
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 23 November 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.597741
 Jianni Wu1,2†
Jianni Wu1,2† Ester Del Duca1,3†
Ester Del Duca1,3† Michael Espino1†
Michael Espino1† Alyssa Gontzes1†Inna Cueto4Ning Zhang1Yeriel D. Estrada1Ana B. Pavel1,5James G. Krueger1,4
Alyssa Gontzes1†Inna Cueto4Ning Zhang1Yeriel D. Estrada1Ana B. Pavel1,5James G. Krueger1,4 Emma Guttman-Yassky1,4*
Emma Guttman-Yassky1,4*Keloids are disfiguring, fibroproliferative growths and their pathogenesis remains unclear, inhibiting therapeutic development. Available treatment options have limited efficacy and harbor safety concerns. Thus, there is a great need to clarify keloid pathomechanisms that may lead to novel treatments. In this study, we aimed to elucidate the profile of lesional and non-lesional keloid skin compared to normal skin. We performed gene (RNAseq, qRT-PCR) and protein (immunohistochemistry) expression analyses on biopsy specimens obtained from lesional and non-lesional skin of African American (AA) keloid patients compared to healthy skin from AA controls. Fold-change≥2 and false-discovery rate (FDR)<0.05 was used to define significance. We found that lesional versus normal skin showed significant up-regulation of markers of T-cell activation/migration (ICOS, CCR7), Th2- (IL-4R, CCL11, TNFSF4/OX40L), Th1- (CXCL9/CXCL10/CXCL11), Th17/Th22- (CCL20, S100As) pathways, and JAK/STAT-signaling (JAK3) (false-discovery rate [FDR]<0.05). Non-lesional skin also exhibited similar trends. We observed increased cellular infiltrates in keloid tissues, including T-cells, dendritic cells, mast cells, as well as greater IL-4rα+, CCR9+, and periostin+ immunostaining. In sum, comprehensive molecular profiling demonstrated that both lesional and non-lesional skin show significant immune alternations, and particularly Th2 and JAK3 expression. This advocates for the investigation of novel treatments targeting the Th2 axis and/or JAK/STAT-signaling in keloid patients.
Keloids are common, benign fibroproliferative overgrowths that manifest as exophytic, hyperpigmented lesions, extending beyond the original wound and result from dysregulated wound-healing secondary to skin injury. Keloids may recur, are associated with pain and pruritus, and are disfiguring (1). Numerous pathophysiologic processes have been investigated (1–3), including hypoxic (4, 5), mechanical tension (6, 7), and inflammatory etiologies (8–12). In addition, genetic studies have identified certain susceptibility loci, including inflammation-related genes, to be significantly associated with keloids (13–18). Yet, the pathogenesis still remains unclear, inhibiting therapeutic development. Moreover, current treatment options have limited efficacy and safety profiles and are associated with high recurrence rates of up to 100% (2, 19). Thus, there is a large unmet need to understand the pathomechanisms of keloids and identify novel therapeutics.
Recently, our group suggested a T-helper (Th)2-skewed pathogenesis of keloids by way of targeting Interleukin(IL)-4Rα with dupilumab, an FDA-approved biologic for moderate-to-severe atopic dermatitis (AD) (20). In this report, treatment of severe AD in an African American patient resulted in both AD improvement and shrinkage of his concomitant keloids (20). Furthermore, a limited analysis demonstrated increased mRNA expression of three Th2-related products (20), suggesting a possible Th2 involvement in mechanisms underlying pathologic fibrosis (21, 22). Clinical associations between keloids and other Th2-skewed diseases (e.g. AD (23) and asthma (24)) have also been reported. However, to our knowledge, a transcriptomic profiling of keloid skin lesions, that attempts to elucidate their immune alterations is lacking.
While a few gene expression studies have been performed, the majority focused on specific genes or limited microarray studies (25–32). Furthermore, these studies did not entirely characterize the inflammatory profile of keloids. Earlier cDNA microarray analyses primarily associated keloids with alterations of the extracellular matrix, growth factors, apoptosis-related molecules, and/or chondrogenic or osteogenic tissue differentiation (25–28). A more recent microarray profiling study analyzed differences in lesional versus non-lesional skin and reported bone and/or cartilage abnormalities but did not evaluate inflammatory pathways (29). Jumper et al. (30) used a laser capture micro-dissection approach, describing dysregulation of a few immune markers in keloid tissues (e.g. IL-13Rα1, IL-1β). Onoufriadis et al. (31) undertook an integrative mRNA and miRNA expression approach, focusing on pathway enrichment analysis and highlighting expression of mitogen-activated protein-kinase signaling pathway in keloid-prone individuals.
Due to a greater prevalence of keloids in individuals of African descent (19), we present a global RNA-seq profiling of skin biopsies obtained from both lesional and non-lesional skin of African American patients compared to healthy controls, complemented with additional validation using quantitative real-time PCR and immunohistochemistry. Our data show immune dysregulation, particularly Th2 and JAK3-skewing, along with Th1 and Th17/Th22 expression, in keloid lesions, extending to uninvolved skin, suggesting the potential for systemic therapies targeting these pathways in keloid patients.
Three African American patients (three females, mean age 47.3 years), with history of severe chronic keloids and no concomitant atopy or other comorbidities (29), and five healthy African American controls (two females, three males, mean age 39.8 years) were recruited under institutional review board-approved protocols. Biopsy specimens (6 mm) were collected from keloid lesions on the upper trunk. Non-lesional skin biopsies were obtained from a similar anatomic region, but at least 10 centimeters away from the keloid lesion. In one of the patients, a biopsy of an emerging keloid lesion was also obtained. Normal skin biopsy samples were collected from the trunk of healthy controls.
RNA was extracted as previously described (33, 34). Libraries were generated using TruSeq Stranded mRNA Library Prep kit (Illumina). Next generation sequencing was performed on Illumina NovaSeq6000 (Illumina Inc., 100 cycles, single-read sequencing). Sample quality was assessed using FastQC. RNA-sequencing data was profiled by Illumina NovaSeq6000 to allow for more global analyses of genomic abnormalities. Data were pre-processed using standard pipeline incorporating quality control metrics, such as FastQC and MultiQC, sequence alignment based on STAR RNA-sequencing aligner, and sequencing reads assignment to genomic features by featureCounts and voom-transformed. A total of 4 keloid lesions obtained from 3 different patients and 6 healthy controls were included in the RNA-sequencing analysis.
RNA was extracted for real-time polymerase chain reaction (RT-PCR) using the miRNAeasy Mini Kit (Qiagen, Hilden, Germany). Reverse transcription to complementary DNA (cDNA) from RNA was carried out using the High Capacity cDNA reverse transcription (Thermo Fisher). Pre-amplification was performed on all samples. Primers are listed in Table S1. Ten nanogram total RNA was used for PreAMP pool. Rplp0 was used as endogenous control. Expression values were normalized to Rplp0.
Immunohistochemistry was performed on frozen lesional and non-lesional skin sections from keloid patients and healthy skin from controls, using monoclonal antibodies as previously described (Table S2) (35, 36). Cell counts were quantified from representative sections in both the epidermis and dermis using ImageJ V1.42 software (National Institutes of Health, Bethesda, Maryland).
Analyses were performed using R-language (R-project.org) and Bioconductor Project packages (www.bioconductor.org). Gene expression profiles were modeled by linear models using R’s lme function. limma framework was used for RNA-sequencing data, and p-values were adjusted for multiple hypotheses using the Benjamini-Hochberg procedure. Fold changes (FCHs)>2.0 and false discovery rates (FDR)<0.05 were considered differentially expressed. We also evaluated a curated immune gene-subset (33, 34, 37) using P-values, due to the small sample size. Mean expressions are displayed in a heatmap, where unsupervised clustering was performed using Euclidean distance and average agglomeration criteria. PCR and immunohistochemistry data were log2 transformed and analyses were performed using Student’s t-test.
We performed RNA-seq global profiling (GSE158395), followed by qRT-PCR and immunohistochemistry, on lesional and non-lesional keloid skin samples obtained from three African American patients (mean age 47.3 years) with history of chronic (present for >10 years), large keloids, and no atopy or other comorbidities (29), and healthy skin samples obtained from five African American controls (mean age 39.8 years). In one of the patients, a biopsy of an emerging keloid lesion was also obtained. Patients did not have any treatments on their biopsied keloids. No significant differences in age or gender were detected between patients and controls.
Using criteria of fold-change (FCH)>2 and false discovery rate (FDR)<0.05 to define differentially expressed genes (DEGs), we detected 3,044 DEGs (1,437 up-regulated and 1,607 down-regulated) in lesional keloid versus normal skin, 2,929 DEGs between lesional and non-lesional skin, and few DEGs between non-lesional and normal skin, as depicted in a heatmap in Figure S1 (Table S3).
The top 50 DEGs in lesional versus normal skin comparison included products related to fibrosis and cartilage/bone-differentiation (ADAM12, ASPN, COL10A1, COL11A1, FBN2), previously reported to be dysregulated in keloids (Table S3) (20, 29). Periostin, implicated both in fibrosis as well as in Th2 inflammation (38–40), was among the top DEGs in lesional versus normal skin (FDR<0.05 for all; Figure 1, Table S3).
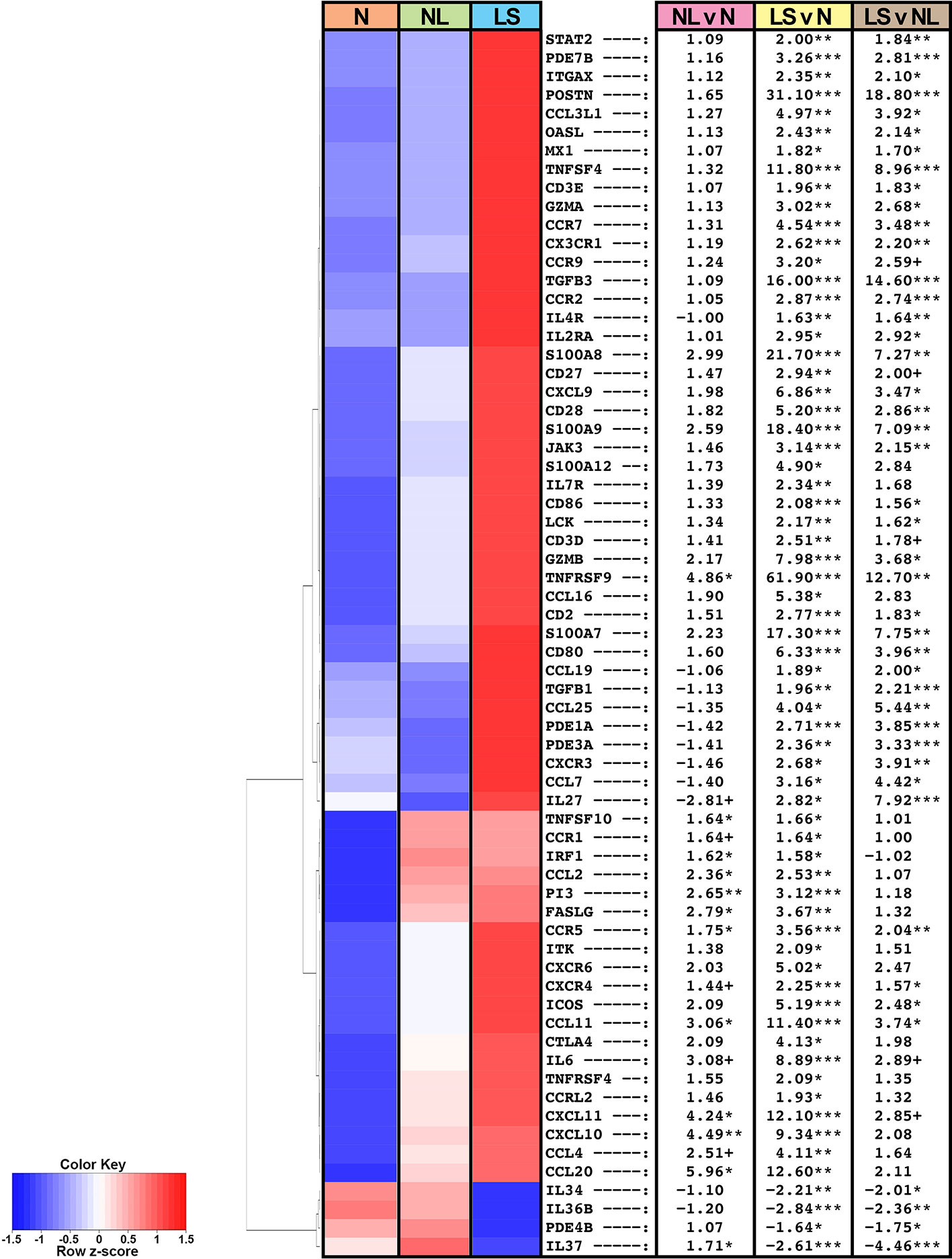
Figure 1 Summary heatmap of immune gene expression of multiple pathways in biopsied lesional and non-lesional keloid skin and normal skin from controls using RNA-seq. Table shows fold-changes in non-lesional versus normal (NL vs N), lesional versus normal (LS vs N), and lesional vs non-lesional (LS vs NL) skin. ***P < 0.001, **P < 0.01, *P < 0.05, +P < 0.1. Red color denotes higher mean expression levels and blue color denotes lower mean expression levels. LS, lesional; NL, non-lesional; N, normal.
Among the significantly modulated genes in keloid lesions compared to normal skin also included markers related to various immune components, as depicted in a heatmap of a curated immune gene-subset (Figure 1) (33, 34, 37). In keloid lesions, we observed significant increases in measures of T-cells (CD2, CD3, CD28), T-cells/NK-cells/T-cell activation and migration (ICOS, LCK, GZMA/B, CCR7, TNFRSF9), and cellular infiltrates, such as dendritic cell markers CD80 and CD86 (FDR<0.05; Figure 1, Table S3). Measures of innate immunity (IL-6), and multiple immune pathways, including the Th2- (IL-4R, CCR5, CCL11, TNFSF4/OX40L), Th1- (CXCL9/CXCL10/CXCL11, OASL), and Th17/Th22- (CCL20, PI3, S100A7/8/9) pathways were also significantly up-regulated (FDR<0.05; Figure 1, Table S3).
Furthermore, keloid lesions displayed significant up-regulation of JAK/STAT signaling molecule, JAK3, as compared to normal skin (FDR<0.05; Figure 1, Table S3). Markers related not only to fibrosis but also Tregs (TGFβ1 and TGFβ3) (41, 42), were highly increased as well (FDR<0.05). Both CCR9, the homing receptor for the small intestine and lung (43, 44), and its ligand CCL25, displayed increased expression in keloid tissues (FDR ≤ 0.1). The CCR9/CCL25 axis has been shown to induce cellular recruitment in early allergic asthma (44). Negative immune regulators, IL-34 (45) and IL-37 (34), were significantly reduced in lesional keloid versus normal skin (FDR<0.05; Figure 1, Table S3).
Similar dysregulation of these immune axes was observed between non-lesional and normal skin comparisons, albeit these did not attain significance by FDR, however some achieved significance using criteria of P<0.05 (e.g. CCL11, CCR5, CXCL10/11, CCL20) (Figure 1, Table S3).
qRT-PCR was performed to validate as well as evaluate key immune molecules that are often below detection limits on RNA-seq (34, 46) (Figure 2). Th2 (CCL11, TSLP, TNFSF4/OX40L, TNFRSF4/OX40) markers showed significant up-regulation in lesional or non-lesional versus normal skin (P<0.05; Figure 2). Expression of CCR9 and its ligand CCL25 showed significant, or trending toward significant, up-regulation in lesional versus normal skin (P<0.05 for CCL25; Figure 2). JAK3 was also highly expressed in keloid lesions compared to normal skin (P<0.01; Figure 2).
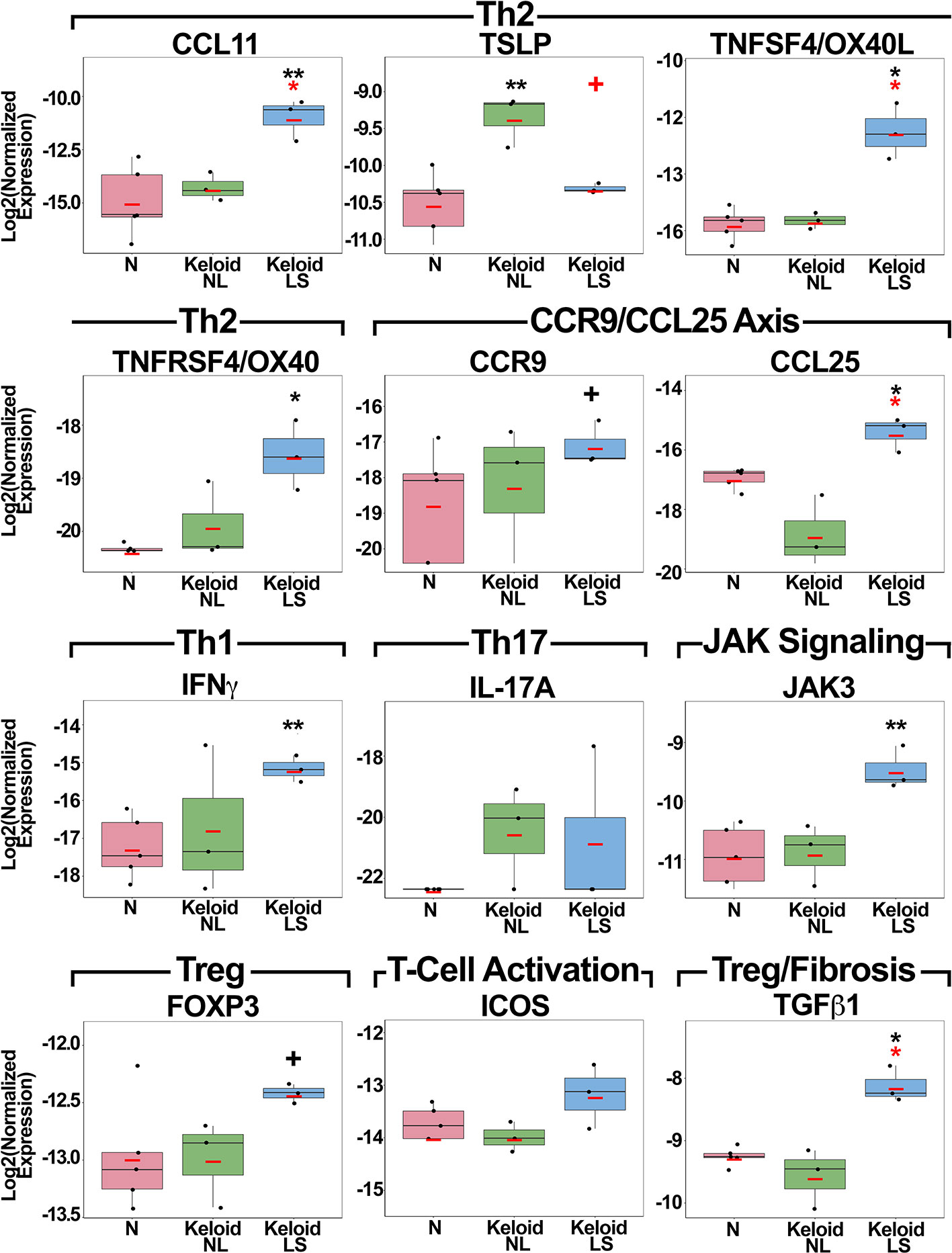
Figure 2 Fold-changes of immune mediators in lesional and non-lesional skin of keloid patients, as well as normal skin, as measured by quantitative real-time PCR. Red bar represents mean. Black symbols: significance of comparison to normal skin; red symbols: significance of comparison between lesional and non-lesional skin. **P < 0.01, *P <0.05, +P < 0.1. PCR, polymerase chain reaction; LS, lesional; NL, non-lesional; N, normal.
While the main Th1 cytokine, IFNγ, showed significant increases in keloid lesions (P<0.01), IL-17A, the Th17 cytokine, only showed a trend for increased expression in both lesional and non-lesional versus normal skin (Figure 2). Treg markers, FOXP3 and TGFβ1, and T-cell activation measure ICOS, were up-regulated in lesional keloid versus normal skin (P<0.05 for TGFβ1; Figure 2). We also evaluated an emerging keloid lesion and obtained similar gene expression results (Figure S2).
Gradual increases were observed for multiple cellular infiltrate measures, such as T-cells (CD3+, CD8+) and dendritic cells (CD11c+), including dendritic cell infiltrates that are characteristic to AD, such as markers typifying “atopic” dendritic cells (OX40L+, FCεR1+) (33, 47, 48) (P<0.05 for CD3+, FCεR1+, OX40L+ in lesional versus normal skin comparisons; Figures 3A–T). OX40L+ immunostaining was also significantly increased in non-lesional keloid versus normal skin (P<0.05; Figures 3Q–T).
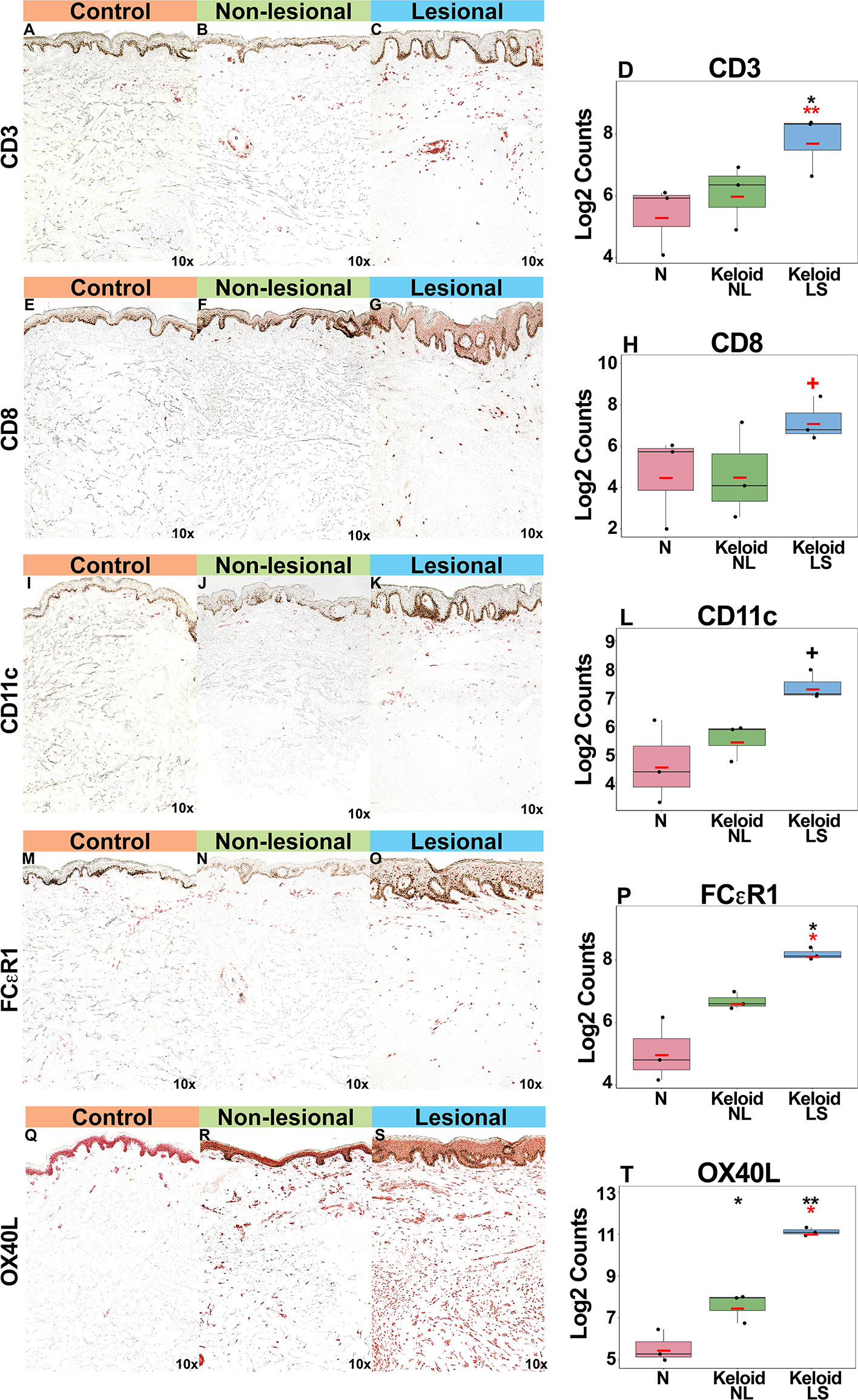
Figure 3 Representative immunohistochemistry images and corresponding cell count quantification of CD3+ T-cells (A–D), CD8+ T-Cells (E–H), CD11c+ dendritic cells (I–L), FCεR1+ dendritic cells (M–P) and OX40L+ dendritic cells (Q–T) in normal skin from controls, and lesional and non-lesional skin of keloid patients, viewed at 10x magnification. Red bar represents mean. Black symbols: significance of comparison to normal; red symbols: significance of comparison between lesional and non-lesional skin. **P < 0.01, *P < 0.05, +P < 0.1. LS, lesional; NL, non-lesional; N, normal.
Supporting our mRNA expression data, we noted increased IL-4Rα+ immunostaining in both lesional and non-lesional keloid skin as compared to normal skin (P<0.05 for lesional versus non-lesional skin comparison; Figures 4A–D). Mast cells (tryptase+), associated with atopy (49, 50), were increased in keloid lesions (P<0.1 for lesional versus non-lesional comparison; Figures 4E–H). Trending toward significantly greater CCR9+ immunostaining was noted in lesional compared to normal skin (P<0.1 Figures 4I–L). Finally, we observed significantly greater periostin immunostaining, concentrating in the lower dermis of keloid lesions, as compared to non-lesional and normal skin (P<0.05; Figures 4M–P). We also evaluated an emerging keloid lesion and obtained similar cellular infiltrates (Figure S3).
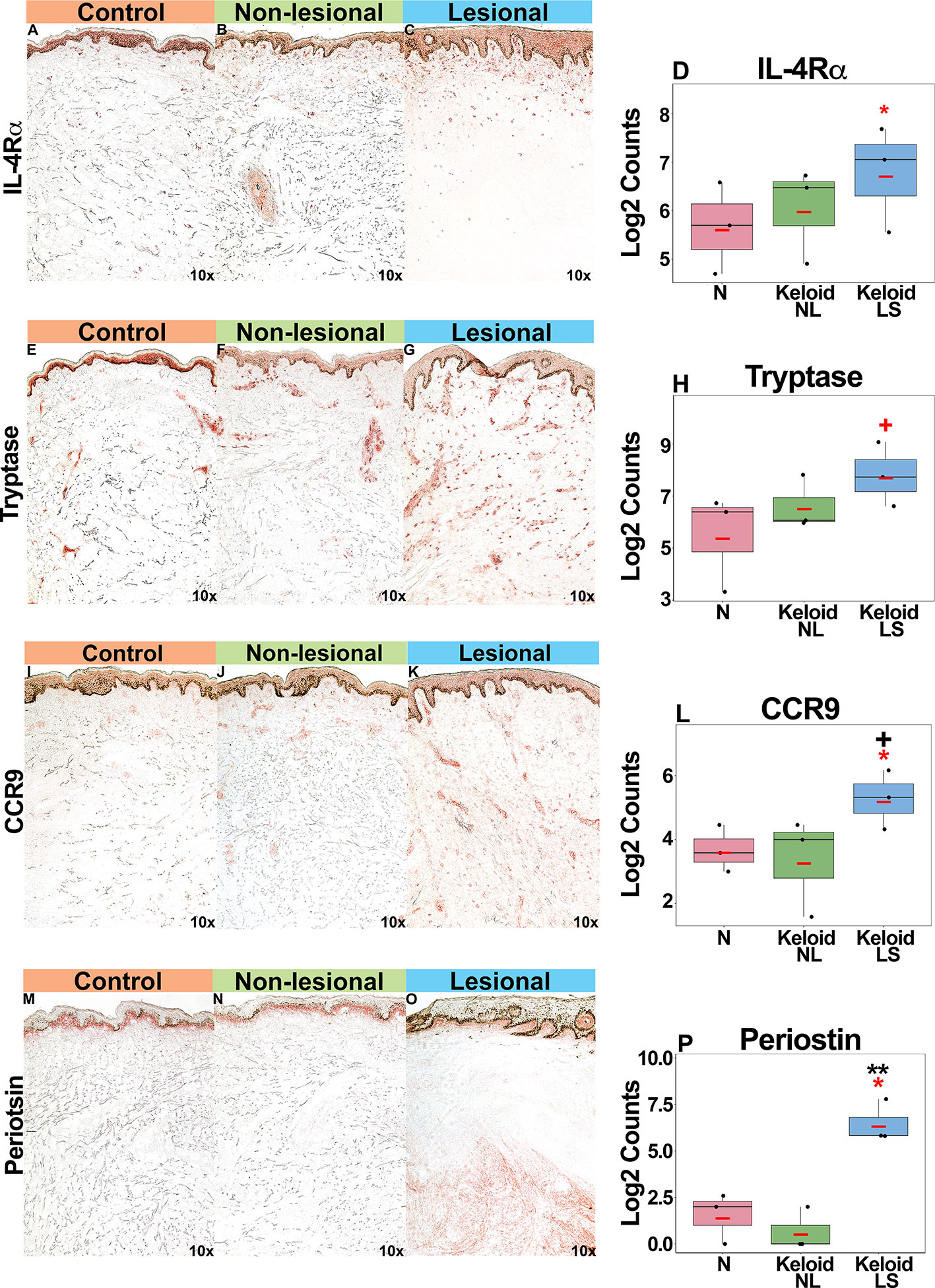
Figure 4 Representative immunohistochemistry images and corresponding cell count quantification of IL-4Rα+ cells (A–D), tryptase+ mast cells (E–H), CCR9+ cells (I–L) and periostin+ cells (M–P) in normal skin from controls, and lesional and non-lesional skin of keloid patients, viewed at 10x magnification. Red bar represents mean. Black symbols: significance of comparison to normal; red symbols: significance of comparison between lesional and non-lesional skin. **P < 0.01, *P < 0.05, +P < 0.1. LS, lesional; NL, non-lesional; N, normal.
Our study combines gene and protein expression analyses, broadly characterizing the molecular and cellular phenotype of lesional and non-lesional keloid skin, highlighting the possible contribution of immune abnormalities to its pathogenesis. While various etiologies for keloid formation have been investigated, the pathogenesis remains to be clearly elucidated, hindering the development of novel therapeutics. Global RNA-seq profiling, as successfully performed in other inflammatory skin diseases, such as AD and psoriasis (34, 51), may uncover further insights into disease mechanisms, and possibly lead to the development of newer, immune-based treatments.
In light of the role of inflammation in normal wound healing, a dysregulated immune component can contribute to an abnormal wound repair process (8, 9, 52). However, to our knowledge, the role of inflammation in the pathogenesis of keloids has not been adequately investigated. Published studies investigating an immune component have largely focused on specific keloidal cell cultures or specific genes. One report identified up-regulation of the proinflammatory cytokines, IL-1α, IL-1β, IL-6, and TNFα, in keloid fibroblasts (10). Our group recently examined a few Th2 markers in keloids (20), while another study found increases for the IL-17/IL-22-induced products, S100A7 and hBD2 (53). Additionally, dysregulation of the IL-17/IL-6 axis (11) and/or JAK/STAT-signaling (54, 55) have also been investigated. The present study expands on prior investigations and provides a comprehensive molecular profiling of keloids, identifying a significant immune component in both lesional and non-lesional skin, as well as linking keloids with fibrosis and cartilage/bone-differentiation.
Our results associate keloid tissues with an inflammatory milieu representing multiple T-helper pathways, including the Th2 (e.g. IL-4R, CCL11, TSLP, TNFSF4/OX40L, TNFRSF4/OX40), Th1 (e.g. IFNγ, CXCL10/11), Th17/Th22 (e.g. PI3, CCL20, S100As) axes, as well as the JAK/STAT signaling molecule JAK3. The up-regulation of Th17 markers expands upon prior reports demonstrating a role of Th17 mediators in keloids (11, 55). Additionally, our results suggest that, similar to other fibrotic diseases (56), the Th1 axis is also involved to keloid pathogenesis. Of note, IL-6 which is associated with innate immunity (57), is highly up-regulated in our study and has also been shown to drive a chronic pro-fibrotic state via a Th1-mediated response (58).
In the present study, we observed a significant Th2 signature in keloids as well as increased cellular infiltrates associated with the Th2 microenvironment, such as tryptase+ mast cells (12, 49, 50), markers of dendritic cells characteristic to atopic dermatitis (AD) (OX40L+ and FCεR1+) (33, 47, 48), IL-4Rα+ cells, and periostin+ cells. In human fibroblasts, it has been found that IL-4 and IL-13 increased TGF-β signaling and enhanced fibrosis via periostin (59). Periostin is a matricellular protein that plays important physiologic and pathogenic roles in skin fibrosis (60, 61) and serves as a biomarker for several known Th2-associated diseases (e.g. AD, asthma, nasal polyps, systemic sclerosis) (38, 39, 62, 63). Its significantly increased upregulation in keloids may represent an important link between immune responses and fibrosis. In fact, one study found periostin to be expressed by a novel subpopulation of Th2-associated fibroblasts in AD (64). These data implicate a cross-talk between Th2 immune mechanisms and fibrosis that is also likely relevant in keloid pathogenesis.
Further reinforcing the concept of immune dysregulation in driving fibrotic processes in keloids is the increased T-cell expression, including Th2-related markers, in several fibrotic conditions, such as frontal fibrosing alopecia and scleroderma (65, 66). In vitro studies have also demonstrated the profibrotic effects of the type 2 cytokines IL-4 and IL-13 (22), including increased collagen production in keloid fibroblasts after IL-13 stimulation (67). These studies strengthen our hypothesis that immune dysregulation, possibly Th2 driven, plays a significant role in keloid formation. Additionally, we found increased expression of CCR9 and its receptor CCL25, which are involved in inflammatory cell recruitment in early allergic asthma (44). Taken together, our results highlight commonalities between keloids and other atopic disorders such as AD and asthma (68), as well as those that exhibit exophytic growth (e.g. nasal polyps) (69, 70). Keloids also display commonalities with connective tissue diseases associated with excessive extracellular matrix deposition, such as scleroderma (65).
The up-regulation of T-cell related genes in non-lesional skin compared to normal skin (e.g. CCL11, CCR5) highlight that the seemingly normal skin of keloid patients is in fact abnormal and predisposed to formation of keloids. These data are also supported by the clinical observation that individuals who develop keloids are prone to future keloid formation after skin wounding (19). These incipient findings in non-lesional skin are important as it might mean that patients with significant keloids may necessitate systemic treatment to target not only lesional but also non-lesional skin to prevent future recurrence of keloids.
While studies in blood can clarify the presence of systemic activation in keloid patients, our results, which demonstrate immune dysregulation extending beyond skin lesions to uninvolved skin, advocate for the investigation of systemic treatments for keloids, such as those targeting the Th2 axis. The potential clinical utility of anti-IL-4Rα antagonism (20) is encouraged by the significant overlap observed between dupilumab-regulated markers in AD and those that are dysregulated in keloids, including markers of the Th2 axis, as well as cellular infiltrates (CD3+, CD11c+, FCεR1+), T-cell activation/migration (GZMB, ICOS, CCR7), and the Th17/Th22 (PI3, S100As) pathway (37, 71). Antagonism of the OX40-TSLP axis, which is known to perpetuate Th2 activation in both AD and asthma (72–74), could also be explored for keloids. GBR830 and KHK4083 are anti-OX40 monoclonal antibodies in clinical trials (75, 76). In one analysis (75), GBR830 significantly decreased measures that are also upregulated in keloid tissues in the present study, including OX40+ T-cells, OX40L+ dendritic cells, and Th2- (CCL11), Th1- (IFNγ, CXCL10), and Th17/Th22- (S100A9/12) related markers. Given the significantly increased expression of TSLP in non-lesional keloid skin, exploration of tezepelumab, an anti-TSLP monoclonal antibody (77), may also be warranted.
Furthermore, biologics targeting IL-6 signaling, such as tocilizumab, sarilumab, and siltuximab, could be explored (78). Indeed, our data showing significant IL-6 up-regulation in keloids expands on previous studies that demonstrated elevated IL-6 expression in keloidal fibroblasts (79, 80). Ghazizadeh et al. (80) further showed that inhibition of IL-6 or IL-6Rα in keloid fibroblasts resulted in a reduction of collagen synthesis, underscoring the potential utility of IL-6 antagonism for this disease.
In contrast to the aforementioned biologics, Janus kinase (JAK) inhibitors comprise a class of broad-acting small molecules. The JAK/STAT signaling and spleen tyrosine kinase (SYK) pathways are implicated in numerous autoimmune and inflammatory diseases (e.g. AD, psoriasis, alopecia areata) (81), modulating a range of immune responses, including the Th2 (IL-4, IL-13, CCL18), Th1 (IFNγ), and Th17/Th22 (CCL20, S100As) pathways (82). Preliminary studies evaluating inhibition of JAK/STAT signaling in human keloid fibroblasts (54, 55), and in a humanized keloid animal model (83), have demonstrated promising results. There are several JAK inhibitors in clinical development showing efficacy for various dermatologic disorders (81, 82, 84) and could be a therapeutic strategy worth investigating in keloids.
This is a preliminary study with a few limitations, including a small sample size. While future studies need to evaluate larger cohorts, we were still able to obtain large differences in lesional and non-lesional skin versus normal skin, as well as a significant signal after adjusting for multiple hypothesis testing by false-discovery rate using a stringent cut-off for significance. Our study group only included African American patients, representing a greater prevalence of keloids in this ethnicity (1, 19). Additional studies should evaluate keloids in other races and ethnicities, such as in Hispanics, Asians, and Caucasians (1, 19). Finally, as our study raises the hypothesis of possible immune-based treatments for keloids, this needs to be proven in future proof of concept studies and clinical trials.
In sum, the current genomic and cellular profiling study of keloids provides a comprehensive molecular fingerprint of the immune alterations in lesional and non-lesional keloid skin compared to normal skin. Our results reveal broad immune dysregulation, including up-regulation of markers of the Th1 and Th17/Th22 axes as well as significant Th2 and JAK3 expression in keloid lesions, extending to uninvolved skin, suggesting the potential use of systemic therapeutics. Future clinical trials for keloid patients may include those targeting the Th2 axis, inhibitors of the TSLP-OX40 axis, antagonists of JAK/STAT-signaling, or IL-6 inhibition.
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/geo with accession number GSE158395.
The studies involving human participants were reviewed and approved by The Rockefeller University IRB. The patients/participants provided their written informed consent to participate in this study.
JW contributed to the study design, data analysis, data interpretation, and writing the manuscript. ED, ME, and AG contributed to the histology stainings, data interpretation, and writing the manuscript. IC, NZ, and YE contributed to the experiments. AP contributed to the study design, data analysis, data interpretation and writing the manuscript. JK edited and improved the manuscript. EG-Y contributed to the study and experimental design, data interpretation, and writing the manuscript. All authors contributed to the article and approved the submitted version.
EG-Y is an employee of Mount Sinai and has received research funds (grants paid to the institution) from: Abbvie, Celgene, Eli Lilly, Janssen, Medimmune/Astra Zeneca, Novartis, Pfizer, Regeneron, Vitae, Glenmark, Galderma, Asana, Innovaderm, Dermira, UCB. EG-Y is also a consultant for Sanofi Aventis, Regeneron, Stiefel/GlaxoSmithKline, MedImmune, Celgene, Anacor, AnaptysBio, Dermira, Galderma, Glenmark, Novartis, Pfizer, Vitae, Leo Pharma, Abbvie, Eli Lilly, Kyowa, Mitsubishi Tanabe, Asana Biosciences, and Promius. JK has received research support (grants paid to his institution) and/or personal fees from Pfizer, Amgen, Janssen, Lilly, Merck, Novartis, Kadmon, Dermira, Boehringer, Innovaderm, Kyowa, BMS, Serono, BiogenIdec, Delenex, AbbVie, Sanofi, Baxter, Paraxel, Xenoport, and Kineta. AP, NZ, and ED are employees of Mount Sinai.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.597741/full#supplementary-material
AD, atopic dermatitis; DC, dendritic cell; FCH, fold-changes; FDR, false discovery rate; IL, interleukin; qRT-PCR, quantitative real time polymerase chain reaction; RNA-seq, RNA-sequencing; Th, T-helper; Treg, T-regulatory cell.
1. Shaffer JJ, Taylor SC, Cook-Bolden F. Keloidal scars: A review with a critical look at therapeutic options. J Am Acad Dermatol (2002) 46:S63–97. doi: 10.1067/mjd.2002.120788
2. Berman B, Maderal A, Raphael B. Keloids and Hypertrophic Scars: Pathophysiology, Classification, and Treatment. Dermatol Surg (2017) 43:S3–s18. doi: 10.1097/DSS.0000000000000819
3. Limandjaja GC, Niessen FB, Scheper RJ, Gibbs S. The Keloid Disorder: Heterogeneity, Histopathology, Mechanisms and Models. Front Cell Dev Biol (2020) 8:360. doi: 10.3389/fcell.2020.00360
4. Kim J, Kim B, Kim SM, Yang CE, Song SY, Lee WJ, et al. Hypoxia-Induced Epithelial-To-Mesenchymal Transition Mediates Fibroblast Abnormalities via ERK Activation in Cutaneous Wound Healing. Int J Mol Sci (2019) 20(10):2546. doi: 10.3390/ijms20102546
5. Ma X, Chen J, Xu B, Long X, Qin H, Zhao RC, et al. Keloid-derived keratinocytes acquire a fibroblast-like appearance and an enhanced invasive capacity in a hypoxic microenvironment in vitro. Int J Mol Med (2015) 35:1246–56. doi: 10.3892/ijmm.2015.2135
6. Suarez E, Syed F, Rasgado TA, Walmsley A, Mandal P, Bayat A. Skin equivalent tensional force alters keloid fibroblast behavior and phenotype. Wound Repair Regener (2014) 22:557–68. doi: 10.1111/wrr.12215
7. Ogawa R, Okai K, Tokumura F, Mori K, Ohmori Y, Huang C, et al. The relationship between skin stretching/contraction and pathologic scarring: The important role of mechanical forces in keloid generation. Wound Repair Regeneration (2012) 20:149–57. doi: 10.1111/j.1524-475X.2012.00766.x
8. Ogawa R. Keloid and Hypertrophic Scars Are the Result of Chronic Inflammation in the Reticular Dermis. Int J Mol Sci (2017) 18(3):606. doi: 10.3390/ijms18030606
9. Dong X, Mao S, Wen H. Upregulation of proinflammatory genes in skin lesions may be the cause of keloid formation (Review). BioMed Rep (2013) 1:833–6. doi: 10.3892/br.2013.169
10. Messadi DV, Doung HS, Zhang Q, Kelly AP, Tuan T-L, Reichenberger E, et al. Activation of NFκB signal pathways in keloid fibroblasts. Arch Dermatol Res (2004) 296:125–33. doi: 10.1007/s00403-004-0487-y
11. Zhang Q, Yamaza T, Kelly AP, Shi S, Wang S, Brown J, et al. Tumor-like stem cells derived from human keloid are governed by the inflammatory niche driven by IL-17/IL-6 axis. PloS One (2009) 4:e7798. doi: 10.1371/journal.pone.0007798
12. Bagabir R, Byers RJ, Chaudhry IH, Müller W, Paus R, Bayat A. Site-specific immunophenotyping of keloid disease demonstrates immune upregulation and the presence of lymphoid aggregates. Br J Dermatol (2012) 167:1053–66. doi: 10.1111/j.1365-2133.2012.11190.x
13. Tsai CH, Ogawa R. Keloid research: current status and future directions. Scars Burn Heal (2019) 5:1–8. doi: 10.1177/2059513119868659
14. Nakashima M, Chung S, Takahashi A, Kamatani N, Kawaguchi T, Tsunoda T, et al. A genome-wide association study identifies four susceptibility loci for keloid in the Japanese population. Nat Genet (2010) 42:768. doi: 10.1038/ng.645
15. Zhu F, Wu B, Li P, Wang J, Tang H, Liu Y, et al. Association study confirmed susceptibility loci with keloid in the Chinese Han population. PloS One (2013) 8:e62377. doi: 10.1371/journal.pone.0062377
16. Fujita M, Yamamoto Y, Jiang JJ, Atsumi T, Tanaka Y, Ohki T, et al. NEDD4 Is Involved in Inflammation Development during Keloid Formation. J Invest Dermatol (2019) 139:333–41. doi: 10.1016/j.jid.2018.07.044
17. Hellwege JN, Russell SB, Williams SM, Edwards TL, Velez Edwards DR. Gene-based evaluation of low-frequency variation and genetically-predicted gene expression impacting risk of keloid formation. Ann Hum Genet (2018) 82:206–15. doi: 10.1111/ahg.12245
18. Velez Edwards DR, Tsosie KS, Williams SM, Edwards TL, Russell SB. Admixture mapping identifies a locus at 15q21.2-22.3 associated with keloid formation in African Americans. Hum Genet (2014) 133:1513–23. doi: 10.1007/s00439-014-1490-9
19. Chike-Obi CJ, Cole PD, Brissett AE. Keloids: pathogenesis, clinical features, and management. Semin Plast Surg (2009) 23:178–84. doi: 10.1055/s-0029-1224797
20. Diaz A, Tan K, He H, Xu H, Cueto I, Pavel AB, et al. Keloid lesions show increased IL-4/IL-13 signaling and respond to Th2-targeting dupilumab therapy. J Eur Acad Dermatol Venereol (2020) 34:e161–e4. doi: 10.1111/jdv.16097
21. Gieseck RL, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol (2018) 18:62–76. doi: 10.1038/nri.2017.90
22. Nguyen JK, Austin E, Huang A, Mamalis A, Jagdeo J. The IL-4/IL-13 axis in skin fibrosis and scarring: mechanistic concepts and therapeutic targets. Arch Dermatol Res (2020) 312:81–92. doi: 10.1007/s00403-019-01972-3
23. Lu Y-Y, Lu C-C, Yu W-W, Zhang L, Wang Q-R, Zhang C-L, et al. Keloid risk in patients with atopic dermatitis: a nationwide retrospective cohort study in Taiwan. BMJ Open (2018) 8:e022865. doi: 10.1136/bmjopen-2018-022865
24. Hajdarbegovic E, Bloem A, Balak D, Thio B, Nijsten T. The Association between Atopic Disorders and Keloids: A Case-control Study. Indian J Dermatol (2015) 60:635–. doi: 10.4103/0019-5154.169144
25. Nassiri M, Woolery-Lloyd H, Ramos S, Jacob SE, Gugic D, Viciana A, et al. Gene expression profiling reveals alteration of caspase 6 and 14 transcripts in normal skin of keloid-prone patients. Arch Dermatol Res (2009) 301:183–8. doi: 10.1007/s00403-008-0880-z
26. Na GY, Seo SK, Lee SJ, Kim DW, Kim MK, Kim JC. Upregulation of the NNP-1 (novel nuclear protein-1, D21S2056E) gene in keloid tissue determined by cDNA microarray and in situ hybridization. Br J Dermatol (2004) 151:1143–9. doi: 10.1111/j.1365-2133.2004.06284.x
27. Naitoh M, Kubota H, Ikeda M, Tanaka T, Shirane H, Suzuki S, et al. Gene expression in human keloids is altered from dermal to chondrocytic and osteogenic lineage. Genes Cells (2005) 10:1081–91. doi: 10.1111/j.1365-2443.2005.00902.x
28. Chen W, Fu X, Sun X, Sun T, Zhao Z, Sheng Z. Analysis of differentially expressed genes in keloids and normal skin with cDNA microarray. J Surg Res (2003) 113:208–16. doi: 10.1016/S0022-4804(03)00188-4
29. Fuentes-Duculan J, Bonifacio KM, Suárez-Fariñas M, Kunjravia N, Garcet S, Cruz T, et al. Aberrant connective tissue differentiation towards cartilage and bone underlies human keloids in African Americans. Exp Dermatol (2017) 26:721–7. doi: 10.1111/exd.13271
30. Jumper N, Hodgkinson T, Paus R, Bayat A. Site-specific gene expression profiling as a novel strategy for unravelling keloid disease pathobiology. PloS One (2017) 12:e0172955. doi: 10.1371/journal.pone.0172955
31. Onoufriadis A, Hsu CK, Ainali C, Ung CY, Rashidghamat E, Yang HS, et al. Time Series Integrative Analysis of RNA Sequencing and MicroRNA Expression Data Reveals Key Biologic Wound Healing Pathways in Keloid-Prone Individuals. J Invest Dermatol (2018) 138:2690–3. doi: 10.1016/j.jid.2018.05.017
32. Shih B, McGrouther DA, Bayat A. Identification of novel keloid biomarkers through profiling of tissue biopsies versus cell cultures in keloid margin specimens compared to adjacent normal skin. Eplasty (2010) 10:e24.
33. Sanyal RD, Pavel AB, Glickman J, Chan TC, Zheng X, Zhang N, et al. Atopic dermatitis in African American patients is TH2/TH22-skewed with TH1/TH17 attenuation. Ann Allergy Asthma Immunol (2019) 122:99–110.e6. doi: 10.1016/j.anai.2018.08.024
34. Suarez-Farinas M, Ungar B, Correa da Rosa J, Ewald DA, Rozenblit M, Gonzalez J, et al. RNA sequencing atopic dermatitis transcriptome profiling provides insights into novel disease mechanisms with potential therapeutic implications. J Allergy Clin Immunol (2015) 135:1218–27. doi: 10.1016/j.jaci.2015.03.003
35. Del Duca E, Pavel AB, Dubin C, Song T, Wallace EB, Peng X, et al. Major Differences in Expression of Inflammatory Pathways in Skin from Different Body Sites of Healthy Individuals. J Invest Dermatol (2019) 139:2228–32.e10. doi: 10.1016/j.jid.2019.04.008
36. Zhou L, Leonard A, Pavel AB, Malik K, Raja A, Glickman J, et al. Age-specific changes in the molecular phenotype of patients with moderate-to-severe atopic dermatitis. J Allergy Clin Immunol (2019) 144:144–56. doi: 10.1016/j.jaci.2019.01.015
37. Guttman-Yassky E, Bissonnette R, Ungar B, Suarez-Farinas M, Ardeleanu M, Esaki H, et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J Allergy Clin Immunol (2019) 143:155–72. doi: 10.1016/j.jaci.2018.08.022
38. Akar-Ghibril N, Casale T, Custovic A, Phipatanakul W. Allergic Endotypes and Phenotypes of Asthma. J Allergy Clin Immunol: Pract (2020) 8:429–40. doi: 10.1016/j.jaip.2019.11.008
39. Kou K, Okawa T, Yamaguchi Y, Ono J, Inoue Y, Kohno M, et al. Periostin levels correlate with disease severity and chronicity in patients with atopic dermatitis. Br J Dermatol (2014) 171:283–91. doi: 10.1111/bjd.12943
40. Conway SJ, Izuhara K, Kudo Y, Litvin J, Markwald R, Ouyang G, et al. The role of periostin in tissue remodeling across health and disease. Cell Mol Life Sci (2014) 71:1279–88. doi: 10.1007/s00018-013-1494-y
41. Wan YY, Flavell RA. TGF-beta and regulatory T cell in immunity and autoimmunity. J Clin Immunol (2008) 28:647–59. doi: 10.1007/s10875-008-9251-y
42. Tran DQ. TGF-beta: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol (2012) 4:29–37. doi: 10.1093/jmcb/mjr033
43. Svensson M, Agace WW. Role of CCL25/CCR9 in immune homeostasis and disease. Expert Rev Clin Immunol (2006) 2:759–73. doi: 10.1586/1744666X.2.5.759
44. López-Pacheco C, Soldevila G, Du Pont G, Hernández-Pando R, García-Zepeda EA. CCR9 Is a Key Regulator of Early Phases of Allergic Airway Inflammation. Mediators Inflammation (2016) 2016:3635809. doi: 10.1155/2016/3635809
45. Esaki H, Ewald DA, Ungar B, Rozenblit M, Zheng X, Xu H, et al. Identification of novel immune and barrier genes in atopic dermatitis by means of laser capture microdissection. J Allergy Clin Immunol (2015) 135:153–63. doi: 10.1016/j.jaci.2014.10.037
46. Tsoi LC, Rodriguez E, Degenhardt F, Baurecht H, Wehkamp U, Volks N, et al. Atopic Dermatitis Is an IL-13-Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J Invest Dermatol (2019) 139:1480–9. doi: 10.1016/j.jid.2018.12.018
47. Wollenberg A, Kraft S, Hanau D, Bieber T. Immunomorphological and Ultrastructural Characterization of Langerhans Cells and a Novel, Inflammatory Dendritic Epidermal Cell (IDEC) Population in Lesional Skin of Atopic Eczema. J Invest Dermatol (1996) 106:446–53. doi: 10.1111/1523-1747.ep12343596
48. Suarez-Farinas M, Dhingra N, Gittler J, Shemer A, Cardinale I, de Guzman Strong C, et al. Intrinsic atopic dermatitis shows similar TH2 and higher TH17 immune activation compared with extrinsic atopic dermatitis. J Allergy Clin Immunol (2013) 132:361–70. doi: 10.1016/j.jaci.2013.04.046
49. Guttman-Yassky E, Nograles KE, Krueger JG. Contrasting pathogenesis of atopic dermatitis and psoriasis–part II: immune cell subsets and therapeutic concepts. J Allergy Clin Immunol (2011) 127:1420–32. doi: 10.1016/j.jaci.2011.01.054
50. Kawakami T, Ando T, Kimura M, Wilson BS, Kawakami Y. Mast cells in atopic dermatitis. Curr Opin Immunol (2009) 21:666–78. doi: 10.1016/j.coi.2009.09.006
51. Jabbari A, Suárez-Fariñas M, Dewell S, Krueger JG. Transcriptional profiling of psoriasis using RNA-seq reveals previously unidentified differentially expressed genes. J Invest Dermatol (2012) 132:246–9. doi: 10.1038/jid.2011.267
52. Nangole FW, Agak GW. Keloid pathophysiology: fibroblast or inflammatory disorders? JPRAS Open (2019) 22:44–54. doi: 10.1016/j.jpra.2019.09.004
53. Huguier V, Giot J-P, Simonneau M, Levillain P, Charreau S, Garcia M, et al. Oncostatin M exerts a protective effect against excessive scarring by counteracting the inductive effect of TGFβ1 on fibrosis markers. Sci Rep (2019) 9:2113. doi: 10.1038/s41598-019-38572-0
54. Zhou Y, Sun Y, Hou W, Ma L, Tao Y, Li D, et al. The JAK2/STAT3 pathway inhibitor, AG490, suppresses the abnormal behavior of keloid fibroblasts in vitro. Int J Mol Med (2020) 46:191–200. doi: 10.3892/ijmm.2020.4592
55. Lee SY, Kim EK, Seo HB, Choi JW, Yoo JH, Jung KA, et al. IL-17 Induced Stromal Cell-Derived Factor-1 and Profibrotic Factor in Keloid-Derived Skin Fibroblasts viathe STAT3 Pathway. Inflammation (2020) 43:664–72. doi: 10.1007/s10753-019-01148-1
56. Kurzinski K, Torok KS. Cytokine profiles in localized scleroderma and relationship to clinical features. Cytokine (2011) 55:157–64. doi: 10.1016/j.cyto.2011.04.001
57. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harbor Perspect Biol (2014) 6:a016295–a. doi: 10.1101/cshperspect.a016295
58. Fielding Ceri A, Jones Gareth W, McLoughlin Rachel M, McLeod L, Hammond Victoria J, Uceda J, et al. Interleukin-6 Signaling Drives Fibrosis in Unresolved Inflammation. Immunity (2014) 40:40–50. doi: 10.1016/j.immuni.2013.10.022
59. Maeda D, Kubo T, Kiya K, Kawai K, Matsuzaki S, Kobayashi D, et al. Periostin is induced by IL-4/IL-13 in dermal fibroblasts and promotes RhoA/ROCK pathway-mediated TGF-beta1 secretion in abnormal scar formation. J Plast Surg Handb Surg (2019) 53:288–94. doi: 10.1080/2000656X.2019.1612752
60. Crawford J, Nygard K, Gan BS, O’Gorman DB. Periostin induces fibroblast proliferation and myofibroblast persistence in hypertrophic scarring. Exp Dermatol (2015) 24:120–6. doi: 10.1111/exd.12601
61. Yamaguchi Y. Periostin in Skin Tissue Skin-Related Diseases. Allergol Int (2014) 63:161–70. doi: 10.2332/allergolint.13-RAI-0685
62. Maxfield AZ, Landegger LD, Brook CD, Lehmann AE, Campbell AP, Bergmark RW, et al. Periostin as a Biomarker for Nasal Polyps in Chronic Rhinosinusitis. Otolaryngol Head Neck Surg (2018) 158:181–6. doi: 10.1177/0194599817737967
63. Yamaguchi Y, Ono J, Masuoka M, Ohta S, Izuhara K, Ikezawa Z, et al. Serum periostin levels are correlated with progressive skin sclerosis in patients with systemic sclerosis. Br J Dermatol (2013) 168:717–25. doi: 10.1111/bjd.12117
64. He H, Suryawanshi H, Morozov P, Gay-Mimbrera J, Del Duca E, Kim HJ, et al. Single-cell transcriptome analysis of human skin identifies novel fibroblast subpopulation and enrichment of immune subsets in atopic dermatitis. J Allergy Clin Immunol (2020) 145:1615–28. doi: 10.1016/j.jaci.2020.01.042
65. Greenblatt MB, Aliprantis AO. The Immune Pathogenesis of Scleroderma: Context Is Everything. Curr Rheumatol Rep (2012) 15:297. doi: 10.1007/s11926-012-0297-8
66. Del Duca E, Ruano Ruiz J, Pavel AB, Sanyal RD, Song T, Gay-Mimbrera J, et al. Frontal fibrosing alopecia shows robust T helper 1 and Janus kinase 3 skewing. Br J Dermatol (2020). doi: 10.1111/bjd.19040
67. Oriente A, Fedarko NS, Pacocha SE, Huang S-K, Lichtenstein LM, Essayan DM. Interleukin-13 Modulates Collagen Homeostasis in Human Skin and Keloid Fibroblasts. J Pharmacol Exp Ther (2000) 292:988.
68. Akdis CA, Arkwright PD, Brüggen MC, Busse W, Gadina M, Guttman-Yassky E, et al. Type 2 immunity in the skin and lungs. Allergy (2020) 75:1582–605. doi: 10.1111/all.14318
69. Avdeeva K, Fokkens W. Precision Medicine in Chronic Rhinosinusitis with Nasal Polyps. Curr Allergy Asthma Rep (2018) 18:25. doi: 10.1007/s11882-018-0776-8
70. Workman AD, Kohanski MA, Cohen NA. Biomarkers in Chronic Rhinosinusitis with Nasal Polyps. Immunol Allergy Clin North Am (2018) 38:679–92. doi: 10.1016/j.iac.2018.06.006
71. Hamilton JD, Suarez-Farinas M, Dhingra N, Cardinale I, Li X, Kostic A, et al. Dupilumab improves the molecular signature in skin of patients with moderate-to-severe atopic dermatitis. J Allergy Clin Immunol (2014) 134:1293–300. doi: 10.1016/j.jaci.2014.10.013
72. Liu YJ. Thymic stromal lymphopoietin and OX40 ligand pathway in the initiation of dendritic cell-mediated allergic inflammation. J Allergy Clin Immunol (2007) 120:238–44; quiz 45-6. doi: 10.1016/j.jaci.2007.06.004
73. Wu J, Guttman-Yassky E. Efficacy of biologics in atopic dermatitis. Expert Opin Biol Ther (2020) 20(5):525–38. doi: 10.1080/14712598.2020.1722998
74. Renert-Yuval Y, Guttman-Yassky E. New treatments for atopic dermatitis targeting beyond IL-4/IL-13 cytokines. Ann Allergy Asthma Immunol (2020) 124:28–35. doi: 10.1016/j.anai.2019.10.005
75. Guttman-Yassky E, Pavel AB, Zhou L, Estrada YD, Zhang N, Xu H, et al. GBR 830, an anti-OX40, improves skin gene signatures and clinical scores in patients with atopic dermatitis. J Allergy Clin Immunol (2019) 144:482–93.e7. doi: 10.1016/j.jaci.2018.11.053
76. Nakagawa H, Iizuka H, Nemoto O, Shimabe M, Furukawa Y, Kikuta N, et al. Safety, tolerability and efficacy of repeated intravenous infusions of KHK4083, a fully human anti-OX40 monoclonal antibody, in Japanese patients with moderate to severe atopic dermatitis. J Dermatol Sci (2020) 99:82–9. doi: 10.1016/j.jdermsci.2020.06.005
77. Simpson EL, Parnes JR, She D, Crouch S, Rees W, Mo M, et al. Tezepelumab, an anti–thymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: A randomized phase 2a clinical trial. J Am Acad Dermatol (2019) 80:1013–21. doi: 10.1016/j.jaad.2018.11.059
78. Kang S, Tanaka T, Narazaki M, Kishimoto T. Targeting Interleukin-6 Signaling in Clinic. Immunity (2019) 50:1007–23. doi: 10.1016/j.immuni.2019.03.026
79. Xue H, McCauley RL, Zhang W. Elevated Interleukin-6 Expression in Keloid Fibroblasts. J Surg Res (2000) 89:74–7. doi: 10.1006/jsre.1999.5805
80. Ghazizadeh M, Tosa M, Shimizu H, Hyakusoku H, Kawanami O. Functional implications of the IL-6 signaling pathway in keloid pathogenesis. J Invest Dermatol (2007) 127:98–105. doi: 10.1038/sj.jid.5700564
81. Solimani F, Meier K, Ghoreschi K. Emerging Topical and Systemic JAK Inhibitors in Dermatology. Front Immunol (2019) 10:2847. doi: 10.3389/fimmu.2019.02847
82. He H, Guttman-Yassky E. JAK Inhibitors for Atopic Dermatitis: An Update. Am J Clin Dermatol (2019) 20:181–92. doi: 10.1007/s40257-018-0413-2
83. Lee YS, Liang YC, Wu P, Kulber DA, Tanabe K, Chuong CM, et al. STAT3 signalling pathway is implicated in keloid pathogenesis by preliminary transcriptome and open chromatin analyses. Exp Dermatol (2019) 28:480–4. doi: 10.1111/exd.13923
Keywords: RNA-seq, keloids, immune, Th2, JAK3, inflammation
Citation: Wu J, Del Duca E, Espino M, Gontzes A, Cueto I, Zhang N, Estrada YD, Pavel AB, Krueger JG and Guttman-Yassky E (2020) RNA Sequencing Keloid Transcriptome Associates Keloids With Th2, Th1, Th17/Th22, and JAK3-Skewing. Front. Immunol. 11:597741. doi: 10.3389/fimmu.2020.597741
Received: 21 August 2020; Accepted: 19 October 2020;
Published: 23 November 2020.
Edited by:
Heiko Mühl, Goethe University Frankfurt, GermanyReviewed by:
Christian David Sadik, University of Lübeck, GermanyCopyright © 2020 Wu, Del Duca, Espino, Gontzes, Cueto, Zhang, Estrada, Pavel, Krueger and Guttman-Yassky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emma Guttman-Yassky, RW1tYS5HdXR0bWFuQG1vdW50c2luYWkub3Jn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.