 Peter C. Hart
Peter C. Hart Ibraheem M. Rajab
Ibraheem M. Rajab May Alebraheem
May Alebraheem Lawrence A. Potempa
Lawrence A. Potempa- Roosevelt University, College of Science, Health and Pharmacy, Schaumburg, IL, United States
Cancer disease describes any pathology involving uncontrolled cell growth. As cells duplicate, they can remain localized in defined tissues, forming tumor masses and altering their microenvironmental niche, or they can disseminate throughout the body in a metastatic process affecting multiple tissues and organs. As tumors grow and metastasize, they affect normal tissue integrity and homeostasis which signals the body to trigger the acute phase inflammatory response. C-reactive protein (CRP) is a predominant protein of the acute phase response; its blood levels have long been used as a minimally invasive index of any ongoing inflammatory response, including that occurring in cancer. Its diagnostic significance in assessing disease progression or remission, however, remains undefined. By considering the recent understanding that CRP exists in multiple isoforms with distinct biological activities, a unified model is advanced that describes the relevance of CRP as a mediator of host defense responses in cancer. CRP in its monomeric, modified isoform (mCRP) modulates inflammatory responses by inserting into activated cell membranes and stimulating platelet and leukocyte responses associated with acute phase responses to tumor growth. It also binds components of the extracellular matrix in involved tissues. Conversely, CRP in its pentameric isoform (pCRP), which is the form quantified in diagnostic measurements of CRP, is notably less bioactive with weak anti-inflammatory bioactivity. Its accumulation in blood is associated with a continuous, low-level inflammatory response and is indicative of unresolved and advancing disease, as occurs in cancer. Herein, a novel interpretation of the diagnostic utility of CRP is presented accounting for the unique properties of the CRP isoforms in the context of the developing pro-metastatic tumor microenvironment.
Introduction
Cancer is a pervasive disease affecting many people across the globe. Worldwide, the prevalence of people living within 5 years of receiving a cancer diagnosis is estimated to be 43.8 million. In 2018, 18.1 million new cancer cases were diagnosed worldwide, and 9.6 million deaths were reported. The most reported new cases involve the lung and breast, with colorectal cancer third, prostate cancer fourth, and stomach cancer fifth (1, 2). Each reported cancer is associated with variable overall life expectancies, years of life lost, and 5-year survival rate (3–6).
The term cancer describes a physiological condition in which body cells grow and replicate in an uncontrolled and unregulated fashion. While cancer is often categorized by the tissue in which the predominant uncontrolled growth originates, the disease is fundamentally defined by a loss of basic cellular processes that regulate proliferation. When cells proliferate, their mass and the mass of ancillary tissues in which the growth occurs increases, leading to localized areas of disrupted tissues which can overwhelm the natural protective immune responses that are designed to identify and destroy aberrantly growing cells, and to repair damaged tissues. Hence, in evaluating cancerous disease, consideration must be given not only aberrant cells, but the tissues, vasculature, and immune responses in which the malignant growth occurs. If cancerous growth is relatively slow and localized, treatment options include surgical resection and radiotherapy. If cancerous growths become unregulated and rapid, involved tissues are compromised and weakened, and tumor cells can break free to metastasize to other tissues. Treatments options for disseminated cancers become limited and prognosis for long term survival decreases. Strategies for treating cancers must therefore include an assessment of how tissue structures involved in cancerous growths may be compromised and how natural barriers might be strengthened to counteract the growth of tumor masses and to better coordinate the immune responses against tumor development.
Both innate and adaptive immune responses exist to recognize and remove diseased cells, primarily involving innate system humoral factors and natural killer cells. Innate responses involve neutrophils, M1 macrophages, natural killer cells, interferons, cytokines, and the acute phase response (7). Adaptive responses involve antibodies developed to specifically recognize malignant cells, and direct cytotoxic T-lymphocytes to specifically recognize intracellular abnormalities associated with uncontrolled cellular duplication that often display tumor specific antigens, leading to cell-mediated tumor cell apoptosis (8). However, a number of cancers present with “cold” tumors, showing limited or no infiltration of activated T cells within the tumor compartment (9). Many cancers are known to produce suppression factors that target effective immune system surveillance, thus giving cancer cells an advantage in the balance between continued proliferation and leukocyte-mediated apoptosis. The exciting development of biotherapeutic reagents known as checkpoint inhibitors, which function by neutralizing the immunosuppressive factors [i.e. by directly binding to Program Death Ligand-1 (PDL-1) or its receptor (PD-1)], shifts the balance back to natural immune function, slowing or stopping tumor cell growth in treated individuals (10). This is likely due to the prevention of PD-1-dependent T cell exhaustion; therefore, blockade increases the presence of cytotoxic T cells in the microenvironment capable of tumor cell lysis through perforin and granenzyme secretions (11). While promoting the cytotoxic capacity of the local immune response can slow or prevent tumor growth in some cases (12), it is notable that certain distributions of leukocytes can also favor a state of chronic inflammation that conversely would promote genomic instability, survival, and proliferation of tumor cells that can confer tumor development and progression [reviewed in (13)]. Thus, the dualistic nature of the immune system in cancer suggests that tight regulation of immune and inflammatory responses is necessary to prevent tumor growth and metastasis.
This report focuses on innate immunity and the acute phase response (APR) to cancerous growth. More specifically, it provides a new perspective on the diagnostic and therapeutic value of the prototypic acute phase reactant, C-reactive protein (CRP), and cancer. The concepts presented herein represent a unifying understanding of the value of CRP as a key protein of the natural inflammatory response and how both beneficial and pathological inflammation contribute to cancerous disease as well as the pathologies that involve any tissue damaged by trauma or disease.
Use of CRP as a Diagnostic Marker in Cancer
The US Food and Drug Administration (FDA) guidance for assessing the relevance of CRP as a diagnostic marker in any disease involving a host defense inflammatory response, describes CRP as a single protein that can be reliably measured from blood using various qualitative, semi-quantitative and quantitative measurements (14). Two concentration thresholds were established: 1.) conventional CRP levels (defined as CRP levels ≥ 10 µg/ml), and 2.) high sensitivity CRP levels (i.e. hsCRP; defined as CRP levels < 10 µg/ml). FDA guidance does not associate either CRP or hsCRP levels with specific diseases or risks for disease, cautioning that any interpretation of CRP levels must include the context of a specific clinical evaluation. However, high levels of CRP were found to be strongly associated with advanced disease severity in numerous cancer types (elaborated below). Hence, CRP measurements have potential utility as a diagnostic tool in assessing disease status and progression, including in cancer.
A comprehensive review of the literature on the diagnostic significance and therapeutic value of CRP blood levels in cancer proved to be problematic. Reported CRP levels varied from <1 μg/ml to more than 175 μg/ml [(15); see Table 1 and Figure 1] and were most often reported with reference to the tissues affected with cancerous growths (e.g. lung, breast, gastrointestinal, esophageal, head and neck, sexual and reproductive organs, renal, pancreas, and blood). One complication to interpreting the value of CRP measurements in any such disease condition is that its relative blood level can change rapidly and in direct relationship to the stage and extent of progressive disease and/or associated complication (e.g. infections) that may accompany the disease progression. Another limitation is that many of these studies did not fully characterize the relationship between CRP and the discrete variables that define cancer staging (e.g., tumor size in TNM staging of breast cancer, etc.); however, the associations drawn between CRP with prognosis and disease severity are consistent among cancers and indicate the potential utility of CRP as a prognostic index. Reports of CRP level in relationship to blood albumin level (i.e. the CRP/albumin ratio) as a novel prognostic index for survival for cervical cancer have appeared (64). In addition, the prognostic value of CRP in relationship to the neutrophil/lymphocyte ratio has also been proposed. Since CRP levels change rapidly during disease, some studies describe a “maximal CRP level” as a relevant diagnostic index. However, such values must be evaluated with reference to how prolonged cancer disease extends, the treatments used during disease, and the levels measured both before and after any surgical intervention. Nonetheless, these maximal CRP values have been discussed as one criterion for assessing the extent of disease and the prognosis for recovery (27, 102).
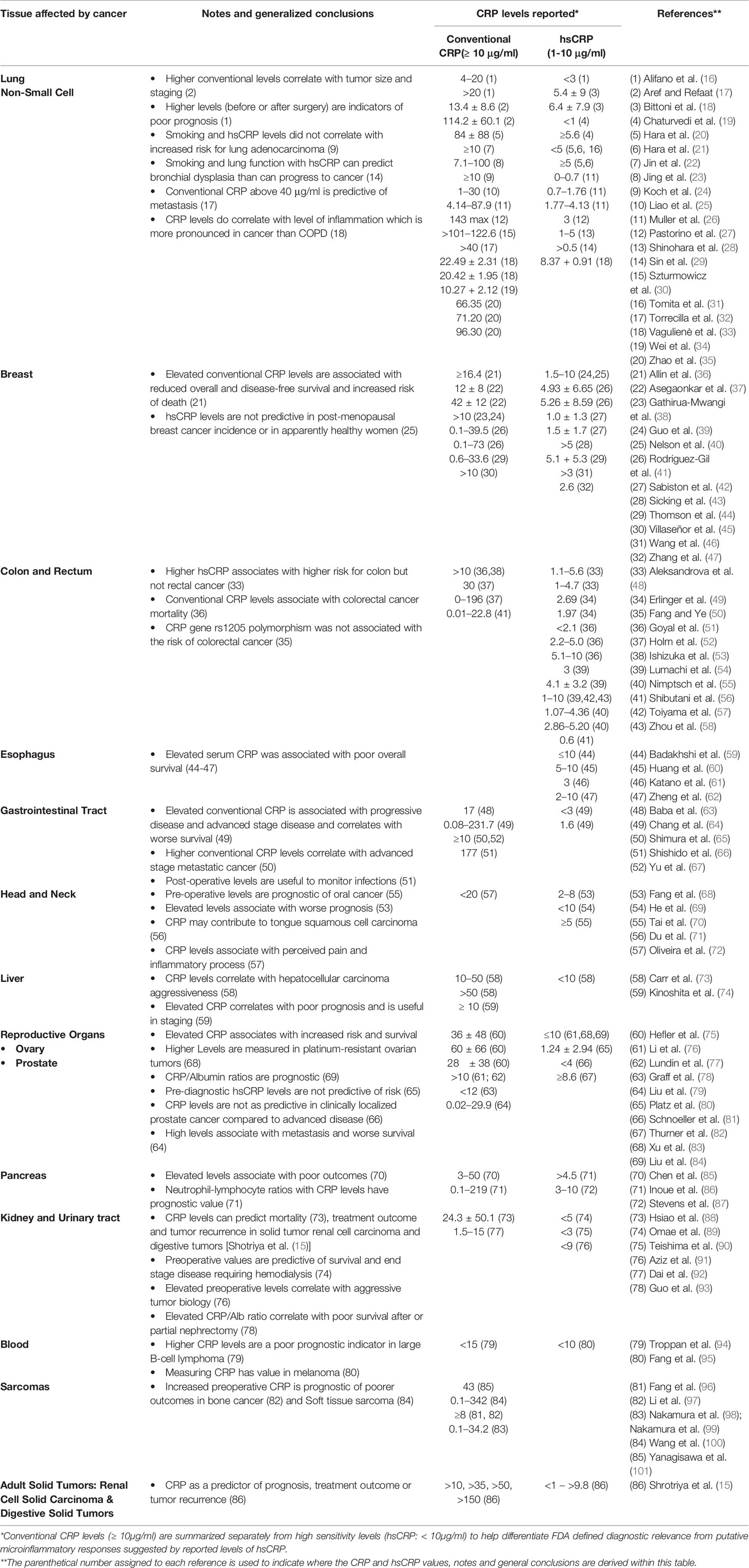
Table 1 Overview of CRP values reported in various cancers distinguishing Conventional CRP levels (≥ 10 μg/ml) from High Sensitivity (hsCRP) levels (< 10 μg/ml).
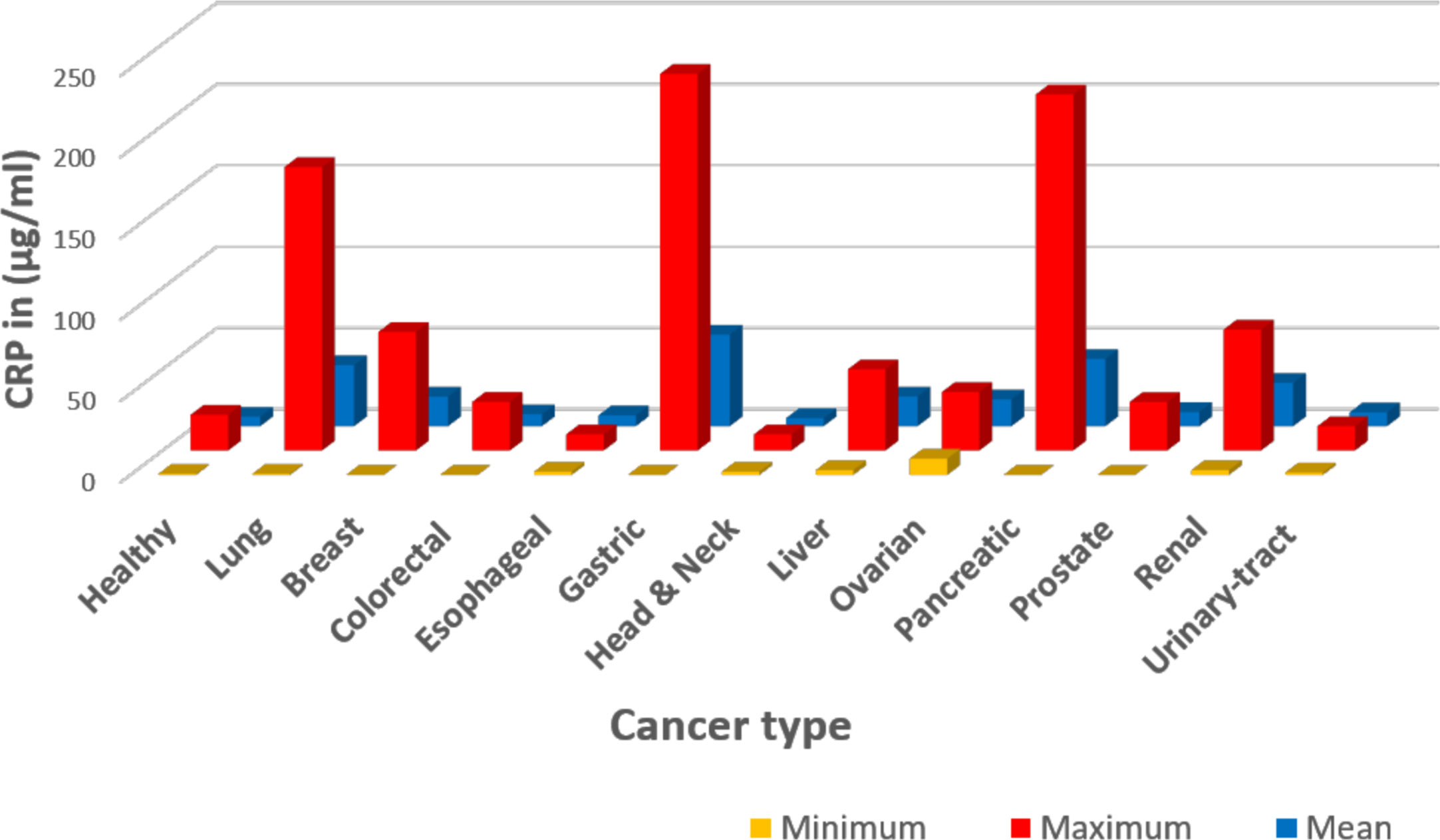
Figure 1 Graphic representation of data summarized in Table 1. The CRP values were extracted from published references as detailed in Table 1. In this presentation, data were tabulated in Microsoft Excel based on cancer type, then the Excel functions were used in calculations: Minimum describes the lowest reported level; Maximum describes the highest reported level; and Mean describes the average of the reported values. One limitation of the reported data summarized here is the lack of specific clinical conditions ongoing when (or how frequent) CRP values were collected and measured.
Another complicating factor in interpreting the diagnostic and therapeutic value of CRP in cancer disease is the current focus on the value and significance of “high sensitivity CRP” levels (i.e. hsCRP). Baseline levels of CRP in purportedly healthy individuals are generally described as being <1–2 μg/ml. Some studies reviewed and included in reports summarized in Table 1 describe and differently interpret cohort groups with hsCRP levels between 1–3 and >3 μg/ml. While FDA offers no guidance on the diagnostic significance of such values in any disease, including cancer, it is important to note that numerous reports have appeared that associate baseline hsCRP levels more generally with populations of individuals grouped by gender, age, ethnicity, degree of fitness, and obesity. Indeed, with sensitive assays for CRP measurement now readily available, including point-of-care measurements where CRP values can be generated from a finger stick drop of blood in minutes, thousands of reports have appeared reporting that slightly elevated hsCRP levels are reflective of increased risk for developing and exacerbating cancerous growth (see Table 1 for a list of references that discuss hsCRP in the defined cancers).
Table 1 offers one literature overview summarizing reported CRP blood levels as a function of cancer type, conventional CRP levels reported (i.e. CRP levels ≥ 10 μg/ml), high sensitivity CRP levels reported [hsCRP levels reported (< 10 μg/ml)] and stage of cancer disease and/or complications. A graphic presentation of reported CRP maximum, minimum and mean (average) values as a function of cancer type is presented in Figure 1. A simplified interpretation of all the data included in Table 1 is presented in Table 2.

Table 2 Most consistent interpretations of the diagnostic significance of CRP in cancer.
CRP—A Marker of Tissue Damage First and Inflammation Second
Systemic CRP exists as a pentameric structure (pCRP) made up of five subunits, each containing calcium dependent bindings sites that interact with exposed phosphocholine ligand (PC) which can be expressed on activated plasma membrane (103). Exposure of PC groups requires phospholipid remodeling such as occurs with Phospholipase A2 activity or oxidation of acyl chains (104). When pCRP binds to membrane anchored PC, juxtaposed apolar bonding energies contribute to the dissociation of the pentameric isoform into the modified, monomeric isoform (i.e. mCRP) which undergoes structural rearrangement to express a cholesterol binding site as is found in lipid rafts (Figure 2) (107, 110).
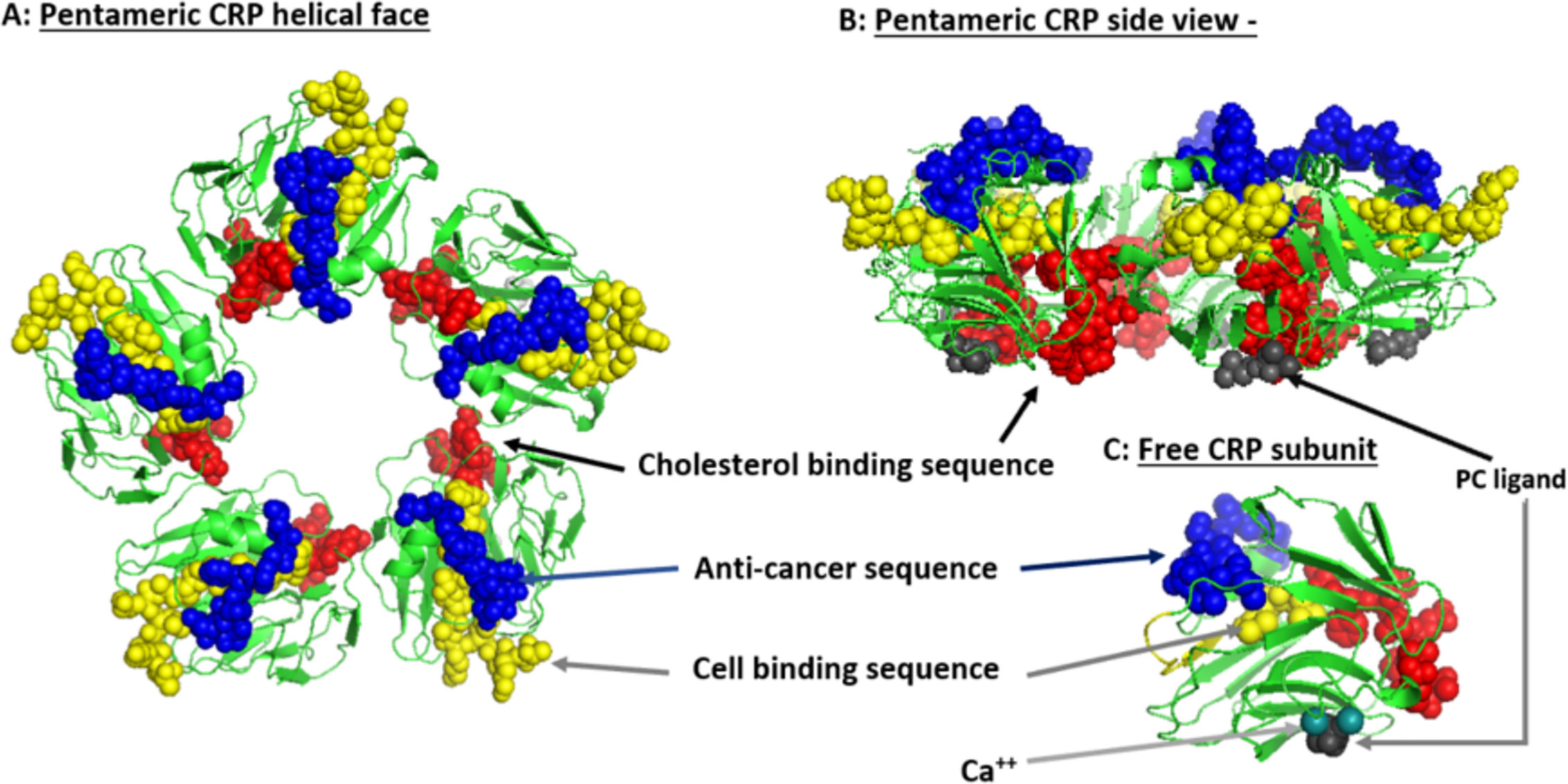
Figure 2 Structural features of serum-soluble pentameric CRP. (A) illustrates the location and orientation peptide sequences in CRP reported to have cell-binding activity (shown in yellow and involving 27TKPLKAFTVCLH38) (105), anti-cancer activity (shown in blue and involving 176LGGPFSPNVL185) (106), cholesterol binding activity (shown in red and involving 35VCLHFYTELSSTR47), and which also controls CRP binding to apolipoprotein B, C1q, fibronectin, and collagen (107), in relationship to the phosphocholine (PC) binding face (PC groups shown in gray and involving residues L64, F66, and T76) and bound calcium ions (two per subunit, juxtaposed to each PC binding sites and involves residue E147) (PDB code: 1B09; PC and calcium residues as defined by Thompson et al. (108) and Shrive et al. (109), respectively). (B) illustrates the orientation of these same residues when the discoid protein is laid flat (i.e. side view). (C) shows the orientation of same sequences on the isolated pCRP subunit (note: the exact orientation of these residues on the conformationally changed mCRP subunit has not been determined). The PC ligand binds in a shallow binding pocket controlled by calcium ions, with all PC sites on one face of the flattened discoid structure. The cholesterol binding sequences are near the PC binding sites so that when pCRP binds membrane associated PC, the cholesterol binding sequence is brought into proximity with intra-membraneous cholesterol (in lipid rafts) contributing to the conversion of pCRP into mCRP. The cell binding and anti-cancer peptides are oriented on the opposite face of the discoid protein where they can interact with effector leukocytes and activated inflammatory responses.
Since the appearance and concentration of pCRP in blood is not specific for cancer types, tissue locations, or stages of disease, and since its appearance correlates with an ongoing inflammatory response, what is the common denominator that triggers this protein to appear? One reflective focus involves evaluating how CRP may affect the fibrinolytic-like responses that are recognized as hallmarks of cancer growth. Indeed, cancer has been described as “a wound that never heals” (111, 112); any discussion of cancer growth and regulation must, therefore, include an understanding of the structure and function of the extracellular matrix and connective tissues within which the cancers are found. As tumors grow, endothelial cells become activated to allow platelets, neutrophils, and blood proteins (e.g. CRP) to enter tissues as part of normal protective inflammatory response. The goal of this early response is to help control the extent of disease growth and return the tissues to healthy homeostasis. Possible pathways by which CRP may participate in this host defense response include its binding reactivity with 1.) PC ligands which become accessible on stimulated endothelial cell membranes (113, 114), 2.) fibronectin (115–117), 3.) laminin (118), and 4.) collagen (107). CRP is also known to activate and regulate complement activity and bind immune complexes (119). Many reports also detail the interactions of CRP with endothelial cells (119), platelets (120), neutrophils (121), monocytes/macrophages (117, 122), epithelial cells (123), and fibroblasts (124).
For many decades, the exact role for CRP in the host defense APR remained undefined and controversial as different groups studying similar systems came to opposite conclusions. More recently, as CRP has been shown to exist in more than just a serum soluble cyclic pentamer disc configuration, it is now apparent that the effects of CRP on cell behavior and the tissue microenvironment are clearly dependent on its structural conformation. The highly soluble circulating pentameric CRP (pCRP) binds to PC on the cell surface (e.g., endothelial cells activated by inflammatory signals), which initiates the dissociation of pCRP into its distinct modified, monomeric isoform (mCRP) which has notably reduced aqueous solubility. mCRP will self-aggregate and deposit in tissues or will internalize into plasma membranes and bind cholesterol. Cells activated by mCRP are known to stimulate intracellular signaling pathways, including activating the NFκB transcription factor which propagates intracellular effects known as hallmarks of inflammation (125). Careful comparison studies have now established that mCRP (rather than pCRP) can interact with integral extracellular matrix (ECM) proteins (e.g., fibronectin, collagen) (98). With the awareness and understanding of both the pCRP and mCRP isoforms (including an understanding of how mCRP can be derived from pCRP), and the different roles each has on both cellular and tissue based components involved in acute inflammatory responses occurring in cancer, a consistent role for CRP as a diagnostic marker becomes apparent. A summary of the interactions of CRP with cell types and components of the extracellular environment is summarized in Figure 3.
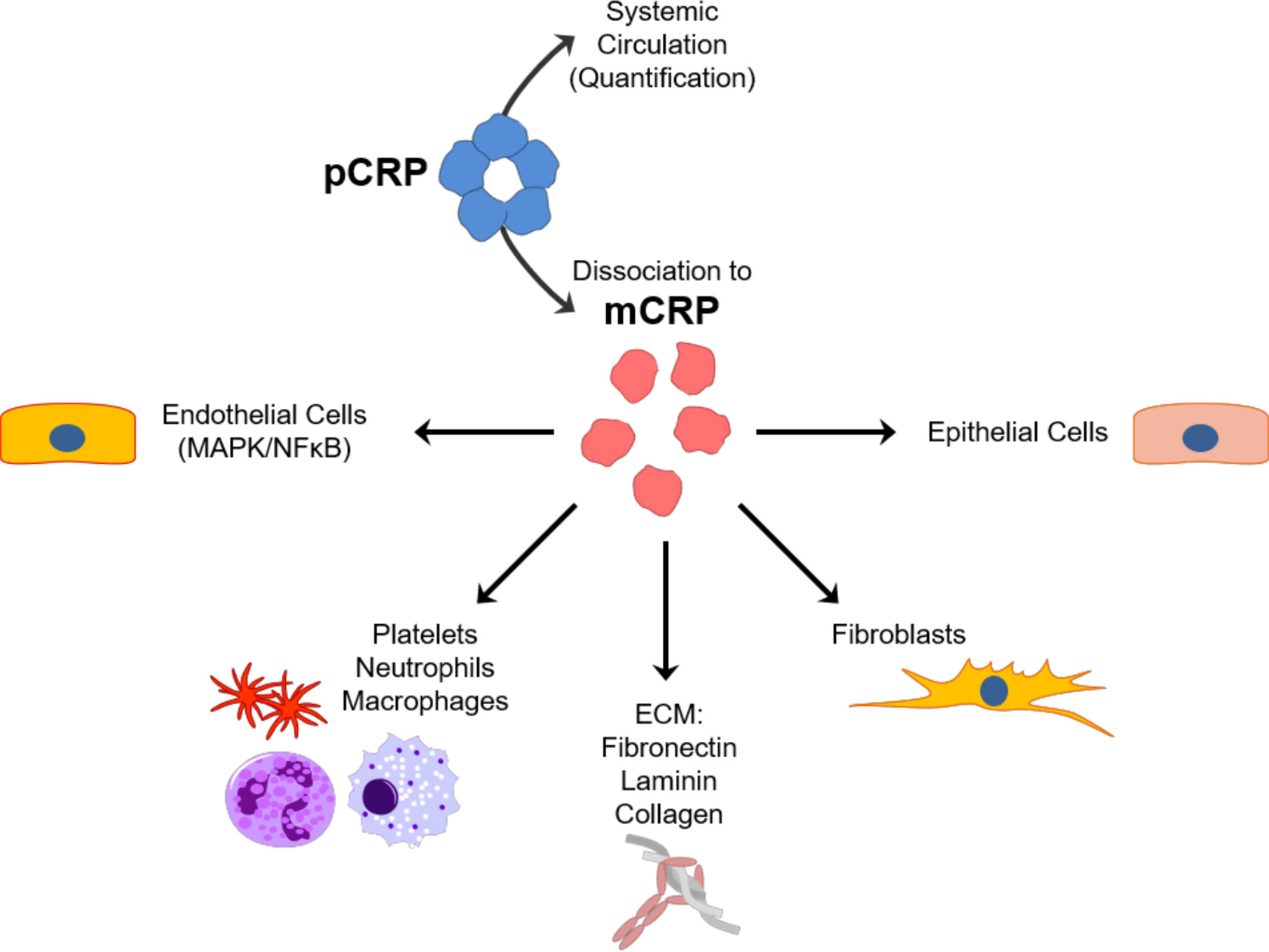
Figure 3 Schematic representation of the predominant interactions of pCRP and mCRP isoforms. Pentameric CRP (pCRP) released from hepatocytes due to inflammation circulates through the systemic vasculature and serves as the pool of quantifiable CRP that is used in diagnostic testing. pCRP, however, once dissociated to monomeric CRP (mCRP) at lipid rafts of cells involved in inflammatory responses instead is highly biologically active. mCRP in turn interacts with a number of different cell types at the sites of inflammation, including endothelial cells, epithelial cells, fibroblasts and immune cells (platelets, neutrophils, macrophages) as well as components of the extracellular matrix (ECM) such as fibronectin, laminin and collagen.
The mCRP isoform can be formed from the pCRP isoform when membranes are activated and cellular responses are stimulated as occurs when tissue are damaged by any means (e.g. trauma, disease, cancerous growth). Once mCRP is formed, it will not reform pCRP; mCRP is readily degraded by proteolytic enzymes and peptides formed by its degradation feedback inhibit many of the acute phase responses stimulated by intact mCRP (126–129). In the earliest minutes of the APR, the rate of conversion of pCRP to mCRP is rapid. Over time, however (hours to days) the rate of conversion of pCRP to mCRP diminishes over time, resulting in quantifiable increases of pCRP in blood [recently reviewed in (130)]. If any injury persists and inflammatory mediators (such as IL-6 and IL-1β) that signal hepatocytes to continue to synthesize acute phase proteins, quantifiable increases in plasma levels of pCRP will result. Increased levels of pCRP, therefore, suggest less pCRP is converted into mCRP. Since mCRP is a potent amplifier of the acute inflammatory response (110, 120, 131–133), any condition that limits its expression will cause a reduction in natural host defense responses. In the case of advancing tumor growth, this would lead to chronic inflammatory signaling that potentiates the “wound that never heals”. As affected tissues are still producing signals that direct the synthesis and release of pCRP, higher blood levels of pCRP are reflective of the persistence and severity of tissue damage associated with cancer growth and progression.
In line with this, CRP levels in blood do indeed correlate with the degree of inflammatory tissue involvement. In soft tissue sarcomas, CRP levels were associated with the degree of tumor infiltration determined by magnetic resonance imaging (MRI) as well as disease-specific survival (99). Similarly, tissue pathologies associated with COVID-19 disease complications identified by computerized tomography (CT) technology were also significantly associated with CRP levels and, importantly, CRP had high sensitivity and specificity to predict severity of the disease (134).
Using this tissue-based perspective as a common denominator, readers are encouraged to interpret CRP diagnostic levels in any clinical situation by first focusing on alterations in tissue structures and second by assessing how it affects or regulates the inflammation that ensues. A brief overview of the extracellular matrix structures and ways that CRP may interact with such structures during inflammatory responses to cancer disease is included below.
The Extracellular Tissue Microenvironment, Acute Phase Response, and Inflammation
In all animals, including humans, the first line of defense against disease is the structural connective tissue that not only presents a barrier to pathogens and toxins, but also contributes an appropriate macromolecular matrix for coordinated biochemical and immunological host defense reactions. Connective tissues include fibrous proteins, various cells, amorphous ground substance (e.g. proteoglycans and glycosaminoglycans), plasma constituents, ions, and water. Connective tissue can be loose or dense, regular or irregular, fibrous or elastic depending on the types of proteins and polysaccharides that are secreted locally or accumulate in the specific space filled by the connective tissue. The organization and interactions of the components, not only within the framework space but also at boundaries and surfaces, define the physical properties and function of each connective tissue. In tissues as wide ranging as skin and bone, connective tissues form an architectural framework (i.e. the extracellular matrix or ECM) that serves not only as an inert space-filling scaffold, but as a physical structure that plays a dynamic role in organizing and regulating the physiological responses that occur within each tissue. While providing mechanical structure through the deposition of numerous molecules (e.g., collagens, laminins), the ECM also acts as an intermediary space in which growth factors, cytokines, metabolites, and other secreted factors can communicate between cells within tissue compartments (135–137).
When connective tissue is injured either by incision, accident, xenobiotic stress or disease, the APR is activated to stimulate innate host defenses to the injury and the wound healing process so to efficiently and effectively repair the injured tissue (138). These processes must work in concert with hematological and immunological mechanisms that are triggered to defend the body from anything that might threaten the homeostasis affecting not only localized tissues, but of the entire organism as well. The APR reaction involves the initial production of signals locally (e.g., IL-6, IL-1β) at the site of injury, which, once secreted into the extracellular milieu, can have extensive physiological effects on cells within the local tissue or act systemically to promote production of acute phase proteins, such as hepatic CRP (138). This frequent early event during wound repair is characterized by such inflammatory responses that in turn facilitate the rapid recruitment and activation of immune cells, such as neutrophils and other leukocytes. Neutrophils migrate into the wound and scavenge for debris and foreign matter that must be removed. In uncomplicated repair, the movement of neutrophils into a wound is transient and resolved prior to the in-growth of vascular tissue (i.e. granulation tissue). If foreign materials are present, the neutrophil response may persist and thus complicate the wound healing process by having the conflicting processes of removal (scavenging) and repair occurring simultaneously. In such an event, the wound healing process is compromised and can lead to a greater amount of scar tissue and regenerated tissue that has only a percentage of the original tissue’s functional activity (139, 140).
Depending on the extent of separation of the edges of the wound, healing can begin from the sides inward, or from the base upward. During the first few days of wound repair, epithelial cells and fibroblasts migrate across the wound surface into the regenerating tissue where they proliferate and differentiate. Such cells are specialized for the synthesis and secretion of the ECM substances and are fundamental to the architectural repair of the tissue framework. Fibroblasts are known to be very versatile cells that can reversibly transform into highly differentiated cells required for the connective tissue within which they are found. Differentiated cells secrete the types of collagen and ground substances needed for the repair and regeneration of that tissue.
Inflammation Can Induce Cancerous Growth
Physiological inflammation that occurs during wound healing involves many of the processes associated with de novo tumor development as well as mechanisms that endow cancers to metastasize (141–143). The exact directionality of whether inflammation causes carcinogenic processes, or that tumor cells induce local inflammatory responses to facilitate their rapid growth and dissemination is unclear. Indeed, it is likely that there are reciprocal interactions between the host and tumor mediated through inflammatory processes that promote tumor initiation and progression. Overall, the current literature suggests that a lack of resolution to inflammation leads to chronic inflammatory signaling that is intimately linked to promoting the development and progression of cancer.
Tissues involved with cancerous growth have long been known to involve inflammation (111, 141). Mechanistic studies have demonstrated that components of inflammation, such as reactive oxygen species (ROS) and growth factors, can promote both the initiation of neoplastic cells and their proliferation (142, 144). While ROS and proteolytic enzymes produced by neutrophils and macrophages are key contributors to a favorable immune response to the stressed tissue, both superoxide anion and hydrogen peroxide have been shown to induce DNA damage that can contribute to mutagenesis, potentially giving rise to neoplastic cells (145). Unresolved (chronic) inflammation, therefore, would not only prolong immune infiltration at the site of injury, but could promote secondary genetic mutations that could exacerbate malignant conversion of otherwise benign neoplastic cells.
Chronic inflammation could also contribute to pro-tumorigenic processes by increased secretions of growth factors (e.g., PDGF, TGFβ) that promote rapid cell proliferation, as well as cytokines that stimulate cell motility (146). Such inflammatory signals have been observed to induce tumor cell epithelial-to-mesenchymal transition (EMT), a process in which epithelial cancer cells dedifferentiate and adopt a fibroblast-like phenotype to promote rapid growth and enhance pro-metastatic signaling (147, 148), in multiple cancer types. Importantly, this mesenchymal phenotype promotes the production of metalloproteinases to breakdown collagen IV and other ECM proteins to facilitate tumor cell invasion through basement membrane (149) and trans-endothelial migration as cancer cells disseminate into tissues (150, 151). EMT is also known to be associated with upregulated secretion of cytokines and chemokines that allow cancer cells to reprogram surrounding stromal cells to provide a conducive environment for growth and metastasis (152). These data suggest that continuous (chronic) inflammatory responses in any tissue could promote the development of de novo malignancies and enhancement of the capacity for tumor cells to metastasize (153). Moreover, inflammatory signaling from the developing tumor could also act systemically to promote acute phase protein production (e.g., hepatic CRP) and provide a positive feedback loop to potentiate this pro-metastatic inflammatory environment (Figure 4).
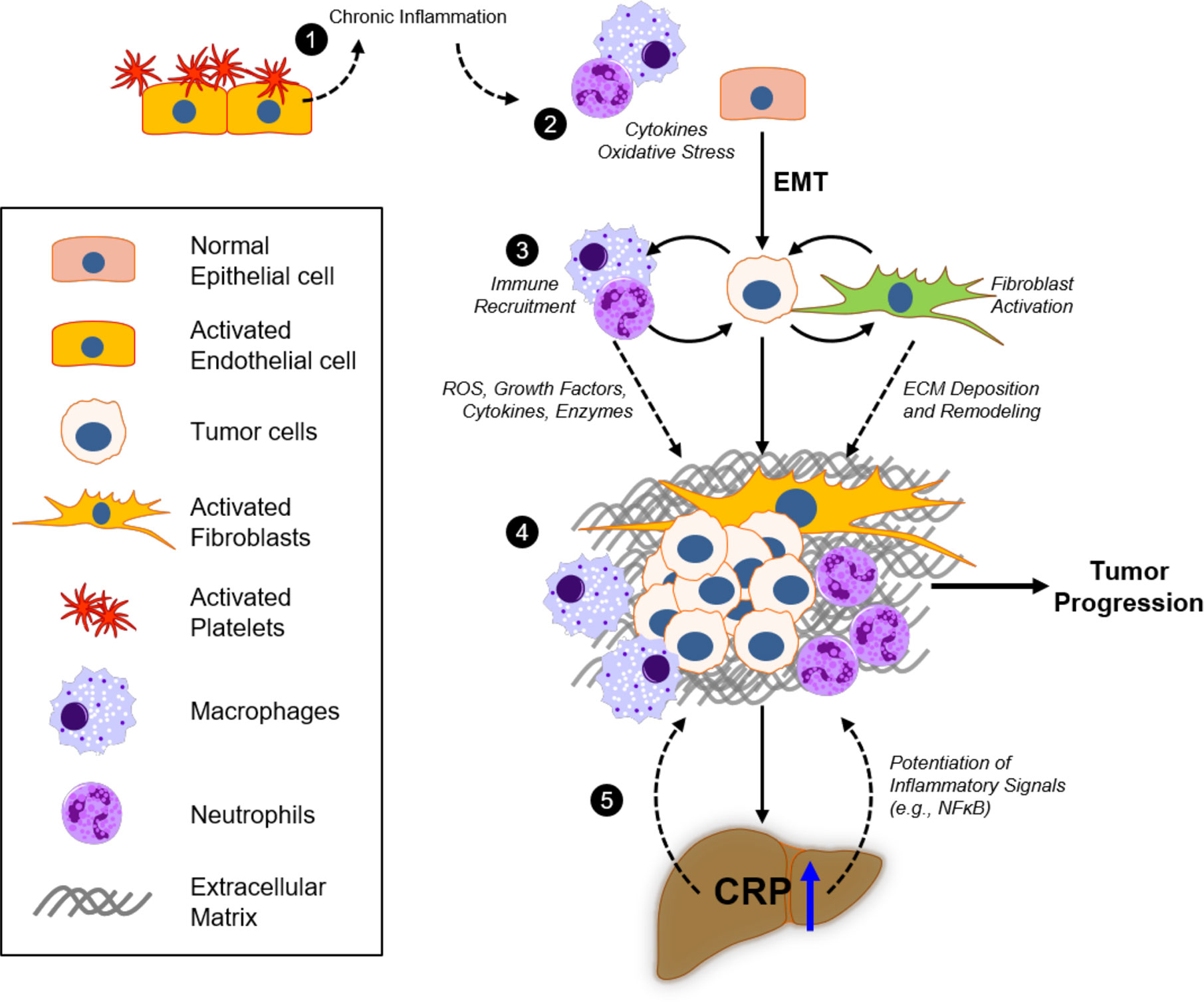
Figure 4 Inflammatory responses of CRP in the extracellular matrix and tumor microenvironment. 1) Platelet recruitment to damaged tissue and fibrin accumulation represent acute phase inflammatory responses that, if injury remains unresolved, will contribute to excessive chronic immunoreactivity. 2) Continuous oxidative stress (reactive oxygen species; ROS) and cytokine production by activated macrophages and neutrophils promote tumorigenicity in epithelial cells, which can promote epithelial-to-mesenchymal transition (EMT) as a result. 3) Deposition of the extracellular matrix (ECM) components, including fibronectin, collagen, laminin, and fibrin, in the tumor microenvironment (TME) by fibroblasts and activated immune cells modulate tumor cell proliferation and invasion. 4) Bidirectional crosstalk in the TME promotes further proliferation of tumor and stromal cells, as well as deposition and remodeling of ECM to promote tumor growth and motility. 5) Excessive cytokine release (e.g., IL-6) from the TME increases systemic circulating levels that promote hepatocyte pCRP production. pCRP secretion and subsequent mCRP-dependent inflammatory signaling (e.g., in involved endothelial cells and neutrophils), as well as its direct action on the ECM, contribute to tumor progression through ROS and cytokine signaling in the TME.
Cancer Can Induce an Inflammatory Response
Tumors that establish and grow in a tissue environment will induce an inflammatory response that will involve secretion of chemokines that promote immune recruitment (154). Tumor cells will also exploit signaling pathways of localized cells that, under non-cancerous situations would promote fibrosis and tissue repair, in order to foster rapid growth and metastatic potential of the growing tumor mass (149, 155, 156). Under normal physiologic conditions, tissue repair occurs in several phases in response to mechanical injury, infection, or irritation from xenobiotics (156–158). Epithelial or endothelial injury stimulates platelet aggregation and subsequent recruitment of neutrophils and mononuclear cells to the site of injury is then followed by the activation and differentiation of monocytes to polarized macrophages, which secrete growth factors and cytokines to facilitate wound healing through stimulating migration and activation of fibroblasts (159). Activated fibroblasts (myofibroblasts) in turn deposit collagen and remodel the extracellular matrix (160), and in concert with immune cells, promote fibrosis and the resulting formation of granulation tissue to resolve tissue damage.
The tumor microenvironment (TME) has been shown to promote tumor cell proliferation, migration, invasion and intravasation (141, 161, 162) through metabolite and cytokine secretion (163) and production of chemokines involved in immune recruitment (154, 164). Further, the TME favors differentiation of naïve monocytes to M2 macrophages which mediate fibrotic-like responses that facilitate tumor progression (165). Tumor-associated macrophages, in concert with activated fibroblasts and other stromal cell types in the TME, secrete laminin, and collagen to promote tumor cell motility as well as fibronectin, a key modulator of integrin-dependent adhesion and invasion (166–168). As observed in multiple cancers, activated stromal cells in the TME notably induce tumor cell EMT to promote not only proliferation, but also key metastatic processes, such as adhesion, migration, invasion, and colonization (163, 169, 170). Thus, the interplay between tissue structure/function and activated inflammatory responses may contribute to both protection from and exacerbation of disease.
Tumor cells that have undergone EMT secrete a number of growth factors observed during physiologic wound healing, including PDGF (171, 172), TGFβ1 (173), and fibrinogen (174), which are associated with immune cell granulation and enhanced tumor growth. Moreover, mesenchymal-like tumor cells and stromal cells common to the TME have been shown to also secrete cytokines involved in the synthesis of CRP [IL-1β (175)] as well as its secretion [IL-6 (176)], indicating a potential for systemic reaction to the progressing tumor. Locally, this crosstalk by these inflammatory mediators between tumor cells and their microenvironment promotes processes involved in metastasis in vitro (163, 169, 177), and markedly enhance the success of tumor growth and metastatic implantation in vivo (178, 179). Taken together, these studies indicate that progressing tumors enhance their development and metastasis in part through processes involved in wound healing and pro-inflammatory signaling, suggesting that cancer cell induced inflammation promotes tumor progression and thus disease severity.
The structural support for the regenerating tissue (e.g. basement membranes, non-damaged adjacent tissue) and the connections to the supporting structure tissues are important factors in the effective repair of wounded tissue. Cells growing into the matrix both influence and are influenced by the macromolecules found in the tissue. This influence is mediated within a cell through the intracellular cytoskeleton, composed primarily of actin, intermediate filament, and microtubule proteins. The cytoskeleton helps orient and organize cells within the tissue matrix for optimized function. Connections between the intracellular spaces and the extracellular matrix provide for dynamic and active interactions. Such connections must be reestablished as part of the wound healing process as cells migrate into a wound to regenerate functional, healthy tissues (136, 180–185).
Proposed Significance of CRP as a Biomarker and as a Biological Response Modifier in Cancer Disease
The clinical studies assessing CRP levels in cancer reviewed above, in light of preclinical data regarding the molecular activity of CRP and its distinctive isoforms in the inflammatory microenvironment, may provide invaluable insight into the contribution of CRP to disease progression of cancers. Moreover, the unique biological activity of mCRP or pCRP could help elaborate the clinical interpretations of CRP levels in patients suffering from cancer, thereby presenting CRP as a potential non-invasive technique to assess the severity of tumor development or progression. In the TME the potential exists for CRP to resolve an inflammatory environment through stimulating retention of monocytes by direct binding to fibronectin (117), which could aid in resolution of a pro-inflammatory (pro-tumorigenic) signaling milieu. Similarly, its ability to limit neutrophil chemotaxis through inhibition of IL-8-dependent migration (121) may also prevent an exacerbation of an inflammatory TME conducive for tumor growth. Conversely, a number of studies suggest the possibility for CRP to positively stimulate leukocyte production of cytokines such as IL-8 and MCP-1 [reviewed in (119)]. Depending on the broader context of stromal cells within the TME, this could indicate that CRP is either 1) enhancing cytotoxic T lymphocyte recruitment and subsequent tumor cell lysis, or 2) prolonging immune recruitment which could potentially lead to a sustained pro-inflammatory, and thus pro-tumorigenic, microenvironment. Therefore, while CRP may facilitate leukocyte retention and subsequent tumor cell lysis early in tumor development, an unresolved neoplastic growth may result in persistent signaling that potentiates both hepatic CRP production and excessive local inflammation, similar to what is observed in other pathologies that fail to resolve following injury and in line with the putative role of CRP in inflammation. While there is tremendous overlap in the matrix characteristics and signaling processes observed in both inflamed tissues and developing tumors, more direct measurement of the physiologic activity of CRP in the TME is both warranted and necessary. Assessing the capacity for CRP to regulate immune cell phenotypes, as well as its ability to modulate behavior of other stromal cells and tumor cells in the TME, can only be accomplished using in vivo studies or sophisticated 3D organotypic models (e.g., organoid cultures) to recapitulate the specific TME. Through these methods it may be possible to fully dissect the impact of CRP on the interactions between tumor and stromal compartments in order to assess its role in tumor development and metastasis.
The emerging relevance of the functional states of CRP isoforms suggest a complex relationship between its response in early inflammation related to de novo tumorigenesis and more advanced disease. The activity of mCRP in acute phase response illustrates its tremendous overlap in requisite components and signaling mechanisms of an actively developing tumor milieu. Further, given the observations that systemic levels of both IL-6 and IL-1β are elevated in multiple advanced cancers (186, 187), it is conceivable that the evolving tumor and its microenvironment may contribute to an exacerbation of CRP de novo synthesis and continuous secretion, potentially in excess of a slowing rate of conversion to mCRP that results in a demonstrable (and quantifiably significant) increase in systemic pCRP levels (Figure 5). This rise in pCRP, and indeed what has been measured in traditional clinical assessments, is thus more likely representative of continued tissue damage resulting from persistent development of neoplasms in situ. However, whether the rate of conversion of pCRP to mCRP during tumorigenesis or metastatic progression is like that observed in other inflammatory diseases remains unknown and requires exhaustive investigation. These potential relationships merit further preclinical assessment of the activity of mCRP in the growing tumor microenvironment and early metastatic niches of cancers in vivo to inform the exact nature of this inflammatory mediator and the significance of pCRP plasma levels throughout disease progression. Moreover, the recent advances in quantifying mCRP through enzyme-linked assays present a potential way forward for not only identifying the significance of mCRP as a diagnostic marker during disease progression per se (188), but could also be adapted to evaluate the molecular role of mCRP in cell-cell communication in the TME. Importantly, further development of such assays that can reliably distinguish between mCRP, pCRP, or possibly membrane-bound CRP will be essential in addressing the limitation of the reviewed clinical studies in that they only measure conventional or hsCRP (Table 1), which does not allow for a deeper appreciation of the contextual biological activity of CRP. While the predominant utility of CRP as a biomarker has traditionally been as a non-invasive diagnostic, it may also be useful to measure CRP by immunohistochemical methods or in tumor explant lysates, especially in the evaluation of CRP under controlled conditions in in vivo xenograft experiments. In combination with molecular techniques to directly identify interactions of mCRP with tumor and stromal cells, as well as other components of the TME (e.g., ECM proteins), these methods provide approaches that may elucidate the exact impact of mCRP on tumor cell proliferation, migration, invasion, and chemoresistance.
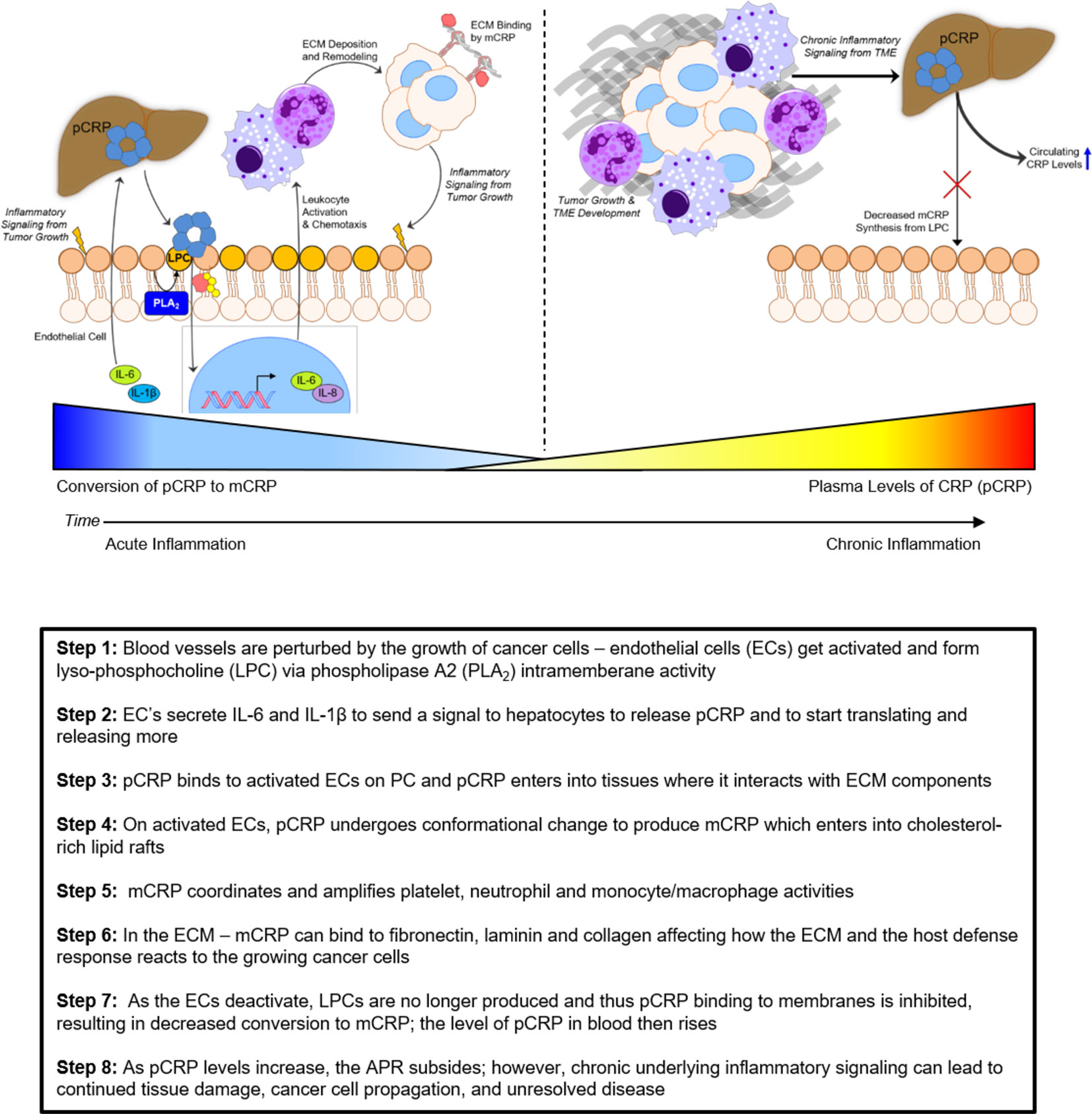
Figure 5 Schematic relationship between pCRP and mCRP as a function of inflammation in cancer.
Taking this diversity in the physiologic activity of CRP isoforms into account in the context of cancer may give further insight into the relationship between inflammation and cancer and, moreover, improve the clinical evaluation of cancer progression using this biomarker in patient assessment. Regardless, there are already several clinically important interpretations from the current preclinical and clinical data that may help refine assessment of CRP as a diagnostic tool in cancer, which are presented in Table 3. Importantly, the data suggest that pCRP levels exceeding 50–100µg/ml indicates pervasive tissue damage and is associated with poor survival. In general, this may relate to the correlation of high CRP levels and metastatic potential of many tumor types, as outlined in Table 1, and is in line with the concept that unresolved inflammation may drive tumor development as well as enhance dissemination and metastasis. This proposed utility of CRP levels to estimate cancer progression are in line with what has been described in assessing disease severity in a number of inflammatory diseases, including analysis of the recent SARS-CoV-2 viral infection (COVID-19) (189). Further evaluation of the role of CRP in cancer will undoubtably improve its ability as a biomarker to indicate disease severity and progression more precisely, and thus may reveal it as an indispensable asset in clinical decision making.

Table 3 Proposed diagnostic significance of CRP as a marker of inflammation associated with tissue damage.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Author Contributions
All authors contributed to the article and approved the submitted version. IR and LP were responsible for the curation of the meta-analysis performed in the review of published works on CRP levels among different cancers.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. WHO: World Health Organization. International Agency for Research on Cancer. Press release N° 263. Geneva, Switzerland (2018). Available at: https://www.who.int/cancer/PRGlobocanFinal.pdf (Accessed on January 2nd, 2020). 12 September.
3. Botta L, Dal Maso L, Guzzinati S, Panato C, Gatta G, Trama A, et al. Changes in life expectancy for cancer patients over time since diagnosis. J Adv Res (2019) 20:153–9. doi: 10.1016/j.jare.2019.07.002
4. Gu X, Zheng R, Xia C, Zeng H, Zhang S, Zou X, et al. Interactions between life expectancy and the incidence and mortality rates of cancer in China: a population−based cluster analysis. Cancer Commun (2018) 38:44. doi: 10.1186/s40880-018-0308-x
5. Cao B, Bray F, Beltrán-Sánchez H, Ginsburg O, Soneji S, Soerjomataram I. Benchmarking life expectancy and cancer mortality: global comparison with cardiovascular disease 1981-2010. BMJ (2017) 357:j2765. doi: 10.1136/bmj.j2765
6. Allemani C, Weir HK, Carreira H, Harewood R, Spika D, Wang X-S, et al. Global surveillance of cancer survival 1995–2009: analysis of individual data for 25 676 887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet (2015) 385:977–1010. doi: 10.1016/S0140-6736(14)62038-9
7. Goldberg JL, Sondel PM. Enhancing Cancer Immunotherapy Via Activation of Innate Immunity. Semin Oncol (2015) 42(4):562–72. doi: 10.1053/j.seminoncol.2015.05.012
8. Yarchoan M, Hopkins A, Jaffee E. Tumor mutational burden and response rate to PD-1 inhibition. N Eng J Med (2017) 377(25):2500–1. doi: 10.1056/NEJMc1713444
9. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold tumors: a therapeutic challenge for immunotherapy. Front Immunol (2019) 10:16. doi: 10.3389/fimmu.2019.0016
10. Sharma P, Allison JP. Dissecting the mechanisms of immune checkpoint therapy. Nat Rev Immunol (2020) 20(2):75–6. doi: 10.1038/s41577-020-0275-8
11. Intlekofer AM, Thompson CB. At the bench: preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J Leukoc Biol (2013) 94(1):25–39. doi: 10.1189/jlb.1212621
12. Martinez-Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res (2015) 21(22):5047–56. doi: 10.1158/1078-0432.CCR-15-0685
13. Gonzalez H, Hagerline C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev (2018) 32(19-20):1267–84. doi: 10.1101/gad.314617.118
14. FDA. US. Department of Health and Human Services. Guidance for Industry and FDA Staff. Review Criteria for Assessment of C-reactive protein (CRP), High Sensitivity C-Reactive Protein (hsCRP) and Cardiac C-Reactive Protein (cCRP) Assays. Document issued on September 22, 2005 Content current as of: 03/13/2018.
15. Shrotriya S, Walsh D, Bennani-Baiti N, Thomas S, Lorton C. C-Reactive Protein Is an Important Biomarker for Prognosis Tumor Recurrence and Treatment Response in Adult Solid Tumors: A Systematic Review. PloS One (2015) 10(12):e0143080. doi: 10.1371/journal.pone.0143080
16. Alifano M, Falcoz PE, Seegers V, Roche N, Schussler O, Younes M, et al. Preresection serum C-reactive protein measurement and survival among patients with resectable non–small cell lung cancer. J Thorac Cardiovasc Surg (2011) 142:1161–7. doi: 10.1016/j.jtcvs.2011.07.021
17. Aref H, Refaat S. CRP evaluation in non-small cell lung cancer. Egyptian J Chest Dis Tuberculosis (2014) 63:717–22. doi: 10.1016/j.ejcdt.2014.02.003
18. Bittoni MA, Focht BC, Clinton SK, Buckworth J, Harris RE. Prospective evaluation of C-reactive protein, smoking and lung cancer death in the Third National Health and Nutrition Examination Survey. Int J Oncol (2015) 47:1537–44. doi: 10.3892/ijo.2015.3141
19. Chaturvedi AK, Caporaso NE, Katki HA, Wong H-L, Chatterjee N, Pine SR, et al. C-Reactive Protein and Risk of Lung Cancer. J Clin Oncol (2010) 28(16):2719–26. doi: 10.1200/JCO.2009.27.0454
20. Hara M, Matsuzaki Y, Shimuzu T, Tomita M, Ayabe T, Enomoto Y, et al. Preoperative Serum C-reactive Protein Level in Non-small Cell Lung Cancer. Anticancer Res (2007) 27:3001–4.
21. Hara M, Yonei A, Ayabe T, Tomita M, Nakamura K, Onitsuka T. Postoperative serum C-Reactive Protein levels in non-small cell lung cancer patients. Ann Thorac Cardiovasc Surg (2010) 16:85–90.
22. Jin Y, Sun Y, Shi X, Zhao J, Shi L, Yu X. Prognostic value of circulating C−reactive protein levels in patients with non-small cell lung cancer: A systematic review with meta−analysis. J Cancer Res Ther (2014) 10(Special Issue 2):Suppl:C160–6. doi: 10.4103/0973-1482.145854
23. Jing X, Huang C, Zhou H, Li C, Fan L, Chen J, et al. Association between serum C-reactive protein value and prognosis of patients with non-small cell lung cancer: a meta-analysis. Int J Clin Exp Med (2015) 8(7):10633–9.
24. Koch A, Fohlin H, Sörenson S. Prognostic Significance of C-reactive protein and Smoking in Patients with Advanced Non-small Cell Lung Cancer Treated with First-Line Palliative Chemotherapy. J Thoracic Oncol (2009) 4(3):326–32. doi: 10.1097/JTO.0b013e31819578c8
25. Liao C, Yu Z, Guo W, Liu Q, Wu Y, Li Y, et al. Prognostic value of circulating inflammatory factors in non-small cell lung cancer: a systematic review and meta-analysis. Cancer Biomark (2014) 14(6):469–81. doi: 10.3233/CBM-140423
26. Muller DC, Larose TL, Hodge A, Guida F, Langhammer A, Grankvist K, et al. Circulating high sensitivity C reactive protein concentrations and risk of lung cancer: nested case-control study within Lung Cancer Cohort Consortium. BMJ (2018) 364:k4981. doi: 10.1136/bmj.k4981
27. Pastorino U, Morelli D, Leuzzi G, Gisabella M, Suatoni P, Taverna F, et al. Baseline and postoperative C-reactive protein levels predict mortality in operable lung cancer. Eur J Cancer (2017) 79:90–7. doi: 10.1016/j.ejca.2017.03.020
28. Shinohara S, Otsuki R, Onitsuka T, Machida K, Matsuo M, Nakagawa M, et al. Postoperative C-reactive Protein Is a Predictive Biomarker for Survival After Non-small Cell Lung Cancer Resection. Anticancer Res (2019) 39:2193–8. doi: 10.21873/anticanres.13334
29. Sin DD, Man SFP, McWilliams A, Lam S. Progression of airway dysplasia and C - reactive protein in smokers at high risk of lung cancer. Am J Respir Crit Care Med (2006) 173:535–9. doi: 10.1164/rccm.200508-1305OC
30. Szturmowicz M, Rudziński P, Kacprzak A, Langfort R, Bestry I, Broniarek-Samson B, et al. Prognostic value of serum C-reactive protein (CRP) and cytokeratin 19 fragments (Cyfra 21-1) but not carcinoembryonic antigen (CEA) in surgically treated patients with non-small cell lung cancer. Pneumonol Alergol Pol (2014) 82:422–9. doi: 10.5603/PiAP.2014.0055
31. Tomita M, Shimizu T, Ayabe T, Onitsuka T. Elevated Preoperative Inflammatory Markers Based on Neutrophil-to-Lymphocyte Ratio and C-Reactive Protein Predict Poor Survival in Resected Non-small Cell Lung Cancer. Anticancer Res (2012) 32:3535–8.
32. Torrecilla JA, Scrimini S, Sauleda J, García-Cosío FB, Noguera A, Iglesias A, et al. Role of C reactive protein in non-small cell lung cancer staging. Eur Respir J (2011) 38(Suppl 55):2806.
33. Vagulienė N, Žemaitis M, Miliauskas S, Urbonienė D, Šitkauskienė B, Sakalauskas R. Comparison of C-reactive Protein Levels in Patients with Lung Cancer and Chronic Obstructive Pulmonary Disease. Medicina (Kaunas) (2011) 47(8):421–7. doi: 10.3390/medicina47080059
34. Wei L, Du Y, Wu W, Li L. Changes of tumor markers and C reactive protein in different status of lung cancer. Int J Clin Exp Pathol (2016) 9(11):11984–8.
35. Zhao Z, Li X, Zhao Y, Wang D, Li Y, Liu L, et al. Role of C-reactive protein and procalcitonin in discriminating between infectious fever and tumor fever in non-neutropenic lung cancer patients. Medicine (2018) 97:33(e11930). doi: 10.1097/MD.0000000000011930
36. Allin KH, Nordestgaard BG, Flyger H, Bojesen SE. Elevated pre-treatment levels of plasma C-reactive protein are associated with poor prognosis after breast cancer: a cohort study. Breast Cancer Res (2011) 13:R55. doi: 10.1186/bcr2891
37. Asegaonkar SB, Takalkar UV, Kodlikeri P, Pagdhune A, Bonduliya V, Thorat AP. Serum high sensitivity C-reactive protein in breast cancer patients. Int J Res Med Sci (2014) 2(4):1408–11. doi: 10.5455/2320-6012.ijrms20141131
38. Gathirua-Mwangi WG, Song Y, Monahan P, Champion VL, Zollinger T. Associations of metabolic syndrome and C-reactive protein with mortality from total cancer, obesity-linked cancers and breast cancer among women in NHANES III. Int J Cancer (2018) 143(3):535–42. doi: 10.1002/ijc.31344
39. Guo L, Liu S, Zhang S, Chen Q, Zhang M, Quan P, et al. C-reactive protein and risk of breast cancer: A systematic review and meta-analysis. Sci Rep (2015) 5:10508. doi: 10.1038/srep10508
40. Nelson SH, Brasky TM, Patterson RE, Laughlin GA, Kritz-Silverstein D, Edwards BJ, et al. The association of the C-reactive protein inflammatory biomarker with breast cancer incidence and mortality in the Women’s Health Initiative. Cancer Epidemiol Biomarkers Prev (2017) 26(7):1100–6. doi: 10.1158/1055-9965.EPI-16-1005
41. Rodriguez-Gil JL, Takita C, Wright J, Reis IM, Zhao W, Brian EL, et al. Inflammatory biomarker C-Reactive Protein and radiotherapy-induced early adverse skin reactions in breast cancer patients. Cancer Epidemiol Biomarkers Prev (2014) 23(9):1873–83. doi: 10.1158/1055-9965.EPI-14-0263
42. Sabiston CM, Wrosch C, Castonguay AL, Sylvester BD. Changes in physical activity behavior and C-reactive protein in breast cancer patients. Ann Behav Med (2018) 52:545–51. doi: 10.1093/abm/kax010
43. Sicking I, Edlund K, Wesbuer E, Weyer V, Battista MJ, Lebrecht A, et al. Prognostic Influence of pre-operative C-Reactive Protein in node-negative breast cancer patients. PloS One (2014) 9(10):e111306. doi: 10.1371/journal.pone.0111306
44. Thomson CA, Thompson PA, Wright-Bea J, Nardi E, Frey GR, Stopeck A. Metabolic Syndrome and Elevated C-Reactive Protein in Breast Cancer Survivors on Adjuvant Hormone Therapy. J Women’s’ Health (2009) 18(12):2041–7. doi: 10.1089/jwh.2009.1365
45. Villaseñor A, Flatt SW, Marinac C, Natarajan L, Pierce JP, Patterson RE. Postdiagnosis C - reactive protein and Breast Cancer Survivorship: Findings from the WHEL Study. Cancer Epidemiol Biomarkers Prev (2013) 23(1):189–99. doi: 10.1158/1055-9965.EPI-13-0852
46. Wang J, Lee IM, Tworoger SS, Buring JE, Ridker PM, Rosner B, et al. Plasma C-reactive protein and risk of breast cancer in two prospective studies and a meta-analysis. Cancer Epidemiol Biomarkers Prev (2015) 24(8):1199–206. doi: 10.1158/1055-9965.EPI-15-0187
47. Zhang SM, Lin J, Cook NR, Lee I-M, Manson JE, Buring JE, et al. C - reactive protein and risk of breast cancer. J Natl Cancer Inst (2007) 99:890–94. doi: 10.1093/jnci/djk202
48. Aleksandrova K, Jenab M, Boeing H, Jansen E, Bueno-de-Mesquita HB, Rinaldi S, et al. Circulating C-Reactive Protein Concentrations and Risks of Colon and Rectal Cancer: A Nested Case-Control Study Within the European Prospective Investigation into Cancer and Nutrition. Am J Epidemiol (2010) 172(4):407–18. doi: 10.1093/aje/kwq135
49. Erlinger TP, Platz EA, Rifai N, Helzlsouer KJ. C - reactive protein and the Risk of Incident Colorectal Cancer. JAMA (2004) 291(5):585–90. doi: 10.1001/jama.291.5.585
50. Fang D, Ye Y. C-reactive protein gene rs1205 polymorphism is not associated with the risk of colorectal cancer. Biosci Rep (2017) 37:BSR20170872. doi: 10.1042/BSR20170872
51. Goyal A, Terry MB, Jin Z, Siegel AB. C-Reactive Protein and Colorectal Cancer Mortality in U.S. Adults Cancer Epidemiol Biomarkers Prev (2014) 23(8):1609–18. doi: 10.1158/1055-9965.EPI-13-0577
52. Holm M, Saraswat M, Joenväärä S, Ristimäki A, Haglund C, Renkonen R. Colorectal cancer patients with different C-reactive protein levels and 5-year survival times can be differentiated with quantitative serum proteomics. PloS One (2018) 13(4):e0195354. doi: 10.1371/journal.pone.0195354
53. Ishizuka M, Nagata H, Takagi K, Kubota K. C-Reactive Protein is Associated with Distant Metastasis of T3 Colorectal Cancer. Anticancer Res (2012) 32:1409–16.
54. Lumachi F, Basso SMM, Santeufemia DA, Ermani M, Lo Re G, Chiara GB. Preoperative Serum C - reactive protein and its Prognostic Significance in Patients with Stage III-IV Colorectal Cancer. Anticancer Res (2014) 34:7263–6.
55. Nimptsch K, Aleksandrova K, Boeing H, Janke J, Lee Y-A, Jenab M, et al. Association of CRP genetic variants with blood concentrations of C-reactive protein and colorectal cancer risk. Int J Cancer (2015) 136:1181–92. doi: 10.1002/ijc.29086
56. Shibutani M, Maeda K, Nagahara H, Ohtani H, Sugano K, Ikeya T, et al. Elevated preoperative serum C-reactive protein levels are associated with poor survival in patients with colorectal cancer. Hepatogastroenterology (2014) 61(136):2236–40.
57. Toiyama Y, Fujikawa H, Koike Y, Saigusa S, Inoue Y, Tanaka K, et al. Evaluation of preoperative C-reactive protein aids in predicting poor survival in patients with curative colorectal cancer with poor lymph node assessment. Oncol Lett (2012) 5:1881–8. doi: 10.3892/ol.2013.1308
58. Zhou B, Shu B, Yang J, Liu J, Xi T, Xing Y. C-reactive protein, interleukin-6 and the risk of colorectal cancer: a meta-analysis. Cancer Causes Control (2014) 25:1397–405. doi: 10.1007/s10552-014-0445-8
59. Badakhshi H, Kaul D, Zhao KL. Association between the inflammatory biomarker, C-reactive protein, and the response to radiochemotherapy in patients with esophageal cancer. Mol Clin Oncol (2016) 4(4):643–7. doi: 10.3892/mco.2016.753
60. Huang Y, Feng JF, Liu JS, Chen QX. Prognostic role of serum C-reactive protein in esophageal cancer: a systematic review and meta-analysis. Ther Clin Risk Manag (2015) 11:89–94. doi: 10.2147/TCRM.S70954
61. Katano A, Takahashi W, Yamashita H, Yamamoto K, Ando M, Yoshida M, et al. The impact of elevated C-reactive protein level on the prognosis for oro-hypopharynx cancer patients treated with radiotherapy. Sci Rep (2017) 7(1):17805. doi: 10.1038/s41598-017-18233-w
62. Zheng TL, Cao K, Liang C, Zhang K, Guo HZ, Li DP, et al. Prognostic value of C-reactive protein in esophageal cancer: a meta-analysis. Asian Pac J Cancer Prev (2014) 15(19):8075–81. doi: 10.7314/apjcp.2014.15.19.8075
63. Baba H, Kuwabara K, Ishiguro T, Hatano S, Matsuzawa T, Fukuchi M, et al. C-reactive Protein as a Significant Prognostic Factor for Stage IV Gastric Cancer Patients. Anticancer Res (2013) 33:5591–6.
64. Chang C-C, Sun C-F, Pai H-J, Wang W-K, Hsieh C-C, Kuo L-M, et al. Preoperative Serum C -reactive protein and Gastric Cancer; Clinical-pathological Correlation and Prognostic Significance. Med J (2010) 33:301–12.
65. Shimura T, Kitagawa M, Yamada T, Ebi M, Mizoshita T, Tanida S, et al. C-reactive Protein is a Potential Prognostic Factor for Metastatic Gastric Cancer. Anticancer Res (2012) 32:491–6.
66. Shishido Y, Fujitani K, Yamamoto K, Hirao M, Tsujinaka T, Sekimoto M. C-reactive protein on postoperative day 3 as a predictor of infectious complications following gastric cancer resection. Gastric Cancer (2016) 19:293–301. doi: 10.1007/s10120-014-0455-y
67. Yu Q, Yu X-F, Zhang S-D, Wang H-H, Wang H-Y, Teng L-S. Prognostic Role of C-reactive protein in Gastric Cancer: A Meta-analysis. Asian Pacific J Cancer Prev (2013) 14(10):5735–40. doi: 10.7314/APJCP.2013.14.10.5735
68. Fang Y, Xu C, Wu P, Zhang L-H, Li D-W, Sun J-H, et al. Prognostic role of C-reactive protein in patients with nasopharyngeal carcinoma A meta-analysis and literature review. Medicine (2017) 96:45(e8463). doi: 10.1097/MD.0000000000008463
69. He X, Li J-P, Liu X-H, Zhang J-P, Zeng Q-Y, Chen H, et al. Prognostic value of C-reactive protein/albumin ratio in predicting overall survival of Chinese cervical cancer patient’s overall survival: comparison among various inflammation-based factors. J Cancer (2018) 9(10):1877–84. doi: 10.7150/jca.23320
70. Tai SF, Chien H-T, Young C-K, Tsao C-K, de Pablo A, Fan K-H, et al. Roles of preoperative C-reactive protein are more relevant in buccal cancer than other subsites. World J Surg Oncol (2017) (2017)15:47. doi: 10.1186/s12957-017-1116-5
71. Du J, Hu W, Yang C, Wang Y, Wang X, Yang P. C-reactive protein is associated with the development of tongue squamous cell carcinoma. Acta Biochim Biophys Sin (2018) 50(3):238–45. doi: 10.1093/abbs/gmy004
72. Oliveira KG, von Zeidler SV, Lamas AZ, de Podesta JRV, Sena A, Souza ED, et al. Relationship of inflammatory markers and pain in patients with head and neck cancer prior to anticancer therapy. Braz J Med Biol Res (2014) 47(7):600–4. doi: 10.1590/1414-431X20143599
73. Carr BI, Akkiz H, Guerra V, Üsküdar O, Kuran S, Karaoğullarından Ü, et al. C-reactive protein and hepatocellular carcinoma: analysis of its relationship to tumor factors. Clin Pract (Lond) (2018) 15(Spec Issue):625–34. doi: 10.4172/clinical-practice.1000409
74. Kinoshita A, Onoda H, Imai N, Iwaku A, Oishi M, Tanaka K, et al. The addition of C-reactive protein to validated staging systems improves their prognostic ability in patients with hepatocellular carcinoma. Oncology (2014) 86:308–17. doi: 10.1159/000360704
75. Hefler L, Concin N, Hofstetter G, Marth C, Mustea A, Sehouli J, et al. Serum C-Reactive Protein as independent prognostic variable in patients with ovarian cancer. Clin Cancer Res (2008) 14(3):710–4. doi: 10.1158/1078-0432.CCR-07-1044
76. Li J, Jiao X, Yuan Z, Qiu H, Guo R. C-reactive protein and risk of ovarian cancer: A systematic review and meta-analysis. Medicine (2017) 96:34(e7822). doi: 10.1097/MD.0000000000007822
77. Lundin E, Dossus L, Clendenen T, Krogh V, Grankvist K, Wulff M, et al. C-reactive protein and ovarian cancer: a prospective study nested in three cohorts (Sweden, USA, Italy). Cancer Causes Control (2009) 20(7):1151–9. doi: 10.1007/s10552-009-9330-2
78. Graff JN, Beer TM, Liu B, Sonpavde G, Taioli E. Pooled analysis of C-Reactive Protein levels and mortality in prostate cancer patients. Clin Genitourin Cancer (2015) 13(4):e217–21. doi: 10.1016/j.clgc.2015.01.011
79. Liu Z-Q, Chu L, Fang J-M, Zhang X, Zhao H-X, Chen Y-J, et al. Prognostic role of C−reactive protein in prostate cancer: a systematic review and meta−analysis. Asian J Androl (2014) 16:467–71. doi: 10.4103/1008-682X.123686
80. Platz EA, De Marzo AM, Erlinger TP, Rifai N, Visvanathan K, Hoffman SC, et al. No association between pre-diagnostic plasma C-reactive protein concentration and subsequent prostate cancer. Prostate (2004) 59(4):393–400. doi: 10.1002/pros.10368
81. Schnoeller TJ, Steinestel J, Steinestel K, Jentzmik F, Schrader AJ. Do preoperative serum C−reactive protein levels predict the definitive pathological stage in patients with clinically localized prostate cancer? Int Urol Nephrol (2015) 47:765–70. doi: 10.1007/s11255-015-0952-x
82. Thurner EM, Krenn-Pilko S, Langsenlehner U, Stojakovic T, Pichler M, Gerger A, et al. The elevated C-reactive protein level is associated with poor prognosis in prostate cancer patients treated with radiotherapy. Eur J Cancer (2015) 51(5):610–9. doi: 10.1016/j.ejca.2015.01.002
83. Xu L, Zhao Q, Huang S, Li S, Wang J, Li Q. Serum C-reactive protein acted as a prognostic biomarker for overall survival in metastatic prostate cancer patients. Tumour Biol (2015) 36(2):669–73. doi: 10.1007/s13277-014-2670-x
84. Liu Y, Chen S, Zheng C, Ding M, Zhang L, Wang L, et al. The prognostic value of the preoperative c-reactive protein/albumin ratio in ovarian cancer. BMC Cancer (2017) 17:285. doi: 10.1186/s12885-017-3220-x
85. Chen J, Jing X, Deng X, Gao F, Wang X, Han D, et al. Prognostic value of serum C-reactive protein in pancreatic cancer: a meta-analysis. Int J Clin Exp Med (2018) 11(11):11789–96.
86. Inoue D, Ozaka M, Matsuyama M, Yamada I, Takano K, Saiura A, et al. Prognostic value of neutrophil–lymphocyte ratio and level of C-reactive protein in a large cohort of pancreatic cancer patients: a retrospective study in a single institute in Japan. Japanese J Clin Oncol (2015) 45(1):61–6. doi: 10.1093/jjco/hyu159
87. Stevens L, Pathak S, Nunes QM, Pandanaboyana S, Macutkiewicz C, Smart N, et al. Prognostic significance of pre-operative C-reactive protein and the neutrophil–lymphocyte ratio in resectable pancreatic cancer: a systematic review. HPB (2015) 17:285–91. doi: 10.1111/hpb.12355
88. Hsiao W, Herrel LA, Yu C, Kattan MW, Canter DJ, Carthon BC, et al. Nomograms incorporating serum C-reactive protein effectively predict mortality before and after surgical treatment of renal cell carcinoma. Int J Urol (2015) 22(3):264–70. doi: 10.1111/iju.12672
89. Omae K, Kondo T, Tanabe K. High preoperative C-reactive protein values predict poor survival in patients on chronic hemodialysis undergoing nephrectomy for renal cancer. Urol Oncol (2015) 33(2):67.e9–13. doi: 10.1016/j.urolonc.2014.07.004
90. Teishima J, Kobatake K, Hayashi T, Seno Y, Ikeda K, Nagamatsu H, et al. Prognostic significance of C-reactive protein in patients with intermediate-risk metastatic renal cell carcinoma treated with molecular targeted therapy. Oncol Lett (2014) 8(2):881–5. doi: 10.3892/ol.2014.2207
91. Aziz A, Rink M, Gakis G, Kluth LA, Dechet C, Miller F, et al. Preoperative C-reactive protein in the serum: a prognostic biomarker for upper urinary tract urothelial carcinoma treated with radical nephroureterectomy. Urol Int (2014) 93(3):352–60. doi: 10.1159/000362248
92. Dai J, Tang K, Xiao W, Yu G, Zeng J, Li W, et al. Prognostic Significance of C -reactive protein in Urological Cancers: A Systematic Review and Meta-analysis. Asian Pac J Cancer Prev (2014) 15(8):3369–75. doi: 10.7314/apjcp.2014.15.8.3369
93. Guo S, He X, Chen Q, Yang G, Yao K, Dong P, et al. The C-reactive protein/albumin ratio, a validated prognostic score, predicts outcome of surgical renal cell carcinoma patients. BMC Cancer (2017) 17:171. doi: 10.1186/s12885-017-3119-6
94. Troppan KT, Schlick K, Deutsch A, Melchardt T, Egle A, Stojakovic T, et al. C-reactive protein level is a prognostic indicator for survival and improves the predictive ability of the R-IPI score in diffuse large B-cell lymphoma patients. Br J Cancer (2014) 111:55–60. doi: 10.1038/bjc.2014.277
95. Fang S, Wang Y, Sui D, Liu H, Ross MI, Gershenwald JE, et al. C-Reactive Protein as a marker of melanoma progression. J Clin Oncol (2015) 33:1389–96. doi: 10.1200/JCO.2014.58.0209
96. Fang E, Wang X, Feng J, Zhao X. The Prognostic Role of Glasgow Prognostic Score and C - reactive protein to Albumin Ratio for Sarcoma: A System Review and Meta-Analysis. Dis Markers (2020) 2020:14 pages. doi: 10.1155/2020/8736509. Article ID 8736509.
97. Li W, Luo X, Liu Z, Chen Y, Li Z. Prognostic value of C-reactive protein levels in patients with bone neoplasms: A meta-analysis. PloS One (2018) 13(4):e0195769. doi: 10.1371/journal.pone.0195769
98. Nakamura T, Grimer R, Gaston C, Francis M, Charman J, Graunt P, et al. The value of C-reactive protein and comorbidity in predicting survival of patients with high grade soft tissue sarcoma. Eur J Cancer (2013) 49(2):377–85. doi: 10.1016/j.ejca.2012.09.004
99. Nakamura T, Matsumine A, Matsubara T, Asanuma K, Yada Y, Hagi T, et al. Infiltrative tumor growth patterns on magnetic resonance imaging associated with systemic inflammation and oncological outcome in patients with high-grade soft-tissue sarcoma. PloS One (2017) 12(7):e0181787. doi: 10.1371/journal.pone.0181787
100. Wang X, Liu S, Zhao X, Fang E, Zhao X. The value of C-reactive protein as an independent prognostic indicator for disease specific survival in patients with soft tissue sarcoma: A meta-analysis. PloS One (2019) 14(7):e0219215. doi: 10.1371/journal.pone.0219215
101. Yanagisawa M, Gingrich AA, Judge S, Li C-S, Wang N, Thorpe SW, et al. Serum C-reactive protein and neutrophil/lymphocyte ratio after neoadjuvant radiotherapy in soft tissue sarcoma. Anticancer Res (2018) 38(3):1491–7. doi: 10.21873/anticanres.12376
102. Shrotriya S, Walsh D, Nowacki AS, Lorton C, Aktas A, Hullihen B, et al. cSerum C-reactive protein is an important and powerful prognostic biomarker in most adult solid tumors. PloS Onev (2018) 13(8):e0202555. doi: 10.1371/journal.pone.0202555
103. Rajab IM, Majerczyk D, Olson ME, Addams JMB, Choe ML, Nelson MS, et al. C-reactive protein in gallbladder diseases: diagnostic and therapeutic insights. Biophysics Rep (2020). doi: 10.1007/s41048-020-00108-9
104. Tyurina YY, St. Croix CM, Watkins SC, Watson AM, Epperly MW, Tamil S, et al. Redox (phospho)lipidomics of signaling in inflammation and programmed cell death. J Leukoc Biol (2019) 106(1):57–81. doi: 10.1002/JLB.3MIR0119-004RR
105. Zen Q, Zhong W, Mortensen RF. Binding site on human C-reactive Protein (CRP) recognized by the Leukocyte CRP-receptor. J Cell Biochem (1997) 64:140–51. doi: 10.1002/(SICI)1097-4644(199701)64:1<140:AIDJBC16>3.0.CO;2-p
106. Thomassen MJ, Meeker DP, Deodhar SD, Wiedemann HP, Barna BP. Activation of human monocytes and alveolar macrophages by a synthetic peptide of C-reactive protein. J Immunother Emphasis Tumor Immunol (1993) 13(1):1–6. doi: 10.1097/00002371-1993010000-00001
107. Li H-Y, Wang J, Meng F, Jia Z-K, Su Y, Bai Q-F, et al. An intrinsically disordered motif mediates diverse actions of monomeric C-reactive protein. J Biol Chem (2016) 291(16):8795–804. doi: 10.1074/jbc.M115.695023
108. Thompson D, Pepys MB, Wood SP. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure (1999) 7(2):169–77. doi: 10.1016/S0969-2126(99)80023-9
109. Shrive AK, Cheetham GMT, Holden D, Myles DAA, Turnell WG, Volanakis JE, et al. Three-dimensional structure of human C-reactive protein. Nat Struc Biol (1996) 3:346–54. doi: 10.1038/nsb0496-346
110. Wu Y, Potempa LA, Kebir DE, Filep JG. C-reactive protein and inflammation: conformational changes affect function. Biol Chem (2015) 396(11):1181–97. doi: 10.1515/hsz-2015-0149
111. Dvorak HF. Tumors: wounds that do not heal. New Engl J Med (1986) 315:1650–9. doi: 10.1056/NEJM198612253152606
112. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet (2001) 357(9255):539–45. doi: 10.1016/S0140-6736(00)04046-0
113. Agrawal A, Xu Y, Ansardi D, Macon KJ, Volanakis JE. Probing the phosphocholine-binding site of human C-reactive protein by site-directed mutagenesis. J Biol Chem (1992) 267:25352–8.
114. Volanakis JE. Human C-reactive protein: expression, structure, and function. Mol Immunol (2001) 38(2-3):189–97. doi: 10.1016/s0161-5890(01)00042-6
115. Salonen E-M, Vartio T, Hedman K, Vaheri A. Binding of fibronectin by the acute phase reactant C-reactive protein. J Biol Chem (1984) 259:1496–501.
116. Tseng J, Mortensen RF. Binding of human C-reactive protein (CRP) to plasma fibronectin occurs via the phosphorylcholine-binding site. Mol Immunol (1988) 25:679–86. doi: 10.1016/0161-5890(88)90103-4
117. Ullah N, Ma F-R, Jin Han J, Liu X-L, Fu Y, Liu Y-T, et al. Monomeric C-reactive Protein Regulate Mediated Monocyte Adhesion. Mol Immunol (2020) 117:122–30. doi: 10.1016/j.molimm.2019.10.013
118. Swanson SJ, McPeek MM, Mortensen RF. Characteristics of the binding of human C-reactive protein (CRP) to laminin. J Cell Biochem (1989) 40:121–32. doi: 10.1002/jcb.240400112
119. Sproston NR, Ashworth JJ. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front Immunol (2018) 9:754. doi: 10.3389/fimmu.2018.00754
120. McFadyen J, Kiefer J, Loseff-Silver J, Braig D, Potempa LA, Eisenhardt SU, et al. Dissociation of C-reactive protein localizes and amplifies inflammation: Evidence for a direct biological role of CRP and its conformational changes. Front Immunol (2018) 9:1351. doi: 10.3389/fimmu.2018.01351.ecollection. Article 1351.
121. Zhong W, Zen Q, Tebo J, Schlottmann K, Coggeshall M, Mortensen RF, et al. Effect of human C-reactive protein on chemokine and chemotactic factor-induced neutrophil chemotaxis and signaling. J Immunol (1998) 161(5):2533–40.
122. Trial J, Potempa LA, Entman ML. The Role of C-reactive Protein in Innate and Acquired Inflammation: New Perspectives. Inflammation Cell Signaling (2016) 3:e1409. doi: 10.14800/ics.1498
123. Zhang L, Shen ZY, Wang K, Li W, Shi J-M, Kombo Orsoro E, et al. C-reactive protein exacerbates epithelial-mesenchymal transition through Wnt/β-catenin and ERK signaling in streptozocin-induced diabetic nephropathy. FASEB J (2019) 33(5):6551–63. doi: 10.1096/fj.201801865RR
124. Nakai K, Tanaka H, Yamanaka K, takahashi Y, Murakami F, Matsuike R, et al. Effects of C-reactive protein on the expression of matrix metalloproteinases and their inhibitors via Fcγ receptors on 3T3-L1 adipocytes. Int J Med Sci (2017) 14(5):484–93. doi: 10.7150/ijms.18059
125. Li H-Y, Wang J, Wu Y-X, Zhang L, Liu Z-P, Filep J, et al. Topological localization of monomeric C-reactive protein determines proinflammatory endothelial cell responses. J Biol Chem (2014) 289(20):14283–90. doi: 10.1074/jbc.M114.555318
126. Shephard EG, Anderson R, Rosen O, Myer MS, Fridkin M, Strachan AF, et al. Peptides generated from C-reactive protein by a neutrophil membrane protease. J Immunol (1990) 145:1469–76.
127. Shephard EG, Anderson R, Rosen O, Fridkin M. C-reactive protein (CRP) peptides inactivate enolase in human neutrophils leading to depletion of intracellular ATP and inhibition of superoxide generation. Immunology (1992a) 76(1):79–85.
128. Shephard EG, Kelly SL, Anderson R, Fridkin M. Characterization of neutrophil-mediated degradation of human C-reactive protein and identification of the protease. Clin Exp Immunol (1992b) 87(3):509–13. doi: 10.1111/j.1365-2249.1992.tb0328.x
129. El Kebir D, Zhang Y, Wang L, Potempa LA, Wu Y, Fournier A, et al. C-reactive protein-derived peptide 201-206 inhibits neutrophil adhesion to endothelial cells and platelets through CD32. J Leukocyte Biol (2011) 90(6):1167–75. doi: 10.1074/jlb.0111032
130. Rajab IM, Hart PC, Potempa LA. How C-reactive protein structural isoforms with distinctive bioactivities affect disease progression. Front Immunol (2020) 11:2126. doi: 10.3389/fimmu.2020.02126
131. Theile JR, Zeller J, Kiefer J, Braig D, Kreuzaler S, Lenz Y, et al. A conformational change in C-reactive protein enhances leukocyte recruitment and reactive oxygen species generation in ischemia/reperfusion injury. Front Immunol (2018) 6:675. doi: 10.3389/fimmu.2018.00675
132. Thiele JR, Zeller J, Bannasch H, Stark GB, Peter K, Eisenhardt SU. Targeting C-Reactive Protein in Inflammatory Disease by Preventing Conformational Changes. Mediators Inflammation (2015) 2015:372432. doi: 10.1155/2015/372432
133. Braig D, Nero TL, Koch H-G, Kaiser B, Wang X, Thiele JR, et al. Characterization of transitional changes in the CRP structure leading to the exposure of pro-inflammatory binding sites. Nat Commun (2017) 23:14188. doi: 10.1038/ncomms14188
134. Tan C, Huang Y, Shi F, Tan K, Ma Q, Chen Y, et al. C-reactive protein correlates with CT findings and predicts severe COVID-19 early. J Med Virol (2020) 92(7):856–62. doi: 10.1002/jmv.25871
135. Birk DE, Silver FH, Trelstad RL. Matrix assembly. In: Hay ED, editor. Cell Biology of Extracellular Matrix, 2nd edition. New York: Plenum Press (1991). p. 221–54.
136. Aumailley M, Gayraud B. Structure and biological activity of the extracellular matrix. J Mol Med (1998) 76:253–65. doi: 10.1007/s001090050215
137. Yue B. Biology of the extracellular matrix: an overview. J Glaucoma (2014) 23(8):S20–3. doi: 10.1097/IJG.0000000000000108
138. Cray C. Acute phase proteins in animals. 2012 Prog Mol Biol Transl Sci (2012) 105:113–50. doi: 10.1016/B978-0-12-394596-9.00005-6
139. Hunt TK. Basic principles of wound healing. J Trauma (1990) 30:S122–128. doi: 10.1097/00005373-199012001-00025
140. Kim WJ, Gittes GK, Longaker MT. Signal transduction in wound pharmacology. Arch Pharm Res (1998) 21:487–95. doi: 10.1007/BF02975363
141. Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol (2012) 22(1):33–40. doi: 10.1016/j.semcancer.2011.12.005
142. Coussens LM, Werb Z. Inflammation and cancer. Nature (2002) 420(6917):860–7. doi: 10.1038/nature01322
143. Murata M. Inflammation and cancer. Environ Health Prev Med (2018) 23(1):50. doi: 10.1186/s12199-018-0740-1
144. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res (2014) 2014:149185. doi: 10.1155/2014/14918
145. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis (2009) 30(7):1073–81. doi: 10.1093/carcin/bgp127
146. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi: 10.1016/j.cell.2010.01.025
147. Giannoni E, Parri M, Chiarugi P. EMT and oxidative stress: a bidirectional interplay affecting tumor malignancy. Antioxid Redox Signal (2012) 16(11):1248–63. doi: 10.1089/ars.2011.4280
148. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res (2009) 19(2):156–72. doi: 10.1038/cr.2009.5
149. Gilles C, Newgreen D, Sato H, Thompson E. Matrix Metalloproteases and Epithelial-to-Mesenchymal Transition: Implications for Carcinoma Metastasis. In: Madame Curie Bioscience Database. (Austin, TX: Landes Bioscience 2000-2013 (2013). Available at: https://www.ncbi.nlm.nih.gov/books/NBK6387.
150. Drake JM, Strohbehn G, Bair TB, Moreland JG, Henry MD. ZEB1 enhances transendothelial migration and represses the epithelial phenotype of prostate cancer cells. Mol Biol Cell (2009) 20(8):2207–17. doi: 10.1091/mbc.e08-10-1076
151. Bergeman J, Caillier A, Houle F, Gagne LM, Huot ME. Localized translation regulates cell adhesion and transendothelial migration. J Cell Sci (2016) 129(21):4105–17. doi: 10.1242/jcs.191320
152. Dominguez C, David JM, Palena C. Epithelial-mesenchymal transition and inflammation at the site of the primary tumor. Semin Cancer Biol (2017) 47:177–84. doi: 10.1016/j.semcancer.2017.08.002
153. Todoric J, Antonucci L, Karin M. Targeting Inflammation in Cancer Prevention and Therapy. Cancer Prev Res (Phila) (2016) 9(12):895–905. doi: 10.1158/1940-6207.CAPR-16-0209
154. Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget (2016) 7(19):28697–710. doi: 10.18632/oncotarget.7376
155. Stone RC, Pastar I, Ojeh N, Chen V, Liu S, Garzon KI, et al. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res (2016) 365(3):495–506. doi: 10.1007/s00441-016-2464-0
156. Shaw TJ, Martin P. Wound repair at a glance. J Cell Sci (2009) 122(Pt 18):3209–13. doi: 10.1242/jcs.031187
157. Gonzalez AC, Costa TF, Andrade ZA, Medrado AR. Wound healing - A literature review. Bras Dermatol (2016) 91(5):614–20. doi: 10.1590/abd1806-4841.20164741
158. Guo S, Dipietro LA. Factors affecting wound healing. J Dent Res (2010) 89(3):219–29. doi: 10.1177/0022034509359125
159. Mack M. Inflammation and fibrosis. Matrix Biol (2018) 68-69:106–21. doi: 10.1016/j.matbio.2017.11.010
160. Darby IA, Laverdet B, Bonte F, Desmouliere A. Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Invest Dermatol (2014) 7:301–11. doi: 10.2147/CCID.S50046
161. Balkwill F, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci (2012) 125(Pt 23):5591–6. doi: 10.1242/jcs.116392
162. Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol Lett (2017) 14(3):2611–20. doi: 10.3892/ol.2017.6497
163. Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer (2019) 18(1):70. doi: 10.1186/s12943-019-0994-2
164. Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev (2018) 63:40–7. doi: 10.1016/j.ctrv.2017.11.007
165. Weagel E, Smith C, Liu PG, Robison R, O’Neill K. Macrophage polarization and its role in cancer. J Clin Cell Immunol (2015) 6(4):1–8. doi: 10.4172/2155-9899.1000338
166. Giannelli G, Antonaci S. Biological and clinical relevance of laminin-5 in cancer. Clin Exp Metastasis (2000) 18(6):439–43.
167. Clark A, Vignjevic DM. Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol (2015) 36:13–22.
168. Wang JP, Hielscher A. Fibronectin: how its aberrant expression in tumors may improve therapeutic targeting. J Cancer (2017) 8(4):674–82.
169. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol (2019) 12(1):76. doi: 10.1186/s13045-019-0760-3
170. Jung HY, Fattet L, Yang J. Molecular pathways: linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin Cancer Res (2015) 21(5):962–8. doi: 10.1158/1078-0432.CCR-13-3173
171. Jechlinger M, Sommer A, Moriggl R, Seither P, Kraut N, Capodiecci P. Autocrine PDGFR signaling promotes mammary cancer metastasis. J Clin Invest (2006) 116(6):1561–70. doi: 10.1172/JCI24652
172. Neri S, Miyashita T, Hashimoto H, Suda Y, Ishibashi M, Kii H, et al. Fibroblast-led cancer cell invasion is activated by epithelial-mesenchymal transition through platelet-derived growth factor BB secretion of lung adenocarcinoma. Cancer Lett (2017) 395:20–30. doi: 10.1016/j.canlet.2017.02.026
173. Lebrun JJ. The Dual Role of TGF beta in Human Cancer: From Tumor Suppression to Cancer Metastasis. ISRN Mol Biol (2012) 2012:381428. doi: 10.5402/2012/381428
174. Simpson-Haidaris PJ, Rybarczyk B. Tumors and fibrinogen. The role of fibrinogen as an extracellular matrix protein. Ann N Y Acad Sci (2001) 936:406–25. doi: 10.1111/j.1749-6632.2001.tb03525.x
175. Tulotta C, Ottewell P. The role of IL-1B in breast cancer bone metastasis. Endocr Relat Cancer (2018) 25(7):R421–34. doi: 10.1530/ERC-17-0309
176. Masjedi A, Hashemi V, Hojjat-Farsangi M, Ghalamfarsa G, Azizi G, Yousefi M, et al. The significant roles of interleukin-6 and it’s signaling pathway in the immunopathogenesis and treatment of breast cancer. BioMed Pharmacother (2018) 108:1415–24. doi: 10.1016/j.biopha.2018.09.177
177. Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol (2017) 11(7):805–23. doi: 10.1002/1878-0261.12095
178. Liu T, Zhou L, Li D, Andl T, Zhang Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front Cell Dev Biol (2019) 7:60. doi: 10.3389/fcell.2019.00060
179. Song W, Mazzieri R, Yang T, Gobe GC. Translational Significance for Tumor Metastasis of Tumor-Associated Macrophages and Epithelial-Mesenchymal Transition. Front Immunol (2017) 8:1106. doi: 10.3389/fimmu.2017.01106
180. Turner CE, Burridge K. Transmembrane molecular assemblies in cell- extracellular matrix interactions. Curr Opin Cell Biol (1991) 5:849–53. doi: 10.1016/0955-0674(91)90059-8
181. Damsky CH, Werb Z. Signal transduction by integrin receptors for extracellular matrix: cooperative processing of extracellular information. Curr Opin Cell Biol (1992) 5:772–81. doi: 10.1016/0955-0674(92)90100-q
182. Roy F, DeBlois C, Doillon CJ. Extracellular matrix analogs as carriers for growth factors: in vitro fibroblast behavior. J BioMed Mat Res (1993) 27:389–97. doi: 10.1002/jbm.820270312
183. Scott JE. Extracellular matrix, supramolecular organization and shape. J Anat (1995) 187:259–69.
184. Yanagishita M. Function of proteoglycans in the extracellular matrix. Acta Pathol Jpn (1993) 43:283–93. doi: 10.1111/j.1440-1827.1993.tb02569.x
185. McCarthy J, Turley EA. Effects of extracellular matrix components on cell locomotion. Crit Rev Oral Biol Med (1993) 4:619–37. doi: 10.1177/10454411930040050101
186. Lippitz BE, Harris RA. Cytokine patterns in cancer patients: A review of the correlation between interleukin 6 and prognosis. Oncoimmunology (2016) 5(5):e1093722. doi: 10.1080/2162402X.2015.1093722
187. Xu J, Yin Z, Cao S, Gao W, Liu L, Yin Y, et al. Systematic review and meta-analysis on the association between IL-1B polymorphisms and cancer risk. PloS One (2013) 8(5):e63654. doi: 10.1371/journal.pone.0063654
188. Zhang L, Li HY, Li W, Shen ZY, Wang YD, Ji SR, et al. An ELISA assay for quantifying monomeric C-reactive protein in plasma. Front Immunol (2018) 9:511. doi: 10.3389/fimmu.2018.00511
Keywords: C-reactive protein, monomeric C-reactive protein, inflammation, tumor microenvironment, acute phase response
Citation: Hart PC, Rajab IM, Alebraheem M and Potempa LA (2020) C-Reactive Protein and Cancer—Diagnostic and Therapeutic Insights. Front. Immunol. 11:595835. doi: 10.3389/fimmu.2020.595835
Received: 17 August 2020; Accepted: 07 October 2020;
Published: 19 November 2020.
Edited by:
Kenji Daigo, Nippon Medical School, JapanReviewed by:
Annette Karen Shrive, Keele University, United KingdomMoises Armides Franco Molina, Autonomous University of Nuevo León, Mexico
Copyright © 2020 Hart, Rajab, Alebraheem and Potempa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter C. Hart, cGhhcnQwMkByb29zZXZlbHQuZWR1; Lawrence A. Potempa, bHBvdGVtcGEwMUByb29zZXZlbHQuZWR1