science.abc8511
62. Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell (2020) 182:59–72.e15. doi: 10.1016/j.cell.2020.05.032
63. Peiris JSM, Chu CM, Cheng VCC, Chan KS, Hung IFN, Poon LLM, et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet (London England) (2003) 361:1767–72. doi: 10.1016/s0140-6736(03)13412-5
64. Xing Y-H, Ni W, Wu Q, Li W-J, Li G-J, Wang W-D, et al. Prolonged viral shedding in feces of pediatric patients with coronavirus disease 2019. J Microbiol Immunol Infect (2020) 53:473–80. doi: 10.1016/j.jmii.2020.03.021
65. Dong L, Tian J, He S, Zhu C, Wang J, Liu C, et al. Possible Vertical Transmission of SARS-CoV-2 From an Infected Mother to Her Newborn. JAMA (2020) 323:1846–8. doi: 10.1001/jama.2020.4621
66. Chen H, Guo J, Wang C, Luo F, Yu X, Zhang W, et al. Clinical characteristics and intrauterine vertical transmission potential of COVID-19 infection in nine pregnant women: a retrospective review of medical records. Lancet (2020) 395:809–15. doi: 10.1016/S0140-6736(20)30360-3
67. Auriti C, De Rose DU, Tzialla C, Caforio L, Ciccia M, Manzoni P, et al. Vertical Transmission of SARS-CoV-2 (COVID-19): Are Hypotheses More than Evidences? Am J Perinatol (2020) 37:S31–8. doi: 10.1055/s-0040-1714346
68. Medzhitov R, Schneider DS, Soares MP. Disease Tolerance as a Defense Strategy. Sci (80-) (2012) 335:936–41. doi: 10.1126/science.1214935
69. Dela Cruz CS, Wunderink RG, Christiani DC, Cormier SA, Crothers K, Doerschuk CM, et al. Future research directions in pneumonia. Am J Respir Crit Care Med (2018) 198:256–63. doi: 10.1164/rccm.201801-0139WS
70. de Alwis R, Chen S, Gan ES, Ooi EE. Impact of immune enhancement on Covid-19 polyclonal hyperimmune globulin therapy and vaccine development. EBioMedicine (2020) 55:102768. doi: 10.1016/j.ebiom.2020.102768
71. Fleming AB, Raabe V. Current studies of convalescent plasma therapy for COVID-19 may underestimate risk of antibody-dependent enhancement. J Clin Virol (2020) 127:104388. doi: 10.1016/j.jcv.2020.104388
72. Arvin AM, Fink K, Schmid MA, Cathcart A, Spreafico R, Havenar-Daughton C, et al. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature (2020) 584:353–63. doi: 10.1038/s41586-020-2538-8
73. Calabrese F, Pezzuto F, Fortarezza F, Hofman P, Kern I, Panizo A, et al. Pulmonary pathology and COVID-19: lessons from autopsy. The experience of European Pulmonary Pathologists. Virchows Arch (2020) 477:359–72. doi: 10.1007/s00428-020-02886-6
74. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
75. Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, et al. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol (2020) 33:1007–14. doi: 10.1038/s41379-020-0536-x
76. Yao XH, He ZC, Li TY, Zhang HR, Wang Y, Mou H, et al. Pathological evidence for residual SARS-CoV-2 in pulmonary tissues of a ready-for-discharge patient. Cell Res (2020) 30:541–3. doi: 10.1038/s41422-020-0318-5
77. Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, Deigendesch N, et al. Post-mortem examination of COVID19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings of lungs and other organs suggesting vascular dysfunction. Histopathology (2020) 77:198–209. doi: 10.1111/his.14134
78. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
79. Desai N, Neyaz A, Szabolcs A, Shih AR, Chen JH, Thapar V, et al. Temporal and Spatial Heterogeneity of Host Response to SARS-CoV-2 Pulmonary Infection. medRxiv Prepr Serv Heal Sci (2020) 1–37. doi: 10.1101/2020.07.30.20165241
80. Nienhold R, Ciani Y, Koelzer VH, Tzankov A, Haslbauer JD, Menter T, et al. Two distinct immunopathological profiles in autopsy lungs of COVID-19. Nat Commun (2020) 11:1–13. doi: 10.1038/s41467-020-18854-2
81. Li S, Zhang Y, Guan Z, Li H, Ye M, Chen X, et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct Target Ther (2020) 5:35. doi: 10.1038/s41392-020-00334-0
82. Leng L, Cao R, Ma J, Mou D, Zhu Y, Li W, et al. Pathological features of COVID-19-associated lung injury: a preliminary proteomics report based on clinical samples. Signal Transduct Target Ther (2020) 5:1–9. doi: 10.1038/s41392-020-00355-9
83. Kerr R, Stirling D, Ludlam CA. Interleukin 6 and Haemostasis. Br J Haematol (2001) 115:3–12. doi: 10.1046/j.1365-2141.2001.03061.x
84. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, et al. Platelet activation and platelet-monocyte aggregates formation trigger tissue factor expression in severe COVID-19 patients. Blood (2020) 136:1330–41. doi: 10.1182/blood.2020007252
85. Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH, et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell (2020) 182:429–446.e14. doi: 10.1016/j.cell.2020.05.042
86. Rodrigues TS, de Sá KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med (2020) 218. doi: 10.1084/jem.20201707
87. Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, et al. Immunology of COVID-19: Current State of the Science. Immunity (2020) 6:910–41. doi: 10.1016/j.immuni.2020.05.002
88. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15:87–103. doi: 10.1038/nri3787
89. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell (2020) 181:1016–35. doi: 10.1016/j.cell.2020.04.035
90. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe (2016) 19:181–93. doi: 10.1016/j.chom.2016.01.007
91. Zhou Y, Fu B, Zheng X, Wang D, Zhao C. Aberrant pathogenic GM-CSF+ T cells and inflammatory CD14+CD16+ monocytes in severe pulmonary syndrome patients of a new coronavirus. bioRxiv (2020). doi: 10.1101/2020.02.12.945576
92. Loutfy MR, Blatt LM, Siminovitch KA, Ward S, Wolff B, Lho H, et al. Interferon Alfacon-1 Plus Corticosteroids in Severe Acute Respiratory Syndrome. JAMA (2003) 290:3222. doi: 10.1001/jama.290.24.3222
93. Cameron MJ, Ran L, Xu L, Danesh A, Bermejo-Martin JF, Cameron CM, et al. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J Virol (2007) 81:8692–706. doi: 10.1128/JVI.00527-07
94. Gabriele L, Fragale A, Romagnoli G, Parlato S, Lapenta C, Santini SM, et al. Type I IFN-dependent antibody response at the basis of sex dimorphism in the outcome of COVID-19. Cytokine Growth Factor Rev (2020) S1359-6101:30215-X. doi: 10.1016/j.cytogfr.2020.10.001
95. Zhou R, To KK-W, Wong Y-C, Liu L, Zhou B, Li X, et al. Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity (2020) 53:864–77.e5. doi: 10.1016/j.immuni.2020.07.026
96. Taneja V. Sex Hormones Determine Immune Response. Front Immunol (2018) 9:1931. doi: 10.3389/fimmu.2018.01931
97. de Souza PR, Sales-Campos H, Basso PJ, Nardini V, Silva A, Banquieri F, et al. Adrenal-Derived Hormones Differentially Modulate Intestinal Immunity in Experimental Colitis. Mediators Inflammation (2016) 2016:4936370. doi: 10.1155/2016/4936370
98. Wilhelmson AS, Lantero Rodriguez M, Stubelius A, Fogelstrand P, Johansson I, Buechler MB, et al. Testosterone is an endogenous regulator of BAFF and splenic B cell number. Nat Commun (2018) 9:2067. doi: 10.1038/s41467-018-04408-0
99. Gubbels Bupp MR, Jorgensen TN. Androgen-Induced Immunosuppression. Front Immunol (2018) 9:794. doi: 10.3389/fimmu.2018.00794
100. Butterworth M, McClellan B, Allansmith M. Influence of sex in immunoglobulin levels. Nature (1967) 214:1224–5. doi: 10.1038/2141224a0
101. Anjo C, Mascaró J-M, Espinosa G, Cervera R. Effectiveness and safety of belimumab in patients with systemic lupus erythematosus in a real-world setting. Scand J Rheumatol (2019) 48:469–73. doi: 10.1080/03009742.2019.1603324
102. Fairweather D, Frisancho-Kiss S, Rose NR. Sex differences in autoimmune disease from a pathological perspective. Am J Pathol (2008) 173:600–9. doi: 10.2353/ajpath.2008.071008
103. Durcan L, O’Dwyer T, Petri M. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet (London England) (2019) 393:2332–43. doi: 10.1016/S0140-6736(19)30237-5
104. Rees F, Doherty M, Grainge M, Davenport G, Lanyon P, Zhang W. The incidence and prevalence of systemic lupus erythematosus in the UK, 1999-2012. Ann Rheum Dis (2016) 75:136–41. doi: 10.1136/annrheumdis-2014-206334
105. Karlberg J, Chong DSY, Lai WYY. Do men have a higher case fatality rate of severe acute respiratory syndrome than women do? Am J Epidemiol (2004) 159:229–31. doi: 10.1093/aje/kwh056
106. Alghamdi IG, Hussain II, Almalki SS, Alghamdi MS, Alghamdi MM, El-Sheemy MA. The pattern of Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive epidemiological analysis of data from the Saudi Ministry of Health. Int J Gen Med (2014) 7:417–23. doi: 10.2147/IJGM.S67061
107. Arima Y, Zu R, Murhekar M, Vong S, Shimada T. Human infections with avian influenza A(H7N9) virus in China: preliminary assessments of the age and sex distribution. West Pacific Surveill Response J (2013) 4:1–3. doi: 10.5365/wpsar.2013.4.2.005
108. Wang XL, Yang L, Chan K-H, Chan K-P, Cao P-H, Lau EH-Y, et al. Age and Sex Differences in Rates of Influenza-Associated Hospitalizations in Hong Kong. Am J Epidemiol (2015) 182:335–44. doi: 10.1093/aje/kwv068
109. Crighton EJ, Elliott SJ, Moineddin R, Kanaroglou P, Upshur REG. An exploratory spatial analysis of pneumonia and influenza hospitalizations in Ontario by age and gender. Epidemiol Infect (2007) 135:253–61. doi: 10.1017/S095026880600690X
110. Furman D, Hejblum BP, Simon N, Jojic V, Dekker CL, Thiébaut R, et al. Systems analysis of sex differences reveals an immunosuppressive role for testosterone in the response to influenza vaccination. Proc Natl Acad Sci USA (2014) 111:869–74. doi: 10.1073/pnas.1321060111
111. Wadman M. Sex hormones signal why virus hits men harder. Science (2020) 368:1038–9. doi: 10.1126/science.368.6495.1038
112. Shi Y, Yu X, Zhao H, Wang H, Zhao R, Sheng J. Host susceptibility to severe COVID-19 and establishment of a host risk score: findings of 487 cases outside Wuhan. Crit Care (2020) 24:108. doi: 10.1186/s13054-020-2833-7
113. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet (London England) (2020) 395:507–13. doi: 10.1016/S0140-6736(20)30211-7
114. Guan W-J, Liang W-H, Zhao Y, Liang H-R, Chen Z-S, Li Y-M, et al. Comorbidity and its impact on 1590 patients with COVID-19 in China: a nationwide analysis. Eur Respir J (2020) 55:2000547. doi: 10.1183/13993003.00547-2020
115. Fish EN. The X-files in immunity: sex-based differences predispose immune responses. Nat Rev Immunol (2008) 8:737–44. doi: 10.1038/nri2394
116. Dalpiaz ELM, Lamas AZ, Caliman IF, Ribeiro RF, Abreu GR, Moyses MR, et al. Correction: Sex Hormones Promote Opposite Effects on ACE and ACE2 Activity, Hypertrophy and Cardiac Contractility in Spontaneously Hypertensive Rats. PloS One (2015) 10:e0133225. doi: 10.1371/journal.pone.0133225
117. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem (2000) 275:33238–43. doi: 10.1074/jbc.M002615200
118. Li Y, Jerkic M, Slutsky AS, Zhang H. Molecular mechanisms of sex bias differences in COVID-19 mortality. Crit Care (2020) 24:405. doi: 10.1186/s13054-020-03118-8
119. Gemmati D, Bramanti B, Serino ML, Secchiero P, Zauli G, Tisato V. COVID-19 and Individual Genetic Susceptibility/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation. Might the Double X-chromosome in Females Be Protective against SARS-CoV-2 Compared to the Single X-Chromosome in Males? Int J Mol Sci (2020) 21:3474. doi: 10.3390/ijms21103474
120. Stelzig KE, Canepa-Escaro F, Schiliro M, Berdnikovs S, Prakash YS, Chiarella SE. Estrogen regulates the expression of SARS-CoV-2 receptor ACE2 in differentiated airway epithelial cells. Am J Physiol Lung Cell Mol Physiol (2020) 318:L1280–1. doi: 10.1152/ajplung.00153.2020
121. Wambier CG, Goren A. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is likely to be androgen mediated. J Am Acad Dermatol (2020) 83:308–9. doi: 10.1016/j.jaad.2020.04.032
122. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181:271–280.e8. doi: 10.1016/j.cell.2020.02.052
123. Lucas JM, Heinlein C, Kim T, Hernandez SA, Malik MS, True LD, et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discovery (2014) 4:1310–25. doi: 10.1158/2159-8290.CD-13-1010
124. Lin B, Ferguson C, White JT, Wang S, Vessella R, True LD, et al. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer Res (1999) 59:4180–4.
125. Mikkonen L, Pihlajamaa P, Sahu B, Zhang F-P, Jänne OA. Androgen receptor and androgen-dependent gene expression in lung. Mol Cell Endocrinol (2010) 317:14–24. doi: 10.1016/j.mce.2009.12.022
126. Montopoli M, Zumerle S, Vettor R, Rugge M, Zorzi M, Catapano CV, et al. Androgen-deprivation therapies for prostate cancer and risk of infection by SARS-CoV-2: a population-based study (N = 4532). Ann Oncol Off J Eur Soc Med Oncol (2020) 31:1040–5. doi: 10.1016/j.annonc.2020.04.479
127. Shaw GL, Whitaker H, Corcoran M, Dunning MJ, Luxton H, Kay J, et al. The Early Effects of Rapid Androgen Deprivation on Human Prostate Cancer. Eur Urol (2016) 70:214–8. doi: 10.1016/j.eururo.2015.10.042
128. Mjaess G, Karam A, Aoun F, Albisinni S, Roumeguère T. COVID-19 and the male susceptibility: the role of ACE2, TMPRSS2 and the androgen receptor. Prog Urol (2020) 30:484–7. doi: 10.1016/j.purol.2020.05.007
129. Wenham C, Smith J, Morgan R. Gender and COVID-19 Working Group. COVID-19: the gendered impacts of the outbreak. Lancet (London England) (2020) 395:846–8. doi: 10.1016/S0140-6736(20)30526-2
130. Roved J, Westerdahl H, Hasselquist D. Sex differences in immune responses: Hormonal effects, antagonistic selection, and evolutionary consequences. Horm Behav (2017) 88:95–105. doi: 10.1016/j.yhbeh.2016.11.017
131. Viselli SM, Reese KR, Fan J, Kovacs WJ, Olsen NJ. Androgens alter B cell development in normal male mice. Cell Immunol (1997) 182:99–104. doi: 10.1006/cimm.1997.1227
132. Laffont S, Blanquart E, Savignac M, Cénac C, Laverny G, Metzger D, et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J Exp Med (2017) 214:1581–92. doi: 10.1084/jem.20161807
133. Consiglio CR, Udartseva O, Ramsey KD, Bush C, Gollnick SO. Enzalutamide, an Androgen Receptor Antagonist, Enhances Myeloid Cell-Mediated Immune Suppression and Tumor Progression. Cancer Immunol Res (2020) 8:1215–27. doi: 10.1158/2326-6066.CIR-19-0371
134. Rastrelli G, Di Stasi V, Inglese F, Beccaria M, Garuti M, Di Costanzo D, et al. Low testosterone levels predict clinical adverse outcomes in SARS-CoV-2 pneumonia patients. Andrology (2020) 00:1–11. doi: 10.1111/andr.12821
135. Samuel RM, Majd H, Richter MN, Ghazizadeh Z, Zekavat SM, Navickas A, et al. Androgen Signaling Regulates SARS-CoV-2 Receptor Levels and Is Associated with Severe COVID-19 Symptoms in Men. Cell Stem Cell (2020) 27:876–889.e12. doi: 10.1016/j.stem.2020.11.009
136. Libert C, Dejager L, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol (2010) 10:594–604. doi: 10.1038/nri2815
137. Conti P, Younes A. Coronavirus COV-19/SARS-CoV-2 affects women less than men: clinical response to viral infection. J Biol Regul Homeost Agents (2020) 34:339–43. doi: 10.23812/Editorial-Conti-3
138. Takahashi T, Ellingson MK, Wong P, Israelow B, Lucas C, Klein J, et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature (2020) 588:315–20. doi: 10.1038/s41586-020-2700-3
139. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science (2001) 294:1871–5. doi: 10.1126/science.294.5548.1871
140. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol (2015) 15:511–23. doi: 10.1038/nri3859
141. Medeiros AI, Sá-Nunes A, Soares EG, Peres CM, Silva CL, Faccioli LH. Blockade of endogenous leukotrienes exacerbates pulmonary histoplasmosis. Infect Immun (2004) 72:1637–44. doi: 10.1128/iai.72.3.1637-1644.2004
142. Machado ER, Ueta MT, Lourenço EV, Anibal FF, Sorgi CA, Soares EG, et al. Leukotrienes play a role in the control of parasite burden in murine strongyloidiasis. J Immunol (2005) 175:3892–9. doi: 10.4049/jimmunol.175.6.3892
143. Lone AM, Taskén K. Proinflammatory and immunoregulatory roles of eicosanoids in T cells. Front Immunol (2013) 4:130. doi: 10.3389/fimmu.2013.00130
144. Zoccal KF, Sorgi CA, Hori JI, Paula-Silva FWG, Arantes EC, Serezani CH, et al. Opposing roles of LTB4 and PGE2 in regulating the inflammasome-dependent scorpion venom-induced mortality. Nat Commun (2016) 7:10760. doi: 10.1038/ncomms10760
145. Martínez-Colón GJ, Moore BB. Prostaglandin E2 as a Regulator of Immunity to Pathogens. Pharmacol Ther (2018) 185:135–46. doi: 10.1016/j.pharmthera.2017.12.008
146. Reis MB, Zoccal KF, Gardinassi LG, Faccioli LH. Scorpion envenomation and inflammation: Beyond neurotoxic effects. Toxicon (2019) 167:174–9. doi: 10.1016/j.toxicon.2019.06.219
147. Sorgi CA, Soares EM, Rosada RS, Bitencourt CS, Zoccal KF, Pereira PAT, et al. Eicosanoid pathway on host resistance and inflammation during Mycobacterium tuberculosis infection is comprised by LTB4 reduction but not PGE2 increment. Biochim Biophys Acta Mol basis Dis (2020) 1866:165574. doi: 10.1016/j.bbadis.2019.165574
148. Volovitz B, Faden H, Ogra PL. Release of leukotriene C4 in respiratory tract during acute viral infection. J Pediatr (1988) 112:218–22. doi: 10.1016/s0022-3476(88)80058-1
149. Peres CM, de Paula L, Medeiros AI, Sorgi CA, Soares EG, Carlos D, et al. Inhibition of leukotriene biosynthesis abrogates the host control of Mycobacterium tuberculosis. Microbes Infect (2007) 9:483–9. doi: 10.1016/j.micinf.2007.01.006
150. Nicolete R, Secatto A, Pereira PAT, Soares EG, Faccioli LH. Leukotriene B4-loaded microspheres as a new approach to enhance antimicrobial responses in Histoplasma capsulatum-infected mice. Int J Antimicrob Agents (2009) 34:365–9. doi: 10.1016/j.ijantimicag.2009.06.009
151. Soares EM, Mason KL, Rogers LM, Serezani CH, Faccioli LH, Aronoff DM. Leukotriene B4 enhances innate immune defense against the puerperal sepsis agent Streptococcus pyogenes. J Immunol (2013) 190:1614–22. doi: 10.4049/jimmunol.1202932
152. Secatto A, Soares EM, Locachevic GA, Assis PA, Paula-Silva FWG, Serezani CH, et al. The leukotriene B₄/BLT₁ axis is a key determinant in susceptibility and resistance to histoplasmosis. PloS One (2014) 9:e85083. doi: 10.1371/journal.pone.0085083
153. Prado MKB, Locachevic GA, Zoccal KF, Paula-Silva FWG, Fontanari C, Ferreira JC, et al. Leukotriene B4 is essential for lung host defence and alpha-defensin-1 production during Achromobacter xylosoxidans infection. Sci Rep (2017) 7:17658. doi: 10.1038/s41598-017-17993-9
154. Medeiros AI, Silva CL, Malheiro A, Maffei CM, Faccioli LH. Leukotrienes are involved in leukocyte recruitment induced by live Histoplasma capsulatum or by the beta-glucan present in their cell wall. Br J Pharmacol (1999) 128:1529–37. doi: 10.1038/sj.bjp.0702912
155. Nicolete R, Lima K de M, Júnior JMR, Baruffi MD, de Medeiros AI, Bentley MVLB, et al. In vitro and in vivo activities of leukotriene B4-loaded biodegradable microspheres. Prostagland Other Lipid Mediat (2007) 83:121–9. doi: 10.1016/j.prostaglandins.2006.10.007
156. McCarthy MK, Weinberg JB. Eicosanoids and respiratory viral infection: coordinators of inflammation and potential therapeutic targets. Mediators Inflammation (2012) 2012:236345. doi: 10.1155/2012/236345
157. Behera AK, Kumar M, Matsuse H, Lockey RF, Mohapatra SS. Respiratory syncytial virus induces the expression of 5-lipoxygenase and endothelin-1 in bronchial epithelial cells. Biochem Biophys Res Commun (1998) 251:704–9. doi: 10.1006/bbrc.1998.9537
158. Le Bel M, Gosselin J. Leukotriene B4 Enhances NOD2-Dependent Innate Response against Influenza Virus Infection. PloS One (2015) 10:e0139856. doi: 10.1371/journal.pone.0139856
159. Trindade BC, Sorgi CA, Nicolete LD de F, Malta TM, Pinto MT, Takayanagui OM, et al. Leukotrienes are upregulated and associated with human T-lymphotropic virus type 1 (HTLV-1)-associated neuroinflammatory disease. PloS One (2012) 7:e51873. doi: 10.1371/journal.pone.0051873
160. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol (2011) 31:986–1000. doi: 10.1161/ATVBAHA.110.207449
161. Pereira PAT, Assis PA, Prado MKB, Ramos SG, Aronoff DM, de Paula-Silva FWG, et al. Prostaglandins D2 and E2 have opposite effects on alveolar macrophages infected with Histoplasma capsulatum. J Lipid Res (2018) 59:195–206. doi: 10.1194/jlr.M078162
162. Zoccal KF, Ferreira GZ, Prado MKB, Gardinassi LG, Sampaio SV, Faccioli LH. LTB4 and PGE2 modulate the release of MIP-1α and IL-1β by cells stimulated with Bothrops snake venoms. Toxicon (2018) 150:289–96. doi: 10.1016/j.toxicon.2018.06.066
163. Sander WJ, O’Neill HG, Pohl CH. Prostaglandin E2 As a Modulator of Viral Infections. Front Physiol (2017) 8:89. doi: 10.3389/fphys.2017.00089
164. Coulombe F, Divangahi M. Targeting eicosanoid pathways in the development of novel anti-influenza drugs. Expert Rev Anti Infect Ther (2014) 12:1337–43. doi: 10.1586/14787210.2014.966082
165. Zhang AJX, Zhu H, Chen Y, Li C, Li C, Chu H, et al. Prostaglandin E2-Mediated Impairment of Innate Immune Response to A(H1N1)pdm09 Infection in Diet-Induced Obese Mice Could Be Restored by Paracetamol. J Infect Dis (2019) 219:795–807. doi: 10.1093/infdis/jiy527
166. Schwarz B, Sharma L, Roberts L, Peng X, Bermejo S, Leighton I, et al. Cutting Edge: Severe SARS-CoV-2 Infection in Humans Is Defined by a Shift in the Serum Lipidome, Resulting in Dysregulation of Eicosanoid Immune Mediators. J Immunol (2020) 206:329–34. doi: 10.4049/jimmunol.2001025
167. Barberis E, Timo S, Amede E, Vanella VV, Puricelli C, Cappellano G, et al. Large-Scale Plasma Analysis Revealed New Mechanisms and Molecules Associated with the Host Response to SARS-CoV-2. Int J Mol Sci (2020) 21:8623. doi: 10.3390/ijms21228623
168. Overmyer KA, Shishkova E, Miller IJ, Balnis J, Bernstein MN, Peters-Clarke TM, et al. Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Syst (2020) S2405-4712:30371–9. doi: 10.1016/j.cels.2020.10.003
169. Das UN. Bioactive Lipids in COVID-19-Further Evidence. Arch Med Res (2020). doi: 10.1016/j.arcmed.2020.09.006
170. Anne-Sophie, Archambault Younes Z, Volatiana, Rakotoarivelo Étienne D, Isabelle D, Cyril M, et al. Lipid storm within the lungs of severe COVID-19 patients: Extensive levels of cyclooxygenase and lipoxygenase-derived inflammatory metabolites. medRxiv Prepr Serv Heal Sci (2020) 1–31. doi: 10.1101/2020.12.04.20242115
171. Chapman SJ, Hill AVS. Human genetic susceptibility to infectious disease. Nat Rev Genet (2012) 13:175–88. doi: 10.1038/nrg3114
172. Chan VSF, Chan KYK, Chen Y, Poon LLM, Cheung ANY, Zheng B, et al. Homozygous L-SIGN (CLEC4M) plays a protective role in SARS coronavirus infection. Nat Genet (2006) 38:38–46. doi: 10.1038/ng1698
173. Gralinski LE, Ferris MT, Aylor DL, Whitmore AC, Green R, Frieman MB, et al. Genome Wide Identification of SARS-CoV Susceptibility Loci Using the Collaborative Cross. PloS Genet (2015) 11:1–21. doi: 10.1371/journal.pgen.1005504
174. Nguyen A, David JK, Maden SK, Wood MA, Weeder BR, Nellore A, et al. Human Leukocyte Antigen Susceptibility Map for Severe Acute Respiratory Syndrome Coronavirus 2. J Virol (2020) 94:e00150-20. doi: 10.1128/JVI.00510-20
175. Ulhaq ZS, Soraya GV. Anti-IL-6 Receptor Antibody Treatment for Severe COVID-19 and the Potential Implication of IL-6 Gene Polymorphisms in Novel Coronavirus Pneumonia. SSRN Electron J (2020) 155:548–56. doi: 10.2139/ssrn.3592878
176. Kirtipal N, Bharadwaj S. Interleukin 6 polymorphisms as an indicator of COVID-19 severity in humans. J Biomol Struct Dyn (2020), 1–3. doi: 10.1080/07391102.2020.1776640
177. Wu Y, Feng Z, Li P, Yu Q. Relationship between ABO blood group distribution and clinical characteristics in patients with COVID-19. Clin Chim Acta (2020) 509:220–3. doi: 10.1016/j.cca.2020.06.026
178. Ramlall V, Thangaraj PM, Meydan C, Foox J, Butler D, Kim J, et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat Med (2020) 26:1609–15. doi: 10.1038/s41591-020-1021-2
179. Sabbagh A, Sonon P, Sadissou I, Mendes-Junior CT, Garcia A, Donadi EA, et al. The role of HLA-G in parasitic diseases. HLA (2018) 91:255–70. doi: 10.1111/tan.13196
180. Hauer V, Risti M, Miranda BLM, da Silva JS, Cidral AL, Pozzi CM, et al. The association of HLA-G polymorphisms and the synergistic effect of sMICA and sHLA-G with chronic kidney disease and allograft acceptance. PloS One (2019) 14:e0212750. doi: 10.1371/journal.pone.0212750
181. Rouas-Freiss N, Gonçalves RM, Menier C, Dausset J, Carosella ED. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc Natl Acad Sci USA (1997) 94:11520–5. doi: 10.1073/pnas.94.21.11520
182. Naji A, Menier C, Morandi F, Agaugué S, Maki G, Ferretti E, et al. Binding of HLA-G to ITIM-bearing Ig-like transcript 2 receptor suppresses B cell responses. J Immunol (2014) 192:1536–46. doi: 10.4049/jimmunol.1300438
183. Donadi EA, Castelli EC, Arnaiz-Villena A, Roger M, Rey D, Moreau P. Implications of the polymorphism of HLA-G on its function, regulation, evolution and disease association. Cell Mol Life Sci (2011) 68:369–95. doi: 10.1007/s00018-010-0580-7
184. Dias FC, Castelli EC, Collares CVA, Moreau P, Donadi EA. The Role of HLA-G Molecule and HLA-G Gene Polymorphisms in Tumors, Viral Hepatitis, and Parasitic Diseases. Front Immunol (2015) 6:9. doi: 10.3389/fimmu.2015.00009
185. Poras I, Yaghi L, Martelli-Palomino G, Mendes-Junior CT, Muniz YCN, Cagnin NF, et al. Haplotypes of the HLA-G 3’ Untranslated Region Respond to Endogenous Factors of HLA-G+ and HLA-G- Cell Lines Differentially. PloS One (2017) 12:e0169032. doi: 10.1371/journal.pone.0169032
186. Brown D, Trowsdale J, Allen R. The LILR family: modulators of innate and adaptive immune pathways in health and disease. Tissue Antigens (2004) 64:215–25. doi: 10.1111/j.0001-2815.2004.00290.x
187. Dietrich J, Cella M, Colonna M. Ig-Like Transcript 2 (ILT2)/Leukocyte Ig-Like Receptor 1 (LIR1) Inhibits TCR Signaling and Actin Cytoskeleton Reorganization. J Immunol (2001) 166:2514–21. doi: 10.4049/jimmunol.166.4.2514
188. Shiroishi M, Tsumoto K, Amano K, Shirakihara Y, Colonna M, Braud VM, et al. Human inhibitory receptors Ig-like transcript 2 (ILT2) and ILT4 compete with CD8 for MHC class I binding and bind preferentially to HLA-G. Proc Natl Acad Sci USA (2003) 100:8856–61. doi: 10.1073/pnas.1431057100
189. Puppo F, Contini P, Ghio M, Indiveri F. Soluble HLA class I molecules/CD8 ligation trigger apoptosis of CD8+ cells by Fas/Fas-ligand interaction. ScientificWorldJournal (2002) 2:421–3. doi: 10.1100/tsw.2002.122
190. Contini P, Ghio M, Poggi A, Filaci G, Indiveri F, Ferrone S, et al. Soluble HLA-A,-B,-C and -G molecules induce apoptosis in T and NK CD8+ cells and inhibit cytotoxic T cell activity through CD8 ligation. Eur J Immunol (2003) 33:125–34. doi: 10.1002/immu.200390015
191. Fainardi E, Castellazzi M, Stignani M, Morandi F, Sana G, Gonzalez R, et al. Emerging topics and new perspectives on HLA-G. Cell Mol Life Sci (2011) 68:433–51. doi: 10.1007/s00018-010-0584-3
192. Hansen TH, Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol (2009) 9:503–13. doi: 10.1038/nri2575
193. Zhang S, Gan J, Chen B, Zheng D, Zhang J, Lin R, et al. Dynamics of peripheral immune cells and their HLA-G and receptor expressions in a patient suffering from critical COVID-19 pneumonia to convalescence. Clin Transl Immunol (2020) 9:e1128. doi: 10.1002/cti2.1128
Keywords: coronavirus disease 2019, peripheral immune response, pulmonary inflammation, lung immunopathology, damage tolerance
Citation: Fraga-Silva TFdC, Maruyama SR, Sorgi CA, Russo EMdS, Fernandes APM, de Barros Cardoso CR, Faccioli LH, Dias-Baruffi M and Bonato VLD (2021) COVID-19: Integrating the Complexity of Systemic and Pulmonary Immunopathology to Identify Biomarkers for Different Outcomes. Front. Immunol. 11:599736. doi: 10.3389/fimmu.2020.599736
Received: 28 August 2020; Accepted: 29 December 2020;
Published: 29 January 2021.
Edited by:
Annalisa Del Prete, University of Brescia, ItalyReviewed by:
Mangai Asokan, Independent Researcher, Bengaluru, IndiaElettra Barberis, Università del Piemonte Orientale, Italy
Copyright © 2021 Fraga-Silva, Maruyama, Sorgi, Russo, Fernandes, de Barros Cardoso, Faccioli, Dias-Baruffi and Bonato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vânia Luiza Deperon Bonato, vlbonato@fmrp.usp.br
†These authors have contributed equally to this work