 Yuan Yuan
Yuan Yuan Zihan Feng
Zihan Feng Jinglin Wang
Jinglin Wang- State Key Laboratory of Pathogen and Biosecurity, Beijing Institute of Microbiology and Epidemiology, Academy of Military Medical Sciences (AMMS), Beijing, China
The Vibrio vulnificus (V. vulnificus) hemolysin (VVH) is a pore-forming cholesterol-dependent cytolysin (CDC). Although there has been some debate surrounding the in vivo virulence effects of the VVH, it is becoming increasingly clear that it drives different cellular outcomes and is involved in the pathogenesis of V. vulnificus. This minireview outlines recent advances in our understanding of the regulation of vvhA gene expression, the biological activity of the VVH and its role in pathogenesis. An in-depth examination of the role of the VVH in V. vulnificus pathogenesis will help reveal the potential targets for therapeutic and preventive interventions to treat fatal V. vulnificus septicemia in humans. Future directions in VVH research will also be discussed.
Introduction
V. vulnificus is an opportunistic human pathogen commonly found in estuarine environments. Human infections usually occur following the consumption of contaminated seafood or via an open wound exposed to a contaminated water source (1). Consumption of contaminated raw oysters can result in rapidly fatal septicemia in susceptible individuals, with V. vulnificus having the highest fatality rate among all food-borne pathogens (2). However, many aspects related to the biology, genomics, and virulence capabilities of V. vulnificus remain elusive or poorly understood (1, 3). During the last decade, research has mainly been focused on the pathogenic mechanisms and virulence factors adopted by V. vulnificus (2, 4). The capsule has proven to be a critical virulence factor, with non-encapsulated V. vulnificus isogenic mutants readily phagocytosed by host immune cells (5). The V. vulnificus multifunctional-auto processing repeats-in-toxin (MARTX) toxin is also likely to be critical to the success of infection. Supporting this, Gavin et al. showed that the MARTX toxin is essential for bacterial dissemination from the intestine (6), while Jones and Oliver demonstrated that the overwhelming tissue destruction that characterizes V. vulnificus infections contracted either via ingestion or wound infection likely results from the powerful collagenase, metalloproteases, and lipases/phospholipases produced by the bacterium (4). Moreover, MARTX is also known to take part in resistance to phagocytosis, cell destruction, and sepsis (7, 8).
Although the VVH belongs to the cytolytic pore-forming family of toxins (PFTs), all of which cause cytolysis in a variety of mammalian cells, VVH as a virulence factor is under debate. An earlier study has shown that disruption of hemolysin gene vvhA had no effect on the virulence of V. vulnificus in a mouse lethality model (9). However, other studies have confirmed that vvhA gene is substantially regulated and expressed in vivo and is likely to play important roles in the pathogenesis of V. vulnificus (10, 11). For example, V. vulnificus is known for a siderophilic bacterium and iron as one of the factors that regulate vvhA expression (12–14). Fan JJ et al. indicate that there was a small difference in mortality when wild type and vvhA-deficient mutant strains were force-fed to mice, but VVH seemed to be important for causing damage in the alimentary tract of the mice (15). Moreover, another study suggested that in addition to the MARTX toxin, the VVH may contribute to bacterial invasion from the intestine into the bloodstream and other organs (16). These results would suggest that VVH may not be responsible for the lethality of V. vulnificus, but may be a contributor to the tissue damage in pathogenesis. Moreover, other proposed virulence factors characterized to date are not sufficient to explain the acute process of V. vulnificus septicemia. The vvhA gene is found in most V. vulnificus isolates, which was often used as a detecting marker for V. vulnificus (17, 18). However, unlike other Vibrio spp. such as V. cholerae and V. parahaemolyticus, where distinct molecular attributes, such as toxin genes, are normally associated with clinical strains (19, 20). More researchers contend that infections may be driven more by factors associated with host susceptibility than the virulence of V. vulnificus (1, 12). Besides V. vulnificus, the hemolysins produced by Vibrio cholerae and Gram-positive species such as Streptococcus pneumoniae, Streptococcus suis, Bacillus Cereus, have been extensively reviewed (21–23). Like most Gram-negative bacteria, the X-ray crystal structure of VVH remains unknown. However, the mechanisms of pore formation by VVH have been studied in crucial amino acid residues and domains related to the activity of VVH (24–27). Molecular architecture and functional analysis of V. cholerae cytolysin (VCC) revealed that VVH has a similar cytolysin domain and a lectin-like domain of VCC (28). However, although these pore-forming cholesterol-dependent cytolysins share structural similarity, they drive divergent cellular outcomes during pathogenesis. In comparison to PFTs in Gram-positive bacteria, more research is needed to clarify the role of VVH in pathogenesis, especially in infections with raw oyster consumption, which can produce rapidly fatal V. vulnificus septicemia.
In this review, we explore the features of VVH in its biological activity, regulation of vvhA expression, and possible roles in pathogenesis. Future directions in VVH research was also discussed in this review. This in-depth evaluation of the contribution of the VVH to V. vulnificus pathogenesis may aid in the development of novel therapies aimed at treating and preventing sepsis in humans.
Effects of the VVH on Eukaryotic Cells
VVH is a 51-kDa water-soluble protein thought to be a member of CDC family of PFT; its hemolytic activity was inhibited by adding cholesterol or divalent cations (29). The VVH causes necrosis, apoptosis, pyroptosis, and lysis in a range of host cell types. First described as a hemolysin, the VVH causes hemolysis of red blood cells in many species, with human erythrocytes being the most susceptible. Although active against erythrocytes from sheep, horses, cows, rabbits, and chickens, the amount of VVH required to cause 50% hemolysis under identical conditions differed between species, suggesting that erythrocyte susceptibility may be closely associated with the binding ability of the VVH and erythrocyte membrane stability (30). In vitro studies have illuminated the effects of the VVH in various host cell types, including human epithelial cells, human umbilical vein endothelial cells (HUVECs), mice macrophages and lymphocytes (Figure 1), and commonly used cell lines, such as Chinese hamster ovary (CHO) cells (26, 27). However, similar cytotoxic effects have not been reported in human platelets or monocytes.
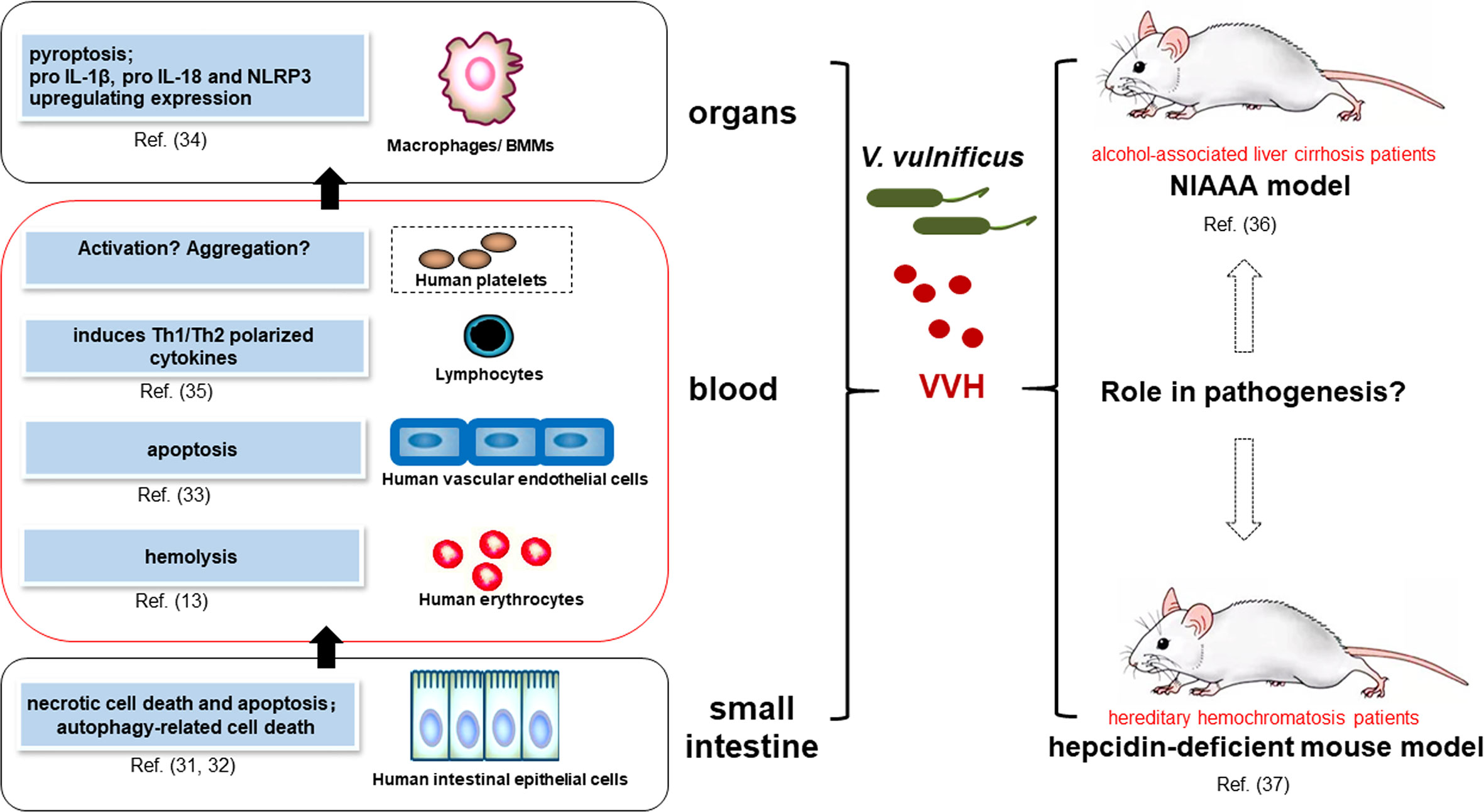
Figure 1 Major activity of VVH’s interactions with host cells and a future perspective of in vivo studies involved in pathogenesis. Major activity and mechanism of VVH’s Interactions with host cells mainly focus on intestinal epithelial cells (31, 32), vascular endothelial cells (33), macrophages, (34) and lymphocytes (35), which are possibly involved in bacterial invasion from intestine to blood stream and other organs. However, the effects of VVH on platelets have not been reported. The animal models that mimick human infection will provide a perspective to elucidate the role of VVH in pathogenesis, mainly including the National Institute on Alcohol Abuse and Alcoholism (NIAAA) model (36) and a hepcidin-deficient mouse model (37).
While in vitro studies have revealed the comprehensive effects of the VVH on eukaryotic cells, researchers have also examined the impact of host effectors on the activity of the VVH. Cholesterol is well known for its ability to inactivate the VVH through oligomerization of the toxin monomer (29). However, there are some reports that the VVH recognizes and binds to certain kinds of carbohydrates (38, 39), which suggested that cellular cholesterol is not a receptor for VVH. It may be a trigger factor of conformational changes from membrane bound form to pore-form (39). Moreover, although two studies indicate VVH induces cell death via lipid raft-mediated signaling pathway in human intestinal epithelial cells (31, 32), there is no evidence that the VVH localizes at lipid raft so far. One study shows that binding of VVH to target cells does not change by the methyl-beta-cyclodextrin (MβCD) treatment (40), and the author subsequently indicates that MβCD induces oligomerization of VVH by binding to VVH directly (41).
Besides that, several other factors have also been reported to affect the cytotoxicity of the VVH. Although albumin affects the activity of many different bacterial toxins, Choi et al. reported that neither human serum albumin (HAS) nor bovine serum albumin (BSA) affected vvhA transcription or the growth of V. vulnificus. However, both HSA and BSA stabilized VVH and delayed its inactivation by oligomerization, thus enhancing VVH activity (42). Blood lipoproteins have also been shown to be an important defense factor against bacterial infection. Park et al. found that low density lipoprotein inactivates the VVH through the oligomerization of the toxin monomer (43). It was widely reported that calcium prevented hemolysis caused by a variety of bacterial hemolysins (44). Jin-Woo Park showed that calcium exerts its major inhibitory effect on V. vulnificus cytolysin-induced hemolysis as an osmotic protectant (45). Consequently, trifluoperazine, a calcium-calmodulin antagonist, was found to block the hyperpermeability induced by V. vulnificus cytolysin in an in vitro modeled endothelium and prevented the deaths of mice (46). Additionally, a recent study showed that melatonin, an endogenous hormone molecule, inhibits apoptotic cell death induced by VVH via melatonin receptor 2 coupling with NCF-1 (47). While promising, these results emphasize the fact that we still have much to learn about how the VVH displays its cytotoxic effects in vivo, knowledge that will provide important insights into the potential for development of therapeutic strategies and agents to combat V. vulnificus infection.
Of special interest is the question of whether the VVH contributes to bacterial invasion from the intestine into the bloodstream and other organs by interacting with host cells. Intestinal epithelial cell death is a host defense response that eliminates damaged cells as well as pathogens to maintain gut homeostasis. However, many bacterial pathogens eventually elicit epithelial cell death and disrupt the gut barrier function to propagate persistent bacterial colonization. A study performed in human intestinal epithelial cells (INT-407) showed that infection with low doses of recombinant VVH protein induces necrotic cell death and apoptosis. The study further demonstrated that (r)VVH induces NF-κB-dependent mitochondrial cell death via lipid raft-mediated reactive oxygen species production by the distinct activation of PKCα and ERK/JNK in intestinal epithelial cells (31). Besides VVH has the ability to induce two general modes of cell death, apoptosis and necrosis mentioned above; another study indicated that the VVH induced autophagy-related cell death through the lipid raft-dependent c-Src/NOX signaling pathway in human intestinal epithelial Caco-2 cells. This study further showed that, in an in vivo model, VVH increased autophagy activation and paracellular permeabilization in the intestinal epithelium, indicating that VVH plays a pivotal role in the pathogenesis and dissemination of V. vulnificus via the upregulation of autophagy, which may provide potential therapeutic targets for strategic modulations of V. vulnificus infections (32).
V. vulnificus has been shown to produce sufficient VVH in the small intestine to accelerate invasion into the bloodstream (16). Once V. vulnificus is in the bloodstream, the VVH interacts with erythrocytes, white blood cells, and vascular endothelial cells. In fact, a recent study has shown that VVH together with MARTX mediates erythrocytes lyses ex vivo and, therefore, could contribute to the bacterial growth in human blood that provokes sepsis (13). Researchers observed in vitro proliferation of lymphocytes upon re-stimulation of recombinant VVH leukocidin domain (rL/VvhA)-primed splenocytes with formalin-inactivated VVH toxin, while co-expression of T-cell-polarizing cytokines (interferon-γ, interleukin (IL)-12, and IL-4) was detected in the cell culture supernatant (35). In an in vitro study, the recombinant VVH induces apoptosis in HUVEC cells via caspase-9/3-dependent pathway (33). The VVH can also spread to other tissues via the bloodstream. Macrophages are large phagocytes found in almost all tissues and play a critical role in increasing inflammation and stimulating the immune system. Claudia Toma et al. indicate that VVH-stimulated NLRP3 Inflammasome activation of bone marrow derived macrophages (BMM), which was induced by TLR and nucleotide-binding oligomerization domain 1/2 ligand-mediated NF-kB activation (34). Recently, analysis of VVH-induced inflammation in mice showed that the VVH induces inflammatory responses in RAW264.7 macrophages via calcium signaling and causes inflammation in vivo (48).
Regulation of VVH Gene (vvhA) Expression
In this review, we will outline the roles of environmental and host factors and global regulators in the regulation of the vvhA in terms of expression and transport (Figure 2). Cyclic-AMP (cAMP) and bacterial cyclic-AMP receptor proteins (CRPs) represent a classic regulatory system that has been adapted to respond to distinct external and internal signals in many bacteria (50). Hemolysin production in V. vulnificus increased after the addition of cAMP but was undetectable in a putative crp mutant, suggesting that vvh expression is positively regulated by cAMP-CRP in V. vulnificus (49). In V. vulnificus, cAMP can be produced from adenylate cyclase-encoding gene cya. Hemolysin and protease production, motility, and cytotoxicity were all negatively affected by mutation of cya (51). CRP activates vvhBA transcription in V. vulnificus by sensing the depletion of specific nutrients, possibly as a result of increased cAMP levels under glucose starvation (52). In Escherichia coli, glucose starvation results in an increase in intracellular cAMP concentrations in response to the altered phosphorylation state of the phosphotransferase system; however, this is difficult to reconcile with observations that the glucose phosphotransferase system remains saturated when intracellular cAMP concentrations increase (53). The regulation of vvhBA expression can be more easily examined in the intestine because the availability of free glucose is quite limited. V. vulnificus is a ferrophilic bacterium that requires high levels of available iron for growth (12, 13). Although iron can repress vvhA transcription via the ferric uptake regulator (Fur), it increases extracellular VVH secretion through increased transcription of pilD, which encodes PilD, a component of the type II general secretion system responsible for extracellular VVH secretion (14). But there are infection models that suggest that high iron levels (susceptible patients) could also increase vvhA transcription (13). So, the regulation of this gene expression should be more complex.
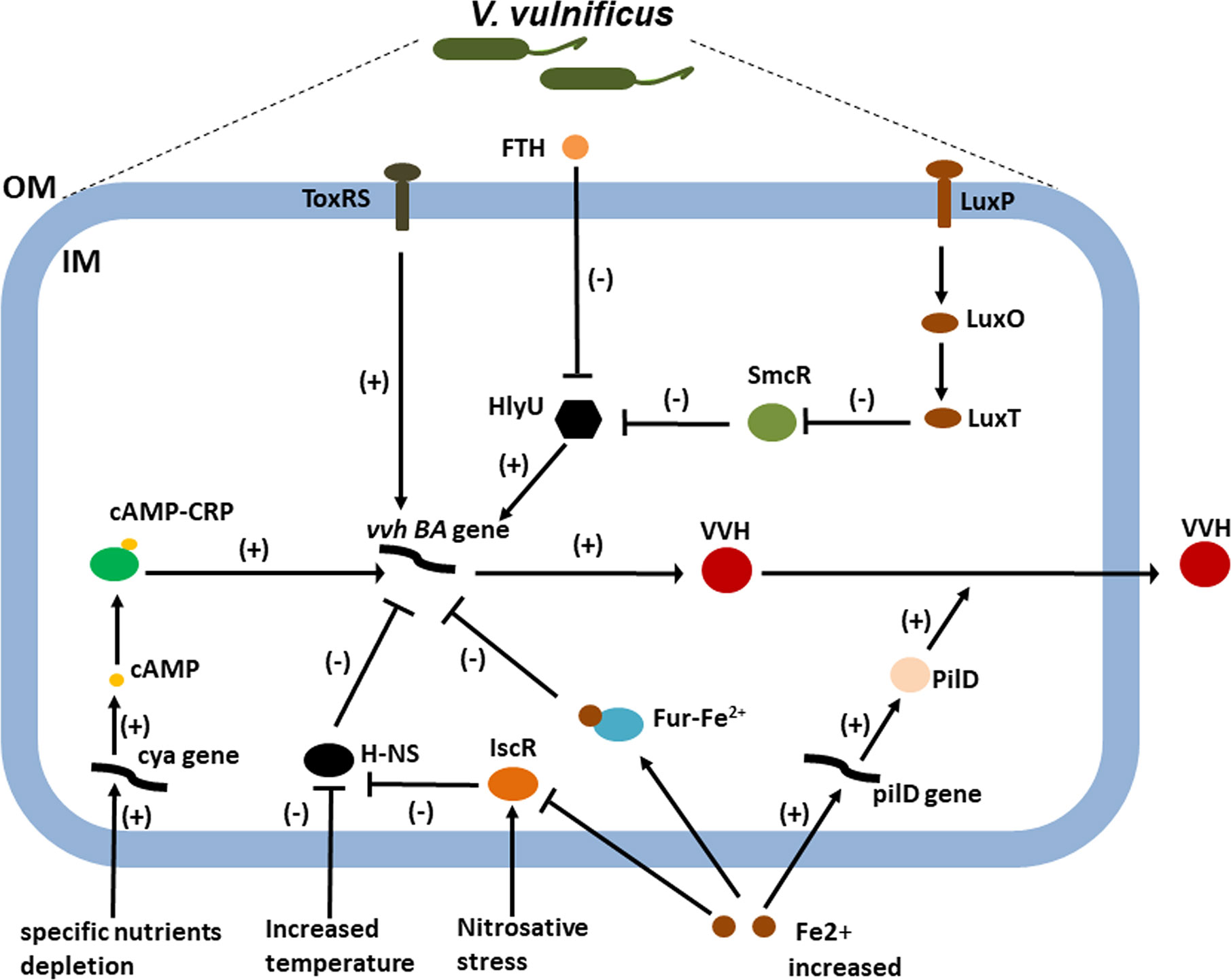
Figure 2 The roles of environmental and host factors and global regulators in the regulation of the VVH expression. CRP activates vvhBA transcription in V. vulnificus by sensing the depletion of specific nutrients, possibly as a result of increased cAMP levels under glucose starvation (32). Increased iron can repress vvhA transcription via the ferric uptake regulator (Fur) and IscR (41, 46). However, it increases extracellular VVH secretion through increased transcription of pilD, which encodes PilD, a component responsible for extracellular VVH secretion (41). IscR activates vvhBA by relieving H-NS repression by sensing nitrosative stress (46). Meanwhile, a repressive interaction of H-NS would be relieved in response to the increase in temperature (39, 49). LuxO is a central response regulator of the QS circuit in V. vulnificus, which negatively regulates vvhA expression via SmcR and HlyU (42, 43). However, the transmembrane transcriptional activator ToxRS positively regulates the expression of the vvhA (47). Taken together, the transcriptional regulators integrate diverse environmental and host signals to collaboratively regulate vvhA transcription during the course of infection. Lastly, FTH, an inhibitor target HlyU, was identified to inhibit the transcription of vvhA along with that of other HlyU-regulated virulence genes.; OM, outer membrane; IM, inner membrane; FTH, fursultiamine hydrochloride; H-NS, histone-like nucleoid structuring protein; cya, gene encoding adenylate cyclase; cAMP, cyclic AMP; CRP, cAMP receptor protein.
In many pathogenic bacteria, including V. vulnificus, quorum sensing (QS) is one of the most important cellular regulatory cascades. QS is responsible for cell–cell communication and is mediated by a small diffusible molecule called autoinducer 2 (AI-2). LuxO is a central response regulator of the QS circuit in V. vulnificus, with disruption of luxO shown to increase the expression of smcR, crp, and luxS, which encodes the autoinducer 2 synthetase (54). In comparison, SmcR regulates cytotoxicity in V. vulnificus via QS signaling by repressing HlyU, which positively regulates vvhA expression (55). Temperature is one of the important host parameters regulating the expression of virulence factors in bacteria. The histone-like nucleoid structuring protein (H-NS) global regulator is known to play a crucial role in the expression of temperature-dependent virulence factors. A study on the role of H-NS in temperature-dependent regulation indicated that hns expression levels were higher at 26 °C than at 37 °C and that vvhA expression and the resulting VVH production were increased following disruption of hns (56). Moreover, H-NS, in its role as a vvhA repressor, competes with HlyU for binding to the vvhA promoter region (57); however, the exact mechanisms of HlyU and H-NS regulation have yet to be fully characterized (56). In addition to cAMP-CRP, Fur, and H-NS, the Fe-S cluster, containing transcriptional regulator IscR, was recently described as an important regulator of V. vulnificus virulence in host environments. IscR activates the vvhBA operon in response to nitrosative stress and iron starvation, thereby aiding successful host infection (58). Lastly, transmembrane transcriptional activator ToxRS, a homolog of the V. cholerae ToxRS transmembrane virulence regulator, may also positively regulate the expression of the vvhA (59). In summary, recognition of the subtle regulation of vvhA gene expression and hemolysin delivery by V. vulnificus has furthered our understanding of how the VVH contributes to disease pathogenesis.
The complicated vvhA regulatory system that emerges from this data suggests that inhibition of global regulators may be a promising approach for the development of alternatives to antibiotic treatment. Recently, an inhibitor-screening reporter platform was used to target HlyU, a master virulence factor transcriptional regulator in V. vulnificus. The study identified a small molecule called fursultiamine hydrochloride that inhibited the transcription of vvhA along with that of other HlyU-regulated virulence genes. Fursultiamine hydrochloride therefore has the potential to inhibit the pathogenesis of V. vulnificus without inducing antimicrobial resistance (60).
The Role of the VVH in Disease and Pathogenesis
V. vulnificus most commonly causes severe gastroenteritis following the consumption of contaminated raw seafood, with sepsis infection mortality rates of 50% (12). Moreover, because V. vulnificus is responsible for >95% of seafood-associated infection deaths in the United States (4), a significant number of studies have focused on the effects of the VVH on human intestinal epithelial cells mentioned above. In addition, small intestine-associated host factors together with mouse models have been used to investigate the role of the VVH in pathogenesis. The human intestine usually secretes cationic antimicrobial peptides to prevent pathogen colonization, with Paneth cells in the small intestine secreting antimicrobial molecule alpha-defensin 5 (HD-5). However, while HD-5 inactivated the Vibrio mimicus hemolysin, it had no effect on VVH. The inability of V. mimicus to penetrate the small intestinal epithelium suggests that the cytolytic activity of the V. mimicus hemolysin is abolished by HD-5 (61). In contrast, V. vulnificus causes intestinal tissue damage and inflammation, which then promotes dissemination of the pathogen from the small intestine into the bloodstream and other organs in infected mice (6, 7). Notably, the small intestine is recognized as the site of the most severe tissue necrosis in humans based on autopsy results from V. vulnificus-infected patients (62). Indeed, VVH and MARTX are the two V. vulnificus virulence factors associated with both enhanced growth in vivo and necrosis of tissue in the small intestine, followed by dissemination into the bloodstream and other tissues. In the absence of these two secreted factors, V. vulnificus is unable to cause intestinal infection in mice (16).
V. vulnificus also causes primary septicemia in patients with underlying liver disease or who are immunocompromised (63). Patients with septicemia tend to die of hypovolemic shock complicated by multi-organ failure. A study in rats found that the VVH dilates the thoracic aorta by activating guanylate cyclase, causing hypotension in vivo and vasodilatation in vitro (64, 65). V. vulnificus can be spreading from the intestine to bloodstream. To survive and proliferate in blood, V. vulnificus requires to overcome the innate immune defenses, including complement-mediated phagocytosis. Recently, capsular polysaccharide and Flp (fimbrial low-molecular-weight protein) pili are reported to play critical roles in evasion of the host innate immune system by resistance to complement-mediated killing (66, 67). Although an earlier work showed that virulent isolates produced high titers of hemolysin, were resistant to inactivation by serum complement (68), further information is needed to uncover the mechanism of VVH-mediated evasion of complement killing, which may help us to better understand the basis of the V. vulnificus infection process in human blood. Being at the crossroads between the immune system, clotting cascade, and endothelial cells, platelets seem to be an appealing central mediator and possible therapeutic target for sepsis (69–71). The mechanism of bacterial-induced platelet activation by pore-forming toxins has been well characterized in other Gram-positive bacteria (72). However, despite the significant fatality rate associated with V. vulnificus-induced sepsis, the interaction between the VVH and platelets is not clear. Because the CDC of Vibrio spp. share structural similarity (28), it is possible that VVH represents a critical molecule of Vibrio spp. involved in pathogenesis by interacting with platelets. Linked to this, efforts should be focused on the mechanisms of VVH-induced platelet activation for future work.
Conclusions and Future Perspective
Cholesterol-dependent cytolysins are a diverse group of proteins that differ between bacterial species. However, it is these differences that have informed much of our understanding of the biological activities of the proteins, as well as their role in pathogenesis. Despite this insight, further studies are needed to determine the structure–function relationships of the VVH. Functionally, the major roles of the VVH are to induce cytotoxicity by binding to the cellular membrane to form pores and activating the host inflammatory response. These functions, along with the subtle regulation of VVH gene expression and other potentially unrecognized activities, contribute to the pathogenesis of V. vulnificus disease. Although the host response to the VVH involves lipid raft-dependent signaling pathway-mediated cell death, it is likely that other mechanisms may also be involved in the host response to the VVH.
V. vulnificus infection can result in severe disease. In fact, most cases occur in patients with underlying conditions resulting in hereditary hemochromatosis, primarily alcohol-associated liver cirrhosis or immuno-compromised males, but it does not cause severe illness in healthy individuals (73). Although there have been many studies on the effects of the VVH on eukaryotic cells in vitro, few animal models that mimick human infection were used to elucidate the role of VVH in pathogenesis. As a result, we still have much to learn about how this toxin contributes to disease pathogenesis in vivo (Figure 1). An interesting study found that hepcidin has a critical role in host defense against V. vulnificus by inducing reactive hypoferremia during early phases of infection (74). Hepcidin is a 25 amino acid peptide secreted by hepatocytes. Hereditary hemochromatosis is caused by deficiency of the iron-regulatory hormone hepcidin (75). Therefore, a hepcidin-deficient mouse model of severe hemochromatosis (37) could be considered for the future work about the role of VVH in the lethal infections by V. vulnificus, a siderophilic bacterium. Additionally, the National Institute on Alcohol Abuse and Alcoholism (NIAAA) model is a mouse model of chronic and binge ethanol feeding, which mimics acute-on-chronic alcoholic liver injury in patients (36). This simple model will be very useful for the study of the function of VVH in vivo, and the underlying mechanisms that contribute to acute infections by V. vulnificus in liver disease patient.
Author Contributions
YY contributed to the research of the literature and the writing and revision of the manuscript. ZF and JW contributed to the revision of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Baker-Austin C, Oliver JD. Vibrio vulnificus: new insights into a deadly opportunistic pathogen. Environ Microbiol (2018) 20(2):423–30. doi: 10.1111/1462-2920.13955
2. Heng SP, Letchumanan V, Deng CY, Ab Mutalib NS, Khan TM, Chuah LH, et al. Vibrio vulnificus: An Environmental and Clinical Burden. Front Microbiol (2017) 8:997. doi: 10.3389/fmicb.2017.00997
3. Rippey SR. Infectious diseases associated with molluscan shellfish consumption. Clin Microbiol Rev (1994) 7(4):419–25. doi: 10.1128/cmr.7.4.419
4. Jones MK, Oliver JD. Vibrio vulnificus: disease and pathogenesis. Infect Immun (2009) 77(5):1723–33. doi: 10.1128/IAI.01046-08
5. Yoshida S, Ogawa M, Mizuguchi Y. Relation of capsular materials and colony opacity to virulence of Vibrio vulnificus. Infect Immun (1985) 47(2):446–51. doi: 10.1128/IAI.47.2.446-451.1985
6. Gavin HE, Beubier NT, Satchell KJ. The Effector Domain Region of the Vibrio vulnificus MARTX Toxin Confers Biphasic Epithelial Barrier Disruption and Is Essential for Systemic Spread from the Intestine. PLoS Pathog (2017) 13(1):e1006119. doi: 10.1371/journal.ppat.1006119
7. Lo HR, Lin JH, Chen YH, Chen CL, Shao CP, Lai YC, et al. RTX toxin enhances the survival of Vibrio vulnificus during infection by protecting the organism from phagocytosis. J Infect Dis (2011) 203(12):1866–74. doi: 10.1093/infdis/jir070
8. Murciano C, Lee CT, Fernandez-Bravo A, Hsieh TH, Fouz B, Hor LI, et al. MARTX Toxin in the Zoonotic Serovar of Vibrio vulnificus Triggers an Early Cytokine Storm in Mice. Front Cell Infect Microbiol (2017) 7:332. doi: 10.3389/fcimb.2017.00332
9. Wright AC, Morris JG Jr. The extracellular cytolysin of Vibrio vulnificus: inactivation and relationship to virulence in mice. Infect Immun (1991) 59(1):192–7. doi: 10.1128/IAI.59.1.192-197.1991
10. Elgaml A, Miyoshi SI. Regulation systems of protease and hemolysin production in Vibrio vulnificus. Microbiol Immunol (2017) 61(1):1–11. doi: 10.1111/1348-0421.12465
11. Lee SE, Ryu PY, Kim SY, Kim YR, Koh JT, Kim OJ, et al. Production of Vibrio vulnificus hemolysin in vivo and its pathogenic significance. Biochem Biophys Res Commun (2004) 324(1):86–91. doi: 10.1016/j.bbrc.2004.09.020
12. Horseman MA, Surani S. A comprehensive review of Vibrio vulnificus: an important cause of severe sepsis and skin and soft-tissue infection. Int J Infect Dis (2011) 15(3):e157–66. doi: 10.1016/j.ijid.2010.11.003
13. Hernandez-Cabanyero C, Lee CT, Tolosa-Enguis V, Sanjuan E, Pajuelo D, Reyes-Lopez F, et al. Adaptation to host in Vibrio vulnificus, a zoonotic pathogen that causes septicemia in fish and humans. Environ Microbiol (2019) 21(8):3118–39. doi: 10.1111/1462-2920.14714
14. Kim CM, Chung YY, Shin SH. Iron differentially regulates gene expression and extracellular secretion of Vibrio vulnificus cytolysin-hemolysin. J Infect Dis (2009) 200(4):582–9. doi: 10.1086/600869
15. Fan JJ, Shao CP, Ho YC, Yu CK, Hor LI. Isolation and characterization of a Vibrio vulnificus mutant deficient in both extracellular metalloprotease and cytolysin. Infect Immun (2001) 69(9):5943–8. doi: 10.1128/iai.69.9.5943-5948.2001
16. Jeong HG, Satchell KJ. Additive function of Vibrio vulnificus MARTX(Vv) and VvhA cytolysins promotes rapid growth and epithelial tissue necrosis during intestinal infection. PLoS Pathog (2012) 8(3):e1002581. doi: 10.1371/journal.ppat.1002581
17. Panicker G, Myers ML, Bej AK. Rapid detection of Vibrio vulnificus in shellfish and Gulf of Mexico water by real-time PCR. Appl Environ Microbiol (2004) 70(1):498–507. doi: 10.1128/aem.70.1.498-507.2004
18. Panicker G, Bej AK. Real-time PCR detection of Vibrio vulnificus in oysters: comparison of oligonucleotide primers and probes targeting vvhA. Appl Environ Microbiol (2005) 71(10):5702–9. doi: 10.1128/AEM.71.10.5702-5709.2005
19. Waldor MK, Mekalanos JJ. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science (1996) 272(5270):1910–4. doi: 10.1126/science.272.5270.1910
20. Bej AK, Patterson DP, Brasher CW, Vickery MC, Jones DD, Kaysner CA. Detection of total and hemolysin-producing Vibrio parahaemolyticus in shellfish using multiplex PCR amplification of tl, tdh and trh. J Microbiol Methods (1999) 36(3):215–25. doi: 10.1016/s0167-7012(99)00037-8
21. Heuck AP, Moe PC, Johnson BB. The cholesterol-dependent cytolysin family of gram-positive bacterial toxins. Subcell Biochem (2010) 51:551–77. doi: 10.1007/978-90-481-8622-8_20
22. Kathuria R, Chattopadhyay K. Vibrio cholerae cytolysin: Multiple facets of the membrane interaction mechanism of a beta-barrel pore-forming toxin. IUBMB Life (2018) 70(4):260–6. doi: 10.1002/iub.1725
23. Ramarao N, Sanchis V. The pore-forming haemolysins of Bacillus cereus: a review. Toxins (Basel) (2013) 5(6):1119–39. doi: 10.3390/toxins5061119
24. Miyoshi S, Abe Y, Senoh M, Mizuno T, Maehara Y, Nakao H. Inactivation of Vibrio vulnificus hemolysin through mutation of the N- or C-terminus of the lectin-like domain. Toxicon (2011) 57(6):904–8. doi: 10.1016/j.toxicon.2011.03.013
25. Senoh M, Okita Y, Shinoda S, Miyoshi S. The crucial amino acid residue related to inactivation of Vibrio vulnificus hemolysin. Microb Pathog (2008) 44(1):78–83. doi: 10.1016/j.micpath.2007.07.002
26. Kashimoto T, Akita T, Kado T, Yamazaki K, Ueno S. Both polarity and aromatic ring in the side chain of tryptophan 246 are involved in binding activity of Vibrio vulnificus hemolysin to target cells. Microb Pathog (2017) 109:71–7. doi: 10.1016/j.micpath.2017.05.029
27. Kashimoto T, Ueno S, Koga T, Fukudome S, Ehara H, Komai M, et al. The aromatic ring of phenylalanine 334 is essential for oligomerization of Vibrio vulnificus hemolysin. J Bacteriol (2010) 192(2):568–74. doi: 10.1128/JB.01049-09
28. Olson R, Gouaux E. Crystal structure of the Vibrio cholerae cytolysin (VCC) pro-toxin and its assembly into a heptameric transmembrane pore. J Mol Biol (2005) 350(5):997–1016. doi: 10.1016/j.jmb.2005.05.045
29. Shinoda S, Miyoshi S, Yamanaka H, Miyoshi-Nakahara N. Some properties of Vibrio vulnificus hemolysin. Microbiol Immunol (1985) 29(7):583–90. doi: 10.1111/j.1348-0421.1985.tb00862.x
30. Yamanaka H, Shimatani S, Tanaka M, Katsu T, Ono B, Shinoda S. Susceptibility of erythrocytes from several animal species to Vibrio vulnificus hemolysin. FEMS Microbiol Lett (1989) 52(3):251–5. doi: 10.1016/0378-1097(89)90206-1
31. Lee SJ, Jung YH, Oh SY, Song EJ, Choi SH, Han HJ. Vibrio vulnificus VvhA induces NF-kappaB-dependent mitochondrial cell death via lipid raft-mediated ROS production in intestinal epithelial cells. Cell Death Dis (2015) 6:1655. doi: 10.1038/cddis.2015.19
32. Song EJ, Lee SJ, Lim HS, Kim JS, Jang KK, Choi SH, et al. Vibrio vulnificus VvhA induces autophagy-related cell death through the lipid raft-dependent c-Src/NOX signaling pathway. Sci Rep (2016) 6:27080. doi: 10.1038/srep27080
33. Zhao JF, Sun AH, Ruan P, Zhao XH, Lu MQ, Yan J. Vibrio vulnificus cytolysin induces apoptosis in HUVEC, SGC-7901 and SMMC-7721 cells via caspase-9/3-dependent pathway. Microb Pathog (2009) 46(4):194–200. doi: 10.1016/j.micpath.2008.12.005
34. Toma C, Higa N, Koizumi Y, Nakasone N, Ogura Y, McCoy AJ, et al. Pathogenic Vibrio activate NLRP3 inflammasome via cytotoxins and TLR/nucleotide-binding oligomerization domain-mediated NF-kappa B signaling. J Immunol (2010) 184(9):5287–97. doi: 10.4049/jimmunol.0903536
35. Lohith GK, Kingston JJ, Singh AK, Murali HS, Batra HV. Evaluation of recombinant leukocidin domain of VvhA exotoxin of Vibrio vulnificus as an effective toxoid in mouse model. Immunol Lett (2015) 167(1):47–53. doi: 10.1016/j.imlet.2015.06.015
36. Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc (2013) 8(3):627–37. doi: 10.1038/nprot.2013.032
37. Lesbordes-Brion JC, Viatte L, Bennoun M, Lou DQ, Ramey G, Houbron C, et al. Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood (2006) 108(4):1402–5. doi: 10.1182/blood-2006-02-003376
38. Kaus K, Lary JW, Cole JL, Olson R. Glycan specificity of the Vibrio vulnificus hemolysin lectin outlines evolutionary history of membrane targeting by a toxin family. J Mol Biol (2014) 426(15):2800–12. doi: 10.1016/j.jmb.2014.05.021
39. Kashimoto T, Sugiyama H, Kawamidori K, Yamazaki K, Kado T, Matsuda K, et al. Vibiro vulnificus hemolysin associates with gangliosides. BMC Microbiol (2020) 20(1):69. doi: 10.1186/s12866-020-01755-1
40. Sugiyama H, Kashimoto T, Ueno S, Ehara H, Kodama T, Iida T, et al. Relationship between localization on cellular membranes and cytotoxicity of Vibrio vulnificus hemolysin. PLoS One (2011) 6(10):e26018. doi: 10.1371/journal.pone.0026018
41. Sugiyama H, Kashimoto T, Ueno S, Susa N. Inhibition of binding of Vibrio vulnificus hemolysin (VVH) by MbetaCD. J Vet Med Sci (2013) 75(5):649–52. doi: 10.1292/jvms.12-0387
42. Choi MH, Sun HY, Park RY, Bai YH, Chung YY, Kim CM, et al. Human serum albumin enhances the hemolytic activity of Vibrio vulnificus. Biol Pharm Bull (2006) 29(1):180–2. doi: 10.1248/bpb.29.180
43. Park KH, Yang HB, Kim HG, Lee YR, Hur H, Kim JS, et al. Low density lipoprotein inactivates Vibrio vulnificus cytolysin through the oligomerization of toxin monomer. Med Microbiol Immunol (2005) 194(3):137–41. doi: 10.1007/s00430-004-0227-0
44. Korchev YE, Bashford CL, Pasternak CA. Differential sensitivity of pneumolysin-induced channels to gating by divalent cations. J Membr Biol (1992) 127(3):195–203. doi: 10.1007/BF00231507
45. Park JW, Jahng TA, Rho HW, Park BH, Kim NH, Kim HR. Inhibitory mechanism of Ca2+ on the hemolysis caused by Vibrio vulnificus cytolysin. Biochim Biophys Acta (1994) 1194(1):166–70. doi: 10.1016/0005-2736(94)90216-x
46. Lee YR, Park KH, Lin ZZ, Kho YJ, Park JW, Rho HW, et al. A calcium-calmodulin antagonist blocks experimental Vibrio vulnificus cytolysin-induced lethality in an experimental mouse model. Infect Immun (2004) 72(10):6157–9. doi: 10.1128/IAI.72.10.6157-6159.2004
47. Lee SJ, Lee HJ, Jung YH, Kim JS, Choi SH, Han HJ. Melatonin inhibits apoptotic cell death induced by Vibrio vulnificus VvhA via melatonin receptor 2 coupling with NCF-1. Cell Death Dis (2018) 9(2):48. doi: 10.1038/s41419-017-0083-7
48. Qin K, Fu K, Liu J, Wu C, Wang Y, Zhou L. Vibrio vulnificus cytolysin induces inflammatory responses in RAW264.7 macrophages through calcium signaling and causes inflammation in vivo. Microb Pathog (2019) 137:103789. doi: 10.1016/j.micpath.2019.103789
49. Bang YB, Lee SE, Rhee JH, Choi SH. Evidence that expression of the Vibrio vulnificus hemolysin gene is dependent on cyclic AMP and cyclic AMP receptor protein. J Bacteriol (1999) 181(24):7639–42. doi: 10.1128/JB.181.24.7639-7642.1999
50. Green J, Stapleton MR, Smith LJ, Artymiuk PJ, Kahramanoglou C, Hunt DM, et al. Cyclic-AMP and bacterial cyclic-AMP receptor proteins revisited: adaptation for different ecological niches. Curr Opin Microbiol (2014) 18:1–7. doi: 10.1016/j.mib.2014.01.003
51. Kim YR, Kim SY, Kim CM, Lee SE, Rhee JH. Essential role of an adenylate cyclase in regulating Vibrio vulnificus virulence. FEMS Microbiol Lett (2005) 243(2):497–503. doi: 10.1016/j.femsle.2005.01.016
52. Choi HK, Park NY, Kim DI, Chung HJ, Ryu S, Choi SH. Promoter analysis and regulatory characteristics of vvhBA encoding cytolytic hemolysin of Vibrio vulnificus. J Biol Chem (2002) 277(49):47292–9. doi: 10.1074/jbc.M206893200
53. Notley-McRobb L, Death A, Ferenci T. The relationship between external glucose concentration and cAMP levels inside Escherichia coli: implications for models of phosphotransferase-mediated regulation of adenylate cyclase. Microbiology (1997) 143( Pt 6):1909–18. doi: 10.1099/00221287-143-6-1909
54. Elgaml A, Higaki K, Miyoshi S. Effects of temperature, growth phase and luxO-disruption on regulation systems of toxin production in Vibrio vulnificus strain L-180, a human clinical isolate. World J Microbiol Biotechnol (2014) 30(2):681–91. doi: 10.1007/s11274-013-1501-3
55. Shao CP, Lo HR, Lin JH, Hor LI. Regulation of cytotoxicity by quorum-sensing signaling in Vibrio vulnificus is mediated by SmcR, a repressor of hlyU. J Bacteriol (2011) 193(10):2557–65. doi: 10.1128/JB.01259-10
56. Elgaml A, Miyoshi S. Role of the Histone-Like Nucleoid Structuring Protein (H-NS) in the Regulation of Virulence Factor Expression and Stress Response in Vibrio vulnificus. Biocontrol Sci (2015) 20(4):263–74. doi: 10.4265/bio.20.263
57. Liu M, Naka H, Crosa JH. HlyU acts as an H-NS antirepressor in the regulation of the RTX toxin gene essential for the virulence of the human pathogen Vibrio vulnificus CMCP6. Mol Microbiol (2009) 72(2):491–505. doi: 10.1111/j.1365-2958.2009.06664.x
58. Choi G, Jang KK, Lim JG, Lee ZW, Im H, Choi SH. The transcriptional regulator IscR integrates host-derived nitrosative stress and iron starvation in activation of the vvhBA operon in Vibrio vulnificus. J Biol Chem (2020) 295(16):5350–61. doi: 10.1074/jbc.RA120.012724
59. Lee SE, Shin SH, Kim SY, Kim YR, Shin DH, Chung SS, et al. Vibrio vulnificus has the transmembrane transcription activator ToxRS stimulating the expression of the hemolysin gene vvhA. J Bacteriol (2000) 182(12):3405–15. doi: 10.1128/jb.182.12.3405-3415.2000
60. Imdad S, Chaurasia AK, Kim KK. Identification and Validation of an Antivirulence Agent Targeting HlyU-Regulated Virulence in Vibrio vulnificus. Front Cell Infect Microbiol (2018) 8:152 doi: 10.3389/fcimb.2018.00152
61. Miyoshi S, Ikehara H, Kumagai M, Mizuno T, Kawase T, Maehara Y. Defensive effects of human intestinal antimicrobial peptides against infectious diseases caused by Vibrio mimicus and V. vulnificus. Biocontrol Sci (2014) 19(4):199–203. doi: 10.4265/bio.19.199
62. Chen Y, Satoh T, Tokunaga O. Vibrio vulnificus infection in patients with liver disease: report of five autopsy cases. Virchows Arch (2002) 441(1):88–92. doi: 10.1007/s00428-002-0613-1
63. Menon MP, Yu PA, Iwamoto M, Painter J. Pre-existing medical conditions associated with Vibrio vulnificus septicaemia. Epidemiol Infect (2014) 142(4):878–81. doi: 10.1017/S0950268813001593
64. Kook H, Lee SE, Baik YH, Chung SS, Rhee JH. Vibrio vulnificus hemolysin dilates rat thoracic aorta by activating guanylate cyclase. Life Sci (1996) 59(3):PL41–7. doi: 10.1016/0024-3205(96)00292-5
65. Kook H, Rhee JH, Lee SE, Kang SY, Chung SS, Cho KW, et al. Activation of particulate guanylyl cyclase by Vibrio vulnificus hemolysin. Eur J Pharmacol (1999) 365(2-3):267–72. doi: 10.1016/s0014-2999(98)00870-x
66. Pettis GS, Mukerji AS. Structure, Function, and Regulation of the Essential Virulence Factor Capsular Polysaccharide of Vibrio vulnificus. Int J Mol Sci (2020) 21(9):3259. doi: 10.3390/ijms21093259
67. Duong-Nu TM, Jeong K, Hong SH, Puth S, Kim SY, Tan W, et al. A stealth adhesion factor contributes to Vibrio vulnificus pathogenicity: Flp pili play roles in host invasion, survival in the blood stream and resistance to complement activation. PLoS Pathog (2019) 15(8):e1007767. doi: 10.1371/journal.ppat.1007767
68. Stelma GN Jr., Reyes AL, Peeler JT, Johnson CH, Spaulding PL. Virulence characteristics of clinical and environmental isolates of Vibrio vulnificus. Appl Environ Microbiol (1992) 58(9):2776–82. doi: 10.1128/AEM.58.9.2776-2782.1992
69. Greco E, Lupia E, Bosco O, Vizio B, Montrucchio G. Platelets and Multi-Organ Failure in Sepsis. Int J Mol Sci (2017) 18(10):2200. doi: 10.3390/ijms18102200
70. Wang Y, Ouyang Y, Liu B, Ma X, Ding R. Platelet activation and antiplatelet therapy in sepsis: A narrative review. Thromb Res (2018) 166:28–36. doi: 10.1016/j.thromres.2018.04.007
71. Kerris EWJ, Hoptay C, Calderon T, Freishtat RJ. Platelets and platelet extracellular vesicles in hemostasis and sepsis. J Invest Med (2020) 68(4):813–20. doi: 10.1136/jim-2019-001195
72. Cox D, Kerrigan SW, Watson SP. Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation. J Thromb Haemost (2011) 9(6):1097–107. doi: 10.1111/j.1538-7836.2011.04264.x
73. Bross MH, Soch K, Morales R, Mitchell RB. Vibrio vulnificus infection: diagnosis and treatment. Am Fam Physician (2007) 76(4):539–44.
74. Arezes J, Jung G, Gabayan V, Valore E, Ruchala P, Gulig PA, et al. Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host Microbe (2015) 17(1):47–57. doi: 10.1016/j.chom.2014.12.001
Keywords: Vibrio vulnificus hemolysin (VVH), cholesterol-dependent cytolysin (CDC), biological activity, gene regulation, sepsis, pathogenesis
Citation: Yuan Y, Feng Z and Wang J (2020) Vibrio vulnificus Hemolysin: Biological Activity, Regulation of vvhA Expression, and Role in Pathogenesis. Front. Immunol. 11:599439. doi: 10.3389/fimmu.2020.599439
Received: 27 August 2020; Accepted: 30 September 2020;
Published: 23 October 2020.
Edited by:
George P. Munson, University of Miami, United StatesReviewed by:
Jessica Jones, United States Food and Drug Administration, United StatesShin-ichi Miyoshi, Okayama University, Japan
Takashige Kashimoto, Kitasato University Japan
Carmen Amaro, University of Valencia, Spain
Copyright © 2020 Yuan, Feng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinglin Wang, d2FuZ2psaW5AYm1pLmFjLmNu; Yuan Yuan, bWluaW1pbml5dWFuQDE2My5jb20=