 Zayda L. Piedra-Quintero
Zayda L. Piedra-Quintero Zachary Wilson
Zachary Wilson Porfirio Nava
Porfirio Nava Mireia Guerau-de-Arellano
Mireia Guerau-de-Arellano- 1School of Health and Rehabilitation Sciences, Division of Medical Laboratory Science, College of Medicine, Wexner Medical Center, The Ohio State University, Columbus, OH, United States
- 2Biomedical Science Undergraduate Program, College of Medicine, Wexner Medical Center, The Ohio State University, Columbus, OH, United States
- 3Departamento de Fisiología, Biofísica y Neurociencias, Centro de Investigación y de Estudios Avanzados (CINVESTAV), México City, México
- 4Institute for Behavioral Medicine Research, The Ohio State University, Columbus, OH, United States
- 5Department of Microbial Infection and Immunity, The Ohio State University, Columbus, OH, United States
- 6Department of Neuroscience, The Ohio State University, Columbus, OH, United States
CD38 is a molecule that can act as an enzyme, with NAD-depleting and intracellular signaling activity, or as a receptor with adhesive functions. CD38 can be found expressed either on the cell surface, where it may face the extracellular milieu or the cytosol, or in intracellular compartments, such as endoplasmic reticulum, nuclear membrane, and mitochondria. The main expression of CD38 is observed in hematopoietic cells, with some cell-type specific differences between mouse and human. The role of CD38 in immune cells ranges from modulating cell differentiation to effector functions during inflammation, where CD38 may regulate cell recruitment, cytokine release, and NAD availability. In line with a role in inflammation, CD38 appears to also play a critical role in inflammatory processes during autoimmunity, although whether CD38 has pathogenic or regulatory effects varies depending on the disease, immune cell, or animal model analyzed. Given the complexity of the physiology of CD38 it has been difficult to completely understand the biology of this molecule during autoimmune inflammation. In this review, we analyze current knowledge and controversies regarding the role of CD38 during inflammation and autoimmunity and novel molecular tools that may clarify current gaps in the field.
Introduction
For the last four decades, the contribution of CD38 to cell biology and CD38’s links to human disease have been the focus of substantial research. CD38 was initially described as a surface protein in T cells capable of inducing cell activation (1, 2). Soon after, CD38 expression was also reported in other immune cells, such as B cells, natural killer (NK) cells, neutrophils, and myeloid cells (1, 3–6). Likewise, roles for CD38 in cell differentiation, cytokine release, migration, and apoptosis processes were revealed (7–11). At the molecular level, the similarity of CD38 with a soluble enzyme purified from the mollusk Aplysia californica led to the characterization of CD38 as an ADP-ribosyl cyclase and a cyclic ADPR (cADPR) hydrolase that utilizes NAD as substrate (12, 13).
Since these findings were reported, many studies have sought to understand the extent to which CD38 contributes to the development of inflammatory and autoimmune disease, via modulation of immune responses. So far, it is known that CD38 expression is robustly induced in immune cells after activation and regulates infection-induced inflammatory processes, from cell recruitment to induction of adaptative immune responses (5, 14). Nonetheless, the mechanisms utilized by CD38 to mediate each stage of inflammation are still poorly understood. The function of CD38 during inflammatory autoimmunity has also been the subject of many studies in several diseases. Depending on the disease type, immune cell population, or animal model analyzed, several reports indicate that CD38 can either suppress or induce autoimmunity. The lack of consensus highlights the need of more research in order to understand the biology of CD38 and its contributions to inflammation and autoimmunity.
In the following sections, we summarize the studies available on the role of CD38 during inflammation and autoimmune disease. In addition, we present a comprehensive summary of molecular tools available for the study of CD38 that should help advance our understanding of CD38’s role in physiology and disease.
CD38: Function, Structure, and Localization
CD38 is a protein of 300 amino acids encoded by homologous genes located on chromosome 4 and 5 in humans and mice, respectively (15). Within the cell, CD38 is often found localized on the cell surface, but it can also be detected in intracellular compartments such as the endoplasmic reticulum, nuclear membrane and mitochondria (16–19). Structurally, CD38 is a single chain glycoprotein with a single transmembrane segment and can topologically behave as a type II or type III membrane protein depending on its membrane orientation. In the most common type II orientation, CD38’s short amino tail faces into the cytosol while CD38’s catalytic domain faces the extracellular environment (20, 21). A type III orientation, with the catalytic domain facing the cytosol, has been also reported (17, 22). These two orientations have functional implications, given that CD38’s enzymatic substrates and products would be consumed and produced in the extracellular or the intracellular compartments. CD38 catalyzes the synthesis of nicotinamide (NAM) and ADPR using nicotinamide adenine dinucleotide (NAD+) as a substrate. NAD+, an essential cofactor that regulates energy metabolism (23), can be converted to cADPR with the release of NAM. Interestingly, cADPR can also be hydrolyzed to ADP-ribose by CD38. Thus, CD38 has both ADP-ribosyl cyclase and cADPR hydrolase enzymatic activities. Both ADPR and cADPR act as second messengers controlling several cell functions through calcium (Ca2+) mobilization (24, 25). Therefore, the implications of these observations in cell physiology have received significant interest. Besides its enzymatic function, CD38 can also act as a receptor to CD31 (26). Through the latter interaction, CD38 could act as an adhesion molecule mediating selectin-like binding of hematopoietic cells to endothelial cells and facilitating their transmigration to tissue (27, 28).
CD38 Distribution in the Immune System
CD38 is a ubiquitous protein expressed in multiple tissues. Non-hematopoietic tissue expression include prostatic epithelial cells, pancreatic islet astrocytes, smooth muscle cells, retinal tubes, kidney, gut, and brain in both mice and humans (Figure 1A) (31–34). However, CD38 is most highly expressed in hematopoietic tissues such as the bone barrow and lymph nodes (35). Within immune cells, CD38 is highly expressed in B cells, macrophages, dendritic cells (DCs), innate lymphoid cells (ILC), natural killer (NK) cells, T cells, neutrophils, and monocytes (36). Nevertheless, the level of CD38 expression among these populations may differ between human and mouse, as observed in a transcriptional comparison between species (29, 30) (Figure 1B).
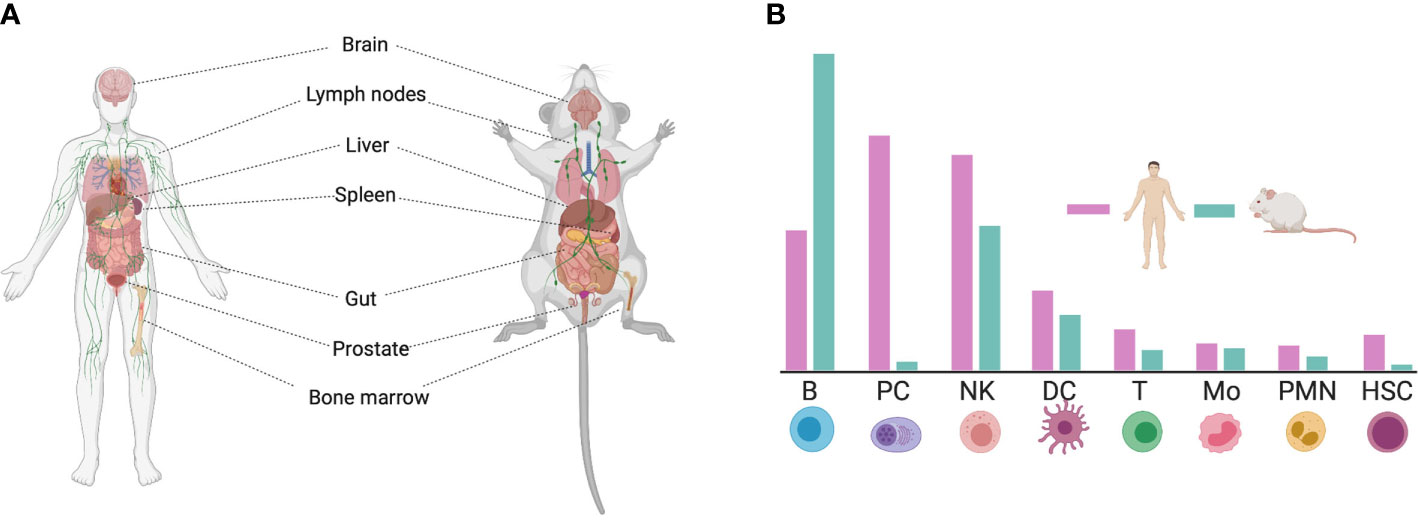
Figure 1 Expression of CD38 in human and mouse tissue. (A) CD38 is ubiquitously expressed in the human and mouse body. (B) Schematic representation of CD38 expression on immune cells. The differences in CD38 expression between human and mouse is highlighted [adapted from (29, 30)]. This figure was created using Biorender (https://biorender.com). B, B cell; PC, Plasma cell; NK, Natural Killer cell; DC, Dendritic Cell; T, T cell; Mo, Monocyte; PMN, Molymorphonuclear cell; HSC, Hematopoietic Stem Cell.
Based on the pattern of CD38 expression during these cells’ life cycle, CD38 may impact a range of processes, from differentiation to effector function. CD38 is expressed in B cell precursors, germinal center B cells, and plasma cells (37, 38). However, some differences in expression have been reported between human and mouse (29, 39). For instance, human progenitor B cells express CD38 on surface but lose this expression after maturation whereas murine B cells express CD38 throughout its entire differentiation process. Also, CD38 is highly expressed on B cells from germinal center while expression on murine B cells is low. In contrast, only human plasma cells have been reported to express CD38. Something similar is observed in early human T cells precursors and double-positive CD4+CD8+ thymocytes (DP), which display high expression of CD38, while mouse DP and resting T cells do not express CD38 (8, 29, 38, 40–43). In contrast, both mature human and mouse T and B cells induce CD38 upon activation. Therefore, this molecule is often used as a cell activation and differentiation marker (7, 8, 14). Likewise, expression of CD38 in NK cells has been linked to activation, induction of cytotoxic activity, and secretion of IFN-γ. It has been demonstrated that CD38 controls these cellular processes through its receptorial activity, a function dependent of CD38’s lateral association with CD16 (3, 44). In the myeloid linage, CD38 expression is induced after activation in inflammatory conditions in both mice and humans. CD38 appears to regulate cytokine release, adhesion, and cellular migration toward sites of inflammation (4, 5, 33, 45, 46). Thus, the prominent expression of CD38 in immune cells suggests important roles in immune responses, ranging from the development of inflammation in response to infection to development or regulation of adaptive immune responses.
Inflammation
Inflammation is a characteristic body response to harmful stimuli aimed at eliminating the source of damage and returning the tissue to functional homeostasis (47). Eliciting stimuli include pathogen-associated molecular patterns (PAMPs) from infectious pathogens and damage-associated molecular patterns (DAMPs) released during sterile tissue/cell injury during traumatic injuries or autoimmune disease (48–52).
Recognition of PAMP/DAMPs by pattern recognition receptors (PRRs), present in innate immune cells, induces the synthesis of lipid inflammatory mediators such as arachidonic acid (AA). AA is then converted to prostaglandins (PG) and leukotrienes (LT) that act as potent neutrophil chemoattractants (53, 54) and trigger inflammation. The inflammatory process is a multistep event encompassing 1) endothelial adhesion, 2) endothelial cell transmigration, 3) chemotaxis, 4) cytokine/chemokine release to recruit additional immune cells (such as monocytes), 5) phagocytosis, and 6) antigen presentation leading to initiation of antigen-specific adaptive immune responses (55, 56). The first wave of cells reaching the tissue is formed by neutrophils, which are recruited in response to PG and LT with robust neutrophil chemoattractant activity. When neutrophils reach the infected/injured tissue, they release inflammatory mediators that recruit a secondary wave of monocytes. Monocytes can differentiate into macrophages (MØ) and dendritic cells. Monocyte derived-macrophages produce additional inflammatory cytokines and, together with neutrophils and DCs, phagocytose pathogens or dying cells (57). After phagocytosis, professional antigen presenting cells (APCs) such as macrophages and DCs process antigens and load them onto Major Histocompatibility Complex (MHC) molecules. These antigen-loaded MHC molecules are presented on the cell surface for recognition by antigen-specific T cells. Thus, this step marks the initiation of antigen-specific adaptive immune responses (58). The combined effects of immune cells and their mediators generally resolve inflammation and initiate tissue repair. However, if the inflammatory process is not resolved, it can lead to chronic inflammation. Lack of inflammation resolution may be due to the continuous presence of self-antigens, as in autoimmune diseases (59).
CD38 is robustly induced during infection and the ensuing inflammation (5, 45, 60). The human CD38 gene promoter includes putative binding sites for the transcription factors Sp1, Retinoic Acid Responsive Elements (RARE) and IRF-1 (61). Thus, has been reported that CD38 expression is under the transcriptional control of IFN type I and II as well as TNF-α/NF-κB stimulation (61–63). Therefore, CD38 has been clearly linked to inflammation and has been the subject of considerable study, particularly in the context of infection. In the sections below, we will summarize our current knowledge on the contribution of CD38 to the various steps of the inflammatory process.
CD38 Regulates Cellular Migration
The role of CD38 during infection-induced inflammation has been the focus of substantial study, revealing a supporting role for CD38 during inflammation (Figure 2). Transmigration through endothelial cells and chemotaxis toward the site of injury is one of the first steps in the inflammatory process (64, 65). CD38 appears to be essential for both of these processes, as deletion of CD38 in mouse impairs the recruitment of immune cell from blood to sites of infection or tissue injury (33, 45, 66). Furthermore, induction of CD38 expression in hematopoietic cells such as neutrophils, monocytes, dendritic cells, and macrophages is observed in response infection or cellular activation (5, 9, 45, 60).

Figure 2 Role of CD38 during inflammation. The figure displays a schematic representation of an inflammatory process and the events impacted by CD38. During inflammation, CD38 can modulate cell recruitment, cytokines and chemokines release, cell activation, phagocytosis, and antigen presentation. CD38 expressing cells consume NAD+ to produce cADPR an event that leads to inflammation. This figure was created using Biorender (https://biorender.com).
The effects of CD38 on transmigration may be mediated by the interaction of CD38 with its cognate receptor CD31 (27). CD31 constitutes the major endothelial adhesion molecule involved in transendothelial migration (67). Thus, immune cells expressing CD38 may adhere to endothelium and mediate transmigration through CD31. This has been demonstrated for neutrophils in the context of infection. Listeria monocytogenes and Streptococcus pneumoniae infections normally result in neutrophil recruitment, but this recruitment is impaired in CD38 deficient mice (45, 68). CD38 also appears to mediate the migration of other immune cells. For instance, CD38 is essential for T cell and macrophage infiltration into the brain in a mouse model of cerebral ischemia (9).
After transmigration into the tissue, chemotaxis will direct infiltrating cells to the site of injury or infection in response to chemoattractant gradients (69). CD38 also appears to contribute to chemotaxis in both mice and humans, given that neutrophils with low levels of CD38 show defects in chemotactic migration to the chemoattractant N-formyl-methionine-leucyl-phenylalanine (fMLF) (6, 68). Moreover, CD38’s calcium mobilization activity has been shown to control chemokine receptor signaling in neutrophils and dendritic cells. CD38’s enzymatic activity produces cADPR/ADPR, thereby triggering Ca2+ release from intracellular stores and Ca2+ influx from the extracellular space. Intracellular Ca2+ signaling induces expression of chemokine receptors such as CXCR4, CCR7 and N-formyl peptide receptor 1 (FPR1), as well as activation of cytoskeletal and adhesion molecules. Thus, cADPR/ADPR levels regulate cellular chemotaxis in a CD38 dependent manner (68, 70, 71). Also, supporting a role for CD38 in chemotaxis, human chronic lymphocytic leukemia (CLL) cells expressing high levels of CD38 exhibit enhanced in vitro migration in response to chemokines. Increased chemotaxis was accompanied by increased intracellular Ca2+ and actin polymerization. These events induced cellular spreading and cell migration via GTPases Rap1/RasGRP2 activation (72).
Overall, multiple lines of evidence support a role for CD38 during transmigration and chemotaxis of neutrophils and monocytes that can next perform their signature phagocytic function.
Phagocytosis and Antigen Presentation
Phagocytosis is the process by which immune cells such as neutrophils, macrophages and dendritic cells, engulf and eliminate invading pathogens, foreign particles, and infected or apoptotic cells (73). Phagocytosis requires calcium signaling for cytoskeleton activation and subsequent phagosome maturation (74, 75). Therefore, one obvious way in which CD38 could contribute to phagocytosis is via cADPR and NAADP-induced Ca2+ signaling (25, 76, 77). Indeed, CD38 is recruited and internalized to the phagocytic cup during macrophage FcγR-mediated phagocytosis. This process is accompanied by increased extracellular cADPR and intracellular Ca2+. More conclusively demonstrating a role, the absence of CD38 in murine macrophages impairs Ca2+ signaling and phagocytosis of Mycobacterium bovis BCG in vivo (76). Similarly, the role of CD38 in controlling Listeria monocytogenes infection appears to stem from its role in phagocytic function. The inability of CD38 deficient macrophages to control Listeria infection in macrophages is due to impaired phagocytosis, as CD38 deficient-murine macrophages maintain their ability to kill L. monocytogenes (45). Thus, CD38 appears to play a key role in phagocytosis at the level of bacterial uptake.
After phagocytosis, internalized protein-antigens are digested into peptide fragments that can be loaded onto MHC molecules (78). Then, the peptide-loaded MHC molecules are transported and displayed on the APC cell surface for T cell recognition and initiation of antigen-specific immune responses (79). The specialized APC-T cell interface formed during antigen presentation is known as the immunological synapse (IS) and involves recruitment and signaling of T cell receptor (TCR), costimulatory and adhesion molecules (80). Remarkably, CD38 has been important for antigen presentation of bacteria in both humans and mice (16, 81) and several reports indicate that CD38 may be regulating the signaling induced at the IS. In support of this idea, the contribution of CD38 to TCR/CD3 complex signaling has been widely demonstrated (2, 82). Further analysis has also revealed that CD38 accumulates through the periphery of the mature T/APC IS, suggesting that this molecule also regulates TCR signaling induced during antigen presentation in a human T cell line (16). Interestingly, surface expression of CD38 in antigen-stimulated T cells correlates with increasing calcium release, consistent with a role of CD38 in antigen induced effector function. Supporting this idea, CD38 blockade with the monoclonal antibody IB6 suppresses antigen-induced IL-12 and IFN-γ production, an effect likely mediated by cADPR (16). As mentioned earlier, adhesion and costimulatory molecules expressed by T cells and/or APCs also intervene for an effective IS. The association of CD38 with adhesion and costimulatory molecules suggests that it could also modulate antigen presentation at this level. For example, in APCs, trafficking of MHC-loaded peptides to the cell surface depends of the tetraspanin CD9 (83). Interestingly, CD38 associates with both MHCII and CD9 in membrane rafts of human monocytes from where it induces intracellular Ca2+ fluxes (81). Also, monocytes loaded with Staphylococcus enterotoxin E require CD38/MHCII/CD9-complex to present the antigen to T cells (81). Similarly, Zilber et al. reported that monocyte surface CD38 acts as an MHCII coreceptor to induce tyrosine phosphorylation of intracellular proteins during superantigen-induced activation (84). In this regard, CD38 has been also reported to associate with CD81 and CCR7, crucial molecules for clustering and signaling activation during T/APC synapses, in human DCs (11, 85, 86). On the other hand, the IS also requires prolonged cell-cell interaction which is dependent of adhesion molecules such as CD11b (87). Co-localization of CD38 and CD11b was also observed in dendritic cells (11). Thus, these observations suggest another important role for CD38 during antigen presentation.
Taken together, these findings indicate that CD38 contributes to phagocytosis and antigen presentation. Through these processes, CD38 may promote antigen-specific adaptive immune responses.
Cytokine Release
During inflammation, immune cells release inflammatory mediators to eliminate pathogens (88), a process modulated by CD38 expression. For instance, CD38 has been shown to limit bacterial infection, as evidenced by increases susceptibility to Salmonella typhimurium infection in CD38-deficient macrophages. Furthermore, the inflammatory response against S. typhimurium was also impacted, with notable reductions in pro-inflammatory cytokines IL-1β, IL-6, IL-12, and TNF-α (89). Similar results have been observed in human primary macrophages, where impaired secretion of IL-12, and IL-6 was observed after CD38 chemical or genetic inhibition (5). The implication of CD38 in cytokine release during viral infections was been also shown (62). During Respiratory Syncytial Virus infection (RSV), CD38 controls RSV-induced type I/III IFNs. This was demonstrated by the inhibition of CD38’s enzymatic activity that resulted in decreases of the expression of IFN-β, IFN-λ1 and ISG15 in human DCs infected in vitro with RSV (62). However, the contribution of CD38 towards cytokine release is still controversial since opposite behaviors have been reported in mouse cells. For instance, CD38 deficient mice had increased levels of TLR4 expression in kidney and enhanced secretion of IL-1β, IL-6, IFN-γ, and TNF-α during kidney injury induced by LPS challenge (90). Moreover, CD38 deficiency in unstimulated Raw264.7 mouse macrophages suppressed expression of TLR2 through deacetylation of NF-κB by Sirtuin (SIRT) 1 in an NAD-dependent manner. This phenomenon was also accompanied by increased secretion of inflammatory mediators, including IL-6, IL-1α, CCL2, CCL5, and G-CSF (91). These contradictory results could be explained by differences in stimuli, environment or the type of receptor or enzymatic functions played by CD38 in various circumstances. However, intrinsic differences between CD38’s role in human and mice should be also considered.
Overall, these data point to an active role for CD38 in regulation of cytokine production and indicate that the specific function of CD38 in these processes must be clarified in the future.
CD38 as a Regulator of NAD+ During Inflammation
Nicotinamide adenine dinucleotide (NAD+) is a pyridine nucleotide that allows the transfer of electrons during enzymatic reactions (92). NAD+ is essential for both glycolysis and oxidative phosphorylation by accepting high-energy electrons from glycolysis and by feeding electrons to oxidative phosphorylation metabolism (93). Fluctuations in NAD+ levels compromise homeostasis and impact cellular processes such as transcription, signaling, and cell survival (92). NAD+ is as co-substrate needed for the activity of signaling enzymes such as poly (ADP-ribose) polymerases (PARPs), sirtuin deacetylases (SIRTs), and CD38. However, CD38 is the main consuming enzyme of NAD+ in mammalian tissues. Supporting this, mice deficient of CD38 have increased NAD+ in brain, liver, and muscle tissues, suggesting that CD38 plays a critical role in maintaining NAD+ homeostasis (94, 95).
Low levels of NAD+ have also been reported during the chronic inflammation associated with aging (96, 97). Aging-associated NAD+ changes induce cellular dysfunction and correlate with increased expression and activity of CD38 (97, 98). Thus, it has been suggested that NAD+ has a protective role during inflammation. For example, it was shown that NAD+ blocks the development of experimental autoimmune encephalomyelitis (EAE) by protecting from axonal degeneration. At the mechanistic level, it was demonstrated that NAD+ regulates the differentiation of CD4+ T cells and induces the production of the anti-inflammatory cytokine IL-10 by Th1 cells (99). Interestingly, CD38 deficient mice also develop less EAE severity compared with WT mice an effect partially induced by a defect in T cell priming. Furthermore, NAD+ precursors (NMN, NAM, and NR) have been linked to anti-inflammatory effects in mouse models of ataxia telangiectasia, another disease characterized by progressive neurodegeneration (100).
Macrophages conduct inflammation through phagocytosis, antigen presentation, and the release of large amounts of pro-inflammatory cytokines and chemokines (101). How NAD+ controls these cellular responses in macrophages is another question that has been partially addressed. For instance, Misawa et al. demonstrated that low concentrations of intracellular NAD+ promote NLRP3 inflammasome activation and IL-1β release in murine macrophages (102). Similarly, low levels of NAD+, in human monocyte-derived macrophages (MDM), induced metabolic changes suppressing oxidative phosphorylation and increasing glycolysis. Furthermore, the reduction of NAD+ was companied by a defect in phagocytosis and by increased surface expression of the inflammatory markers CD86 and CD64 while the anti-inflammatory markers CD206 and CD23 were decreased (93). Although these reports did not demonstrate the contribution of CD38 to control NAD+ concentration, our laboratory has shown that CD38 is highly expressed in inflammatory MDM and that CD38 controls the release of IL-6 and IL-12. Also, inhibition of CD38 led to a decrease in lactate production, indicating a glycolysis defect (5). Overall, current evidence supports a link between CD38 and NAD+ and the idea that CD38-mediated NAD+ depletion contributes to inflammation.
It is also important to note that NAD+ consumption by CD38 also leads to synthesis of cADPR which can act as an inflammatory molecule by promoting calcium signaling and the activation of several signaling pathways (24). CD38 expression and subsequent cADPR synthesis induced upregulation of COX-2 and prostaglandin E2, a potent chemoattractant (54), in human mesenchymal stem cells (103). Similarly, cADPR controlled murine neutrophils migration toward sites of inflammation, a process dependent on CD38 expression (70, 104). The role of cADPR promoting inflammation is also supported by evidence showing that 8-Br-cADPR, an antagonist of cADPR, suppressed CCL2, reactive oxygen species (ROS), and apoptosis in human retinal pigment epithelium (24, 105).
CD38 in Autoimmunity
During the last 40 years, CD38 has been shown to regulate multiple components of the inflammatory process, at the level of cell migration, activation, antigen presentation, and cytokine release. Thus, it is not surprising that links between CD38 and chronic inflammation or autoimmunity may exist (106). Autoimmune disorders are a heterogeneous group of diseases that appear as a result of loss of tolerance to self-antigens (107). Autoimmunity is often characterized by lymphocytic infiltration in target tissue and/or circulating autoantibodies (108). Several reports have demonstrated that the absence or expression of CD38 modulates the development of autoimmunity (Table 1) (10, 33, 113, 120). The most important findings and controversies on our current understanding on the role of CD38 in autoimmunity are summarized below.

Table 1 Role of CD38 during autoimmunity.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a systemic autoimmune disorder characterized by autoantibodies, joint inflammation and destruction (121). Autoantibodies are antibodies that react against self-antigens found in cells and tissues. These autoantibodies are produced by plasma cells and are highly specific for target organs in autoimmune diseases (122). In RA, autoantibodies contribute to the pathogenesis by binding to tissue antigens or forming immune complexes that deposit within tissues (123). Therefore, antibody-producing plasma cells and their B cell precursors are thought to play important pathogenesis roles in RA. The pivotal role of B cells in RA has been demonstrated by the fact that rituximab (anti-CD20) proved to be an effective therapy for RA (124). During early rheumatoid and chronic septic arthritis, increases in infiltrating CD38+ plasma cells were observed in the synovium of patients, suggesting a role for CD38 in RA pathogenesis (116, 117, 125, 126). Indeed, treatment with plasma cell/plasmablast-depleting anti-CD38 monoclonal antibodies Daratumumab or TAK-079 decreased RA symptoms and disease progression in humans and primates respectively (118, 120). Therefore, CD38 targeting has been proposed as a therapy for RA (120).
A role for CD38 in RA pathology is also suggested by increased CD38 expression in RA patients synovial tissues, high percentages of circulating CD38+CD3+ and CD38+CD56+ cells, and high levels of rheumatic factor (120, 127, 128). Furthermore, the expression of CD38 in NK cells has been shown to contribute to RA development through the release of the pro-inflammatory cytokines TNF-α and IFN-γ (119). Using a rat model of RA, Wang et al. demonstrated that cyanidin-3-O-glucoside (C3G), a competitive inhibitor of CD38 cyclase activity, decreases the percentage of CD38+ NK cells, IL-6 and IFN-γ levels in rat peripheral blood and rat synovial fluid. Furthermore, C3G also decreases RA synovial fibroblast proliferation and increases the T regulatory cells proportion in these compartments (119). Similarly, treatment with TAK-079 prevented arthritis development accompanied by a decrease of NK cells, T cells and B cells in blood of cynomolgus monkeys (118) These data support a pathogenic role for CD38 in RA via humoral and cellular responses. However, other findings suggest instead that CD38 can also play regulatory roles in RA. For instance, reduced CD38hiPDL1+CD24hi circulating regulatory transitional B cells have been reported in RA patients (129). In summary, many CD38+ pathogenic populations appear to increase during RA. However, the exact impact and mechanism of CD38 during autoreactive immune responses in RA remains obscure.
Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) is a chronic disorder of the gastrointestinal mucosa encompassing different pathologies, such as Crohn’s disease (CD) or ulcerative colitis (UC) (130). IBD development is thought to be due to unbalanced interactions between genes, environment, immune system and microbiota that result in chronic gut inflammation (131–133). Inflammatory Th1 and Th17 effector T cells are observed infiltrating the gut of IBD patients and IBD animal models (134, 135), suggesting that dysregulated T cell responses may be a pathogenic mechanism (136). In addition, circulating T regulatory cells are reduced in IBD (136).
Several lines of evidence support the involvement of CD38 in the pathogenesis of IBD. Firstly, CD38 is highly expressed in resident and infiltrating immune cells in the colonic mucosa of human and mouse (33, 34). Also, the presence of CD38 has been detected in human T lymphocytes that reside within the lamina propria in the gut (137). Furthermore, the expression of CD38 on intestinal antigen-specific peripheral blood CD4+ effector cells has been reported, suggesting a pathogenic role of the molecule (109). In that context, the proportion of CD38+ CD4+ T cells in humans positively correlate with the mucosal damage marker serum LPS-binding protein (110). These findings suggest that phenotypic changes in circulating CD38+ effector T cells are linked to the severity of the disease or its course (138). Further, Schneider et al. showed that CD38 deletion decreases immune cell infiltration and ameliorates DSS-induced colitis (39). However, as has been reported for other autoimmune diseases, CD38 may also actively suppress other pathogenic processes during IBD. In support of this idea, CD38 expression was observed in a population of T cells with regulatory properties in peripheral blood of active IBD patients (138).
At the mechanistic level, an interesting question is how CD38 contributes to IBD development. One possibility is through metabolic regulation of NAD+ since increased levels of NAD+ have been reported to modulate inflammation in the gut (139). In this sense, a recent proteomic analysis of intestinal tissue from healthy controls and patients with IBD, revealed upregulation of proteins related to NAD+ metabolism, including CD38, suggesting that CD38’s NADase activity contributes to modulate NAD+ levels during IBD. In this study, they also observed enrichment of CD38 protein expression in inflamed regions of colon mucosa from patients with UC as well as a colocalization of this protein with the marker of macrophages F4/80 (34). Although these findings point CD38 as a regulator of NAD+ during IBD, it is not clear how this may impact the development of disease or whether its activity as ADPR cyclase or cADPR hydrolase are also involved. Similarly, the receptor activity of CD38 may also modulate cell responses to induce inflammation however the vast absence of evidence doesn’t allow us to analyze this fact and indicates that more research is required to identify the mechanism driving inflammation by CD38 in the gut.
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disorder characterized by a relapsing and remitting clinical course (140) and autoantibodies against nuclear and cytoplasmic antigens (141, 142). Immunecomplex deposition in multiple organs such as the kidney and skin results in inflammation and tissue damage (143). An increase of CD38+ T cells, B cells and monocytes in the circulation of patients with SLE is observed early in SLE (114, 144–146). However, it has been difficult to assign a positive or negative role for CD38 in SLE development, since data supporting both pathogenic and regulatory functions have emerged.
Supporting a pathogenic role, CD38 expression in non-classical monocytes was recently linked to severe active SLE disease in a small group of patients (5), suggesting a potential role as a biomarker and/or a pathogenic role in the development of SLE. Some evidence of a pathogenic role has also been obtained from animal models. For example, CD38 deficiency suppressed SLE in the pristane-induced, type I IFN-dependent, murine lupus model. Clinical benefits were associated with reduction of anti-single-stranded DNA, anti-nuclear ribonucleoprotein (RNP) antibodies, glomerulonephritis, and type I IFN-stimulated gene expression. In this model, CD38 loss also decreased recruitment of-Ly6Clo/monocytes/macrophages and Ly6G+ neutrophils in the peritoneal cavity (10). This leads to the question of how CD38 may mediate these effects? One possibility is that these effects are mediated by activation of transient receptor potential melastatin (TRPM) 2. ADPR synthetized by CD38 can bind to and active the calcium-permeable channel TRPM2. After activation, the Ca2+ flux through TRPM2 induces signaling of cell death pathways that could lead to pristane-induced apoptosis of peritoneal cells, the primary source of autoantigens in this model (10). Another possibility is that CD38 deficiency improved lupus disease through an increase of IL-10-producing splenic B cells and reduction of plasmacytoid dendritic cells and IFN-α production in the peritoneal cavity (147).
While several lines of evidence suggest CD38 may promote SLE disease, evidence to the contrary has also emerged. For instance, CD38 deficiency in the Faslpr/Faslpr mouse model accelerated disease development while increasing kidney damage and circulating IgG. While the mechanism for disease protection is unclear, altered CD4+CD25+ and CD4+ single-positive thymocyte and CD8- dendritic cell proportions were observed in CD38 deficient mice. These observations suggest that CD38 modulation of T-cell and dendritic cell differentiation may suppress lupus autoimmunity (113). Another line of evidence suggesting regulatory roles of CD38 in B cells again originates from the lpr SLE model. High CD38 expression is observed in CD1dhi CD5+ regulatory B cells in this model. Finally, treatment with an agonistic anti-CD38 stimulus during LPS activation increased the percentage of regulatory B cells and, of relevance to regulatory function, their ability to produce IL-10 (115).
In summary, controversial data supporting both beneficial or pathogenic roles for CD38 in SLE disease and or animal models have arisen. These reports, however, clearly indicate that CD38 plays important roles in SLE and indicates the need for a better understanding of how CD38 contributes to immune cell physiology and SLE pathobiology.
Multiple Sclerosis
Multiple sclerosis (MS) is a chronic autoimmune disease of the central nervous system (CNS) that leads to multiple neurological signs, from sensory/cognitive deficits to motor disability (111, 148). MS is characterized by axonal demyelination as a consequence of the infiltration of circulating inflammatory cells into the CNS, including T cells, B cells, and myeloid cells (149, 150). Together, these cells induce tissue damage through secretion of soluble mediators and oxidative stressors (151, 152). The first evidence that CD38 may be involved in this disease resulted from efforts to identify pathogenic factors in the experimental autoimmune encephalomyelitis (EAE) model. CD38 was specifically induced during active myelin oligodendrocyte glycoprotein (MOG) in a rat model of EAE disease (111). To demonstrate the contribution of CD38 to pathogenesis, EAE disease was evaluated in CD38 deficient mice. CD38 deficiency reduced EAE severity, and this effect appears to be mediated by T and B cells (111). Supporting this idea, MOG-specific T cell responses were reduced in absence of CD38, indicating T-cell priming/proliferation defects. Humoral responses are also affected, as evidenced by decreased anti-MOG IgG autoantibody release in CD38 deficient mice (111).
The molecular mechanisms behind disease modulation are unclear, albeit NAD+ consumption by CD38 may be a contributing factor. During chronic CNS inflammation in mice, NAD+ levels are altered (153–155), promoting neuron demyelination (99, 153). Furthermore, it has been shown that the absence of CD38 significant increase NAD levels in mice brain (156, 157). Thus, as one of the main regulators of tissue NAD content, CD38 could have a profound impact on neuronal damage. Recently, the bis-cyclohexanone oxaldihydrazone (CPZ)-induced demyelination model revealed that CD38 deficiency suppresses glial activation, axonal damage, and demyelination. This effect was hypothesized to be mediated via enhanced levels of NAD+ (112). Under homeostatic conditions, astrocytic CD38 regulates murine astrocytes maturation and oligodendrocytes (OL) differentiation (158). However, under CPZ-induced demyelination, absence of CD38 impaired OL repopulation (112).
The expression of CD38 in other cells that contribute to MS pathogenesis has also been demonstrated (156). For instance CD38’s expression in human astrocytes positively regulates IL-6 and CCL2 production while CD38 deficiency in rat astrocytes increases H2O2-induced cell death (156, 159, 160). Similarly, activated microglial cells also express CD38. Here, CD38 positively regulates release of the proinflammatory cytokines IL-1β, TNF-α and IL-6 as it was demonstrated in a murine model of LPS-activated microglia (161).
Overall, these studies support a role for CD38 in MS physiopathology. However, more studies are needed to understand the cellular and molecular mechanisms behind CD38-mediated disease pathology.
Tools for CD38 Study: Successes and Remaining Challenges
Better understanding of the specific effects and mechanisms by which CD38 modulates inflammation and autoimmune disease will require specialized tools and resources. Fortunately, several tools have already been generated (Figure 3), with the potential to provide novel insights into CD38’s biology and mechanism in physiology and disease. These tools include recombinant proteins, monoclonal antibodies, inhibitors, transgenic and knockout mouse strains (12, 162–167). Currently available tools and models, as well as remaining challenges, are discussed below

Figure 3 Molecular tools to analyze CD38’s functions.
Recombinant Proteins
A number of CD38 molecular tools have been instrumental in our understanding of how CD38 expression is controlled or specific aspects of CD38’s enzymatic function/s. These tools include reporter systems carrying CD38 promoter sequences and recombinant wild-type or mutated CD38 proteins/peptides fragments (12, 168, 169).
Mutated CD38 proteins and fragments have provided insights into the CD38 domains mediating crucial enzymatic and CD31 binding activities. For example, in an effort to modulate CD38 enzymatic activity, site-directed mutagenesis studies of human CD38 revealed E226 as the essential residue for CD38’s catalytic activity. Substitution of E226 with D, N, Q, L, or G completely eliminates NADase and cADPR hydrolase activity. Instead, residue D155 was found to be crucial for cyclic ADP-ribosyl synthesis (12, 170). On the other hand, early evidence documented the intracellular localization of CD38. To determine whether intracellular CD38 has enzymatic activity, a soluble form of human CD38 that cannot localize to the membrane was engineered. Interestingly, this protein synthesized intracellular cADPR using cytosolic NAD as substrate. Also, mutations at E226, E146 and T221 in soluble CD38 confirmed that E226 is essential for cADPR production and revealed that E146A and T221F mutations in turn increase cADPR synthesis (168). Together these findings support the idea that intracellular CD38 is functionally active. As mention earlier, CD38 can take up type II or type III orientations on the cell membrane, depending on whether the C-terminal catalytic site of CD38 faces the extracellular or cytosolic compartment cytosol (171). However, the functional significance of type III orientation was not confirmed until recently thanks to a mouse type III CD38 construct. This tool revealed hydrolase/cyclase/enzymatic activity and the requirement of H2O2/Nox4 for intracellular cADPR generation. Additional type III CD38 mutant constructs revealed that C164 is an essential aa for cADPR synthesis and Ca2+ signaling (172). Defining the functional activity of type III CD38 solved the topological paradox of CD38, namely why CD38’s catalytic domain faces extracellularly while its NAD substrate is cytosolic and its cADPR product targets intracellular pathways. These are some examples of how recombinant protein strategies have substantially advanced our understanding of CD38 biology.
Luciferase CD38 reporters have also been useful to dissect the pathways and transcription factors controlling the transcriptional regulation of CD38 (173). The induction of CD38 following cell stimulation was analyzed following this approach using a putative CD38 gene promoter fragment cloned into the pGL3 luciferase vector. This approach identified the TNF-α-dependent binding of NF-κb, AP-1 and glucocorticoid receptor (GR) to the CD38 promoter, linking these transcription factors to CD38 expression (169). Without a doubt, this strategy is a powerful tool since identifying the elements controlling or inducing CD38 expression may shed light into why CD38 is linked to heterogeneous responses under similar immunological contexts.
CD38 Pharmacologic Inhibitors
Several molecules with CD38 inhibitor activity have been developed (98, 167, 174). These compounds bind to CD38’s active site, modulating its activity, and can be classified in three classes, namely NAD+ analogs, flavonoids, and heterocyclic compounds (25).
NAD+ analogs are thought to act as CD38 inhibitors via substrate competition. Specifically, the mechanism of action of these molecules involves competitive inhibition of NAD glycohydrolase activity (175). NAD+ analogs have low affinity to CD38 and can be subclassified into covalent and non-covalent inhibitors. Examples of NAD analogs include Ara-F-NAD, Ara-F-NFM, Ara-F-NMM, Carba-NAD, etc (167, 176). Unfortunately, these NAD+ derivatives are charged molecules with limited cell permeability, reducing their usefulness as CD38 modulatory tools (167).
Flavonoids are CD38 inhibitor compounds naturally produced by plants (174, 177). Flavonoid inhibitors include apigenin, quercetin, luteolinidin and 4,5-dihydroxyanthrquinone-2-carboxyl acid (Rhein) (174). Some CD38-flavonoid inhibitors act as competitive agonists with NAD, what results in increased NAD+ and NAD-dependent sirtuin activation (178). However, the mechanism of action of most flavonoid inhibitors is yet unknown.
Finally, several heterocyclic compounds act either as competitive or uncompetitive inhibitors of NADase activity via non-covalent reversible binding to CD38. These compounds are derived from 4-amino-quinoline and include 78c, 1ai, and Iah (19). Furthermore, they are positioned as better candidates than flavonoids and NAD+ analogs due to their specificity, improved pharmacokinetics, oral bioavailability and biological stability. The lead inhibitor 78c has demonstrated potent, specific and uncompetitive CD38 inhibitory activity. 78c preferentially inhibits CD38 NADase activity and is 10-fold less potent against cyclase than hydrolase activity. Interestingly, 78c rescued age-related NAD+ levels in tissues improving metabolism and physiological characteristics related to aging as glucose homeostasis, age-related skeletal muscle or cardiac dysfunction (98).
Regulation of CD38 Enzymatic Activity by Antibodies
CD38 is a diagnostic and prognostic biomarker in hematologic malignancies and multiple myeloma (MM) (179). As a consequence, several anti-CD38 monoclonal antibody (mAbs) therapies have been developed and are at either preclinical or clinical stages of drug development (18, 166, 180). Daratumumab (DARA) was the first fully humanized immunoglobulin G1 kappa mAb targeting CD38 but additional therapeutic antibodies such as isatuximab, MOR202 and TAK-079 have been developed. Although the anti-tumor activity mechanism is still under investigation, there is evidence that anti-CD38 mAbs can kill CD38-overexpressing cancer cells by multiple processes. These processes include complement-dependent cytotoxicity, antibody-dependent cellular cytotoxicity, antibody-dependent phagocytosis, tumor cells apoptosis and regulation of enzymatic activity of CD38 (181).
Besides their therapeutic activity in cancer, anti-CD38 mAbs may also provide interesting experimental tools to dissect CD38’s functions in other contexts. In vitro studies suggest that DARA could modulate CD38’s enzymatic activity (166, 180). In fact, a report indicates that DARA decreases cyclase activity while stimulating hydrolase function (166). However, these observations need further validation. Stronger evidence is available for Isatuximab as an inhibitor of CD38 enzymatic activity (182). In vitro, Isatuximab suppressed the production of cyclic GDPRibose (cGDPR) from NGD+ (183). In addition, Isatuximab treatment inhibited the hydrolase activity of CD38 in a dose-dependent manner (183).
Moreover, classical antibodies, nanobodies, and single domain antibodies against CD38 have been generated. These tools have improved CD38 binding and blocking capabilities while providing improved tissue penetration over conventional antibodies. Interestingly, several nanobodies modulate CD38’s catalytic activity. Fumey W et al, identified three nanobodies (JK2, MU1067, and MU523) able to decrease CD38-dependent cGDPR synthesis while two others (WF14 and MU738) conversely enhance CD38-catalyzed synthesis of cADPR (165). Although the evidence is still limited, these findings suggest that nanobodies could be also used to target enzymatic activity of CD38. Nanobodies have also allowed to study type II vs Type III membrane orientation of CD38. Nanobodies recognizing two different epitopes within CD38’s C-terminal domain were fused to distinct luciferase fragments and expressed in the cellular cytosol. When both epitopes are recognized by the nanobodies, the two luciferase fragments are sufficiently close to produce luminescence. This strategy demonstrated that type III CD38 exists as a transmembrane protein with ADPR cyclase activity. In addition, it showed that type II CD38 activation is dependent on the interaction with cytosolic Ca2+ and integrin-binding protein 1 (CIB1) (17).
CD38 Animal Models
In 1998, the groups of Howard and Okamoto developed the first two CD38 deficient (CD38-/-) mouse models, allowing to analyze the in vivo role of CD38 (162, 163). Both models were generated on a C57Bl/6J genetic background, albeit through different genetic approaches. Okamoto’s model used a homologous recombination strategy that deleted CD38’s exon 1, resulting in loss of CD38 transcript and protein. On the other hand, Howard’s model was designed to delete exons 2 and 3 encoding CD38’s putative active site. Both CD38 deficient mice revealed alterations consistent with the loss of CD38’s enzymatic activity. For instance, Okamoto’s model revealed impaired intracellular calcium and cADPR in pancreatic cells (163) while Howard’s model shown reduced NAD+ glycohydrolase activity in liver, spleen, and brain (162).
CD38-/- mice have been instrumental to our understanding of the enzymatic activity of CD38, reveling its multiple roles as NADase, ADPR cyclase, and cADPR hydrolase, as well as its contribution to regulate intracellular calcium mobilization/signaling through the second messenger cADPR (70). Furthermore, additional functions of CD38 in signaling, adhesion and cell migration have been also identified through these models (70, 71). CD38-/- mice have also been excellent tools to demonstrate the crucial role of CD38 in immunity to L. monocytogenes (45), S. pneumoniae (68), M. avium (184), S. Thyphimurium (89), and E. histolytica (66). Although most of these findings have been reported in immune cells the contribution of CD38 to physiology of non-immune cells have been also target of study utilizing these constitutive CD38-/- mice. For instance, CD38 deficient pancreatic beta cells displayed impaired glucose-induced cADPR production and Ca2+ signaling, resulting in impaired insulin secretion. Similarly, CD38 was also reported to mediate Ca2+ signaling and activation of hepatic stellate cells that contribute to liver fibrosis (185).
Besides single CD38 knock-out (KO) mice on the B6 background, the CD38 mutation has been combined with other genetic backgrounds or other gene KOs. For a variety of reasons, these models have provided information on CD38’s impact on inflammatory and autoimmune processes. Due to the observed effects of CD38 on pancreatic beta cell biology, the CD38 KO mutation was crossed onto the autoimmune diabetes-prone NOD/Lt background. The resulting NOD-CD38-/- mice suffer from accelerated type 1 diabetes development as consequence of NAD-induced apoptosis in T cells (164). ADP-ribosyltransferase 2 (ART2) is an ectoenzyme that catalyzes the transfer of ADP-ribose group from NAD+ to target proteins such as P2X7, a purinoreceptor that elicits T cell apoptosis (186). Hence, the double deficient NOD-CD38-/-ART2-/- mice were generated demonstrating that accelerated diabetes observed in NOD-CD38-/- mice was dependent of ART2 activity to mediate T cell apoptosis through P2X7 (164). Similarly, ART2-/-CD38-/- and TRPM2-/-CD38-/- double KO mice were used to study the contribution of CD38 to pristane-induced lupus. This approach demonstrated that Ly6Chi monocyte recruitment to inflamed tissues requires CD38 expression. In addition, these experiments provided evidence that CD38 promotes apoptosis of monocyte/macrophages in a TRMP2-dependent manner during experimental lupus (10).
Double-KO strategies have also shed light on the contribution of CD38 to aging-associated inflammation and immune responses against pathogens. During aging, low-grade inflammation, known as inflammaging, and a decline in NAD+ levels is observed (23). Interestingly, NAD levels are restored in absence of CD38 and this induces an increase of SIRT3 activity, a mitochondrial enzyme that uses NAD+ as a substrate. In order to determine to what extent CD38 contributes to inflammaging, a CD38-/-SIRT3-/- double KO mice model was developed. This model revealed that CD38’s NADase activity induces an age-related NAD decline that drives mitochondrial dysfunction through SIRT3, as SIRT3 deletion in CD38-/- mice reversed the effects observed in this model (97). Similarly, the contribution of CD38 to immune responses against pathogens was analyzed by Partida-Sanchez et al. taking advantage of crossed CD38-/- mice onto the lymphocyte-deficient Rag2-/- background. This strategy demonstrated that myeloid cells expressing CD38 are responsible for controlling S. pneumoniae infection and dissemination (70).
Constitutive CD38-/- mouse models have provided really valuable information on CD38 biology. However, given the ubiquitous expression and multifunctional nature of CD38 molecule, these models have the disadvantage of impacting many tissues and CD38 functions at the same time. Thereby, the ability to generate tissue specific CD38-deficient models or suppress specifically one of CD38 function is essential to understanding CD38 physiology in vivo. With this in mind, mice with LoxP flanking of exon 2 and 3 of the CD38 gene were recently generated to induce specific deletions of CD38 in airway smooth muscle cells (187). This approach helped describe how CD38 regulates hypoxia-induced apoptosis by SIRT1 and the p53 signaling pathway (187). At the same time, this conditional KO model opens many opportunities to analyze CD38’s tissue-specific roles. Regarding functional activity of CD38, Tarragó et al. developed a promising Knock-in mouse to elucidate the role of CD38 in age-related metabolic dysfunction mediated by NAD+ decline. This model eliminated CD38 catalytic activity by the punctual mutation E230Q in the CD38 gene. Catalytically inactive CD38 (CD38-CI) mice preserved CD38 protein expression but had null CD38 NADase activity thus ensuring specific analysis of CD38 at the enzymatic level (98).
Overall, the animal models discussed in this section provide evidence of the ever-expanding array of biological tools available to analyze the function of CD38 in vivo and its impact in autoimmune and immune mediated diseases.
Concluding Remarks
Constant efforts to understand CD38’s biology have allowed us to improve our knowledge of this complex molecule. CD38 was originally identified as a protein expressed on the surface of T cells but nowadays, we know that CD38’s function goes beyond a single cell or serving as a simple molecular marker. Indeed, CD38 plays critical roles in human physiology by modulating homeostasis, inflammation, or autoimmune responses in our body. Hence, CD38 has been proposed as a prognostic marker in some pathologies and several therapeutic anti-CD38 monoclonal antibodies are currently being developed for the treatment of malignancies such as melanoma. However, the ubiquitous localization of CD38, its multiple functions, and its dual membrane orientation represent challenges for mechanistically understanding the contribution of CD38 to health and disease. As a consequence, several outstanding questions remain. For example, it is unknown how CD38 is able to mediate regulatory and pathogenic responses in the same disease. Similarly, it is not completely understood which cellular events are triggered by each of CD38’s functions as well as what are their biological consequences. Some molecular tools have been developed that could help to partially answer these questions; however, more novel strategies are needed to be able to analyze and modulate each of CD38’s functions independently. Moreover, there are several immune cells, tissues and diseases where the role of CD38 has not been characterized. In summary, we have witnessed great progress in our knowledge of CD38 biology, and there is still much more to learn about this fascinating protein.
Author Contributions
ZP-Q and MG-d-A conceptualized the manuscript, reviewed literature, and wrote the review. PN contributed ideas and wrote the review. ZW reviewed the literature. ZP-Q and ZW designed figures and table. All authors contributed to the article and approved the submitted version.
Funding
MG-d-A is supported by funds from the NIH National Institute of Allergy and Infectious Diseases grants 1R21AI127354 by R21AI127354; 1R03AI151769 by R03AI151769; American Association of Immunologists, Careers in immunology by Association of Immunologists-Careers in immunology; subaward GRT00050231 by R01AI137525.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Albert Zlotnik for the critical comments on the review.
References
1. Deterre P, Berthelier V, Bauvois B, Dalloul A, Schuber F, Lund F. CD38 in T- and B-Cell Functions. Hum CD38 Relat Mol (2000) 75:146–68. doi: 10.1159/000058767
2. Morra M, Zubiaur M, Terhorst C, Sancho J, Malavasi F. CD38 is functionally dependent on the TCR/CD3 complex in human T cells. FASEB J (1998) 12:581–92. doi: 10.1096/fasebj.12.7.581
3. Zambello R, Barilà G, Manni S, Piazza F, Semenzato G. NK cells and CD38: Implication for (Immuno)Therapy in Plasma Cell Dyscrasias. Cells (2020) 9:768. doi: 10.3390/cells9030768
4. Fedele G, Frasca L, Palazzo R, Ferrero E, Malavasi F, Ausiello CM. CD38 is expressed on human mature monocyte-derived dendritic cells and is functionally involved in CD83 expression and IL-12 induction. Eur J Immunol (2004) 34:1342–50. doi: 10.1002/eji.200324728
5. Amici SA, Young NA, Narvaez-Miranda J, Jablonski KA, Arcos J, Rosas L, et al. CD38 Is Robustly Induced in Human Macrophages and Monocytes in Inflammatory Conditions. Front Immunol (2018) 9:1–13. doi: 10.3389/fimmu.2018.01593
6. Fujita T, Kantarci A, Warbington ML, Zawawi KH, Hasturk H, Kurihara H, et al. CD38 Expression in Neutrophils From Patients With Localized Aggressive Periodontitis. J Periodontol (2005) 76:1960–5. doi: 10.1902/jop.2005.76.11.1960
7. Shubinsky G, Schlesinger M. The CD38 Lymphocyte Differentiation Marker: New Insight into Its Ectoenzymatic Activity and Its Role as a Signal Transducer. Immunity (1997) 7:315–24. doi: 10.1016/S1074-7613(00)80353-2
8. Sandoval-Montes C, Santos-Argumedo L. CD38 is expressed selectively during the activation of a subset of mature T cells with reduced proliferation but improved potential to produce cytokines. J Leukoc Biol (2005) 77:513–21. doi: 10.1189/jlb.0404262
9. Choe C, Lardong K, Gelderblom M, Ludewig P, Leypoldt F, Koch-Nolte F, et al. CD38 Exacerbates Focal Cytokine Production, Postischemic Inflammation and Brain Injury after Focal Cerebral Ischemia. PloS One (2011) 6:e19046. doi: 10.1371/journal.pone.0019046
10. García-Rodríguez S, Rosal-Vela A, Botta D, Cumba Garcia LM, Zumaquero E, Prados-Maniviesa V, et al. CD38 promotes pristane-induced chronic inflammation and increases susceptibility to experimental lupus by an apoptosis-driven and TRPM2-dependent mechanism. Sci Rep (2018) 8:3357. doi: 10.1038/s41598-018-21337-6
11. Frasca L, Fedele G, Deaglio S, Capuano C, Palazzo R, Vaisitti T, et al. CD38 orchestrates migration, survival, and Th1 immune response of human mature dendritic cells. Blood (2006) 107:2392–9. doi: 10.1182/blood-2005-07-2913
12. Munshi C, Aarhus R, Graeff R, Walseth TF, Levitt D, Lee HC. Identification of the enzymatic active site of CD38 by site-directed mutagenesis. J Biol Chem (2000) 275:21566–71. doi: 10.1074/jbc.M909365199
13. States DJ, Walseth TF, Lee HC. Similarities in amino acid sequences of Aplysia ADP-ribosyl cyclase and human lymphocyte antigen CD38. Trends Biochem Sci (1992) 17:495. doi: 10.1016/0968-0004(92)90337-9
14. Glaría E, Valledor AF. Roles of CD38 in the Immune Response to Infection. Cells (2020) 9:1–15. doi: 10.3390/cells9010228
15. Santos-Argumedo L. CD38. In: Choi S, editor. Encyclopedia of Signaling Molecules. Cham: Springer International Publishing (2018). p. 869–77. doi: 10.1007/978-3-319-67199-4_278
16. Muñoz P, Mittelbrunn M, de la Fuente H, Pérez-Martínez M, García-Pérez A, Ariza-Veguillas A, et al. Antigen-induced clustering of surface CD38 and recruitment of intracellular CD38 to the immunologic synapse. Blood (2008) 111:3653–64. doi: 10.1182/blood-2007-07-101600
17. Liu J, Zhao YJ, Li WH, Hou YN, Li T, Zhao ZY, et al. Cytosolic interaction of type III human CD38 with CIB1 modulates cellular cyclic ADP-ribose levels. Proc Natl Acad Sci (2017) 114:8283–8. doi: 10.1073/pnas.1703718114
18. van de Donk NWCJ, Richardson PG, Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood (2018) 131:13–29. doi: 10.1182/blood-2017-06-740944
19. Chini EN, Chini CCS, Netto JME, de Oliveira GC, van Schooten W. The Pharmacology of CD38/NADase: An emerging target for cancer and aging diseases. Trends Pharmacol Sci (2018) 39:424–36. doi: 10.1016/j.tips.2018.02.001
20. Zhao YJ, Lam CMC, Lee HC. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci Signal (2012) 5:ra67. doi: 10.1126/scisignal.2002700
21. Franco L, Guida L, Bruzzone S, Zocchi E, Usai C, De Flora A. The transmembrane glycoprotein CD38 is a catalytically active transporter responsible for generation and influx of the second messenger cyclic ADP-ribose across membranes. FASEB J Off Publ Fed Am Soc Exp Biol (1998) 12:1507–20. doi: 10.1096/fasebj.12.14.1507
22. Lee HC, Zhao YJ. Resolving the topological enigma in Ca2+ signaling by cyclic ADP-ribose and NAADP. J Biol Chem (2019) 294:19831–43. doi: 10.1074/jbc.REV119.009635
23. Okabe K, Yaku K, Tobe K, Nakagawa T. Implications of altered NAD metabolism in metabolic disorders. J BioMed Sci (2019) 26:1–13. doi: 10.1186/s12929-019-0527-8
24. Wei W, Graeff R, Yue J. Roles and mechanisms of the CD38/cyclic adenosine diphosphate ribose/Ca2+ signaling pathway. World J Biol Chem (2014) 5:58–67. doi: 10.4331/wjbc.v5.i1.58
25. Hogan KA, Chini CCS, Chini EN. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front Immunol (2019) 10:1–12. doi: 10.3389/fimmu.2019.01187
26. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and Function of the ADP Ribosyl Cyclase/CD38 Gene Family in Physiology and Pathology. Physiol Rev (2008) 88:841–86. doi: 10.1152/physrev.00035.2007
27. Deaglio S, Morra M, Mallone R, Ausiello CM, Prager E, Garbarino G, et al. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J Immunol Baltim Md 1950 (1998) 160:395–402.
28. Deaglio S, Dianzani U, Horenstein AL, Fernández JE, van Kooten C, Bragardo M, et al. Human CD38 ligand. A 120-KDA protein predominantly expressed on endothelial cells. J Immunol (1996) 156:727–34.
29. Shay T, Jojic V, Zuk O, Rothamel K, Puyraimond-Zemmour D, Feng T, et al. Conservation and divergence in the transcriptional programs of the human and mouse immune systems. Proc Natl Acad Sci (2013) 110:2946–51. doi: 10.1073/pnas.1222738110
30. Brynjolfsson SF, Berg LP, Ekerhult TO, Rimkute I, Wick M-J, Mårtensson I-L, et al. Long-Lived Plasma Cells in Mice and Men. Front Immunol (2018) 9:1–7. doi: 10.3389/fimmu.2018.02673
31. Deaglio S, Mehta K, Malavasi F. Human CD38: a (r)evolutionary story of enzymes and receptors. Leuk Res (2001) 25:1–12. doi: 10.1016/S0145-2126(00)00093-X
32. Fernàndez JE, Deaglio S, Donati D, Beusan IS, Corno F, Aranega A, et al. Analysis of the distribution of human CD38 and of its ligand CD31 in normal tissues. J Biol Regul Homeost Agents (1998) 12:81–91.
33. Schneider M, Schumacher V, Lischke T, Lücke K, Meyer-Schwesinger C, Velden J, et al. CD38 Is Expressed on Inflammatory Cells of the Intestine and Promotes Intestinal Inflammation. PloS One (2015) 10:e0126007. doi: 10.1371/journal.pone.0126007
34. Ning L, Shan G, Sun Z, Zhang F, Xu C, Lou X, et al. Quantitative Proteomic Analysis Reveals the Deregulation of Nicotinamide Adenine Dinucleotide Metabolism and CD38 in Inflammatory Bowel Disease. BioMed Res Int (2019) 2019:3950628. doi: 10.1155/2019/3950628
35. Tissue expression of CD38 - Summary - The Human Protein Atlas. Available at: https://www.proteinatlas.org/ENSG00000004468-CD38/tissue (Accessed August 4, 2020).
36. Gene Skyline. Available at: http://rstats.immgen.org/Skyline_microarray/skyline.html (Accessed August 4, 2020).
37. Clavarino G, Delouche N, Vettier C, Laurin D, Pernollet M, Raskovalova T, et al. Novel Strategy for Phenotypic Characterization of Human B Lymphocytes from Precursors to Effector Cells by Flow Cytometry. PloS One (2016) 11:1–16. doi: 10.1371/journal.pone.0162209
38. Vences-Catalán F, Santos-Argumedo L. CD38 through the life of a murine B lymphocyte. IUBMB Life (2011) 63:840–6. doi: 10.1002/iub.549
39. Mestas J, Hughes CCW. Of Mice and Not Men: Differences between Mouse and Human Immunology. J Immunol (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
40. Bean AGD, Godfrey DI, Ferlin WG, Santos-Argumedo L, Parkhouse RME, Howard MC, et al. CD38 expression on mouse T cells: CD38 defines functionally distinct subsets of αβ TCR+CD4–CD8–thymocytes. Int Immunol (1995) 7:213–21. doi: 10.1093/intimm/7.2.213
41. Tenca C, Merlo A, Zarcone D, Saverino D, Bruno S, De Santanna A, et al. Death of T cell precursors in the human thymus: a role for CD38. Int Immunol (2003) 15:1105–16. doi: 10.1093/intimm/dxg111
42. Bahri R, Bollinger A, Bollinger T, Orinska Z, Bulfone-Paus S. Ectonucleotidase CD38 Demarcates Regulatory, Memory-Like CD8+ T Cells with IFN-γ-Mediated Suppressor Activities. PloS One (2012) 7:e45234. doi: 10.1371/journal.pone.0045234
43. Funaro A, Morra M, Calosso L, Zini MG, Ausiello CM, Malavasi F. Role of the human CD38 molecule in B cell activation and proliferation. Tissue Antigens (1997) 49:7–15. doi: 10.1111/j.1399-0039.1997.tb02703.x
44. Deaglio S, Zubiaur M, Gregorini A, Bottarel F, Ausiello CM, Dianzani U, et al. Human CD38 and CD16 are functionally dependent and physically associated in natural killer cells. Blood (2002) 99:2490–8. doi: 10.1182/blood.v99.7.2490
45. Lischke T, Heesch K, Schumacher V, Schneider M, Haag F, Koch-Nolte F, et al. CD38 Controls the Innate Immune Response against Listeria monocytogenes. Infect Immun (2013) 81:4091–9. doi: 10.1128/IAI.00340-13
46. Wykes MN, Beattie L, MacPherson GG, Hart DN. Dendritic cells and follicular dendritic cells express a novel ligand for CD38 which influences their maturation and antibody responses. Immunology (2004) 113:318–27. doi: 10.1111/j.1365-2567.2004.01968.x
47. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget (2017) 9:7204–18. doi: 10.18632/oncotarget.23208
48. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
49. Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol (2010) 10:826–37. doi: 10.1038/nri2873
50. Ibrahim IBM, Pidaparti R. Influence of Pathogens and Mechanical Stimuli in Inflammation. Bioengineering (2019) 6:1–12. doi: 10.3390/bioengineering6020055
51. Roh JS, Sohn DH. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw (2018) 18:1–14. doi: 10.4110/in.2018.18.e27
52. Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front Immunol (2018) 9:1–19. doi: 10.3389/fimmu.2018.02379
53. Astudillo AM, Pérez-Chacón G, Balboa MA, Balsinde J. Arachidonic acid mobilization by stimuli of the innate immune response. Inmunología (2009) 28:182–92. doi: 10.1016/S0213-9626(09)70041-1
54. Ricciotti E, FitzGerald GA. Prostaglandins and Inflammation. Arterioscler Thromb Vasc Biol (2011) 31:986–1000. doi: 10.1161/ATVBAHA.110.207449
55. Newton K, Dixit VM. Signaling in Innate Immunity and Inflammation. Cold Spring Harb Perspect Biol (2012) 4:1–19. doi: 10.1101/cshperspect.a006049
56. Nowarski R, Gagliani N, Huber S, Flavell RA. Innate immune cells in inflammation and cancer. Cancer Immunol Res (2013) 1:77–84. doi: 10.1158/2326-6066.CIR-13-0081
57. Prame Kumar K, Nicholls AJ. Wong CHY. Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res (2018) 371:551–65. doi: 10.1007/s00441-017-2753-2
58. Gaudino SJ, Kumar P. Cross-Talk Between Antigen Presenting Cells and T Cells Impacts Intestinal Homeostasis, Bacterial Infections, and Tumorigenesis. Front Immunol (2019) 10:360. doi: 10.3389/fimmu.2019.00360
59. Duan L, Rao X, Sigdel KR. Regulation of Inflammation in Autoimmune Disease. J Immunol Res (2019) 2019:1–2. doi: 10.1155/2019/7403796
60. Musso T, Deaglio S, Franco L, Calosso L, Badolato R, Garbarino G, et al. CD38 expression and functional activities are up-regulated by IFN-gamma on human monocytes and monocytic cell lines. J Leukoc Biol (2001) 69:605–12. doi: 10.1189/jlb.69.4.605
61. Bauvois B, Durant L, Laboureau J, Barthelemy E, Rouillard D, Boulla G, et al. Upregulation of CD38 Gene Expression in Leukemic B Cells by Interferon Types I and II. J Interferon Cytokine Res (1999) 19:1059–66. doi: 10.1089/107999099313299
62. Schiavoni I, Scagnolari C, Horenstein AL, Leone P, Pierangeli A, Malavasi F, et al. CD38 modulates respiratory syncytial virus-driven proinflammatory processes in human monocyte-derived dendritic cells. Immunology (2018) 154:122–31. doi: 10.1111/imm.12873
63. Kang B-N, Tirumurugaan KG, Deshpande DA, Amrani Y, Panettieri RA, Walseth TF, et al. Transcriptional regulation of CD38 expression by tumor necrosis factor-alpha in human airway smooth muscle cells: role of NF-kappaB and sensitivity to glucocorticoids. FASEB J Off Publ Fed Am Soc Exp Biol (2006) 20:1000–2. doi: 10.1096/fj.05-4585fje
64. Muller WA. Getting Leukocytes to the Site of Inflammation. Vet Pathol (2013) 50:7–22. doi: 10.1177/0300985812469883
65. Teixidó J, Hidalgo A, Fagerholm S. Editorial: Leukocyte Trafficking in Homeostasis and Disease. Front Immunol (2019) 10:2560. doi: 10.3389/fimmu.2019.02560
66. Estrada-Figueroa LA, Ramírez-Jiménez Y, Osorio-Trujillo C, Shibayama M, Navarro-García F, García-Tovar C, et al. Absence of CD38 delays arrival of neutrophils to the liver and innate immune response development during hepatic amoebiasis by Entamoeba histolytica. Parasite Immunol (2011) 33:661–8. doi: 10.1111/j.1365-3024.2011.01333.x
67. Petri B, Bixel MG. Molecular events during leukocyte diapedesis. FEBS J (2006) 273:4399–407. doi: 10.1111/j.1742-4658.2006.05439.x
68. Partida-Sánchez S, Randall TD, Lund FE. Innate immunity is regulated by CD38, an ecto-enzyme with ADP-ribosyl cyclase activity. Microbes Infect (2003) 5:49–58. doi: 10.1016/s1286-4579(02)00055-2
69. Cremer J, Honda T, Tang Y, Wong-Ng J, Vergassola M, Hwa T. Chemotaxis as a navigation strategy to boost range expansion. Nature (2019) 575:658–63. doi: 10.1038/s41586-019-1733-y
70. Partida-Sánchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med (2001) 7:1209–16. doi: 10.1038/nm1101-1209
71. Partida-Sánchez S, Goodrich S, Kusser K, Oppenheimer N, Randall TD, Lund FE. Regulation of dendritic cell trafficking by the ADP-ribosyl cyclase CD38: impact on the development of humoral immunity. Immunity (2004) 20:279–91. doi: 10.1016/s1074-7613(04)00048-2
72. Mele S, Devereux S, Pepper AG, Infante E, Ridley AJ. Calcium-RasGRP2-Rap1 signaling mediates CD38-induced migration of chronic lymphocytic leukemia cells. Blood Adv (2018) 2:1551–61. doi: 10.1182/bloodadvances.2017014506
73. Rosales C, Uribe-Querol E. Phagocytosis: A Fundamental Process in Immunity. BioMed Res Int (2017) 2017:1–18. doi: 10.1155/2017/9042851
74. Nunes P, Demaurex N. The role of calcium signaling in phagocytosis. J Leukoc Biol (2010) 88:57–68. doi: 10.1189/jlb.0110028
75. Pradhan G, Raj Abraham P, Shrivastava R, Mukhopadhyay S. Calcium Signaling Commands Phagosome Maturation Process. Int Rev Immunol (2019) 38:57–69. doi: 10.1080/08830185.2019.1592169
76. Kang J, Park K-H, Kim J-J, Jo E-K, Han M-K, Kim U-H. The Role of CD38 in Fcγ Receptor (FcγR)-mediated Phagocytosis in Murine Macrophages. J Biol Chem (2012) 287:14502–14. doi: 10.1074/jbc.M111.329003
77. Schulz D, Severin Y, Zanotelli VRT, Bodenmiller B. In-Depth Characterization of Monocyte-Derived Macrophages using a Mass Cytometry-Based Phagocytosis Assay. Sci Rep (2019) 9:1925. doi: 10.1038/s41598-018-38127-9
78. Kotsias F, Cebrian I, Alloatti A. Antigen processing and presentation. Int Rev Cell Mol Biol (2019) 348:69–121. doi: 10.1016/bs.ircmb.2019.07.005
79. Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol (2015) 15:203–16. doi: 10.1038/nri3818
80. Onnis A, Baldari CT. Orchestration of Immunological Synapse Assembly by Vesicular Trafficking. Front Cell Dev Biol (2019) 7:110:110. doi: 10.3389/fcell.2019.00110
81. Zilber M-T, Setterblad N, Vasselon T, Doliger C, Charron D, Mooney N, et al. MHC class II/CD38/CD9: a lipid-raft-dependent signaling complex in human monocytes. Blood (2005) 106:3074–81. doi: 10.1182/blood-2004-10-4094
82. Reinis M, Morra M, Funaro A, Di Primio R, Malavasi F. Functional associations of CD38 with CD3 on the T-cell membrane. J Biol Regul Homeost Agents (1997) 11:137–42.
83. Rocha-Perugini V, Martínez del Hoyo G, González-Granado JM, Ramírez-Huesca M, Zorita V, Rubinstein E, et al. CD9 Regulates Major Histocompatibility Complex Class II Trafficking in Monocyte-Derived Dendritic Cells. Mol Cell Biol (2017) 37:1–17. doi: 10.1128/MCB.00202-17
84. Zilber M-T, Gregory S, Mallone R, Deaglio S, Malavasi F, Charron D, et al. CD38 expressed on human monocytes: A coaccessory molecule in the superantigen-induced proliferation. Proc Natl Acad Sci U.S.A. (2000) 97:2840–5. doi: 10.1073/pnas.050583197
85. Laufer JM, Kindinger I, Artinger M, Pauli A, Legler DF. CCR7 Is Recruited to the Immunological Synapse, Acts as Co-stimulatory Molecule and Drives LFA-1 Clustering for Efficient T Cell Adhesion Through ZAP70. Front Immunol (2018) 9:3115. doi: 10.3389/fimmu.2018.03115
86. Rocha-Perugini V, Zamai M, González-Granado JM, Barreiro O, Tejera E, Yañez-Mó M, et al. CD81 controls sustained T cell activation signaling and defines the maturation stages of cognate immunological synapses. Mol Cell Biol (2013) 33:3644–58. doi: 10.1128/MCB.00302-13
87. van Spriel AB, Leusen JH, van Egmond M, Dijkman HB, Assmann KJ, Mayadas TN, et al. Mac-1 (CD11b/CD18) is essential for Fc receptor-mediated neutrophil cytotoxicity and immunologic synapse formation. Blood (2001) 97:2478–86. doi: 10.1182/blood.v97.8.2478
88. Lacy P, Stow JL. Cytokine release from innate immune cells: association with diverse membrane trafficking pathways. Blood (2011) 118:9–18. doi: 10.1182/blood-2010-08-265892
89. Matalonga J, Glaria E, Bresque M, Escande C, Carbó JM, Kiefer K, et al. The Nuclear Receptor LXR Limits Bacterial Infection of Host Macrophages through a Mechanism that Impacts Cellular NAD Metabolism. Cell Rep (2017) 18:1241–55. doi: 10.1016/j.celrep.2017.01.007
90. Li Q, Wu C, Liu Z, Zhang H, Du Y, Liu Y, et al. Increased TLR4 Expression Aggravates Sepsis by Promoting IFN- γ Expression in CD38 –/– Mice. J Immunol Res (2019) 2019:1–12. doi: 10.1155/2019/3737890
91. Qian Y, Chen C, Ma L, Wang Z, Wang L-F, Zuo L, et al. CD38 Deficiency Promotes Inflammatory Response through Activating Sirt1/NF- κ B-Mediated Inhibition of TLR2 Expression in Macrophages. Mediators Inflammation (2018) 2018:1–13. doi: 10.1155/2018/8736949
92. Sultani G, Samsudeen AF, Osborne B, Turner N. NAD+: A key metabolic regulator with great therapeutic potential. J Neuroendocrinol (2017) 29:e12508. doi: 10.1111/jne.12508
93. Minhas PS, Liu L, Moon PK, Joshi AU, Dove C, Mhatre S, et al. Macrophage de novo NAD + synthesis specifies immune function in aging and inflammation. Nat Immunol (2019) 20:50–63. doi: 10.1038/s41590-018-0255-3
94. Chini CCS, Tarragó MG, Chini EN. NAD and the aging process: Role in life, death and everything in between. Mol Cell Endocrinol (2017) 455:62–74. doi: 10.1016/j.mce.2016.11.003
95. Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science (2015) 350:1208–13. doi: 10.1126/science.aac4854
96. Johnson S, Imai S. NAD + biosynthesis, aging, and disease. F1000Research (2018) 7:1–10. doi: 10.12688/f1000research.12120.1
97. Camacho-Pereira J, Tarragó MG, Chini CCS, Nin V, Escande C, Warner GM, et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab (2016) 23:1127–39. doi: 10.1016/j.cmet.2016.05.006
98. Tarragó MG, Chini CCS, Kanamori KS, Warner GM, Caride A, de Oliveira GC, et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD+ Decline. Cell Metab (2018) 27:1081–95.e10. doi: 10.1016/j.cmet.2018.03.016
99. Tullius SG, Biefer HRC, Li S, Trachtenberg AJ, Edtinger K, Quante M, et al. NAD+ protects against EAE by regulating CD4+ T-cell differentiation. Nat Commun (2014) 5:5101. doi: 10.1038/ncomms6101
100. Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H, et al. NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab (2016) 24:566–81. doi: 10.1016/j.cmet.2016.09.004
101. Atri C, Guerfali FZ, Laouini D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int J Mol Sci (2018) 19:1–15. doi: 10.3390/ijms19061801
102. Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol (2013) 14:454–60. doi: 10.1038/ni.2550
103. Scarfì S, Ferraris C, Fruscione F, Fresia C, Guida L, Bruzzone S, et al. Cyclic ADP-ribose-mediated expansion and stimulation of human mesenchymal stem cells by the plant hormone abscisic acid. Stem Cells Dayt Ohio (2008) 26:2855–64. doi: 10.1634/stemcells.2008-0488
104. Ng LG, Qin JS, Roediger B, Wang Y, Jain R, Cavanagh LL, et al. Visualizing the Neutrophil Response to Sterile Tissue Injury in Mouse Dermis Reveals a Three-Phase Cascade of Events. J Invest Dermatol (2011) 131:2058–68. doi: 10.1038/jid.2011.179
105. Yang D, Elner SG, Chen X, Field MG, Petty HR, Elner VM. MCP-1–Activated Monocytes Induce Apoptosis in Human Retinal Pigment Epithelium. Invest Ophthalmol Vis Sci (2011) 52:6026–34. doi: 10.1167/iovs.10-7023
106. Antonelli A, Ferrannini E. CD38 autoimmunity: recent advances and relevance to human diabetes. J Endocrinol Invest (2004) 27:695–707. doi: 10.1007/BF03347507
107. Anaya J-M, Ramirez-Santana C, Alzate MA, Molano-Gonzalez N, Rojas-Villarraga A. The Autoimmune Ecology. Front Immunol (2016) 7:1–31. doi: 10.3389/fimmu.2016.00139
108. Rahman S, Archana A, Jan AT, Dutta D, Shankar A, Kim J, et al. Molecular Insights Into the Relationship Between Autoimmune Thyroid Diseases and Breast Cancer: A Critical Perspective on Autoimmunity and ER Stress. Front Immunol (2019) 10:1–18. doi: 10.3389/fimmu.2019.00344
109. du Pré MF, van Berkel LA, Ráki M, van Leeuwen MA, de Ruiter LF, Broere F, et al. CD62LnegCD38+ expression on circulating CD4+ T cells identifies mucosally differentiated cells in protein fed mice and in human celiac disease patients and controls. Am J Gastroenterol (2011) 106:1147–59. doi: 10.1038/ajg.2011.24
110. Funderburg NT, Stubblefield Park SR, Sung HC, Hardy G, Clagett B, Ignatz-Hoover J, et al. Circulating CD4+ and CD8+ T cells are activated in inflammatory bowel disease and are associated with plasma markers of inflammation. Immunology (2013) 140:87–97. doi: 10.1111/imm.12114
111. Herrmann MM, Barth S, Greve B, Schumann KM, Bartels A, Weissert R. Identification of gene expression patterns crucially involved in experimental autoimmune encephalomyelitis and multiple sclerosis. Dis Model Mech (2016) 9:1211–20. doi: 10.1242/dmm.025536
112. Roboon J, Hattori T, Ishii H, Takarada-Iemata M, Le TM, Shiraishi Y, et al. Deletion of CD38 Suppresses Glial Activation and Neuroinflammation in a Mouse Model of Demyelination. Front Cell Neurosci (2019) 13:1–13. doi: 10.3389/fncel.2019.00258
113. Viegas MS, Silva T, Monteiro MM, do Carmo A, Martins TC. Knocking out of CD38 accelerates development of a lupus-like disease in lpr mice. Rheumatology (2011) 50:1569–77. doi: 10.1093/rheumatology/ker178
114. Katsuyama E, Suarez-Fueyo A, Bradley SJ, Mizui M, Marin AV, Mulki L, et al. The CD38/NAD/SIRTUIN1/EZH2 Axis Mitigates Cytotoxic CD8 T Cell Function and Identifies Patients with SLE Prone to Infections. Cell Rep (2020) 30:112–23. doi: 10.1016/j.celrep.2019.12.014
115. Domínguez-Pantoja M, López-Herrera G, Romero-Ramírez H, Santos-Argumedo L, Chávez-Rueda AK, Hernández-Cueto Á, et al. CD38 protein deficiency induces autoimmune characteristics and its activation enhances IL-10 production by regulatory B cells. Scand J Immunol (2018) 87:e12664. doi: 10.1111/sji.12664
116. Pessler F, Dai L, Diaz-Torne C, Ogdie A, Gomez-Vaquero C, Paessler ME, et al. Increased angiogenesis and cellular proliferation as hallmarks of the synovium in chronic septic arthritis. Arthritis Care Res (2008) 59:1137–46. doi: 10.1002/art.23915
117. Smeets TJ, Dolhain RJ, Breedveld FC, Tak PP. Analysis of the cellular infiltrates and expression of cytokines in synovial tissue from patients with rheumatoid arthritis and reactive arthritis. J Pathol (1998) 186:75–81. doi: 10.1002/(SICI)1096-9896(199809)186:1<75::AID-PATH142>3.0.CO;2-B
118. Korver W, Carsillo M, Yuan J, Idamakanti N, Wagoner M, Shi P, et al. A Reduction in B, T, and Natural Killer Cells Expressing CD38 by TAK-079 Inhibits the Induction and Progression of Collagen-Induced Arthritis in Cynomolgus Monkeys. J Pharmacol Exp Ther (2019) 370:182–96. doi: 10.1124/jpet.119.256602
119. Wang H, Li S, Zhang G, Wu H, Chang X. Potential therapeutic effects of cyanidin-3-O-glucoside on rheumatoid arthritis by relieving inhibition of CD38+ NK cells on Treg cell differentiation. Arthritis Res Ther (2019) 21:220. doi: 10.1186/s13075-019-2001-0
120. Cole S, Walsh A, Yin X, Wechalekar MD, Smith MD, Proudman SM, et al. Integrative analysis reveals CD38 as a therapeutic target for plasma cell-rich pre-disease and established rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res Ther (2018) 20:85. doi: 10.1186/s13075-018-1578-z
121. Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res (2018) 6:1–14. doi: 10.1038/s41413-018-0016-9
122. Elkon K, Casali P. Nature and functions of autoantibodies. Nat Clin Pract Rheumatol (2008) 4:491–8. doi: 10.1038/ncprheum0895
123. Silverman GJ, Carson DA. Roles of B cells in rheumatoid arthritis. Arthritis Res Ther (2003) 5:S1–6. doi: 10.1186/ar1010
124. Mok CC. Rituximab for the treatment of rheumatoid arthritis: an update. Drug Des Devel Ther (2013) 8:87–100. doi: 10.2147/DDDT.S41645
125. Kraan MC, Haringman JJ, Post WJ, Versendaal J, Breedveld FC, Tak PP. Immunohistological analysis of synovial tissue for differential diagnosis in early arthritis. Rheumatology (1999) 38:1074–80. doi: 10.1093/rheumatology/38.11.1074
126. Soto H, Hevezi P, Roth RB, Pahuja A, Alleva D, Acosta HM, et al. Gene Array Analysis Comparison between Rat Collagen-induced Arthritis and Human Rheumatoid Arthritis. Scand J Immunol (2008) 68:43–57. doi: 10.1111/j.1365-3083.2008.02117.x
127. Pan W, Yue L, Wang Q, Wang Z, Wang X, Li S, et al. CD38 positive natural killer cells may be involved in progression of inflammation in rheumatoid arthritis patients. Int J Clin Exp Med (2017) 10(3):4696–704.
128. Chang X, Yue L, Liu W, Wang Y, Wang L, Xu B, et al. CD38 and E2F transcription factor 2 have uniquely increased expression in rheumatoid arthritis synovial tissues. Clin Exp Immunol (2014) 176:222–31. doi: 10.1111/cei.12268
129. Zacca ER, Onofrio LI, Acosta CDV, Ferrero PV, Alonso SM, Ramello MC, et al. PD-L1+ Regulatory B Cells Are Significantly Decreased in Rheumatoid Arthritis Patients and Increase After Successful Treatment. Front Immunol (2018) 9:1–13. doi: 10.3389/fimmu.2018.02241
130. Guan Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J Immunol Res (2019) 2019:1–16. doi: 10.1155/2019/7247238
131. Gassler N, Rohr C, Schneider A, Kartenbeck J, Bach A, Obermüller N, et al. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol-Gastrointest Liver Physiol (2001) 281:G216–28. doi: 10.1152/ajpgi.2001.281.1.G216
132. Balmus IM, Ciobica A, Trifan A, Stanciu C. The implications of oxidative stress and antioxidant therapies in Inflammatory Bowel Disease: Clinical aspects and animal models. Saudi J Gastroenterol (2016) 22:3. doi: 10.4103/1319-3767.173753
133. Laukoetter MG, Nava P, Nusrat A. Role of the intestinal barrier in inflammatory bowel disease. World J Gastroenterol WJG (2008) 14:401–7. doi: 10.3748/wjg.14.401
134. Gálvez J. Role of Th17 Cells in the Pathogenesis of Human IBD. ISRN Inflammation (2014) 2014:928461. doi: 10.1155/2014/928461
135. Feng T, Qin H, Wang L, Benveniste EN, Elson CO, Cong Y. Th17 Cells Induce Colitis and Promote Th1 Cell Responses through IL-17 Induction of Innate IL-12 and IL-23 Production. J Immunol (2011) 186:6313–8. doi: 10.4049/jimmunol.1001454
136. Sun X, He S, Lv C, Sun X, Wang J, Zheng W, et al. Analysis of murine and human Treg subsets in inflammatory bowel disease. Mol Med Rep (2017) 16:2893–8. doi: 10.3892/mmr.2017.6912
137. Deaglio S, Mallone R, Baj G, Donati D, Giraudo G, Corno F, et al. Human CD38 and its ligand CD31 define a unique lamina propria T lymphocyte signaling pathway. FASEB J (2001) 15:580–2. doi: 10.1096/fj.00-0522fje
138. Joosse ME, Menckeberg CL, de Ruiter LF, Raatgeep HRC, van Berkel LA, Simons-Oosterhuis Y, et al. Frequencies of circulating regulatory TIGIT+CD38+ effector T cells correlate with the course of inflammatory bowel disease. Mucosal Immunol (2019) 12:154–63. doi: 10.1038/s41385-018-0078-4
139. Gerner RR, Klepsch V, Macheiner S, Arnhard K, Adolph TE, Grander C, et al. NAD metabolism fuels human and mouse intestinal inflammation. Gut (2018) 67:1813–23. doi: 10.1136/gutjnl-2017-314241
140. Gergianaki I, Bertsias G. Systemic Lupus Erythematosus in Primary Care: An Update and Practical Messages for the General Practitioner. Front Med (2018) 5:1–12. doi: 10.3389/fmed.2018.00161
141. Fortuna G, Brennan MT. Systemic Lupus Erythematosus. Dent Clin North Am (2013) 57:631–55. doi: 10.1016/j.cden.2013.06.003
142. Tsokos GC. Systemic Lupus Erythematosus. N Engl J Med (2011) 12:2110–21. doi: 10.1056/NEJMra1100359
143. Moulton VR, Suarez-Fueyo A, Meidan E, Li H, Mizui M, Tsokos GC. Pathogenesis of Human Systemic Lupus Erythematosus: A Cellular Perspective. Trends Mol Med (2017) 23:615–35. doi: 10.1016/j.molmed.2017.05.006
144. al-Janadi M, Raziuddin S. B cell hyperactivity is a function of T cell derived cytokines in systemic lupus erythematosus. J Rheumatol (1993) 20:1885–91.
145. Wardowska A, Komorniczak M, Skoniecka A, Bułło-Piontecka B, Lisowska KA, Dębska-Ślizień MA, et al. Alterations in peripheral blood B cells in systemic lupus erythematosus patients with renal insufficiency. Int Immunopharmacol (2020) 83:106451. doi: 10.1016/j.intimp.2020.106451
146. Lugar PL, Love C, Grammer AC, Dave SS, Lipsky PE. Molecular characterization of circulating plasma cells in patients with active systemic lupus erythematosus. PloS One (2012) 7:e44362. doi: 10.1371/journal.pone.0044362
147. Burlock B, Richardson G, García-Rodríguez S, Guerrero S, Zubiaur M, Sancho J. The Role of CD38 on the Function of Regulatory B Cells in a Murine Model of Lupus. Int J Mol Sci (2018) 19:1–14. doi: 10.3390/ijms19102906
149. Dargahi N, Katsara M, Tselios T, Androutsou M-E, de Courten M, Matsoukas J, et al. Multiple Sclerosis: Immunopathology and Treatment Update. Brain Sci (2017) 7:1–27. doi: 10.3390/brainsci7070078
150. Wu GF, Alvarez E. The immunopathophysiology of multiple sclerosis. Neurol Clin (2011) 29:257–78. doi: 10.1016/j.ncl.2010.12.009
151. Ortiz GG, Pacheco-Moisés FP, Bitzer-Quintero OK, Ramírez-Anguiano AC, Flores-Alvarado LJ, Ramírez-Ramírez V, et al. Immunology and Oxidative Stress in Multiple Sclerosis: Clinical and Basic Approach. Clin Dev Immunol (2013) 2013:1–14. doi: 10.1155/2013/708659
152. Palle P, Monaghan KL, Milne SM, Wan ECK. Cytokine Signaling in Multiple Sclerosis and Its Therapeutic Applications. Med Sci (2017) 5:1–17. doi: 10.3390/medsci5040023
153. Penberthy WT, Tsunoda I. The Importance of NAD in Multiple Sclerosis. Curr Pharm Des (2009) 15:64–99. doi: 10.2174/138161209787185751
154. Nimmagadda VK, Bever CT, Vattikunta NR, Talat S, Ahmad V, Nagalla NK, et al. Overexpression of SIRT1 protein in neurons protects against experimental autoimmune encephalomyelitis through activation of multiple SIRT1 targets. J Immunol Baltim Md 1950 (2013) 190:4595–607. doi: 10.4049/jimmunol.1202584
155. Rajman L, Chwalek K, Sinclair DA. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab (2018) 27:529–47. doi: 10.1016/j.cmet.2018.02.011
156. Guerreiro S, Privat A-L, Bressac L, Toulorge D. CD38 in Neurodegeneration and Neuroinflammation. Cells (2020) 9:1–10. doi: 10.3390/cells9020471
157. Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem Biophys Res Commun (2006) 345:1386–92. doi: 10.1016/j.bbrc.2006.05.042
158. Hattori T, Kaji M, Ishii H, Jureepon R, Takarada-Iemata M, Minh Ta H, et al. CD38 positively regulates postnatal development of astrocytes cell-autonomously and oligodendrocytes non-cell-autonomously. Glia (2017) 65:974–89. doi: 10.1002/glia.23139
159. Kou W, Banerjee S, Eudy J, Smith LM, Persidsky R, Borgmann K, et al. CD38 regulation in activated astrocytes: Implications for neuroinflammation and HIV-1 brain infection. J Neurosci Res (2009) 87:2326–39. doi: 10.1002/jnr.22060
160. Ma Y, Wu D, Ding X, Ying W. CD38 plays key roles in both antioxidation and cell survival of H2O2-treated primary rodent astrocytes. Int J Physiol Pathophysiol Pharmacol (2014) 6:102–8.
161. Wang Y-M, Liu Z-Y, Ai Y-H, Zhang L-N, Zou Y, Peng Q-Y. Blocking the CD38/cADPR pathway plays a double-edged role in LPS stimulated microglia. Neuroscience (2017) 361:34–42. doi: 10.1016/j.neuroscience.2017.08.010
162. Cockayne DA, Muchamuel T, Grimaldi JC, Muller-Steffner H, Randall TD, Lund FE, et al. Mice deficient for the ecto-nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood (1998) 92:1324–33. doi: 10.1182/blood.V92.4.1324
163. Kato I, Yamamoto Y, Fujimura M, Noguchi N, Takasawa S, Okamoto H. CD38 Disruption Impairs Glucose-induced Increases in Cyclic ADP-ribose, [Ca2+] i, and Insulin Secretion. J Biol Chem (1999) 274:1869–72. doi: 10.1074/jbc.274.4.1869
164. Chen J, Chen Y-G, Reifsnyder PC, Schott WH, Lee C-H, Osborne M, et al. Targeted Disruption of CD38 Accelerates Autoimmune Diabetes in NOD/Lt Mice by Enhancing Autoimmunity in an ADP-Ribosyltransferase 2-Dependent Fashion. J Immunol (2006) 176:4590–9. doi: 10.4049/jimmunol.176.8.4590
165. Fumey W, Koenigsdorf J, Kunick V, Menzel S, Schütze K, Unger M, et al. Nanobodies effectively modulate the enzymatic activity of CD38 and allow specific imaging of CD38+ tumors in mouse models in vivo. Sci Rep (2017) 7:14289. doi: 10.1038/s41598-017-14112-6
166. van de Donk NWCJ, Janmaat ML, Mutis T, van Bueren JJL, Ahmadi T, Sasser AK, et al. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol Rev (2016) 270:95–112. doi: 10.1111/imr.12389
167. Dong M, Si Y-Q, Sun S-Y, Pu X-P, Yang Z-J, Zhang L-R, et al. Design, synthesis and biological characterization of novel inhibitors of CD38. Org Biomol Chem (2011) 9:3246–57. doi: 10.1039/C0OB00768D
168. Zhao YJ, Zhang HM, Lam CMC, Hao Q, Lee HC. Cytosolic CD38 Protein Forms Intact Disulfides and Is Active in Elevating Intracellular Cyclic ADP-ribose. J Biol Chem (2011) 286:22170–7. doi: 10.1074/jbc.M111.228379
169. Tirumurugaan KG, Kang BN, Panettieri RA, Foster DN, Walseth TF, Kannan MS. Regulation of the cd38 promoter in human airway smooth muscle cells by TNF-alpha and dexamethasone. Respir Res (2008) 9:26. doi: 10.1186/1465-9921-9-26
170. Graeff RM, Mehta K, Lee HC. GDP-ribosyl cyclase activity as a measure of CD38 induction by retinoic acid in HL-60 cells. Biochem Biophys Res Commun (1994) 205:722–7. doi: 10.1006/bbrc.1994.2725
171. Zhao YJ, Zhu WJ, Wang XW, Zhang L-H, Lee HC. Determinants of the membrane orientation of a calcium signaling enzyme CD38. Biochim Biophys Acta (2015) 1853:2095–103. doi: 10.1016/j.bbamcr.2014.10.028
172. Park D-R, Nam T-S, Kim Y-W, Bae YS, Kim U-H. Oxidative activation of type III CD38 by NADPH oxidase–derived hydrogen peroxide in Ca2+ signaling. FASEB J (2018) 33:3404–19. doi: 10.1096/fj.201800235R
173. Cevenini L, Calabretta MM, Calabria D, Roda A, Michelini E. Luciferase Genes as Reporter Reactions: How to Use Them in Molecular Biology? Adv Biochem Eng Biotechnol (2016) 154:3–17. doi: 10.1007/10_2015_325
174. Kellenberger E, Kuhn I, Schuber F, Muller-Steffner H. Flavonoids as inhibitors of human CD38. Bioorg Med Chem Lett (2011) 21:3939–42. doi: 10.1016/j.bmcl.2011.05.022
175. Wang S, Zhu W, Wang X, Li J, Zhang K, Zhang L, et al. Design, synthesis and SAR studies of NAD analogues as potent inhibitors towards CD38 NADase. Mol Basel Switz (2014) 19:15754–67. doi: 10.3390/molecules191015754
176. Muller-Steffner HM, Malver O, Hosie L, Oppenheimer NJ, Schuber F. Slow-binding inhibition of NAD+ glycohydrolase by arabino analogues of beta-NAD. J Biol Chem (1992) 267:9606–11.
177. Panche AN, Diwan AD, Chandra SR. Flavonoids: an overview. J Nutr Sci (2016) 5:1–15. doi: 10.1017/jns.2016.41
178. Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, et al. Flavonoid Apigenin Is an Inhibitor of the NAD+ase CD38: Implications for Cellular NAD+ Metabolism, Protein Acetylation, and Treatment of Metabolic Syndrome. Diabetes (2013) 62:1084–93. doi: 10.2337/db12-1139
179. Atanackovic D, Steinbach M, Radhakrishnan SV, Luetkens T. Immunotherapies targeting CD38 in Multiple Myeloma. Oncoimmunology (2016) 5:1–11. doi: 10.1080/2162402X.2016.1217374
180. Lammerts van Bueren J, Jakobs D, Kaldenhoven N, Roza M, Hiddingh S, Meesters J, et al. Direct in Vitro Comparison of Daratumumab with Surrogate Analogs of CD38 Antibodies MOR03087, SAR650984 and Ab79. Blood (2014) 124:3474–4. doi: 10.1182/blood.V124.21.3474.3474
181. van de Donk NWCJ, Usmani SZ. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front Immunol (2018) 9:1–12. doi: 10.3389/fimmu.2018.02134
182. Martin TG, Corzo K, Chiron M, van de Velde H, Abbadessa G, Campana F, et al. Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab. Cells (2019) 8:1–26. doi: 10.3390/cells8121522
183. Deckert J, Wetzel M-C, Bartle LM, Skaletskaya A, Goldmacher VS, Vallée F, et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res Off J Am Assoc Cancer Res (2014) 20:4574–83. doi: 10.1158/1078-0432.CCR-14-0695
184. Viegas MS, do Carmo A, Silva T, Seco F, Serra V, Lacerda M, et al. CD38 plays a role in effective containment of mycobacteria within granulomata and polarization of Th1 immune responses against Mycobacterium avium. Microbes Infect (2007) 9:847–54. doi: 10.1016/j.micinf.2007.03.003
185. Kim S-Y, Cho BH, Kim U-H. CD38-mediated Ca2+ signaling contributes to angiotensin II-induced activation of hepatic stellate cells: attenuation of hepatic fibrosis by CD38 ablation. J Biol Chem (2010) 285:576–82. doi: 10.1074/jbc.M109.076216
186. Seman M, Adriouch S, Scheuplein F, Krebs C, Freese D, Glowacki G, et al. NAD-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity (2003) 19:571–82. doi: 10.1016/s1074-7613(03)00266-8
Keywords: CD38, ADP-ribosyl cyclase, cADPR hydrolase, inflammation, autoimmunity, NADase
Citation: Piedra-Quintero ZL, Wilson Z, Nava P and Guerau-de-Arellano M (2020) CD38: An Immunomodulatory Molecule in Inflammation and Autoimmunity. Front. Immunol. 11:597959. doi: 10.3389/fimmu.2020.597959
Received: 23 August 2020; Accepted: 02 November 2020;
Published: 30 November 2020.
Edited by:
Teun J. De Vries, VU University Amsterdam, NetherlandsCopyright © 2020 Piedra-Quintero, Wilson, Nava and Guerau-de-Arellano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mireia Guerau-de-Arellano, bWlyZWlhLmd1ZXJhdUBvc3VtYy5lZHU=