
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 10 February 2021
Sec. Cytokines and Soluble Mediators in Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.590749
This article is part of the Research TopicRole of the IL-23/IL-17 Pathway in Chronic Immune-Mediated Inflammatory Diseases: Mechanisms and Targeted TherapiesView all 21 articles
 Sabrina Porta1
Sabrina Porta1 Matilde Otero-Losada2
Matilde Otero-Losada2 Rodolfo A. Kölliker Frers1,2
Rodolfo A. Kölliker Frers1,2 Vanesa Cosentino1
Vanesa Cosentino1 Eduardo Kerzberg1
Eduardo Kerzberg1 Francisco Capani2,3,4*
Francisco Capani2,3,4*Psoriatic arthritis is a chronic inflammatory disease with skin and joint pathology as the dominant characteristics. Scientific evidence supports its systemic nature and relevant relationship with obesity, metabolic syndrome, and associated conditions. Metabolic syndrome and obesity share common signaling pathways with joint inflammation, reinforcing the idea that adipose tissue is a major contributor to disease development and severity. The adipose tissue is not a mere energy store but also an endocrine organ participating in the immune response. In the search for the best therapeutic strategy for a patient, we should appraise the adipose tissue as an endocrine and immune organ responsible for mild chronic inflammation. Today, our challenge is not only to achieve disease remission but to control the associated comorbidities as well. In light of the high prevalence of obesity in psoriatic arthritis patients and the importance of the adipose tissue in the development of chronic inflammation, we aimed to identify the most relevant articles in this regard published in English until June 2020 using the PubMed database. Search terms included psoriatic arthritis, in combination with metabolic syndrome, obesity, adipokines, cardiovascular disease, and treatment. This review summarizes the current evidence regarding the role of adipose tissue as an adipokine-secreting endocrine organ, discussing its influence on disease development and severity, and ultimately in meeting successful disease management.
Psoriasis is a chronic inflammatory disease mainly compromising skin with systemic involvement associated with various comorbidities (1). With 6% to 40% percent of psoriatic patients experiencing psoriatic arthritis (PsA) (2), it equally affects men and women, starting usually at the age of 40 (3). The first signs appear on the skin usually 10 years before arthritis is diagnosed (4, 5).
Rheumatoid factor and cyclic citrullinated anti-peptide are absent, so PsA is considered a seronegative arthritis. Five PsA phenotypes may be distinguished based on the articular compromise pattern: predominantly axial, symmetric polyarthritis, asymmetric oligoarthritis, predominantly distal interphalangeal damage, and mutilating arthritis (6, 7).
Two distinguishing features of the disease are inflammation at the tendon and ligament insertions in the bone or enthesitis, and dactylitis when inflammation affects one finger entirely, risking the tendon sheath (8, 9).
The diagnosis of PsA is mainly clinical, reckoning on the classification criteria for psoriatic arthritis (CASPAR) (10, 11) (CASPAR) (10, 11).
Besides synovitis, enthesitis, dactylitis, and spondylitis, PsA typically presents non-musculoskeletal manifestations as anterior uveitis and inflammatory bowel disease. Comorbidities like obesity, cardiovascular disease, metabolic syndrome (MetS), and its components (obesity, diabetes mellitus, hypertension, and dyslipidemia) are also more frequent than in the general population (12).
Although the etiopathogenesis of PsA is multifactorial and not entirely known, there is an interaction between genetic susceptibility factors, mainly human leukocyte antigens (HLA) B27, together with other HLA-B loci and the HLA-Cw * 0602 allele) and environmental triggers (such as mechanical stress, dysbiosis, trauma, smoking, or infection), leading to dysregulation of immunoinflammatory pathways and the development of the disease (3).
In the last decades, a substantial body of evidence indicates that cutaneous psoriasis and psoriatic arthritis patients are at higher risk of developing cardiovascular disease. It is known that adipose tissue is metabolically active and an important source of inflammatory adipokines. Obesity and atherogenesis are low-grade inflammatory diseases and a link with associated PsA. Inflammatory mediators of obesity may represent one added key cardio-metabolic risk factor in PsA subjects (13).
The prevalence of immune-mediated diseases like psoriasis has increased in industrialized countries in the last decades, likely accountable to environmental factors, considering that the genetic predisposition has not changed (14). Western lifestyle, sedentary, with high fat and carbohydrate diets, and excessive sodium consumption favor the development of overweight and obesity (15).
In light of the high prevalence of obesity in psoriatic arthritis patients (16) and the importance of the adipose tissue in the development of chronic inflammation, we aimed to identify the most relevant articles in this regard published in English until June 2020 using the PubMed database. Search terms included psoriatic arthritis, in combination with metabolic syndrome, obesity, adipokines, cardiovascular disease, and treatment.
This review summarizes the current understanding of the role of the adipose tissue as an adipokine-secreting endocrine organ, discussing its influence on disease development and severity. Ultimately, robust evidence acknowledges considering the relevance of the adipose tissue in meeting successful disease management.
Multiple factors participate in PsA development in an as yet not fully understood fashion. Some environmental factors like infections or mechanical stress give way to a genetically predisposed terrain, activating both the innate and adaptive immune systems. Genetic variants the major histocompatibility complex molecules and inflammatory mediators like the IL-12B gene, which encodes the p40 subunit of IL-23 and IL-12, the IL-23A gene, encoding for the p19 subunit of IL-23, and the IL-23 receptor gene (IL-23R), which encodes a common domain of IL-23R and IL-12R could favor the IL-23/IL17 axis activation, predisposing to the disease (17).
Chronic infections have raised great interest. In dysbiosis, microbiome proliferation, distorting its balance with the immune system, aberrant activation of the latter could favor cells recruitment, T helper (Th) 17 lymphocytes, in particular (17, 18). Activated Th17 lymphocytes might migrate from the intestinal mucosa and lymph nodes to the skin and joint tissue, causing local inflammation.
Repeated trauma causes tissue damage at the enthesis, i.e., tendon, ligament, and joint capsule insertion to the bone, activating the innate immune system by Damage-Associated-Molecular-Patterns (DAMPs) recognition (17, 19).
The growing importance of entheses in PsA pathophysiology has encouraged research studies. The enthesis is no longer considered a simple focal insertion but referred to as the ‘organ of enthesis’ (20). A healthy enthesis is usually anti-inflammatory, unlike the synovial membrane, which is proinflammatory regarding cellular composition and structure. When a mechanically stressed enthesis is injured, the associated inflammatory reaction manifests prominently within the juxtaposed synovium. The “synovio-entheseal complex” concept supports that specific factors at the joint level are relevant as danger signals, activating innate immune responses (20).
Regarding the adaptive immune system, IL17 A and IL23 appear key in PsA development.
Interleukin-17 not only activates innate-immunity cells but epithelial and endothelial cells, keratinocytes, chondrocytes, osteoblasts, and osteoclasts as well. Its ability to increase the level of IL1-IL6, IL8, TNF-α, matrix metalloproteinase-9 (MMP-9), granulocyte-macrophage colony-stimulating factor (GM-CSF), inducible nitric oxide synthase (iNOS), and receptor activator of nuclear factor-kappa B (RANK) applies to its participation in the inflammatory cascade (17).
In psoriatic patients, IL-17 handles CCL2 aberrant expression on keratinocytes, which recruits inflammatory cells, leading to hyperkeratosis and cell dysfunction (17).
Interleukin-17 and IL-23 act synergically in joint injury development, playing complementary roles in PsA pathogenesis. The respective intracellular signaling cascades are different, although IL-23 activates Th17 lymphocytes.
Antibodies against the keratinocyte cytoskeleton and enthesis sites have been identified, posing new theories as to disease development (21), while TH2-type response participation in seronegative arthritis has not been classically considered.
Patients with PsA have increased cardiovascular risk, while the debate about the compromised metabolic pathways remains open (22). The so-called traditional cardiovascular risk factors, as hypertension, obesity, smoking, dyslipidemia, and metabolic syndrome, and hence the cardiovascular risk are increased in PsA patients (23, 24). Psoriasis is a systemic chronic inflammatory condition, where a high level of pro-inflammatory cytokines spreads beyond skin limits. Then, endothelial damage might be responsible for the increase of atherosclerotic disease in PsA patients (22). The ‘two plaques for one syndrome’ theory has been put forward. It states that a similar cytokine profile and inflammatory infiltrate is found in plaques both in skin psoriasis and atheroma (25).
We reported evidence supporting the contribution of classic cardiovascular risk factors and systemic inflammatory milieu in disease development. We found an increased level of C-reactive protein (CRP) and soluble intercellular adhesion molecule-1 (sICAM-1)in PsA patients without cardiovascular history or traditional risk factors compared with healthy subjects, the same as in recent-onset PsA patients, in the absence of cardiovascular risk factors (13).
Psoriatic arthritis is undeniably a systemic disease, well expressed by the concept of psoriatic disease, having joint compromise as the central and most relevant characteristic (26). Within the spectrum of extra-articular manifestations, MetS stands out. Over the years, the diagnostic criterion for MetS, a cluster of coexistent hypertension, obesity, insulin resistance, and dyslipidemia, has undergone changes, each of which posing per se cardiovascular risk (27) Joint inflammation and MetS share common signaling pathways, which result in cardiovascular disease (3, 28). Atherosclerosis, MetS, and PsA show a common pattern of T-cell activation. They increase cytokine production with a Th-1 profile as tumor necrosis alpha (TNF-α), interleukin 1 beta (IL1-β), interleukin 10 (IL-10), and interferon (IFN) (3, 29). These cytokines induce insulin resistance in skeletal muscle, increase hepatic synthesis of pro-coagulant factors, inhibit lipoprotein lipase, and increase fatty acid oxidation, and atherogenic lipoprotein serum level. Cytokine production, insulin resistance, and endothelial dysfunction favor the formation and deposition of atheroma plaques in a deleterious positive feedback cycle (30). Proinflammatory cytokines not only promote vascular wall recruitment of macrophages and T lymphocytes precipitating atheroma plaque formation and deposition but induce foam cell lysis. This rupture or plaque accident releases cellular debris and thrombi to circulation with the consequent at distance damage often presented as acute myocardial infarction and cerebrovascular accident. The relationship between joint inflammation and atherosclerosis has led to considering PsA a cardiovascular risk factor on its own (31). Furthermore, PsA patients having no cardiovascular risk factors showed increased carotid intima-media thickness (IMTc) compared with control subjects (32, 33). In 2015, Di Minno et al.’s meta-analysis reported a higher carotid IMT and prevalence of carotid plaques, and reduced flow-mediated dilatation in 898 PsA patients compared with 1140 control (34). Although lifestyle changes are the fundamental pillar in MetS management, pharmacological intervention is often necessary. However, confirmed PsA diagnosis should refocus the attention to the inflammatory activity since psoriasis intrinsically favors endothelial dysfunction, insulin resistance, and a prothrombotic environmental background. Accordingly, the adequate MetS control should also consider joint disease remission.
The complex, multifactorial relationship between obesity and psoriasis is not clear. Common pathophysiological mechanisms might account for pinning a vicious cycle that results in both. Whether one causes the other or they express different aspects of a same disorder is still undetermined.
While well-known factors may be accountable for arthritis development in PsA patients like severe skin compromise, nail compromise, genetic predisposition, and obesity, the latter has the advantage that can be changed.
Functional disability in PsA reduces physical activity, favoring weight gain, and, conversely, obesity prior to arthritis development is also true. This supports that chronic inflammation is likely involved in PsA development.
Two hypotheses explain how obesity favors PsA development in genetically predisposed subjects (35). One hypothesis proposes that adipose tissue acts as a source of inflammatory mediators like adipokines and proinflammatory cytokines, including TNF-alpha and IL-6 (30). The other one puts forward that overweight might would stress the enthesis due to the increase in the mechanical load with the following microtrauma, enticing an aberrant inflammatory response and PsA development (20). Obese patients had a higher prevalence of tuft resorption, Achilles and calcaneal spurs, and pelvic enthesitis compared with normal-weight subjects, reinforcing this hypothesis. Obesity was related to a late PsA onset, while normal-weight was associated with the HLA-B27 allele and an earlier onset of the disease (36).
Proinflammatory cytokines increased in obese patients are particularly involved in the pathophysiology of PsA and other inflammatory diseases.
Obesity is related to systemic inflammation and shares several pathways with PsA. Obesity, inflammatory status, and PsA are definitely related. A high level of inflammatory adipokines as found in obese patients may favor PsA expression in predisposed people.
Table 1 shows most of the few publications addressing the role of adipokines in PsA.
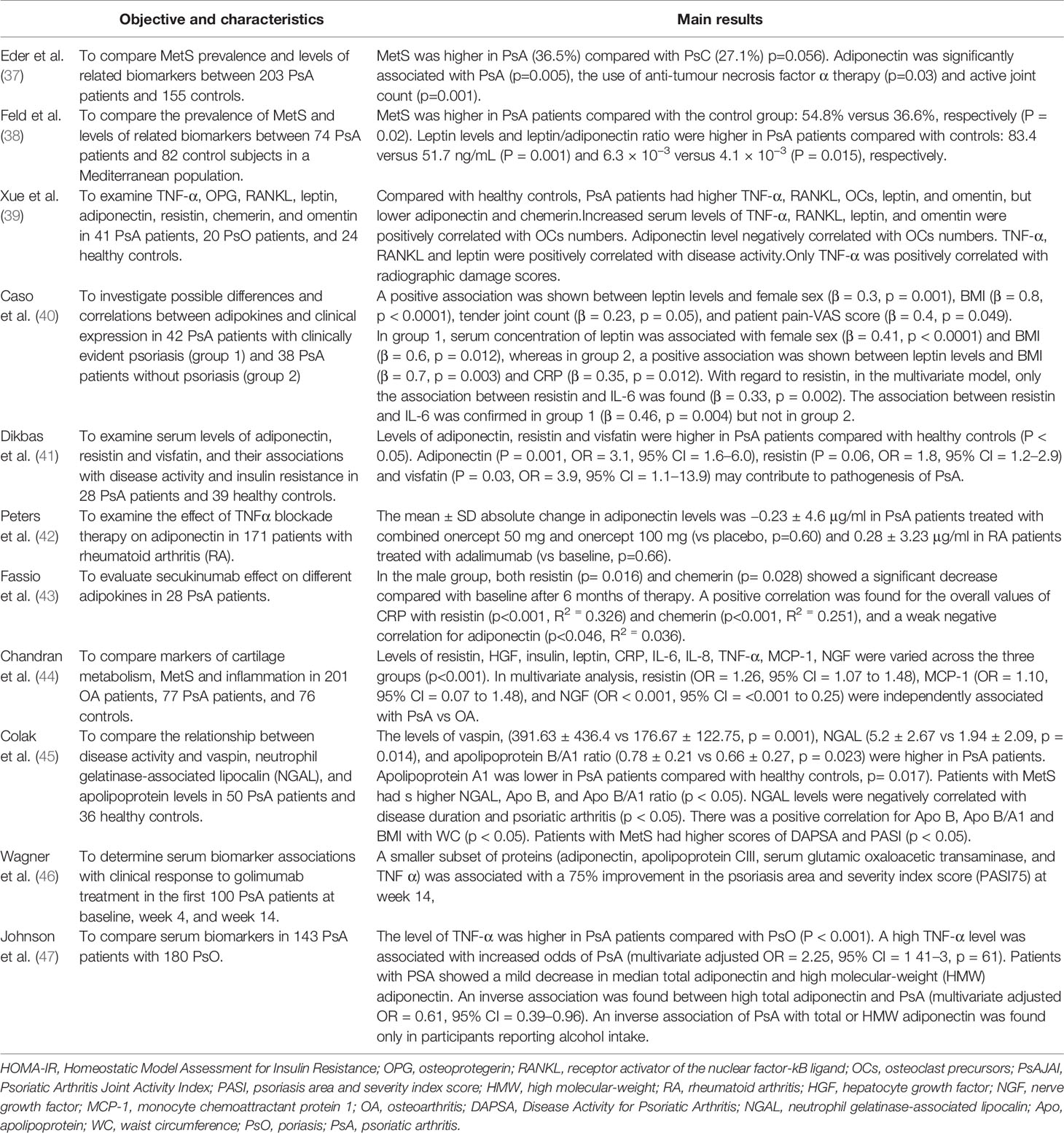
Table 1 Representative studies linking PsA with adipokines published between 2009 and 2019.
Despite certain aspects to be clarified, we can affirm that not only psoriasis increases the risk of developing obesity but also obesity is associated with higher prevalence and severity of psoriasis, even in the pediatric population (48, 49).
Studying 943 patients diagnosed with cutaneous psoriasis (CPs) showed BMI at 18 years was predictive of PsA. In the United Kingdom, the risk of developing PsA was studied based on the relationship between the BMI measured after the diagnosis of PsC and PsA development.
Compared with PsC patients with a BMI <25, those with a BMI of 25–29.9, 30.0–34.9 and ≥35.0 had age and sex-adjusted RRs of 1.1, 1.24, and 1.52, (p for trend <0.001) (50). One 14-year follow-up prospective study showed that BMI, weight change from early adulthood, waist circumference, hip circumference, and waist-hip ratio were associated with an increased risk of developing PsA in all the participants and in PsA women in particular (51).
Figure 1 shows the interaction between obesity, adipokines, in the development of psoriasis and psoriatic arthritis and cardiovascular disease.
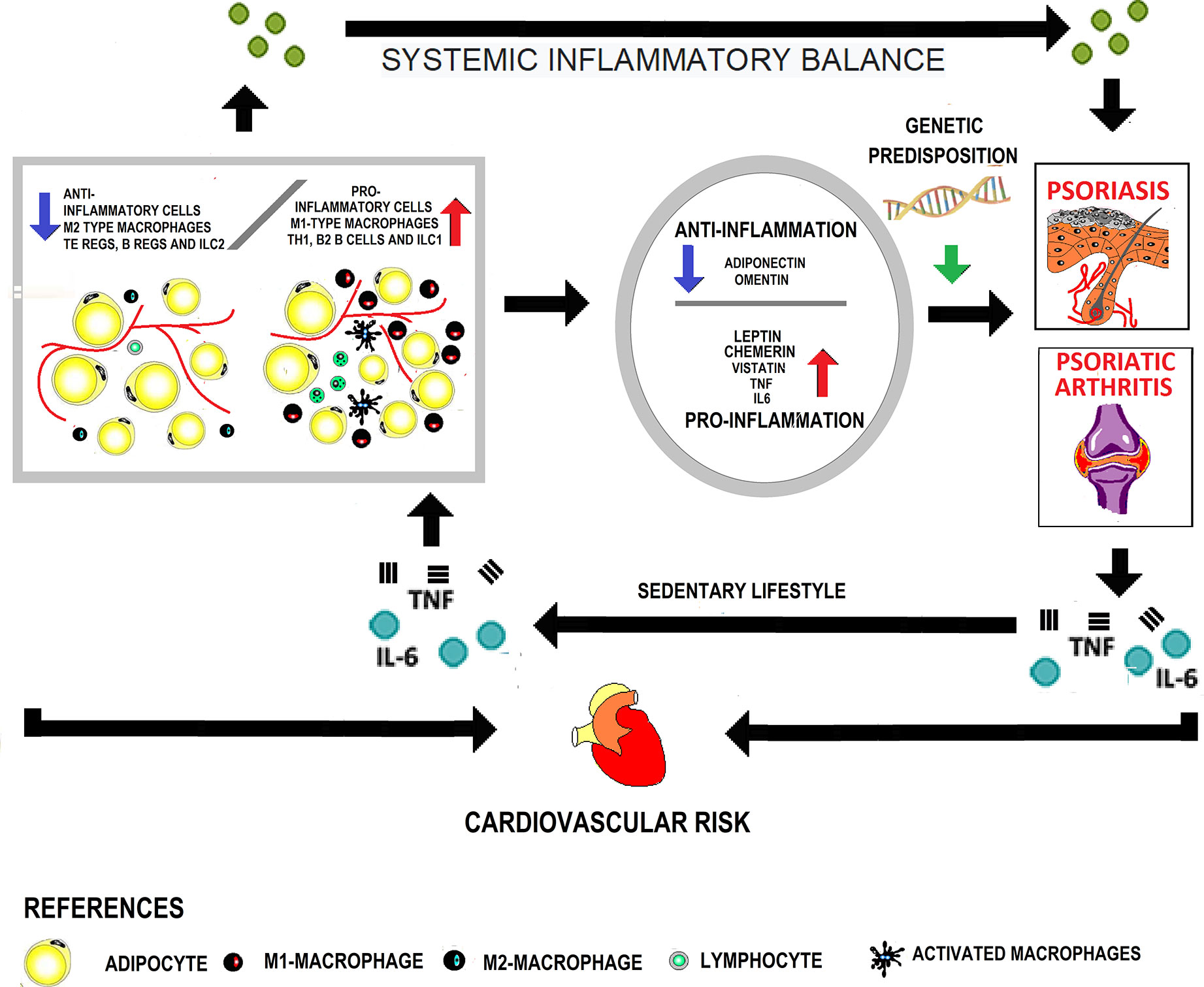
Figure 1 Interaction between obesity and adipokines in the development of psoriasis, psoriatic arthritis, and cardiovascular disease. In obese patients, the adipose tissue presents cellular changes due to greater infiltration of pro-inflammatory cells with a decrease in the anti-inflammatory cell population. In this way, it becomes dysfunctional and acts as an endocrine organ, increasing the secretion of pro-inflammatory adipokines. A systemic inflammatory state is generated that, in genetically predisposed subjects, favors the appearance of skin psoriasis. On the other hand, the increased mechanical load at the enthesis sites contributes to the onset of psoriatic arthritis. The inflammation characteristic of the disease added to the sedentary lifestyle secondary to joint involvement perpetuates the growth of adipose tissue. Finally, both due to the presence of classical and non-classical factors associated with obesity, cardiovascular risk increases.
Obesity is considered as having a body mass index (BMI) above 30 kg/m2 (52). Its deleterious impact on health has been widely studied, often embedded in the well-known MetS. Metabolic syndrome is a cluster of risk factors leading to microvascular dysfunction and chronic cerebral hypoperfusion, brought by the coexistence of diabetes type 2 or insulin resistance, abdominal obesity, hypertension, and dyslipidemia, mainly a low level of high-density lipoprotein-cholesterol, (HDL) (53–55).
Although weight gain and a high BMI imply an increase in adiposity, studying fat distribution is crucial given the many functions of adipose tissue.
Two types of adipose tissue may be recognized based on cellular structure, location, color, vascularity, and function. Newborns have brown adipose tissue, which has multilocular adipocytes with a large number of mitochondria. Brown adipose tissue is vital at this life stage when heat production is essential for survival. Adults show only white adipose tissue, which is basically an energy store made up of triglycerides within adipocytes (54).
The white adipose tissue presents different distribution patterns, traditionally named as android and gynoid. Android phenotype typically has fat centrally distributed mainly in the abdomen, chest, shoulder, and nape of the neck. Gynoid fat distribution refers to fat peripherally accumulated mainly at the buttocks and hips (54).
The android or gynoid distribution pattern depends on the subcutaneous adipose tissue which contributes to regulate body temperature acting as a thermal insulator.
However, there is another fraction of the white adipose tissue, the visceral adipose tissue, which occupies the space between the internal organs, allowing their proper juxtaposition.
There are important differences between subcutaneous and visceral fatty tissue, because of the expression of different genes related to insulin resistance, inflammation, and a different pattern of adipokines secretion (56).
These different secretion patterns have local repercussions involving autocrine or paracrine mechanisms and systemic ones. In this way visceral fat directly affects the liver, its increase being responsible for the development of insulin resistance, dyslipidemia, glucose intolerance and cardiovascular hypertension (54).
Adipose tissue is made up not only of adipocytes which represent one-third of total fat. Adipose tissue also comprises fibroblasts, macrophages, stroma cells, and pre-adipocytes (54) Then, adipose tissue is not a mere energy store but also an endocrine organ participating in the immune response (57).
Adipose tissue is responsible for synthesizing adipokines which form a heterogeneous group including hormones like leptin and adiponectin, cytokines like TNF-α, IL -6, omentin, visfatin, and other proteins like the plasminogen activator (PAI) -1, angiotensinogen and resistin.
The adipokines family includes anti-inflammatory and proinflammatory adipokines. Under normal metabolic conditions, they are balanced. However, their plasma profile changes in response to different challenges, like metabolic variations. A deficit in calorie supply during starvation causes that proinflammatory adipokines decrease, and anti-inflammatory adipokines increase.
The hormone leptin is produced mainly by adipocytes and regulates whole-body energy homeostasis. Fat mass and inflammatory status determine leptin levels (30).It reduces food intake and increases energy expenditure, acting directly on the hypothalamus and other brain regions (58). Obese patients typically show a high circulating level of leptin and leptin resistance (57) Leptin pro-angiogenic properties have been described directly related to endothelial dysfunction and atherosclerosis development (59). Leptin also modulates the immune response as an inflammatory molecule capable of activating adaptive immunity cells (57).
High level of leptin found in PsA patients, considering leptin properties, has suggested that leptin might be involved in bone erosion, activating osteoclasts in these patients (22) perhaps due to its cytokine-like structure (30). Leptin correlates with metabolic syndrome features and inflammation in patients with moderate-to-severe psoriasis (60).
Chemerin is involved in inflammation, adipogenesis, angiogenesis, and dyslipidemia. It participates in preadipocyte differentiation into adipocytes and in the hyperplasia and hypertrophy of mature adipocytes (61).
Visfatin is another adipokine related to abdominal obesity that increases pro-inflammatory factors in monocytes, promoting T lymphocyte activation. Correlation between visfatin concentration and insulin resistance, measured by the HOMA (Homeostatic Model Assessment for Insulin Resistance)-index was found in patients with ankylosing spondylitis, chronic inflammatory arthritis closely related to psoriatic arthritis (62). At the vascular level, visfatin upregulates vascular endothelial growth factor (VEGF) secretion and downregulates metalloproteins expression (63).
The increased cardiovascular risk in obese patients may be related to altered fibrinolysis. Adipocytes produce PAI-1, and obese patients have a high PAI level correlated with insulin resistance (64).
Both IL-6 and TNF-α promote insulin resistance inhibiting lipoprotein lipase (LPL), decreasing triglyceride hydrolysis into free fatty acids, and increasing triglyceride storage in adipocytes, resulting in larger fat cells (57). Besides, TNF-α inhibits preadipocytes conversion to mature adipocytes (65) and promotes atherosclerotic plaque formation, favoring leukocyte recruitment and activation, endothelial cell expression of adhesion molecules, and triggering arterial wall inflammatory cascade (30).
Unlike TNF-α, whose change in local production has not been confirmed, the expression of IL-6 in visceral fat was higher than in subcutaneous fat (30).
Adiponectin is exclusively synthesized and secreted by adipose tissue. Its protective role in the development of MetS and atherosclerosis has been suggested as it improves insulin sensitivity and fatty acid oxidation (63). Patients with chronic diseases show low adiponectin levels resulting from a decreased synthesis in adipose tissue after the increase in IL-6 and TNFα cytokines. A low adiponectin level is associated with insulin resistance, vasodilation, endothelial damage, and diastolic failure and with a high CRP level in obese patients (30). In addition, adiponectin receptor mRNA (AdipoR1/R2) expresses to a lesser extent as well (66). Adiponectin concentration was inversely correlated with triglycerides/HDL and total cholesterol/HDL cholesterol ratios, and with high fasting plasma glucose level in rheumatoid arthritis, the prototypical inflammatory arthritis. Therefore, in rheumatoid arthritis, low adiponectin level clustered with metabolic syndrome features implicated in accelerated atherosclerosis development (67).
Omentin, produced by the vascular stroma cells of the visceral adiposity, increases insulin sensitivity, stimulating insulin-mediated glucose uptake in human adipocytes (68).
In obese patients, adipokines synthesis and secretion are altered. Adipocytes are dysfunctional and increase the secretion of pro-inflammatory adipokines (69), triggering systemic mild chronic inflammation in the adipose tissue at first, then spreading away all through (57). Leptin secretion increases secondary to the increase in adipocyte lipid content. Though aimed to reduce caloric intake by acting at the central nervous system, leptin secretion leads to an imbalance at the adipose tissue. Accordingly, infiltration and accumulation of proinflammatory M1-type macrophages increase, replacing the normally found type 2 macrophages. Not only IL6 and TNF-α increase but insulin resistance develops as well. Accompanying M1 macrophages, proinflammatory cells (Th1, B2 B cells and ILC1) increase and anti-inflammatory immune cells (Tregs, Bregs, and ILC2) decrease in adipose tissue, perpetuating chronic inflammation (57). In sum, the adipose tissue drives an imbalance between anti-inflammatory and pro-inflammatory adipokines favoring the latter, increasing insulin resistance, propitiating diabetes, MetS, and cardiovascular disease.
Faced with this scene, two ways to improve the inflammatory state in adipose tissue and insulin sensitivity appear: weight loss and white adipose tissue gaining characteristics of brown adipose tissue in the so-called beiging process. The resulting beige or brite (brown in white) adipocytes (59) can burn lipids and restore normal insulin sensitivity, reducing the proinflammatory environment. Exposure to cold, β-adrenergic stimulation and some cytokines like IL33, IL13 and IL4 activate this mechanism.
In obesity, this process is slowed down, hampering insulin sensitivity restoration. Interleukin-17 is postulated to inhibit adipocyte differentiation and glucose absorption, and stem cell differentiation into adipocytes (57).
Although IL-17–producing T cells represent only 10% of the total immune cells in adipose tissue, the increase in fatty acids regulates the differentiation of CD4 + T cells into IL-17 T cells through acetyl Co-A carboxylase 1, increasing IL-17 T cells in obese patients, limiting the ability of fatty tissue to turn into brown tissue, decrease the inflammatory state and improve insulin sensitivity (57).
Obese patients have higher levels of IL-17 and IL-23 compared with normal weight counterparts unrelated to abdominal fat, insulin-resistance, or leptin level (70).
Presumably, IL-17 synthesized by visceral fat might favor vascular smooth muscle cells expression of eotaxin, related to carotid intima-media thickening as a sign of subclinical atherosclerosis (71).
Bone marrow is a substantial reservoir of white adipose tissue alongside visceral and subcutaneous white fat. Adipocytes stand for up to 70% of total human bone marrow (72). Marrow adipose tissue (MAT) contributes to local and systemic metabolic processes with adipokine secretion (73). Its relevance as a paracrine organ, and its increase when bone mass decreases, have encouraged hypotheses regarding its role in bone health and metabolism. Bone MAT releases adiponectin in response to caloric restriction, highlighting its relevance as an endocrine organ (74).
Contrary to expectations, PsA with high BMI values show high adiponectin levels. Far from improving metabolism as in other tissues, adiponectin seems to have pro-inflammatory properties at the joints, increasing damage. Adiponectin proved a biomarker of radiographic progression independent of metabolic status in rheumatoid arthritis patients (75–77).
Regarding the involved mechanisms, adiponectin might act on osteoblasts, reducing the new bone formation and on osteoclasts, increasing bone resorption (73). Adiponectin and leptin receptors are found in osteoblasts, osteoclasts, and osteoclast precursors. Ultimately, the real impact of these adipokines in bone remodeling is still controversial (73).
In addition, adiponectin induces the release of proinflammatory cytokines like IL-6, monocytic chemotactic protein 1 (MCP-1), prodegradative enzymes like metalloproteinases, and nitric oxide in chondrocytes. Synovial fibroblasts have adiponectin receptors whose stimulation also contributes to promoting a prodegradative microenvironment (78–80).
Different adiponectin isoforms might account for the anti-inflammatory and pro-inflammatory properties of adiponectin (81).
Currently, peripheral joint compromise is initially treated with synthetic disease-modifying drugs like methotrexate, leflunomide, sulfasalazine, and cyclosporine. Acute episodes are often controlled with non-steroidal anti-inflammatory drugs (NSAIDs), which should be used with caution in patients with cardiovascular disease or cardiovascular risk factors. Similarly, small corticosteroids dose for as short as possible should be used (82).
Disease-modifying antirheumatic drugs (DMARDs) should have a positive impact on cardiovascular disease in PsA patients since their pharmacological action reduces systemic inflammation.
Methotrexate (MTX) is a folate antagonist that inhibits the activity of dihydrofolate reductase activity and is one of the most widely used synthetic drugs in PsA treatment. Unfortunately, its effects on the cardiovascular are diametrically opposed, and, eventually, its prolonged administration may increase serum homocysteine level (83) and cardiovascular risk in patients with previous hyperhomocysteinemia (84). Fortunately, increasing homocysteine levels by MTZ can be prevented with folic acid administration (83) while keeping the anti-inflammatory properties that reduce cardiovascular risk. Large observational studies have shown that MTX reduces cardiovascular disease incidence in PsA and rheumatoid arthritis patients. Low or moderate cumulative doses and concomitant use of folic acid have shown more beneficial than high cumulative doses (85). However, the risk of hospitalization for ischemic heart disease among people who received MTX was comparable to that of people receiving other non-biologic antipsoriatic drugs like oral retinoids, cyclosporine, azathioprine, and mycophenolate (86).
Regarding liver disease, it may beiatrogenic or due to the disease itself. Leflunomide and MTX can induce nonalcoholic steatohepatitis (NASH). NASH was more frequent in MTX-treated patients and in patients with PsA compared with rheumatoid arthritis (87). However, PsA patients treated with an anti–TNF-α/MTX association had a lower risk of liver fibrosis compared with those treated with MTX alone (87).
The presence of liver disease should be considered when selecting the PsA treatment (16). Hepatic steatosis, found in 28.1% of the studied sample, was an independent predictor for not achieving the MDA (hazard ratio [HR], 1.91; 95% confidence interval [CI], 1.04–3.38) as reported by a prospective study (88). Patients with liver disease should not be treated with MTX, leflunomide, sulfasalazine, and NSAIDs. Whenever these drugs are administered, careful monitoring of transaminases and liver function should be performed.
Proteins and/or their derivatives targeting specific molecular cascade steps involved in the pathophysiology of different diseases are defined as biological agents (89). According to their molecular structure, they are classified as recombinant human cytokines and growth factors, and fusion antibody proteins and monoclonal antibodies (89).
Fusion proteins are molecules formed by a naturally occurring receptor bound to an immunoglobulin structure. Monoclonal antibodies may be (a) chimeric antibodies, having 30% murine genes fused with human antibodies, (b) humanized antibodies, with 10% murine sequences, and (c) human antibodies derived solely from human immunoglobulin genes.
The development of biological drugs and small molecules pharmacologically active has expanded the therapeutic arsenal in PsA management. This has improved life quality and prognosis in patients refractory to methotrexate, sulfasalazine, leflunomide, or cyclosporin A, conventional immunosuppressive agents.
The complexity and systemic characteristics of PsA demand considering both the ongoing disease activity and extra-articular manifestations, and comorbidities before coming to a drug choice.
Evidence-based, obesity reflects mild chronic systemic inflammation. Worse, visceral fat might add to PsA intrinsic inflammation, secreting pro-inflammatory cytokines. As a result, it affects the therapeutic response and increases cardiovascular risk and the likelihood of expressing the disease in genetically predisposed subjects.
Abdominal obesity not only increases systemic inflammation in a positive feed-back cycle but is relevant to the therapeutic response.
The TNF-α cytokine is key in PsA pathophysiology. Currently, five anti-TNFα molecules are available. Infliximab, adalimumab, and golimumab are monoclonal antibodies. Etanercept is a genetically manufactured dimeric protein resulting from the fusion of the soluble extracellular domain of human TNF-2α (TNFR2/p75) bound to the human IgG1 Fc domain. Certolizumab pegol is a humanized murine monoclonal antibody Fab fragment bound to two polyethylene glycol molecules.
Psoriatic arthritis patients treated with TNF-α underwent a larger decrease in IMTc, a marker of subclinical atherosclerotic disease, compared with those treated with synthetic disease-modifying drugs (6). However, obesity is a negative predictive factor for achieving minimum disease activity (MDA) in patients treated with biological therapies. Treatment with TNF-α inhibitors may induce weight gain in PsA patients (90).
In 2012, Di Minno et al. found that obesity was prevalent in patients not reaching the MDA end-point compared with those who did (64.0% versus 25.5%, p <0.001). Obesity was associated with an increased risk of not reaching MDA (HR = 4.90, 95% CI = 3.04–7.87, p<0.001). The HR for not reaching MDA was 3.98 (95% CI = 1.96–8.06, p< 0.001) and 5.40 (95% CI = 3.09–9.43, p<0.001) in subjects with BMI <30 kg/m2 and <30 to 35 kg/m2. In 98 subjects achieving MDA at 12 months of study, obesity was associated with a low probability of maintaining MDA at 24 months of follow-up (HR = 2.04, 95% CI = 1,015–3.61, p = 0.014) (91). In psoriatic arthritis, the disease activity index for PsA (DAPSA) score was associated with increased risk of subclinical atherosclerotic disease (92).
Assuming the negative effect of obesity in the disease’s control, the same group of researchers studied the effect of a low-calorie diet and weight loss in 126 obese patients starting anti–TNF-α administration. After adjusting for any other clinical and laboratory factors, the dietary intervention proved a strong and independent predictor of MDA achievement (93).
This has probably been related to drug elimination. Patients weighing over100 kg eliminate the drug 55% faster and have a 35% larger distribution volume, reaching lower minimum levels at the time of dosing (94).
In obese patients with metabolic syndrome, the TNF-α blockade may be less effective. The circulating T helper (Th) 17 level and IL-1 cells, elevated in obese patients, can predict the response inadequate to Anti TNF-α (95).
The Danish and Icelandic biological treatment registry included 1943 patients with PsA. Of these, 1271 (65%) had a BMI, with 408 (32%) obese. Obese patients had a higher disease activity and poor adherence to treatment, men in particular, with the median duration of Anti-TNF-α treatment of 2.5 years (95% CI = 1.7–3.2) in the obese vs 5.9 (4.1–7.7) in the non-obese. (P <0.01). Obesity came out as a risk factor for abandoning treatment [HR = 1.6 (95% CI = 1.3–2.0)] (96).
Six-month treatment with adalimumab increased insulin sensitivity and reduced CRP and retinol-binding protein-4 (RBP-4) in a prospective study of 29 patients with moderate-to-severe psoriasis (97, 98). These results were supported by a meta-analysis that included 38 randomized controlled trials (RCTs) with 18024 patients reporting adverse events in adults with plaque psoriasis who received at least one dose of biological therapy, conventional systematic therapy, or placebo. No risk differences for major cardiovascular events were associated with the use of AntiTNF-α (odds ratio [OR] = 0.67, 95% CI = 0. 10–4.63) (99).
Among the biological agents acting through IL-17 inhibition, we find secukinumab and ixekizumab, which are anti–IL-17A monoclonal antibodies. Secukinumab efficacy in obese patients is controversial. In a retrospective observational study including 136 patients with cutaneous psoriasis from 10 dermatology centers in Spain with a 52-week follow-up, secukinumab efficacy was lower in patients with a BMI above 30 (100).
There is scarce information on secukinumab efficacy in obese patients with psoriatic arthritis.
Recently, Pantano et al., carried out a prospective analysis of 100 patients with PsA, divided into those with a BMI above or below 25. After 6 months of treatment, overweight and obese patients had an even better response to secukinumab compared with normal weight patients. Analyzing serum IL-17 levels in 20 obese and 20 non-obese patients, higher serum levels of IL-17 were found in the former (101). This finding was in line with the literature since obesity promotes the expansion of IL17-producing T cells in adipose tissue, and higher levels of IL17 and IL23 were found in this study.
A recent retrospective study of 290 PsA patients, 310 PsC patients and 600 healthy controls found that obesity was more frequent in either PsC (36.5% vs. 22%, OR = 2.1, 95% CI = 1.5–2.8, p <0.01) or PsA (27.6% vs. 22%, OR = 1.4, 95% CI = 1.0–1.9, p <0.05) compared with controls with no inflammatory disease. Curiously, obesity was more frequent in PsC (36.5%) than in PsA (27.6%) (OR = 1.5, 95% CI = 1.1 to 2.1, p <0.05). A family history of PsA (OR = 3.6, 95% CI = 1.1–12.4), axial compromise (OR = 4.4, 95% CI = 1.0–15.4), and dyslipidemia (OR = 3.5, 95% CI = 1.5–8.6) were independently associated with obesity after correcting for probable confounding factors (102).
The effect of IL-17 on cardiovascular risk has been the subject of debate, likely due to its dual anti-atherogenic and pro-atherogenic action, depending on the inflammatory context (22).
An observational study including 195 secukinumab-treated patients with a 2-year follow-up reported that 2% of patients experienced a CV event (103). However, the safety data of the published meta-analysis did not risk differences in the risk of major cardiovascular events associated with the use of anti-IL-17A agents (secukinumab and ixekizumab) (OR = 1·00, 95% CI = 0·09–11·09) (99).
Ustekinumab is the first biological drug specifically targeted at IL-12/IL-23 approved to treat PsA. It binds to the p40 subunit, which is shared by IL-12 and IL-23, interfering with Th1 and Th17-dependent proinflammatory cytokines´ production (95).
Recent studies have shown that PsA patients´ response to ustekinumab was effective and sustained, regardless of body weight (104).
In a study of 79 patients diagnosed with cutaneous psoriasis, patients treated for 7 months with infliximab had an increase (P <0.001) in the mean BMI (2.1 ± 4.5%) and body weight. (2.5 ± 3.3 kg) compared with those treated with ustekinumab (0.1 ± 3.3%; 0.6 ± 1.1 kg) (105).
The pivotal, randomized, placebo-controlled, phase III PSUMMIT I trial not only confirmed the efficacy of ustexinumab in PsA but, unlike other biological agents, those with a bodyweight below or equal to 100 kg had higher response rates than those weighing over 100 kg. In a post hoc analysis by weight group, patients over 100 kg treated with the 90 mg dose showed a trend to higher American College of Rheumatology (ACR) and Psoriasis Area Severity Index (PASI) response rates than those treated with the 45-mg dose (106). Whether these differences are due to suboptimal dosing is to be determined (95).
An observational study of 50 patients with PsA, of whom 28% were obese, studied the efficacy of using ≥3 doses of ustekinumab. Fifty-four percent of patients treated with ustekinumab achieved an MDA response (107).
Regarding ustekinumab on cardiovascular risk, the debate is still open. Contrary to expectations, as IL-12 and IL-23 are found in atheroma plaques, the first data obtained from clinical studies using inhibitors reported a higher rate of major cardiovascular events in the ustekinumab group (108). However, later on, there was no increase in the number of events in the study, so the risk was even lower than in the general population (108).
Two meta-analyses have been carried out to assess the risk of major cardiovascular events (109, 110). Although one possible explanation has been that the methods used for data analysis were different, the results of both meta-analyses turned out contradictory (22). One of them reported no statistically significant differences in the risk of major cardiovascular events compared with placebo (OR = 4.48, 95% CI = 0.24–84.77, p = 0.32) (99)
Randomized long-term follow-up controlled trials are necessary to make recommendations (22).
Psoriasis, a systemic disease entangling MetS, cardiovascular disease, and joint involvement, puts forth obesity and PsA as indirect indicators of disease severity (111). The high prevalence of MetS and its components in PsA patients stresses the importance of performing anamnesis, thorough physical examination, and carotid arteries’ Doppler evaluation, screening subclinical atherosclerotic disease. Counseling on lifestyle changes pointing to a well-balanced, healthy diet, smoking cessation, increasing physical activity, sleep care, and so on, is essential and should take part in their comprehensive assessment. Ultimately, non-classical factors’ influence on cardiovascular disease development should not be underestimated, demanding rigorous control of disease activity.
Provided the large available therapeutic arsenal, the choice of treatment should consider the individual characteristics, obesity, and metabolic syndrome, in particular.
Optimal care of PsA patients is a huge challenge for the treating physicians. Therapeutic management should not be limited to the care of the skin and joints. The systemic and inflammatory nature of the disease makes the search for comorbidities imperative. Adequate cardiovascular risk assessment and strict bodyweight control should be considered therapeutic objectives. These will not only modify the patient’s prognosis but influence therapeutic efficacy and have to be considered. In this way, the comprehensive management of the patient will be decisive in the choice of the therapeutic scheme.
SP, RK: writing—original draft. VC, EK: supervision. MO-L: review and editing—grammar, style, and language. FC: funding acquisition, writing—review. All authors contributed to the article and approved the submitted version.
This work was supported by grant PICD 0031 (2016–2020) from FONCyT. Argentina.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Takeshita J, Grewal S, Langan SM, Mehta NN, Ogdie A, Van Voorhees AS, et al. Psoriasis and comorbid diseases: Epidemiology. J Am Acad Dermatol (2017) 76(3):377–90. doi: 10.1016/j.jaad.2016.07.064
2. Gladman DD. Natural history of psoriatic arthritis. Baillieres Clin Rheumatol (1994) 8(2):379–94. doi: 10.1016/S0950-3579(94)80024-3
3. Caso F, Chimenti MS. Metabolic Syndrome and psoriatic arthritis: considerations for the clinician. Expert Rev Clin Immunol (2020) 16: (4):409–20. doi: 10.1080/1744666X.2020.1740593
4. Gladman DD, Antoni C, Mease P, Clegg DO, Nash P. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis (2005) 64 Suppl 2(Suppl 2):ii14–7. doi: 10.1136/ard.2004.032482
5. Raimondo A, Lembo S, Di Caprio R, Donnarumma G, Monfrecola G, Balato N, et al. Psoriatic cutaneous inflammation promotes human monocyte differentiation into active osteoclasts, facilitating bone damage. Eur J Immunol (2017) 47(6):1062–74. doi: 10.1002/eji.201646774
6. Ritchlin CT, Colbert RA, Gladman DD. Psoriatic Arthritis. N Engl J Med (2017) 376(10):957–70. doi: 10.1056/NEJMra1505557
7. Giacomelli R, Afeltra A, Alunno A, Bartoloni-Bocci E, Berardicurti O, Bombardieri M, et al. Guidelines for biomarkers in autoimmune rheumatic diseases - evidence based analysis. Autoimmun Rev (2019) 18(1):93–106. doi: 10.1016/j.autrev.2018.08.003
8. Kehl AS, Corr M, Weisman MH. Review: Enthesitis: New Insights Into Pathogenesis, Diagnostic Modalities, and Treatment. Arthritis Rheumatol (2016) 68(2):312–22. doi: 10.1002/art.39458
9. Gladman DD, Ziouzina O, Thavaneswaran A, Chandran V, et al. Dactylitis in psoriatic arthritis: prevalence and response to therapy in the biologic era. J Rheumatol (2013) 40(8):1357–9. doi: 10.3899/jrheum.130163
10. Taylor W, Gladman D, Helliwell P, Marchesoni A, Mease P, Mielants H, CASPAR Study Group. Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum (2006) 54(8):2665–73. doi: 10.1002/art.21972
11. Tillett W, Costa L, Jadon D, Wallis D, Cavill C, McHugh J, et al. The ClASsification for Psoriatic ARthritis (CASPAR) criteria–a retrospective feasibility, sensitivity, and specificity study. J Rheumatol (2012) 39(1):154–6. doi: 10.3899/jrheum.110845
12. Cañete JD, Mease P. The link between obesity and psoriatic arthritis. Ann Rheum Dis (2012) 71(8):1265–6. doi: 10.1136/annrheumdis-2012-201632
13. Kolliker Frers RA, Cosentino V, Tau J, Kerzberg EM, Urdapilleta A, Chiocconi M, et al. Immune-Mediated Inflammation Promotes Subclinical Atherosclerosis in Recent-Onset Psoriatic Arthritis Patients without Conventional Cardiovascular Risk Factors. Front Immunol (2018) 9:139. doi: 10.3389/fimmu.2018.00139
14. Icen M, Crowson CS, McEvoy MT, Dann FJ, Gabriel SE, Maradit Kremers H, et al. Trends in incidence of adult-onset psoriasis over three decades: a population-based study. J Am Acad Dermatol (2009) 60(3):394–401. doi: 10.1016/j.jaad.2008.10.062
15. Jensen P, Skov L. Psoriasis and Obesity. Dermatology (2016) 232(6):633–9. doi: 10.1159/000455840
16. Husni ME. Comorbidities in Psoriatic Arthritis. Rheum Dis Clin North Am (2015) 41(4):677–98. doi: 10.1016/j.rdc.2015.07.008
17. Talotta R, Atzeni F, Sarzi-Puttini P, Masala IF. Psoriatic arthritis: From pathogenesis to pharmacologic management. Pharmacol Res (2019) 148:104394. doi: 10.1016/j.phrs.2019.104394
18. Rehaume LM, Mondot S, Aguirre de Cárcer D, Velasco J, Benham H, Hasnain SZ, et al. ZAP-70 Genotype Disrupts the Relationship Between Microbiota and Host, Leading to Spondyloarthritis and Ileitis in SKG Mice. Arthritis Rheumatol (2014) 66(10):2780–92. doi: 10.1002/art.38773
19. McGonagle D, Aydin SZ, Tan AL. The synovio-entheseal complex and its role in tendon and capsular associated inflammation. J Rheumatol Suppl (2012) 89:11–4. doi: 10.3899/jrheum.120233
20. McGonagle D, Lories RJ, Tan AL, Benjamin M. The concept of a “synovio-entheseal complex” and its implications for understanding joint inflammation and damage in psoriatic arthritis and beyond. Arthritis Rheum (2007) 56(8):2482–91. doi: 10.1002/art.22758
21. Dolcino M, Lunardi C, Ottria A, Tinazzi E, Patuzzo G, Puccetti A. Crossreactive autoantibodies directed against cutaneous and joint antigens are present in psoriatic arthritis. PLoS One (2014) 9(12):e115424. doi: 10.1371/journal.pone.0115424
22. Caiazzo G, Fabbrocini G, Di Caprio R, Raimondo A, Scala E, Balato N, et al. Psoriasis, Cardiovascular Events, and Biologics: Lights and Shadows. Front Immunol (2018) 9:1668. doi: 10.3389/fimmu.2018.01668
23. Naldi L, Chatenoud L, Linder D, Belloni Fortina A, Peserico A, Virgili AR, et al. Cigarette smoking, body mass index, and stressful life events as risk factors for psoriasis: results from an Italian case-control study. J Invest Dermatol (2005) 125(1):61–7. doi: 10.1111/j.0022-202X.2005.23681.x
24. Sommer DM, Jenisch S, Suchan M, Christophers E, Weichenthal M. Increased prevalence of the metabolic syndrome in patients with moderate to severe psoriasis. Arch Dermatol Res (2006) 298(7):321–8. doi: 10.1007/s00403-006-0703-z
25. Flammer AJ, Ruschitzka F. Psoriasis and atherosclerosis: two plaques, one syndrome? Eur Heart J (2012) 33(16):1989–91. doi: 10.1093/eurheartj/ehr425
26. Scarpa R, Caso F, Costa L, Peluso R, Spanò A, Lubrano E, et al. Psoriatic Disease: Clinical Staging. J Rheumatol Suppl (2015) 93:24–6. doi: 10.3899/jrheum.150629
27. Otero-Losada M, Llambí HG, Ottaviano G, Cao G, Müller A, Azzato F, et al. Cardiorenal Involvement in Metabolic Syndrome Induced by Cola Drinking in Rats: Proinflammatory Cytokines and Impaired Antioxidative Protection. Mediators Inflamm (2016) 2016:5613056. doi: 10.1155/2016/5613056
28. Boehncke WH, Boehncke S, Tobin A-M, Kirby B. The ‘psoriatic march’: a concept of how severe psoriasis may drive cardiovascular comorbidity. Exp Dermatol (2011) 20(4):303–7. doi: 10.1111/j.1600-0625.2011.01261.x
29. Kölliker Frers RA, Bisoendial RJ, Montoya SF, Kerzkerg E, Castilla R, Takc PP, et al. Psoriasis and cardiovascular risk: Immune-mediated crosstalk between metabolic, vascular and autoimmune inflammation. IJC Metab Endocr (2015) 6:43–54. doi: 10.1016/j.ijcme.2015.01.005
30. Russolillo A, Iervolino S, Peluso R, Lupoli R, Di Minno A, Pappone N, et al. Obesity and psoriatic arthritis: from pathogenesis to clinical outcome and management. Rheumatol (Oxford) (2013) 52(1):62–7. doi: 10.1093/rheumatology/kes242
31. Zhu TY, Li EK, Tam LS. Cardiovascular risk in patients with psoriatic arthritis. Int J Rheumatol (2012) 2012:714321. doi: 10.1155/2012/714321
32. Kimhi O, Caspi D, Bornstein NM, Maharshak N, Gur A, Arbel Y, et al. Prevalence and risk factors of atherosclerosis in patients with psoriatic arthritis. Semin Arthritis Rheum (2007) 36(4):203–9. doi: 10.1016/j.semarthrit.2006.09.001
33. Gonzalez-Juanatey C, Llorca J, Amigo-Diaz E, Dierssen T, Martin J, Gonzalez-Gay MA. High prevalence of subclinical atherosclerosis in psoriatic arthritis patients without clinically evident cardiovascular disease or classic atherosclerosis risk factors. Arthritis Rheum (2007) 57(6):1074–80. doi: 10.1002/art.22884
34. Di Minno MN, Ambrosino P, Lupoli R, Di Minno A, Tasso M, Peluso R, et al. Cardiovascular risk markers in patients with psoriatic arthritis: A meta-analysis of literature studies. Ann Med (2015) 47(4):346–53. doi: 10.3109/07853890.2015.1031822
35. Green A, Shaddick G, Charlton R, Snowball J, Nightingale A, Smith C, et al. Modifiable risk factors and the development of psoriatic arthritis in people with psoriasis. Br J Dermatol (2020) 182(3):714–20. doi: 10.1111/bjd.18227
36. Eder L, Abji F, Rosen CF, Chandran V, Gladman DD. The Association Between Obesity and Clinical Features of Psoriatic Arthritis: A Case-control Study. J Rheumatol (2017) 44(4):437–43. doi: 10.3899/jrheum.160532
37. Eder L, Jayakar J, Pollock R, Pellett F, Thavaneswaran A, Chandran V, et al. Serum adipokines in patients with psoriatic arthritis and psoriasis alone and their correlation with disease activity. Ann Rheum Dis (2013) 72(12):1956–61. doi: 10.1136/annrheumdis-2012-202325
38. Feld J, Nissan S, Eder L, Rahat MA, Elias M, Rimar D, et al. Increased Prevalence of Metabolic Syndrome and Adipocytokine Levels in a Psoriatic Arthritis Cohort. J Clin Rheumatol (2018) 24(6):302–7. doi: 10.1097/RHU.0000000000000721
39. Xue Y, Jiang L, Cheng Q, Chen H, Yu Y, Lin Y, et al. Adipokines in psoriatic arthritis patients: the correlations with osteoclast precursors and bone erosions. PLoS One (2012) 7(10):e46740. doi: 10.1371/journal.pone.0046740
40. Caso F, Postiglione L, Covelli B, Ricciardone M, Di Spigna G, Formisano P, et al. Pro-inflammatory adipokine profile in psoriatic arthritis: results from a cross-sectional study comparing PsA subset with evident cutaneous involvement and subset “sine psoriasis”. Clin Rheumatol (2019) 38(9):2547–52. doi: 10.1007/s10067-019-04619-w
41. Dikbas O, Tosun M, Bes C, Tonuk SB, Aksehirli OY, Soy M. Serum levels of visfatin, resistin and adiponectin in patients with psoriatic arthritis and associations with disease severity. Int J Rheum Dis (2016) 19(7):672–7. doi: 10.1111/1756-185X.12444
42. Peters MJL, Watt P, Cherry L, Welsh P, Henninger E, Dijkmans BAC, et al. Lack of effect of TNFalpha blockade therapy on circulating adiponectin levels in patients with autoimmune disease: results from two independent prospective studies. Ann Rheum Dis (2010) 69(9):1687–90. doi: 10.1136/ard.2009.114207
43. Fassio A, Gatti D, Gisondi P, Girolomoni G, Viapiana O, Giollo A, et al. Effects of secukinumab on serum adipocytokines: preliminary data. Reumatismo (2017) 69(3):105–10. doi: 10.4081/reumatismo.2017.953
44. Chandran V, Abji F, Perruccio AV, Gandhi R, Li S, Cook RJ, et al. Serum-based soluble markers differentiate psoriatic arthritis from osteoarthritis. Ann Rheum Dis (2019) 78(6):796–801. doi: 10.1136/annrheumdis-2018-214737
45. Colak S, Omma A, Sandikci SC, Yucel C, Omma T, Turhan T. Vaspin, neutrophil gelatinase-associated lipocalin and apolipoprotein levels in patients with psoriatic arthritis. Bratisl Lek Listy (2019) 120(1):65–9. doi: 10.4149/BLL_2019_010
46. Wagner CL, Visvanathan S, Elashoff M, McInnes IB, Mease PJ, Krueger GG, et al. Markers of inflammation and bone remodelling associated with improvement in clinical response measures in psoriatic arthritis patients treated with golimumab. Ann Rheum Dis (2013) 72(1):83–8. doi: 10.1136/annrheumdis-2012-201697
47. Johnson CM, Fitch K, Merola JF, Han J, Qureshi AA, Li WQ. Plasma levels of tumour necrosis factor-α and adiponectin can differentiate patients with psoriatic arthritis from those with psoriasis. Br J Dermatol (2019) 181(2):379–80. doi: 10.1111/bjd.17700
48. Chiricozzi A, Raimondo A, Lembo S, Fausti F, Dini V, Costanzo A, et al. Crosstalk between skin inflammation and adipose tissue-derived products: pathogenic evidence linking psoriasis to increased adiposity. Expert Rev Clin Immunol (2016) 12(12):1299–308. doi: 10.1080/1744666X.2016.1201423
49. Paller AS, Mercy K, Kwasny MJ, Choon SE, Cordoro KM, Girolomoni G, et al. Association of pediatric psoriasis severity with excess and central adiposity: an international cross-sectional study. JAMA Dermatol (2013) 149(2):166–76. doi: 10.1001/jamadermatol.2013.1078
50. Love TJ, Zhu Y, Zhang Y, Wall-Burns L, Ogdie A, Gelfand JM, et al. Obesity and the risk of psoriatic arthritis: a population-based study. Ann Rheum Dis (2012) 71(8):1273–7. doi: 10.1136/annrheumdis-2012-201299
51. Li W, Han J, Qureshi AA. Obesity and risk of incident psoriatic arthritis in US women. Ann Rheum Dis (2012) 71(8):1267–72. doi: 10.1136/annrheumdis-2011-201273
52. Organization WH. Overweight/obesity: overweight by country. Global Health Observatory Data Reposit (2008) 2013:2013.
53. Otero-Losada ME, Mc Loughlin S, Rodríguez-Granillo G, Müller A, Ottaviano G, Moriondo M, et al. Metabolic disturbances and worsening of atherosclerotic lesions in ApoE-/- mice after cola beverages drinking. Cardiovasc Diabetol (2013) 12:57. doi: 10.1186/1475-2840-12-57
54. Kumari R, Kumar S, Kant R. An update on metabolic syndrome: Metabolic risk markers and adipokines in the development of metabolic syndrome. Diabetes Metab Syndr (2019) 13(4):2409–17. doi: 10.1016/j.dsx.2019.06.005
55. Herrera MI, Udovin LD, Toro-Urrego N, Kusnier CF, Luaces JP, Otero-Losada M, et al. Neuroprotection Targeting Protein Misfolding on Chronic Cerebral Hypoperfusion in the Context of Metabolic Syndrome. Front Neurosci (2018) 12:339. doi: 10.3389/fnins.2018.00339
56. Lafontan M, Berlan M. Do regional differences in adipocyte biology provide new pathophysiological insights? Trends Pharmacol Sci (2003) 24(6):276–83. doi: 10.1016/S0165-6147(03)00132-9
57. Stolarczyk E. Adipose tissue inflammation in obesity: a metabolic or immune response? Curr Opin Pharmacol (2017) 37:35–40. doi: 10.1016/j.coph.2017.08.006
58. Elmquist JK, Ahima RS, Maratos-Flier E, Flier JS, Saper CB. Leptin activates neurons in ventrobasal hypothalamus and brainstem. Endocrinology (1997) 138(2):839–42. doi: 10.1210/endo.138.2.5033
59. Sanchez-Gurmaches J, Guertin DA. Adipocyte lineages: tracing back the origins of fat. Biochim Biophys Acta (2014) 1842(3):340–51. doi: 10.1016/j.bbadis.2013.05.027
60. Pina T, Genre F, Lopez-Mejias R, Armesto S, Ubilla B, Mijares V, et al. Relationship of leptin with adiposity and inflammation and resistin with disease severity in psoriatic patients undergoing anti-TNF-alpha therapy. J Eur Acad Dermatol Venereol (2015) 29(10):1995–2001. doi: 10.1111/jdv.13131
61. Helfer G, Wu QF. Chemerin: a multifaceted adipokine involved in metabolic disorders. J Endocrinol (2018) 238(2):R79–r94. doi: 10.1530/JOE-18-0174
62. Miranda-Filloy JA, López-Mejias R, Genre F, Carnero-López B, Ochoa R, Diaz de Terán T, et al. Leptin and visfatin serum levels in non-diabetic ankylosing spondylitis patients undergoing TNF-α antagonist therapy. Clin Exp Rheumatol (2013) 31(4):538–45.
63. Kong Y, Zhang S, Wu R, Su X, Peng D, Zhao M, et al. New insights into different adipokines in linking the pathophysiology of obesity and psoriasis. Lipids Health Dis (2019) 18(1):171. doi: 10.1186/s12944-019-1115-3
64. Mertens I, Van Gaal LF. Obesity, haemostasis and the fibrinolytic system. Obes Rev (2002) 3(2):85–101. doi: 10.1046/j.1467-789X.2002.00056.x
65. Xu H, Sethi JK, Hotamisligil GS. Transmembrane tumor necrosis factor (TNF)-alpha inhibits adipocyte differentiation by selectively activating TNF receptor 1. J Biol Chem (1999) 274(37):26287–95. doi: 10.1074/jbc.274.37.26287
66. Engin A. Adiponectin-Resistance in Obesity. Adv Exp Med Biol (2017) 960:415–41. doi: 10.1007/978-3-319-48382-5_18
67. Gonzalez-Gay MA, Llorca J, Garcia-Unzueta MT, Gonzalez-Juanatey C, De Matias JM, Martin J, et al. High-grade inflammation, circulating adiponectin concentrations and cardiovascular risk factors in severe rheumatoid arthritis. Clin Exp Rheumatol (2008) 26(4):596–603.
68. Toussirot E, Aubin F, Dumoulin G. Relationships between Adipose Tissue and Psoriasis, with or without Arthritis. Front Immunol (2014) 5:368. doi: 10.3389/fimmu.2014.00368
69. Tang QQ, Lane MD. Adipogenesis: from stem cell to adipocyte. Annu Rev Biochem (2012) 81:715–36. doi: 10.1146/annurev-biochem-052110-115718
70. Sumarac-Dumanovic M, Stevanovic D, Ljubic A, Jorga J, Simic M, Stamenkovic-Pejkovic D, et al. Increased activity of interleukin-23/interleukin-17 proinflammatory axis in obese women. Int J Obes (Lond) (2009) 33(1):151–6. doi: 10.1038/ijo.2008.216
71. Tarantino G, Costantini S, Finelli C, Capone F, Guerriero E, La Sala N, et al. Is serum Interleukin-17 associated with early atherosclerosis in obese patients? J Transl Med (2014) 12:214. doi: 10.1186/s12967-014-0214-1
72. Fazeli PK, Horowitz MC, MacDougald OA, Scheller EL, Rodeheffer MS, Rosen CJ, et al. Marrow fat and bone–new perspectives. J Clin Endocrinol Metab (2013) 98(3):935–45. doi: 10.1210/jc.2012-3634
73. Sulston RJ, Cawthorn WP. Bone marrow adipose tissue as an endocrine organ: close to the bone? Horm Mol Biol Clin Invest (2016) 28(1):21–38. doi: 10.1515/hmbci-2016-0012
74. Cawthorn WP, Scheller EL, Learman BS, Parlee SD, Simon BR, Mori H, et al. Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab (2014) 20(2):368–75. doi: 10.1016/j.cmet.2014.06.003
75. Meyer M, Sellam J, Fellahi S, Kotti S, Bastard JP, Meyer O, et al. Serum level of adiponectin is a surrogate independent biomarker of radiographic disease progression in early rheumatoid arthritis: results from the ESPOIR cohort. Arthritis Res Ther (2013) 15(6):R210. doi: 10.1186/ar4404
76. Giles JT, van der Heijde DM, Bathon JM. Association of circulating adiponectin levels with progression of radiographic joint destruction in rheumatoid arthritis. Ann Rheum Dis (2011) 70(9):1562–8. doi: 10.1136/ard.2011.150813
77. Klein-Wieringa IR, van der Linden MP, Knevel R, Kwekkeboom JC, van Beelen E, Huizinga TW, et al. Baseline serum adipokine levels predict radiographic progression in early rheumatoid arthritis. Arthritis Rheum (2011) 63(9):2567–74. doi: 10.1002/art.30449
78. Lago R, Gomez R, Otero M, Lago F, Gallego R, Dieguez C, et al. A new player in cartilage homeostasis: adiponectin induces nitric oxide synthase type II and pro-inflammatory cytokines in chondrocytes. Osteoarthritis Cartilage (2008) 16(9):1101–9. doi: 10.1016/j.joca.2007.12.008
79. Kusunoki N, Kitahara K, Kojima F, Tanaka N, Kaneko K, Endo H, et al. Adiponectin stimulates prostaglandin E(2) production in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum (2010) 62(6):1641–9. doi: 10.1002/art.27450
80. Ehling A, Schäffler A, Herfarth H, Tarner IH, Anders S, Distler O, et al. The potential of adiponectin in driving arthritis. J Immunol (2006) 176(7):4468–78. doi: 10.4049/jimmunol.176.7.4468
81. Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol (2006) 6(10):772–83. doi: 10.1038/nri1937
82. Agca R, Heslinga SC. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheum Dis (2017) 76: (1):17–28. doi: 10.1136/annrheumdis-2016-209775
83. Tiftikci A, Ozdemir A, Tarcin O, Tarcin O, Inanc N, Sahinoglu S, et al. Influence of serum folic acid levels on plasma homocysteine concentrations in patients with rheumatoid arthritis. Rheumatol Int (2006) 26(3):191–4. doi: 10.1007/s00296-004-0546-x
84. Lin X, Meng X, Song Z. Homocysteine and psoriasis. Biosci Rep (2019) 39(11):BSR20190867. doi: 10.1042/BSR20190867
85. Prodanovich S, Ma F, Taylor JR, Pezon C, Fasihi T, Kirsner RS. Methotrexate reduces incidence of vascular diseases in veterans with psoriasis or rheumatoid arthritis. J Am Acad Dermatol (2005) 52(2):262–7. doi: 10.1016/j.jaad.2005.02.002
86. Chen YJ, Chang Y-T, Shen J-L, Chen T-T, Wang C-B, Chen C-M, et al. Association between systemic antipsoriatic drugs and cardiovascular risk in patients with psoriasis with or without psoriatic arthritis: a nationwide cohort study. Arthritis Rheum (2012) 64(6):1879–87. doi: 10.1002/art.34335
87. Seitz M, Reichenbach S, Möller B, Zwahlen M, Villiger PM, Dufour J-F. Hepatoprotective effect of tumour necrosis factor α blockade in psoriatic arthritis: a cross-sectional study. Ann Rheum Dis (2010) 69(6):1148–50. doi: 10.1136/ard.2009.116194
88. Di Minno MND, Iervolino S, Peluso R, Russolillo A, Lupoli R, Scarpa R, et al. Hepatic Steatosis and Disease Activity in Subjects with Psoriatic Arthritis Receiving Tumor Necrosis Factor-α Blockers. J Rheumatol (2012) 39(5):1042–6. doi: 10.3899/jrheum.111391
89. Balato A, Scala E, Balato N, Caiazzo G, Di Caprio R, Monfrecola G, et al. Biologics that inhibit the Th17 pathway and related cytokines to treat inflammatory disorders. Expert Opin Biol Ther (2017) 17(11):1363–74. doi: 10.1080/14712598.2017.1363884
90. Renzo LD, Saraceno R, Schipani C, Rizzo M, Bianchi A, Noce A, et al. Prospective assessment of body weight and body composition changes in patients with psoriasis receiving anti-TNF-α treatment. Dermatologic Ther (2011) 24(4):446–51. doi: 10.1111/j.1529-8019.2011.01439.x
91. di Minno MN, Peluso R, Iervolino S, Lupoli R, Russolillo A, Scarpa R, et al. Obesity and the prediction of minimal disease activity: a prospective study in psoriatic arthritis. Arthritis Care Res (Hoboken) (2013) 65(1):141–7. doi: 10.1002/acr.21711
92. Palmou-Fontana N, Martínez-Lopez D, Corrales A, Rueda-Gotor J, Genre F, Armesto S, et al. Disease Activity Influences Cardiovascular Risk Reclassification Based on Carotid Ultrasound in Patients with Psoriatic Arthritis. J Rheumatol (2020) 47(9):1344–53. doi: 10.3899/jrheum.190729
93. Di Minno MN, Peluso R, Iervolino S, Russolillo A, Lupoli R, Scarpa R, et al. Weight loss and achievement of minimal disease activity in patients with psoriatic arthritis starting treatment with tumour necrosis factor α blockers. Ann Rheum Dis (2014) 73(6):1157–62. doi: 10.1136/annrheumdis-2012-202812
94. Tiberio R, Graziola F, Miglino B, Veronese F, Annali G, Savoia P. Secukinumab for Psoriasis in Obese Patients: Minireview and Clinical Experience. Case Rep Dermatol (2019) 11(Suppl 1):29–36. doi: 10.1159/000501990
95. Costa L, Ramonda R, Ortolan A, Favero M, Foti R, Visalli E, et al. Psoriatic arthritis and obesity: the role of anti-IL-12/IL-23 treatment. Clin Rheumatol (2019) 38(9):2355–62. doi: 10.1007/s10067-019-04663-6
96. Højgaard P, Glintborg B, Kristensen LE, Gudbjornsson B, Love TJ, Dreyer L. The influence of obesity on response to tumour necrosis factor-α inhibitors in psoriatic arthritis: results from the DANBIO and ICEBIO registries. Rheumatol (Oxford) (2016) 55(12):2191–9. doi: 10.1093/rheumatology/kew326
97. Pina T, Armesto S, Lopez-Mejias R, Genre F, Ubilla B, Gonzalez-Lopez MA, et al. Anti-TNF-α therapy improves insulin sensitivity in non-diabetic patients with psoriasis: a 6-month prospective study. J Eur Acad Dermatol Venereol (2015) 29(7):1325–30. doi: 10.1111/jdv.12814
98. Pina T, Genre F, Lopez-Mejias R, Armesto S, Ubilla B, Mijares V, et al. Anti-TNF-α therapy reduces retinol-binding protein 4 serum levels in non-diabetic patients with psoriasis: a 6-month prospective study. J Eur Acad Dermatol Venereol (2016) 30(1):92–5. doi: 10.1111/jdv.13005
99. Rungapiromnan W, Yiu ZZN, Warren RB, Griffiths CEM, Ashcroft DM. Impact of biologic therapies on risk of major adverse cardiovascular events in patients with psoriasis: systematic review and meta-analysis of randomized controlled trials. Br J Dermatol (2017) 176(4):890–901. doi: 10.1111/bjd.14964
100. Notario J, Deza G, Vilarrasa E, Valentí F, Muñoz C, Mollet J, et al. Treatment of patients with plaque psoriasis with secukinumab in a real-life setting: a 52-week, multicenter, retrospective study in Spain. J Dermatolog Treat (2019) 30(5):424–9. doi: 10.1080/09546634.2018.1528000
101. Pantano I, Iacono D, Favalli EG, Scalise G, Costa L, Caso F, et al. Secukinumab efficacy in patients with PsA is not dependent on patients’ body mass index. Ann Rheum Dis (2020), pii:annrheumdis-2020-217251. doi: 10.1136/annrheumdis-2020-217251
102. Queiro R, Lorenzo A, Tejón P, Coto P, Pardo E. Obesity in psoriatic arthritis: Comparative prevalence and associated factors. Medicine (Baltimore) (2019) 98(28):e16400. doi: 10.1097/MD.0000000000016400
103. Egeberg A, Ottosen MB, Gniadecki R, Broesby-Olsen S, Dam TN, Bryld LE, et al. Safety, efficacy and drug survival of biologics and biosimilars for moderate-to-severe plaque psoriasis. Br J Dermatol (2018) 178(2):509–19. doi: 10.1111/bjd.16102
104. Iannone F, Santo L, Bucci R, Semeraro A, Carlino G, Paoletti F, et al. Drug survival and effectiveness of ustekinumab in patients with psoriatic arthritis. Real-life data from the biologic Apulian registry (BIOPURE). Clin Rheumatol (2018) 37(3):667–75. doi: 10.1007/s10067-018-3989-2
105. Gisondi P, Conti A, Galdo G, Piaserico S, De Simone C, Girolomoni G, et al. Ustekinumab does not increase body mass index in patients with chronic plaque psoriasis: a prospective cohort study. Br J Dermatol (2013) 168(5):1124–7. doi: 10.1111/bjd.12235
106. Kavanaugh A, Puig L, Gottlieb AB, Ritchlin C, Li S, Wang Y, et al. Maintenance of Clinical Efficacy and Radiographic Benefit Through Two Years of Ustekinumab Therapy in Patients With Active Psoriatic Arthritis: Results From a Randomized, Placebo-Controlled Phase III Trial. Arthritis Care Res (2015) 67(12):1739–49. doi: 10.1002/acr.22645
107. Queiro R, Brandy A, Rosado MC, Lorenzo A, Coto P, Carriles C, et al. Minimal Disease Activity and Patient-Acceptable Symptom State in Psoriatic Arthritis: A Real-World Evidence Study With Ustekinumab. JCR: J Clin Rheumatol (2018) 24(7):381–4. doi: 10.1097/RHU.0000000000000751
108. Reich K, Langley RG, Lebwohl M, Szapary P, Guzzo C, Yeilding N, et al. Cardiovascular safety of ustekinumab in patients with moderate to severe psoriasis: results of integrated analyses of data from phase II and III clinical studies. Br J Dermatol (2011) 164(4):862–72. doi: 10.1111/j.1365-2133.2011.10257.x
109. Tzellos T, Kyrgidis A, Zouboulis CC. Re-evaluation of the risk for major adverse cardiovascular events in patients treated with anti-IL-12/23 biological agents for chronic plaque psoriasis: a meta-analysis of randomized controlled trials. J Eur Acad Dermatol Venereol (2013) 27(5):622–7. doi: 10.1111/j.1468-3083.2012.04500.x
110. Ryan C, Leonardi CL, Krueger JG, Kimball AB, Strober BE, Gordon KB, et al. Association between biologic therapies for chronic plaque psoriasis and cardiovascular events: a meta-analysis of randomized controlled trials. JAMA (2011) 306(8):864–71. doi: 10.1001/jama.2011.1211
Keywords: psoriatic arthritis, metabolic syndrome, cardiovascular risk, obesity, adipokines, pathophysiology, treatment
Citation: Porta S, Otero-Losada M, Kölliker Frers RA, Cosentino V, Kerzberg E and Capani F (2021) Adipokines, Cardiovascular Risk, and Therapeutic Management in Obesity and Psoriatic Arthritis. Front. Immunol. 11:590749. doi: 10.3389/fimmu.2020.590749
Received: 02 August 2020; Accepted: 21 December 2020;
Published: 10 February 2021.
Edited by:
Matteo Vecellio, University of Oxford, United KingdomReviewed by:
Emanuele Scala, Karolinska Institutet, SwedenCopyright © 2021 Porta, Otero-Losada, Kölliker Frers, Cosentino, Kerzberg and Capani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francisco Capani, ZnJhbmNpc2NvY2FwYW5pQGhvdG1haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.