 Francesca Velotti
Francesca Velotti Ilaria Barchetta
Ilaria Barchetta Flavia Agata Cimini
Flavia Agata Cimini Maria Gisella Cavallo2
Maria Gisella Cavallo2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 11 November 2020
Sec. Cytokines and Soluble Mediators in Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.587581
This article is part of the Research Topic Cellular Stress and Inflammation: How the Immune System Drives Tissue Homeostasis View all 11 articles
Inflammation is strictly interconnected to anti-inflammatory mechanisms to maintain tissue homeostasis. The disruption of immune homeostasis can lead to acute and chronic inflammatory diseases, as cardiovascular, pulmonary, metabolic diseases and cancer. The knowledge of the mechanisms involved in the development and progression of these pathological conditions is important to find effective therapies. Granzyme B (GrB) is a serine protease produced by a variety of immune, non-immune and tumor cells. Apoptotic intracellular and multiple extracellular functions of GrB have been recently identified. Its capability of cleaving extracellular matrix (ECM) components, cytokines, cell receptors and clotting proteins, revealed GrB as a potential multifunctional pro-inflammatory molecule with the capability of contributing to the pathogenesis of different inflammatory conditions, including inflammaging, acute and chronic inflammatory diseases and cancer. Here we give an overview of recent data concerning GrB activity on multiple targets, potentially allowing this enzyme to regulate a wide range of crucial biological processes that play a role in the development, progression and/or severity of inflammatory diseases. We focus our attention on the promotion by GrB of perforin-dependent and perforin-independent (anoikis) apoptosis, inflammation derived by the activation of some cytokines belonging to the IL-1 cytokine family, ECM remodeling, epithelial-to-mesenchymal transition (EMT) and fibrosis. A greater comprehension of the pathophysiological consequences of GrB-mediated multiple activities may favor the design of new therapies aim to inhibit different inflammatory pathological conditions such as inflammaging and age-related diseases, EMT and organ fibrosis.
Inflammation is a physiological response to infections or tissue injury and is essential for survival, having beneficial effects towards the neutralization of dangerous or harmful agents. Inflammation is strictly interconnected with anti-inflammatory mechanisms, which control and resolve the inflammatory process to maintain immune homeostasis (1). Under some circumstances, this immune homeostasis is disrupted and inflammation becomes excessive and/or persistent, leading to the development of inflammatory diseases (1). In this context, aging can be characterized by an uncontrolled and unresolved chronic, low-grade inflammation, the so-called ‘‘inflamm-aging”, which can lead to inflammatory age-related diseases, as cardiovascular, pulmonary, metabolic diseases (as type 2 diabetes, T2D) and cancer (2). Multiple factors underlie the pathogenesis of inflammatory diseases and can lead to tissue fibrosis and organ dysfunction, associated with high morbidity and mortality (3). Therefore, the knowledge of mechanisms involved in the development and/or progression of these pathological conditions is important to find specific and effective therapies.
Granzyme B (GrB) is a serine protease traditionally known for its perforin-dependent pro-apoptotic function underlying the capability of cytotoxic immune cells, as cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, to kill tumor and virus-infected target cells (4–7). GrB expression has been recently demonstrated also in non-tumor or tumor immune and non-immune cells (8). Indeed, GrB is produced and secreted by immune cells, like T and B cell subpopulations, monocyte/macrophages, mast cells, and basophils (8–13), by non-immune cells, like vascular smooth muscle cells (V-SMCs), pneumocytes, keratinocytes, and chondrocytes (12, 14–16), as well as by tumor cells, like leukemia cells and breast, urothelial, prostate, pancreatic and colorectal cancer cells (17–21) (Table 1). GrB not only exerts a perforin-dependent intracellular activity, but also an extracellular perforin-independent function, consisting in the cleavage of multiple extracellular substrates, as extracellular matrix (ECM) components, cytokines, cell receptors, angiogenic and clotting proteins (28, 49, 50). Hence, the pathophysiological function of GrB has been redefined and a putative role for GrB in the pathogenesis of inflammatory and age-related diseases has emerged (8, 29) (Table 1).

Table 1 GrB in Inflammatory Diseases: GrB producing cells, GrB cellular and molecular targets, GrB-associated organ-specific diseases.
In this review, we discuss data concerning GrB activity on multiple targets involved in inflammation, potentially allowing this enzyme to regulate a wide range of crucial processes that play a role in inflammatory disease development, progression and severity. We focus our attention on the possible impact of GrB on inflammatory events leading to tissue fibrosis in both acute and age-related inflammatory diseases.
The recent discovery of multiple intracellular and extracellular substrates for GrB has revealed this protease as a potential multifunctional pro-inflammatory molecule, contributing to the pathogenesis of multiple pathological inflammatory conditions.
Elevated extracellular GrB levels were found in biological fluids, as in plasma from patients with acute myocardial infarction (36), atherosclerosis (37), obesity and T2D (51, 52), in broncho-alveolar lavage (BAL) in chronic obstructive pulmonary disease (COPD), pneumonia, and asthma (8), and in the synovial fluid in rheumatoid arthritis (53).
Elevated GrB levels were also found in inflamed tissues, including V-SMCs and atherosclerotic plaque in cardiovascular diseases (14), CTLs, pneumocytes and alveolar macrophages in pulmonary diseases (12, 27), adipose tissue-T cells in obesity (33) and in skin diseases (49). Moreover, according to a putative contribution of GrB in inflammaging, increased GrB expression levels were found in the elderly affected by obesity, cardiovascular and skin diseases (29, 49, 54, 55).
GrB extracellular substrates include cytokines and ECM components (29, 31, 50). The potential pathophysiological consequences of their cleavage constitute the basis to envisage a crucial pro-inflammatory role for GrB in the pathogenesis of inflammatory diseases (29).
GrB has the ability to process and activate pro-inflammatory, pro-fibrotic and aging mediators belonging to the IL-1 cytokine family (31, 56). Indeed, GrB processes IL-18 from its inactive to its active form and IL-1α into a significantly more potent pro-inflammatory fragment. IL-1α enhances persistent inflammation and stimulates fibroblasts to produce more interstitial collagenase and ECM remodeling, regulating normal and aberrant tissue repair (56, 57). IL-1α fragments, similar to those produced by GrB, were found in BAL in human airway inflammatory diseases, as COPD, cystic fibrosis and bronchiectasis (8), while GrB activity on IL-1α was demonstrated in vivo in GrB knockout mice (29), strongly suggesting that this activity also exists in vivo.
GrB has also the ability to degrade several ECM components, including proteins, as fibronectin, vitronectin, laminin, SMC matrix, VE-cadherin, and fibrillin-1, as well as proteoglycan, as biglycan and decorin, indicating GrB as a crucial player in ECM remodeling (28–30). Indeed, ECM undergoes remodeling, that is degradation by proteases and renewal and repair by fibroblasts, thus regulating tissue homeostasis and acting on tissue healing. ECM components assist cell attachment, ligate receptors and store growth factors, regulating cell survival, proliferation, differentiation, and migration. Therefore, abnormal ECM remodeling can result in cell detachment-dependent apoptosis and alterations in cell proliferation, differentiation and migration, as observed in several inflammatory conditions, such as cardiovascular, pulmonary and metabolic diseases, obesity, and cancer progression and metastasis (58, 59). Hence, GrB capability of targeting multiple ECM components, might allow this enzyme to regulate several fundamental biological processes involved in the development and/or progression of inflammatory diseases.
Thus, considering the extracellular and intracellular GrB function and the context in which GrB is produced, this molecule has the potential to contribute to the pathogenesis of non-neoplastic and neoplastic inflammatory diseases through a multitude of mechanisms ranging from the induction of perforin-dependent and/or –independent apoptosis and the promotion of epithelial-to-mesenchymal transition (EMT) and/or fibrosis, as illustrated below (Figure 1).
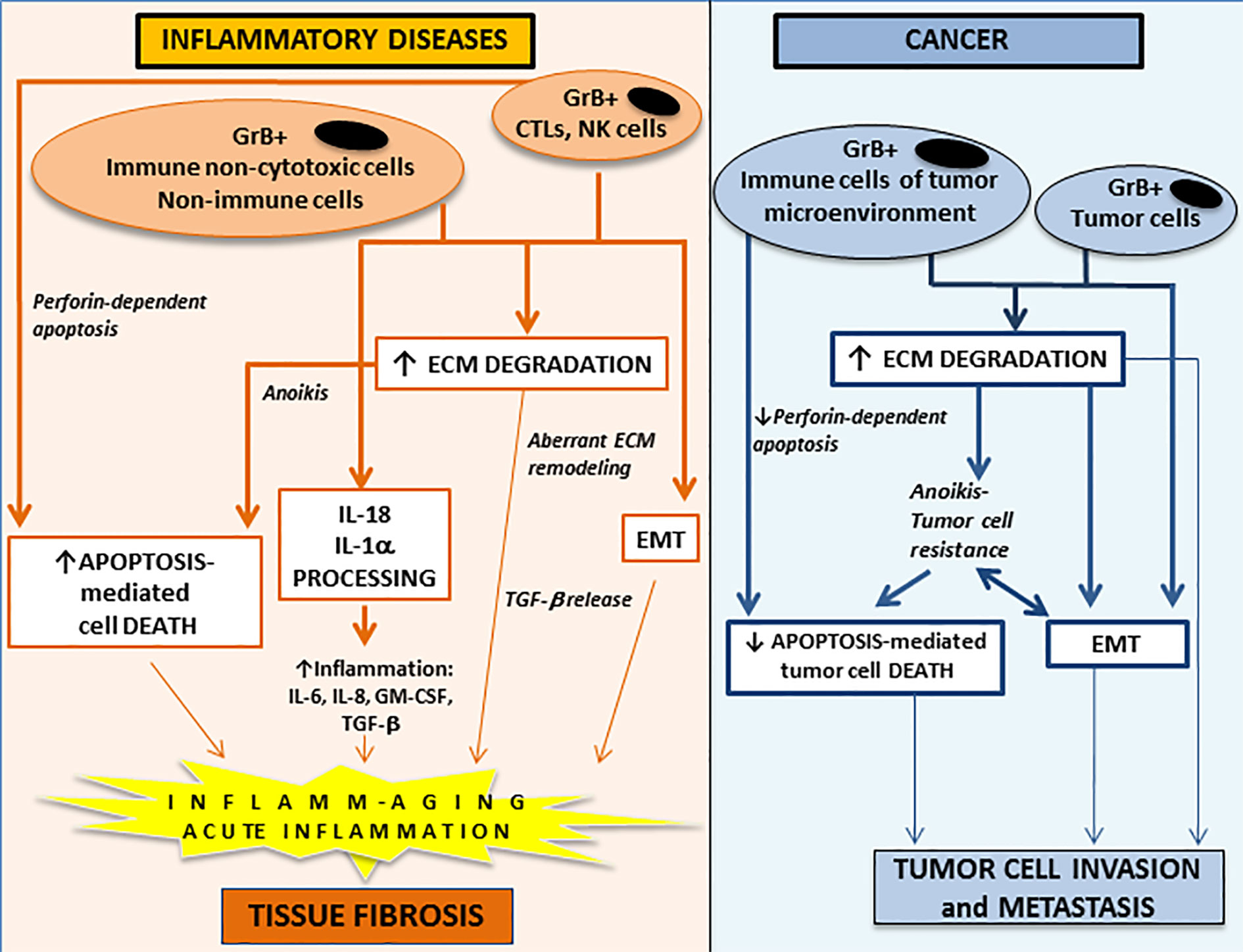
Figure 1 The potential contribution of extracellular and intracellular GrB functions to the development and/or the progression of acute and chronic inflammatory diseases (left panel) and to cancer invasion and metastasis (right panel). GrB is a multifunctional pro-inflammatory molecule regulating a wide range of inflammatory events. GrB produced by perforin-expressing immune cells (CTL and NK cells) can induce perforin-dependent cell apoptosis, while GrB produced by perforin-deficient immune (e.g. non-cytotoxic T and B cell subpopulations, monocyte/macrophages/myeloid-derived suppressor cells, mast cells, basophils, neutrophils), non-immune (e.g. vascular smooth muscle cells, pneumocytes) and tumor (e.g. breast, urothelial, prostate, pancreatic, colorectal) cells can induce anoikis (anchorage-dependent cell death). Extracellular GrB can promote activation of pro-inflammatory cytokines (IL-18 and IL-1α), ECM degradation/remodeling, pathologic EMT and tissue fibrosis. GrB, granzyme B; CTL, cytotoxic T lymphocytes; NK, natural killer; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; IL, interleukin; TGF-β, transforming growth factor-β.
Apoptosis promotes tissue injury during inflammation and is involved in the pathogenesis of acute and chronic inflammatory diseases, as cardiovascular and pulmonary diseases and metabolic syndrome (60–62). The capability of immune and non-immune cell-derived GrB of inducing apoptosis makes GrB a potential important player of apoptosis-mediated tissue damage in inflammation. GrB can induce two kinds of apoptotic cell death, the intracellular perforin-dependent apoptosis (7) and the extracellular perforin-independent apoptosis, named anoikis (63). Anoikis is due to the detachment of cells from ECM and from neighboring cells, playing a role in preventing inappropriate cell translocation and attachment, and assisting appropriate tissue renewal (62). In cancer, anoikis resistance characterizes cancer cell anchorage-independent growth and EMT, contributing to cancer cell invasion and metastasis (62, 64, 65). In inflammatory diseases, as cardiovascular (66), pulmonary (67) and skin (49) diseases, and diabetes-related cardiovascular complications and retinopathy (62), aberrant anoikis is involved in excessive cell death and tissue injury.
A role for GrB-mediated apoptosis -either perforin-dependent apoptosis or anoikis- has been reported in inflammatory pulmonary diseases, including age-related diseases, as COPD (23, 38), and acute severe lung inflammatory diseases, as respiratory syncytial virus (RSV) pulmonary infections (24, 25). In COPD patients, GrB was identified in type II pneumocytes, alveolar macrophages and in bronchial and alveolar wall-infiltrating CTLs, suggesting a role for GrB in bronchial and alveolar cell apoptosis (8, 12, 22, 38). Of note, in COPD, increased GrB- and perforin-expressing CTLs were found in BAL and blood, and GrB-expressing T cells in BAL positively correlated with bronchial epithelial cell apoptosis (22, 23, 38). In spite of these findings, the evidence of a causative role for GrB-mediated apoptosis in the pathogenesis of COPD is lacking. In vivo animal studies are made difficult, because of the lack of appropriate mouse COPD models. A role for GrB has been proposed also in acute pulmonary pathologies. There is evidence of high GrB expression by CD8+T, CD4+T, and NK cells in human RSV-induced acute severe lung injury (24), suggesting a role for GrB in amplifying pro-apoptotic and pro-inflammatory activities. Supporting this hypothesis, Bem et al. (25) showed GrB contribution to acute lung injury in pneumovirus-infected mice; GrB deficiency in pneumovirus-infected mice significantly delayed clinical response to fatal pneumovirus infection and this effect was associated with delayed neutrophil recruitment, decreased caspase-3 activation and reduced lung permeability, suggesting a role for GrB in acute disease progression due to alveolar injury.
In the last years, a putative role for GrB-mediated apoptosis in atherosclerosis is also emerged in both the elderly and insulin resistant young individuals (8, 26, 39–41). Elevated plasma GrB levels were found in patients with myocardial infarction (36, 42) and unstable carotid plaques associated with increased cerebrovascular events (37). GrB was absent in normal vessels and its expression appeared during atherosclerosis; studies on mild and advanced atherosclerotic human coronary arteries showed higher GrB expression in V-SMCs, CTLs and macrophages in advanced lesions (11, 14). GrB expression co-localized with V-SMCs and macrophages undergoing apoptosis, suggesting that GrB may mediate apoptosis in these cells (11, 14). Furthermore, peripheral blood mononuclear cells (PBMCs) from patients with unstable angina produced higher GrB levels than PBMCs from patients with stable angina, and PBMC-derived conditioned media induced apoptosis in cultured endothelial cells, supporting a possible role for GrB in atherosclerosis severity, possibly inducing vascular apoptosis in unstable angina (43). Finally, the proteinase inhibitor-9, the GrB endogenous inhibitor, was reduced in unstable atherosclerotic lesions compared to stable lesions (44), according to the hypothesis of a role for GrB in plaque instability and suggesting that GrB activity in atherosclerosis may be regulated by an imbalance between GrB and its inhibitor. Although these findings do not allow to definitively establish an in vivo role for GrB in the induction of apoptosis in atherosclerotic plaque instability and rupture, in vitro and animal studies support this hypothesis (14, 26, 45, 46). Indeed, GrB mediates anoikis of cultured human coronary artery SMCs and endothelial cells (14). Moreover, in angiotensin II-treated apolipoprotein E (ApoE), GrB deficiency was associated with decreased abdominal aortic aneurysms and increased survival (because of rare aneurism rupture) compared to perforin-deficient or control mice (26, 45). Finally, a role for NK and NKT cells in the promotion of atherosclerosis has also been proposed (47, 48). Increased atherosclerosis was observed when NK cells were transferred into ApoE(-/-)Rag2(-/-)IL2rγ(-/-) mice, whereas decreased atherosclerotic lesions were found in NK cell depleted ApoE(-/-) or when GrB/perforin-deficient NK cells were transferred (47). Transfer of CD4+NKT cells into T-, B- and NK-cell-deficient ApoE mice augmented aortic root atherosclerosis; this effect reversed when GrB/perforin-deficient NKT cells were transferred (48).
Inflammation, characterized by excessive apoptosis and abnormal ECM remodeling, can lead to tissue fibrosis, which impairs the affected organ’s function (3). Fibrosis is triggered by inflammatory cytokines and growth factors signaling abnormal ECM regulation; this leads to an imbalance between ECM degradation by proteases and excessive ECM deposition by different cells, mainly myofibroblast (3) derived by mesenchymal cells and by epithelial cells undergoing EMT (EMT-derived myofibroblasts) (68). Noteworthy, fibrosis and EMT share one of their major inducer that is transforming growth factor-β (TGF-β) (69).
Recent studies have proposed a role for GrB in heart, lung, adipose tissue and skin fibrosis (27, 29, 32, 49, 61, 69).
Elevated GrB expression was detected in human and murine fibrotic hearts (32). Moreover, a perforin-independent role for GrB in the pathogenesis of cardiac fibrosis was suggested in vivo, showing that GrB deficiency in mice protected against angiotensin II-induced cardiac fibrosis, reducing microhemorrhage, inflammation, and fibroblast accumulation (32).
In COPD, GrB–expressing monocytes and granulocytes were identified, and CD8+T infiltrating cells and apoptosis increased in airway epithelial cells, while soluble GrB levels and GrB-expressing T cells increased in BAL, suggesting that GrB upregulation in CD8+ and CD8- cells may be involved in small airway wall remodeling (27).
In obesity, increased CD8+T cells and GrB expression were found in adipose tissue in vivo, suggesting a role for GrB in adipose tissue fibrosis (33, 61). Furthermore, CD8+T-cell-depletion in overfed mice improved obesity-induced insulin resistance and decreased adipose tissue pro-inflammatory macrophages; these effects were reversed when mice were reconstituted with CD8+T cells (34). These findings suggest that, in obesity, adipose tissue CD8+T cells induce the recruitment of macrophages and that both may induce adipose tissue dysfunction and insulin resistance.
A role for GrB has also been indicated in skin fibrosis, as extensively discuss elsewhere (29, 49).
The mechanisms by which GrB induces fibrosis have not been completely elucidated and multiple GrB-mediated activities have been proposed.
GrB can cleave the ECM proteoglycan decorin, a potent anti-fibrotic (30) and a pro-autophagic (35) molecule. Indeed, decorin, by attaching to cell surface receptor and ECM molecules, regulates signal transduction pathways controlling genes involved in ECM organization (30). In addition, by attaching to cell receptors, decorin promotes autophagy in endothelial cells leading to inhibition of angiogenesis (35). Therefore, decorin cleavage by GrB might underlie aberrant ECM and/or vascular remodeling, involved in the initiation and/or the progression of various fibroproliferative disorders. Note also that decreased autophagy is involved in the pathogenesis of inflammaging, fibrotic diseases and tumors. Studies have shown a reduction of decorin in different fibrotic organs, as in cardiac fibrosis following myocardial infarction and in acute exacerbation-idiopathic pulmonary fibrosis (IPF) (70–72). Animal experiments in decorin-null mice with myocardial infarction (70) or in hamster and mice models of lung fibrosis (73–75) showed both decorin requirement for proper fibrotic evolution of tissue injury and the potential therapeutic anti-fibrotic effect of decorin administration. Evidence also exists for a role of decorin in maintaining glucose tolerance in obesity (76).
Moreover, GrB, cleaving decorin and other ECM substrates as biglycan, beta-glycan and fibrillin-1 which act as reservoir of cytokines and growth factors as TGF-β, induces the release of active TGF-β, a key regulator of fibrosis (30, 69). Therefore, the aberrant release of sequestered TGF-β by GrB-mediated cleavage of ECM components represents another potential mechanism by which GrB may contribute to fibrosis.
Noteworthy, GrB (18, 21, 77), as some other granzymes (78–80), has been recently proposed as promoters of EMT, an important process linked the stimulation of the three following events: 1) tissue and organ formation during embryogenesis; 2) tissue and organ physiologic repair and pathologic fibrosis; 3) tumor cell invasion and metastasis (81). EMT is a process in which epithelial cells lose E-cadherin-mediated cell-cell adhesion and acquire some mesenchymal features, as N-cadherin expression and the capability of invasion, migration, and production of ECM. Inflammatory molecules, mainly TGF-β, trigger intracellular signaling cascades, activating EMT-transcription factors like Snail, ZEB, and TWIST (81). EMT is involved in multiple organ fibrosis, as those occurring in cardiovascular and pulmonary (COPD and IPF) diseases (81–88). Now interestingly, EMT-derived fibrosis has been also called to possibly account to pulmonary fibrosis in SARS-CoV-2 infection (89), suggesting a possible contribution of GrB in the severe pulmonary damage in COVID-19 (89, 90). A possible role for GrB in EMT promotion has emerged in human tumor models (18, 21, 77). Enzymatically active GrB was expressed, in absence of perforin, by tumor cells in vitro and in tissues (ex vivo) (17–21). Although GrB in cancer tissues is widely used as activation marker for cytotoxic lymphocytes, and lymphocyte-derived GrB-positive tumor immunostaining is associated with a favorable clinical outcome in a large spectrum of cancers, in some cases, GrB expression in tumors correlates to the severity of the disease, poor prognosis and therapy resistance (91–96). It has been documented GrB expression by urothelial carcinoma cells in primary urothelial cancer tissues and its expression was associated to EMT (analyzed by Snail-1, E- and N-cadherin expression) (18). Significantly, GrB expression was concentrated in urothelial neoplastic cells undergoing EMT at the cancer invasion front, suggesting that the expression of GrB and EMT molecules might be functionally related (18). A further support to the hypothesis of considering GrB as an EMT promoter, derives from the association that existed between GrB expression in tumor tissues and the pathological tumor spreading, in particular, the increasing invasiveness status of urothelial carcinomas (18). In addition, in vitro experiments of loss and gain of GrB function performed in CRC (including also CRC patient-derived Cancer Stem Cells), bladder and pancreatic carcinoma cells showed that GrB deficiency was associated to the loss of the EMT phenotype and the inhibition of invasion through matrigel, further supporting a role for GrB in tumor EMT promotion and cancer cell invasion (18, 21). Finally, GrB function in EMT was further supported by data indicating a contribution of GrB in the induction of TGF-β1-driven EMT in CRC cells (21). Indeed, TGF-β1 enhanced GrB expression while inducing EMT in CRC cells, whereas GrB depletion resulted in the inhibition of TGF-β1-driven EMT (21). However, research is needed to identify GrB targets involved in the mechanisms underlying EMT modulation by GrB. It should also be taken into account the possible regulation of GrB activity and function by the GrB-bound proteoglycan serglycin, considering that its intracellular activity consists in the promotion of secretory granule maturation and GrB storage, while its extracellular activity is implicated in the regulation of tumorigenesis, driving inflammation, EMT and tumor progression (97). Lastly, the examination of GrB expression in a large number of cancers in relation to the clinical outcome is needed, together with the evaluation of EMT in murine tumor and non-tumor models.
GrB is emerging as a multifunctional pro-inflammatory protease, acting with tissue and context dependence on multiple targets, thus representing a putative powerful regulator of a wide range of crucial processes involved in the pathogenicity and/or in the severity of inflammatory diseases, either acute or age-related. The major limitation of this assumption is the paucity of in vivo direct evidence for the multiple GrB pro-inflammatory activities. It should be considered that the in vivo function of human GrB is a challenging problem and difficult to deal with, in that, although few mechanistic animal studies connecting clinical observations with in vitro data exist, animal experiments might generate false interspecies functions of GrB, because of GrB interspecies structural and functional diversity (98–100). Therefore, further research is required to explore the multiple activities of GrB potentially occurring in the inflammatory events underlying acute and chronic inflammatory diseases.
A greater comprehension of GrB function may favor the design of new therapies aimed to inhibit and regulate GrB pro-inflammatory activities, counteracting excessive inflammation, fibrosis and abnormal EMT-derived processes. Current research is considering the development and the use of pharmacological GrB inhibitors as potential therapeutic options for the prevention and/or treatment of GrB-associated inflammatory pathological conditions (101–106). Progress in this field might be even more urgent if we consider the possibility to develop therapies that have an impact on inflammaging and chronic age-related diseases, as well as on excessive acute inflammatory reactions, as they occur in COVID-19, especially in aged individuals tending to excessive inflammatory responses resulting in lethal lung damage (89, 107, 108).
FV made substantial contributions to conception and design of the review. IB, FAC, and MGC contributed to the manuscript revision, read, and approved the submitted version. All authors contributed to the article and approved the submitted version.
IB is supported by a research fellowship from Eli Lilly Foundation. FAC was supported by research funding from Sapienza University, Rome, Italy. This work was supported by research funding from Department of Experimental Medicine (Rome, Italy) to MGC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation with several of the authors IB, FAC, MGC at time of review.
1. Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, et al. A guiding map for inflammation. Nat Immunol (2017) 18:826–31. doi: 10.1038/ni.3790
2. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann NY Acad Sci (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
3. Mack M. Inflammation and fibrosis. Matrix Biol (2018) 68-69:106–21. doi: 10.1016/j.matbio.2017.11.010
4. Masson D, Nabholz C, Estrade J, Tschopp J. Granules of cytolytic T-lymphocytes contain two serine esterases. EMBO J (1986) 5:1595–600.
5. Garcia-Sanz JA, Velotti F, MacDonald HR, Masson D, Tschopp J, Nabholz M. Appearance of granule-associated molecules during activation of cytolytic T-lymphocyte precursors by defined stimuli. Immunology (1988) 64:129–34.
6. Velotti F, Palmieri G, D’Ambrosio D, Piccoli M, Frati L, Santoni A. Differential expression of granzyme A and granzyme B proteases and their secretion by fresh rat natural killer cells (NK) and lymphokine-activated killer cells with NK phenotype (LAK-NK). Eur J Immunol (1992) 22:1049–53. doi: 10.1002/eji.1830220426
7. Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol (2008) 26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404
8. Hendel A, Hiebert PR, Boivin WA, Williams SJ, Granville DJ. Granzymes in age-related cardiovascular and pulmonary diseases. Cell Death Differ (2010) 17:596–606. doi: 10.1038/cdd.2010.5
9. Jin J, Li X, Hu B, Kim C, Cao W, Zhang H, et al. FOXO1 deficiency impairs proteostasis in aged T cells. Sci Adv (2020) 6:eaba1808. doi: 10.1126/sciadv.aba1808
10. Bulati M, Buffa S, Martorana A, Candore G, Lio D, Caruso C, et al. Trafficking phenotype and production of granzyme B by double negative B cells (IgG(+)IgD(-)CD27(-)) in the elderly. Exp Gerontol (2014) 54:123–9. doi: 10.1016/j.exger.2013.12.011
11. Kim WJ, Kim H, Suk K, Lee WH. Macrophages express granzyme B in the lesion areas of atherosclerosis and rheumatoid arthritis. Immunol Lett (2007) 111:57–65. doi: 10.1016/j.imlet.2007.05.004
12. Vernooy JH, Moller GM, van Suylen RJ. Increased granzyme A expression in type II pneumocytes of patients with severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2007) 175:464–72. doi: 10.1164/rccm.200602-169OC
13. Pardo J, Wallich R, Ebnet K. Granzyme B is expressed in mouse mast cells in vivo and in vitro and causes delayed cell death independent of perforin. Cell Death Differ (2007) 14:1768–79. doi: 10.1038/sj.cdd.4402183
14. Choy JC, McDonald PC, Suarez AC. Granzyme B in atherosclerosis and transplant vascular disease: association with cell death and atherosclerotic disease severity. Mod Pathol (2003) 16:460–70. doi: 10.1097/01.MP.0000067424.12280.BC
15. Hernandez-Pigeon H, Jean C, Charruyer A. Human keratinocytes acquire cellular cytotoxicity under UV-B irradiation. Implication of granzyme B and perforin. J Biol Chem (2006) 281:13525–32. doi: 10.1074/jbc.M512694200
16. Horiuchi K, Saito S, Sasaki R. Expression of granzyme B in human articular chondrocytes. J Rheumatol (2003) 30:1799–810.
17. Hu SX, Wang S, Wang JP, Mills GB, Zhou Y, Xu HJ. Expression of endogenous granzyme B in a subset of human primary breast carcinomas. Br J Cancer (2003) 89(1):135–9. doi: 10.1038/sj.bjc.6601051
18. D’Eliseo D, Pisu P, Romano C, Tubaro A, De Nunzio C, Morrone S, et al. Granzyme B is expressed in urothelial carcinoma and promotes cancer cell invasion. Int J Cancer (2010) 127:1283–94. doi: 10.1002/ijc.25135
19. D’Eliseo D, Manzi L, Merendino N, Velotti F. Docosahexaenoic acid inhibits invasion of human RT112 urinary bladder and PT45 pancreatic carcinoma cells via down-modulation of granzyme B expression. J Nutr Biochem (2012) 23:452–7. doi: 10.1016/j.jnutbio.2011.01.010
20. Fang Y, Herrick EJ, Nicholl MB. A possible role for perforin and granzyme B in resveratrol-enhanced radiosensitivity of prostate cancer. J Androl (2012) 33:752–60. doi: 10.2164/jandrol.111.015164
21. D’Eliseo D, Di Rocco G, Loria R, Soddu S, Santoni A, Velotti F. Epitelial-to-mesenchimal transition and invasion are upmodulated by tumor-expressed granzyme B and inhibited by docosahexaenoic acid in human colorectal cancer cells. J Exp Clin Cancer Res (2016) 35:24. doi: 10.1186/s13046-016-0302-6
22. Ngan DA, Vickerman SV, Granville DJ, Man SF, Sin DD. The possible role of granzyme B in the pathogenesis of chronic obstructive pulmonary disease. Ther Adv Respir Dis (2009) 3:113–29. doi: 10.1177/1753465809341965
23. Hodge S, Hodge G, Nairn J, Holmes M, Reynolds PN. Increased airway granzyme b and perforin in current and ex-smoking COPD subjects. COPD (2006) 3:179–87. doi: 10.1080/15412550600976868
24. Bem RA, Bos AP, Bots M, Wolbink AM, van Ham SM, Medema JP, et al. Activation of the granzyme pathway in children with severe respiratory syncytial virus infection. Pediatr Res (2008) 63:650–5. doi: 10.1203/PDR.0b013e31816fdc32
25. Bem RA, van Woensel JB, Lutter R, Domachowske JB, Medema JP, Rosenberg HF, et al. Granzyme A- and B-cluster deficiency delays acute lung injury in pneumovirus-infected mice. J Immunol (2010) 184:931–8. doi: 10.4049/jimmunol.0903029
26. Chamberlain CM, Ang LS, Boivin WA, Cooper DM, Williams SJ, Zhao H, et al. Perforin-independent extracellular granzyme B activity contributes to abdominal aortic aneurysm. Am J Pathol (2010) 176:1038–49. doi: 10.2353/ajpath.2010.090700
27. Kim WD, Chi HS, Choe KH, Oh YM, Lee SD, Kim KR, et al. A possible role for CD8+ and non-CD8+ cell granzyme B in early small airway wall remodelling in centrilobular emphysema. Respirology (2013) 18:688–96. doi: 10.1111/resp.12069
28. Buzza MS, Zamurs L, Sun J, Bird CH, Smith AI, Trapani JA, et al. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J Biol Chem (2005) 280:23549–58. doi: 10.1074/jbc.M412001200
29. Hiebert PR, Granville DJ. Granzyme B in injury, inflammation, and repair. Trends Mol Med (2012) 18:732–41. doi: 10.1016/j.molmed.2012.09.009
30. Boivin WA, Shackleford M, Vanden Hoek A, Zhao H, Hackett TL, Knight DA, et al. Granzyme B cleaves decorin, biglycan and soluble betaglycan, releasing active transforming growth factor-β1. PLoS One (2012) 7:e33163. doi: 10.1371/journal.pone.0033163
31. Wensink AC, Hack CE, Bovenschen N. Granzymes regulate proinflammatory cytokine responses. J Immunol (2015) 194:491–7. doi: 10.4049/jimmunol.1401214
32. Shen Y, Cheng F, Sharma M, Merkulova Y, Raithatha SA, Parkinson LG, et al. Granzyme B Deficiency Protects against Angiotensin II-Induced Cardiac Fibrosis. Am J Pathol (2016) 186:87–100. doi: 10.1016/j.ajpath.2015.09.010
33. Yang H, Youm YH, Vandanmagsar B, Ravussin A, Gimble JM, Greenway F, et al. Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: implications for systemic inflammation and insulin resistance. J Immunol (2010) 185:1836–45. doi: 10.4049/jimmunol.1000021
34. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med (2009) 15:914–20. doi: 10.1038/nm.1964
35. Buraschi S, Neill T, Iozzo RV. Decorin is a devouring proteoglycan: Remodeling of intracellular catabolism via autophagy and mitophagy. Matrix Biol (2019) 75-76:260–70. doi: 10.1016/j.matbio.2017.10.005
36. Kondo H, Hojo Y, Tsuru R, Nishimura Y, Shimizu H, Takahashi N, et al. Elevation of plasma granzyme B levels after acute myocardial infarction. Circ J (2009) 73:503–7. doi: 10.1253/circj.cj-08-0668
37. Skjelland M, Michelsen AE, Krohg-Sørensen K, Tennøe B, Dahl A, Bakke S, et al. Plasma levels of granzyme B are increased in patients with lipid-rich carotid plaques as determined by echogenicity. Atherosclerosis (2007) 195:e142–6. doi: 10.1016/j.atherosclerosis.2007.05.001
38. Freeman CM, Han MK, Martinez FJ, Murray S, Liu LX, Chensue SW, et al. Cytotoxic potential of lung CD8(+) T cells increases with chronic obstructive pulmonary disease severity and with in vitro stimulation by IL-18 or IL-15. J Immunol (2010) 184:6504–13. doi: 10.4049/jimmunol.1000006
39. Saito Y, Kondo H, Hojo Y. Granzyme B as a novel factor involved in cardiovascular diseases. J Cardiol (2011) 57:141–7. doi: 10.1016/j.jjcc.2010.10.001
40. Ikemoto T, Hojo Y, Kondo H, Takahashi N, Hirose M, Nishimura Y, et al. Plasma granzyme B as a predicting factor of coronary artery disease–clinical significance in patients with chronic renal failure. J Cardiol (2009) 54:409–15. doi: 10.1016/j.jjcc.2009.06.009
41. Oztas E, Ozler S, Tokmak A, Yilmaz N, Celik HT, Kazancı FH, et al. Increased levels of serum granzyme-B is associated with insulin resistance and increased cardiovascular risk in adolescent polycystic ovary syndrome patients. Eur J Obstet Gynecol Reprod Biol (2016) 198:89–93. doi: 10.1016/j.ejogrb.2016.01.009
42. El-Mesallamy HO, Hamdy NM, El-Etriby AK, Wasfey EF. Plasma granzyme B in ST elevation myocardial infarction versus non-ST elevation acute coronary syndrome: comparisons with IL-18 and fractalkine. Mediators Inflamm (2013) 2013:343268. doi: 10.1155/2013/343268
43. Tsuru R, Kondo H, Hojo Y, Gama M, Mizuno O, Katsuki T, et al. Increased granzyme B production from peripheral blood mononuclear cells in patients with acute coronary syndrome. Heart (2008) 94:305–10. doi: 10.1136/hrt.2006.110023
44. Hendel A, Cooper D, Abraham T, Zhao H, Allard MF, Granville DJ. Proteinase inhibitor 9 is reduced in human atherosclerotic lesion development. Cardiovasc Pathol (2012) 21:28–38. doi: 10.1016/j.carpath.2010.12.002
45. Hiebert PR, Boivin WA, Zhao H, McManus BM, Granville DJ. Perforin and granzyme B have separate and distinct roles during atherosclerotic plaque development in apolipoprotein E knockout mice. PLoS One (2013) 8:e78939. doi: 10.1371/journal.pone.0078939
46. Selathurai A, Deswaerte V, Kanellakis P, Tipping P, Toh BH, Bobik A, et al. Natural killer (NK) cells augment atherosclerosis by cytotoxic-dependent mechanisms. Cardiovasc Res (2014) 102:128–37. doi: 10.1093/cvr/cvu016
47. Garzón-Tituaña M, Arias MA, Sierra-Monzón JL, Morte-Romea E, Santiago L, Ramirez-Labrada A, et al. The Multifaceted Function of Granzymes in Sepsis: Some Facts and a Lot to Discover. Front Immunol (2020) 11:1054. doi: 10.3389/fimmu.2020.01054
48. Li Y, To K, Kanellakis P, Hosseini H, Deswaerte V, Tipping P, et al. CD4+ natural killer T cells potently augment aortic root atherosclerosis by perforin- and granzyme B-dependent cytotoxicity. Circ Res (2015) 116:245–54. doi: 10.1161/CIRCRESAHA.116.304734
49. Turner CT, Lim D, Granville DJ. Granzyme B in skin inflammation and disease. Matrix Biol (2019) 75-76:126–40. doi: 10.1016/j.matbio.2017.12.005
50. Boivin WA, Cooper DM, Hiebert PR, Granville DJ. Intracellular versus extracellular granzyme B in immunity and disease: challenging the dogma. Lab Invest (2009) 89:1195–220. doi: 10.1038/labinvest.2009.91
51. El Mesallamy HO, Hamdy NM, Mostafa DM, Amin AI. The serine protease granzyme B as an inflammatory marker, in relation to the insulin receptor cleavage in human obesity and type 2 diabetes mellitus. J Interferon Cytokine Res (2014) 34:179–86. doi: 10.1089/jir.2013.0059
52. Cimini FA, D’Eliseo D, Barchetta I, Bertoccini L, Velotti F, Cavallo MG. Increased circulating granzyme B in type 2 diabetes patients with low-grade systemic inflammation. Cytokine (2019) 115:104–8. doi: 10.1016/j.cyto.2018.11.019
53. Tak PP, Spaeny-Dekking L, Kraan MC, Breedveld FC, Froelich CJ, Hack CE. The levels of soluble granzyme A and B are elevated in plasma and synovial fluid of patients with rheumatoid arthritis (RA). Clin Exp Immunol (1999) 116:366–70. doi: 10.1046/j.1365-2249.1999.00881.x
54. Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, et al. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension (2013) 62:126–33. doi: 10.1161/HYPERTENSIONAHA.113.00689
55. Garg SK, Delaney C, Shi H, Yung R. Changes in adipose tissue macrophages and T cells during aging. Crit Rev Immunol (2014) 34:1–14. doi: 10.1615/critrevimmunol.2013006833
56. Dinarello CA. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr (2006) 83:447S–55S. doi: 10.1093/ajcn/83.2.447S
57. Borthwick LA. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin Immunopathol (2016) 38:517–34. doi: 10.1007/s00281-016-0559-z
58. Theocharis AD, Manou D, Karamanos NK. The extracellular matrix as a multitasking player in disease. FEBS J (2019) 286(15):2830–69. doi: 10.1111/febs.14818
59. Scott LE, Weinberg SH, Lemmon CA. Mechanochemical Signaling of the Extracellular Matrix in Epithelial-Mesenchymal Transition. Front Cell Dev Biol (2019) 7:135. doi: 10.3389/fcell.2019.00135
60. Miyazaki H, Kuwano K, Yoshida K, Maeyama T, Yoshimi M, Fujita M, et al. The perforin mediated apoptotic pathway in lung injury and fibrosis. J Clin Pathol (2004) 57:1292–8. doi: 10.1136/jcp.2003.015495.42
61. Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta (2014) 1842:446–62. doi: 10.1016/j.bbadis.2013.05.017
62. Taddei ML, Giannoni E, Fiaschi T, Chiarugi P. Anoikis: an emerging hallmark in health and diseases. J Pathol (2012) 226:380–93. doi: 10.1002/path.3000
63. Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol (1994) 124:619–26. doi: 10.1083/jcb.124.4.619
64. Steven M, Frisch M, Schaller B. Mechanisms that link the oncogenic epithelial–mesenchymal transition to suppression of anoikis. J Cell Sci (2013) 126:21–9. doi: 10.1242/jcs.120907
65. Zheng C, Livas T, Kyprianou. N. Anoikis and EMT: Lethal “Liaisons” during Cancer Progression. Crit Rev Oncog (2016) 21(3-4):155–68. doi: 10.1615/CritRevOncog.2016016955
66. Michel JB. Anoikis in the cardiovascular system: known and unknown extracellular mediators. Arterioscler Thromb Vasc Biol (2003) 23(12):2146–54. doi: 10.1161/01.ATV.0000099882.52647.E4
67. Fitzgerald ES, Luz NF, Jamieson AM. Competitive Cell Death Interactions in Pulmonary Infection: Host Modulation Versus Pathogen Manipulation. Front Immunol (2020) 11:814. doi: 10.3389/fimmu.2020.00814
68. Li M, Luan F, Zhao Y, Hao H, Zhou Y, Han W, et al. Epithelial-mesenchymal transition: An emerging target in tissue fibrosis. Exp Biol Med (Maywood) (2016) 241:1–13. doi: 10.1177/1535370215597194
69. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF -β: the master regulator of fibrosis. Nat Rev Nephrol (2016) 12:325–38. doi: 10.1038/nrneph.2016.48
70. Weis SM, Zimmerman SD, Shah M, Covell JW, Omens JH, Ross J, et al. A role for decorin in the remodeling of myocardial infarction. Matrix Biol (2005) 24:313–24. doi: 10.1016/j.matbio.2005.05.003
71. Vu TT, Marquez J, Le LT, Nguyen ATT, Kim HK, Han J. The role of decorin in cardiovascular diseases: more than just a decoration. Free Radic Res (2018) 52:1210–9. doi: 10.1080/10715762.2018.1516285
72. Nikaido T, Tanino Y, Wang X, Sato Y, Togawa R, Kikuchi M, et al. Serum decorin is a potential prognostic biomarker in patients with acute exacerbation of idiopathic pulmonary fibrosis. J Thorac Dis (2018) 10:5346–58. doi: 10.21037/jtd.2018.08.60
73. Giri SN, Hyde DM, Braun RK, Gaarde W, Harper JR, Pierschbacher MD. Antifibrotic effect of decorin in a bleomycin hamster model of lung fibrosis. Biochem Pharmacol (1997) 54:1205–16. doi: 10.1016/s0006-2952(97)00343-2
74. Kolb M, Margetts PJ, Sime PJ, Gauldie J. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am J Physiol Lung Cell Mol Physiol (2001) 280:L1327–34. doi: 10.1152/ajplung.2001.280.6.L1327
75. Kolb M, Margetts PJ, Galt T, Sime PJ, Xing Z, Schmidt M, et al. Transient transgene expression of decorin in the lung reduces the fibrotic response to bleomycin. Am J Respir Crit Care Med (2001) 163:770–7. doi: 10.1164/ajrccm.163.3.2006084
76. Svärd J, Røst TH, Sommervoll CEN, Haugen C, Gudbrandsen OA, Mellgren AE. Absence of the proteoglycan decorin reduces glucose tolerance in overfed male mice. Sci Rep (2019) 9:4614. doi: 10.1038/s41598-018-37501-x
77. Dufait I, Pardo J, Escors D, De Vlaeminck Y, Jiang H, Keyaerts M, et al. Perforin and Granzyme B Expressed by Murine Myeloid-Derived Suppressor Cells: A Study on Their Role in Outgrowth of Cancer Cells. Cancers (Basel) (2019) 11:808. doi: 10.3390/cancers11060808
78. Wang H, Sun Q, Wu Y, Wang L, Zhou C, Ma W, et al. Granzyme M expressed by tumor cells promotes chemoresistance and EMT in vitro and metastasis in vivo associated with STAT3 activation. Oncotarget (2015) 6:5818–31. doi: 10.18632/oncotarget.3461
79. Santiago L, Castro M, Sanz-Pamplona R, Garzòn M, Ramirez-Labrada A, Tapia E, et al. Extracellular Granzyme A Promotes Colorectal Cancer Development by Enhancing Gut Inflammation. Cell Rep (2020) 32:107847. doi: 10.1016/j.celrep.2020.10784777
80. Arias M, Martínez-Lostao L, Santiago L, Ferrandez A, Granville DJ, Pardo J. The Untold Story of Granzymes in Oncoimmunology: Novel Opportunities with Old Acquaintances. Trends Cancer (2017) 3:407–22. doi: 10.1016/j.trecan.2017.04.001
81. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest (2009) 119:1420–8. doi: 10.1172/JCI39104
82. Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G, et al. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J Am Coll Cardiol (2019) 73:190–209. doi: 10.1016/j.jacc.2018.09.089
83. Salton F, Volpe MC, Confalonieri M. Epithelial⁻Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina (Kaunas) (2019) 55:83. doi: 10.3390/medicina55040083
84. Rout-Pitt N, Farrow N, Parsons D, Donnelley M. Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir Res (2018) 19:136. doi: 10.1186/s12931-018-0834-8
85. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A (2006) 103:13180–5. doi: 10.1073/pnas.0605669103
86. Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforminggrowth factor-ss 1 – potential role in idiopathic pulmonary fibrosis. Am J Pathol (2005) 166:1321–32. doi: 10.1016/s0002-9440(10)62351-6
87. Hewlett JC, Kropski JA, Blackwell TS. Idiopathic pulmonary fibrosis: Epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol (2018) 71–72:112–27. doi: 10.1016/j.matbio.2018.03.021
88. Wu Z, Yang LL, Cai L, Zhang M, Cheng X, Yang X, et al. Detection of epithelial tomesenchymal transition in airways of a bleomycin induced pulmonary fibrosis model derivedfrom an alpha-smooth muscle actin-Cre transgenic mouse. Respir Res (2007) 8:1. doi: 10.1186/1465-9921-8-1
89. George PM, Wells AU, Jenkins RG. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir Med (2020) 15:S2213–2600(20)30225-3. doi: 10.1016/S2213-2600(20)30225-3
90. Agrati C, Sacchi A, Bordoni V, Cimini E, Notari S, Grassi G, et al. Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19). Cell Death Differ (2020) 8:1–12. doi: 10.1038/s41418-020-0572-6
91. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer (2012) 12:298–306. doi: 10.1038/nrc3245
92. Ten Berge RL, Oudejans JJ, Dukers DF, Meijer JW, Ossenkoppele GJ, Meijer CJ. Percentage of activated cytotoxic T-lymphocytes in anaplastic large cell lymphoma and Hodgkin’s disease: an independent biological prognostic marker. Leukemia (2001) 15:458–64. doi: 10.1038/sj.leu.2402045
93. Oudejans JJ, Harijadi H, Kummer JA, Tan IB, Bloemena E, Middeldorp JM, et al. High numbers of granzyme B/CD8-positive tumour-infiltrating lymphocytes in nasopharyngeal carcinoma biopsies predict rapid fatal outcome in patients treated with curative intent. J Pathol (2002) 198:468–75. doi: 10.1002/path.1236
94. Asano N, Oshiro A, Matsuo K, Kagami Y, Ishida F, Suzuki R, et al. Prognostic significance of T-cell or cytotoxic molecules phenotype in classical Hodgkin’s lymphoma: a clinicopathologic study. J Clin Oncol (2006) 24:4626–33. doi: 10.1200/JCO.2006.06.5342
95. Guzman VB, Silva ID, Brenna SM, Carvalho CR, Ribalta JC, Gerbase-Delima M. High levels of granzyme B expression in invasive cervical carcinoma correlates to poor response to treatment. Cancer Invest (2008) 26:499–503. doi: 10.1080/07357900701805678
96. Belfort-Mattos PN, Focchi GR, Speck NM, Taha NS, Carvalho CR, Ribalta JC. Immunohistochemical expression of granzyme B and vascular endothelial growth factor (VEGF) in normal uterine cervices and low and high grade squamous intraepithelial lesions. Eur J Gynaecol Oncol (2010) 31:459–61.
97. Manou D, Karamanos NK, Theocharis AD. Tumorigenic functions of serglycin: Regulatory roles in epithelial to mesenchymal transition and oncogenic signaling. Semin Cancer Biol (2020) 62:108–15. doi: 10.1016/j.semcancer.2019.07.004
98. Kaiserman D, Bird CH, Sun J, Matthews A, Ung K, Whisstock JC, et al. The major human and mouse granzymes are structurally and functionally divergent. J Cell Biol (2006) 175:619–30. doi: 10.1083/jcb.200606073
99. Casciola-Rosen L, Garcia-Calvo M, Bull HG, Becker JW, Hines T, Thornberry NA, et al. Mouse and human granzyme B have distinct tetrapeptide specificities and abilities to recruit the bid pathway. Biol Chem (2007) 282:4545–52. doi: 10.1074/jbc.M606564200
100. Cullen SP, Adrain C, Lüthi AU, Duriez PJ, Martin SJ. Human and murine granzyme B exhibit divergent substrate preferences. J Cell Biol (2007) 176:435–44. doi: 10.1083/jcb.200612025
101. Meschini R, D’Eliseo D, Filippi S, Bertini L, Bizzarri BM, Botta L, et al. Tyrosinase-Treated Hydroxytyrosol-Enriched Olive Vegetation Waste with Increased Antioxidant Activity Promotes Autophagy and Inhibits the Inflammatory Response in Human THP-1 Monocytes. J Agric Food Chem (2018) 66(46):12274–84. doi: 10.1021/acs.jafc.8b03630
102. D’Eliseo D, Pannucci E, Bernini R, Campo M, Romani A, Santi L, et al. In vitro studies on anti-inflammatory activities of kiwifruit peel extract in human THP-1 monocytes. J Ethnopharmacol (2019) 233:41–6. doi: 10.1016/j.jep.2018.12.044
103. Turner CT, Hiroyasu S, Granville DJ. Granzyme B as a therapeutic target for wound healing. Expert Opin Ther Targets (2019) 23:745–54. doi: 10.1080/14728222.2019.1661380
104. Shen Y, Zeglinski MR, Turner CT, Raithatha SA, Wu Z, Russo V, et al. Topical small molecule granzyme B inhibitor improves remodeling in a murine model of impaired burn wound healing. Exp Mol Med (2018) 50:1–11. doi: 10.1038/s12276-018-0095-0
105. Ikram S, Ahmad J, Durdagi SJ. Screening of FDA approved drugs for finding potential inhibitors against Granzyme B as a potent drug-repurposing target. Mol Graph Model (2020) 95:107462. doi: 10.1016/j.jmgm.2019.107462
106. Aslam MS, Yuan L. Serpina3n: Potential drug and challenges, mini review. J Drug Targeting (2020) 28:368–78. doi: 10.1080/1061186X.2019.1693576
107. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
Keywords: granzyme B, inflammatory cytokines, inflammaging, extracellular matrix remodeling, anoikis, apoptosis, epithelial-to-mesenchymal transition, fibrosis
Citation: Velotti F, Barchetta I, Cimini FA and Cavallo MG (2020) Granzyme B in Inflammatory Diseases: Apoptosis, Inflammation, Extracellular Matrix Remodeling, Epithelial-to-Mesenchymal Transition and Fibrosis. Front. Immunol. 11:587581. doi: 10.3389/fimmu.2020.587581
Received: 26 July 2020; Accepted: 09 October 2020;
Published: 11 November 2020.
Edited by:
Helena Stabile, Sapienza University of Rome, ItalyReviewed by:
Steven Frisch, West Virginia University, United StatesCopyright © 2020 Velotti, Barchetta, Cimini and Cavallo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Velotti, dmVsb3R0aUB1bml0dXMuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.