 Wan-Jung H. Wu
Wan-Jung H. Wu Daniel F. Zegarra-Ruiz
Daniel F. Zegarra-Ruiz Gretchen E. Diehl
Gretchen E. Diehl- 1Immunology Graduate Program, Baylor College of Medicine, Houston, TX, United States
- 2Immunology Program of the Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, New York, NY, United States
Autoimmune diseases and chronic inflammatory disorders are characterized by dysregulated immune responses resulting in excessive and uncontrolled tissue inflammation. Multiple factors including genetic variation, environmental stimuli, and infection are all thought to contribute to continued inflammation and pathology. Current evidence supports the microbiota as one such factor with emerging data linking commensal organisms to the onset and progression of disease. In this review, we will discuss links between the microbiota and specific diseases as well as highlight common pathways that link intestinal microbes with multiple autoimmune and inflammatory diseases.
Introduction
Increases in autoimmune and inflammatory diseases are a major health problem currently affecting over 200 million people worldwide and represent a leading cause of death for women under 65 (1, 2). Better understanding of factors that affect disease progression and initiation will lead to new ways to address these important health issues.
In the human body, the microbiota dynamically interacts with the host at all barrier sites with the largest load of microbes residing within the intestine (3). Commensals coevolved with humans and provide multiple benefits including facilitating nutrition and xenobiotic metabolism, enhancing barrier function, inhibiting pathogens, and modulating immunity (3). Alteration in the microbiota composition is linked to dysregulated immunity and is associated with inflammatory and autoimmune diseases (4–9).
While individual studies find a number of disease-associated changes, how these changes relate to disease initiation or amplification are still being elucidated. Importantly, understanding host regulation by intestinal microbes or of microbial physiology have led to greater understanding of a number of diseases. For example, microbial factors such as metabolites can play an important role in modulating intestinal and systemic inflammation and a subset of metabolites are linked to multiple diseases (10). Short-chain fatty acids (SCFAs), which are converted from dietary fiber and as the main energy source for colonocytes, directly support intestinal epithelial health (11). SCFAs also promote differentiation of regulatory T cells (Tregs) supporting an anti-inflammatory environment within the gut and at distal sites (12–15). While many types of microbes can generate SCFA, the main producers are Firmicutes and Bacteroidetes and increased proportion of these organisms is associated with human health (16). Dietary factors such as fiber can also shape the microbial community by modifying the metabolic landscape resulting in microbial compositional changes that can modulate diseases (17, 18). Common associations with metabolites highlight how common metabolic pathways utilized by distinct microbes could modulate disease. They also give clues to common pathways that could be manipulated to treat these diseases.
Additionally, in many of these diseases, increased microbes or microbial products can be found in the blood indicating that changes to the intestinal barrier may be a common feature (19–21). However, whether these changes are causative or a consequence of disease development remains to be seen (22).
On the host side, several pathways associated with microbiota-regulated immune responses are linked to autoimmune and inflammatory diseases (23–25). Mutations in HLA-DR, toll like receptors (TLRs), inflammasome, and autophagy components are associated with multiple diseases where they lead to dysregulated immune responses and increased inflammation (26–29).
In this review, we will discuss association of the microbiota with pathways involved in the pathogenesis of inflammatory bowel disease (IBD), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), multiple sclerosis (MS), and type I diabetes (T1D) to highlight commonalities between diseases as well as point out disease specific associations.
Inflammatory Bowel Disease
IBD is characterized by dysregulated immune responses against the microbiota leading to chronic inflammation in the gastrointestinal (GI) tract. The major forms of IBD are ulcerative colitis (UC), which is limited to the colon, and Crohn’s disease (CD), which can affect tissue throughout the GI tract (30). In IBD patients, there are reductions in potentially anti-inflammatory microbes such as Bacteroidetes, Lachnospiraceae (16), and Faecalibacterium prausnitzii (31, 32) alongside increases in potentially inflammatory microbes such as Proteobacteria and Ruminococcus gnavus (30, 33–39). Further, increased mucosa-associated bacteria (16, 40) results in greater contact between gut microbes and immune system and leads to anti-bacterial immunity associated with IBD pathogenesis (41–45).
In humans, over 240 genetic loci are associated with risk for IBD (23, 46–49). Gene mutations in pathways related to interactions with the microbiota highlight common mechanisms for disease development (23). Mutations are found in genes associated with microbial recognition including nucleotide-binding oligomerization domain-containing protein 2 (NOD2); anti-inflammatory mechanisms including IL-10 and IL-10 receptor (50–53) and barrier repair including IL-22 (54, 55). Many have found these pathways are induced by the microbiota and the microbiota is important for barrier repair in mouse models of disease (56–62). However, microbes also drive pathology and rederivation to germfree is protective in T cell dependent models (59, 63).
In addition, diet and dietary metabolites are critical factors in IBD pathogenesis (64). In IBD patients, specific bacteria, such as butyrate producers Faecalibacterium prausnitzii and Roseburia hominis are decreased (32, 65). The crucial roles of diet and dietary metabolites are shown in multiple mouse models where high fiber diets or direct administration of SCFA are beneficial while loss of the SCFA receptor, Gpr43, is pathogenic (12–15). Tryptophan metabolites can also mitigate colitis severity. These are ligands for the aryl hydrocarbon receptor (AhR), which activates IL-22 and IL-10 production and is negatively associated with colitis (66–69). A tryptophan-free diet exacerbates pathology in colitis models (70), whereas Lactobacillus bulgaricus, an AhR-activating bacterium, ameliorates pathology (66, 71). Secondary bile acids are additional metabolites with both pro- and anti-inflammatory functions that can promote differentiation of Tregs or Th17 cells within the intestine and in peripheral sites (72, 73). Bile acids can also regulate intestinal bacterial growth by enhancing biofilm formation thereby increasing colonization by pathogens such as vancomycin-resistant Enterococcus in mice (74). These studies together highlight the complex interaction between the host, diet, and intestinal microbes that can underlie alterations in disease pathology.
As evidence supports the potential for the microbiota in maintaining intestinal homeostasis and preventing inflammation, there is great interest in utilizing microbes as treatment for IBD patients. The administration of probiotics shows success in animal models (75, 76) and some patients (77, 78). However, broad scale benefits are yet to emerge (78). This may be due to the genetic complexity or other environmental factors associated with IBD. Another alternative is fecal microbiota transplants (FMT), which are utilized successfully to treat C. difficile infection (79). Several trials demonstrate success in some UC patients (80, 81). FMT increases microbiota diversity in responders and non-responders (80), demonstrating that increased diversity alone is not sufficient for benefit. Interestingly, recent work found expanded intestinal bacteriophages in patients who did not respond after FMT with bacteriophages exacerbating colitis in animal models (82). More work needs to be done to understand how FMT can shape the recipient’s microbial community to define if this method can broadly ameliorate diseases.
Together, work in IBD demonstrates myriad ways the microbiota interacts with the host to regulate local inflammation and suggests a number of microbiota-related pathways to target for treating this disease. Understanding affected pathways in IBD have also improved understanding of how microbes impact other inflammatory and autoimmune diseases and will lead to a broader understanding of how to utilize the microbiome to improve patient outcomes.
Systemic Lupus Erythematosus
SLE patients suffer from production of autoantibodies and proinflammatory cytokines that cause disease in multiple organs including skin, blood, and kidneys with many environmental influences, including the gut microbiota (83). SLE patients exhibit intestinal and oral dysbiosis. As with other autoimmune diseases, studies find decreased bacterial diversity correlated with disease activity (6). Oral and gut microbiota from SLE patients are enriched in the family Lactobacillaceae, with Bifidobacteria and Clostridiales decreased in the intestine (84–86).
Further, antibodies and T cells from SLE patients recognize bacterial antigens from the oral, intestinal, and skin microbiota including Propionibacterium propionicum and Bacteroides thetaiotaomicron (6, 84, 85, 87). Molecular mimicry is a possible link between the microbiota and SLE. One of the most common autoantibodies associated with SLE targets the broadly expressed RNA binding protein Ro60 (88). Antibodies against Ro60 are commonly found before SLE symptoms develop (89). Some human commensals produce proteins similar to human Ro60 and, although these bacteria are found in both healthy donors and lupus patients, only lupus patients have antibodies and T cells reactive with human Ro60 and microbial Ro60 orthologs (87). In addition, in SLE patients, disease severity correlated with R. gnavus enrichment. Further, SLE patients with severe disease had IgG antibodies that recognized cell wall lipoglycans from a subset of R. gnavus strains. Importantly, auto-DNA antibodies from SLE patients with lupus nephritis were cross-reactive with R. gnavus lipoglycans (6).
Multiple spontaneous and inducible mouse lupus models have given great insight into how microbiota changes regulate pathology. Intercross of mouse strains NZW with BXSB results in spontaneous lupus-related antiphospholipid syndrome and liver damage, predominately in male mice, due to an extra copy of the TLR7 gene. In these mice, Enterococcus gallinarum translocates to the liver and triggers autoimmune responses. Depletion of this pathobiont with vancomycin suppressed bacterial translocation, autoreactive T cells, and autoantibodies. Monocolonization of germfree mice with E. gallinarum increased gut permeability, plasmacytoid dendritic cells (pDCs) and Th17 cells in the intestine lamina propria and mesenteric lymph nodes, exacerbating disease and mortality (90).
Bacterial metabolites also modulate SLE as seen with Lactobacillus which, as discussed above can modulate intestinal inflammation by producing AhR-activating ligands (66, 71). In mouse models, and in contrast with IBD, a high protein diet with a high tryptophan content is associated with increased pathology by promoting anti-double stranded DNA autoantibody production and increased T follicular helper (Tfh) cells (91). A metabolic screening from feces of lupus prone mice homozygous for the NZM2410 lupus susceptibility quantitative trait loci (Sle1, Sle2, and Sle3) showed increased intestinal tryptophan-derived bacterial metabolites with enriched fecal Lactobacillus (91).
In contrast, and similar to IBD, a high fiber diet is associated with improved outcomes in mouse lupus models (86). In a TLR7-dependent model, there was outgrowth of Lactobacillus reuteri, which then translocated to the mesenteric lymph node, spleen, and liver. Translocation led to increased pDC production of type I interferon (IFN-I), exacerbating disease pathogenesis and mortality. Treatment with SCFAs or a high fiber diet suppressed L. reuteri outgrowth and translocation, reducing excess IFN-I and ameliorating disease (86). This example shows both direct and indirect effects of gut commensals on disease progression.
Together, these results demonstrate that the gut microbiota can modulate lupus pathogenesis by molecular mimicry, changes in bacterial translocation, metabolites, or microbe-microbe competition. Each can result in a dysregulated immune response in distal tissues including Th17 cell and pDC recruitment and activation of IFN-I pathways that together amplify disease.
Rheumatoid Arthritis
RA is a chronic synovial inflammation characterized by immune infiltration in the joints due to lost tolerance including B and T cell responses against self-proteins with a citrulline residue leading to cartilage degradation and bone erosion (92). In a subset of RA patients, bacterial DNA and peptidoglycan–polysaccharide complexes are found in the synovium (93). RA patients exhibit oral dysbiosis, characterized by enrichment of Porphyromonas gingivalis and Lactobacillus salivarius and intestinal dysbiosis with increased Gram-positive bacteria (94, 95). These changes in the oral and gut microbiota are linked to clinical variations in RA (93). Increased abundance of Lactobacillus correlated with increased total IgG titers, while other oral microbes such as Prevotella spp. correlated with rheumatoid factor (95). Prevotella copri is enriched in fecal samples of patients and individuals at risk for RA. A subset of RA patients has P. copri-specific Th1 and Th17 cells along with IgG and IgA antibodies which correlates with increased proinflammatory cytokine levels and more severe disease (95–97). Interestingly, RA therapies partially restores the microbiota to more closely resemble one found in healthy controls (95).
In mice, TLR2 and TLR4 engagement modulates autoimmune arthritis (98). IL-1 receptor antagonist–knockout (Il1rn-/-) mice spontaneously develop autoimmune arthritis due to uncontrolled IL-1 signaling (99). Disease progression is delayed in germfree Il1rn-/- mice (98). A single injection of a TLR2 agonist or monocolonization with Lactobacillus bifidus was sufficient to restore pathogenesis (98). However, as with other diseases, there are complex interactions between these pathways. Il1rn-/- mice lacking TLR2 exhibited exacerbated disease with increased bone destruction mediated by Th1 cells, suggesting a dual role for TLR2 in disease (98). BALB/c ZAP-70(W163C)-mutant (SKG) mice spontaneously develop chronic arthritis due to a naturally occurring mutation of the ZAP-70 gene, a signal transduction molecule downstream of the T cell receptor (100, 101). Germfree SKG mice do not develop disease (100). Conventionalization with altered Schaedler flora (ASF), a defined community of eight bacteria including Lactobacillus species, was sufficient to induce arthritis, supporting the role of gut microbes in pathogenesis (100). Further supporting microbiota shifts found in RA as amplifying disease, conventionalization of SKG germfree mice with fecal samples from RA patients elicited more severe arthritis with higher levels of IL-17A as compared to fecal samples from healthy controls (97). Similarly, P. copri-monocolonized SKG mice have exacerbated disease with increased Th17 cells (97). Colonization with Segmented filamentous bacteria (SFB), a Th17 cell inducing mouse commensal, exacerbates a K/BxN autoimmune arthritis model (in which KRN T cells recognize glucose-6-phosphate isomerase) by expanding Tfh cells, which promote the production of autoantibodies involved in RA (102). These data show that gut microbes can modulate immune responses involved in RA such as Th1 and Th17 cells recruitment and expansion exacerbating the inflamed tissue environment.
Multiple Sclerosis
MS patients suffer from autoimmune responses against the brain and spinal cord due to T cell targeting of oligodendrocytes resulting in demyelination and axonal loss (103). MS patients exhibit intestinal dysbiosis with increases in the Euryarchaeota and Verrucomicrobia phyla. Specifically, Methanobrevibacter smithii and Akkermansia muciniphila are enriched in the stool of patients and their abundance decreased after treatment (8). In addition, reduced levels are found of bacteria belonging to the Clostridia clusters XIVa and IV and Bacteroidetes, microbes well known to produce SCFA and induce Treg cells (104, 105).
As with other diseases, in the mouse model of MS, experimental autoimmune encephalitis (EAE), pathology is ameliorated in germfree mice with lower levels of IFN-γ and IL-17A and increased Treg cells (106). Interestingly, in the relapsing remitting MS mouse model, in which CD4+ T cells are specific for myelin oligodendrocyte glycoprotein (MOG), transfer of intestinal microbes from MS patients but not from healthy monozygotic twins increased incidence of disease due to decreased T cell IL-10 production (107). A. muciniphila also affects T cell differentiation by inducing Th1 differentiation in PBMCs from both healthy donors and MS patients, potentially contributing to the proinflammatory environment in MS (108).
Similar to IBD and in contrast with SLE, in mouse models tryptophan can protect from pathogenesis. Colonization of mice in the EAE model with Lactobacillus reuteri through its conversion of tryptophan into AhR agonists, activates IFN-I responses in astrocytes and limits disease severity (109). Another Lactobacillius species, L. murinus reduces EAE severity by inhibiting Th17 cell differentiation (110). Intestinal colonization by L. murinus is suppressed by a high salt diet, which also amplifies disease (110). Together these studies demonstrate a gut/brain axis in which gut microbes and metabolites modulate immune responses including innate and adaptive immunity at distal sites to influence disease onset and severity.
Type 1 Diabetes
Immune destruction of pancreatic β-cells by islet-specific autoreactive CD8+ T cells results in lost insulin production and T1D (111). A longitudinal human study analyzing stool samples from the Environmental Determinants of Diabetes in the Young (TEDDY) cohort identified reduced microbial pathways related to fermentation and synthesis of SCFAs as well as decreased microbial diversity as well as reduced Bifidobacteria and Lachnospiraceae and overabundance of Blautia, Rikenellaceae, and Ruminococcus in patients who progressed to T1D (9, 112). In a similar cohort, children who progress to T1D show changes in the Bacteroidetes/Firmicutes ratio and increased Bacteroides ovatus (9, 112–115).
In contrast with other disease models, germfree non-obese diabetic (NOD) mice have increased islet destruction demonstrating that in diabetes, microbes may limit disease severity (116). However, some microbes are likely pathogenic as depletion of Gram-negative gut microbes in neonatal mice results in decreased diabetes incidence with fewer IFN-γ producing T cells (117). Supporting the complicated pro- and anti-inflammatory signals downstream of microbes, in contrast with IBD, loss of MyD88 protects specific pathogen free (SPF) or ASF colonized NOD mice from diabetes however rederivation to germfree, restores disease incidence (116). As in SLE, bacterial translocation can be a factor in T1D pathogenesis. In a model of streptozotocin (STZ)-induced T1D, gut microbial translocation to the pancreatic lymph node led to recognition of bacterial MDP by the intracellular NOD2 receptor resulting in increased number of Th1 and Th17 cells and increased islet destruction (118). Similar to the enrichment seen in children that develop T1D, STZ-treated mice also had increased intestinal Bacteroides (114, 118). Gut microbiota also plays a role in sex differences in autoimmune diseases. In SPF NOD mice, female mice have a higher incidence of disease than male mice with no differences between the sexes in germfree mice (119). Cecal microbiome transplants from male to female mice reduced islet inflammation and autoantibody levels due to microbiome changes along with hormonal and metabolic changes downstream of elevated testosterone (119).
In NOD mice, as with IBD and MS, SCFAs, notably butyrate, decreased the incidence and severity of diabetes with reduced frequency of autoimmune CD8+ T cells and B cells and increased Tregs and IL-10 production (120). Treatment with SCFAs increased the abundance of Bacteroides, which protected against disease when transplanted to germfree NOD mice (120).
Together, in mouse models, gut microbes and gut microbial metabolites can modulate immune responses involved in T1D including pancreas T cell infiltration as well as shaping the balance between pro- and anti-inflammatory T cell responses, thereby influencing disease onset and severity.
Conclusion
In this review, we provided examples of mechanistic ways microbes can alter disease pathology in IBD, SLE, RA, MS, and T1D models with microbes playing a role in pathology of additional autoimmune diseases (121–124). While we focused on bacteria, emerging data suggests potential roles for yeast and enteric viruses in modulating immune responses and autoimmune and inflammatory disease (125–128).
We have highlighted disease specific interactions as well as numerous common links between the microbiota and human disease (Table 1). Common associations relate to microbial behaviors such as translocation or microbial metabolites that are shared between multiple microbes. Understanding these common functions and as the host pathways regulated by the microbiota will enable for identification of targetable pathways to treat multiple autoimmune and inflammatory disease.
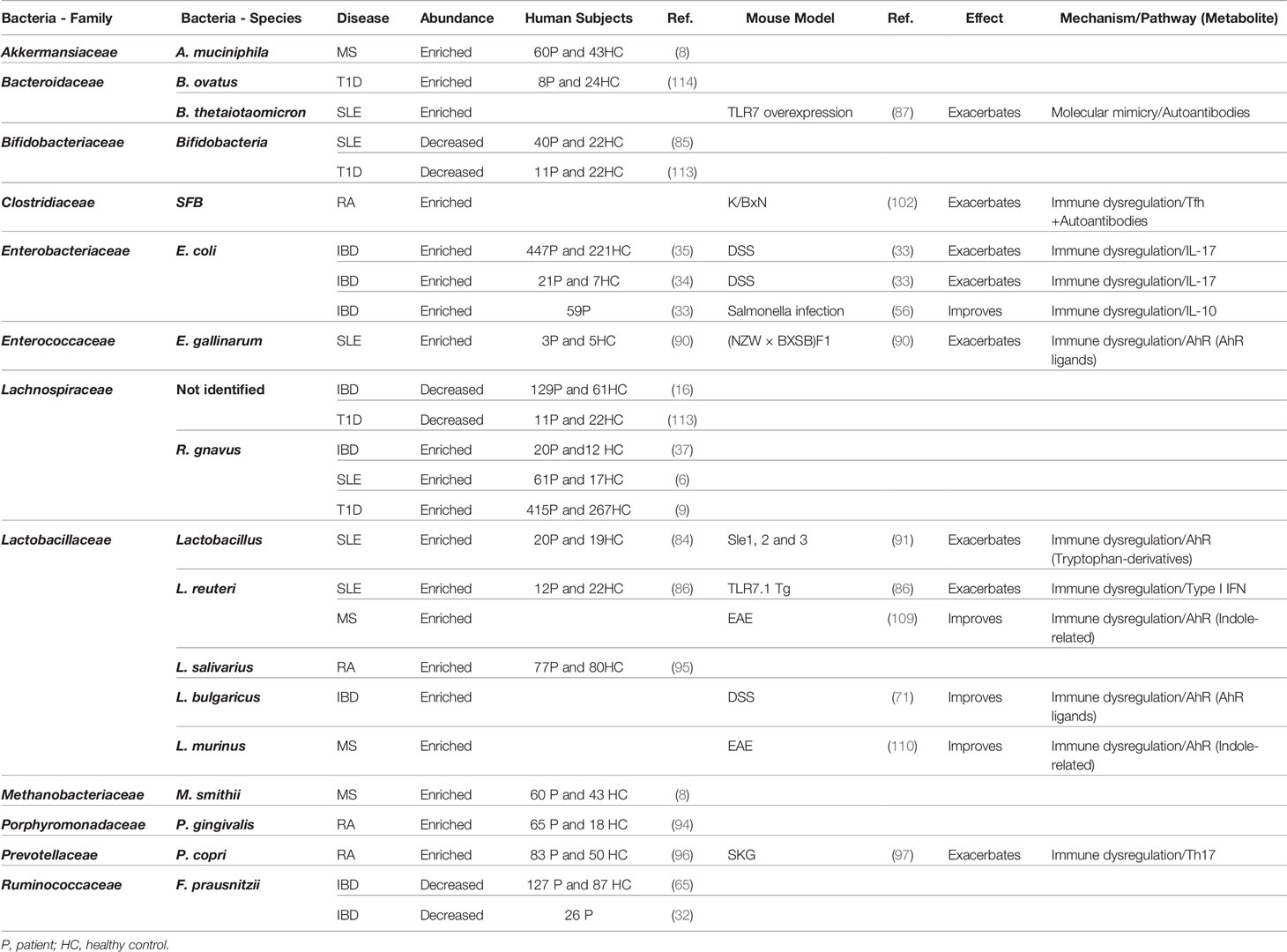
Table 1 Summary table for the relationship between bacteria and autoinflammatory and autoimmune diseases.
Author Contributions
W-JW and DZ-R performed literature searches and with GD wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH AI125264 and Rainin Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Youinou P, Pers J-O, Gershwin ME, Shoenfeld Y. Geo-epidemiology and autoimmunity. J Autoimmun (2010) 34:J163–7. doi: 10.1016/j.jaut.2009.12.005
2. Cooper GS, Stroehla BC. The epidemiology of autoimmune diseases. Autoimmun Rev (2003) 2:119–25. doi: 10.1016/s1568-9972(03)00006-5
3. Sommer F, Bäckhed F. The gut microbiota–masters of host development and physiology. Nat Rev Microbiol (2013) 11:227–38. doi: 10.1038/nrmicro2974
4. Khan MF, Wang H. Environmental Exposures and Autoimmune Diseases: Contribution of Gut Microbiome. Front Immunol (2020) 10:3094. doi: 10.3389/fimmu.2019.03094
5. Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut (2004) 53:1–4. doi: 10.1136/gut.53.1.1
6. Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, et al. Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann Rheum Dis (2019) 78:947–56. doi: 10.1136/annrheumdis-2018-214856
7. Pianta A, Arvikar S, Strle K, Drouin EE, Wang Q, Costello CE, et al. Evidence of the Immune Relevance of Prevotella copri, a Gut Microbe, in Patients with Rheumatoid Arthritis. Arthritis Rheumatol (Hoboken NJ) (2017) 69:964–75. doi: 10.1002/art.40003
8. Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun (2016) 7:12015–11. doi: 10.1038/ncomms12015
9. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature (2018) 562:589–94. doi: 10.1038/s41586-018-0620-2
10. Garrett WS. Immune recognition of microbial metabolites. Nat Rev Immunol (2020) 20:91–2. doi: 10.1038/s41577-019-0252-2
11. Venegas DP, la Fuente MKD, Landskron G, González MJ, Quera R, Dijkstra G, et al. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front Immunol (2019) 10:277. doi: 10.3389/fimmu.2019.00277
12. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science (2013) 341:569–73. doi: 10.1126/science.1241165
13. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi: 10.1038/nature12726
14. Bhaskaran N, Quigley C, Paw C, Butala S, Schneider E, Pandiyan P. Role of Short Chain Fatty Acids in Controlling Tregs and Immunopathology During Mucosal Infection. Front Microbiol (2018) 9:1995. doi: 10.3389/fmicb.2018.01995
15. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504:446–50. doi: 10.1038/nature12721
16. Frank DN, Amand ALS, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci (2007) 104:13780–5. doi: 10.1073/pnas.0706625104
17. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature (2014) 505:559–63. doi: 10.1038/nature12820
18. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y-Y, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science (2011) 334:105–8. doi: 10.1126/science.1208344
19. Groschwitz KR, Hogan SP. Intestinal barrier function: Molecular regulation and disease pathogenesis. J Allergy Clin Immun (2009) 124:3–20. doi: 10.1016/j.jaci.2009.05.038
20. Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe (2017) 21:455–66.e4. doi: 10.1016/j.chom.2017.03.002
21. Mu Q, Kirby J, Reilly CM, Luo XM. Leaky Gut As a Danger Signal for Autoimmune Diseases. Front Immunol (2017) 8:598. doi: 10.3389/fimmu.2017.00598
22. Ajamian M, Steer D, Rosella G, Gibson PR. Serum zonulin as a marker of intestinal mucosal barrier function: May not be what it seems. PloS One (2019) 14:e0210728. doi: 10.1371/journal.pone.0210728
23. Graham DB, Xavier RJ. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature (2020) 578:527–39. doi: 10.1038/s41586-020-2025-2
24. Gregersen PK, Olsson LM. Recent advances in the genetics of autoimmune disease. Annu Rev Immunol (2009) 27:363–91. doi: 10.1146/annurev.immunol.021908.132653
25. Kyogoku C, Tsuchiya N. A compass that points to lupus: genetic studies on type I interferon pathway. Genes Immun (2007) 8:445–55. doi: 10.1038/sj.gene.6364409
26. Busch R, Kollnberger S, Mellins ED. HLA associations in inflammatory arthritis: emerging mechanisms and clinical implications. Nat Rev Rheumatol (2019) 15:364–81. doi: 10.1038/s41584-019-0219-5
27. Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol (2006) 6:823–35. doi: 10.1038/nri1957
28. Shaw PJ, McDermott MF, Kanneganti T-D. Inflammasomes and autoimmunity. Trends Mol Med (2011) 17:57–64. doi: 10.1016/j.molmed.2010.11.001
29. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469:323–35. doi: 10.1038/nature09782
30. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol (2010) 28:573–621. doi: 10.1146/annurev-immunol-030409-101225
31. Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic Approaches for Defining the Pathogenesis of Inflammatory Bowel Diseases. Cell Host Microbe (2008) 3:417–27. doi: 10.1016/j.chom.2008.05.001
32. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux J-J, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U States America (2008) 105:16731–6. doi: 10.1073/pnas.0804812105
33. Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, et al. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci Trans Med (2017) 9(376):eaaf9655. doi: 10.1126/scitranslmed.aaf9655
34. Baumgart M, Dogan B, Rishniw M, Weitzman G, Bosworth B, Yantiss R, et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J (2007) 1:403–18. doi: 10.1038/ismej.2007.52
35. Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Treuren WV, Ren B, et al. The Treatment-Naive Microbiome in New-Onset Crohn’s Disease. Cell Host Microbe (2014) 15:382–92. doi: 10.1016/j.chom.2014.02.005
36. Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis (2011) 17:179–84. doi: 10.1002/ibd.21339
37. Hall AB, Yassour M, Sauk J, Garner A, Jiang X, Arthur T, et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med (2017) 9:103. doi: 10.1186/s13073-017-0490-5
38. Henke MT, Kenny DJ, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc Natl Acad Sci (2019) 116:12672–7. doi: 10.1073/pnas.1904099116
39. Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature (2019) 569:655–62. doi: 10.1038/s41586-019-1237-9
40. Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology (2002) 122:44–54. doi: 10.1053/gast.2002.30294
41. Zholudev A, Zurakowski D, Young W, Leichtner A, Bousvaros A, Serologic Testing with ANCA. ASCA, and Anti-OmpC in Children and Young Adults with Crohn’s Disease and Ulcerative Colitis: Diagnostic Value and Correlation with Disease Phenotype. Am J Gastroenterol (2004) 99:2235–41. doi: 10.1111/j.1572-0241.2004.40369.x
42. Duarte-Silva M, Afonso PC, de Souza PR, Peghini BC, Rodrigues-Júnior V, Cardoso CR de B. Reappraisal of antibodies against Saccharomyces cerevisiae (ASCA) as persistent biomarkers in quiescent Crohn’s disease. Autoimmunity (2019) 52:37–47. doi: 10.1080/08916934.2019.1588889
43. Sitaraman SV, Klapproth J-M, Moore DA, Landers C, Targan S, Williams IR, et al. Elevated flagellin-specific immunoglobulins in Crohn’s disease. Am J Physiol Gastr L (2005) 288:G403–6. doi: 10.1152/ajpgi.00357.2004
44. Iltanen S, Tervo L, Halttunen T, Wei B, Braun J, Rantala I, et al. Elevated serum anti-I2 and anti-OmpW antibody levels in children with IBD. Inflammation Bowel Dis (2006) 12:389–94. doi: 10.1097/01.mib.0000218765.84087.42
45. Coufal S, Galanova N, Bajer L, Gajdarova Z, Schierova D, Zakostelska ZJ, et al. Inflammatory Bowel Disease Types Differ in Markers of Inflammation, Gut Barrier and in Specific Anti-Bacterial Response. Cells (2019) 8:719. doi: 10.3390/cells8070719
46. Huang H, Fang M, Jostins L, Mirkov MU, Boucher G, Anderson CA, et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature (2017) 547:173–8. doi: 10.1038/nature22969
47. Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet (2011) 43:1066–73. doi: 10.1038/ng.952
48. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet (2007) 39:207–11. doi: 10.1038/ng1954
49. Liu T-C, Stappenbeck TS. Genetics and Pathogenesis of Inflammatory Bowel Disease. Annu Rev Pathol (2016) 11:127–48. doi: 10.1146/annurev-pathol-012615-044152
50. Shouval DS, Biswas A, Goettel JA, McCann K, Conaway E, Redhu NS, et al. Interleukin-10 Receptor Signaling in Innate Immune Cells Regulates Mucosal Immune Tolerance and Anti-Inflammatory Macrophage Function. Immunity (2014) 40:706–19. doi: 10.1016/j.immuni.2014.03.011
51. Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology (2012) 143:347–55. doi: 10.1053/j.gastro.2012.04.045
52. Glocker E-O, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. New Engl J Med (2009) 361:2033–45. doi: 10.1056/nejmoa0907206
53. Shim JO, Seo JK. Very early-onset inflammatory bowel disease (IBD) in infancy is a different disease entity from adult-onset IBD; one form of interleukin-10 receptor mutations. J Hum Genet (2014) 59:337–41. doi: 10.1038/jhg.2014.32
54. Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. Clinical importance of IL-22 cascade in IBD. J Gastroenterol (2018) 53:465–74. doi: 10.1007/s00535-017-1401-7
55. Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y, et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology (2005) 129:969–84. doi: 10.1053/j.gastro.2005.06.071
56. Kim M, Galan C, Hill AA, Wu W-J, Fehlner-Peach H, Song HW, et al. Critical Role for the Microbiota in CX3CR1+ Intestinal Mononuclear Phagocyte Regulation of Intestinal T Cell Responses. Immunity (2018) 49:151–63.e5. doi: 10.1016/j.immuni.2018.05.009
57. Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, et al. CX3CR1+ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med (2014) 211:1571–83. doi: 10.1084/jem.20140678
58. Morhardt TL, Hayashi A, Ochi T, Quirós M, Kitamoto S, Nagao-Kitamoto H, et al. IL-10 produced by macrophages regulates epithelial integrity in the small intestine. Sci Rep uk (2019) 9:1223. doi: 10.1038/s41598-018-38125-x
59. Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish ED, et al. Resident Enteric Bacteria Are Necessary for Development of Spontaneous Colitis and Immune System Activation in Interleukin-10-Deficient Mice. Infect Immun (1998) 66:5224–31. doi: 10.1128/IAI.66.11.5224-5231.1998
60. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med (2008) 14:282–9. doi: 10.1038/nm1720
61. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and Adaptive Interleukin-22 Protects Mice from Inflammatory Bowel Disease. Immunity (2008) 29:947–57. doi: 10.1016/j.immuni.2008.11.003
62. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of Commensal Microflora by Toll-Like Receptors Is Required for Intestinal Homeostasis. Cell (2004) 118:229–41. doi: 10.1016/j.cell.2004.07.002
63. Feng T, Wang L, Schoeb TR, Elson CO, Cong Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J Exp Med (2010) 207:1321–2. doi: 10.1084/jem.20092253
64. Sugihara K, Morhardt TL, Kamada N. The Role of Dietary Nutrients in Inflammatory Bowel Disease. Front Immunol (2018) 9:3183. doi: 10.3389/fimmu.2018.03183
65. Machiels K, Joossens M, Sabino J, Preter VD, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut (2014) 63:1275–83. doi: 10.1136/gutjnl-2013-304833
66. Lamas B, Richard ML, Leducq V, Pham H-P, Michel M-L, Costa GD, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med (2019) 22:1–12. doi: 10.1038/nm.4102
67. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol (Baltimore Md: 1950) (2010) 185:3190–8. doi: 10.4049/jimmunol.0903670
68. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453:65–71. doi: 10.1038/nature06880
69. Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun (2018) 9:3294–10. doi: 10.1038/s41467-018-05470-4
70. Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature (2012) 487:1–7. doi: 10.1038/nature11228
71. Takamura T, Harama D, Fukumoto S, Nakamura Y, Shimokawa N, Ishimaru K, et al. Lactobacillus bulgaricus OLL1181 activates the aryl hydrocarbon receptor pathway and inhibits colitis. Immunol Cell Biol (2011) 89:817–22. doi: 10.1038/icb.2010.165
72. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature (2019) 576:143–8. doi: 10.1038/s41586-019-1785-z
73. Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature (2020) 581:475–9. doi: 10.1038/s41586-020-2193-0
74. McKenney PT, Yan J, Vaubourgeix J, Becattini S, Lampen N, Motzer A, et al. Intestinal Bile Acids Induce a Morphotype Switch in Vancomycin-Resistant Enterococcus that Facilitates Intestinal Colonization. Cell Host Microbe (2019) 25:695–705. doi: 10.1016/j.chom.2019.03.008
75. Nagpal R, Wang S, Ahmadi S, Hayes J, Gagliano J, Subashchandrabose S, et al. Human-origin probiotic cocktail increases short-chain fatty acid production via modulation of mice and human gut microbiome. Sci Rep (2018) 8:12649–15. doi: 10.1038/s41598-018-30114-4
76. Ukena SN, Singh A, Dringenberg U, Engelhardt R, Seidler U, Hansen W, et al. Probiotic Escherichia coli Nissle 1917 inhibits leaky gut by enhancing mucosal integrity. PloS One (2007) 2:e1308. doi: 10.1371/journal.pone.0001308
77. Mardini HE, Grigorian AY. Probiotic Mix VSL3 Is Effective Adjunctive Therapy for Mild to Moderately Active Ulcerative Colitis. Inflamm Bowel Dis (2014) 20:1562–7. doi: 10.1097/mib.0000000000000084
78. Derwa Y, Gracie DJ, Hamlin PJ, Ford AC. Systematic review with meta-analysis: the efficacy of probiotics in inflammatory bowel disease. Aliment Pharmacol Ther (2017) 46:389–400. doi: 10.1111/apt.14203
79. Giles EM, D’Adamo GL, Forster SC. The future of faecal transplants. Nat Rev Microbiol (2019) 17:719–9. doi: 10.1038/s41579-019-0271-9
80. Jacob V, Crawford C, Cohen-Mekelburg S, Viladomiu M, Putzel GG, Schneider Y, et al. Single Delivery of High-Diversity Fecal Microbiota Preparation by Colonoscopy Is Safe and Effective in Increasing Microbial Diversity in Active Ulcerative Colitis. Inflamm Bowel Dis (2017) 23:903–11. doi: 10.1097/mib.0000000000001132
81. Moayyedi P, Surette MG, Kim PT, Libertucci J, Wolfe M, Onischi C, et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology (2015) 149:102–9.e6. doi: 10.1053/j.gastro.2015.04.001
82. Gogokhia L, Buhrke K, Bell R, Hoffman B, Brown DG, Hanke-Gogokhia C, et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe (2019) 25:285–99.e8. doi: 10.1016/j.chom.2019.01.008
83. Tsokos GC. Autoimmunity and organ damage in systemic lupus erythematosus. Nat Immunol (2020) 21:605–14. doi: 10.1038/s41590-020-0677-6
84. Li B-Z, Zhou H-Y, Guo B, Chen W-J, Tao J-H, Cao N-W, et al. Dysbiosis of oral microbiota is associated with systemic lupus erythematosus. Arch Oral Biol (2020) 113:104708. doi: 10.1016/j.archoralbio.2020.104708
85. Li Y, Wang H-F, Li X, Li H-X, Zhang Q, Zhou H-W, et al. Disordered intestinal microbes are associated with the activity of Systemic Lupus Erythematosus. Clin Sci (London England: 1979) (2019) 133:821–38. doi: 10.1042/cs20180841
86. Zegarra-Ruiz DF, Beidaq AE, Iñiguez AJ, Ricco MLD, Vieira SM, Ruff WE, et al. A Diet-Sensitive Commensal Lactobacillus Strain Mediates TLR7-Dependent Systemic Autoimmunity. Cell Host Microbe (2019) 25:113–27.e6. doi: 10.1016/j.chom.2018.11.009
87. Greiling TM, Dehner C, Chen X, Hughes K, Iñiguez AJ, Boccitto M, et al. Commensal orthologs of the human autoantigen Ro60 as triggers of autoimmunity in lupus. Sci Trans Med (2018) 10:eaan2306. doi: 10.1126/scitranslmed.aan2306
88. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. New Engl J Med (2003) 349:1526–33. doi: 10.1056/nejmoa021933
89. Heinlen LD, McClain MT, Ritterhouse LL, Bruner BF, Edgerton CC, Keith MP, et al. 60 kD Ro and nRNP A frequently initiate human lupus autoimmunity. PloS One (2010) 5:e9599. doi: 10.1371/journal.pone.0009599
90. Vieira SM, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science (2018) 359:1156–61. doi: 10.1126/science.aar7201
91. Choi S-C, Brown J, Gong M, Ge Y, Zadeh M, Li W, et al. Gut microbiota dysbiosis and altered tryptophan catabolism contribute to autoimmunity in lupus-susceptible mice. Sci Trans Med (2020) 12:eaax2220. doi: 10.1126/scitranslmed.aax2220
92. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. New Engl J Med (2011) 365:2205–19. doi: 10.1056/nejmra1004965
93. van der Heijden IM, Wilbrink B, Tchetverikov I, Schrijver IA, Schouls LM, Hazenberg MP, et al. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum (2000) 43:593–8. doi: 10.1002/1529-0131(200003)43:3<593::aid-anr16>3.0.co;2-1
94. Scher JU, Ubeda C, Equinda M, Khanin R, Buischi Y, Viale A, et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum (2012) 64:3083–94. doi: 10.1002/art.34539
95. Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med (2015) 21:895–905. doi: 10.1038/nm.3914
96. Alpizar-Rodriguez D, Lesker TR, Gronow A, Gilbert B, Raemy E, Lamacchia C, et al. Prevotella copri in individuals at risk for rheumatoid arthritis. Ann Rheum Dis (2019) 78:590–3. doi: 10.1136/annrheumdis-2018-214514
97. Maeda Y, Kurakawa T, Umemoto E, Motooka D, Ito Y, Gotoh K, et al. Dysbiosis Contributes to Arthritis Development via Activation of Autoreactive T Cells in the Intestine. Arthritis Rheumatol (Hoboken NJ) (2016) 68:2646–61. doi: 10.1002/art.39783
98. Abdollahi-Roodsaz S, Joosten LAB, Koenders MI, Devesa I, Roelofs MF, Radstake TRDJ, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest (2008) 118:205–16. doi: 10.1172/jci32639
99. Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med (2000) 191:313–20. doi: 10.1084/jem.191.2.313
100. Rehaume LM, Mondot S, de Cárcer DA, Velasco J, Benham H, Hasnain SZ, et al. ZAP-70 genotype disrupts the relationship between microbiota and host, leading to spondyloarthritis and ileitis in SKG mice. Arthritis Rheumatol (Hoboken NJ) (2014) 66:2780–92. doi: 10.1002/art.38773
101. Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature (2003) 426:454–60. doi: 10.1038/nature02119
102. Teng F, Klinger CN, Felix KM, Bradley CP, Wu E, Tran NL, et al. Gut Microbiota Drive Autoimmune Arthritis by Promoting Differentiation and Migration of Peyer’s Patch T Follicular Helper Cells. Immunity (2016) 44:875–88. doi: 10.1016/j.immuni.2016.03.013
103. Compston A, Coles A. Multiple sclerosis. Lancet (London England) (2002) 359:1221–31. doi: 10.1016/s0140-6736(02)08220-x
104. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature (2013) 500:232–6. doi: 10.1038/nature12331
105. Miyake S, Kim S, Suda W, Oshima K, Nakamura M, Matsuoka T, et al. Dysbiosis in the Gut Microbiota of Patients with Multiple Sclerosis, with a Striking Depletion of Species Belonging to Clostridia XIVa and IV Clusters. PloS One (2015) 10:e0137429. doi: 10.1371/journal.pone.0137429
106. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A (2011) 108 Suppl 1:4615–22. doi: 10.1073/pnas.1000082107
107. Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z, et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci U S A (2017) 114:10719–24. doi: 10.1073/pnas.1711233114
108. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci U States America (2017) 114:10713–8. doi: 10.1073/pnas.1711235114
109. Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med (2016) 22:586–97. doi: 10.1038/nm.4106
110. Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature (2017) 551:585–9. doi: 10.1038/nature24628
111. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet (London England) (2018) 391:2449–62. doi: 10.1016/s0140-6736(18)31320-5
112. Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature (2018) 562:583–8. doi: 10.1038/s41586-018-0617-x
113. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyötyläinen T, Hämäläinen A-M, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe (2015) 17:260–73. doi: 10.1016/j.chom.2015.01.001
114. Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J (2011) 5:82–91. doi: 10.1038/ismej.2010.92
115. Pozzilli P, Signore A, Williams AJ, Beales PE. NOD mouse colonies around the world–recent facts and figures. Immunol Today (1993) 14:193–6. doi: 10.1016/0167-5699(93)90160-m
116. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature (2008) 455:1109–13. doi: 10.1038/nature07336
117. Hu Y, Peng J, Tai N, Hu C, Zhang X, Wong FS, et al. Maternal Antibiotic Treatment Protects Offspring from Diabetes Development in Nonobese Diabetic Mice by Generation of Tolerogenic APCs. J Immunol (Baltimore Md: 1950) (2015) 195:4176–84. doi: 10.4049/jimmunol.1500884
118. Costa FRC, Françozo MCS, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med (2016) 213:1223–39. doi: 10.1084/jem.20150744
119. Markle JGM, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science (2013) 339:1084–8. doi: 10.1126/science.1233521
120. Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol (2017) 18:552–62. doi: 10.1038/ni.3713
121. Masetti G, Moshkelgosha S, Köhling H-L, Covelli D, Banga JP, Berchner-Pfannschmidt U, et al. Gut microbiota in experimental murine model of Graves’ orbitopathy established in different environments may modulate clinical presentation of disease. Microbiome (2018) 6:1–15. doi: 10.1186/s40168-018-0478-4
122. Horai R, Zárate-Bladés CR, Dillenburg-Pilla P, Chen J, Kielczewski JL, Silver PB, et al. Microbiota-Dependent Activation of an Autoreactive T Cell Receptor Provokes Autoimmunity in an Immunologically Privileged Site. Immunity (2015) 43:343–53. doi: 10.1016/j.immuni.2015.07.014
123. Rinaldi E, Consonni A, Cordiglieri C, Sacco G, Crasà C, Fontana A, et al. Therapeutic Effect of Bifidobacterium Administration on Experimental Autoimmune Myasthenia Gravis in Lewis Rats. Front Immunol (2019) 10:2949. doi: 10.3389/fimmu.2019.02949
124. Moon J, Choi SH, Yoon CH, Kim MK. Gut dysbiosis is prevailing in Sjögren’s syndrome and is related to dry eye severity. PloS One (2020) 15:e0229029. doi: 10.1371/journal.pone.0229029
125. Limon JJ, Skalski JH, Underhill DM. Commensal Fungi in Health and Disease. Cell Host Microbe (2017) 22:156–65. doi: 10.1016/j.chom.2017.07.002
126. Iliev ID, Leonardi I. Fungal dysbiosis: immunity and interactions at mucosal barriers. Nat Rev Immunol (2017) 17:635–46. doi: 10.1038/nri.2017.55
127. Underhill DM, Iliev ID. The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol (2014) 14:405–16. doi: 10.1038/nri3684
Keywords: multiple sclerosis, autoimmunity, inflammation, inflammatory bowel disease, type 1 diabetes, rheumathoid arthritis, systemic lupus erythematosus, intestinal microbiota
Citation: Wu W-JH, Zegarra-Ruiz DF and Diehl GE (2020) Intestinal Microbes in Autoimmune and Inflammatory Disease. Front. Immunol. 11:597966. doi: 10.3389/fimmu.2020.597966
Received: 23 August 2020; Accepted: 17 November 2020;
Published: 23 December 2020.
Edited by:
Gregg Joshua Silverman, New York University, United StatesReviewed by:
Trine N. Jorgensen, Case Western Reserve University, United StatesRichard David Leslie, Queen Mary University of London, United Kingdom
Copyright © 2020 Wu, Zegarra-Ruiz and Diehl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gretchen E. Diehl, ZGllaGxnMUBtc2tjYy5vcmc=