 Vivian Lei
Vivian Lei Amy J. Petty
Amy J. Petty Amber R. Atwater1
Amber R. Atwater1 Amanda S. MacLeod
Amanda S. MacLeod- 1Department of Dermatology, Duke University, Durham, NC, United States
- 2School of Medicine, Duke University, Durham, NC, United States
- 3Department of Immunology, Duke University, Durham, NC, United States
- 4Pinnell Center for Investigative Dermatology, Duke University, Durham, NC, United States
- 5Department of Molecular Genetics and Microbiology, Duke University, Durham, NC, United States
The skin is an active immune organ that functions as the first and largest site of defense to the outside environment. Serving as the primary interface between host and pathogen, the skin’s early immune responses to viral invaders often determine the course and severity of infection. We review the current literature pertaining to the mechanisms of cutaneous viral invasion for classical skin-tropic, oncogenic, and vector-borne skin viruses. We discuss the skin’s evolved mechanisms for innate immune viral defense against these invading pathogens, as well as unique strategies utilized by the viruses to escape immune detection. We additionally explore the roles that demographic and environmental factors, such as age, biological sex, and the cutaneous microbiome, play in altering the host immune response to viral threats.
Introduction
The skin is a dynamic barrier organ that establishes a clear boundary between the host and the outside world. As an immune organ, the skin actively surveils the surrounding environment and establishes an appropriate barrier and immune response to commensal microbiota including bacteria, fungi, and viruses. However, upon disruption of the skin barrier, the skin must orchestrate complex immune signals to protect against infiltration and attack by pathogenic invaders. Importantly, responses by the cutaneous innate immune system and its effectors play essential roles in early destruction of pathogens as well as establishment of an immune barrier to prevent systemic infection. This is accomplished via phagocytic cells (i.e. macrophages, neutrophils, and dendritic cells), leukocytes (i.e. natural killer (NK) cells, mast cells, basophils, and eosinophils), as well as epidermal keratinocytes. The introduction of pathogens activates these innate immune cells’ pathogen recognition receptors (PRRs), including toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors, retinoic acid-inducible gene 1 (RIG-I)-like helicase receptors, and c-type lectin receptors. PRRs recognize different pathogen-associated molecular patterns (PAMPs) on microbes and damage-associated molecular patterns (DAMPs) that arise from damaged host cells, which subsequently leads to the induction of pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α and interferon (IFN)-γ, as well as chemokines that recruit phagocytic cells. Keratinocytes and infiltrating immune cells further the hostile environment to pathogens by generating peptides and proteins with distinct antibacterial, antifungal, antiviral capabilities (1).
Cutaneous viral infection presents a unique challenge to the skin’s immune system, as viruses have the ability to hijack host machinery to advance viral replication. As such, early abrogation of viral pathogenicity by the innate immune response establishes a protective antiviral state and limits the potential for systemic spread. Here, we provide an overview of viral entry mechanisms by various viruses with differing infection propensities, i.e. classically skin-tropic and oncogenic skin viruses, as well as vector-introduced skin viruses. We review how these viruses uniquely interact with different aspects of the cutaneous innate immune system, and we further explore some evolved viral mechanisms that directly interfere with the host innate immune response. Lastly, we provide insights on how demographic and environmental factors, such as host age, biological sex, and the commensal microbiome, contribute to various aspects of innate antiviral immunity in the skin (Figure 1, Table 1).
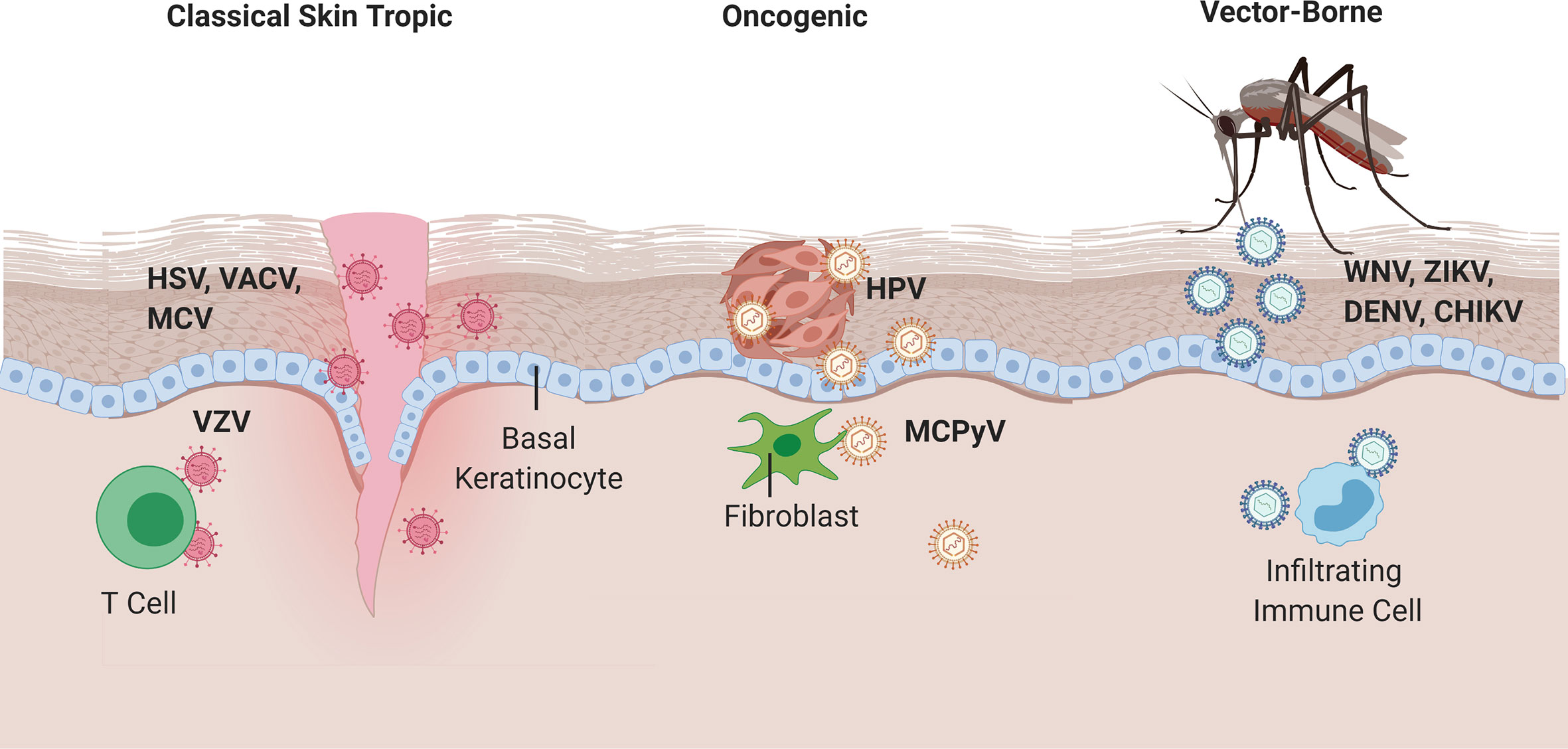
Figure 1 Viral entry of classical skin tropic, oncogenic, and vector-borne viruses. Classical skin tropic viruses such as herpes simplex virus (HSV), vaccinia virus (VACV), molluscum contagiosum virus (MCV), and varicella zoster virus (VZV) have tropism to skin epidermis where keratinocytes are the predominant cell type. HSV and MCV can enter the skin via defects in the skin barrier, which provide viruses with direct contact to the basal epidermal layers. VACV is introduced iatrogenically via vaccination needles. VZV inoculation occurs in the respiratory epithelia and hematogenously spreads to epidermis via infected T cells. Oncogenic viruses such as human papillomaviruses (HPV) and merkel cell polyomavirus (MCPyV) commonly take on their neoplastic potential in immunocompromised patients where the barrier to overcome immune defenses are significantly lower. HPV enters via micro-lesions and replicates in keratinocytes, whereas MCPyV has proclivities toward replication in dermal fibroblasts and CD4+ T cells, respectively. West Nile, Zika, Dengue, and Chikungunya viruses are introduced into the skin via mosquito vectors and cause a local inflammatory response that homes immune cells to the skin infection site, which allows for subsequent infection of migratory immune cells and potential for systemic spread.
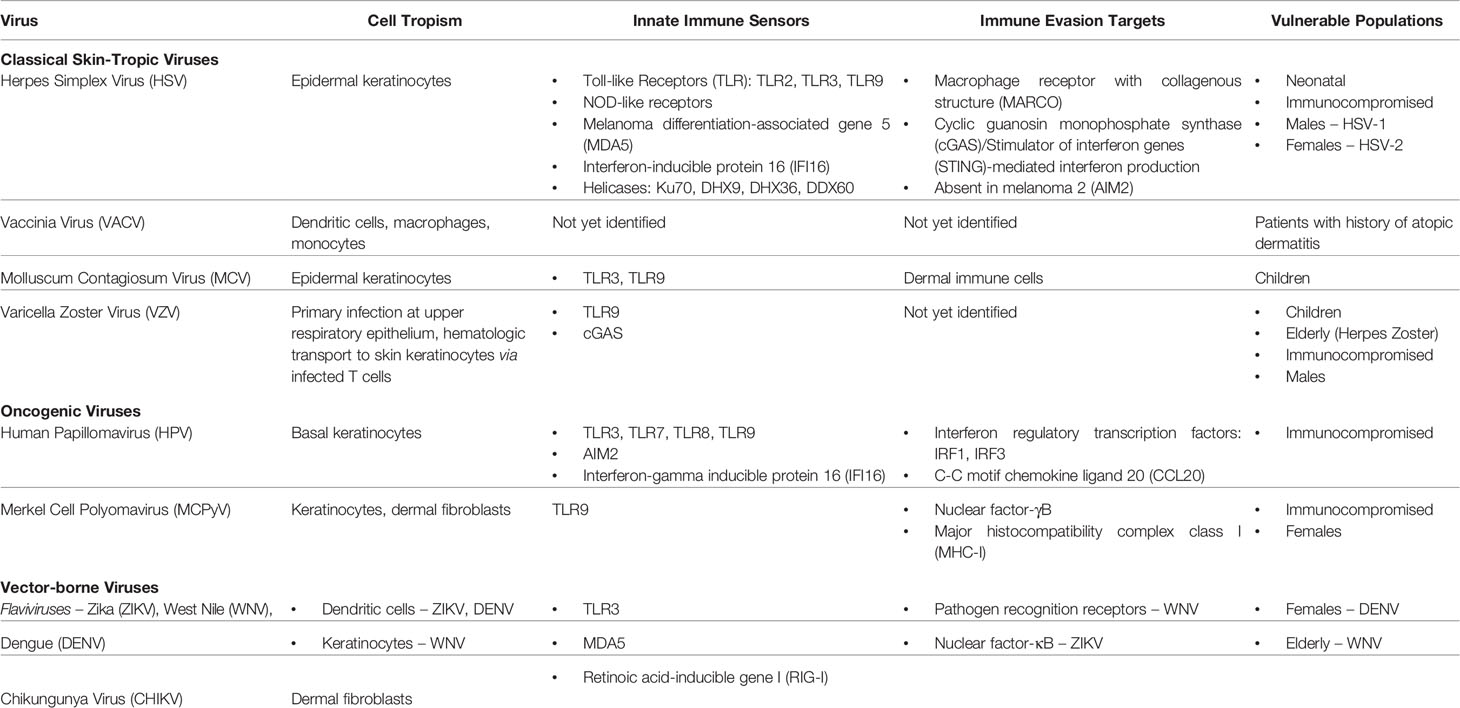
Table 1 Summary of cutaneous viruses, their cell tropism, their innate immune sensors and evasion targets, and populations vulnerable to viral infection.
Classical Skin-Tropic Viruses
Herpes Simplex Virus (HSV)
Herpes simplex virus (HSV) type-1 and 2, of the Herpesviridae family, are enveloped double-stranded DNA viruses that are notable for their neurotropism to the dorsal root ganglia and trigeminal ganglia after primary infection at a mucocutaneous site (2). Primary and reactivated infections are marked by tender grouped erythematous vesicles with varying presentations and degrees of severity (3). HSV-1 is typically characterized by oro-facial lesions with primary infection most often occurring in childhood, whereas HSV-2 is traditionally known as a sexually transmitted infection producing genital lesions, although both types can be found at either site (4). In immunocompromised and neonatal patients, HSV has the potential to disseminate and cause severe morbidity and mortality (3).
In both primary and reactivated infections, viral entry and replication largely occur in the epidermis, where keratinocytes are the predominant cell type. Host cell entry is coordinated by seven HSV glycoproteins; however, four glycoproteins (gB, gD, gH, and gL) are necessary and sufficient for complete viral fusion (5). Viral entry steps start with initial attachment to heparan sulfate proteoglycans (HSPGs) on keratinocytes via gB and gC. Subsequent fusion of the viral envelope with the plasma membrane is mediated by gB and heterodimer gH/gL (6, 7). Envelope glycoprotein gD additionally interacts with cell surface receptors nectin-1, nectin-2, and herpesvirus entry mediator (HVEM) to aid in viral envelope fusion with the plasma membrane (8, 9). After fusion, HSV viral spread relies on the trans-Golgi network for delivery of viral glycoproteins and particles with resultant infection of nearby cells via cell–cell junctions (10, 11).
At the cell surface, Toll-like receptor (TLR) 2 senses viral gB and gH/gL and activates the nuclear factor κB (NF-κB) pathway to induce expression of pro-inflammatory cytokines (e.g. tumor necrosis factor (TNF)-α, interleukin (IL)-6 and IL-12) and chemokines (e.g. CC chemokine ligand 2 (CCL2)) (12–15). Once within the cell, HSV nucleic acids activate TLR3 and TLR9 in the endosomes, while a slew of PRRs (i.e. NOD-like receptors, melanoma differentiation-associated gene 5 (MDA5), interferon-inducible protein 16 (IFI16), and several helicases (Ku70, DHX9, DHX36, DDX60)) sense HSV DNA and RNA in the cytoplasm (16). Together, PRR activation confers type I and III interferon signaling in both human keratinocytes and infiltrating monocyte-lineage cells (17–21). Several induced interferon stimulated gene (ISG) products, such as myxovirus (Mx) A and double-stranded RNA-activated protein kinase (PKR), have direct antiviral properties against HSV, such as limiting viral replication and initiating autophagy to limit cell–cell spread (22). The importance of these many facets of the innate immune antiviral response are highlighted in observations that patients with tyrosine kinase 2 (TYK2) deficiency, who have impaired type I IFN, IL-6, and IL-12 responses, have increased frequency of recurrent HSV infections (23).
Additional innate host defense regulators acting prior to the canonical IFN signaling pathways have also been discovered to play roles in the battle against HSV. For example, promyelocytic leukemia nuclear bodies associate with histone chaperones to capture viral DNA and block HSV replication (24, 25). Keratinocytes were also found to release IL-1α and IL-36 to bolster the antiviral state by acting as early alarm signals for leukocyte recruitment and increasing cellular sensitivity to type I IFN signaling, respectively (26, 27).
Recent discoveries have also identified novel potential roles of NK cells to contribute directly to innate protection against HSV infection. A 2003 study in mice identified that NK cells provided a critical source of early IFNs to control HSV-2 infection and that mice deficient in NK cells had enhanced susceptibility to HSV (28). Corroborating these observations is a case report in 2004 of two individuals with NK cell deficiency who were observed to have severe disseminated HSV-2 infection (29). Absence of NK cells resulted in a diminished CD4+ and CD8+ T cell responses, and the presence of NK cells alone were identified to be able to rescue dysmorphic CD8+ T cells to mount an effective CD8+ T cell response even in the absence of CD4+ T helper cells (30). These findings propose a potential role of NK cells to mediate and bridge innate and adaptive immune responses. Further investigations can be conducted to elucidate the specific mechanisms utilized by NK cells to enhance T cell responses and determine whether NK cells exposed to HSV confer a ‘memory’ response to more readily bolster both innate and adaptive immune functions upon HSV reactivation. These discoveries may present NK cells as attractive targets to enhance both arms of the immune response against HSV infection. The role of innate lymphoid cells (ILCs) has been additionally studied in the context of HSV infection, though in vivo mouse studies showed that ILC-deficiency showed no difference in survival or disease severity (31).
Despite the many innate immune players against HSV, the virus has evolved mechanisms to usurp host machinery and enhance infectivity. For example, HSV was discovered to use scavenger receptors to increase affinity of surface protein interactions (32), inhibit intracellular viral DNA sensing (33, 34), dampen pro-inflammatory cytokine production and inflammasome formation (35), and directly abrogate type I IFN signaling (36). These mechanisms have rendered HSV to be one of the most successful viruses capable of infecting other cell types, including fibroblasts, lymphocytes, and leukocytes (8). Unsurprisingly, HSV’s ability to counteract multiple facets of the early, innate cutaneous immune response helps to explain its capacity to successfully infect beyond the initial infection site and cause latent disease. Given the plethora of studies of viral mechanisms and viral targets for immune evasion, HSV is primed as a viable target to study ways to strengthen innate antiviral immune responses, both IFN-dependent and IFN-independent, to provide different avenues of attenuating disease severity.
Vaccinia Virus
Vaccinia viruses are large, enveloped double-stranded DNA viruses of the Poxviridae family. Due to highly conserved structural proteins across orthopoxviruses, VACV is often used to immunize against smallpox caused by variola virus (37). All human orthopoxvirus infections are zoonoses and typically present as localized or disseminated papules, vesicles, or scabs that may be accompanied by fever, lymphadenopathy, malaise, and myalgia (38).
VACV replication preferentially occurs in cutaneous sites with compromised barrier function (39), where there is increased access to the basolateral membrane (40). Viral entry begins with attachment of four viral proteins (A26, A27, D8 and H3) of the mature virion to cell surface glycosaminoglycans (GAGs), extracellular matrix proteins, and, at lipid rafts, integrin membrane receptors (41, 42). Following attachment is an intricate synchrony of twelve entry proteins that compose the fusion complex, which introduces viral DNA into the cell (reviewed in 43).
Infection with VACV is uncommon when exposure occurs in a healthy cutaneous environment where innate immune responses effectively suppress viral pathogenicity. In fact, a study by Rice and colleagues showed that enhancement of early pro-inflammatory signals using a scarification model of viral delivery significantly decreased lethality of VACV. The group proposed that scarification allowed keratinocytes to actively produce an antiviral state through secretion of chemokines and cytokines (44). These findings are corroborated by discoveries that TNF-receptor knockout and IL-1 receptor type 1 knockout mice had larger cutaneous lesions and higher viral copies compared to their wild type counterparts (45, 46). In vitro, VACV viral infection of epidermal Langerhans cells (LC) and plasmacytoid dendritic cells (pDCs) resulted in inhibition of their ability to elicit cytokine production, including IFN-α and IFN-γ (47, 48). Activated NK cells also secrete necessary IFN-γ to attenuate early infection and promote VACV clearance (49–51). Together, these findings suggest a key role in early innate immune signaling in preventing viral lethality; these signals are essential for VACV vaccine efficacy.
Though typically regarded as safe, VACV vaccination has the potential to cause eczema vaccinatum or progressive vaccinia, both severe and potentially lethal complications (52, 53). Occurring mostly in individuals with a history of atopic dermatitis (AD), a disease that is distinguished by barrier defects resulting from disrupted terminal epidermal differentiation, disseminated VACV includes a generalized vesiculopustular eruption that can progress to large non-healing lesions and predispose individuals to sepsis (54, 55). Viral progression is theorized to be due to reduced capability of AD skin’s innate immune mechanisms to subvert viral attack. With VACV’s preferential infection of dendritic cells, macrophages, and monocytes (56), infection of epidermal antigen-presenting LCs at the early stage impairs release of pro-inflammatory cytokines and IFNs (48). Next, attempts to limit viral spread via programmed cell death are offset by AD skin’s hyper-proliferative state, which presents the virus with many new targets (57). Moreover, the skew towards Th2 responses in AD, with increased IL-4 and IL-13 expression in particular, further decreases antiviral cytokines and type I and II IFNs (58, 59). Consequently, this results in reduced expression of antimicrobial proteins such as human β-defensin (hBD) 3 and human cathelicidin LL-37, which have been shown to directly deter VACV pathogenicity (60, 61). Together, the compromised immune landscape in AD skin provides fertile ground for VACV spread. Given the strong association between VACV (and also HSV) dissemination and AD, future studies are warranted regarding how alterations in terminal epidermal differentiation affect innate antiviral immune signatures at homeostasis as well as upon viral challenge.
Molluscum Contagiosum Virus
Molluscum contagiosum virus, an enveloped linear double-stranded DNA virus of the Poxviridae family, is introduced via direct contact with infected skin or fomites (62). Although MCV infection is common, specific studies on viral entry mechanisms have been limited due to lack of working in vivo and in vitro models. Early electron microscopy of MCV showed preferential infection of keratinocytes in the basal layers at the outset of primary infection (63, 64). Similar to other viruses with tropism to the basal layer, micro-abrasions in the skin provide MCV a direct pathway of entry, and it has been well documented that individuals with skin barrier defects have increased susceptibility (65, 66). Viral proliferation then continues in mitotically active keratinocytes and expands apically, giving rise to distinct dome-shaped papules called molluscum bodies. Viral dissemination occurs as viral particles exit via a keratinized tunnel at the umbilicated center of the lesion (67).
MCV is notable for its ability to evade immune detection as it replicates within epidermal keratinocytes; it forms enclosed molluscum bodies that effectively evade dermal immune detection (68, 69). Interestingly, reports that physical manipulation of molluscum bodies results in local inflammation and ultimate resolution of the infection posit the notion of viral clearance by nearby dermal immune cells (70, 71). Although studies of specific innate immune responses to MCV are limited, one study suggests that MCV activates TLR3 and TLR9 in epidermal keratinocytes. They additionally observed upregulation of IFN-β and TNF-α in the environment surrounding molluscum bodies (72). Work by Vermi et al. further identified plasmacytoid and type I IFN-induced dendritic cells as key effectors in spontaneous regression of MCV in the aforementioned inflammatory setting (73). While MCV’s preference toward epidermal replication allows it to escape dermal immune detection, it remains unclear whether and how epidermal Langerhans cells contribute to immune responses to MCV infection and whether MCV has evolved mechanisms to silence LC contributions to immune surveillance.
Varicella Zoster Virus
Varicella zoster virus is another neurotropic enveloped, double-stranded DNA virus of the Herpesviridae family with primary infection consisting of a generalized pruritic vesicular eruption along with fever, headache and malaise (74). Unlike the previously discussed skin-tropic viruses, infection of epidermal keratinocytes is introduced via hematologic transport of infected T cells after primary inoculation in the upper respiratory epithelium (75, 76). VZV utilizes gB and heterodimer gH/gL, conserved fusion machinery of herpesviruses, for attachment and entry into keratinocytes. Within the skin, cell–cell fusion generates multinucleated infected cells that reside within the vesicular skin lesions. Studies show that VZV fusion protein gB possesses components on both its ecto- and cytoplasmic domains that are essential for infectivity: gB drives VZV’s replication, cell–cell fusion, and characteristic syncytial formation (77, 78). However, additional studies suggest that VZV virulence requires careful regulation of gB, as gain-of-function mutations in gB have been shown to limit viral spread in human skin (79).
Given the poor outcomes in VZV-infected individuals with adaptive immune deficiencies, early establishment of an antiviral state in the skin is vital. These responses work effectively to limit disease severity and activate cell-mediated immunity. Cytosolic sensing activates stimulator of interferon genes (STING)-mediated IFN-γ production to upregulate antiviral genes, like MxA and OAS. TLR9 dependent sensing of VZV is also noted to trigger massive IFN-α release by pDCs (80, 81). Exogenous treatment with IFN-α has been shown to abrogate VZV severity through inhibition of viral replication via interferon regulator factor (IRF) protein 9 (82, 83). However, IFN-α signaling was not sufficient to completely terminate VZV transmission due to down-regulation of this pathway by viral gene products (75). Natural killer cells also prevent viral spread by killing infected cells, and their absence has been linked to severe infection (84, 85). Given the discoveries of the important role of NK cells during innate immune signaling and priming adaptive responses in other skin viruses, studies of the specific functions of NK cells in the context of VZV can provide promising avenues of discovery into establishment of an early antiviral state.
Oncogenic Viruses
Human Papillomavirus
Human papillomaviruses are non-enveloped double-stranded DNA viruses that can be transmitted through direct skin-to-skin contact (86). There are more than 200 described HPV types. The alpha HPVs (i.e. HPV16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58 and 59) are considered high risk or carcinogenic and have been identified as etiologic agents of a multitude of cancers, including cervical, oropharyngeal, vaginal, vulvar, penile, and anal cancers (87, 88). Beta and gamma types are considered possibly carcinogenic or non-carcinogenic. Several studies have identified potential contributory roles of beta HPVs to non-melanoma skin cancer when associated with ultraviolet radiation (89). The low risk non-carcinogenic HPVs are known to cause benign lesions such as anogenital, palmar, and plantar warts (90).
Viral penetration into the epidermis is facilitated via microlesions and HPV’s replication cycle starts at the mitotically active basal layer (91). Once within the basal layer, viruses gain entry into the cells through endocytosis, which are enabled by viral proteins L1 and L2 that help the virus interact with the cell surface. Molecules such as HSPGs and syndecan-1 are putative targets of HPV that enable viral trafficking into the host cell (92). After internalization, HPV virions reach the nucleus through the clathrin-mediated endocytic pathway (93, 94).
Within the basal layers, HPV DNA copy number is low and viral replication is slow. As viral replication speeds up and the virus leaves the basal layer to reach the upper layers of the epidermis, innate and adaptive immune responses become more important in surveilling and controlling viral spread (95). HPV DNA within a host cell is recognized by innate pathogen sensors, including absence in melanoma 2 (AIM2), interferon-gamma inducible protein 16 (IFI16), and cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS) (96–98). AIM2 inflammasome activation results in maturation of caspase-1 and IL-1β in HPV16-infected keratinocytes (99). TLR activation in keratinocytes by HPV also results in release of pro-inflammatory cytokines such as TNF-α, IL-8, C-X-C motif chemokine ligand 9 (CXCL9), and type I interferon (IFN-α and -β) (100). In fact, higher expression of TLRs was found to be correlative with clearance of initial HPV16 infection in women (101).
HPV-infected keratinocytes additionally recruit macrophages, Langerhans cells (LCs), natural killer (NK) cells, and T lymphocytes in the initial antiviral response. TLR activation in macrophages and LCs through NF-γB and interferon response factor (IRF)-3 further promotes the release of TNF-α, IFN-γ, IL-1β, IL-12 and IL-18, which can in turn activate other inflammatory cells through paracrine signaling. IL-1 and TNF-α have also been shown to downregulate the transcription of viral oncoproteins E6 and E7 (100). Though there is limited evidence on the role NK cells play in controlling HPV infections, it was reported that patients with functional NK deficiencies were more susceptible to HPV infection and HPV-associated cancer (102). Together, these studies highlight the importance of host innate immunity during the initial antiviral responses against HPV in cutaneous tissues.
Many studies provide evidence that HPV has evolved mechanisms to counter host immune responses. HPV-infected cells can reprogram the local immune milieu to promote chronic inflammation and subsequently carcinogenesis. HPV E6 protein can directly target IRF3 while E7 protein interferes with the antiviral and pro-apoptotic functions of IRF1 via protein–protein interactions, leading to suppressed IFN signaling and downstream responses (103–105). Additionally, HPV infection was found to interfere with LC homeostasis due to the suppression of C-C motif chemokine ligand 20 (CCL20), a chemokine critical for the repopulation of CD1a+ LC precursor cells in the epidermis (106). It was shown that viral E7 protein abrogates the binding of CCAAT/enhancer-binding protein beta (C/EBPβ) in the promoter region of CCL20. As a result, CCL20-directed migration of LCs and subsequent antigen-presentation in the epithelium is suppressed, allowing for viral persistence (106). In summary, HPV modulates several host cellular pathways to evade immune responses, leading to virus-mediated immunosuppression and neoplastic development. However, given the diversity of HPV types and their various neoplastic or benign propensities, further investigations are needed to identify differential mechanisms utilized by the host to respond to various HPV types, as well as how certain specific HPVs are able to subvert host immune signaling to impart immunosuppression and impart neoplastic potential.
Merkel Cell Polyomavirus
Merkel cell polyomavirus belongs to the Polyomaviridae family which consists of non-enveloped, double-stranded DNA viruses that have infectious and tumorigenic potential (107). Since the initial identification in Merkel cell carcinoma (MCC) in 2008, many reports have provided additional evidence of the causal relationship between MCPyV and MCC (108–112). MCC is an aggressive cancer that is characterized by a rapidly expanding, asymptomatic, erythematous dome-shaped tumor that presents often on sun-exposed areas of the skin (113).
It remains under debate which cutaneous cell type(s) MCPyV primarily infects due to poor replication of MCPyV in in vitro cultures (114). Keratinocytes were thought to be the primary target due to chronic cutaneous shedding of MCPyV (115). However, a recent report showed that MCPyV preferentially infects human dermal fibroblasts (116). Viral attachment relies on recognition of sulfated GAGs and interaction with sialylated oligosaccharides containing the Neu5Acα2-3Gal linear motif by viral capsid protein, VP1 (117, 118). MCPyV eventually enters target cells through caveolar/lipid raft-mediated endocytosis (119).
Many recent reports suggest the important role the host immune system plays in MCPyV infection and MCC development. First of all, immunocompromised patients are more likely to develop MCC (120). Secondly, high intratumoral CD8+ T cell counts and immune transcripts are associated with more favorable outcomes in MCC patients (121, 122). Innate immune responses were thought to play a critical role in the initial sensing and clearance of MCPyV virions. Shadzad et al. reported that TLR9, a critical sensor for viral and bacterial dsDNA, is downregulated by MCPyV large T antigen during infection (123). Additionally, MCPyV small T antigen negatively regulates NF-γB-mediated inflammatory signaling by inhibiting IKKα/IKKβ-induced IγB phosphorylation, further dampening host antiviral responses (124). Lastly, MCPyV-positive MCC tumors were discovered to have lower expression of major histocompatibility complex class I (MHC-I) compared to MCPyV-negative MCC samples, suggesting another potential mechanism by which MCPyV-infection cells escape immune destruction (125). However, precise interactions between MCPyV and the host immune system are largely unknown. Further work is needed to elucidate the various mechanisms by which MCPyV subverts host immune surveillance to establish persistence.
Vector-Borne Skin Viruses
Mosquitos infect hundreds of millions of people around the world annually, introducing individuals to pathogenic bacteria, parasites, and viruses that have the potential to cause severe systemic illness in the host and, with Zika virus, their offspring (126, 127). Despite the prevalence of mosquito-borne illnesses and their threat to global human health, little is known about the early stages of cutaneous infection.
Zika virus (ZIKV), West Nile virus (WNV), and Dengue virus (DENV) belong to the Flaviviridae family and are enveloped RNA viruses. ZIKV and DENV have a predisposition to infect cutaneous dendritic cells, whose migratory characteristic allows for rapid dissemination and viremia (128, 129). WNV has been shown to preferentially infect keratinocytes, though it is capable of infecting dendritic cells as well (130, 131). Flaviviral envelope (E) glycoprotein is key to initial viral entry via low-affinity attachment to GAGs on the target cell surface (132, 133). More specific attachment to a wide array of entry receptors that help facilitate internalization into dendritic cells has been identified, including C-type lectin receptors, αvβ3 integrins, T-cell immunoglobulin and mucin domain (TIM) and TYRO3, AXL and MER (TAM) receptors (134). Clathrin-dependent endocytosis then allows for viral fusion into the target cell (135).
Chikungunya virus (CHIKV) is also an enveloped RNA virus but belongs to the Togaviridae family with tropism to dermal fibroblasts (136). CHIKV viral glycoprotein E2 interaction with cell surface GAGs, TIM family receptors, and prohibitins has been shown to assist with early interactions of CHIKV with the target cell, although CHIKV is able to infect in the absence of these proteins (137). Similar to flaviviruses, CHIKV utilizes clathrin-dependent endocytosis to generate a low pH environment to cause conformational changes in glyocoprotein E1 and permit fusion (138).
Once in the skin, ZIKV, WNV, DENV, and CHIKV all trigger PRRs retinoic acid-inducible gene I (RIG-I), TLR3, and melanoma differentiation associated gene-5 (MDA-5). Next, pro-inflammatory cytokine and chemokine signaling is coupled with activation of IFN-β and antiviral proteins, including members of the OAS, Mx, interferon stimulated gene (ISG), and interferon induced proteins with tetratricopeptide repeats (IFIT) families, in keratinocytes and dermal myeloid cells (81, 129, 139–142). Specific to ZIKV, our group recently identified a novel IFN-independent pathway of antiviral protein induction via IL-27. Uniquely, signal transducer and activator of transcription (STAT) 1- and interferon regulatory factor (IRF) 3-dependent IL-27 signaling was able to induce antiviral proteins OAS1, OAS2, OASL2, and MX1 in keratinocytes and reduce ZIKV pathogenicity when the virus was introduced via a cutaneous, and not intravenous, route (143). These results suggest a potential avenue to distinctly upregulate cutaneous antiviral proteins independent of interferon signaling, although whether this pathway confers similar resistance to other vector-borne viruses remains to be discovered.
Given arboviruses’ predilection to infect immune cells, the recruitment of distal immune cells to the dermis may not be as advantageous to the host as is the case for many other pathogens. After an early infection in the epidermis, a second round occurs when the immune response homes arbovirus-susceptible monocytes and monocyte-derived dendritic cells to the site (144). These infected immune cells then travel to the draining lymph nodes to continue systemic spread. This begs the question of whether pathogenicity can be reduced or limited to the epidermis by dampening inflammatory signaling. One group observed a 75–90% reduction in infection of LCs, macrophages, and dermal dendritic cells when cytokine IL-1β expression was inhibited (128).
An additional non-viral factor also contributes to the immune picture. Intriguingly, mosquito saliva has been shown to significantly alter the early innate signatures to enhance viral spread. Mosquito saliva protein D7 inhibits DENV virions and envelope proteins (145). ZIKV-activated NF-ĸB signaling is inhibited by salival protein LTRIN (146). In CHIKV, mosquito saliva suppresses Th1 cytokine (IFN-γ and IL-2), TLR3, and chemokine expression while simultaneously pushing toward a Th2 polarity—which, as we have discussed, is a less advantageous antiviral profile from the host perspective (147, 148). Decreases in expression of PRRs and antiviral proteins with specific targeting of flaviviruses (OAS1, MX1, and ISG20) were also observed in WNV-infected keratinocytes (149, 150).
The unique mode of inoculation of vector-borne viruses at the skin presents an alluring rationale to study potential methods of undermining viral pathogenicity when the infection is still local and while innate immune responses predominate. However, the frequency of mosquito bites and the lack of urgency to seek medical attention prior to systemic infection may pose a conceivable difficulty for translation into clinical practice.
Demographic and Environmental Contributors To Host Antiviral Responses
As a frontline organ of defense against the outside world, maintaining integrity of the skin barrier and function is critical to the organ’s success in combating potential invaders. However, increasing studies show that, like other regenerating organs, the skin is constantly adapting in response to a multitude of environmental factors (Figure 2).
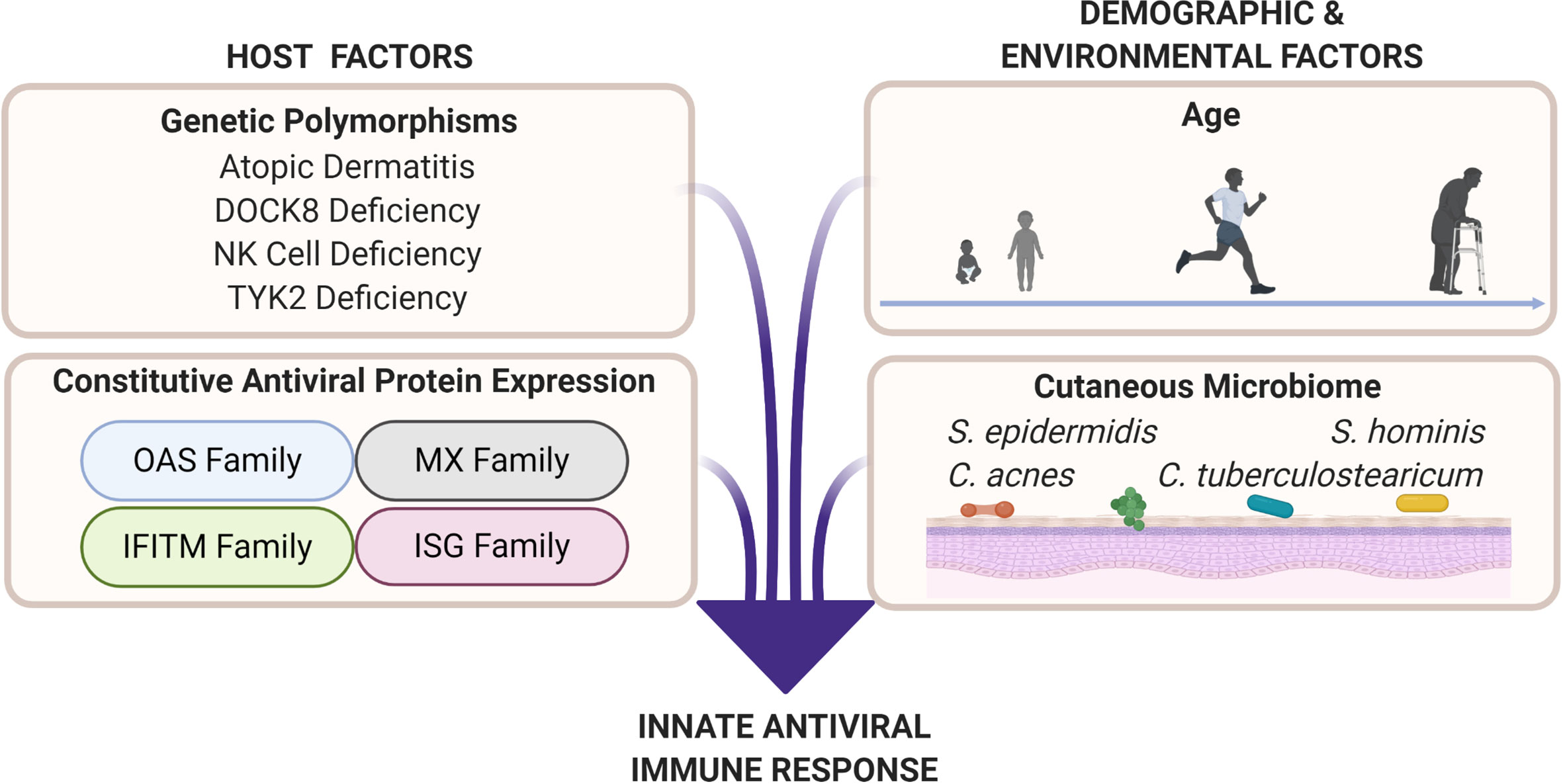
Figure 2 Cutaneous antiviral immune responses are influenced by host as well as demographic and environmental factors. Genetic polymorphisms that result in atopic dermatitis, dedicator of cytokinesis 8 (DOCK8) deficiency, natural killer (NK) cell deficiency, and tyrosine kinase 2 (TYK2) deficiency produce unique immune profiles that are disadvantageous for viral protection. Professional antiviral proteins such as those in the oligoadenylate synthetase (OAS), myxovirus resistance (MX), interferon-induced transmembrane (IFITM), and interferon-stimulated gene (ISG) families are part of the innate antiviral response. These proteins exert their antiviral abilities by inhibiting various parts of the viral replication cycle (151). Factors such as age (see Figure 3), biological sex, and cutaneous microbiome have potential to deter or enhance innate antiviral responses. Microbial interactions, such as bacteria–viral, viral–viral, and fungal–viral, can possibly produce antiviral effectors or influence host antiviral responses.
Age
Given the skin’s constant contact with potential pathogens, the susceptibility of certain patient populations to skin viruses is an interesting area of investigation. Notably, age appears to play a role in the host immune defenses against viral invaders (Figure 3). Some trends are more obvious: MCV and VACV show increased incidence and more severe effects in children as prevalence of AD is highest in this age group, and as previously discussed, the AD milieu contributes to increased viral pathogenicity and impaired antiviral responses (152–154). However, there is less clarity on why certain age groups are more afflicted with other cutaneous viral infections. Intriguingly, prenatal, neonatal, and elderly populations have demonstrated increased susceptibility to systemic malaise and higher risk of mortality compared to young and mature adults. For example, whereas only mild symptoms would typically result from primary HSV infection in children and adults, preterm and neonatal infants, if untreated, only have a 40% chance of survival (155). Similarly, in the elderly population, reports have emerged suggesting that HSV increases the risk for development of neurological diseases like Alzheimer’s and may be a direct infectious etiology (156). Moreover, while VZV dissemination occurs most commonly in children due to primary infection, suppression of the virus is maintained throughout adulthood. However, reactivation, which only occurs after VZV overthrows immune safeguards and presents in the form of herpes zoster, occurs most frequently in elderly individuals or upon immunosuppresion (157).
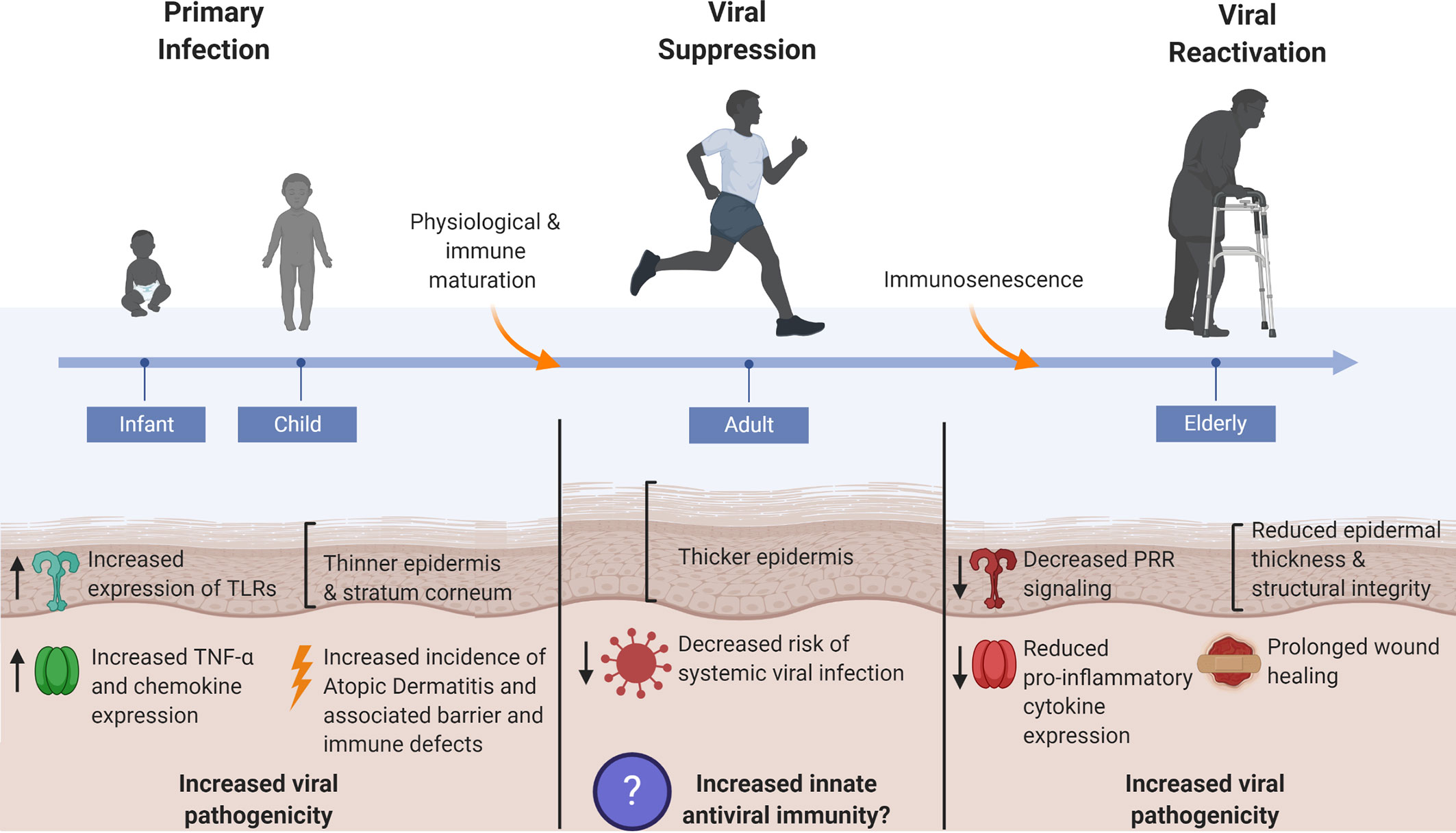
Figure 3 Skin’s antiviral protection changes throughout age. Systemic viral infections are most prevalent at the young and elderly ages where factors such as epidermal thickness and cutaneous innate immunity are markedly different from healthy adult human skin. Thin skin leads to increased susceptibility to micro-injuries and abrasions, thereby providing direct avenues for viral entry. Dysregulated innate immune signaling, consequent to immunological immaturity or immunosenescence in the young and elderly, respectively, furthers the risk of systemic viral infection as immune defenses cannot adequately control early viral propagation. The young and elderly are also at increased risk for viral pathogen exposure due to compromises in skin barrier integrity that manifest in the form of atopic dermatitis in the young and chronic non-healing wounds in the elderly.
One potential explanation for these observations is alteration in the skin’s physical barrier with aging. Preterm and neonatal infants have a thinner epidermis and stratum corneum, and a similar observation applies to the elderly population where cutaneous structural integrity deteriorates and skin thickness is once again reduced (158, 159). Such changes to skin integrity may render it more susceptible to micro-injuries and therefore subsequent pathogen exposure and infection. Functional studies on whether the rate of viral infectivity is enhanced in the setting of thin, fragile skin barriers are limited. Theoretically, decreased epidermal thickness may provide for earlier access to deeper skin layers, which could potentially lessen the time the virus spends replicating at the initial infection site prior to systemic spread, and therefore limit the time available for propagation of early innate immune responses as well as initiation of adaptive immune responses. Additional concerns are warranted in the elderly where the skin’s wound healing capabilities are also reduced, thereby allowing for increased pathogen exposure (159).
Age also has profound effects on certain aspects of the skin’s innate antiviral defenses. For example, in WNV infection, which usually afflicts individuals >60 years of age, worse outcomes were identified in mice with dysregulated TLR7 and STING signaling, both with critical roles in initiating antiviral signaling cascades (160, 161). Generally, older individuals exemplified decreased PRR signaling and decreased induction of pro-inflammatory cytokines and chemokines in several cutaneous compartments, including sebaceous glands, sweat glands, and epidermis (162). Surprisingly, prenatal skin actually exhibited higher levels of TLRs (1–5) compared to adults, and neonatal keratinocytes demonstrated greater secretion of TNF-α and several chemokines when stimulated with poly (I:C), a synthetic dsRNA used to mimic viral nucleic acids (163). It is unclear how this dichotomy corresponds to viral preference and susceptibility at different age groups, although similar outcomes of greater morbidity and mortality in both age groups highlight the importance of better understanding the regulators and effectors of innate antiviral immunity.
Studies have additionally identified discrepancies in the expression levels of cutaneous antimicrobial peptides and proteins at the extremes of age. For example, neonatal skin was observed to express increased levels of antimicrobial peptides LL-37 and hBD2 compared to adults in both mice and humans (164). Contrastingly, reduced levels of antimicrobial peptides were observed in aged skin compared to adult skin (165). While these studies begin to point toward differing antimicrobial signatures across age groups, investigations specifically looking at antiviral proteins and their functional implications are currently lacking.
Biological Sex
Biological sex poses another important variable when considering immune defenses against viral pathogens. Sex differences in innate and adaptive immunity have been well characterized in humans; known to us is that infant and adult males mount weaker innate and adaptive immune responses to pathogens compared to females and are, therefore, theorized to be more susceptible to viral infections. Particularly in the context of innate immunity, varied responses to pathogens can be explained by differential expression in TLR and type I IFN signaling between sexes, wherein females exhibit higher basal and inducible expression levels of TLR7, TLR9, IRF5, and IFN-α (166, 167). The sex differential expression of these pathways confers greater pro-inflammatory responses in peripheral blood mononuclear cells (PBMCs), neutrophils, and macrophages in males, whereas higher anti-inflammatory and cytokine signaling for type I IFN responses are seen in females (168, 169). Further, studies in rodents have shown that expression of signaling molecules associated with antiviral sensing and immunity (Myd88, IRF7, IFN-β, IFNAR1, JAK2, and STAT3) as well as antiviral protein Mx is higher in females compared to males (169). These dimorphic effects are posited to be mediated by gonadal hormones, with possible androgen- and estrogen-specific response elements driving different effector cells’ signaling and expression.
Despite these findings, studies directly looking at the sex differential contribution to viral susceptibility and disease outcome in humans are complicated by various behavioral and environmental differences associated with biological sex as well as gender. Several of the previously discussed viruses show preferential responses between females versus males, though whether biological differences are the cause of these observations is more difficult to tease out. Studies show that males have higher relative incidence of more serious illness and susceptibility to VZV and HSV-1, which may be explained by the aforementioned weakened immune response and pro-inflammatory cytokine profile (170, 171). However, interestingly, epidemiological studies show that females infected with Dengue virus in endemic areas have the same susceptibility to infection though exhibit more severe symptoms, such as hemorrhagic fever, compared to male counterparts (172). Females with Merkel cell carcinomas also have higher prevalence of MCPyV-positive tumors than male patients (173, 174). Additionally, HSV-2 shows a higher prevalence in females compared to males in humans (175, 176). These observations may appear to contradict immunological findings that females show a greater anti-inflammatory signature as well as an enhanced innate and adaptive immune profile compared to males. However, particularly in human studies, direct correlations of biological sex and viral susceptibility and disease outcome not only have to take into account sex hormones and chromosomal/genetic differences, they must also consider the differential effects that arise as a result of behaviors associated with gender and host environment, which may have direct consequences of increasing risk and susceptibility to certain viral pathogens. Murine studies have attempted to control for these confounding factors, although findings do not directly translate to humans. For example, increased progesterone levels are theorized to reduce immune-protective effects and therefore increase HSV-2 susceptibility in females. Female mice that underwent ovariectomy and had estradiol hormone injected showed reduced pathology compared to counterparts injected with progesterone or placebo (177). However, HSV-2 infection is increased in ex vivo human endometrial epithelial cells treated with estradiol (178). These divergent discoveries highlight the immense difficulty of using biological sex as a method of predicting viral susceptibility as well as disease outcome, although knowledge of sexual preferences of pathogens can be utilized to focus clinical efforts to provide better care to at-risk populations.
Cutaneous Microbiome
The skin is home to a highly diverse collection of commensal bacteria, fungi and viruses that form the cutaneous microbiome. The makeup of these colonizers varies across individuals, skin compartments (e.g. hair follicle versus sebaceous gland), body location (e.g. axillary versus facial skin), and even age (179–181). This diversity is mirrored in the varying relationships between host skin and commensal microbiota, ranging from opportunistic to mutualistic interactions. For example, the Cutibacteria family (formerly known as Propionibacteria) of bacteria is a major component of normal skin flora that colonizes preferentially to skin sites that are rich in sebaceous glands. The presence of cutibacteria has been observed to impart protective benefits to the host in common skin pathologies including atopic dermatitis and psoriasis (182, 183). Conversely, Cutibacterium acnes often causes opportunistic infections and is a common etiologic agent in diseases such as acne vulgaris (184). These disparate consequences imply a necessity for the skin to maintain a healthy balance between itself and its surrounding microbiome. Furthermore, the predisposition for viral infection in populations with dysbiosis, such as those with atopic dermatitis, proposes the question of how microbial interactions influence skin responses to viral challenges (185).
Recent studies have begun to identify various antimicrobial roles of skin microbiota. Skin bacterial commensal Staphylococcus epidermidis was observed to produce peptides called bacteriocins that have direct antimicrobial properties against Staphylococcus aureus and Group A Streptococcus (186). Additionally, S. epidermidis was noted to augment the antimicrobial actions of cathelicidin LL-37 (187). C. acnes is also reported to secrete bacteriocins with bactericidal properties toward other cutibacteria (188). This work indicates that commensal bacteria actively participate in maintaining cutaneous microbial homeostasis; however, there is a current lack of understanding of antifungal and antiviral contributions from the cutaneous resident microbiota, including fungi and viruses.
Evidence of how the skin microbiome directly influences cutaneous antiviral immunity is also limited, although studies in patients with primary immunodeficiency, such as dedicator of cytokinesis 8 (DOCK8) deficiency who have altered cutaneous microbiomes compared to healthy patients, reveal that changes in the cutaneous virome lead to increased colonization of DNA viruses like HPVs, HSVs, polyomaviruses, and MCV (189). Inferences can additionally be drawn from studies in other barrier organs and their commensal microbiome. For instance, germ-free mice, i.e. lacking intestinal commensal microbiota, were observed to be more susceptible to influenza A virus, coxsackie B virus, Friend leukemia virus, and murine cytomegalovirus (190, 191). In the respiratory epithelium, S. epidermidis produced an extracellular matrix-binding protein that exhibited anti-influenza activity (192). Further, probiotic colonization of resident Corynebacteria improved resistance to respiratory syncytial virus (193). At the vaginal surface, lack of Lactobacillus bacteria, a dominant colonizer of the vaginal mucosa, led to increased susceptibility of HSV-2 due to abrogated IFN-γ signaling (194). Together, these findings suggest that commensal microbiota contribute directly to antiviral immunity via secretion of antiviral effectors and through enhancement of host immune signaling at their resident sites.
Conclusion
The skin is an active immune organ with immune capabilities that are constantly challenged by friendly commensal and pathogenic microorganisms. Consequently, it has evolved effective defense strategies to combat a wide range of threats, ranging from overpopulation of opportunistic commensal bacteria to pathogenic viruses. Particularly in the scenario of viral infection, the skin’s complex multi-layered defense strategies, even within the innate immune system alone, are highlighted as different viruses’ attempt to hijack and suppress various aspects of its immune machinery. Given the severity of primary infections to many cutaneously introduced viruses, early antiviral responses are critical in the attempt to prevent further viral propagation and to allow time for adaptive immune responses to take effect. Recent advances in understanding specific viral targets of innate immunity begin to provide opportunities for further exploration into bolstering areas of vulnerability, including weaknesses that arise throughout age and between females and males. Additionally, insights to antiviral contributions from the commensal microbiome obtained from studies in other barrier organs suggest potential for future study in the skin.
Author Contributions
VL and AM contributed to conception and design of the review. VL and AP performed initial literature search and wrote the first draft of the manuscript. AM supervised all aspects of the review and manuscript writing and is the corresponding author. All authors contributed to the article and approved the submitted version.
Funding
VL is supported by the Burroughs-Wellcome and Poindexter Medical Student Research Fellowships. AM is supported by R01AI139207 and received a Duke Physician-Scientist Strong Start Award. AM is also supported by a Silab company partnership. The funding sources did not have any control over the content nor results of the review. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
AM consults for Silab and is on the scientific evaluation committee of the LEO Foundation and receives honoraria. AM’s spouse is employed by Precision BioSiences and holds stock and stock options. AA received the Pfizer Independent Grant for Learning and Change and has consulted for Henkel.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Coates M, Blanchard S, MacLeod AS. Innate antimicrobial immunity in the skin: A protective barrier against bacteria, viruses, and fungi. PLoS Pathog (2018) 14(12):e1007353. doi: 10.1371/journal.ppat.1007353
2. Davison AJ. Herpesvirus systematics. Vet Microbiol (2010) 143(1):52–69. doi: 10.1016/j.vetmic.2010.02.014
3. Cunningham AL, Diefenbach RJ, Miranda-Saksena M, Bosnjak L, Kim M, Jones C, et al. The cycle of human herpes simplex virus infection: virus transport and immune control. J Infect Dis (2006) 194(Suppl 1):S11–8. doi: 10.1086/505359
4. Smith JS, Robinson NJ. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis (2002) 186(Suppl 1):S3–S28. doi: 10.1086/343739
5. Karasneh GA, Shukla D. Herpes simplex virus infects most cell types in vitro: clues to its success. Virol J (2011) 8:481. doi: 10.1186/1743-422X-8-481
6. Shukla D, Spear PG. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J Clin Invest (2001) Aug108(4):503–10. doi: 10.1172/JCI200113799
7. Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol (2011) 9(5):369–81. doi: 10.1038/nrmicro2548
8. Spear PG. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol (2004) 6(5):401–10. doi: 10.1111/j.1462-5822.2004.00389.x
9. Petermann P, Rahn E, Their K, Hsu MJ, Rixon FJ, Kopp SJ, et al. Role of nectin-1 and herpesvirus entry mediator as cellular erceptors for herpes simplex virus 1 on primary murine dermal fibroblasts. J Virol (2015) 89(18):9407–16. doi: 10.1128/JVI.01415-15
10. Farnsworth A, Johnson DC. Herpes simplex virus gE/gI must accumulate in the trans-Golgi network at early times and then redistribute to cell junctions to promote cell-cell spread. J Virol (2006) 80(7):3167–79. doi: 10.1128/JVI.80.7.3167-3179.2006
11. Akhtar J, Shukla D. Viral entry mechanisms: cellular and viral mediators of herpes simplex virus entry. FEBS J (2009) 276(24):7228–36. doi: 10.1111/j.1742-4658.2009.07402.x
12. Sato A, Linehan MM, Iwasaki A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc Natl Acad Sci U S A (2006) 103(46):17343–8. doi: 10.1073/pnas.0605102103
13. Ma Y, He B. Recognition of herpes simplex viruses: toll-like receptors and beyond. J Mol Biol (2014) 426(6):1133–47. doi: 10.1016/j.jmb.2013.11.012
14. Gianni T, Leoni V, Campadelli-Fiume G. Type I interferon and NF-κB activation elicited by herpes simplex virus gH/gL via αvβ3 integrin in epithelial and neuronal cell lines. J Virol (2013) 87(24):13911–6. doi: 10.1128/JVI.01894-13
15. Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, et al. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A (2004) 101(5):1315–20. doi: 10.1073/pnas.0308057100
16. Melchjorsen J. Sensing herpes: more than toll. Rev Med Virol (2012) 22(2):106–21. doi: 10.1002/rmv.716
17. Zhou L, Li JL, Zhou Y, Liu JB, Zhuang K, Gao JF, et al. Induction of interferon-λ contributes to TLR3 and RIG-I activation-mediated inhibition of herpes simplex virus type 2 replication in human cervical epithelial cells. Mol Hum Reprod (2015) 21(12):917–29. doi: 10.1093/molehr/gav058
18. Donaghy H, Bosnjak L, Harman AN, Marsden V, Tyring SK, Meng TC, et al. Role for plasmacytoid dendritic cells in the immune control of recurrent human herpes simplex virus infection. J Virol (2009) 83(4):1952–61. doi: 10.1128/JVI.01578-08
19. Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol (2006) 80(9):4501–9. doi: 10.1128/JVI.80.9.4501-4509.2006
20. Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med (2003) 198(3):513–20. doi: 10.1084/jem.20030162
21. Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. Recognition of herpesviruses by the innate immune system. Nat Rev Immunol (2011) 11(2):143–54. doi: 10.1038/nri2937
22. Handfield C, Kwock J, MacLeod AS. Innate Antiviral Immunity in the Skin. Trends Immunol (2018) 39(4):328–40. doi: 10.1016/j.it.2018.02.003
23. Kreins AY, Ciancanelli MJ, Okada S, Kong XF, Ramirez-Alejo N, Kilic SS, et al. Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med (2015) 212(10):1641–62. doi: 10.1084/jem.20140280
24. Alandijany T, Roberts APE, Conn KL, Loney C, McFarlane S, Orr A, et al. Distinct temporal roles for the promyelocytic leukaemia (PML) protein in the sequential regulation of intracellular host immunity to HSV-1 infection. PLoS Pathog (2018) 14(1):e1006769. doi: 10.1371/journal.ppat.1006769. [published correction appears in PLoS Pathog. 2018;14 (2):e1006927].
25. McFarlane S, Orr A, Roberts APE, Conn KL, Iliev V, Loney C, et al. The histone chaperone HIRA promotes the induction of host innate immune defences in response to HSV-1 infection. PLoS Pathog (2019) 15(3):e1007667. doi: 10.1371/journal.ppat.1007667
26. Milora KA, Miller SL, Sanmiguel JC, Jensen LE. Interleukin-1α released from HSV-1-infected keratinocytes acts as a functional alarmin in the skin. Nat Commun (2014) 5:5230. doi: 10.1038/ncomms6230
27. Wang P, Gamero AM, Jensen LE. IL-36 promotes anti-viral immunity by boosting sensitivity to IFN-α/β in IRF1 dependent and independent manners. Nat Commun (2019) 10(1):4700. doi: 10.1038/s41467-019-12318-y
28. Ashkar AA, Rosenthal KL. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J Virol (2003) 77(18):10168–71. doi: 10.1128/jvi.77.18.10168-10171.2003
29. Dalloul A, Oksenhendler E, Chosidow O, Ribaud P, Carcelain G, Louvet S, et al. Severe herpes virus (HSV-2) infection in two patients with myelodysplasia and undetectable NK cells and plasmacytoid dendritic cells in the blood. J Clin Virol (2004) 30(4):329–36. doi: 10.1016/j.jcv.2003.11.014
30. Nandakumar S, Woolard SN, Yuan D, Rouse BT, Kumaraguru U. Natural killer cells as novel helpers in anti-herpes simplex virus immune response. J Virol (2008) 82(21):10820–31. doi: 10.1128/JVI.00365-08
31. Hirose S, Wang S, Tormanen K, Wang Y, Tang J, Akbari O, et al. Roles of Type 1, 2, and 3 Innate Lymphoid Cells in Herpes Simplex Virus 1 Infection In Vitro and In Vivo. J Virol (2019) 93(13):e00523–19. doi: 10.1128/JVI.00523-19
32. MacLeod DT, Nakatsuji T, Yamasaki K, Kobzik L, Gallo RL. HSV-1 exploits the innate immune scavenger receptor MARCO to enhance epithelial adsorption and infection. Nat Commun (2013) 4:1963. doi: 10.1038/ncomms2963
33. Huang J, You H, Su C, Li Y, Chen S, Zheng C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. J Virol (2018) 92(15):e00841–18. doi: 10.1128/JVI.00841-18
34. Pan S, Liu X, Ma Y, Cao Y, He B. Herpes Simplex Virus 1 γ134.5 Protein Inhibits STING Activation That Restricts Viral Replication. J Virol (2018) 92(20):e01015–18. doi: 10.1128/JVI.01015-18
35. Maruzuru Y, Ichinohe T, Sato R, Miyake K, Okano T, Suzuki T, et al. Herpes Simplex Virus 1 VP22 Inhibits AIM2-Dependent Inflammasome Activation to Enable Efficient Viral Replication. Cell Host Microbe (2018) 23(2):254–265.e7. doi: 10.1016/j.chom.2017.12.014
36. Yuan H, You J, You H, Zheng C. Herpes Simplex Virus 1 UL36USP Antagonizes Type I Interferon-Mediated Antiviral Innate Immunity. J Virol (2018) 92(19):e01161–18. doi: 10.1128/JVI.01161-18
37. Jacobs BL, Langland JO, Kibler KV, Denzler KL, White SD, Holechek SA, et al. Vaccinia virus vaccines: past, present and future. Antiviral Res (2009) 84(1):1–13. doi: 10.1016/j.antiviral.2009.06.006
38. Moss B. Smallpox vaccines: targets of protective immunity. Immunol Rev (2011) 239(1):8–26. doi: 10.1111/j.1600-065X.2010.00975.x
39. Oyoshi MK, Beaupré J, Venturelli N, Lewis CN, Iwakura Y, Geha RS. Filaggrin deficiency promotes the dissemination of cutaneously inoculated vaccinia virus. J Allergy Clin Immunol (2015) 135(6):1511–8. doi: 10.1016/j.jaci.2014.12.1923
40. Vermeer PD, McHugh J, Rokhlina T, Vermeer DW, Zabner J, Welsh MJ. Vaccinia virus entry, exit, and interaction with differentiated human airway epithelia. J Virol (2007) 81(18):9891–9. doi: 10.1128/JVI.00601-07. [published correction appears in J Virol. 2007 Dec;81(23):13278].
41. Izmailyan R, Hsao JC, Chung CS, Chen CH, Hsu PWC, Liao CL, et al. Integrin β1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J Virol (2012) 86(12):6677–87. doi: 10.1128/JVI.06860-11
42. White JM, Delos SE, Brecher M, Schornberg K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol (2008) 43(3):189–219. doi: 10.1080/10409230802058320. [published correction appears in Crit Rev Biochem Mol Biol. 2008 Jul-Aug;43(4):287-8].
43. Moss B. Poxvirus cell entry: how many proteins does it take? Viruses (2012) 4(5):688–707. doi: 10.3390/v4050688
44. Rice AD, Adams MM, Lindsey SF, Swetnam DM, Manning BR, Smith AJ, et al. Protective properties of vaccinia virus-based vaccines: skin scarification promotes a nonspecific immune response that protects against orthopoxvirus disease. J Virol (2014) 88(14):7753–63. doi: 10.1128/JVI.00185-14
45. Tian T, Dubin K, Jin Q, Qureshi A, King SL, Liu L, et al. Disruption of TNF-α/TNFR1 function in resident skin cells impairs host immune response against cutaneous vaccinia virus infection. J Invest Dermatol (2012) 132(5):1425–34. doi: 10.1038/jid.2011.489
46. Tian T, Jin MQ, Dubin K, et al. IL-1R Type 1-Deficient Mice Demonstrate an Impaired Host Immune Response against Cutaneous Vaccinia Virus Infection. J Immunol (2017) 198(11):4341–51. doi: 10.4049/jimmunol.1500106
47. Cao H, Dai P, Wang W, Li H, Yuan J, Wang F, et al. Innate immune response of human plasmacytoid dendritic cells to poxvirus infection is subverted by vaccinia E3 via its Z-DNA/RNA binding domain. PLoS One (2012) 7(5):e36823. doi: 10.1371/journal.pone.0036823
48. Deng L, Dai P, Ding W, Granstein RD, Shuman S. Vaccinia virus infection attenuates innate immune responses and antigen presentation by epidermal dendritic cells. J Virol (2006) 80(20):9977–87. doi: 10.1128/JVI.00354-06
49. Abboud G, Tahiliani V, Desai P, Varkoly K, Driver J, Hutchinson TE, et al. Natural Killer Cells and Innate Interferon Gamma Participate in the Host Defense against Respiratory Vaccinia Virus Infection. J Virol (2015) 90(1):129–41. doi: 10.1128/JVI.01894-15
50. Martinez J, Huang X, Yang Y. Direct action of type I IFN on NK cells is required for their activation in response to vaccinia viral infection in vivo. J Immunol (2008) 180(3):1592–7. doi: 10.4049/jimmunol.180.3.1592
51. Borst K, Flindt S, Blank P, Larsen PK, Chhatbar C, Skerra J, et al. Selective reconstitution of IFN−γ gene function in Ncr1+ NK cells is sufficient to control systemic vaccinia virus infection. PLoS Pathog (2020) 16(2):e1008279. doi: 10.1371/journal.ppat.1008279
52. Lane JM, Goldstein J. Adverse events occurring after smallpox vaccination. Semin Pediatr Infect Dis (2003) 14(3):189–95. doi: 10.1016/s1045-1870(03)00032-3
53. Vellozzi C, Lane JM, Averhoff F, Maure T, Norton S, Damon I, et al. Generalized vaccinia, progressive vaccinia, and eczema vaccinatum are rare following smallpox (vaccinia) vaccination: United States surveillance 2003. Clin Infect Dis (2005) 41(5):689–97. doi: 10.1086/432584
54. Czarnowicki T, Krueger JG, Guttman-Yassky E. Skin barrier and immune dysregulation in atopic dermatitis: an evolving story with important clinical implications. J Allergy Clin Immunol Pract (2014) 2(4):371–81. doi: 10.1016/j.jaip.2014.03.006
55. Vellozzi C, Lane JM, Averhoff F, Maurer T, Norton S, Damon I, et al. Generalized vaccinia, progressive vaccinia, and eczema vaccinatum are rare following smallpox (vaccinia) vaccination: United States surveillance, 2003. Clin Infect Dis (2005) 41(5):689–97. doi: 10.1086/432584
56. Yu Q, Jones B, Hu N, Chang H, Ahmad S, Liu J, et al. Comparative analysis of tropism between canarypox (ALVAC) and vaccinia viruses reveals a more restricted and preferential tropism of ALVAC for human cells of the monocytic lineage. Vaccine (2006) 24(40-41):6376–91. doi: 10.1016/j.vaccine.2006.06.011
57. He Y, Fisher R, Chowdhury S, Sultana I, Pereira CP, Bray M, et al. Vaccinia virus induces rapid necrosis in keratinocytes by a STAT3-dependent mechanism. PLoS One (2014) 9(11):e113690. doi: 10.1371/journal.pone.0113690
58. Freyschmidt EJ, Mathias CB, Diaz N, MacArthur DH, Laouar A, Manjunath N, et al. Skin inflammation arising from cutaneous regulatory T cell deficiency leads to impaired viral immune responses. J Immunol (2010) 185(2):1295–302. doi: 10.4049/jimmunol.0903144
59. Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med (2002) 347(15):1151–60. doi: 10.1056/NEJMoa021481
60. Howell MD, Jones JF, Kisich KO, Streib JE, Gallo RL, Leung DY. Selective killing of vaccinia virus by LL-37: implications for eczema vaccinatum. J Immunol (2004) 172(3):1763–7. doi: 10.4049/jimmunol.172.3.1763
61. Grigoryev DN, Howell MD, Watkins TN, Chen YC, Cheadle C, Boguniewicz M, et al. Vaccinia virus-specific molecular signature in atopic dermatitis skin. J Allergy Clin Immunol (2010) 125(1):153–159.e28. doi: 10.1016/j.jaci.2009.10.024
62. Senkevich TG, Koonin EV, Bugert JJ, Darai G, Moss B. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology (1997) 233(1):19–42. doi: 10.1006/viro.1997.8607
63. Dourmashkin R, Bernhard W. A study with the electron microscope of the skin tumour of molluscum contagiosum. J Ultrastruct Res (1959) 3:11–38. doi: 10.1016/S0022-5320(59)80011-3
64. Vreeswijk J, Leene W, Kalsbeek GL. Early interactions of the virus Molluscum contagiosum with its host cell. Virus-induced alterations in the basal and suprabasal layers of the epidermis. J Ultrastruct Res (1976) 54(1):37–52. doi: 10.1016/s0022-5320(76)80006-8
65. Manti S, Amorini M, Cuppari C, Sapietro A, Procino F, Leonardi S, et al. Filaggrin mutations and Molluscum contagiosum skin infection in patients with atopic dermatitis. Ann Allergy Asthma Immunol (2017) 119(5):446–51. doi: 10.1016/j.anai.2017.07.019
66. Olsen JR, Piguet V, Gallacher J, Francis NA. Molluscum contagiosum and associations with atopic eczema in children: a retrospective longitudinal study in primary care. Br J Gen Pract (2016) 66(642):e53–8. doi: 10.3399/bjgp15X688093
67. Almeida HL Jr, Abuchaim MO, Schneide MA, Marques L, Castro LA. Scanning electron microscopy of molluscum contagiosum. Bras Dermatol (2013) 88(1):90–3. doi: 10.1590/s0365-05962013000100011
68. Bhawan J, Dayal Y, Bhan AK. Langerhans cells in molluscum contagiosum, verruca vulgaris, plantar wart, and condyloma acuminatum. J Am Acad Dermatol (1986) 15(4 Pt 1):645–9. doi: 10.1016/s0190-9622(86)70219-3
69. Yamauchi-Yamada A, Yamamoto T, Nakayama Y, Ikedo K, Miyao T, Yamaguchi M, et al. Immune escape phenomenon in molluscum contagiosum and the induction of apoptosis. J Dermatol (2014) 41(12):1058–64. doi: 10.1111/1346-8138.12695
71. Steffen C, Markman JA. Spontaneous disappearance of molluscum contagiosum. Report of a case. Arch Dermatol (1980) 116(8):923–4. doi: 10.1001/archderm.116.8.923
72. Ku JK, Kwon HJ, Kim MY, Kang H, Song PI, Armstrong CA, et al. Expression of Toll-like receptors in verruca and molluscum contagiosum. J Korean Med Sci (2008) 23(2):307–14. doi: 10.3346/jkms.2008.23.2.307
73. Vermi W, Fisogni S, Salogni L, Schärer L, Kutzner H, Sozzani S, et al. Spontaneous regression of highly immunogenic Molluscum contagiosum virus (MCV)-induced skin lesions is associated with plasmacytoid dendritic cells and IFN-DC infiltration. J Invest Dermatol (2011) 131(2):426–34. doi: 10.1038/jid.2010.256
74. Cohen JI. The varicella-zoster virus genome. Curr Top Microbiol Immunol (2010) 342:1–14. doi: 10.1007/82_2010_10
75. Ku CC, Zerboni L, Ito H, Graham BS, Wallace M, Arvin AM. Varicella-zoster virus transfer to skin by T Cells and modulation of viral replication by epidermal cell interferon-alpha. J Exp Med (2004) 200(7):917–25. doi: 10.1084/jem.20040634
76. Moffat JF, Stein MD, Kaneshima H, Arvin AM. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J Virol (1995) 69(9):5236–42. doi: 10.1128/JVI.69.9.5236-5242.1995
77. Vleck SE, Oliver SL, Brady JJ, Blau HM, Rajamani J, Sommer MH, et al. Structure-function analysis of varicella-zoster virus glycoprotein H identifies domain-specific roles for fusion and skin tropism. Proc Natl Acad Sci U S A (2011) 108(45):18412–7. doi: 10.1073/pnas.1111333108
78. Yang E, Arvin AM, Oliver SL. The Glycoprotein B Cytoplasmic Domain Lysine Cluster Is Critical for Varicella-Zoster Virus Cell-Cell Fusion Regulation and Infection. J Virol (2016) 91(1):e01707–16. doi: 10.1128/JVI.01707-16
79. Oliver SL, Brady JJ, Sommer MH, Richelt M, Sung P, Blau HM, et al. An immunoreceptor tyrosine-based inhibition motif in varicella-zoster virus glycoprotein B regulates cell fusion and skin pathogenesis. Proc Natl Acad Sci U S A (2013) 110(5):1911–6. doi: 10.1073/pnas.1216985110
80. Yu HR, Huang HC, Kuo HC, Sheen JM, Ou CY, Hsu TY, et al. IFN-α production by human mononuclear cells infected with varicella-zoster virus through TLR9-dependent and -independent pathways. Cell Mol Immunol (2011) 8(2):181–8. doi: 10.1038/cmi.2010.84
81. Kim JA, Park SK, Seo SW, Lee CH, Shin OS. STING Is Involved in Antiviral Immune Response against VZV Infection via the Induction of Type I and III IFN Pathways. J Invest Dermatol (2017) 137(10):2101–9. doi: 10.1016/j.jid.2017.03.041
82. Arvin AM, Kushner JH, Feldman S, Baehner RL, Hammond D, Merigan TC. Human leukocyte interferon for treatment of varicella in children with cancer. N Engl J Med (1982) 306:761–7. doi: 10.1056/NEJM198204013061301
83. Sen N, Sung P, Panda A, Arvin AM. Distinctive Roles for Type I and Type II Interferons and Interferon Regulatory Factors in the Host Cell Defense against Varicella-Zoster Virus. J Virol (2018) 92(21):e01151–18. doi: 10.1128/JVI.01151-18
84. Erdemli N, Ünal Ş, Okur H, Seçmeer G, Kara A, Gürgey A. Transient depletion of innate immunity in varicella infections in otherwise healthy children. Turk J Haematol (2009) 26(1):12–6.
85. Notarangelo LD, Mazzolari E. Natural killer cell deficiencies and severe varicella infection. J Pediatr (2006) 148(4):563–4. doi: 10.1016/j.jpeds.2005.06.028
86. Smola S. Immunopathogenesis of HPV-Associated Cancers and Prospects for Immunotherapy. Viruses (2017) 9(9):254. doi: 10.3390/v9090254
87. Tampa M, Mitran CI, Mitran MI, Ilinca N, Dumitru A, Matei C, et al. The Role of Beta HPV Types and HPV-Associated Inflammatory Processes in Cutaneous Squamous Cell Carcinoma. J Immunol Res (2020) 2020:5701639. doi: 10.1155/2020/5701639
88. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol (2017) 40(2):80–5.
89. Tommasino M. HPV and skin carcinogenesis. Papillomavirus Res (2019) 7:129–31. doi: 10.1016/j.pvr.2019.04.003
90. Gheit T. Mucosal and Cutaneous Human Papillomavirus Infections and Cancer Biology. Front Oncol (2019) 9:355. doi: 10.3389/fonc.2019.00355
91. Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog (2009) 5(2):e1000318. doi: 10.1371/journal.ppat.1000318
92. Giroglou T, Florin L, Schäfer F, Streeck RE, Sapp M. Human papillomavirus infection requires cell surface heparan sulfate. J Virol (2001) 75(3):1565–70. doi: 10.1128/JVI.75.3.1565-1570.2001
93. Florin L, Sapp M, Spoden GA. Host-cell factors involved in papillomavirus entry. Med Microbiol Immunol (2012) 201(4):437–48. doi: 10.1007/s00430-012-0270-1
94. Sapp M, Bienkowska-Haba M. Viral entry mechanisms: human papillomavirus and a long journey from extracellular matrix to the nucleus. FEBS J (2009) 276(24):7206–16. doi: 10.1111/j.1742-4658.2009.07400.x
95. Paaso A, Jaakola A, Syrjänen S, Louvanto K. From HPV Infection to Lesion Progression: The Role of HLA Alleles and Host Immunity. Acta Cytol (2019) 63(2):148–58. doi: 10.1159/000494985
96. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458(7237):514–8. doi: 10.1038/nature07725
97. Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity (2013) 39(6):1019–31. doi: 10.1016/j.immuni.2013.10.019
98. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol (2010) 11(11):997–1004. doi: 10.1038/ni.1932
99. Reinholz M, Kawakami Y, Salzer S, Kreter A, Dombrowski Y, Koglin S, et al. HPV16 activates the AIM2 inflammasome in keratinocytes. Arch Dermatol Res (2013) 305(8):723–32. doi: 10.1007/s00403-013-1375-0
100. Moerman-Herzog A, Nakagawa M. Early Defensive Mechanisms against Human Papillomavirus Infection. Clin Vaccine Immunol (2015) 22(8):850–7. doi: 10.1128/CVI.00223-15
101. Daud II, Scott ME, Ma Y, Shiboski S, Farhat S, Moscicki AB. Association between toll-like receptor expression and human papillomavirus type 16 persistence. Int J Cancer (2011) 128(4):879–86. doi: 10.1002/ijc.25400
102. Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol (2013) 132(3):515–25. doi: 10.1016/j.jaci.2013.07.020
103. Zhou F, Chen J, Zhao KN. Human papillomavirus 16-encoded E7 protein inhibits IFN-γ-mediated MHC class I antigen presentation and CTL-induced lysis by blocking IRF-1 expression in mouse keratinocytes. J Gen Virol (2013) 94(Pt 11):2504–14. doi: 10.1099/vir.0.054486-0
104. Um SJ, Rhyu JW, Kim EJ, Jeon KC, Hwang ES, Park JS. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett (2002) 179(2):205–12. doi: 10.1016/s0304-3835(01)00871-0
105. Ronco LV, Karpova AY, Vidal M, Howley PM. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev (1998) 12(13):2061–72. doi: 10.1101/gad.12.13.2061
106. Sperling T, Ołdak M, Walch-Rückheim B, Wickenhauser C, Doorbar J, Pfister H, et al. Human papillomavirus type 8 interferes with a novel C/EBPβ-mediated mechanism of keratinocyte CCL20 chemokine expression and Langerhans cell migration. PLoS Pathog (2012) 8(7):e1002833. doi: 10.1371/journal.ppat.1002833
107. Chang Y, Moore PS. Merkel cell carcinoma: a virus-induced human cancer. Annu Rev Pathol (2012) 7:123–44. doi: 10.1146/annurev-pathol-011110-130227
108. Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science (2008) 319(5866):1096–100. doi: 10.1126/science.1152586
109. Shuda M, Kwun HJ, Feng H, Chang Y, Moore PS. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J Clin Invest (2011) 121(9):3623–34. doi: 10.1172/JCI46323
110. Andres C, Puchta U, Sander CA, Ruzicka T, Flaig MJ. Prevalence of Merkel cell polyomavirus DNA in cutaneous lymphomas, pseudolymphomas, and inflammatory skin diseases. Am J Dermatopathol (2010) 32(6):593–8. doi: 10.1097/DAD.0b013e3181ce8beb
111. Kassem A, Schöpflin A, Diaz C, Weyers W, Stickeler E, Werner M, et al. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer Res (2008) 68(13):5009–13. doi: 10.1158/0008-5472.CAN-08-0949
112. Laude HC, Jonchère B, Maubec E, Carlotti A, Marinho E, Couturaud B, et al. Distinct merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with merkel cell carcinoma. PLoS Pathog (2010) 6(8):e1001076. doi: 10.1371/journal.ppat.1001076
113. Becker JC, Stang A, DeCaprio JA, DeCaprio JA, Cerroni L, Lebbé C, et al. Merkel cell carcinoma. Nat Rev Dis Primers (2017) 3:17077. doi: 10.1038/nrdp.2017.77
114. Liu W, MacDonald M, You J. Merkel cell polyomavirus infection and Merkel cell carcinoma. Curr Opin Virol (2016a) 20:20–7. doi: 10.1016/j.coviro.2016.07.011
115. Schowalter RM, Pastrana DV, Pumphrey KA, Moyer AL, Buck CB. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe (2010) 7(6):509–15. doi: 10.1016/j.chom.2010.05.006
116. Liu W, Yang R, Payne AS, Schowalter RM, Spurgeon ME, Lambert PF, et al. Identifying the Target Cells and Mechanisms of Merkel Cell Polyomavirus Infection. Cell Host Microbe (2016b) 19(6):775–87. doi: 10.1016/j.chom.2016.04.024
117. Schowalter RM, Pastrana DV, Buck CB. Glycosaminoglycans and sialylated glycans sequentially facilitate Merkel cell polyomavirus infectious entry. PLoS Pathog (2011) 7(7):e1002161. doi: 10.1371/journal.ppat.1002161
118. Neu U, Hengel H, Blaum BS, Schowalter RM, Macejak D, Gilbert M, et al. Structures of Merkel cell polyomavirus VP1 complexes define a sialic acid binding site required for infection. PLoS Pathog (2012) 8(7):e1002738. doi: 10.1371/journal.ppat.1002738
119. Becker M, Dominguez M, Greune L, Soria-Martinez L, Pfleiderer MM, Schowalter R, et al. Infectious Entry of Merkel Cell Polyomavirus. J Virol (2019) 93(6):e02004–18. doi: 10.1128/JVI.02004-18
120. Ma JE, Brewer JD. Merkel cell carcinoma in immunosuppressed patients. Cancers (Basel) (2014) 6(3):1328–50. doi: 10.3390/cancers6031328
121. Sihto H, Böhling T, Kavola H, Koljonen V, Salmi M, Jalkanen S, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res (2012) 18(10):2872–81. doi: 10.1158/1078-0432.CCR-11-3020
122. Paulson KG, Iyer JG, Tegeder AR, Thibodeau R, Schelter J, Koba S, et al. Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral CD8+ lymphocyte invasion as an independent predictor of survival. J Clin Oncol (2011) 29(12):1539–46. doi: 10.1200/JCO.2010.30.6308
123. Shahzad N, Shuda M, Gheit T, Kwun HJ, Cornet I, Saidj D, et al. The T antigen locus of Merkel cell polyomavirus downregulates human Toll-like receptor 9 expression. J Virol (2013) 87(23):13009–19. doi: 10.1128/JVI.01786-13
124. Griffiths DA, Abdul-Sada H, Knight LM, Jackson BR, Richards K, Prescott EL, et al. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J Virol (2013) 87(24):13853–67. doi: 10.1128/JVI.02159-13
125. Paulson KG, Tegeder A, Willmes C, Iyer JG, Afanasiev OK, Schrama D, et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol Res (2014) 2(11):1071–9. doi: 10.1158/2326-6066.CIR-14-0005
126. Caraballo H, King K. Emergency department management of mosquito-borne illness: malaria, dengue, and West Nile virus. Emerg Med Pract (2014) 16(5):1–24.
127. Chibueze EC, Tirado V, Lopes KD, Balogun OO, Takemoto Y, Swa T, et al. Zika virus infection in pregnancy: a systematic review of disease course and complications. Reprod Health (2017) 14(1):28. doi: 10.1186/s12978-017-0285-6
128. Duangkhae P, Erdos G, Ryman KD, Watkins SC, Falo LD Jr, Marques ETA Jr, et al. Interplay between Keratinocytes and Myeloid Cells Drives Dengue Virus Spread in Human Skin. J Invest Dermatol (2018) 138(3):618–26. doi: 10.1016/j.jid.2017.10.018
129. Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, et al. Biology of Zika Virus Infection in Human Skin Cells. J Virol (2015) 89(17):8880–96. doi: 10.1128/JVI.00354-15
130. Lim PY, Behr MJ, Chadwick CM, Shi PY, Bernard KA. Keratinocytes are cell targets of West Nile virus in vivo. J Virol (2011) 85(10):5197–201. doi: 10.1128/JVI.02692-10
131. Kovats S, Turner S, Simmons A, Powe T, Chakravarty E, Alberola-Ila J. West Nile virus-infected human dendritic cells fail to fully activate invariant natural killer T cells. Clin Exp Immunol (2016) 186(2):214–26. doi: 10.1111/cei.12850
132. Chen Y, Maguire T, Hileman RE, Fromm JR, Esko JD, Linhardt J, et al. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat Med (1997) 3(8):866–71. doi: 10.1038/nm0897-866
133. Stiasny K, Fritz R, Pangerl K, Heinz FX. Molecular mechanisms of flavivirus membrane fusion. Amino Acids (2011) 41(5):1159–63. doi: 10.1007/s00726-009-0370-4
134. Laureti M, Narayanan D, Rodriguez-Andres J, Fazakerley JK, Kedzierski L. Flavivirus Receptors: Diversity, Identity, and Cell Entry. Front Immunol (2018) 9:2180. doi: 10.3389/fimmu.2018.02180
135. Hackett BA, Cherry S. Flavivirus internalization is regulated by a size-dependent endocytic pathway. Proc Natl Acad Sci U S A (2018) 115(16):4246–51. doi: 10.1073/pnas.1720032115
136. Matusali G, Colavita F, Bordi L, Lalle E, Ippolito G, Capobianchi MR, et al. Tropism of the Chikungunya VirusPublished 2019 Feb 20. Viruses (2019) 11(2):175. doi: 10.3390/v11020175
137. van Duijl-Richter MK, Hoornweg TE, Rodenhuis-Zybert IA, Smit JM. Early Events in Chikungunya Virus Infection-From Virus Cell Binding to Membrane Fusion. Viruses (2015) 7(7):3647–74. doi: 10.3390/v7072792
138. Selvarajah S, Sexton NR, Kahle KM, Fong RH, Mattia KA, Gardner J, et al. A neutralizing monoclonal antibody targeting the acid-sensitive region in chikungunya virus E2 protects from disease. PLoS Negl Trop Dis (2013) 7(9):e2423. doi: 10.1371/journal.pntd.0002423
139. Nasirudeen AM, Wong HH, Thien P, Xu S, Lam KP, Liu DX. RIG-I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLoS Negl Trop Dis (2011) 5(1):e926. doi: 10.1371/journal.pntd.0000926
140. Ekchariyawat P, Hamel R, Bernard E, Wichit S, Surasombatpattana P, Talignani L, et al. Inflammasome signaling pathways exert antiviral effect against Chikungunya virus in human dermal fibroblasts. Infect Genet Evol (2015) 32:401–8. doi: 10.1016/j.meegid.2015.03.025
141. Priya R, Patro IK, Parida MM. TLR3 mediated innate immune response in mice brain following infection with Chikungunya virus. Virus Res (2014) 189:194–205. doi: 10.1016/j.virusres.2014.05.010
142. Wichit S, Hamel R, Yainoy S, Gumpangseth N, Panich S, Phuadraksa T, et al. Interferon-inducible protein (IFI) 16 regulates Chikungunya and Zika virus infection in human skin fibroblasts. EXCLI J (2019) 18:467–76. doi: 10.17179/excli2019-1271
143. Kwock JT, Handfield C, Suwanpradid J, Hoang P, McFadden MJ, Labagnara KF, et al. IL-27 signaling activates skin cells to induce innate antiviral proteins and protects against Zika virus infection. Sci Adv (2020) 6(14):eaay3245. doi: 10.1126/sciadv.aay3245
144. Schmid MA, Harris E. Monocyte recruitment to the dermis and differentiation to dendritic cells increases the targets for dengue virus replication. PLoS Pathog (2014) 10(12):e1004541. doi: 10.1371/journal.ppat.1004541
145. Conway MJ, Londono-Renteria B, Troupin A, Watson AM, Klimstra WB, Fikrig E, et al. Aedes aegypti D7 Saliva Protein Inhibits Dengue Virus Infection. PLoS Negl Trop Dis (2016) 10(9):e0004941. doi: 10.1371/journal.pntd.0004941
146. Jin L, Guo X, Shen C, Hao X, Sun P, Li P, et al. Salivary factor LTRIN from Aedes aegypti facilitates the transmission of Zika virus by interfering with the lymphotoxin-β receptor. Nat Immunol (2018) 19(4):342–53. doi: 10.1038/s41590-018-0063-9
147. Thangamani S, Higgs S, Ziegler S, Vanlandingham D, Tesh R, Wikel S. Host immune response to mosquito-transmitted chikungunya virus differs from that elicited by needle inoculated virus. PLoS One (2010) 5(8):e12137. doi: 10.1371/journal.pone.0012137
148. Puiprom O, Morales Vargas RE, Potiwat R, Chaichana P, Ikuta K, Ramasoota P, et al. Characterization of chikungunya virus infection of a human keratinocyte cell line: role of mosquito salivary gland protein in suppressing the host immune response. Infect Genet Evol (2013) 17:210–5. doi: 10.1016/j.meegid.2013.04.005
149. Schneider BS, Soong L, Coffey LL, Stevenson HL, McGee CE, Higgs S. Aedes aegypti saliva alters leukocyte recruitment and cytokine signaling by antigen-presenting cells during West Nile virus infection. PLoS One (2010) 5(7):e11704. doi: 10.1371/journal.pone.0011704
150. Garcia M, Alout H, Diop F, Damour A, Bengue M, Weill M, et al. Innate Immune Response of Primary Human Keratinocytes to West Nile Virus Infection and Its Modulation by Mosquito Saliva. Front Cell Infect Microbiol (2018) 8:387. doi: 10.3389/fcimb.2018.00387
151. Metz P, Reuter A, Bender S, Bartenschlager R. Interferon-stimulated genes and their role in controlling hepatitis C virus. J Hepatol (2013) 59(6):1331–41. doi: 10.1016/j.jhep.2013.07.033
152. Olsen JR, Gallacher J, Piguet V, Francis NA. Epidemiology of molluscum contagiosum in children: a systematic review. Fam Pract (2014) 31(2):130–6. doi: 10.1093/fampra/cmt075
153. Walsh SR, Dolin R. Vaccinia viruses: vaccines against smallpox and vectors against infectious diseases and tumors. Expert Rev Vaccines (2011) 10(8):1221–40. doi: 10.1586/erv.11.79
154. Eichenfield LF, Ellis CN, Mancini AJ, Paller AS, Simpson EL. Atopic dermatitis: epidemiology and pathogenesis update. Semin Cutan Med Surg (2012) 31(3 Suppl):S3–5. doi: 10.1016/j.sder.2012.07.002
155. Corey L, Wald A. Maternal and neonatal herpes simplex virus infections. N Engl J Med (2009) 361(14):1376–85. doi: 10.1056/NEJMra0807633. [published correction appears in N Engl J Med. 2009 Dec 31;361(27):2681].
156. Itzhaki RF. Corroboration of a Major Role for Herpes Simplex Virus Type 1 in Alzheimer’s Disease. Front Aging Neurosci (2018) 10:324. doi: 10.3389/fnagi.2018.00324
157. John AR, Canaday DH. Herpes Zoster in the Older Adult. Infect Dis Clin North Am (2017) 31(4):811–26. doi: 10.1016/j.idc.2017.07.016
158. Stamatas GN, Nikolovski J, Luedtke MA, Kollias N, Wiegand BC. Infant skin microstructure assessed in vivo differs from adult skin in organization and at the cellular level. Pediatr Dermatol (2010) 27(2):125–31. doi: 10.1111/j.1525-1470.2009.00973.x
159. Farage MA, Miller KW, Elsner P, Maibach HI. Characteristics of the Aging Skin. Adv Wound Care (N Rochelle) (2013) 2(1):5–10. doi: 10.1089/wound.2011.0356
160. Xie G, Luo H, Pang L, Peng BH, Winkelmann E, McGruder B, et al. Dysregulation of Toll-Like Receptor 7 Compromises Innate and Adaptive T Cell Responses and Host Resistance to an Attenuated West Nile Virus Infection in Old Mice. J Virol (2016) 90(3):1333–44. doi: 10.1128/JVI.02488-15
161. McGuckin Wuertz K, Treuting PM, Hemann EA, Esser-Nobis K, Snyder AG, Graham JB, et al. STING is required for host defense against neuropathological West Nile virus infection. PLoS Pathog (2019) 15(8):e1007899. doi: 10.1371/journal.ppat.1007899
162. Elewa RM, Abdallah MA, Zouboulis CC. Age-associated skin changes in innate immunity markers reflect a complex interaction between aging mechanisms in the sebaceous gland. J Dermatol (2015) 42(5):467–76. doi: 10.1111/1346-8138.12793
163. Iram N, Mildner M, Prior M, Petzelbauer P, Fiala C, Hacker S, et al. Age-related changes in expression and function of Toll-like receptors in human skin. Development (2012) 139(22):4210–9. doi: 10.1242/dev.083477
164. Dorschner RA, Lin KH, Murakami M, Gallo RL. Neonatal skin in mice and humans expresses increased levels of antimicrobial peptides: innate immunity during development of the adaptive response. Pediatr Res (2003) 53(4):566–72. doi: 10.1203/01.PDR.0000057205.64451.B7
165. Zhang LJ, Chen SX, Guerrero-Juarez CF, Li F, Yong Y, Liang Y, et al. Age-Related Loss of Innate Immune Antimicrobial Function of Dermal Fat Is Mediated by Transforming Growth Factor Beta. Immunity (2019) 50(1):121–136.e5. doi: 10.1016/j.immuni.2018.11.003
166. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol (2016) 16(10):626–38. doi: 10.1038/nri.2016.90
167. Giefing-Kroll C, Berger P, Lepperdinger G, Grubeck-Loebenstein B. How sex and age affect immune responses, susceptibility to infections, and response to vaccination. Aging Cell (2015) 14:309–21. doi: 10.1111/acel.12326
168. Berghofer B, Frommer T, Haley G, Fink L, Bein G, Hackstein H. TLR7 ligands induce higher IFN-alpha production in females. J Immunol (2006) 177(4):2088–96. doi: 10.4.4049/jimmunol.177.4.2088
169. Hannah MF, Bajic VB, Kelin SL. Sex differences in the recognition of and innate antiviral responses to Seoul virus in Norway rats. Brain Behav Immun (2008) 22(4):503–16. doi: 10.1016/j.bbi.2007.10.005
170. Puchhammer-Stöckl E, Aberle SW, Heinzl H. Association of age and gender with alphaherpesvirus infections of the central nervous system in the immunocompetent host. J Clin Virol (2012) 53(4):356–9. doi: 10.1016/j.jcv.2011.12.015
171. McClelland EE, Smith JM. Gender specific differences in the immune response to infection. Arch Immunol Ther Exp (Warsz) (2011) 59(3):203–13. doi: 10.1007/x00005-11-0124-3
172. Kabra SK, Jain Y, Pandey RM, Madhulika, Singhal T, Tripathi P, et al. Dengue haemorrhagic fever in children in the 1996 Delhi epidemic. Trans R Soc Trop Med Hyg (1999) 93(3):294–8. doi: 10.1016/s0035-9203(99)90027-5
173. Schrama D, Peitsch WK, Zapatka M, Kneitz H, Houben R, Eib S, et al. Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J Invest Dermatol (2011) 131(8):1631–8. doi: 10.1038/jid.2011.115
174. Wang L, Harms PW, Palanisamy N, Carskadon S, Cao X, Siddiqui J, et al. Age and Gender Associations of Virus Positivity in Merkel Cell Carcinoma Characterized Using a Novel RNA In Situ Hybridization Assay. Clin Cancer Res (2017) 23(18):5622–30. doi: 10.1158/1078-0432.CCR-17-0299
175. Rabenau HF, Buxbaum S, Preiser W, Weber B, Doerr HW. Seroprevalence of herpes simplex virus types 1 and type 2 in the Frankfurt am Main area, Germany. Med Microbiol Immunol (2002) 190(4):153–60. doi: 10.1007/s00430-001-0102-1
176. Glynn JR, Crampin AC, Ngwira BM, Ndhlovu R, Mwanyongo O, Fine PE. Herpes simplex virus type 2 trends in relation to the HIV epidemic in northern Malawi. Sex Transm Infect (2008) 84(5):356–60. doi: 10.1136/sti.2008.030056
177. Bhavanam S, Snider DP, Kaushic C. Intranasal and subcutaneous immunization under the effect of estradiol leads to better protection against genital HSV-2 challenge compared to progesterone. Vaccine (2008) 26(48):6165–72. doi: 10.1016/j.vaccine.2008.08.045
178. MacDonald EM, Savoy A, Gillgrass A, Fernandez S, Smieja M, Rosenthal KL, et al. Susceptibility of human female primary genital epithelial cells to herpes simplex virus, type-2 and the effect of TLR3 ligand and sex hormones on infection. Biol Reprod (2007) 77(6):1049–59. doi: 10.1095/biolreprod.107.063933.jmb.2013.11.012
179. Grice EA, Kong HH, Conlan S, Deming CB, David J, Young AC, et al. Topographical and temporal diversity of the human skin microbiome. Science (2009) 324(5931):1190–2. doi: 10.1126/science.1171700
180. Younge NE, Araújo-Pérez F, Brandon D, Seed PC. Early-life skin microbiota in hospitalized preterm and full-term infants. Microbiome (2018) 6(1):98. doi: 10.1186/s40168-018-0486-4
181. Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, et al. Compartmentalized control of skin immunity by resident commensals. Science (2012) 337(6098):1115–9. doi: 10.1126/science.1225152
182. Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res (2012) 22(5):850–9. doi: 10.1101/gr.131029.111
183. Wang WM, Jin HZ. Skin Microbiome: An Actor in the Pathogenesis of Psoriasis. Chin Med J (Engl) (2018) 131(1):95–8. doi: 10.4103/0366-6999.221269
184. Beylot C, Auffret N, Poli F, Claudel JP, Leccia MT, Del Giudice P, et al. Propionibacterium acnes: an update on its role in the pathogenesis of acne. J Eur Acad Dermatol Venereol (2014) 28(3):271–8. doi: 10.1111/jdv.12224
185. Paller AS, Kong HH, Seed P, Naik S, Scharschmidt TC, Gallo RL, et al. The microbiome in patients with atopic dermatitis. J Allergy Clin Immunol (2019) 143(1):26–35. doi: 10.1016/j.jaci.2018.11.015. [published correction appears in J Allergy Clin Immunol. 2019 Apr;143(4):1660].
186. Bastos MC, Ceotto H, Coelho ML, Nascimento JS. Staphylococcal antimicrobial peptides: relevant properties and potential biotechnological applications. Curr Pharm Biotechnol (2009) 10(1):38–61. doi: 10.2174/138920109787048580
187. Cogen AL, Yamasaki K, Sanchez KM, Dorschner RA, Lai Y, MacLeod DT, et al. Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J Invest Dermatol (2010) 130(1):192–200. doi: 10.1038/jid.2009.243
188. Faye T, Holo H, Langsrud T, Nes IF, Brede DA. The unconventional antimicrobial peptides of the classical propionibacteria. Appl Microbiol Biotechnol (2011) 89(3):549–54. doi: 10.1007/s00253-010-2967-7
189. Tirosh O, Conlan S, Deming C, Lee-lin SQ, Huang X, Su HC, et al. Expanded skin virome in DOCK8-deficient patients. Nat Med (2018) 24(12):1815–21. doi: 10.1038/s41591-018-0211-7
190. Pfeiffer JK, Sonnenburg JL. The intestinal microbiota and viral susceptibility. Front Microbiol (2011) 2:92. doi: 10.3389/fmicb.2011.00092
191. Tanaka K, Sawamura S, Satoh T, Kobayashi K, Noda S. Role of the indigenous microbiota in maintaining the virus-specific CD8 memory T cells in the lung of mice infected with murine cytomegalovirus. J Immunol (2007) 178(8):5209–16. doi: 10.4049/jimmunol.178.8.5209
192. Chen HW, Liu PF, Liu YT, Kuo S, Zhang XQ, Schooley RT, et al. Nasal commensal Staphylococcus epidermidis counteracts influenza virus. Sci Rep (2016) 6:27870. doi: 10.1038/srep27870
193. Kanmani P, Clua P, Vizoso-Pinto MG, Rodriguez C, Alvarez S, Melnikov V, et al. Respiratory Commensal Bacteria Corynebacterium pseudodiphtheriticum Improves Resistance of Infant Mice to Respiratory Syncytial Virus and Streptococcus pneumoniae Superinfection. Front Microbiol (2017) 8:1613. doi: 10.3389/fmicb.2017.01613
Keywords: cutaneous innate immunity, skin viruses, antiviral proteins, skin antiviral response, cutaneous microbiome, skin aging
Citation: Lei V, Petty AJ, Atwater AR, Wolfe SA and MacLeod AS (2020) Skin Viral Infections: Host Antiviral Innate Immunity and Viral Immune Evasion. Front. Immunol. 11:593901. doi: 10.3389/fimmu.2020.593901
Received: 11 August 2020; Accepted: 06 October 2020;
Published: 06 November 2020.
Edited by:
Fabienne Tacchini-Cottier, University of Lausanne, SwitzerlandReviewed by:
Irah L. King, McGill University, CanadaWalderez Ornelas Dutra, Federal University of Minas Gerais, Brazil
Copyright © 2020 Lei, Petty, Atwater, Wolfe and MacLeod. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda S. MacLeod, YW1hbmRhLm1hY2xlb2RAZHVrZS5lZHU=