 Lutz Menzel
Lutz Menzel Uta E. Höpken2
Uta E. Höpken2 Armin Rehm
Armin Rehm
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 03 December 2020
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.591741
This article is part of the Research Topic New Insights into the Complexity of Tumor Immunology in B-cell Malignancies: Prognostic and Predictive Biomarkers and Therapy View all 11 articles
Tumor-induced remodeling of the microenvironment in lymph nodes (LNs) includes the formation of blood vessels, which goes beyond the regulation of metabolism, and shaping a survival niche for tumor cells. In contrast to solid tumors, which primarily rely on neo-angiogenesis, hematopoietic malignancies usually grow within pre-vascularized autochthonous niches in secondary lymphatic organs or the bone marrow. The mechanisms of vascular remodeling in expanding LNs during infection-induced responses have been studied in more detail; in contrast, insights into the conditions of lymphoma growth and lodging remain enigmatic. Based on previous murine studies and clinical trials in human, we conclude that there is not a universal LN-specific angiogenic program applicable. Instead, signaling pathways that are tightly connected to autochthonous and infiltrating cell types contribute variably to LN vascular expansion. Inflammation related angiogenesis within LNs relies on dendritic cell derived pro-inflammatory cytokines stimulating vascular endothelial growth factor-A (VEGF-A) expression in fibroblastic reticular cells, which in turn triggers vessel growth. In high-grade B cell lymphoma, angiogenesis correlates with poor prognosis. Lymphoma cells immigrate and grow in LNs and provide pro-angiogenic growth factors themselves. In contrast to infectious stimuli that impact on LN vasculature, they do not trigger the typical inflammatory and hypoxia-related stroma-remodeling cascade. Blood vessels in LNs are unique in selective recruitment of lymphocytes via high endothelial venules (HEVs). The dissemination routes of neoplastic lymphocytes are usually disease stage dependent. Early seeding via the blood stream requires the expression of the homeostatic chemokine receptor CCR7 and of L-selectin, both cooperate to facilitate transmigration of tumor and also of protective tumor-reactive lymphocytes via HEV structures. In this view, the HEV route is not only relevant for lymphoma cell homing, but also for a continuous immunosurveillance. We envision that HEV functional and structural alterations during lymphomagenesis are not only key to vascular remodeling, but also impact on tumor cell accessibility when targeted by T cell–mediated immunotherapies.
Lymph nodes (LNs) are strategically positioned hubs of the immune system, connecting the lymphatic system with the blood circulation, filtering antigens and organizing the encounter of lymphocytes with antigen presenting cells (APCs). The LN parenchyma is tightly packed with numerous types of immune cells and susceptible for their immigration and release during conditions of homeostasis, inflammation and tumor transformation. The complex reciprocal interactions of stromal cells and immune cells in LNs shape an adapted microenvironment that supports angiogenesis and increased LN vascularization (1). Although numerous studies reported vascular remodeling and expansion in LNs upon pathogen or tumor cell encounter, the detailed mechanisms and the participating cells of these angiogenic processes are not yet identified. In this review, we delineate the current state of knowledge and propose probable cellular interactions that participate in vascular growth in LNs. In particular, we will focus on the intricate relationship between immune cells and vascular cells as a major pillar of the tumor microenvironment (TME).
B cell non-Hodgkin lymphoma (B-NHL) is a heterogenous group of hematological malignancies that arise from B lymphocytes at various stages of differentiation. Lymphomas grow in the bone marrow and in the secondary lymphatic organs (SLOs), with a predominance of LNs and spleen, but they can also manifest in non-lymphoid tissues (2). The genetic and epigenetic alterations and the intracellular pathway dysregulations responsible for the pathogenesis and progression of lymphomas have been extensively studied and led to tremendous advancements in therapeutic intervention strategies (3). The idea of tumor dependency on cells in the surrounding of a a priori benign environment and on adapted organ properties goes back to Rudolph Virchow in the 19th century (4). The crucial influence of the cellular context in which lymphoma cells arise and lodge attracts growing interest, and the investigation of the TME became an increasingly appreciated field in cancer research (5, 6). The TME constitutes about half of the tumor mass in indolent follicular lymphoma (FL) and marginal zone lymphoma (MCL), whereas the proportion in aggressive diffuse large B cell lymphoma (DLBCL) is generally lower and scarce in Burkitt’s lymphoma (BL) (7). On the extreme, in classical Hodgkin lymphoma (cHL) only about 2%–3% of the cells comprise the malignant Hodgkin-Reed-Sternberg cells (8). Hence, the composition and the dependency of the different B-NHL and cHL on the TME differ substantially between the entities (7). What distinguishes solid tumors and their metastasis most from lymphoma is that within SLOs, transformed B cells encounter a TME infrastructure that genuinely supports survival of benign B cells. These tissues undergo refinement to the needs of the tumor cells induced by a continuous reciprocal crosstalk of tumor, immune and mesenchymal stromal cells (9).
The complex interactions of transformed B cells and the TME lead to extensive changes of the vasculature within the affected organs, which is considered to have a substantial prognostic impact on the patients’ disease outcome (7, 10). The stromal compartment, mainly comprised of blood vessels, lymphatic sinuses and the fibroblastic reticular network is tightly interconnected and regulated. In some respect, it can be considered to represent a joint structural compartment in which its distinct subcompartments grow and remodel in a synchronized manner (11, 12). While the crucial role of lymphatic vessels during lymphoma growth and dissemination is undisputed (13, 14), here we will highlight the influence of blood endothelial cells (BECs) and the blood vasculature, which comprise the main provider of nutrition for proliferating and differentiating immune and tumor cells. In addition, the blood vasculature shapes a major dissemination route for benign immune and transformed cells (15, 16).
Tumors often recapitulate developmental traits of tissues in which they arise. The stem cell-like phenotype of many tumors is characterized by gene expression signatures that are associated with embryonic stem cell identity and underlines the close transcriptional relationship between neoplastic and developmental tissue (17, 18). Similar to rapidly developing and growing organs, tumors require blood vessels to access oxygen and nutrients. The initiation of blood vessel expansion, referred to as angiogenic switch, occurs at different stages during tumorigenesis, depending on the tumor type and the respective TME. The onset of neo-vascularization and vascular remodeling is a multifactorial processes orchestrated by activating and inhibiting factors whose balance determines whether BECs stay quiescent or get activated (19).
Therefore, it is useful to recapitulate the essential steps during development to understand the basal mechanisms of the microenvironmental remodeling in LNs. Blood vessels in LNs reside within the stromal scaffold and are crucial for the delivery of oxygen, nutrients, and cells. The critical delivery function was demonstrated by the rapid occurrence of necrosis in LNs upon ablation of the arterial feeding vessel in rats (20). During development (Figure 1), LNs evolve from budding lymphatic veins that form a primordial lymph sac, also known as LN anlagen. Studies with transgenic mice lacking lymphatic vessels due to the deficiency for the transcription factor (TF) Prox1 or appropriate lymphangiogenesis factors, e.g., vascular endothelial growth factor-c (Vegfc+/−), revealed a compromised LN development (21, 22). LN anlagen recruit hematopoietic lymphoid tissue-inducer (LTi) cells, which in turn stimulate local mesenchymal cell differentiation into lymphoid tissue-organizer (LTo) cells. The accumulation and interaction of lymphotoxin (LT) α1β2 on LTi cells and LTβ receptor (LTβR) expressing LTo cells results in a self-amplifying loop of LTi recruitment and LTo differentiation that drives the LN development (23). The lymphoid organogenesis is accompanied by the maturation of blood vasculature driven by locally generated retinoic acid (RA) (24). RA is presumably provided by neurons localized adjacent to the developing LN. It directly regulates the proliferation of endothelial cells, but also induces CXCL13 expression in LTo cells via binding to the RA receptor-related orphan receptor (RORγt). CXCL13 in cooperation with its receptor CXCR5 is the exclusive inducer of the initial clustering of LTi cells in LN anlagen independently of LT-LTβR signaling (25). A ubiquitous expression of the mucosal addressin cell adhesion molecule-1 (MadCAM-1) on developing venous blood vessels in the LN mediates the directed immigration of the α4β7 integrin expressing LTi cells (26–28). Notably, the expression of MadCAM-1 in peripheral LNs of newborns switches during the formation and maturation of high endothelial venules (HEVs) into the expression of the peripheral node addressin (PNAd). PNAd expression marks the completion of the maturation of the postcappillary vessels to highly differentiated HEVs that provide all prerequisites for the functional transmigration of blood-borne lymphocytes into the developing and homeostatic LN (Figure 1) (26).
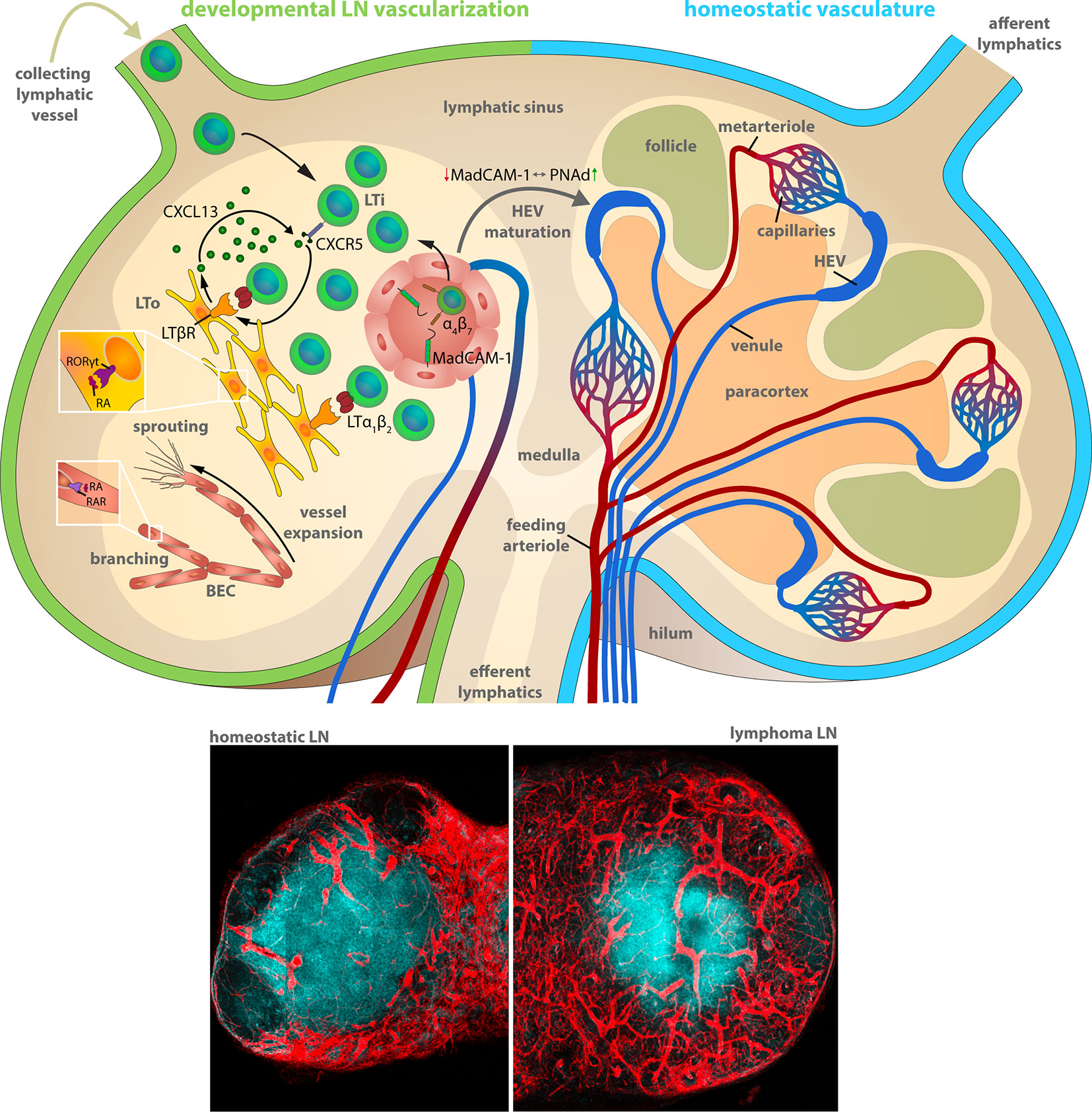
Figure 1 Lymph node vascularization in development and under homeostatic conditions. The LN compartments during LN development (left) and homeostatic conditions (right). Left: Lymphoid organogenesis is driven by recruitment of Lymphoid tissue-inducer (LTi) cells that stimulate lymphoid-organizer (LTo) cells via lymphotoxin (LT) α1β2 - LT β receptor signaling, which secrete LTi-recruiting CXCL13 in turn. LTi recruitment from the blood circulation and the afferent lymphatics accumulates LTi cells within the LN anlagen resulting in a self-amplifying process of LN development. α4β7 integrin-expressing LTi recruitment and extravasation utilizes the mucosal vascular addressin cell adhesion molecule-1 (MadCAM-1) on the luminal surface of blood vessels. MadCAM-1 switches to peripheral node addressin (PNAd) expression during differentiation of mature high endothelial venules (HEVs) within peripheral LNs. The formation of the blood vessel network comprises sprouting and branching of expanding blood vessels driven by retinoic acid (RA) stimulation of the RA receptor (RAR) on blood endothelial cells (BECs). Right: The blood circulation enters the LN during homeostatic conditions via the feeding arteriole at the LN hilum, proceeds along the medullary cord and branches into metarterioles that feed the capillary networks around the medulla and at the subcapsular sinus. HEVs are post-capillary venules with a characteristically enlarged vessel diameter. The venous backflow leaves the LN in a bundle of venules at the hilum. Bottom: Representative histochemistry sections (vessels: Cadherin5fluoresent_reporter, red) of murine LNs during homeostasis and during progression of a murine high-grade B cell lymphoma.
The vascular system of LNs in adult mammals is composed of arteries, capillaries, post-capillary venules and veins (29). Arteries are characteristically located at the periphery of the LN. The feeding arteriole enters the LN at the hilum and exhibits a gradual decrease in diameter at its few branching points alongside the medullary cords until it reaches the subcapsular capillary network. Capillaries form a dense network under the subcapsular sinus and around the medullary cords, whereas they are markedly less dense in cortex regions and sparse within the paracortex under homeostatic conditions. The vessel diameter abruptly increases at the capillary to post-capillary transitions. These post-capillary venules, referred to as HEVs, are primarily located within the cortex in the interfollicular space. HEVs form loop-like structures following a centripetal course that ends in transitions to veins at the corticomedullary junctions. Finally, a bundle of larger main veins leave the LN though the hilum (Figure 1) (29, 30).
Tumor growth is often accompanied by the ingrowth of blood vessels and the formation of a vascular network, consistent with the need for malignant cells to have access to the circulation system. Tumor vascularization occurs either through co-option of the pre-existing vasculature, or by induction of neovascularization. Vessel co-option is a non-angiogenic process in which tumor cells utilize pre-existing blood vessels of surrounding tissue to support tumor growth, survival and metastasis (31). In contrast, neovascularization involves a series of complex and sequential events: (I) activation of microvascular endothelial cells, (II) enzymatic degradation of the vascular basal membrane, (III) gradual degradation of other extracellular matrix (ECM) components, (IV) endothelial cell migration and proliferation, (V) lumen formation within neo-sprouts, (VI) branching of the neo-vessel, and (VII) formation of a functional vessel network by fusion with neighboring vessels to initiate blood flow (32, 33). Located at the leading edge of the vascular sprout, tip cells form cellular protrusions or filopodia to guide migration toward a source of angiogenic growth factors. Simultaneously, they signal to adjacent endothelial cells via Delta-like ligand (DLL)-Notch interactions not to adapt the tip cell phenotype, but to maintain the proliferative stalk cell phenotype and to form a vascular lumen (34, 35). The vascular endothelial growth factors (VEGFs) are the major contributors to angiogenesis. The local secretion of VEGF-A and its gradient forming deposition on the ECM triggers endothelial tip cell formation via binding to VEGFR2, resulting in endothelial cell proliferation and migration and eventually, formation of tube structures resembling new capillaries (35–38). VEGF-B, VEGF-C, and VEGF-D are other members of the VEGF family of which VEGF-C plays a critical role upon LN remodeling because it is the most potent inducer of lymphangiogenesis as a ligand of VEGFR3. VEGFR3 is known for its involvement in physiological and tumor-associated lymphangiogenesis and lymphatic metastasis (39, 40). Apart from lymphatics, VEGFR3 is highly expressed at the leading-edge of BECs that undergo sprouting (41) and was recently shown to coregulate the expansion of the blood vessel network in LNs in a Myc-driven high-grade B cell lymphoma mouse model (42). Fibroblast growth factors (FGFs) stimulate endothelial cell migration and proliferation in a very potent manner, which in in vitro experiments even exceeds the stimulation capacity of VEGF-A (43, 44). FGF-1 stimulates proliferation and differentiation of all cell types necessary for the formation of arterial vessels, including endothelial and smooth muscle cells. The angiogenic potency of FGFs extends to prompt fibroblastic cells (e.g., pericytes, smooth muscle cells, and mural cells) and recruits them for vessel formation and maturation during tumorigenesis (45). FGF-2, the second most abundant growth factor of the FGF family, promotes endothelial cell proliferation and the physical organization of the endothelial cell tube-like formation during developmental vessel assembly (46, 47).
The integral investigation of the highly complex vascular network and the unique features of its parts in context of the compartmentalized architecture of the LN has long been a challenge for microscopic image analysis. Because higher order anatomical data sets were obtained from such advanced optical imaging approaches, algorithms for data handling were also demanding to generate. Over the last couple of years, novel tissue preparation methods (48, 49), imaging systems and computational rendering strategies evolved, which enable contextual and organ-wide topological analyses in three-dimensional spaces and over time. In particular, optical projection tomography (OPT) and light sheet microscopy have been established to study anatomical and functional features of LN, e.g., to quantify capillary and HEV structures and their contextual relationship to B cell follicles and dendritic cells (DCs) throughout the organ (50–52). A combination of microscopic imaging and computational modulation of the hydrodynamic properties of vessels in LNs revealed a tight connection of the hydraulic conductivity between lymphatic and blood vessels and the respective hydrodynamic conditions within the LN. These biophysical conditions are vital for inter- and intra-LN transport mechanisms and immunological functions, and most likely for lymphoma B cell dissemination and immunosurveillance as well (53, 54). Up to date, these dynamic conditions are not easy to mimic in organoid models. However, in an early 3D organoid model mimicking a LN exposed to tissue injury or inflammation, the interstitial flow affected the fibroblastic reticular cells (FRCs) that enwrap conduits transporting fluid from the subcapsular sinus to HEVs. Blocking this flow led to CCL21 downregulation, indicating that increased lymph flow as a hydrodynamic factor acts on the paracortex and thus, affects the remodeling and functionality of conduits and FRCs (55). In line, mechanosensing of conduit flow deprivation by FRCs in Peyer’s patches resulted in dysfunctional HEVs and disturbed mucosal immune responses (56). Similar processes are also conceivable during lymphoma growth within LNs, where a gradual loss of HEVs in numerous B-NHL was described many years ago (57). A comprehensive and continuous blood vessel network of LNs under homeostatic conditions has been revealed (54, 58) and brought up an analysis pipeline for detailed and whole-organ investigations of the LN vasculature upon perturbations through inflammation, lymphoma homing and LN solid tumor metastasis. Recently, utilization of single cell transcriptome analysis methods revealed a broad overview of the heterogeneity of ECs throughout several different murine organs, including the spleen and LN as representatives for SLOs (59, 60).
Inflammation, vessel reorganization and angiogenesis are intimately connected processes. In adults, angiogenesis usually occurs during pathological settings such as infection, wound healing and cancer. Notably, hematopoietic cells and endothelial precursors share common CD34+ stem and progenitor cells (61).
Growth of solid tumors is typically associated with inflammation that triggers tissue-protective and pro-tumorigenic mechanisms. Inflammatory responses in normal tissue and cancer are initiated and maintained by local tissue or cancer associated macrophages (TAMs) and DCs. Sustained inflammation further leads to recruitment of bone marrow–derived monocytes, neutrophilic granulocytes, myeloid-derived suppressor cells (MDSC), and tissue or tumor infiltration of lymphocytes from the SLOs. Especially cytokines and chemokines, transcriptionally regulated downstream of NF-κB signaling pathways in immune cells, promote cell survival and proliferation, recruit more immune cells and re-shape the TME. Pro-inflammatory cytokines like IL-6, TNFα and IL-17, increase the proliferation rate of other inflammatory immune cells and prime the tumor to overcome suboptimal microenvironmental conditions including lack of nutrients, growth factors and hypoxia (62). Inflamed tissue and solid tumors are often characterized by insufficient oxygen supply that triggers angiogenesis. Hypoxia, which is the major driver of vascular alterations in solid tumors, stabilizes the TF HIF-1α, the master regulator of pro-angiogenic factor expression such as VEGFs, CXCL12, and COX-2 (63–65). The presence of a constant pro-angiogenic milieu in solid tumors often causes a disturbed maturation and pruning of blood vessels. The division in arterioles, capillaries and venules can be deficient and results in an aberrant distribution of vessel caliber, influencing the blood flow. Morphologically, a poorly organized, malformed vessel network develops under these conditions (66, 67). The endothelial junctions in such malformed networks are often defective and lead to enhanced permeability and elevated interstitial fluid pressure (68). Pericytes can be partially detached and newly build blood vessels often fail to recruit sufficient pericyte coverage, causing an unevenly distributed basement membrane, vessel fragility, and risk of hemorrhage (69, 70). Besides the structural and functional defects, the specific transcriptional response of tumor vasculature is not only related to angiogenesis and vessel integrity, but affects endothelial activation and recruitment of leukocytes as well. Pro-angiogenic signaling leads to endothelial anergy, reduced response to pro-inflammatory signaling and decreased expression of adhesion molecules and chemokines necessary for capture and trans-endothelial migration of leukocytes (71, 72).
In LNs, which are the authochthonous environment for most B-NHL, the pre-existing vasculature takes part in the massive remodeling process during immune responses, best studied for strong inflammatory stimuli in mice (50, 73, 74). LNs are plastic organs able to expand to a multiple of their normal size within days including an extensive remodeling of the vascular-stromal compartment. The rapid expansion of the LN size and cellularity includes early events of remodeling of the feeding artery, causing an increased blood flow and LN hypertension accompanied by an increase of the vascular permeability (75, 76) and increased interstitial pressure. The capillary network within the cortex and medulla expands toward the paracortex, and post-capillary venules are reorganized (30). Skin allograft-draining LNs in rats exhibited a progressive elongation and branching of HEVs resulting from focal proliferation of endothelial cells in the transition zone from high to low endothelium (77). Several years later, Bajénoff and colleagues revisited these observations and investigated the BEC proliferation applying a multicolor fluorescence fate-mapping mouse model. They found similar proliferation foci in post-capillary venules as proposed by Anderson and Anderson. In addition, an extensive expansion of the LN vasculature relying on the sequential assembly of endothelial cell proliferative units upon inflammation was observed. Clonally proliferating HEV cells (73) and capillary resident precursors (60) comprised local progenitors for HEV elongation and capillary neo-vessels during BEC turnover and vessel sprouting. Interestingly, recruitment of bone marrow–derived endothelial cell progenitors did not contribute to the local LN vascular alterations in this model. LN expansion stimulated by several immunization strategies in mouse experiments, e.g., bone marrow–derived DCs (78), ovalbumin/complete Freund’s adjuvant (OVA/CFA) (79), OVA/alum (80), oxazalone (11), and lymphocytic choriomeningitis virus (LCMV) infection (50) indicated similar courses of vessel expansion, starting with early proliferation events that last for up to 5–8 days. The remodeling eventually ends with a gradual re-establishment of the vascular endothelial cell quiescence, a normalization of the vascular bed and restoration of the normal LN size (30, 73, 78).
Both innate and adaptive immune cells have an intricate relationship with angiogenesis. They are involved in regulation of BEC proliferation, migration and activation and they provide a large spectrum of pro-angiogenic mediators apart from their genuine immunological function. Hence, immune cells induce, support or antagonize angiogenic processes during inflammation and tumor growth (Figure 2 and Table 1) (124, 125). Angiogenesis is also important for the progression of B cell lymphoma, however the role of angiogenic factors and the composition of pro-angiogenic immune cells within LNs varies between different entities.
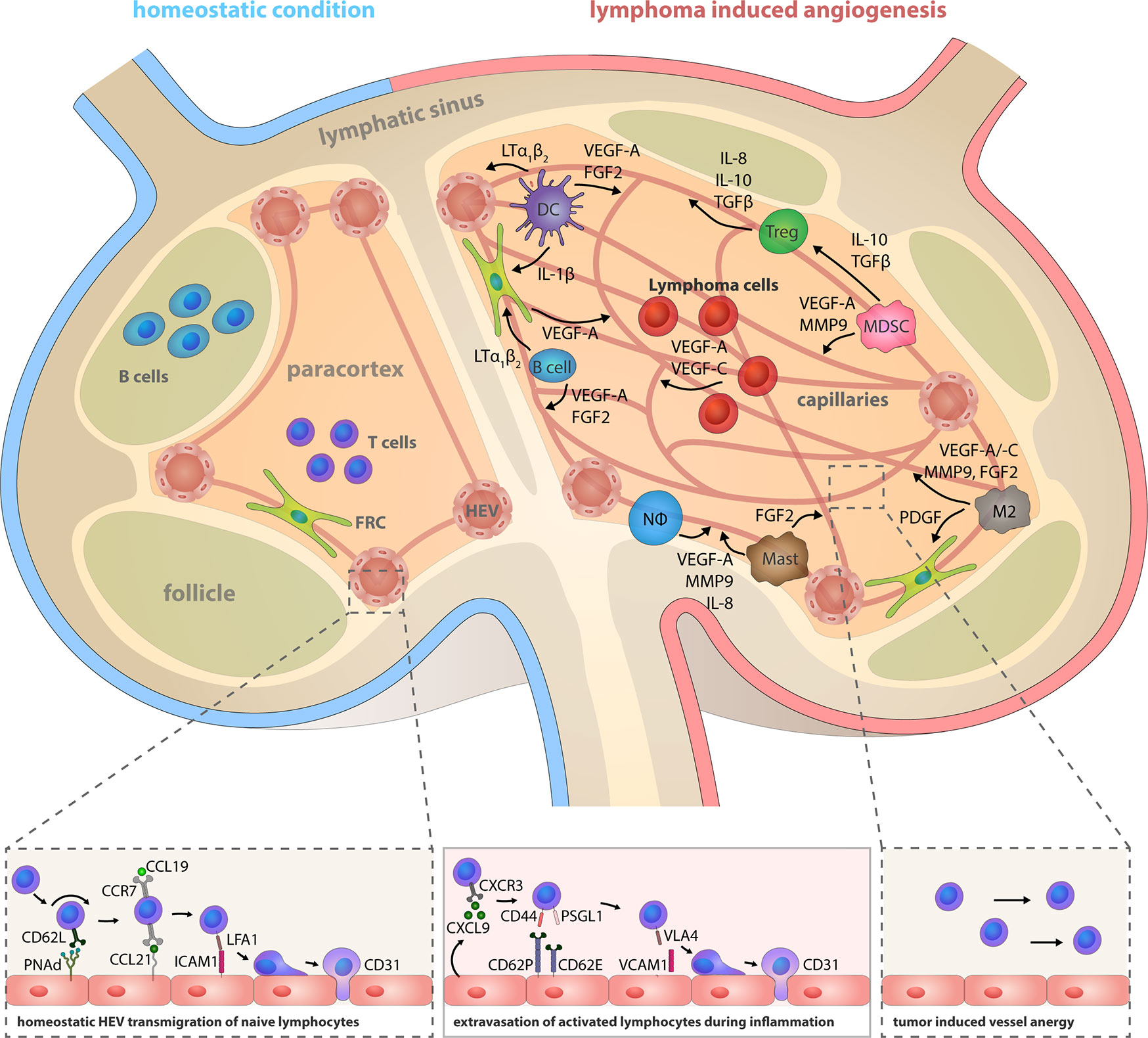
Figure 2 Lymphoma induced angiogenesis in LNs and participating immune cells. Top: The LN compartments represented under homeostatic conditions (left) and lymphoma-activated angiogenesis (right). Lymphoma growth is characterized by a strong LN volume expansion and blood vasculature growth. Remodeling of the stromal infrastructure involves an increase of the microvessel density (MVD), as effectuated by direct angiogenic stimulation through lymphoma B cells cells, but concomitantly also through reciprocal crosstalk of cells in the TME and recruited immune cells. Notably, the initiation of the angiogenic switch in lymphoma is independent from hypoxia-induced HIF1α pathway activation. Tumor polarized DCs (CEBP/βhigh) control the HEV differentiation status via LTα1β2 and LIGHT presentation; they release IL-1β and hereby take part in the blood vessel growth by inducing VEGF-A expression in FRCs. They also secrete the angiogenic factors VEGF-A and FGF2. B cells express LTα1β2, which exerts minor effects on HEVs, but a predominating stimulatory effect on FRCs. Expression of the chemokines CCL2, CXCL12, and MIF recruits additional immune cells into the LN. Regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), M2-polarized macrophages, neutrophils and mast cells are capable of producing the pro-angiogenic factors VEGF-A, VEGF-B, VEGF-C, MMP9, IL-8, IL-10, TGFβ, and FGF1/2. Bottom left: HEVs express PNAd, CCL21, and ICAM1 and thereby constitute the transmigration routes for lymphocytes under homeostatic conditions. Interaction of CD62L, CCR7, and LFA-1 on naïve lymphocytes with these HEV-associated surface receptors and chemokines initiates lymphocyte rolling, HEV wall adhesion and eventually, transmigration into the LN parenchyma. Bottom middle: Inflammatory vessels in reactive LNs recruit activated lymphocytes by CXCL9 secretion and replace the homeostatic receptors on endothelial cells with CD62P, CD63E, and VCAM1 that are interaction partners of leukocyte-expressed CD44, PSGL1, and VLA4. Bottom right: The lymphoma induced expansion of the blood vessel network favors the assembly of smaller anergic endothelium that is insufficiently equipped for lymphocyte extravasation.
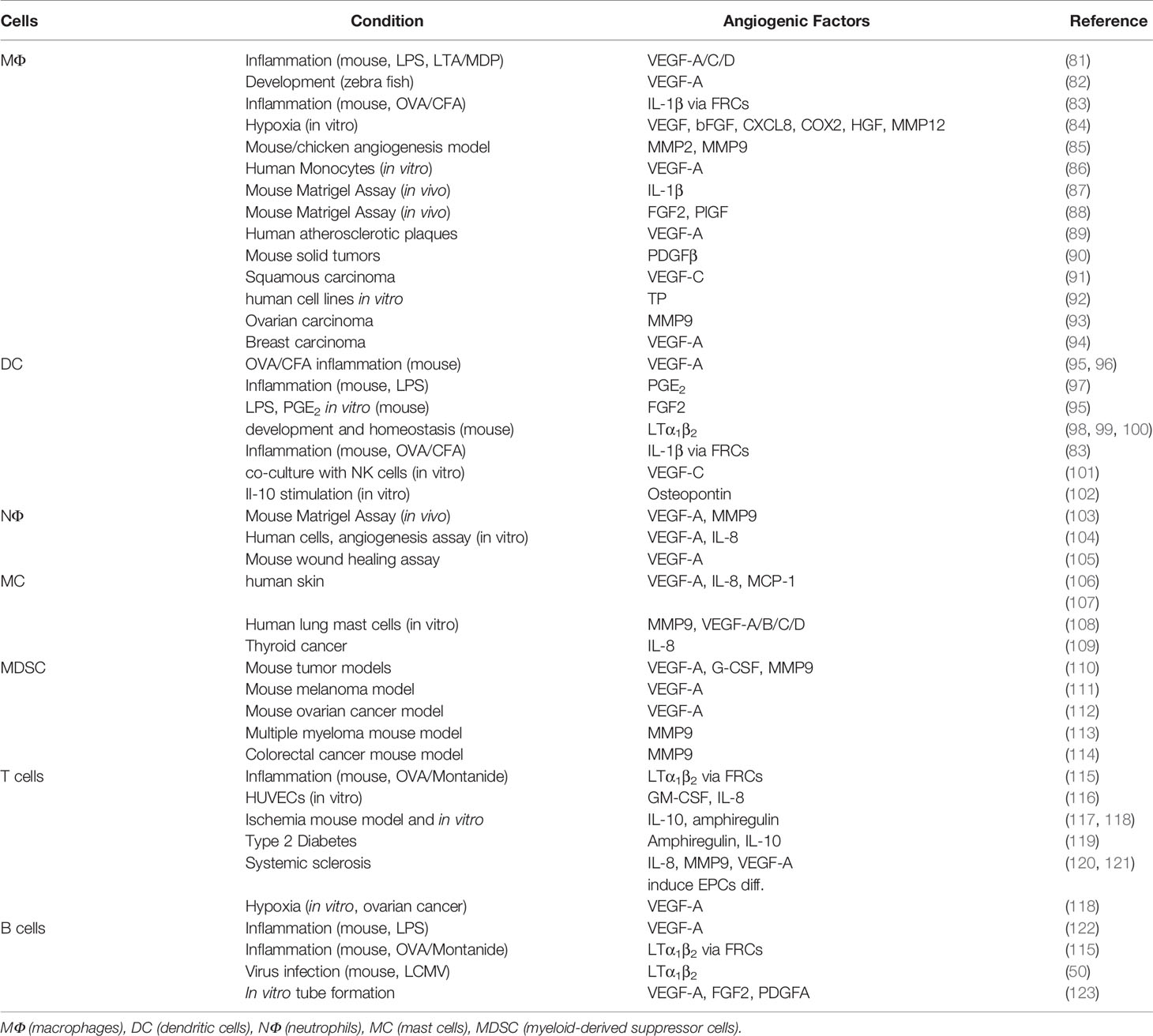
Table 1 Immune cells derived pro-angiogenic factors.
A leading immune cell source for growth factors and chemokines to promote angiogenesis under inflammatory and tumorous conditions are myeloid cells (126). Macrophages are phagocytic immune cells and important regulators of tissue homeostasis, morphogenesis and repair. In LNs, macrophages are an abundant immune cell population that is divided into subcapsular sinus macrophages (SSM), medullary sinus macrophages (MSMs), and medullary cord macrophages (MCMs) (127). Monocytes from the blood stream and macrophages from LN remote tissues (e.g., bone marrow) infiltrate the LN attracted by a variety of chemotactic factors, among others CCL2, CXCL12, and the macrophage migration inhibitory factor (MIF) (128–130). Tumor-associated macrophages (TAMs) play a prominent role during progression of chronic lymphocytic leukemia (CLL) by supporting tumor cell survival (131) and regulation of the TME (132). The presence and polarization of macrophages during CLL is critical for the tumor progression, as indicated by a CLL-associated skewing of T cells toward antigen-experienced phenotypes and T cell exhaustion, which could be reversed by monocyte and macrophage depletion. Thus, interference with macrophage polarization in CLL turned out to be a promising target for immunotherapy (133). Similar to TAMs in leukemia, macrophages likely support angiogenesis in lymphoma as well, both in cHL and B-NHL. M2-polarized macrophages induce an immunosuppressive milieu in cHL, comprising the majority of the PD-L1 expressing cells, located in close proximity to the Hodgkin Reed Sternberg (HRS) cells (134). In this disease-defining tumor cell population, high frequencies of alterations on chromosome 9p24.1, involving copy number gain and amplifications, have been shown to increase the abundance of the PD-1 ligands, PD-L1 and PD-L2 (135). Furthermore, Epstein-Barr virus (EBV) infection can increase expression of PD-1 ligands in cHL as well (136). The TAM derived PD-L1 in conjunction with the HRS-cell derived PD-1 ligands PD-L1 and PD-L2 may neutralize the anticancer activity of PD-1+ T cells and natural killer cells, a process that can be reversed by utilizing PD-1 blocking antibodies (137). TAMs were also frequently found in FL and DLBCL, among them often polarized and pro-angiogenic M2-like macrophages, which secrete angiogenic factors and re-arrange the ECM by matrix metalloproteinase (MMP) release for vascular expansion (138, 139).
Regarding angiogenesis, macrophages play a crucial role at each step of the angiogenic cascade, starting from blood vessel sprouting to vessel maturation and remodeling of the vascular network. Pro-inflammatory conditions polarize classical activated macrophages (M1), whereas anti-inflammatory conditions give rise to alternatively activated macrophages (M2) including angiogenesis associated macrophages (140–142). For example, in cHL HRS-cell derived TGF-β, IL-13 and M-CSF educate monocytes or TAMs to become immunosuppressive M2-polarized TAMs (Aldinucci D, Casagrande N, 2016, Cancer Letters; Skinnidder and Tak Mak, 2002, Blood).
Macrophage-derived MMP2 and MMP9 proteases cleave the ECM to break open matrix remodeling to pave the way for endothelial sprout migration (85). Pro-angiogenic growth factors, such as VEGF-A, MMPs, IL-1β, FGF2, and transforming growth factor beta 1 (TGFβ1), are part of the repertoire secreted by macrophages in a pro-angiogenic milieu (85–89). Upon hypoxic conditions that lead to HIF1α activation, macrophages are able to establish capillary-like networks in which they line a vessel micro-tunnel and express lineage aberrant endothelial markers such as CD31, von Willebrand factor and Cadherin-5, leading to the assumption that macrophages may transdifferentiate into ECs under specific conditions (143, 144). Macrophages also function as cellular chaperones during anastomosis of vascular sprouts by guiding endothelial tip cells to undergo sprout fusion (82). Such macrophages expressed the markers tyrosine kinase with immunoglobulin-like and EGF-like domains (TIE2) and neuropilin-1 (Nrp1), indicating that they are M2 polarized cells with properties similar to TAMs (82, 145). New blood vessels need to undergo maturation to become functionally stable. A crucial step in this process is the integration of new blood vessels into the established stromal environment and the recruitment of pericytes to strengthen vascular junctions. Macrophages are highly abundant around new blood vessels and help to recruit pericytes by secretion of the platelet-derived growth factor β (PDGFβ) (90, 146).
Dendritic cells (DCs) are sentinel cells that connect the innate and adaptive branches of the immune system wherein they have important roles in host defense against pathogens and in generating anti-tumor immune responses. The classical DC compartment of the spleen is comprised of lymphoid tissue-resident DCs, whereas LNs also include non-lymphoid tissue-migratory DCs (147). Especially CD11cmediumMHCIIhigh DCs are associated with the initiation of vascular expansion after bone marrow–derived DC (BMDC) transfer, whereas CD11chighMHCIImedium DCs accumulate later in the process and promote the re-establishment of vascular quiescence (78). Apart from their predominant immunologic role as professional APC population, DCs carry a wide range of angiogenic mediators to modulate vascularization. They do so by engaging cognate signaling receptors, such as VEGFR2 on endothelial cells or by recruiting and stimulating adjacent cells or cells of the TME (96). LN-resident DCs are closely associated with FRCs and sample conduit-conveyed antigens within the paracortical and interfollicular zone, where they are located in the proximity of HEVs (148). This spatial proximity suggests that DCs are likely to be a link between immune cells, vasculature and mesenchymal stromal cells. The development, maturation and lineage commitment of DC subsets is differentially regulated by acomplex TF network, depending on homeostatic, inflammatory, and tumorous conditions. We recently demonstrated that the TF C/EBPβ plays a crucial role in murine DC maturation and immunogenic functionality under homeostatic and lymphoma-transformed conditions (149). In the presence of lymphoma cells, enhanced expression of C/EBPβ in DCs was observed which transformed them into an immature, tolerogenic and pro-tumorigenic subtype (150). Such aberrant maturation stages may potentially affect the angiogenic capacities of the DCs as well. The crucial role of DCs for the LN vasculature was elaborated in several studies (12, 78, 79, 96) and revealed the DC-coordinated remodeling mechanisms of blood- and lymph-vasculature, and the FRC network as well. The DC associated increase of VEGF-A in reactive LNs further includes stimulation of a pro-angiogenic program in FRCs and the recruitment of blood-borne cells that participate in the angiogenic process. IL-1β expression by recruited CCR7+ CD11c+ DCs is associated with the enrichment of VEGF-A expressing FRCs at the border of the LN paracortex (83). The angiogenic role of DCs in lymphoma LNs has not been investigated yet; however, in reactive LNs, resident classic DCs produce biologically active VEGF-A downstream of the inflammation-associated TFs HIF-1α, STAT3, and CREB. HIF-1α and STAT3 are generally related to hypoxic conditions, whereas CREB phosphorylation is the consequence of autocrine and paracrine prostaglandin E2 (PGE2) signaling (151). The PGE2 production is directly connected to pathogen induced toll like receptor-4 (TLR4) signaling and therefore, delineates the connection of infection induced LN reactivity with angiogenic vessel formation (152). DCs also release other classical angiogenic growth factors like FGF2, endothelin-1 (ET-1), CXCL12, and COX-2. FGF2 activates endothelial cells and induces VEGF-A expression in mesenchymal cells, but also recruits and activates macrophages and mast cells that in turn exhibit angiogenic properties (152, 153). DCs further have the capacity to modulate angiogenesis in an indirect manner through secretion of the monocyte and granulocyte attracting chemokines CXCL8, CXCL1, CXCL2, CXCL3, and CXCL5 (154). The recruited myeloid cells can be triggered to secrete the pro-angiogenic IL-1β by a signaling pathway that includes classical DC-derived osteopontin (155). DCs are not only associated with vascular expansion, but also with the re-establishment of vascular quiescence and stability in the process of reinstallation of the LN homeostasis (78).
Neutrophilic granulocytes are the most abundant type of leukocytes throughout the body, representing the pioneering cells that are recruited to injuries and thus, they are frontline defenders against pathogens. Neutrophils infiltrate LNs guided by inflammatory cytokines like IL-1β and TNFα, the complement factors C3a and C5a, along the CXCR4-CXCL12 axis, and eventually they are also attracted by a plethora of inflammatory chemokines (156, 157) (Capucetti, Albano, Bonecchi, Frontiers in Immunology, 2020). Neutrophils are a source of soluble mediators that exert important angiogenic functions. VEGF-A, IL-8, hepatocyte growth factor (HGF), granulocyte colony-stimulating factor (G-CSF), and MMP9 are the most important activators of angiogenesis produced by these cells (103, 158). Interestingly, neutrophils are able to release VEGF-A-enriched granules upon TNFα stimulation and thus, promote vessel growth during inflammation (105). This can become an self-amplifying process since neutrophil-derived VEGF stimulates neutrophil migration via an autocrine amplification mechanism, a process that likely contributes to pathological angiogenesis during inflammation and cancer (159). Human polymorphonuclear granulocytes have been demonstrated to directly induce the sprouting of capillary-like structures in an in vitro angiogenesis assay, mediated by secretion of both pre-formed VEGF from cell stores and de novo synthesized IL-8 (104). In the murine Eμ-Tcl1 model, mimicking CLL, a tumor-associated neutrophil (TAN) population with a B cell helper-like polarization was identified. Selective depletion of these TANs retarded leukemia progression in SLOs substantially (160).
Mast cells (MC) are hematopoietic tissue resident immune cells that are classically recognized as the main effector cell type of IgE-mediated immediate allergic reactions, however they are also frequently associated with tumorigenesis (161–163). According to their protease expression, mast cells are divided in two phenotypical populations: the trypase+ chymase− (MCT) and the trypase+ and chymase+ (MCTC) cells (164, 165). MC produce several proangiogenic factors, among them VEGF-A, VEGF-B, MMP9, and FGF-2. In addition, mast cells chemotactically respond to VEGF-A and FGF2, indicating that a connection between mast cell accumulation at tumor sites, angiogenesis and tumor growth exists (166, 167).
Myeloid derived suppressor cells (MDSCs) contribute to the induction of an immune suppressive and tumor permissive microenvironment. They are frequently found in SLOs like spleen, but they are rare in LNs (168). However, they are able to modulate the L-selectin expression of naïve T and B cells, preventing efficient HEV adhesion, transmigration, and subsequent antigen encounter within LN parenchyma (169, 170). MDSCs promote the formation of T regulatory cells (Tregs), the secretion of immunosuppressive IL-10 and TGF-β, and inhibit the activity of cytotoxic CD8 T cells via expression of arginase-1 (Arg1) and inducible nitric oxidase (iNOS) within the TME of several tumor entities including B cell lymphoma (171, 172). Moreover, MDSCs directly influence the tumor stroma by inducing differentiation of cancer-associated fibroblasts (CAFs) (173, 174). Pro-angiogenic properties of MDSCs during tumor progression have been reviewed recently (175). MDSCs and their progenitors, immature myeloid cells are usually not present in LNs during steady state conditions. However, inflammatory conditions and tumor-derived factors (e.g., CXCL12, GM-CSF, and CCL2) induce the activation and accumulation of MDSC in SLOs (176–178). MDSCs exhibit numerous immunomodulatory properties that have considerable potential to influence angiogenic processes in LNs, either through direct triggering of ECs, or by stimulating leukocyte and stromal cells to establish an angiogenic milieu (175). MDSCs are able to promote tumor angiogenesis through releasing VEGF-A and MMP9. Mouse models suggest that MDSCs integrate into the line of vessel-decorating endothelial cells (179). In mouse melanoma, MDSC contribute to A2B adenosine receptor-induced VEGF-A production (111, 180). VEGF-A in turn stimulates MDSC recruitment from the bone marrow, creating a self-enhancing feedback loop that promotes immunosuppression and vessel growth (112). One of the reasons why several angiogenic tumors occur to be insensitive to VEGF-A-targeted therapy is the presence and recruitment of MDSCs. These cells secrete high amounts of VEGF-A which might lead to neutralization of the VEGF-inhibition and additionally, they establish pro-angiogenic signaling pathways involving several other cells of the TME (110, 181). Moreover, MDSCs limit T cell adhesion and extravasation by VEGF-A stimulated suppression of endothelial ICAM-1 and VCAM-1 expression during tumor angiogenesis (71, 182).
Lymphocytes, the major regulatory and executive cell subset of the adaptive immune response are also able to influence angiogenesis during inflammation and cancer, although their specific implications are still enigmatic.
T cells comprise different subsets involved in lymphomagenesis, including naive T cells, memory T cells, and Treg cells (183). Several negative regulators of T cell activation act as checkpoints to fine-tune the immune response and regulate hyperactivation. Cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed cell death 1 (PD-1) are the most potent examples of T cell immune checkpoint molecules (ICB) (184). Cancer patients often display dysfunctional antitumor T cell responses because of the signaling pathways downstream of these receptors. PD-1 and CTLA-4 inhibition are subject of extended clinical studies and led already to impressive response rates in some tumor entities, among them melanoma, non–small cell lung cancer and for hematopoietic tumors, (184–186), in cHL as well (187, 188). By targeting abnormal formation of tumor vessels, anti-angiogenic agents potentially result in an enhanced infiltration of anti-tumor effector cells, making the combination of immune checkpoint inhibitors and anti-angiogenic agents a promising and complementary approach in cancer adoptive T cell therapy (189). On the other hand, as a result of IFNγ and IL-12 stimulation, microvascular endothelial cells express checkpoint molecules like PD-L1 (190, 191). In line, arterial vessels express PD-L1 and PD-L2 after toll like receptor (TLR)-3 activation upon bacterial infection (192). The regulatory and angiogenic effects of CD4+ T helper cells (Th cells) are strictly associated with their differentiation. Cytotoxic CD8+ T cells and CD4+ Th1 cells produce IFNγ that restrains endothelial cell proliferation and induces expression of the angiostatic chemokines CXCL9/10/11 in TAMs (126, 193). In vitro studies revealed that Th2- and Th17 cell-conditioned medium triggered endothelial sprouting, whereas medium of Th1 cultures induced vascular regression. Conditioned medium from Tregs had a minor or no effect (116). In vivo, CD4+ T cells display opposing effects on vascularization depending on their subset differentiation. Th1 cell-derived IFNγ impairs angiogenesis in ischemic tissue, an effect that is counteracted by regulatory CD4+ T cells (Tregs) that antagonize the immunologic Th1 cell response by secreting anti-inflammatory IL-10 and TGFβ. Thus, Tregs display rather indirect pro-angiogenic properties, most likely by paracrine effects on other potentially pro-angiogenic immune cells (e.g., macrophages, DCs, mast cells) (119, 194). T cell recruitment, survival and functionality are highly dependent on tumor-polarized myeloid cells and tumor-derived factors. The typical immuno-suppressive milieu of the TME is characterized by polarizing factors, shifting CD4+ T cell differentiation toward CD4+CD25+FOXP3+ Tregs. In the aggressive Myc-driven murine lymphoma model, this polarization process is promoted by DCs expressing increased amounts of the TF C/EBPβ (144).
In ovarian cancer, Tregs were selectively recruited into the tumor tissue via CCL22 and CCL28 production by the tumor cells and subsequently, Treg-induced secretion of high amounts of VEGF-A to promote endothelial cell proliferation (118, 195). A striking example for Treg recruitment represents cHL; here, Tregs are attracted via the Hodgkin-Reed-Sternberg cell-secreted chemokines CCL17 and CCL22, which engage the Treg-expressed chemokine receptor CCR4 (196), or by the chemokine CCL20 that binds to CCR6 (Baumforth, Birgersdotter, Machado, Am J Pathol, 2008). Th cells and cytotoxic T cells are required to mediate the anti-angiogenic effect of IL-12. IL-12–activated lymphocytes effectuate inhibition of tumor growth and function as anti-vascular agents that release higher amounts of IFNγ while they down-regulate VEGF in neighboring cells (197, 198). Noteworthy, the presence of IFNγ comes at the expense of an induction of PD-L1 on numerous stromal cell types, among them endothelial cells (199, 200); this process is likely to counteract the beneficial effects of IFNγ-secreting effector T cells which may be rendered dysfunctional (201). The infiltration of tumor sites by cytotoxic CD8+ T cells is usually correlated with a favorable clinical prognosis, however immunosuppressive conditions can polarize these cells to CD8+FOXP3+ regulatory cells with similar immunomodulatory and angiogenic properties as CD4+ Tregs (202–205). Studies of coronary artery disease and systemic sclerosis found T cells with angiogenic potential in blood samples of patients and demonstrated that these CD3+CD31+CXCR4+ cells (referred to as angiogenic T cells) play a vital role for the colony formation and differentiation of endothelial progenitor cells (EPCs) in the bone marrow (110, 206). Such EPCs have been detected in the circulation and in LN samples from patients with B-NHL as well, although their influence on lymphoma-induced vessel growth is still elusive (207, 208). However, inflammation models argue against a significant functional role of BM-recruited EPCs in LN vascularization (73).
B cells are frequently found to be part of the TME (209); however, their role in tumor progression and vascularization is still unclear. They can directly promote angiogenesis by secreting pro-angiogenic factors such as VEGF-A, FGF2, and MMP9 (210), or indirectly by polarizing macrophages to the M2 pro-angiogenic phenotype (211). Transgenic mice (CD19Cre/hVEGF-Afl) overexpressing human VEGF-A in murine B cells exhibited a VEGF-A induced lymphangiogenesis and an expansion of HEVs in LNs. The authors of the study speculated that the unphysiologically high levels of human VEGF-A might not directly influence the LN lymph- and blood vasculature, but may rather cause an accumulation of pro-angiogenic macrophages (122). In a mouse model of LCMV infection, B cells were shown to be required for LN tissue remodeling and vessel expansion. Surprisingly, the latter was independent of VEGF-A signaling pathways, but required LTα1β2-expressing B cells (50). A recent study emphasized the angiogenic capacity of a B cell subset during eosinophilic esophagitis and in patients with melanoma. These pro-angiogenic B cells were identified by the surface markers IgG4+CD49b+CD73+ and shown to promote vascular tube formation in vitro through VEGF-A, FGF2, and PDGFA expression (123). Taken together, although B cells express VEGF-A and LTα1β2 during certain conditions, their role in LN angiogenesis is not well understood. Potentially, B cells may exert pro-angiogenic effects themselves, but also through stimulation of other stromal cell types, such as FRCs and macrophages (Figure 2).
The clinical importance of angiogenic processes and mechanisms for the growth of solid tumors is well recognized (212, 213). Therapeutic concepts from solid tumors targeting the VEGF-A/VEGFR1/2 axis have been adopted for combinatorial therapies of B-NHL, resulting in rather disappointing clinical outcomes (214, 215). We recently showed that angiogenic processes in LNs in a mouse model of high-grade B cell lymphoma are induced by signaling pathways distinct from solid tumors. In sharp contrast to most solid tumors, lymphoma growth in LNs was not associated with hypoxic conditions or inflammation. Instead, lymphoma affected vessel expansion via the VEGF-C/VEGFR3 and LTα1β2/LTβR signaling axes (42). In patients, the growth of tumor cells in low-grade B-NHLs is usually exponential for a few months and remains in a steady state as indolently growing tumor mass for years. This indolent lymphoma is considered to be avascular with dormant endothelial cells within the TME. In contrast, high-grade B-NHL progression is often accompanied by a so called “vascular phase”, which represents extensive vascularization of LNs (216, 217). Such intermediate- and high-grade B-NHLs grow exponentially without intermission phase until they reach a mass critical for a patient’s survival. As a clinical indicator of the vascularization, B-NHLs are usually quantified by terms of the microvessel density (MVD). Immunohistology using anti-CD31 antibody staining is still considered the “gold standard” of blood vessel detection, even though there is substantial variation between different studies due to the heterogeneity of the lymphoma stroma and different scoring methodologies. In some cases, the marker CD34 is used to detect the blood vasculature. Notably, lymphatic vasculature also expresses CD31, but at much lower levels (42, 218, 219).
Non-invasive assessment of tumor vascularization in vivo is possible by using Doppler sonography, contrast-enhanced dynamic magnetic resonance imaging (dMRI) and positron emission tomography-computer tomography (PET-CT). These techniques do not allow a direct quantification of the blood vessel density but provide information on the functional status of the blood vessels, e.g., vessel integrity, permeability, perfusion and metabolism (220). Another diagnostic approach to detect ongoing angiogenesis in vivo is the serological quantification of growth factors. VEGF-A levels in the serum of patients with progressive NHL were significantly elevated in comparison to patients in complete remission (221, 222). Elevated VEGF-A levels have been found in aggressive B cell lymphoma subtypes including MCL, DLBCL, but also in indolent lymphoma, such as CLL and small lymphocytic lymphoma (SLL), respectively (223–225). A variety of commonly used B-NHL cell lines secrete measurable VEGF amounts under serum starvation conditions, whereas other angiogenic factors like the placental growth factor (PlGF) and FGF-2 are not expressed (226). However, the detection of angiogenic factors in clinical serum samples gives no information on the cellular source of these molecules and is not a reliable indicator of angiogenesis in the compartment of interest. Previously, a group of angiogenesis experts published consensus guidelines for the use and interpretation of angiogenesis assays, which involve in vivo, ex vivo explantation, and in vitro bioassays. They explicitly highlighted critical aspects that are relevant for the execution of angiogenesis detection and proper interpretation (227).
Mantle cell lymphoma (MCL) is an aggressive B cell neoplasm that comprises 6% of all NHL cases (228, 229). It is susceptible to paracrine signaling from the microenvironment and in turn shapes the microenvironment by secreting soluble factors (230). MCL is genetically characterized by overexpression of the CCND1gene, encoding for cyclin D1 (231). Recent studies identified a subgroup of MCL that has a more indolent behavior with a clinical presentation as leukemic disease, exhibiting minimal LN distribution and a frequent splenomegaly. These tumors also overexpress cyclin D1 but lack expression of the sex determining region-Y-box11 (SOX11), a TF specifically expressed in conventional MCL and associated with an aggressive and angiogenic phenotype (232). These results have been confirmed in MCL patient samples by using immunohistochemistry, demonstrating a correlation between an increased MVD and high levels of SOX11 expression (233). Experiments with MCL tumor xenotransplants in mice, in cell lines, and in primary MCL samples revealed that SOX11 actively modulates angiogenesis by up-regulation of the platelet-derived growth factor α (PDGFα), which is a competent inducer of an FRC-associated pro-angiogenic program (234, 235). Moreover, SOX11 overexpression promotes B cell receptor signaling represses Bcl6 transcription and upregulates PAX5 to avoid B cell differentiation into memory B cells or plasma cells. PAX5 supports tumor cell homing and invasion via up-regulation of CXCR4 and the focal adhesion kinase (FAK) (236–238). The absolute monocyte count in MCL correlated with the prognosis and supports the hypothesis that the TME is relevant for MCL tumor progression (239). CD68+ and CD163+ macrophages were found in MCL LNs without exception. Substantial numbers of VEGF-C expressing macrophages were found in a mouse xenotransplantation model as well (240). Treatment with the immunomodulator lenalidomide depleted monocytes and VEGF-C expressing macrophages, resulted in impaired functional lymphangiogenesis. However, a relevant impact on lymphoma-associated blood vessel growth in MCL was not investigated in this study. Of note, in human MCL anti-inflammatory and pro-angiogenic CD163+ cells (M2-like) outnumbered the more inflammatory CD68+CD163- macrophages (233), indicating a propensity to stimulate angiogenesis. This M2-like polarization of macrophages is actively driven by MCL derived CSF-1 and IL-10 (241). MCL cells exhibit increased expression of the T cell, B cell, and monocyte recruiting chemokines CCL4 and CCL5 compared to normal B cells (242). T cell infiltration has been considered as a prognostic marker in MCL in which CD8+, and particularly CD4+ T cell frequencies are higher in indolent MCL and decrease with more aggressive histological and clinical presentation (243). In contrast, a recent study reported an expanded vascularization of MCL associated with a high infiltration of CD4+ and CD8+ T lymphocytes (233). The differences might be explained by a weak comparability of data that were either correlated with the clinical outcome, or with the SOX11 expression level in MCL, two hallmarks that are not always correlated. A more detailed T cell characterization of CD4/CD8 T cell subsets is required for a more reliable assessment of the T cell–related influence on angiogenesis and the clinical outcome in MCL. Interestingly, MCL cells itself express the VEGFR-1, providing a strong rationale to target VEGF in order to interfere with angiogenic processes and concomitantly, with autocrine survival signals (230, 244).
Angiogenesis is likely a part of MCL progression, driven by MCL derived PDGFα. Therapeutical interference with PDGFR-β signaling, the receptor for PDGFα, can be achieved with receptor tyrosine kinase (RTK) inhibitors. Some PDGFR-β targeted drugs have been tested in clinical trials for B-NHL but failed to bring significant benefit (217). In contrast, immunomodulating drugs (IMiD) like thalidomide and lenalidomide have anti-angiogenic properties and showed great potential in combination with rituximab for the treatment of untreated or relapsed MCL patients (245, 246).
Follicular lymphoma (FL) is the second most common B-NHL, accounting for 20% of all B-NHL cases (247). The disease affects LNs, spleen and frequently also the bone marrow. Neoplastic follicles in FL have a lower proliferative index in comparison to reactive germinal centers. However, the proliferative capacity of FL cells increases gradually with the FL grade. FL progression requires the supporting infrastructure of the follicular TME to maintain survival, a requirement that gets progressively lost in the process of transformation to aggressive DLBCL (248, 249). Follicular dendritic cells (FDCs) are one branch of this supporting infrastructure. They are of mesenchymal origin and represent a crucial stromal cell population supporting the germinal center reaction and maintenance of the B cell follicle in LN and spleen (250, 251). FDC secreted B cell survival factors such as Indian hedgehog (HH), the B cell activating factor (BAFF), and IL-15 are potentially pro-tumorigenic (252, 253). CXCR5-controlled access to FDCs conferred survival and proliferation stimuli to CLL B cells in the murine Eμ-Tcl1 model, which mimics some aspects of indolent tumor growth (253). Similar to reactive LN follicles, neoplastic follicles in FL preserve the organized FDC network structure at least in early stages of the disease progression (254). FL-FDC cross-talk induces a pro-angiogenic expression pattern in FL cells, including secretion of VEGF-A and VEGF-C (255). This cross-talk is crucially dependent on the phosphoinositide-3-kinase δ (PI3Kδ), providing therapeutic intervention options with PI3K specific inhibitors like idelalisib, which is approved for the treatment of FL, CLL, and SLL (256). The second branch of the supportive infrastructure in follicles are the CD4+CXCR5+PD1+ T follicular helper (Tfh) cells, which provide vital survival signals for FL cells by secreting IL2, IL4, IFNγ, and by CD40L presentation (9, 257). FL cells are further dependent on proliferation and survival signals of the B cell receptor (BCR) in interaction with FDCs and TAMs (258). Elevated numbers of M2-like TAMs are found in the immediate microenvironment of FL cells and neo-vascular sprouts within the follicle (138). However, the prognostic value of CD163+ TAMs remains controversial and is highly dependent on the prior course of treatment (259). In sum, FL appeared to be less prone to induce relevant vascular changes, whereas LNs of high-grade B-NHLs exhibited a dense and aberrantly distributed vasculature within the paracortical zone. In contrast to most other B-NHL malignancies in which high levels of pro-angiogenic factors and an increased MVD is associated with an adverse prognosis, high level FL vascularization correlates with a beneficial disease course (260–262). The improved clinical outcome apparently correlated with the increased vascularization, but was surprisingly independent of follicular VEGF-A expression (223, 263). Some studies stated a minor vascular remodeling in FL compared to reactive LN or follicular hyperplasia, or even vascular regression constraining the growth of reactive and neoplastic follicles (260, 264). Therefore, the clinical significance of angiogenesis in FL remains uncertain. In one clinical trial, addition of the anti-VEGF bevacizumab during rituximab treatment of relapsed FL significantly improved the progression-free survival (265). The potential of angiogenesis inhibition upon treatment of FL requires further evaluation in larger clinical trials.
Collectively, according to the data currently available it seems that angiogenesis is important for high-grade lymphoma, but has less impact on indolent FL growth.
Diffuse large B-cell lymphoma (DLBCL) is the most common type of lymphoid tumors worldwide accounting for 30% of all diagnosed NHL and characterized by the large size of neoplastic B cells and usually a very aggressive clinical presentation (266). Lenz et al. identified gene expression profiles in LN from patients pre-treated with the combination therapy anti-CD20 antibody, cyclophosphamide, doxorubicin, vincristin, prednisolone (R-CHOP), dividing DLBCL in two distinct subgroups that are predictive of the clinical outcome (267). The “stromal-1” signature includes expression of extracellular matrix (ECM) elements, ECM remodeling factors (MMP2, MMP9, M1-MMP, PLAU, TIMP2) and is associated with a favorable prognosis. The “stromal-2” signature” was found in tumors with an increased MVD and is characterized by markers of endothelial cells (Pecam1, Vwf, Kdr, Tek). The latter signature is associated with a poor clinical outcome, emphasizing the critical impact of angiogenic processes on aggressive B-NHL progression. Several studies investigated the clinical consequences of the “stromal-2” signature and confirmed the correlation of a high MVD with an adverse outcome and a shorter overall survival rate (268–271). The relationship between MVD and DLBCL behavior was the object of many studies and was found to be associated with poor prognostic parameters such as splenic involvement, high mitotic rate, and capsular invasion (268–272). Gomez-Gelvez et al. reported contradictory results, showing that high MVD is associated with rather better progression-free survival (PFS) and event-free survival (EFS) (273). Several other studies also failed to draw a connection between the MVD, tumor grade and prognostic outcome (274–277). A DLBCL mouse xenotransplantation model demonstrated that the inhibition of the paracrine VEGFR-2 pathway reduced growth of an established lymphoma and correlated with decreased tumor angiogenesis (226). DLBCL cells often overexpress the phosphodiesterase 4B (PDE4B), which intracellularly catalyzes the hydrolysis of cyclic-AMP (cAMP). The cAMP-PDE4B axis modulates signaling of PI3K and AKT and therefore acts upstream of VEGF-A expression. Experiments with genetically or pharmacologically inhibited PDE4B resulted in decreased VEGF-A expression in lymphoma cells and reduced angiogenesis in the Eµ-Myc high-grade lymphoma mouse model (278).
In a gene expression study on relapsed or refractory DLBCL, patients with the ABC-like DLBCL subtype that had low VEGF121 isoform expression, exhibited a significantly better overal survival than those with high VEGF121 gene expression levels (279). Interestingly, VEGF121 low transcript levels were associated to a gene signature reflecting immune response and T cell activation.
DCs are likely a major source of VEGF-A in LNs with DLBCL. Functionally, DCs could be involved in lymphoma TME remodeling, but their number in DLBCL LNs is significantly lower than in reactive LNs. Lower expression levels of the LN homing receptors CD62L and CCR7 in DCs in LNs of DLBCL patients were thought to result in reduced DC immigration. However, it remains elusive if the DCs lose the receptor expression upon arrival in the LN, or whether these cells are recruited via alternative routes (280). In an aggressive Myc-driven lymphoma model in mice a tumor-specific DC differentiation occurs that promotes tumor cell survival and favors the maturation of monocytic-derived DCs (MHCIImedium) (149, 150). Alongside tumor repressing M1 macrophages, “alternatively” activated M2 macrophages exhibit angiogenic capacities, they are frequently found in DLBCL and often correlate with a poor prognosis (281–283). Although numerous studies reported an association between TAMs and MVD in DLBCL, others could not find a correlation between CD68+ macrophages and an increased MVD (270). Such controversies can probably be best explained by variabilities in the methodological approaches. The macrophage marker CD68 represents M1 and M2 macrophages and therefore, produces inaccuracies in the interpretation of studies concerning the macrophage-MVD correlation. The addition of the marker CD163, which rather recognizes M2 activated macrophages, including angiogenic macrophages, provides a more reliable view on the role of macrophages in DLBCL (284). Elevated numbers of macrophages have been correlated with poor prognosis in DLBCL (282). However, in therapeutic setting macrophages are required to confer treatment effects when patients were treated with anti-CD20 antibody (e.g., Rituximab). Here, macrophages mediate tumor cell depletion via the macrophage Fc-gamma receptor (FcγR) expression (215). Another abundant immune cell population in LNs of DLBCL patients is mast cells with a predominance of MCT-type (tryptase-positive) cells. MCTC-type (tryptase-positive and chymase‐positive) and CD4+ Th2 were shown to express IL-4 in DLBCL and therefore, they may actively promote survival of the tumor cells (285). Hedström and coworkers examined 154 DLBCL cases and suggested that the infiltration of mast cells reflects the inflammatory immune response of the endogenous anti-tumor defense and is therefore related to a favorable outcome (286). The gradual increase of the MVD was correlated with an increasing number of mast cells. Although mast cells are considered to be bystanders in tumor immunology, additional pro-angiogenic effects of these cells are likely as they secrete relevant amounts of different VEGFs, FGF-2, trypase, and granzyme B. The latter has a pro-angiogenic effect via the enzymatic mobilization of ECM-bound FGF-1 (287, 288). The wide range of physiological conditions and tumor entities that include mast cell-supported angiogenesis and the respective recruitment and signaling pathways were excellently reviewed by Ribatti et al. (289).
Apart from the direct effect on immune and tumor cells, surprisingly, the application of the VEGF-A inhibiting antibody bevacizumab to R-CHOP therapy increased adverse cardiac events, yet without increasing the therapeutic efficacy in DLBCL patients (214, 215). From these studies it can be inferred that the increased MVD in DLBCL patients may be simply a correlation with minor importance for the disease course, or that other non-VEGF-A angiogenic pathways prevail and cause enhanced vascular assembly instead. In a study of Pazgal et al., VEGF-C, VEGF-D, and VEGFR3 were expressed in both lymphoma cells and endothelial cells of the blood and lymphatic vasculature. They reported a significant correlation of the VEGF-C expression and the presence of blood vessels. VEGF-D expression correlated with the patient International Prognostic Index (IPI) Score and the patients’ overall survival (14). These results may indicate that apart from its role as primary signaling pathway for lymphatic vessels, the VEGF-C–VEGFR3 axis also has implications on angiogenic processes of LN blood vessels. A study in breast cancer demonstrated that VEGFR3 is significantly upregulated in the endothelium of new blood vessels. The results also suggested that VEGF-C secreted by the intraductal carcinoma cells acts predominantly as an angiogenic growth factor for blood vessels, although other immune or stromal cells might be involved in this paracrine signaling network as well (290). An experimental study using a Myc-driven aggressive lymphoma mouse model, which resembles important aspects of aggressive B-NHL, supported this hypothesis, showing that the MVD expansion was triggered by lymphoma-provided VEGF-C, in a synergistic activity with LTα1β2 (42).
Representing a high-grade and angiogenesis-associated lymphoma type, multiple clinical trials with anti-angiogenic agents for the treatment of DLBCL have been conducted. Most of the treatment approaches using single agent angiogenesis inhibitors failed to prove a beneficial effect. However, combinatorial treatment strategies such as R2-CHOP (lenalidomide, R-CHOP) (291, 292), brought encouraging results. Such oberservations emphasize that anti-angiogenesis therapies might not be effective when applied alone, even in highly vascularized lymphoma, but are valuable components in combination with other drugs.
Burkitt’s lymphoma (BL) represents around half of all malignant non-Hodgkin lymphoma in children and around 2% in adults (293). The BL pathogenicity is usually associated with the infection of B cells with the Epstein-Barr virus (EBV). EBV gene products induce BL cell-derived soluble factors that result in inhibition of neo-vascularization and eventually tumor necrosis and regression (294). However, in in vivo experiments, EBV-positive cells induced massive recruitment of leukocytes at the tumor border and the development of granulation tissue with large numbers of blood and lymphatic vessels (295). Surprisingly, aggressive BL displayed the highest MVD in comparison to intermediate DLBCL and indolent B-NHL (42, 262, 287). In support of this observation, BL showed increased vascularization relative to benign lymphadenopathies and can produce several angiogenic factors, although it is not yet known whether this is due to Myc gene overexpression or the EBV transformation (296–298). BL were found to be closely associated with VEGF-producing CD68+VEGFR1+ myeloid cells located around the neo-vasculature. The newly formed blood vessels were identified by the absence of pericyte coverage as result of the rapid vessel growth (299, 300). Genetic depletion of this subpopulation of CD68+VEGFR1+ myeloid cells was sufficient to inhibit angiogenesis in experimental lymphoma (301). To our knowledge, to date there are no clinical data or published treatment strategies of BL that target angiogenesis specifically.
Classical Hodgkin Lymphoma (cHL) is characterized by mono-nucleated Hodgkin and multi-nucleated Reed-Sternberg (HRS) cells, which comprises tumors with mixed cellularity, nodular sclerosis and lymphocyte-rich or lymphocyte-depleted subtypes. Different from other lymphoma, HRS cells are the minority of cells within the affected LN. Most of the cells in cHL tumors are cells of the TME, indicating a prominent role of benign immune cells and the LN stroma (302). A crucial role of angiogenesis and increased MVD have been reported for cHL and correlate with a poor prognosis (303). Similar to observations in highly vascularized LNs in an aggressive B-NHL mouse model (42) and in immunohistochemically characterized B-NHL patient specimen (277), in cHL HIF-1α was only moderately expressed (304), suggesting that angiogenesis in cHL is not hypoxia-driven and may utilize other angiogenic pathways instead. In childhood cHL, HRS cells express VEGF, MMP-2 and MMP-9. However, the expression of these factors did not correlate with the MVD and neovascularization level (305, 306). On the other hand, VEGF-D, a ligand for VEGFR3 and usually associated with lymphangiogenesis, is expressed in HRS cells at high abundance and correlated with high numbers of microvessels (307). Moreover, in vitro HRS cell-derived TGF-β, FGF-2, and VEGF supported HUVEC tubulogenesis (308, 309). Secretion of Ltα by HRS cells activated endothelial cells, which enhances adhesion molecule expression and consequently, recruitment of T cells. This mechanism amplifies the inflammatory milieu in the cHL TME through conditioning of the blood vasculature (310).
Commonly attributed to the occurrence of angiogenic M2 macrophages, TAMs are linked to poor outcome in HL. Interestingly, lack of macrophages, but also high numbers of macrophages is associated with a poorer disease-free survival and overall survival, whereas intermediate numbers are associated with a better outcome. This macrophage paradox suggests that a lack of TAMs is beneficial for HL growth, while TAMs have an inhibitory effect with increasing numbers (311). The inhibitory effect seems to be displaced by an adverse effect of TAM-induced angiogenesis, supposedly predominated by CD163+ M2-like TAMs (312). High numbers of CD163+ TAMs correlate with elevated VEGF-A levels and an increased MVD, indicating that CD163 is an independent prognostic marker in cHL (313). Interestingly, although the particular signaling pathways within TAMs remain elusive, pre-clinical experiments with PI3K-Akt pathway inhibition suggested a connection to macrophage M2-polarization (314, 315), which could be a promising anti-angiogenic intervention clue by prevention of pro-angiogenic activity of M2-like TAMs.
Cancer therapy earlier than the 1970s was solely focused on targeting the actual cancer cells. Judah Folkman’s discovery that tumor growth is angiogenesis-dependent led to a profound paradigm shift in cancer therapy (316, 317). Sprouting angiogenesis plays an essential role in tumor growth, invasion, progression, and metastasis, targeting this process is a promising strategy to inhibit growth and spread of solid tumors. Clinical trials and treatment strategies of anti-angiogenesis therapy in B-NHL were recently reviewed (217). Angiogenesis inhibitors are classified into direct and indirect agents. Direct inhibitors target vascular ECs and include endostatin, arrestin, and tumstatin. Indirect angiogenesis inhibitors target tumor cells or cells of the TME to prevent the expression of pro-angiogenic factors or block their activity (318). The anti-VEGF monoclonal antibody Bevacizumab was the first anti-angiogenesis drug approved by the FDA for the treatment of metastatic colon, ovarian, renal, non-squamous cell lung cancer, and glioblastoma multiforme. Unfortunately, clinical significance was only reached in glioblastoma multiforme treatment (319, 320), a result that could not be confirmed in other studies (321). In contrast to Bevacizumab, treatment with tyrosine kinase inhibitors (e.g., Sorafenib) that interfere with the signal transmission of VEGFRs resulted in remarkable effects throughout several cancer entities. Combination of tyrosine kinase inhibitors and conventional chemotherapy have not been beneficial (322). Conventional chemotherapy can cause direct cytotoxicity of endothelial cells, but this effect is non-selective and only observed upon the maximal tolerated dose (MTD). Insufficient tumor and vascular bed destruction can effectuate a strong hypoxic condition, which results in release of chemoattractant CXCL12. Accordingly, MTD chemotherapy potentially increases systemic CXCL12, which recruits bone marrow–derived EPCs. These cells can cause recurring angiogenesis in mouse models of solid tumors (323, 324). Therefore, an anti-angiogenesis therapy that is complementary to chemo- or immunotherapy is aimed at restricting pro-angiogenic bystander effects of the tumor treatment. In addition, instead of aiming for a complete vascular eradication, the paradigm in anti-angiogenic therapies shifted to vascular normalization (325, 326).
Rituximab has become an essential part of first-line treatment of several B cell lymphoma entities, foremost of DLBCL. However, ongoing research aims to improve the therapeutic efficiency and the reduction of the relapse rate of drug-resistant lymphoma cells. Tumor anti-angiogenesis therapy approaches are one branch of such research, in which Bevacizumab and Endostatin were the most promising representatives for lymphoma treatment (327, 328). VEGF-A has a crucial role in promoting vessel growth, but is also considered to be an immunosuppressive factor that modulates the migration and function of several immune cells, e.g., DCs and mast cells. The potential pharmaceutical targeting of the VEGF/VEGFR axis to modulate anti-tumor immunity has been reviewed recently (329).
An important challenge of anti-angiogenic therapy in solid tumors as well as in lymphoma is the identification of the particular angioactive receptors throughout different tumor entities and individual patients. The inhibition of intracellular signaling hubs is a strategy to overcome the targeting of distinct angiogenic tyrosine-kinase receptors. Class I PI3Ks are involved in the signal transduction of many pro-angiogenic signals and control cell growth, survival, motility, and metabolism (330). PI3Kδ inhibition in lymphoma potentially also interferes with tonic signaling in tumor cells, e.g., via the BCR signaling pathway (331), or breaks the Treg-mediated immune tolerance (332). Interestingly, PI3K activity is essential for macrophage M2 polarization (333) and therefore, a potential target to hamper M2-like angiogenic macrophages. Inhibition of PI3K signaling represents a valuable therapeutic strategy to target different indolent B cell lymphoma entities, among them FL, CLL, SLL, and more recently, they showed promise in T cell lymphomas as well (334, 335). The combinatorial treatment of the first generation PI3K inhibitor idelalisib with rituximab or bendamustine revealed favorable response rates in FL patients (334), but serious adverse effects due to bacterial and viral infections were observed. Additionally, immune-mediated and hematologic adverse events occurred. Beyond that first generation PI3K inhibitor, newer PI3K inhibitors such as copanlisib and duvelisib were introduced for patients with relapsed and progressive FL, CLL, SLL, respectively. These inhibitors differ in their preference for PI3K isoforms which are expressed differentially in various tissues (336). Despite relevant side effects of PI3K inhibitors, they have been judged clinically manageable and thus, prompted an FDA approval for relapsed and refractory indolent B-NHL (335, 337). Published reports on anti-angiogenic therapies in B-NHL allow the conclusion that the complex mechanisms of angiogenesis in lymphoma are incompletely understood and require further pre-clinical and translational research to develop reliable and effective anti-angiogenic treatment strategies. Moreover, new anti-angiogenic treatment regimens need to be validated regarding an actual reduction of tumor growth, since sole targeting of angiogenic factors often fail to cause substantial tumor regression (Figure 3) (340).
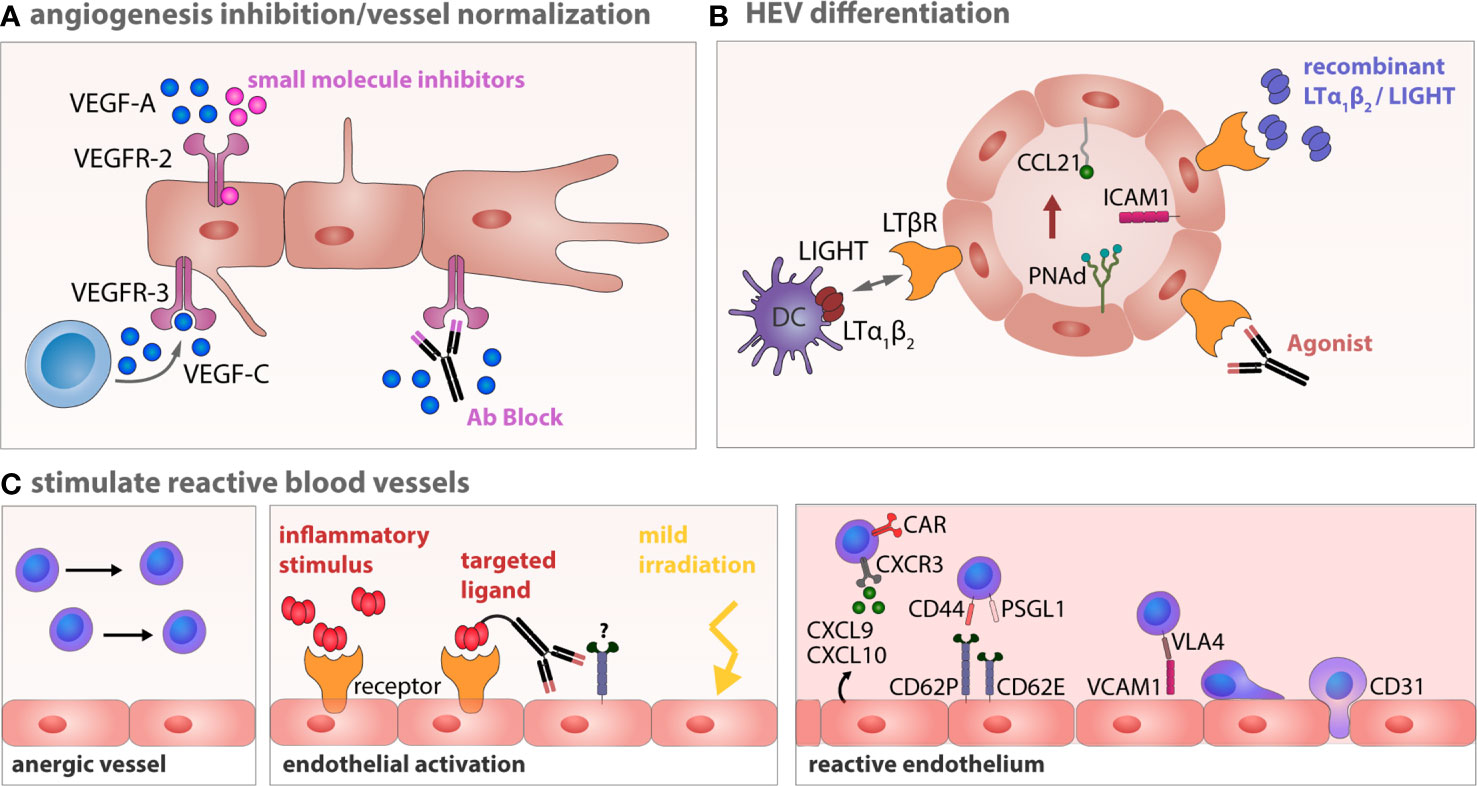
Figure 3 Therapeutic strategies to induce vessel normalization and revert endothelial anergy in B-NHL. (A) Anti-angiogenesis therapy tageted at VEGFR-2 or VEGFR-3 can restore a normalized vessel network. (B) Targeting of the LTβR with LTα1β2 and LIGHT expressing DCs, agonistic antibodies or recombinant factors potentially circumvents impaired lymphocyte homing by establishing or stabilizing HEV integrity within the lymphoma TME (338). (C) Vessel anergy can be changed by a targeted conversion of the endothelium toward a reactive endothelium using inflammatory cytokines, which might be site directed to avoid unintended systemic effects. Normalization of aberrant vessels and activation of the endothelium can also be achieved by locally applied low-dose gamma irradiation (339). Reactive endothelium within LNs is a prerequisite for an effective infiltration of effector T cells during cellular immunotherapy.
Vascular remodeling and angiogenesis have been increasingly recognized as crucial factors in the pathophysiology of B-NHLs. We here present an integrated concept that includes angiogenic processes of the LN TME beyond the proliferation and survival of endothelial cells stimulated by the VEGF-VEGFR axis. In human, the angiogenic properties of LN resident and recruited immune cells are still insufficiently understood. Studies to address such functional states are limited because tissues from appropriate human patients are rarely available. Notably, most of the human data available so far are observations on clinically progressed and even terminal stage lymphoma LNs. Flow cytometry analysis of blood samples is usually limited to a few entities (e.g., FL, CLL, and MCL), common markers and cannot readily be correlated with pathohistological observations due to the lack of tissue specimen. Availability of LN tissue from progressed disease stages is often limited to scarce material from fine-needle biopsies. As a useful surrogate, mouse models of reactive LNs and LNs with lymphoma growth demonstrated that the angiogenic processes are regulated by a timely complex interplay of immune, tumor, and stromal cells (42).
In the future, modern methods like single-cell RNA sequencing alone or with spatial resolution, or single-cell analysis in combination with proteomics will help to resolve the complexity of participating cells and their heterogenous differentiation status. This technique requires much less input material for a high resolution analysis at the genome, protein, or epigenome level (341–343). Even patient-derived specimen from fine needle biopsies seem amenable to such analysis, allowing then a kinetic description of LN remodeling in the course of diagnosis, treatment response, and eventually relapse. Single-cell RNA sequencing will further enable the discrimination of different endothelial cell subtypes and their differentiation traits (344). The compartment of BECs is comprised of several functionally distinguishable subpopulations that further differentiate during angiogenesis. To date, we know little about the role, differentiation conditions and distribution of these subpopulations in LNs. Such transcriptional observations need to be correlated with the topology of the single cells and cell networks within the complex LN (345). Modern imaging methods, e.g., light sheet microscopy and intravital 2-photon microscopy enable the complex spatial integration and the investigation of dynamic processes in situ, which have long been restricted to snapshot observations. Very recently, a new generation of flow cytometry devices became available that, based on a spectrum wide detection of fluorophores, allow a simultaneous detection of a multi-fold higher number of cell markers. The possibility to determine extended marker panels with small sample sizes will not only improve basic knowledge in the pre-clinical context, but will also provide innovative approaches for clinical diagnostics (346, 347).
Improving insight into angiogenesis is also of considerable relevance for the emerging immunotherapies using chimeric antigen receptor (CAR)- and T cell receptor (TCR)-transgenic T cells and NK cells. It is reasonable to suggest that tumor blood vessels have a leading role in granting effector T cell access to the LN and the tumor niche formed therein. For example, solid tumors condition an endothelial activation status that can be considered immunologically “silent” (348). However, reactivation of such vessel-lining endothelial layers is a prerequisite for the adhesion and transmigration cascade of naive and therapeutic T cell populations. We envision that this endothelial tuning is not only applicable to solid tumors, but also to LN-localized lymphatic neoplasm. Except for cHL, immune checkpoint blockade (ICB) targeting PD-1 or CTLA-4 has not shown relevant benefit in other B cell neoplasm. Because ICB efficacy depends on the presence of a repertoire of antigen-specific T cells, a rational sequence of immunotherapeutic interference in B-NHL would start with a vessel induction toward a more activated or even inflammatory state. It seems not even necessary to overactivate local endothelial cells, as shown by the application of a modified TNFα cytokine that upregulates adhesion molecules, but then even eradicates solid tumors through rapid destruction of the tumor neovasculature (349). Enhanced adhesion, e.g., involving ICAM-1 and VCAM-1 up-regulation, may be sufficient to allow T cells to get access to the primary lymphoma site in the deep parenchyma. Finally, in a time window to be defined, application of ICB might then unleash the activity of effector T cells that already invaded the tumor site.
Collectively, efforts to target tumor cells only or single lymphoma-promoting cellular stromal elements in the TME are unlikely to confer long lasting remissions. For example, although anti-CD19 CAR T-cell therapies have proven remarkable efficacy in B cell malignancies, they become ineffective due to CD19 antigen loss or downregulation (350, 351). Other contributing factors to substantial rates of treatment failure might be the nodal immunosuppressive microenvironments in B-NHL (9, 352).What is needed is an integrative concept that blocks vicious feedback cycles in lymphoma. We suggest that combinatorial targeting of aberrantly polarized myeloid cell populations, blood endothelial activation, angiogenesis, and effector T cell dysfunction is a rational stepwise strategy (Figure 3). Advanced CAR T cell technologies try to integrate a few of these demands, for example by deleting the functionality of PD-1 (353, 354) and secretion of immune-stimulatory cytokines such as IL-12, IL-21, or IL-18 (355–357). We envision that the vasculature is important for control of lymphoma relapse. In this process, mutual stimulation of residual tumor cells, mesenchymal and hematopoietic stromal cells, and endothelial cells might favor neo-angiogenesis and eventually, re-shaping a growth supporting niche for lymphoma B cells.
LM: conceived the general idea, wrote the manuscript, and created the figures and tables. AR: conceived the general idea, co-wrote the manuscript, and edited figures and tables. UH: provided expert opinion/knowledge input and edited manuscript, figures, and tables. All authors contributed to the article and approved the submitted version.
This work was funded by the Wilhelm Sander-Stiftung (grant number 213.100.02), and by the Deutsche Krebshilfe (grant number 107749) awarded to AR and UH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Krishnamurty AT, Turley SJ. Lymph node stromal cells: cartographers of the immune system. Nat Immunol (2020). doi: 10.1038/s41590-020-0635-3
2. Till KJ, Coupland SE, Pettitt AR. Motility and trafficking in B-cell non-Hodgkin’s lymphoma (Review). Int J Oncol (2014). doi: 10.3892/ijo.2014.2395
3. Wang L, Qin W, Huo YJ, Li X, Shi Q, Rasko JEJ, et al. Advances in targeted therapy for malignant lymphoma. Signal Transduct Target Ther (2020). doi: 10.1038/s41392-020-0113-2
4. Maman S, Witz IP. A history of exploring cancer in context. Nat Rev Cancer (2018). doi: 10.1038/s41568-018-0006-7
5. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell (2011). doi: 10.1016/j.cell.2011.02.013
6. Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood (2009). doi: 10.1182/blood-2009-06-225326
7. Menter T, Tzankov A. Lymphomas and their microenvironment: A multifaceted relationship. Pathobiology (2019). doi: 10.1159/000502912
9. Höpken UE, Rehm A. Targeting the Tumor Microenvironment of Leukemia and Lymphoma. Trends Cancer (2019). doi: 10.1016/j.trecan.2019.05.001
10. Cao Z, Ding BS, Guo P, Lee SB, Butler JM, Casey SC, et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell (2014). doi: 10.1016/j.ccr.2014.02.005
11. Liao S, Ruddle NH. Synchrony of High Endothelial Venules and Lymphatic Vessels Revealed by Immunization. J Immunol (2006). doi: 10.4049/jimmunol.177.5.3369
12. Dasoveanu DC, Shipman WD, Chia JJ, Chyou S, Lu TT. Regulation of Lymph Node Vascular–Stromal Compartment by Dendritic Cells. Trends Immunol (2016). doi: 10.1016/j.it.2016.08.013
13. Habenicht LM, Kirschbaum SB, Furuya M, Harrell MI, Ruddell A. Tumor regulation of lymph node lymphatic sinus growth and lymph flow in mice and in humans. Yale J Biol Med (2017).
14. Pazgal I, Boycov O, Shpilberg O, Okon E, Bairey O. Expression of VEGF-C, VEGF-D and their receptor VEGFR-3 in diffuse large B-cell lymphomas. Leuk Lymphoma (2007). doi: 10.1080/10428190701632822
15. Girard JP, Moussion C, Förster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol (2012). doi: 10.1038/nri3298
16. Ito K, Smith BR, Parashurama N, Yoon JK, Song SY, Miething C, et al. Unexpected dissemination patterns in lymphoma progression revealed by serial imaging within a murine lymph node. Cancer Res (2012). doi: 10.1158/0008-5472.CAN-12-2579
17. Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet (2008). doi: 10.1038/ng.127
18. Naxerova K, Bult CJ, Peaston A, Fancher K, Knowles BB, Kasif S, et al. Analysis of gene expression in a developmental context emphasizes distinct biological leitmotifs in human cancers. Genome Biol (2008). doi: 10.1186/gb-2008-9-7-r108
19. Mazzone M, Bergers G. Regulation of Blood and Lymphatic Vessels by Immune Cells in Tumors and Metastasis. Annu Rev Physiol (2019). doi: 10.1146/annurev-physiol-020518-114721
20. Osogoe B, Courtice FC. The effects of occlusion of the blood supply to the popliteal lymph node of the rabbit on the cell and protein content of the lymph and on the histology of the node. Aust J Exp Biol Med Sci (1968). doi: 10.1038/icb.1968.159
21. Lee Y-e, Koh GY. Coordinated lymphangiogenesis is critical in lymph node development and maturation. Dev Dyn (2016). doi: 10.1002/dvdy.24456
22. Vondenhoff MF, van de Pavert SA, Dillard ME, Greuter M, Goverse G, Oliver G, et al. Lymph sacs are not required for the initiation of lymph node formation. Development (2009). doi: 10.1242/dev.028456
23. Bovay E, Sabine A, Prat-Luri B, Kim S, Son K, Willrodt AH, et al. Multiple roles of lymphatic vessels in peripheral lymph node development. J Exp Med (2018). doi: 10.1084/jem.20180217
24. Lai L, Bohnsack BL, Niederreither K, Hirschi KK. Retinoic acid regulates endothelial cell proliferation during vasculogenesis. Development (2003). doi: 10.1242/dev.00887
25. Van De Pavert SA, Olivier BJ, Goverse G, Vondenhoff MF, Greuter M, Beke P, et al. Chemokine cxcl13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat Immunol (2009). doi: 10.1038/ni.1789
26. Mebius RE, Streeter PR, Michie S, Butcher EC, Weissman IL. A developmental switch in lymphocyte homing receptor and endothelial vascular addressin expression regulates lymphocyte homing and permits CD4+CD3- cells to colonize lymph nodes. Proc Natl Acad Sci U S A (1996). doi: 10.1073/pnas.93.20.11019
27. Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+CD3- LTβ+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity (1997). doi: 10.1016/S1074-7613(00)80371-4
28. Mebius RE, Dowbenko D, Williams A, Fennie C, Lasky LA, Watson SR. Expression of GlyCAM-1, an endothelial ligand for L-selectin, is affected by afferent lymphatic flow. J Immunol (1993).
29. Anderson AO, Anderson ND. Studies on the structure and permeability of the microvasculature in normal rat lymph nodes. Am J Pathol (1975).
30. Herman PG, Yamamoto I, Mellins HZ. Blood microcirculation in the lymph node during the primary immune response. J Exp Med (1972). doi: 10.1097/00004424-197209000-00038
31. Kuczynski EA, Vermeulen PB, Pezzella F, Kerbel RS, Reynolds AR. Vessel co-option in cancer. Nat Rev Clin Oncol (2019). doi: 10.1038/s41571-019-0181-9
32. Krishnan L, Chang CC, Nunes SS, Williams SK, Weiss JA, Hoying JB. Manipulating the microvasculature and its microenvironment. Crit Rev BioMed Eng (2013). doi: 10.1615/CritRevBiomedEng.2013008077
33. Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res (2007). doi: 10.1161/01.RES.0000259593.07661.1e
34. Hellström M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature (2007). doi: 10.1038/nature05571
35. Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol (2003). doi: 10.1083/jcb.200302047
36. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell (2011). doi: 10.1016/j.cell.2011.08.039
37. Akeson A, Herman A, Wiginton D, Greenberg J. Endothelial cell activation in a VEGF-A gradient: Relevance to cell fate decisions. Microvasc Res (2010). doi: 10.1016/j.mvr.2010.02.001
38. Bautch VL. VEGF-directed blood vessel patterning: From cells to organism. Cold Spring Harb Perspect Med (2012). doi: 10.1101/cshperspect.a006452
39. Zhang L, Zhou F, Han W, Shen B, Luo J, Shibuya M, et al. VEGFR-3 ligand-binding and kinase activity are required for lymphangiogenesis but not for angiogenesis. Cell Res (2010). doi: 10.1038/cr.2010.116
40. Matsumoto M, Roufail S, Inder R, Caesar C, Karnezis T, Shayan R, et al. Signaling for lymphangiogenesis via VEGFR-3 is required for the early events of metastasis. Clin Exp Metastasis (2013). doi: 10.1007/s10585-013-9581-x
41. Tammela T, Zarkada G, Wallgard E, Murtomäki A, Suchting S, Wirzenius M, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature (2008). doi: 10.1038/nature07083
42. Gloger M, Menzel L, Grau M, Vion A-C, Anagnostopoulos I, Zapukhlyak M, et al. Lymphoma Angiogenesis Is Orchestrated by Noncanonical Signaling Pathways. Cancer Res (2020). doi: 10.1158/0008-5472.CAN-19-1493
43. Fahmy RG, Dass CR, Sun LQ, Chesterman CN, Khachigian LM. Transcription factor Egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nat Med (2003). doi: 10.1038/nm905
44. Yu P, Wilhelm K, Dubrac A, Tung JK, Alves TC, Fang JS, et al. FGF-dependent metabolic control of vascular development. Nature (2017). doi: 10.1016/j.jvs.2017.07.055
45. Turner N, Grose R. Fibroblast growth factor signalling: From development to cancer. Nat Rev Cancer (2010). doi: 10.1038/nrc2780
46. Kroon ME, Koolwijk P, Van Der Vecht B, Van Hinsbergh VWM. Hypoxia in combination with FGF-2 induces tube formation by human microvascular endothelial cells in a fibrin matrix: Involvement of at least two signal transduction pathways. J Cell Sci (2001).
47. Tsuda S, Ohtsuru A, Yamashita S, Kanetake H, Kanda S. Role of c-Fyn in FGF-2-mediated tube-like structure formation by murine brain capillary endothelial cells. Biochem Biophys Res Commun (2002). doi: 10.1006/bbrc.2002.6345
48. Susaki EA, Tainaka K, Perrin D, Kishino F, Tawara T, Watanabe TM, et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell (2014). doi: 10.1016/j.cell.2014.03.042
49. Susaki EA, Tainaka K, Perrin D, Yukinaga H, Kuno A, Ueda HR. Advanced CUBIC protocols for whole-brain and whole-body clearing and imaging. Nat Protoc (2015). doi: 10.1038/nprot.2015.085
50. Kumar V, Scandella E, Danuser R, Onder L, Nitschké M, Fukui Y, et al. Global lymphoid tissue remodeling during a viral infection is orchestrated by a B cell-lymphotoxin-dependent pathway. Blood (2010). doi: 10.1182/blood-2009-10-250118
51. Mayer J, Swoger J, Ozga AJ, Stein JV, Sharpe J. Quantitative measurements in 3-dimensional datasets of mouse lymph nodes resolve organ-wide functional dependencies. Comput Math Methods Med (2012). doi: 10.1155/2012/128431
52. Abe J, Ozga AJ, Swoger J, Sharpe J, Ripoll J, Stein JV. Light sheet fluorescence microscopy for in situ cell interaction analysis in mouse lymph nodes. J Immunol Methods (2016). doi: 10.1016/j.jim.2016.01.015
53. Jafarnejad M, Woodruff MC, Zawieja DC, Carroll MC, Moore JE. Modeling lymph flow and fluid exchange with blood vessels in lymph nodes. Lymphat Res Biol (2015). doi: 10.1089/lrb.2015.0028
54. Jafarnejad M, Ismail AZ, Duarte D, Vyas C, Ghahramani A, Zawieja DC, et al. Quantification of the Whole Lymph Node Vasculature Based on Tomography of the Vessel Corrosion Casts. Sci Rep (2019). doi: 10.1038/s41598-019-49055-7
55. Tomei AA, Siegert S, Britschgi MR, Luther SA, Swartz MA. Fluid Flow Regulates Stromal Cell Organization and CCL21 Expression in a Tissue-Engineered Lymph Node Microenvironment. J Immunol (2009). doi: 10.4049/jimmunol.0900835
56. Chang JE, Buechler MB, Gressier E, Turley SJ, Carroll MC. Mechanosensing by Peyer’s patch stroma regulates lymphocyte migration and mucosal antibody responses. Nat Immunol (2019). doi: 10.1038/s41590-019-0505-z
57. Pajor K, Pap T, Csanaky G, Kalász V. High endothelial venules in B-cell non-Hodgkin malignant lymphomas. J Pathol (1990). doi: 10.1002/path.1711610213
58. Kelch ID, Bogle G, Sands GB, Phillips ARJ, Legrice IJ, Rod Dunbar P. Organ-wide 3D-imaging and topological analysis of the continuous microvascular network in a murine lymph node. Sci Rep (2015). doi: 10.1038/srep16534
59. Kalucka J, de Rooij LPMH, Goveia J, Rohlenova K, Dumas SJ, Meta E, et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell (2020). doi: 10.1016/j.cell.2020.01.015
60. Brulois K, Rajaraman A, Szade A, Nordling S, Bogoslowski A, Dermadi D, et al. A molecular map of murine lymph node blood vascular endothelium at single cell resolution. Nat Commun (2020) 11(1):3798. doi: 10.1038/s41467-020-17291-5
61. Wood HB, May G, Healy L, Enver T, Morriss-Kay GM. Cd34 expression patterns during early mouse development are related to modes of blood vessel formation and reveal additional sites of hematopoiesis. Blood (1997). doi: 10.1182/blood.V90.6.2300.2300_2300_2311
62. Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity (2019). doi: 10.1016/j.immuni.2019.06.025
63. Oladipupo S, Hu S, Kovalski J, Yao J, Santeford A, Sohn RE, et al. VEGF is essential for hypoxia-inducible factor-mediated neovascularization but dispensable for endothelial sprouting. Proc Natl Acad Sci U S A (2011). doi: 10.1073/pnas.1101321108
64. Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheumatol (2002). doi: 10.1002/art.10520
65. Semenza GL. Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis. Oncogene (2013). doi: 10.1038/onc.2012.578
66. McDonald DM, Baluk P. Imaging of angiogenesis in inflamed airways and tumors: Newly formed blood vessels are not alike and may be wildly abnormal. Parker B. Francis lecture. Chest (2005). doi: 10.1378/chest.128.6_suppl.602S-a
67. Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat Rev (2011). doi: 10.1016/j.ctrv.2010.05.001
68. Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol (2000). doi: 10.1016/S0002-9440(10)65006-7
69. Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol (2002). doi: 10.1016/S0002-9440(10)64920-6
70. Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM. Abnormalities of Basement Membrane on Blood Vessels and Endothelial Sprouts in Tumors. Am J Pathol (2003). doi: 10.1016/S0002-9440(10)63540-7
71. Dirkx AEM, Oude Egbrink MGA, Kuijpers MJE, Van der Niet ST, Heijnen VVT, Bouma-ter Steege JCA, et al. Tumor angiogenesis modulates leukocyte-vessel wall interactions in Vivo by reducing endothelial adhesion molecule expression. Cancer Res (2003).
72. Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, et al. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation. FASEB J (2015). doi: 10.1096/fj.14-250985
73. Mondor I, Jorquera A, Sene C, Adriouch S, Adams RH, Zhou B, et al. Clonal Proliferation and Stochastic Pruning Orchestrate Lymph Node Vasculature Remodeling. Immunity (2016). doi: 10.1016/j.immuni.2016.09.017
74. Martinez VG, Pankova V, Krasny L, Singh T, Makris S, White IJ, et al. Fibroblastic Reticular Cells Control Conduit Matrix Deposition during Lymph Node Expansion. Cell Rep (2019). doi: 10.1016/j.celrep.2019.10.103
75. Anderson ND, Anderson AO, Wyllie RG. Microvascular changes in lymph nodes draining skin allografts. Am J Pathol (1975).
76. Soderberg KA, Payne GW, Sato A, Medzhitov R, Segal SS, Iwasaki A. Innate control of adaptive immunity via remodeling of lymnh node feed arteriole. Proc Natl Acad Sci U S A (2005). doi: 10.1073/pnas.0506190102
77. Anderson AO, Anderson ND. Lymphocyte emigration from high endothelial venules in rat lymph nodes. Immunol Commun (1976).
78. Tzeng T-C, Chyou S, Tian S, Webster B, Carpenter AC, Guaiquil VH, et al. CD11c hi Dendritic Cells Regulate the Re-establishment of Vascular Quiescence and Stabilization after Immune Stimulation of Lymph Nodes. J Immunol (2010). doi: 10.4049/jimmunol.0902914
79. Chyou S, Benahmed F, Chen J, Kumar V, Tian S, Lipp M, et al. Coordinated Regulation of Lymph Node Vascular–Stromal Growth First by CD11c + Cells and Then by T and B Cells. J Immunol (2011). doi: 10.4049/jimmunol.1101724
80. Kumar V, Dasoveanu DC, Chyou S, Tzeng TC, Rozo C, Liang Y, et al. A Dendritic-Cell-Stromal Axis Maintains Immune Responses in Lymph Nodes. Immunity (2015). doi: 10.1016/j.immuni.2015.03.015
81. Kataru RP, Jung K, Jang C, Yang H, Schwendener RA, Jung EB, et al. Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood (2009). doi: 10.1182/blood-2008-09-176776
82. Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S, et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood (2010). doi: 10.1182/blood-2009-12-257832
83. Benahmed F, Chyou S, Dasoveanu D, Chen J, Kumar V, Iwakura Y, et al. Multiple CD11c + Cells Collaboratively Express IL-1β To Modulate Stromal Vascular Endothelial Growth Factor and Lymph Node Vascular–Stromal Growth. J Immunol (2014). doi: 10.4049/jimmunol.1301765
84. White JR, Harris RA, Lee SR, Craigon MH, Binley K, Price T, et al. Genetic amplification of the transcriptional response to hypoxia as a novel means of identifying regulators of angiogenesis. Genomics (2004). doi: 10.1016/S0888-7543(03)00215-5
85. Zajac E, Schweighofer B, Kupriyanova TA, Juncker-Jensen A, Minder P, Quigley JP, et al. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood (2013). doi: 10.1182/blood-2013-05-501494
86. Eubank TD, Galloway M, Montague CM, Waldman WJ, Marsh CB. M-CSF Induces Vascular Endothelial Growth Factor Production and Angiogenic Activity From Human Monocytes. J Immunol (2003). doi: 10.4049/jimmunol.171.5.2637
87. Carmi Y, Voronov E, Dotan S, Lahat N, Rahat MA, Fogel M, et al. The Role of Macrophage-Derived IL-1 in Induction and Maintenance of Angiogenesis. J Immunol (2009). doi: 10.4049/jimmunol.0901511
88. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MPJ, Donners MMPC. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis (2014). doi: 10.1007/s10456-013-9381-6
89. Guo L, Akahori H, Harari E, Smith SL, Polavarapu R, Karmali V, et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J Clin Invest (2018). doi: 10.1172/JCI93025doi:10.1172/JCI93025
90. Abramsson A, Lindblom P, Betsholtz C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest (2003). doi: 10.1172/JCI200318549
91. Birner P, Stöckl J, Kalt R, Ullrich R, Caucig C. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am J Pathol (2002). doi: 10.1016/S0002-9440(10)64255-1
92. Huang S. Contributions of Stromal Metalloproteinase-9 to Angiogenesis and Growth of Human Ovarian Carcinoma in Mice. CancerSpectrum Knowl Environ (2002). doi: 10.1093/jnci/94.15.1134
93. Lewis JS, Landers RJ, Underwood JCE, Harris AL, Lewis CE. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J Pathol (2000). doi: 10.1002/1096-9896(2000)9999:9999::AID-PATH6873.0.CO;2-G
94. Tsutsui S, Yasuda K, Suzuki K, Tahara K, Higashi H, Era S. Macrophage infiltration and its prognostic implications in breast cancer: The relationship with VEGF expression and microvessel density. Oncol Rep (2005). doi: 10.3892/or.14.2.425
95. Riboldi E, Musso T, Moroni E, Urbinati C, Bernasconi S, Rusnati M, et al. Proangiogenic Properties of Alternatively Activated Dendritic Cells. J Immunol (2005). doi: 10.4049/jimmunol.175.5.2788
96. Webster B, Ekland EH, Agle LM, Chyou S, Ruggieri R, Lu TT. Regulation of lymph node vascular growth by dendritic cells. J Exp Med (2006). doi: 10.1084/jem.20052272
97. Salvi V, Vaira X, Gianello V, Vermi W, Bugatti M, Sozzani S, et al. TLR Signalling Pathways Diverge in Their Ability to Induce PGE2. Mediators Inflammation (2016). doi: 10.1155/2016/5678046
98. Browning JL, Allaire N, Ngam-Ek A, Notidis E, Hunt J, Perrin S, et al. Lymphotoxin-β receptor signaling is required for the homeostatic control of HEV differentiation and function. Immunity (2005). doi: 10.1016/j.immuni.2005.10.002
99. Moussion C, Girard JP. Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature (2011). doi: 10.1038/nature10540
100. Onder L, Danuser R, Scandella E, Firner S, Chai Q, Hehlgans T, et al. Endothelial cell–specific lymphotoxin-β receptor signaling is critical for lymph node and high endothelial venule formation. J Exp Med (2013). doi: 10.1084/jem.20121462
101. Gagliostro V, Seeger P, Garrafa E, Salvi V, Bresciani R, Bosisio D, et al. Pro-lymphangiogenic properties of IFN-γ-activated human dendritic cells. Immunol Lett (2016). doi: 10.1016/j.imlet.2016.03.008
102. Konno S, Eckman JA, Plunkett B, Li X, Berman JS, Schroeder J, et al. Interleukin-10 and Th2 cytokines differentially regulate osteopontin expression in human monocytes and dendritic cells. J Interf Cytokine Res (2006). doi: 10.1089/jir.2006.26.562
103. Ohki Y, Heissig B, Sato Y, Akiyama H, Zhu Z, Hicklin DJ, et al. Granulocyte colony-stimulating factor promotes neovascularization by releasing vascular endothelial growth factor from neutrophils. FASEB J (2005). doi: 10.1096/fj.04-3496fje
104. Schruefer R, Lutze N, Schymeinsky J, Walzog B. Human neutrophils promote angiogenesis by a paracrine feedforward mechanism involving endothelial interleukin-8. Am J Physiol Heart Circ Physiol (2005). doi: 10.1152/ajpheart.00237.2004
105. Gaudry M, Brégerie O, Andrieu V, El Benna J, Pocidalo MA, Hakim J. Intracellular pool of vascular endothelial growth factor in human neutrophils. Blood (1997). doi: 10.1182/blood.V90.10.4153
106. McHale C, Mohammed Z, Gomez G. Human skin-derived mast cells spontaneously secrete several angiogenesis-related factors. Front Immunol (2019). doi: 10.3389/fimmu.2019.01445
107. Tanaka A, Arai K, Kitamura Y, Matsuda H. Matrix metalloproteinase-9 production, a newly identified function of mast cell progenitors, is downregulated by c-kit receptor activation. Blood (1999). doi: 10.1182/blood.v94.7.2390.419k16_2390_2395
108. Detoraki A, Staiano RI, Granata F, Giannattasio G, Prevete N, de Paulis A, et al. Vascular endothelial growth factors synthesized by human lung mast cells exert angiogenic effects. J Allergy Clin Immunol (2009). doi: 10.1016/j.jaci.2009.01.044
109. Melillo RM, Guarino V, Avilla E, Galdiero MR, Liotti F, Prevete N, et al. Mast cells have a protumorigenic role in human thyroid cancer. Oncogene (2010). doi: 10.1038/onc.2010.348
110. Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A (2009). doi: 10.1073/pnas.0902280106
111. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia (United States) (2013). doi: 10.1593/neo.131748
112. Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, et al. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin Cancer Res (2017). doi: 10.1158/1078-0432.CCR-16-0387
113. Binsfeld M, Muller J, Lamour V, De Veirman K, De Raeve H, Bellahcène A, et al. Granulocytic myeloid-derived suppressor cells promote angiogenesis in the context of multiple myeloma. Oncotarget (2016). doi: 10.18632/oncotarget.9270
114. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell (2004). doi: 10.1016/j.ccr.2004.08.031
115. Yang CY, Vogt TK, Favre S, Scarpellino L, Huang HY, Tacchini-Cottier F, et al. Trapping of naive lymphocytes triggers rapid growth and remodeling of the fibroblast network in reactive murine lymph nodes. Proc Natl Acad Sci U S A (2014). doi: 10.1073/pnas.1312585111
116. Kwee BJ, Budina E, Najibi AJ, Mooney DJ. CD4 T-cells regulate angiogenesis and myogenesis. Biomaterials (2018). doi: 10.1016/j.biomaterials.2018.06.003
117. Freeman MR, Schneck FX, Niknejad K, Gagnon ML, Soker S, Klagsbrun M, et al. Peripheral Blood T Lymphocytes and Lymphocytes Infiltrating Human Cancers Express Vascular Endothelial Growth Factor: A Potential Role for T Cells in Angiogenesis. Cancer Res (1995).
118. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T reg cells. Nature (2011). doi: 10.1158/1538-7445.AM2012-308
119. Leung OM, Li J, Li X, Chan VW, Yang KY, Ku M, et al. Regulatory T Cells Promote Apelin-Mediated Sprouting Angiogenesis in Type 2 Diabetes. Cell Rep (2018). doi: 10.1016/j.celrep.2018.07.019
120. Hur J, Yang HM, Yoon CH, Lee CS, Park KW, Kim JH, et al. Identification of a novel role of T cells in postnatal vasculogenesis: Characterization of endothelial progenitor cell colonies. Circulation (2007). doi: 10.1161/CIRCULATIONAHA.107.694778
121. Manetti M, Pratesi S, Romano E, Bellando-Randone S, Rosa I, Guiducci S, et al. Angiogenic T cell expansion correlates with severity of peripheral vascular damage in systemic sclerosis. PLoS One (2017). doi: 10.1371/journal.pone.0183102
122. Shrestha B, Hashiguchi T, Ito T, Miura N, Takenouchi K, Oyama Y, et al. B Cell-Derived Vascular Endothelial Growth Factor A Promotes Lymphangiogenesis and High Endothelial Venule Expansion in Lymph Nodes. J Immunol (2010). doi: 10.4049/jimmunol.0903063
123. van de Veen W, Globinska A, Jansen K, Straumann A, Kubo T, Verschoor D, et al. A novel proangiogenic B cell subset is increased in cancer and chronic inflammation. Sci Adv (2020). doi: 10.1126/sciadv.aaz3559
124. Missiaen R, Mazzone M, Bergers G. The reciprocal function and regulation of tumor vessels and immune cells offers new therapeutic opportunities in cancer. Semin Cancer Biol (2018). doi: 10.1016/j.semcancer.2018.06.002
125. Hua Y, Bergers G. Tumors vs. Chronic Wounds: An Immune Cell’s Perspective. Front Immunol (2019). doi: 10.3389/fimmu.2019.02178
126. Lewis CE, Harney AS, Pollard JW. The Multifaceted Role of Perivascular Macrophages in Tumors. Cancer Cell (2016). doi: 10.1016/j.ccell.2016.07.009
128. Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, Yang YH, et al. Macrophage Migration Inhibitory Factor Induces Macrophage Recruitment via CC Chemokine Ligand 2. J Immunol (2006). doi: 10.4049/jimmunol.177.11.8072
129. Argyle D, Kitamura T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front Immunol (2018). doi: 10.3389/fimmu.2018.02629
130. Reinart N, Nguyen PH, Boucas J, Rosen N, Kvasnicka HM, Heukamp L, et al. Delayed development of chronic lymphocytic leukemia in the absence of macrophage migration inhibitory factor. Blood (2013). doi: 10.1182/blood-2012-05-431452
131. Hanna BS, Öztürk S, Seiffert M. Beyond bystanders: Myeloid cells in chronic lymphocytic leukemia. Mol Immunol (2019). doi: 10.1016/j.molimm.2017.11.014
132. Haderk F, Schulz R, Iskar M, Cid LL, Worst T, Willmund KV, et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci Immunol (2017). doi: 10.1126/sciimmunol.aah5509
133. Hanna BS, Mcclanahan F, Yazdanparast H, Zaborsky N, Kalter V, Rößner PM, et al. Depletion of CLL-associated patrolling monocytes and macrophages controls disease development and repairs immune dysfunction in vivo. Leukemia (2016). doi: 10.1038/leu.2015.305
134. Carey CD, Gusenleitner D, Lipschitz M, Roemer MGM, Stack EC, Gjini E, et al. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood (2017). doi: 10.1182/blood-2017-03-770719
135. Roemer MGM, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 genetic alterations define classical hodgkin lymphoma and predict outcome. J Clin Oncol (2016). doi: 10.1200/JCO.2016.66.4482
136. Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O’Donnell E, et al. Constitutive AP-1 activity and EBV infection induce PD-l1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin Cancer Res (2012). doi: 10.1158/1078-0432.CCR-11-1942
137. Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood (2018). doi: 10.1182/blood-2017-07-796342
138. Clear AJ, Lee AM, Calaminici M, Ramsay AG, Morris KJ, Hallam S, et al. Increased angiogenic sprouting in poor prognosis FL is associated with elevated numbers of CD163+ macrophages within the immediate sprouting microenvironment. Blood (2010). doi: 10.1182/blood-2009-11-253260
139. Shen L, Li H, Shi Y, Wang D, Gong J, Xun J, et al. M2 tumour-associated macrophages contribute to tumour progression via legumain remodelling the extracellular matrix in diffuse large B cell lymphoma. Sci Rep (2016). doi: 10.1038/srep30347
140. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2017). doi: 10.1146/annurev-physiol-022516-034339
141. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014). doi: 10.1016/j.immuni.2014.07.009
142. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer (2017). doi: 10.1038/nrc.2017.51
143. Barnett FH, Rosenfeld M, Wood M, Kiosses WB, Usui Y, Marchetti V, et al. Macrophages form functional vascular mimicry channels in vivo. Sci Rep (2016). doi: 10.1038/srep36659
144. Schmeisser A, Garlichs CD, Zhang H, Eskafi S, Graffy C, Ludwig J, et al. Monocytes coexpress endothelial and macrophagocytic lineage markers and form cord-like structures in Matrigel® under angiogenic conditions. Cardiovasc Res (2001). doi: 10.1016/S0008-6363(00)00270-4
145. Lewis CE, De Palma M, Naldini L. Tie2-expressing monocytes and tumor angiogenesis: Regulation by hypoxia and angiopoietin-2. Cancer Res (2007). doi: 10.1158/0008-5472.CAN-07-1684
146. He H, Mack JJ, Güç E, Warren CM, Squadrito ML, Kilarski WW, et al. Perivascular Macrophages Limit Permeability. Arterioscler Thromb Vasc Biol (2016). doi: 10.1161/ATVBAHA.116.307592
147. Acton SE, Reis e Sousa C. Dendritic cells in remodeling of lymph nodes during immune responses. Immunol Rev (2016). doi: 10.1111/imr.12414
148. Bajénoff M, Granjeaud S, Guerder S. The strategy of T cell antigen-presenting cell encounter in antigen-draining lymph nodes revealed by imaging of initial T cell activation. J Exp Med (2003). doi: 10.1084/jem.20030167
149. Scholz F, Grau M, Menzel L, Graband A, Zapukhlyak M, Leutz A, et al. The transcription factor C/EBPβ orchestrates dendritic cell maturation and functionality under homeostatic and malignant conditions. Proc Natl Acad Sci (2020). http://www.pnas.org/content/early/2020/09/30/2008883117.abstract.
150. Rehm A, Gätjen M, Gerlach K, Scholz F, Mensen A, Gloger M, et al. Dendritic cell-mediated survival signals in EI 1/4-Myc B-cell lymphoma depend on the transcription factor C/EBPI 2. Nat Commun (2014). doi: 10.1038/ncomms6057
151. Kocieda VP, Adhikary S, Emig F, Yen JH, Toscano MG, Ganea D. Prostaglandin E2-induced IL-23p19 subunit is regulated by cAMP-responsive element-binding protein and C/AATT enhancer-binding protein β in bone marrow-derived dendritic cells. J Biol Chem (2012). doi: 10.1074/jbc.M112.402958
152. Salvi V, Vermi W, Gianello V, Lonardi S, Gagliostro V, Naldini A, et al. Dendritic cell-derived VEGF-A plays a role in inflammatory angiogenesis of human secondary lymphoid organs and is driven by the coordinated activation of multiple transcription factors. Oncotarget (2016). doi: 10.18632/oncotarget.9684
153. Fainaru O, Adini A, Benny O, Adini I, Short S, Bazinet L, et al. Dendritic cells support angiogenesis and promote lesion growth in a murine model of endometriosis. FASEB J (2008). doi: 10.1096/fj.07-9034com
154. Sozzani S, Rusnati M, Riboldi E, Mitola S, Presta M. Dendritic cell-endothelial cell cross-talk in angiogenesis. Trends Immunol (2007). doi: 10.1016/j.it.2007.07.006
155. Scimone ML, Lutzky VP, Zittermann SI, Maffia P, Jancic C, Buzzola F, et al. Migration of polymorphonuclear leucocytes is influenced by dendritic cells. Immunology (2005). doi: 10.1111/j.1365-2567.2005.02104.x
156. Hampton HR, Chtanova T. The lymph node neutrophil. Semin Immunol (2016). doi: 10.1016/j.smim.2016.03.008
157. Wang HW, Tedla N, Lloyd AR, Wakefield D, McNeil HP. Mast cell activation and migration to lymph nodes during induction of an immune response in mice. J Clin Invest (1998). doi: 10.1172/JCI3704
158. Schruefer R, Sulyok S, Schymeinsky J, Peters T, Scharffetter-Kochanek K, Walzog B. The proangiogenic capacity of polymorphonuclear neutrophils delineated by microarray technique and by measurement of neovascularization in wounded skin of CD18-deficient mice. J Vasc Res (2005). doi: 10.1159/000088975
159. Ancelin M, Chollet-Martin S, Hervé MA, Legrand C, El Benna J, Perrot-Applanat M. Vascular endothelial growth factor VEGF189 induces human neutrophil chemotaxis in extravascular tissue via an autocrine amplification mechanism. Lab Investig (2004). doi: 10.1038/labinvest.3700053
160. Gätjen M, Brand F, Grau M, Gerlach K, Kettritz R, Westermann J, et al. Splenic marginal zone granulocytes acquire an accentuated neutrophil B-cell helper phenotype in chronic lymphocytic leukemia. Cancer Res (2016). doi: 10.1158/0008-5472.CAN-15-3486
161. Rabenhorst A, Schlaak M, Heukamp LC, Förster A, Theurich S, Von Bergwelt-Baildon M, et al. Mast cells play a protumorigenic role in primary cutaneous lymphoma. Blood (2012). doi: 10.1182/blood-2012-03-415638
162. Vyzoukaki R, Tsirakis G, Pappa CA, Devetzoglou M, Tzardi M, Alexandrakis MG. The Impact of Mast Cell Density on the Progression of Bone Disease in Multiple Myeloma Patients. Int Arch Allergy Immunol (2016). doi: 10.1159/000443275
163. Franco G, Guarnotta C, Frossi B, Piccaluga PP, Boveri E, Gulino A, et al. Bone marrow stroma CD40 expression correlates with inflammatory mast cell infiltration and disease progression in splenic marginal zone lymphoma. Blood (2014). doi: 10.1182/blood-2013-04-497271
164. Bradding P, Okayama Y, Howarth PH, Church MK, Holgate ST. Heterogeneity of human mast cells based on cytokine content. J Immunol (1995).
165. Irani AA, Schechter NM, Craig SS, DeBlois G, Schwartz LB. Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci U S A (1986). doi: 10.1073/pnas.83.12.4464
166. Ronca R, Tamma R, Coltrini D, Ruggieri S, Presta M, Ribatti D. Fibroblast growth factor modulates mast cell recruitment in a murine model of prostate cancer. Oncotarget (2017). doi: 10.18632/oncotarget.19773
167. Varricchi G, Granata F, Loffredo S, Genovese A, Marone G. Angiogenesis and lymphangiogenesis in inflammatory skin disorders. J Am Acad Dermatol (2015). doi: 10.1016/j.jaad.2015.03.041
168. Ostrand-Rosenberg S, Sinha P. Myeloid-Derived Suppressor Cells: Linking Inflammation and Cancer. J Immunol (2009). doi: 10.4049/jimmunol.0802740
169. Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells Down-Regulate L-Selectin Expression on CD4 + and CD8 + T Cells. J Immunol (2009). doi: 10.4049/jimmunol.0804253
170. Parker KH, Sinha P, Horn LA, Clements VK, Yang H, Li J, et al. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res (2014). doi: 10.1158/0008-5472.CAN-13-2347
171. MacDonald KPA, Rowe V, Clouston AD, Welply JK, Kuns RD, Ferrara JLM, et al. Cytokine Expanded Myeloid Precursors Function as Regulatory Antigen-Presenting Cells and Promote Tolerance through IL-10-Producing Regulatory T Cells. J Immunol (2005). doi: 10.4049/jimmunol.174.4.1841
172. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res (2008). doi: 10.1158/0008-5472.CAN-07-6621
173. Alkasalias T, Moyano-Galceran L, Arsenian-Henriksson M, Lehti K. Fibroblasts in the tumor microenvironment: Shield or spear? Int J Mol Sci (2018). doi: 10.3390/ijms19051532
174. Gunes EG, Rosen ST, Querfeld C. The role of myeloid-derived suppressor cells in hematologic malignancies. Curr Opin Oncol (2020). doi: 10.1097/CCO.0000000000000662
175. Bruno A, Mortara L, Baci D, Noonan DM, Albini A. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front Immunol (2019). doi: 10.3389/fimmu.2019.00771
176. Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res (2004). doi: 10.1158/0008-5472.CAN-04-0757
177. Huang B, Lei Z, Zhao J, Gong W, Liu J, Chen Z, et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett (2007). doi: 10.1016/j.canlet.2006.12.012
178. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009). doi: 10.1038/nri2506
179. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer (2008). doi: 10.1038/nrc2444
180. Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget (2015). doi: 10.18632/oncotarget.4393
181. Ferrara N. Role of myeloid cells in vascular endothelial growth factor-independent tumor angiogenesis. Curr Opin Hematol (2010). doi: 10.1097/MOH.0b013e3283386660
182. Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol (2018). doi: 10.1038/nrclinonc.2018.9
183. Kumar BV, Connors TJ, Farber DL. Human T Cell Development, Localization, and Function throughout Life. Immunity (2018). doi: 10.1016/j.immuni.2018.01.007
184. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol (2020). doi: 10.1038/s41577-020-0306-5
185. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (2016). doi: 10.1126/science.aaf1490
186. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med (2019). doi: 10.1101/648899
187. Sasse S, Reddemann K, Diepstra A, Oschlies I, Schnitter A, Borchmann S, et al. Programmed cell death protein-1 (PD-1)-expression in the microenvironment of classical hodgkin lymphoma at relapse during anti-PD-1-treatment. Haematologica (2019). doi: 10.3324/haematol.2018.196279
188. Chen Y-B, Armand P, Redd RA, Bsat J, Merryman RW, Coleman K, et al. PD-1 Blockade for Diffuse Large B-Cell Lymphoma after Autologous Stem Cell Transplantation. Blood (2018). doi: 10.1182/blood-2018-99-114914
189. Ciciola P, Cascetta P, Bianco C, Formisano L, Bianco R. Combining Immune Checkpoint Inhibitors with Anti-Angiogenic Agents. J Clin Med (2020). doi: 10.3390/jcm9030675
190. Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, Erickson J, et al. Expression and regulation of the PD-L1 immunoinhibitory molecule on micro vascular endothelial cells. Microcirculation (2002). doi: 10.1080/713774061
191. Mazanet MM, Hughes CCW. B7-H1 Is Expressed by Human Endothelial Cells and Suppresses T Cell Cytokine Synthesis. J Immunol (2002). doi: 10.4049/jimmunol.169.7.3581
192. Cole JE, Navin TJ, Cross AJ, Goddard ME, Alexopoulou L, Mitra AT, et al. Unexpected protective role for Toll-like receptor 3 in the arterial wall. Proc Natl Acad Sci U S A (2011). doi: 10.1073/pnas.1018515108
193. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4+ T Cells Regulate Pulmonary Metastasis of Mammary Carcinomas by Enhancing Protumor Properties of Macrophages. Cancer Cell (2009). doi: 10.1016/j.ccr.2009.06.018
194. Hata T, Takahashi M, Hida S, Kawaguchi M, Kashima Y, Usui F, et al. Critical role of Th17 cells in inflammation and neovascularization after ischaemia. Cardiovasc Res (2011). doi: 10.1093/cvr/cvq397
195. Hoechst B, Gamrekelashvili J, Manns MP, Greten TF, Korangy F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood (2011). doi: 10.1182/blood-2010-11-317321
196. Ishida T, Ishii T, Inagaki A, Yano H, Komatsu H, Iida S, et al. Specific recruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res (2006). doi: 10.1158/0008-5472.CAN-06-0261
197. Cavallo F, Quaglino E, Cifaldi L, Di Carlo E, André A, Bernabei P, et al. Interleukin 12-activated lymphocytes influence tumor genetic programs. Cancer Res (2001).
198. Strasly M, Cavallo F, Geuna M, Mitola S, Colombo MP, Forni G, et al. IL-12 Inhibition of Endothelial Cell Functions and Angiogenesis Depends on Lymphocyte-Endothelial Cell Cross-Talk. J Immunol (2001). doi: 10.4049/jimmunol.166.6.3890
199. Lucas ED, Finlon JM, Burchill MA, McCarthy MK, Morrison TE, Colpitts TM, et al. Type 1 IFN and PD-L1 Coordinate Lymphatic Endothelial Cell Expansion and Contraction during an Inflammatory Immune Response. J Immunol (2018). doi: 10.4049/jimmunol.1800271
200. Lucas ED, Tamburini BAJ. Lymph node lymphatic endothelial cell expansion and contraction and the programming of the immune response. Front Immunol (2019). doi: 10.3389/fimmu.2019.00036
201. Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, et al. Tumour ischaemia by interferon-γ resembles physiological blood vessel regression. Nature (2017). doi: 10.1038/nature22311
202. Shafer-Weaver KA, Anderson MJ, Stagliano K, Malyguine A, Greenberg NM, Hurwitz AA. Cutting Edge: Tumor-Specific CD8 + T Cells Infiltrating Prostatic Tumors Are Induced to Become Suppressor Cells. J Immunol (2009). doi: 10.4049/jimmunol.0900848
203. Yu Y, Ma X, Gong R, Zhu J, Wei L, Yao J. Recent advances in CD8+ regulatory t cell research (Review). Oncol Lett (2018). doi: 10.3892/ol.2018.8378
204. Kasakovski D, Zeng X, Xu L, Li Y. Increasing CD28-FOXP3+CD8+ Treg and Senescent CD8+NK2GA+Eomes+ NK-like T Cells in Peripheral Blood of Patients with Multiple Myeloma. Blood (2018). doi: 10.1182/blood-2018-99-118088
205. Machicote A, Belén S, Baz P, Billordo LA, Fainboim L. Human CD8+HLA-DR+Regulatory T Cells, similarly to classical CD4+Foxp3+cells, suppress immune responses via PD-1/PD-L1 axis. Front Immunol (2018). doi: 10.3389/fimmu.2018.02788
206. Manetti M, Pratesi S, Romano E, Bellando-Randone S, Rosa I, Guiducci S, et al. Angiogenic T cell expansion correlates with severity of peripheral vascular damage in systemic sclerosis. PloS One (2017). doi: 10.1136/annrheumdis-2017-eular.3017
207. Igreja C, Courinha M, Cachaço AS, Pereira T, Cabeçadas J, Da Silva MG, et al. Characterization and clinical relevance of circulating and biopsy-derived endothelial progenitor cells in lymphoma patients. Haematologica (2007). doi: 10.3324/haematol.10723
208. Wiessman M, Leshem D, Yeshurun M, Yavin H, Iakobishvilli Z, Raanani P, et al. Dysfunctional endothelial progenitor cells in patients with Hodgkin’s lymphoma in complete remission. Cancer Med (2019). doi: 10.1002/cam4.1914
209. Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV, Chudakov DM. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat Rev Immunol (2020). doi: 10.1038/s41577-019-0257-x
210. Yang C, Lee H, Pal S, Jove V, Deng J, Zhang W, et al. B Cells Promote Tumor Progression via STAT3 Regulated-Angiogenesis. PloS One (2013). doi: 10.1371/journal.pone.0064159
211. Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S, et al. FcRγ Activation Regulates Inflammation-Associated Squamous Carcinogenesis. Cancer Cell (2010). doi: 10.1016/j.ccr.2009.12.019
212. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature (2011). doi: 10.1038/nature10144
213. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci (2020). doi: 10.1007/s00018-019-03351-7
214. Ganjoo K, An C, Robertson M, Gordon L, Sen J, Weisenbach J, et al. Rituximab, Bevacizumab and CHOP (RA-CHOP) in untreated diffuse large B-cell lymphoma: Safety, biomarker and pharmacokinetic analysis. Leuk Lymphoma (2006). doi: 10.1080/10428190600563821
215. Seymour JF, Pfreundschuh M, Trněný M, Sehn LH, Catalano J, Csinady E, et al. R-CHOP with or without bevacizumab in patients with previously untreated diffuse large B-cell lymphoma: Final MAIN study outcomes. Haematologica (2014). doi: 10.3324/haematol.2013.100818
216. Vacca A, Ribatti D, Roncali L, Dammacco F. Angiogenesis in b cell lymphoproliferative diseases. Biological and clinical studies. Leuk Lymphoma (1995). doi: 10.3109/10428199509054750
217. Jiang L, Li N. B-cell non-Hodgkin lymphoma: importance of angiogenesis and antiangiogenic therapy. Angiogenesis (2020). doi: 10.1007/s10456-020-09729-7
218. Hattori H. Caution should be taken in using CD31 for distinguishing the vasculature of lymph nodes. J Clin Pathol (2003). doi: 10.1136/jcp.56.8.638-a
219. Rakocevic J, Orlic D, Mitrovic-Ajtic O, Tomasevic M, Dobric M, Zlatic N, et al. Endothelial cell markers from clinician’s perspective. Exp Mol Pathol (2017). doi: 10.1016/j.yexmp.2017.02.005
220. Asabella AN, Di Palo A, Altini C, Ferrari C, Rubini G. Multimodality imaging in tumor angiogenesis: Present status and perspectives. Int J Mol Sci (2017). doi: 10.3390/ijms18091864
221. Yang J, Li W, He X, Zhang G, Yue L, Chai Y. VEGF overexpression is a valuable prognostic factor for non-Hodgkin’s lymphoma evidence from a systemic meta-analysis. Dis Markers (2015). doi: 10.1155/2015/786790
222. Paydas S, Seydaoglu G, Ergin M, Erdogan S, Yavuz S. The prognostic significance of VEGF-C and VEGF-A in non-Hodgkin lymphomas. Leuk Lymphoma (2009). doi: 10.1080/10428190802706665
223. Koster A, Van Krieken JHJM, MacKenzie MA, Schraders M, Borm GF, Van Der Laak JAWM, et al. Increased vascularization predicts favorable outcome in follicular lymphoma. Clin Cancer Res (2005).
224. Chen H, Treweeke AT, West DC, Till KJ, Cawley JC, Zuzel M, et al. In vitro and in vivo production of vascular endothelial growth factor by chronic lymphocytic leukemia cells. Blood (2000). doi: 10.1182/blood.V96.9.3181.h8003181_3181_3187
225. Salven P, Orpana A, Teerenhovi L, Joensuu H. Simultaneous elevation in the serum concentrations of the angiogenic growth factors VEGF and bFGF is an independent predictor of poor prognosis in non-Hodgkin lymphoma: A single-institution study of 200 patients. Blood (2000). doi: 10.1182/blood.V96.12.3712.h8003712_3712_3718
226. Wang ES, Teruya-Feldstein J, Wu Y, Zhu Z, Hicklin DJ, Moore MAS. Targeting autocrine and paracrine VEGF receptor pathways inhibits human lymphoma xenografts in vivo. Blood (2004). doi: 10.1182/blood-2004-01-0226
227. Nowak-Sliwinska P, Alitalo K, Allen E, Anisimov A, Aplin AC, Auerbach R, et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis (2018). doi: 10.1007/s10456-018-9613-x
230. Chiron D, Bellanger C, Papin A, Tessoulin B, Dousset C, Maiga S, et al. Rational targeted therapies to overcome microenvironment-dependent expansion of mantle cell lymphoma. Blood (2016). doi: 10.1182/blood-2016-06-720490
231. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H TJ. “WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues.” in WHO Classification of Tumours, Revised 4th Edition, vol. 2. (2017). doi:10.1038/jhg.2010.42
232. Petrakis G, Veloza L, Clot G, Gine E, Gonzalez-Farre B, Navarro A, et al. Increased tumour angiogenesis in SOX11-positive mantle cell lymphoma. Histopathology (2019). doi: 10.1111/his.13935
233. Annese T, Ingravallo G, Tamma R, De Giorgis M, Maiorano E, Perrone T, et al. Inflammatory Infiltrate and Angiogenesis in Mantle Cell Lymphoma. Transl Oncol (2020). doi: 10.1016/j.tranon.2020.100744
234. Palomero J, Vegliante MC, Rodríguez M, Planas E, Ribera I, Cid MC, et al. SOX11 Regulates Angiogenesis In Mantle Cell Lymphoma. Blood (2013). doi: 10.1182/blood.V122.21.246.246
235. Palomero J, Vegliante MC, Rodríguez ML, Eguileor Á, Castellano G, Planas-Rigol E, et al. SOX11 promotes tumor angiogenesis through transcriptional regulation of PDGFA in mantle cell lymphoma. Blood (2014). doi: 10.1182/blood-2014-04-569566
236. Kuo PY, Jatiani SS, Rahman AH, Edwards D, Jiang Z, Ahr K, et al. Sox11 augments bcr signaling to drive mcl-like tumor development. Blood (2018). doi: 10.1182/blood-2018-02-832535
237. Vegliante MC, Palomero J, Pérez-Galán P, Roué G, Castellano G, Navarro A, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood (2013). doi: 10.1182/blood-2012-06-438937
238. Balsas P, Palomero J, Eguileor Á, Rodriguez ML, Vegliante MC, Planas-Rigol E, et al. SOX11 promotes tumor protective microenvironment interactions through CXCR4 and FAK regulation in mantle cell lymphoma. Blood (2017). doi: 10.1182/blood-2017-04-776740
239. Koh YW, Shin SJ, Park C, Yoon DH, Suh C, Huh J. Absolute monocyte count predicts overall survival in mantle cell lymphomas: Correlation with tumour-associated macrophages. Hematol Oncol (2014). doi: 10.1002/hon.2106
240. Song K, Herzog BH, Sheng M, Fu J, McDaniel M, Ruan J, et al. Lenalidomide inhibits lymphangiogenesis in preclinical models of mantle cell lymphoma. Cancer Res (2013). doi: 10.1158/0008-5472.CAN-13-0750
241. Papin A, Tessoulin B, Bellanger C, Moreau A, Le Bris Y, Maisonneuve H, et al. CSF1R and BTK inhibitions as novel strategies to disrupt the dialog between mantle cell lymphoma and macrophages. Leukemia (2019). doi: 10.1038/s41375-019-0463-3
242. Ek S, Björck E, Högerkorp CM, Nordenskjöld M, Porwit-MacDonald A, Borrebaeck CAK. Mantle cell lymphomas acquire increased expression of CCL4, CCL5 and 4-1BB-L implicated in cell survival. Int J Cancer (2006). doi: 10.1002/ijc.21579
243. Nygren L, Wasik AM, Baumgartner-Wennerholm S, Jeppsson-Ahlberg Å, Klimkowska M, Andersson P, et al. T-cell levels are prognostic in mantle cell lymphoma. Clin Cancer Res (2014). doi: 10.1158/1078-0432.CCR-14-0889
244. Ruan J, Coleman M, Furman RR, Glynn P, Joyce M, Ketas J, et al. Targeting Angiogenesis in Mantle Cell Lymphoma: Clinical Efficacy and Correlative Studies of a Phase II Trial of RT-PEPC (Rituximab, Thalidomide and Metronomic Oral Chemotherapy with Prednisone, Etoposide, Procarbazine and Cyclophosphamide) in Relapsed/Re. Blood (2006). doi: 10.1182/blood.V108.11.2751.2751
245. Ruan J, Martin P, Shah B, Schuster SJ, Smith SM, Furman RR, et al. Lenalidomide plus rituximab as initial treatment for mantle-cell Lymphoma. N Engl J Med (2015). doi: 10.1056/NEJMoa1505237
246. Wang M, Fayad L, Wagner-Bartak N, Zhang L, Hagemeister F, Neelapu SS, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: A phase 1/2 clinical trial. Lancet Oncol (2012). doi: 10.1016/S1470-2045(12)70200-0
247. Monga N, Nastoupil L, Garside J, Quigley J, Hudson M, O’Donovan P, et al. Burden of illness of follicular lymphoma and marginal zone lymphoma. Ann Hematol (2019). doi: 10.1007/s00277-018-3501-8
248. Muppidi JR, Schmitz R, Green JA, Xiao W, Larsen AB, Braun SE, et al. Loss of signalling via G α 13 in germinal centre B-cell-derived lymphoma. Nature (2014). doi: 10.1038/nature13765
249. González-Rincón J, Méndez M, Gómez S, García JF, Martín P, Bellas C, et al. Unraveling transformation of follicular lymphoma to diffuse large B-cell lymphoma. PloS One (2019). doi: 10.1371/journal.pone.0212813
250. Aguzzi A, Kranich J, Krautler NJ. Follicular dendritic cells: Origin, phenotype, and function in health and disease. Trends Immunol (2014). doi: 10.1016/j.it.2013.11.001
251. Pikor NB, Mörbe U, Lütge M, Gil-Cruz C, Perez-Shibayama C, Novkovic M, et al. Remodeling of light and dark zone follicular dendritic cells governs germinal center responses. Nat Immunol (2020). doi: 10.1038/s41590-020-0672-y
252. He B, Chadburn A, Jou E, Schattner EJ, Knowles DM, Cerutti A. Lymphoma B Cells Evade Apoptosis through the TNF Family Members BAFF/BLyS and APRIL. J Immunol (2004). doi: 10.4049/jimmunol.172.8.5128-c
253. Heinig K, Gätjen M, Grau M, Stache V, Anagnostopoulos I, Gerlach K, et al. Access to follicular dendritic cells is a pivotal step in murine chronic lymphocytic leukemia b-cell activation and proliferation. Cancer Discovery (2014). doi: 10.1158/2159-8290.CD-14-0096
254. Kurshumliu F, Sadiku-Zehri F, Qerimi A, Vela Z, Jashari F, Bytyci S, et al. Divergent immunohistochemical expression of CD21 and CD23 by follicular dendritic cells with increasing grade of follicular lymphoma. World J Surg Oncol (2019). doi: 10.1186/s12957-019-1659-8
255. Matas-Céspedes A, Rodriguez V, Kalko SG, Vidal-Crespo A, Rosich L, Casserras T, et al. Disruption of follicular dendritic cells-follicular lymphoma cross-talk by the pan-PI3K inhibitor BKM120 (buparlisib). Clin Cancer Res (2014). doi: 10.1158/1078-0432.CCR-14-0154
256. Serrat N, Guerrero-Hernández M, Matas-Céspedes A, Yahiaoui A, Valero JG, Nadeu F, et al. PI3Kδ inhibition reshapes follicular lymphoma-immune microenvironment cross talk and unleashes the activity of venetoclax. Blood Adv (2020) 4(17):4217–31. doi: 10.1182/bloodadvances.2020001584
257. Amé-Thomas P, Le Priol J, Yssel H, Caron G, Pangault C, Jean R, et al. Characterization of intratumoral follicular helper T cells in follicular lymphoma: Role in the survival of malignant B cells. Leukemia (2012). doi: 10.1038/leu.2011.301
258. Amin R, Mourcin F, Uhel F, Guirriec M, Lamy T, Tarte K. DC-SIGN Binds Preferentially Highly Glycosylated IgM to Trigger Classical BCR Signaling in Follicular Lymphoma. Blood (2014). doi: 10.1182/blood.V124.21.2968.2968
259. Kridel R, Xerri L, Gelas-Dore B, Tan K, Feugier P, Vawda A, et al. The prognostic impact of CD163-positive macrophages in follicular Lymphoma: A study from the BC cancer agency and the lymphoma study association. Clin Cancer Res (2015). doi: 10.1158/1078-0432.CCR-14-3253
260. Arias V, Soares FA. Vascular density (tumor angiogenesis) in non-Hodgkin’s lymphomas and florid follicular hyperplasia: A morphometric study. Leuk Lymphoma (2001). doi: 10.3109/10428190009054893
261. Crivellato E, Nico B, Vacca A, Ribatti D. B-cell non-Hodgkin’s lymphomas express heterogeneous patterns of neovascularization. Haematologica (2003).
262. Ribatti D, Vacca A, Nico B, Fanelli M, Roncali L, Dammacco F. Angiogenesis spectrum in the stroma of B-cell non-Hodgkin’s lymphomas. An immunohistochemical and ultrastructural study. Eur J Haematol (2009). doi: 10.1111/j.1600-0609.1996.tb00293.x
263. Shahini L, Gašparov S, Petruševska G, Kerliu SM, Veselaj F, Kurshumliu F, et al. Clinical significance of VEGF-A and microvessel density in diffuse large B-cell lymphoma and low-grade follicular lymphoma. Acta Clin Croat (2017). doi: 10.20471/acc.2017.56.04.02
264. Passalidou E, Stewart M, Trivella M, Steers G, Pillai G, Dogan A, et al. Vascular patterns in reactive lymphoid tissue and in non-Hodgkin’s lymphoma. Br J Cancer (2003). doi: 10.1038/sj.bjc.6600742
265. Hainsworth JD, Greco FA, Raefsky EL, Thompson DS, Lunin S, Reeves J, et al. Rituximab with or without bevacizumab for the treatment of patients with relapsed follicular lymphoma. Clin Lymphoma Myeloma Leuk (2014). doi: 10.1016/j.clml.2014.02.010
266. Abramson JS, Shipp MA. Advances in the biology and therapy of diffuse large B-cell lymphoma: Moving toward a molecularly targeted approach. Blood (2005). doi: 10.1182/blood-2005-02-0687
267. Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med (2008). doi: 10.1056/NEJMoa0802885
268. Abdou AG, Asaad N, Kandil M, Shabaan M, Shams A. Significance of stromal-1 and stromal-2 signatures and biologic prognostic model in diffuse large B-cell lymphoma. Cancer Biol Med (2017). doi: 10.20892/j.issn.2095-3941.2017.0007
269. Perry AM, Cardesa-Salzmann TM, Meyer PN, Colomo L, Smith LM, Fu K, et al. A new biologic prognostic model based on immunohistochemistry predicts survival in patients with diffuse large B-cell lymphoma. Blood (2012). doi: 10.1182/blood-2012-05-430389
270. Marinaccio C, Ingravallo G, Gaudio F, Perrone T, Nico B, Maoirano E, et al. Microvascular density, CD68 and tryptase expression in human Diffuse Large B-Cell Lymphoma. Leuk Res (2014). doi: 10.1016/j.leukres.2014.09.007
271. Cardesa-Salzmann TM, Colomo L, Gutierrez G, Chan WC, Weisenburger D, Climent F, et al. High microvessel density determines a poor outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus chemotherapy. Haematologica (2011). doi: 10.3324/haematol.2010.037408
272. Tzankov A, Heiss S, Ebner S, Sterlacci W, Schaefer G, Augustin F, et al. Angiogenesis in nodal B cell lymphomas: A high throughput study. J Clin Pathol (2007). doi: 10.1136/jcp.2006.038661
273. Gomez-Gelvez JC, Salama ME, Perkins SL, Leavitt M, Inamdar KV. Prognostic impact of tumor microenvironment in diffuse large B-cell lymphoma uniformly treated with R-CHOP chemotherapy. Am J Clin Pathol (2016). doi: 10.1093/ajcp/aqw034
274. Hazar B, Paydas S, Zorludemir S, Sahin B, Tuncer I. Prognostic significance of microvessel density and vascular endothelial growth factor (VEGF) expression in non-Hodgkin’s lymphoma. Leuk Lymphoma (2003). doi: 10.1080/1042819031000123519
275. Foss HD, Araujo I, Demel G, Klotzbach H, Hummel M, Stein H. Expression of vascular endothelial growth factor in lymphomas and Castleman’s disease. J Pathol (1997). doi: 10.1002/(SICI)1096-9896(199709)183:1<44::AID-PATH1103>3.0.CO;2-I
276. Ribatti D, Vacca A, Bertossi M, De Benedictis G, Roncali L, Dammacco F. Angiogenesis induced by B-cell non-Hodgkin’s lymphomas. Lack of correlation with tumor malignancy and immunologic phenotype. Anticancer Res (1990).
277. Stewart M, Talks K, Leek R, Turley H, Pezzella F, Harris A, et al. Expression of angiogenic factors and hypoxia inducible factors HIF 1, HIF 2 and CA IX in non-Hodgkin’s lymphoma. Histopathology (2002). doi: 10.1046/j.1365-2559.2002.01357.x
278. Suhasini AN, Wang L, Holder KN, Lin AP, Bhatnagar H, Kim SW, et al. A phosphodiesterase 4B-dependent interplay between tumor cells and the microenvironment regulates angiogenesis in B-cell lymphoma. Leukemia (2016). doi: 10.1038/leu.2015.302
279. Broséus J, Mourah S, Ramstein G, Bernard S, Mounier N, Cuccuini W, et al. VEGF121, is predictor for survival in activated B-cell-like diffuse large B-cell lymphoma and is related to an immune response gene signature conserved in cancers. Oncotarget (2017). doi: 10.18632/oncotarget.19385
280. Fiore F, von Bergwelt-Baildon MS, Drebber U, Beyer M, Popov A, Manzke O, et al. Dendritic cells are significantly reduced in non-Hodgkin’s lymphoma and express less CCR7 and CD62L. Leuk Lymphoma (2006). doi: 10.1080/10428190500360971
281. Cai QC, Liao H, Lin SX, Xia Y, Wang XX, Gao Y, et al. High expression of tumor-infiltrating macrophages correlates with poor prognosis in patients with diffuse large B-cell lymphoma. Med Oncol (2012). doi: 10.1007/s12032-011-0123-6
282. Li YL, Shi ZH, Wang X, Gu KS, Zhai ZM. Tumor-associated macrophages predict prognosis in diffuse large B-cell lymphoma and correlation with peripheral absolute monocyte count. BMC Cancer (2019). doi: 10.1186/s12885-019-6208-x
283. Hasselblom S, Hansson U, Sigurdardottir M, Nilsson-Ehle H, Ridell B, Andersson PO. Expression of CD68+ tumor-associated macrophages in patients with diffuse large B-cell lymphoma and its relation to prognosis. Pathol Int (2008). doi: 10.1111/j.1440-1827.2008.02268.x
284. Marchesi F, Cirillo M, Bianchi A, Gately M, Olimpieri OM, Cerchiara E, et al. High density of CD68+/CD163+ tumour-associated macrophages (M2-TAM) at diagnosis is significantly correlated to unfavorable prognostic factors and to poor clinical outcomes in patients with diffuse large B-cell lymphoma. Hematol Oncol (2015). doi: 10.1002/hon.2142
285. Fukushima H, Ohsawa M, Ikura Y, Naruko T, Sugama Y, Suekane T, et al. Mast cells in diffuse large B-cell lymphoma; their role in fibrosis. Histopathology (2006). doi: 10.1111/j.1365-2559.2006.02534.x
286. Hedström G, Berglund M, Molin D, Fischer M, Nilsson G, Thunberg U, et al. Mast cell infiltration is a favourable prognostic factor in diffuse large B-cell lymphoma. Br J Haematol (2007). doi: 10.1111/j.1365-2141.2007.06612.x
287. Ribatti D, Vacca A, Marzullo A, Nico B, Ria R, Roncali L, et al. Angiogenesis and mast cell density with tryptase activity increase simultaneously with pathological progression in B-cell non-Hodgkin’s lymphomas. Int J Cancer (2000). doi: 10.1002/(SICI)1097-0215(20000115)85:2%3C171::AID-IJC4%3E3.0.CO;2-W
288. Wroblewski M, Bauer R, Cubas Córdova M, Udonta F, Ben-Batalla I, Legler K, et al. Mast cells decrease efficacy of anti-angiogenic therapy by secreting matrix-degrading granzyme B. Nat Commun (2017). doi: 10.1038/s41467-017-00327-8
289. Ribatti D, Crivellato E. Mast cells, angiogenesis, and tumour growth. Biochim Biophys Acta - Mol Basis Dis (2012). doi: 10.1007/978-1-4614-5857-9_7
290. Valtola R, Salven P, Heikkilä P, Taipale J, Joensuu H, Rehn M, et al. VEGFR-3 and its ligand VEGF-C are associated with angiogenesis in breast cancer. Am J Pathol (1999). doi: 10.1016/S0002-9440(10)65392-8
291. Nowakowski GS, LaPlant B, Macon WR, Reeder CB, Foran JM, Nelson GD, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-cell lymphoma: A phase II study. J Clin Oncol (2015). doi: 10.1200/JCO.2014.55.5714
292. Hartert KT, Wenzl K, Krull JE, Manske M, Sarangi V, Asmann Y, et al. Targeting of inflammatory pathways with R2CHOP in high-risk DLBCL. Leukemia (2020). doi: 10.1038/s41375-020-0766-4
293. Kalisz K, Alessandrino F, Beck R, Smith D, Kikano E, Ramaiya NH, et al. An update on Burkitt lymphoma: a review of pathogenesis and multimodality imaging assessment of disease presentation, treatment response, and recurrence. Insights Imaging (2019). doi: 10.1186/s13244-019-0733-7
294. Teruya-Feldstein J, Jaffe ES, Burd PR, Kanegane H, Kingma DW, Wilson WH, et al. The role of Mig, the monokine induced by interferon-γ, and IP-10, the interferon-γ-inducible protein-10, in tissue necrosis and vascular damage associated with Epstein-Barr virus-positive lymphoproliferative disease. Blood (1997). doi: 10.1182/blood.V90.10.4099
295. Becker J, Covelo-Fernandez A, Von Bonin F, Kube D, Wilting J. Specific tumor-stroma interactions of EBV-positive Burkitt’s lymphoma cells in the chick chorioallantoic membrane. Vasc Cell (2012). doi: 10.1186/2045-824X-4-3
296. Vacca A, Ribatti D, Ruco L, Giacchetta F, Nico B, Quondamatteo F, et al. Angiogenesis extent and macrophage density increase simultaneously with pathological progression in B-cell non-Hodgkin’s lymphomas. Br J Cancer (1999). doi: 10.1038/sj.bjc.6690154
297. Ribatti D, Nico B, Vacca A, Marzullo A, Calvi N, Roncali L, et al. Do mast cells help to induce angiogenesis in B-cell non-Hodgkin’s lymphomas? Br J Cancer (1998). doi: 10.1038/bjc.1998.316
298. Brandvold KA, Neiman P, Ruddell A. Angiogenesis is an early event in the generation of myc-induced lymphomas. Oncogene (2000). doi: 10.1038/sj.onc.1203589
299. Ruan J, Hyjek E, Kermani P, Christos PJ, Hooper AT, Coleman M, et al. Magnitude of stromal hemangiogenesis correlates with histologic subtype of non-Hodgkin’s lymphoma. Clin Cancer Res (2006). doi: 10.1158/1078-0432.CCR-06-1204
300. Ruan J, Hyjek E, Hooper AT, Vincent L, Kermani P, Christos PJ, et al. Stromal Incorporation of VEGFR-1+, CD68+ and α-SMA+ Hemangiogenic Cells Correlates with Histologic Subtype in Non-Hodgkin’s Lymphoma. Blood (2005). doi: 10.1182/blood.V106.11.1930.1930
301. De Palma M, Coukos G, Hanahan D. A New Twist on Radiation Oncology: Low-Dose Irradiation Elicits Immunostimulatory Macrophages that Unlock Barriers to Tumor Immunotherapy. Cancer Cell (2013). doi: 10.1016/j.ccr.2013.10.019
302. Marinaccio C, Nico B, Maiorano E, Specchia G, Ribatti D. Insights in Hodgkin Lymphoma angiogenesis. Leukemia Res (2014). doi: 10.1016/j.leukres.2014.05.023
303. Korkolopoulou P, Thymara I, Kavantzas N, Vassilakopoulos TP, Angelopoulou MK, Kokoris SI, et al. Angiogenesis in Hodgkin’s lymphoma: A morphometric approach in 286 patients with prognostic implications. Leukemia (2005). doi: 10.1038/sj.leu.2403690
304. Niitsu N, Okamoto M, Nakamine H, Yoshino T, Tamaru JI, Nakamura S, et al. Simultaneous elevation of the serum concentrations of vascular endothelial growth factor and interleukin-6 as independent predictors of prognosis in aggressive non-Hodgkin’s lymphoma. Eur J Haematol (2002). doi: 10.1034/j.1600-0609.2002.01609.x
305. Ben Arush MW, Ben Barak A, Maurice S, Livne E. Serum VEGF as a significant marker of treatment response in Hodgkin lymphoma. Pediatr Hematol Oncol (2007). doi: 10.1080/08880010601052381
306. Kuittinen O, Soini Y, Turpeenniemi-Hujanen T. Diverse role of MMP-2 and MMP-9 in the clinicopathological behavior of Hodgkin’s lymphoma. Eur J Haematol (2002). doi: 10.1034/j.1600-0609.2002.02751.x
307. Bardelli M, Leucci E, Schürfeld K, Bellan C, Passiatore G, Rocchigiani M, et al. VEGF-D is expressed in activated lymphoid cells and in tumors of hematopoietic and lymphoid tissues. Leuk Lymphoma (2007). doi: 10.1080/10428190701540975
308. Linke F, Harenberg M, Nietert MM, Zaunig S, Von Bonin F, Arlt A, et al. Microenvironmental interactions between endothelial and lymphoma cells: A role for the canonical WNT pathway in Hodgkin lymphoma. Leukemia (2017). doi: 10.1038/leu.2016.232
309. Celegato M, Borghese C, Casagrande N, Mongiat M, Kahle XU, Paulitti A, et al. Preclinical activity of the repurposed drug auranofin in classical Hodgkin lymphoma. Blood (2015). doi: 10.1182/blood-2015-07-660365
310. Fhu CW, Graham AM, Yap CT, Al-Salam S, Castella A, Chong SM, et al. Reed-Sternberg cell-derived lymphotoxin-a activates endothelial cells to enhance T-cell recruitment in classical Hodgkin lymphoma. Blood (2014). doi: 10.1182/blood-2014-05-576140
311. Werner L, Dreyer JH, Hartmann D, Barros MHM, Büttner-Herold M, Grittner U, et al. Tumor-associated macrophages in classical Hodgkin lymphoma: hormetic relationship to outcome. Sci Rep (2020). doi: 10.1038/s41598-020-66010-z
312. Al Sayed Ahmed H, Raslan WF, Deifalla AHS, Fathallah MD. CD163 is a predictive biomarker for prognosis of classical Hodgkin’s lymphoma in Saudi patients. Mol Clin Oncol (2019). doi: 10.3892/mco.2019.1850
313. Koh YW, Park CS, Yoon DH, Suh C, Huh J. CD163 Expression was associated with angiogenesis and shortened survival in patients with uniformly treated classical Hodgkin lymphoma. PloS One (2014). doi: 10.1371/journal.pone.0087066
314. Locatelli SL, Careddu G, Serio S, Consonni FM, Maeda A, Viswanadha S, et al. Dual PI3K/inhibition suppresses M2 macrophage polarization in hodgkin lymphoma through PKM2 downregulation. Haematologica (2018). doi: 10.1158/1078-0432.CCR-18-1133
315. Locatelli SL, Careddu G, Serio S, Consonni FM, Maeda A, Viswanadha S, et al. Targeting cancer cells and tumor microenvironment in preclinical and clinical models of Hodgkin lymphoma using the dual PI3Kd/G inhibitor RP6530. Clin Cancer Res (2019). doi: 10.1158/1078-0432.CCR-18-1133
316. Cao Y, Langer R. A review of Judah Folkman’s remarkable achievements in biomedicine. Proc Natl Acad Sci U S A (2008). doi: 10.1073/pnas.0806582105
317. Sherwood LM, Parris EE, Folkman J. Tumor Angiogenesis: Therapeutic Implications. New Engl J Med (1971). doi: 10.1056/NEJM197111182852108
318. Haibe Y, Kreidieh M, El Hajj H, Khalifeh I, Mukherji D, Temraz S, et al. Resistance Mechanisms to Anti-angiogenic Therapies in Cancer. Front Oncol (2020). doi: 10.3389/fonc.2020.00221
319. Rosen LS, Jacobs IA, Burkes RL. Bevacizumab in Colorectal Cancer: Current Role in Treatment and the Potential of Biosimilars. Target Oncol (2017). doi: 10.1007/s11523-017-0518-1
320. Summers J, Cohen MH, Keegan P, Pazdur R. FDA Drug Approval Summary: Bevacizumab plus Interferon for Advanced Renal Cell Carcinoma. Oncologist (2010). doi: 10.1634/theoncologist.2009-0250
321. Winkler F, Osswald M, Wick W. Anti-Angiogenics: Their Role in the Treatment of Glioblastoma. Oncol Res Treat (2018). doi: 10.1159/000488258
322. Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. Lancet (2016). doi: 10.1016/S0140-6736(15)01088-0
323. Shaked Y, Henke E, Roodhart JML, Mancuso P, Langenberg MHG, Colleoni M, et al. Rapid Chemotherapy-Induced Acute Endothelial Progenitor Cell Mobilization: Implications for Antiangiogenic Drugs as Chemosensitizing Agents. Cancer Cell (2008). doi: 10.1016/j.ccr.2008.08.001
324. Shaked Y, Ciarrocchi A, Franco M, Lee CR, Man S, Cheung AM, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science (2006). doi: 10.1126/science.1127592
325. Park JS, Kim IK, Han S, Park I, Kim C, Bae J, et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell (2016). doi: 10.1016/j.ccell.2016.10.018
326. Mattheolabakis G, Mikelis CM. Nanoparticle Delivery and Tumor Vascular Normalization: The Chicken or The Egg? Front Oncol (2019). doi: 10.3389/fonc.2019.01227
327. Wang L, Peng S, Sun W, Liu X. Bevacizumab synergises with the BCL 2 inhibitor venetoclax to effectively treat B-cell non-Hodgkin’s lymphoma. Eur J Haematol (2019). doi: 10.1111/ejh.13279
328. Zhang Q, Cao J, Xue K, Liu X, Ji D, Guo Y, et al. Recombinant human endostatin in combination with CHOP regimen for peripheral T cell lymphoma. Onco Targets Ther (2017). doi: 10.2147/OTT.S117007
329. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol (2018). doi: 10.3389/fimmu.2018.00978
330. Azad AK, Zhabyeyev P, Vanhaesebroeck B, Eitzen G, Oudit GY, Moore RB, et al. Inactivation of endothelial cell phosphoinositide 3-kinase β inhibits tumor angiogenesis and tumor growth. Oncogene (2020). doi: 10.1038/s41388-020-01444-3
331. Lannutti BJ, Meadows SA, Herman SEM, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood (2011). doi: 10.1182/blood-2010-03-275305
332. Ali K, Soond DR, Piñeiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature (2014). doi: 10.1038/nature13444
333. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J Immunol (2017). doi: 10.4049/jimmunol.1601515
334. von Keudell G, Moskowitz AJ. The Role of PI3K Inhibition in Lymphoid Malignancies. Curr Hematol Malig Rep (2019). doi: 10.1007/s11899-019-00540-w
335. Wullenkord R, Friedrichs B, Erdmann T, Lenz G. Therapeutic potential of PI3K signaling in distinct entities of B-cell lymphoma. Expert Rev Hematol (2019). doi: 10.1080/17474086.2019.1676716
336. Yang Q, Modi P, Newcomb T, Quéva C, Gandhi V. Idelalisib: First-in-class PI3K delta inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma. Clin Cancer Res (2015). doi: 10.1158/1078-0432.CCR-14-2034
337. Sapon-Cousineau V, Sapon-Cousineau S, Assouline S. PI3K Inhibitors and Their Role as Novel Agents for Targeted Therapy in Lymphoma. Curr Treat Options Oncol (2020). doi: 10.1007/s11864-020-00746-8
338. Tang H, Zhu M, Qiao J, Fu Y-X. Lymphotoxin signalling in tertiary lymphoid structures and immunotherapy. Cell Mol Immunol (2017) 14(10):809–18. doi: 10.1038/cmi.2017.13
339. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-Dose Irradiation Programs Macrophage Differentiation to an iNOS+/M1 Phenotype that Orchestrates Effective T Cell Immunotherapy. Cancer Cell (2013). doi: 10.1016/j.ccr.2013.09.014
340. Ruan J. Antiangiogenic Therapies in Non-Hodgkin’s Lymphoma. Curr Cancer Drug Targets (2011). doi: 10.2174/156800911798073014
341. Vieth B, Parekh S, Ziegenhain C, Enard W, Hellmann I. A systematic evaluation of single cell RNA-seq analysis pipelines. Nat Commun (2019). doi: 10.1101/583013
342. Tang X, Huang Y, Lei J, Luo H, Zhu X. The single-cell sequencing: New developments and medical applications. Cell Biosci (2019). doi: 10.1186/s13578-019-0314-y
343. Strzelecka PM, Ranzoni AM, Cvejic A. Dissecting human disease with single-cell omics: Application in model systems and in the clinic. Dis Models Mech (2018). doi: 10.1242/dmm.036525
344. Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol (2019). doi: 10.15252/msb.20188746
345. Arnol D, Schapiro D, Bodenmiller B, Saez-Rodriguez J, Stegle O. Modeling Cell-Cell Interactions from Spatial Molecular Data with Spatial Variance Component Analysis. Cell Rep (2019). doi: 10.1101/265256
346. Solomon M, DeLay M, Reynaud D. Phenotypic Analysis of the Mouse Hematopoietic Hierarchy Using Spectral Cytometry: From Stem Cell Subsets to Early Progenitor Compartments. Cytometry A (2020). doi: 10.1002/cyto.a.24041
347. Ferrer-Font L, Pellefigues C, Mayer JU, Small SJ, Jaimes MC, Price KM. Panel Design and Optimization for High-Dimensional Immunophenotyping Assays Using Spectral Flow Cytometry. Curr Protoc Cytom (2020). doi: 10.1101/784884
348. Corey DM, Rinkevich Y, Weissman IL. Dynamic patterns of clonal evolution in tumor vasculature underlie alterations in lymphocyte-endothelial recognition to foster tumor immune escape. Cancer Res (2016). doi: 10.1158/0008-5472.CAN-15-1150
349. Huyghe L, Van Parys A, Cauwels A, Van Lint S, De Munter S, Bultinck J, et al. Safe eradication of large established tumors using neovasculature-targeted tumor necrosis factor-based therapies. EMBO Mol Med (2020). doi: 10.15252/emmm.201911223
350. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov (2015). doi: 10.1158/2159-8290.CD-15-1020
351. Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood (2016). doi: 10.1182/blood-2015-08-665547
352. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol (2019). doi: 10.1038/s41571-019-0184-6
353. John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res (2013). doi: 10.1158/1078-0432.CCR-13-0458
354. Wei J, Luo C, Wang Y, Guo Y, Dai H, Tong C, et al. PD-1 silencing impairs the anti-tumor function of chimeric antigen receptor modified T cells by inhibiting proliferation activity. J Immunother Cancer (2019). doi: 10.1186/s40425-019-0685-y
355. Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Mol Ther Oncolytics (2018). doi: 10.1016/j.omto.2017.12.003
356. Štach M, Musil J, Cetkovsky P, Otahal P. Interleukin 21 Enhances Survival and Expansion of CAR T Cells Via Inhibition of Their Terminal Differentiation during Interaction with Tumor Target Cells. Blood (2018). doi: 10.1182/blood-2018-99-116294
Keywords: lymphoma, B cell malignancy, angiogenesis, lymph node, tumor microenvironment, reactive endothelium, lymphocyte trafficking, high endothelial venule
Citation: Menzel L, Höpken UE and Rehm A (2020) Angiogenesis in Lymph Nodes Is a Critical Regulator of Immune Response and Lymphoma Growth. Front. Immunol. 11:591741. doi: 10.3389/fimmu.2020.591741
Received: 05 August 2020; Accepted: 19 October 2020;
Published: 03 December 2020.
Edited by:
Martina Seiffert, German Cancer Research Center (DKFZ), GermanyReviewed by:
Tanja Nicole Hartmann, University of Freiburg Medical Center, GermanyCopyright © 2020 Menzel, Höpken and Rehm. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lutz Menzel, bHV0ei5tZW56ZWxAbWRjLWJlcmxpbi5kZQ==; Armin Rehm, YXJlaG1AbWRjLWJlcmxpbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.