 Jason T. Weinfurter1
Jason T. Weinfurter1 Michael E. Graham2Adam J. Ericsen2
Michael E. Graham2Adam J. Ericsen2 Lea M. Matschke1Sian Llewellyn-Lacey3David A. Price3,4Roger W. Wiseman2
Lea M. Matschke1Sian Llewellyn-Lacey3David A. Price3,4Roger W. Wiseman2 Matthew R. Reynolds1,2*
Matthew R. Reynolds1,2*- 1Department of Pathobiological Sciences, School of Veterinary Medicine, University of Wisconsin-Madison, Madison, WI, United States
- 2Wisconsin National Primate Research Center, University of Wisconsin-Madison, Madison, WI, United States
- 3Division of Infection and Immunity, Cardiff University School of Medicine, Cardiff, United Kingdom
- 4Systems Immunity Research Institute, Cardiff University School of Medicine, Cardiff, United Kingdom
Allogeneic hematopoietic stem cell transplants can lead to dramatic reductions in human immunodeficiency virus (HIV) reservoirs. This effect is partially mediated by donor T cells recognizing lymphocyte-expressed minor histocompatibility antigens (mHAgs). The potential to mark malignant and latently infected cells for destruction makes mHAgs attractive targets for cellular immunotherapies. However, testing such HIV reservoir reduction strategies will likely require preclinical studies in non-human primates (NHPs). In this study, we used a combination of alloimmunization, whole exome sequencing, and bioinformatics to identify an mHAg in Mauritian cynomolgus macaques (MCMs). We mapped the minimal optimal epitope to a 10-mer peptide (SW10) in apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3C (APOBEC3C) and determined the major histocompatibility complex class I restriction element as Mafa-A1∗063, which is expressed in almost 90% of MCMs. APOBEC3C SW10-specific CD8+ T cells recognized immortalized B cells but not fibroblasts from an mHAg-positive MCM. These results provide a framework for identifying mHAgs in a non-transplant setting and suggest that APOBEC3C SW10 could be used as a model antigen to test mHAg-targeted therapies in NHPs.
Introduction
The establishment of long-lived viral reservoirs is a major obstacle to developing curative therapies for human immunodeficiency virus (HIV) (1–4). These latent reservoirs are unaffected by antiretroviral therapy (ART) and readily reactivate upon cessation of treatment (5, 6). Moreover, it has been estimated that over 60 years of continuous ART would be needed to decay latently infected cells to extinction (2, 7). Strategies are therefore being developed to reduce or eliminate viral reservoirs to attain sustained ART-free remission (8, 9).
Human immunodeficiency virus-infected patients who develop hematological malignancies are commonly treated with allogeneic hematopoietic stem cell transplants (allo-HSCTs), which frequently lead to dramatic decreases in latent viral reservoirs. In two widely publicized cases, the “Berlin” and “London” patients achieved ART-free remission after receiving allo-HSCTs from donors homozygous for the CCR5Δ32 gene mutation (10, 11). The absence of functional CCR5 co-receptors on donor-derived immune cells was undoubtedly a crucial factor in these remarkable successes, preventing the HIV reservoir from reseeding the new hematopoietic system. However, graft-versus-host (GvH) responses likely also contributed to the elimination of endogenous viral reservoirs (10, 12). Interestingly, the London patient received reduced-intensity conditioning prior to allo-HSCT, leaving a residual pool of cancerous and latently HIV-infected cells (10, 13). In this setting, allogeneic T cell responses potentially eliminated both malignant and HIV-infected cells by simultaneously mediating “graft-versus-leukemia” and “graft-versus-HIV” effects. Intriguingly, similar reductions in latent viral reservoirs have also been achieved with HSCs from donors expressing wild-type CCR5 (14–16). The most notable examples are the “Boston” patients, who received allo-HSCTs under the cover of ART and exhibited prolonged ART-free remission (14). Collectively, these anecdotal cases demonstrate that alloreactive T cells can attack and destroy endogenous cells latently infected with HIV.
Alloreactive T cells frequently recognize minor histocompatibility antigens (mHAgs). mHAgs are polymorphic peptides presented by major histocompatibility complex (MHC) molecules and recognized as “foreign” by allogeneic T cells (17). The expression profile of mHAg-encoding genes across tissues is known to affect the outcome of GvH responses (18, 19). Expression of mHAgs across a broad range of tissues can result in toxic GvH reactions and potentially fatal GvH disease (GvHD). Conversely, mHAgs presented exclusively by recipient leukocytes, including malignant and latently infected cells, can elicit beneficial GvH responses that eliminate tumors and latently infected cells alike. Immunotherapies targeting leukocyte-derived mHAgs may therefore provide a novel approach to eliminating cells latently infected with HIV. Further studies are required to advance this concept, however, and the potential efficacy of graft-versus-HIV responses can only realistically be assessed in non-human primates (NHPs) (20).
Non-human primates are common preclinical organ transplant and infectious disease models. Despite their ubiquitous use in research, no mHAgs have been identified in NHPs. In contrast, multiple human mHAgs have been identified using peptide elution, cDNA library screens, and genetic linkage analyses incorporating large panels of immortalized B cells (21). Recently, these labor-intensive methods have been complemented by advances in next-generation sequencing technologies, accelerating the pace of mHAg discovery (22, 23).
Most outbred NHP populations are genetically diverse, making it challenging to identify cohorts of animals with one or more matches at the MHC class I (MHC-I) locus. In contrast, Mauritian-origin cynomolgus macaques (MCMs) descended from a small founder population approximately 400 years ago (24), resulting in limited genetic diversity, even within highly polymorphic loci (25–27). Indeed, only seven major MHC haplotypes, designated M1 through M7, have been identified in MCMs.
The unique population genetics of MCMs provides an opportunity to map mHAgs in a relevant NHP transplant and HIV cure model. In this report, we describe the identification and characterization of an mHAg, apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3C (APOBEC3C) SW10, in MCMs. This epitope is restricted by the MHC- I molecule Mafa-A1∗063, which is expressed by almost 90% of MCMs (28). Our data suggest that the APOBEC3C SW10 mHAg may serve as a useful model antigen for interventional studies of mHAg-targeted therapies in NHPs.
Materials and Methods
Ethics Statement and Animal Care
Cynomolgus macaques (Macaca fascicularis) used in this study were cared for by the staff at the Wisconsin National Primate Research Center according to the regulations and guidelines of the University of Wisconsin Institutional Animal Care and Use Committee, which approved this study (protocol g00695) in accordance with the recommendations of the Weatherall Report and the principles described in the National Research Council’s Guide for the Care and Use of Laboratory Animals. Macaques were housed in enclosures with at least 4.3, 6.0, or 8.0 sq. ft. of floor space, measuring 30, 32, or 36 inches high and containing a tubular polyvinyl-chloride or stainless-steel perch. Each enclosure was equipped with a horizontal or vertical sliding door, an automatic water lixit, and a stainless-steel feed hopper. Macaques were fed twice daily using a nutritional plan based on recommendations published by the National Research Council. Feeding strategies were individually tailored to the age and physical condition of each animal. Carbohydrate, energy, fat, fiber (10%), mineral, protein, and vitamin requirements were provided in an extruded dry diet (2050 Teklad Global 20% Protein Primate Diet). Dry diets were supplemented with fruits, vegetables, and other edible objects (e.g., nuts, cereals, seed mixtures, yogurt, peanut butter, popcorn, and marshmallows) to provide variety and to inspire foraging and other species-specific behaviors. To further promote psychological well-being, macaques were provided with food enrichment, human-to-monkey interaction, structural enrichment, and manipulanda. Environmental enrichment objects were selected to minimize the chances of pathogen transmission from one animal to another and from animals to care staff. Macaques were evaluated by trained animal care staff at least twice daily for signs of pain, distress, and illness by observing appetite, stool quality, activity level, and physical condition. Animals presenting abnormally for any of these clinical parameters were provided with appropriate care by attending veterinarians. Macaques were sedated with ketamine before each experimental procedure and reversed with atipamezole after each experimental procedure. Animals were monitored regularly until fully recovered from anesthesia.
Immunization of Cynomolgus Macaques
Peripheral blood mononuclear cells (PBMCs) were separated from EDTA-anticoagulated blood by density gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare). Freshly isolated PBMCs from four donor MCMs heterozygous for the M1 and M2 MHC haplotypes were pooled and resuspended in RPMI-1640 containing 10% fetal calf serum (FCS; R10) and 5 μg/ml phytohemagglutinin-P (Sigma-Aldrich). The culture was incubated overnight at 37°C in a 5% CO2 atmosphere. Activated PBMCs were harvested the following morning, washed twice with phosphate-buffered saline (PBS), resuspended in PBS, and loaded into tuberculin syringes. Each macaque was then immunized with up to 4 × 107 activated PBMCs as described previously (29).
Growing Bulk T Cell Lines to Potential mHAgs
Minor histocompatibility antigens-specific T cell lines were generated from cryopreserved PBMCs isolated from the alloimmunized macaques and thawed as described previously (30). Briefly, 5 × 106 PBMCs were combined with 5 × 106 irradiated immortalized B lymphoblastoid cells (BLCs) from MHC-identical donor MCMs in 5 ml of RPMI-1640 containing 15% FCS (R15) and 10 ng/ml recombinant human IL-7 (R&D Systems). Cultures were supplemented 2 days later with 1.25 ml of R15 containing 100 U/ml recombinant human IL-2 (R&D Systems; R15-100). R15-100 was added every 2–3 days thereafter, and T cell lines were stimulated weekly with equal numbers of irradiated BLCs from MHC-identical donor MCMs. Peptide-specific T cell lines were grown similarly using autologous irradiated BLCs pulsed with 15-mers A3C-A and A3C-B (GenScript).
Limiting Dilution Cloning
Bulk T cell lines were counted with Trypan Blue exclusion dye and resuspended at 1 cell/ml in R15-100. Each cell line was distributed across three 96-well plates at a mean of 0.2 cells/well and placed at 37°C in a 5% CO2 incubator. T cell clones were fed twice weekly by replacing half of the media volume with R15-100 and restimulated periodically with a mixture of irradiated MHC-identical donor BLCs.
ELISpot Assays
ELISpot assays were performed using precoated monkey IFN-γ ELISpotPLUS Kits according to the manufacturer’s instructions (Mabtech). Briefly, 5,000 clonal T cells and 5,000 fibroblasts or BLCs derived from individual MHC-identical MCMs were added to each well in 200 μl of R10. Positive control wells contained concanavalin A (Sigma-Aldrich). Alternatively, 5,000 bulk T cells and 5,000 fibroblasts or BLCs derived from individual MHC-identical MCMs were added to each well in 200 μl of R10. Positive control wells contained autologous BLCs and the APOBEC3C SW10 peptide. All tests were performed in triplicate. Plates were incubated overnight at 37°C in a 5% CO2 atmosphere and imaged using an AID ELISpot Reader (Autoimmun Diagnostika GmbH). Differences in the number of IFN-γ spot-forming cells (SFCs) after stimulation with allogeneic versus autologous fibroblasts or BLCs were assessed for significance using a one-way ANOVA with Tukey’s test for multiple comparisons in Prism version 5.0 (GraphPad Software Inc.).
Mapping the APOBEC3C SW10 Epitope
The mHAg epitope in APOBEC3C was mapped using a T cell line generated from an alloimmunized MCM and autologous irradiated BLCs pulsed with the 15-mer peptides A3C-A and A3C-B (GenScript). The minimal optimal epitope was determined using overlapping 8-mer, 9-mer, 10-mer, and 11-mer peptides (GenScript). Briefly, 100,000 mHAgneg BLCs were incubated for 1 h with each peptide at a final concentration of 1 μM and then washed twice with R10. Next, 100,000 APOBEC3C-specific T cells were added to each tube containing R10 with 10 μg/ml brefeldin A (Sigma-Aldrich). Cells were then incubated for 5 h at 37°C in a 5% CO2 atmosphere, washed twice with PBS, and stained with LIVE/DEAD Fixable Near-IR dye according to the manufacturer’s instructions (Thermo Fisher Scientific). After a further wash with R10, cells were stained for 30 min at room temperature with anti-CD3 Alexa 700 (clone SP34-2; BD Biosciences) and anti-CD8 Pacific Blue (clone RPA-T8; BD Biosciences). Cells were then washed twice with PBS containing 2% FCS (FACS buffer), fixed with 1% paraformaldehyde (PFA), permeabilized with FACS buffer containing 0.1% saponin, and incubated for 1 h at room temperature with IFN-γ FITC (clone 4S.B3; BD Biosciences). Stained cells were washed twice with FACS buffer and resuspended in 1% PFA. Data were acquired using an LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software version 10 (FlowJo LLC).
Exome Sequencing
Exome sequencing was performed as described previously (31, 32). Briefly, genomic DNA was isolated from the blood of 21 MCMs and enriched for exon-containing fragments using a custom target-capture probe set, SeqCap EZ HGSC VCRome2.1 (Roche). After enrichment, Illumina barcodes were appended using standard procedures (33), and tagged samples were sequenced using an Illumina HiSeq 2500. The exome sequences are available in the sequence read archive (SRA) under accession numbers SAMN12325046–SAMN12325066.
Variant Segregation Analysis
Segregation analysis was performed using a custom-built OSX application entitled Variant Segregator. This tool provides a user interface to define comparison groups and generate filter variant call format (VCF) files, identifying genetic variants that segregate within the designated groups. Variant Segregator was used to identify candidate single-nucleotide polymorphisms (SNPs) through simple comparative scoring of each animal in the mHAgpos and mHAgneg groups, relative to the Mmul-8.0.1 rhesus macaque reference genome, which was the most inclusive macaque reference genome available. The scoring system assigned 5 points for each macaque with a homozygous SNP mismatch relative to the reference genome, 3 points for heterozygous SNP mismatches, and zero points for homozygous SNP matches. The final segregation score was defined as the point sum in the mHAgpos group minus the point sum in the mHAgneg group. Segregation scores were used to identify SNPs exclusively present in mHAgpos MCMs and prioritize SNPs in polymorphic genes. Variant Segregator is available upon request from the corresponding author.
Results
Generation of mHAg-Specific CD8+ T Cell Clones
We identified 21 MCMs heterozygous for the M1 and M2 MHC haplotypes (M1/M2). mHAg-reactive T cells were stimulated in vivo by alloimmunizing four M1/M2 MCMs (vaccinees) with a mixture of PBMCs from four MHC-identical MCMs (donors). Alloreactive T cells were then isolated by incubating PBMCs from the vaccinees with a mixture of irradiated immortalized donor BLCs. After 3 weeks, these T cell lines reacted to allogeneic but not autologous BLCs in IFN-γ ELISpot assays, indicating specific recognition of donor-restricted mHAgs.
To identify individual mHAgs, we isolated T cell clones via limiting dilution of the alloreactive T cell lines. After 8 weeks in culture, these T cell clones (n = 69) displayed unique patterns of binary reactivity against panels of MHC-matched allogeneic BLCs (n ≤ 10) in IFN-γ ELISpot assays, indicating specific recognition of distinct mHAgs (Figure 1 and Supplementary Table 1). These reactivity patterns were used to identify genomic polymorphisms associated with potential mHAgs.
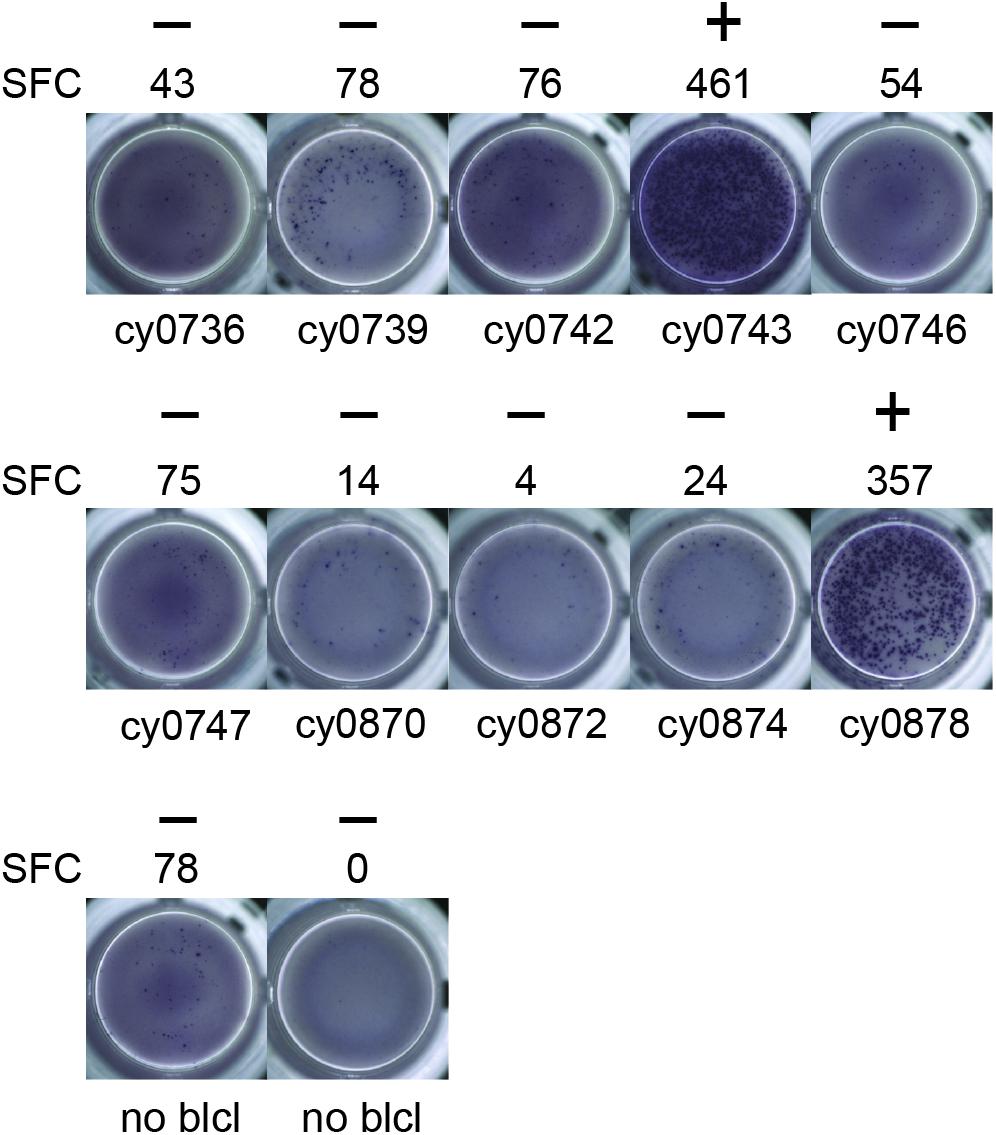
Figure 1. Alloreactive T cell clones differentially recognize allogeneic BLCs. Representative data from T cell clone 11 demonstrating strong reactivity against two of the ten allogeneic BLCs. Equal numbers of T cells and BLCs were incubated overnight in an IFN-γ ELISpot assay. (+) and (−) signify animals that were placed into mHAgpos and mHAgneg groups for WES segregation analysis, respectively. SFC, spot-forming cell.
Identification of Minor Histocompatibility Antigen-Associated SNPs in APOBEC3C
We reasoned that comparing the genomic sequences of MCMs whose BLCs were or were not recognized by alloreactive T cell clones would simplify the winnowing of germline polymorphisms associated with mHAgs. Accordingly, we performed whole exome sequencing (WES) using a probe set specially designed to capture MCM coding sequences, which enabled the generation of SNP profiles for each MCM. Using a custom-built analysis tool, we then parsed the WES data for non-synonymous SNPs (ns-SNPs) that distinguished T cell clone-specific mHAgpos and mHAgneg groups of BLCs. We prioritized ns-SNPs present only in the mHAgpos group for further analysis, surmising these ns-SNPs likely encoded the relevant mHAgs.
We focused our mHAg mapping efforts on the ns-SNPs associated with T cell clones 2, 4, 11, and 14. The segregation analysis identified SNPs encoding amino acid polymorphisms in the IGHM, OR4K3, APOBEC3C, and IL20RA or MAP3K5 genes (Supplementary Table 2). For each candidate mHAg, we identified the 11 amino acids upstream and the 11 amino acids downstream of the corresponding SNPs and synthesized 15-mer peptides overlapping by 11 amino acids. Each 15-mer peptide was then pulsed onto mHAgneg BLCs at a concentration of 1 μM and incubated with the respective T cell clone. In IFN-γ ELISpot assays, T cell clone 11 (T11) strongly recognized two of the three 15-mers containing an arginine-to-leucine amino acid change, relative to the mHAgneg sequence, in APOBEC3C (Figure 2A). The other 15-mer peptides failed to elicit IFN-γ production in parallel assays with T2, T4, or T14. We therefore concentrated on identifying the minimal optimal epitope recognized by T11.
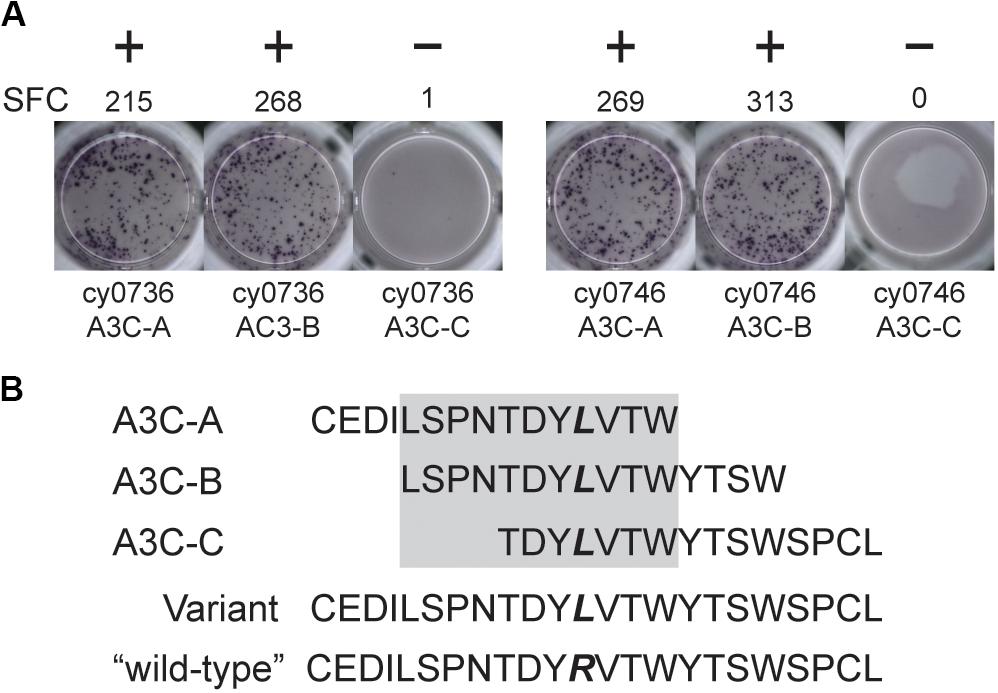
Figure 2. An mHAg-reactive T cell clone recognizes an amino acid variant in APOBEC3C. (A) T cell clone T11 was incubated with mHAgneg BLCs from cy0736 (left) or cy0746 (right) pulsed with overlapping 15-mer peptides containing an arginine-to-leucine amino acid change relative to the mHAgneg sequence in an IFN-γ ELISpot assay. (B) Sequences of the APOBEC3C (A3C) peptides used to pulse the mHAgneg BLCs in panel (A). The variant amino acid is highlighted in bold. The gray box corresponds to the putative region containing the mHAg epitope. (+) and (−) signify wells determined to be positive or negative, respectively. SFC, spot-forming cell.
Identification of the Minimal Optimal mHAg Epitope in APOBEC3C
We reasoned that the mHAg epitope recognized by T11 was present in each of the two stimulatory APOBEC3C 15-mer peptides, denoted as A3C-A and A3C-B, but not in the non-stimulatory APOBEC3C 15-mer peptide, denoted as A3C-C (Figure 2B). Accordingly, we synthesized a series of overlapping 8-mer, 9-mer, 10-mer, and 11-mer peptides spanning the 11 amino acids present in A3C-A and A3C-B to map the minimal optimal epitope in APOBEC3C. We also synthesized an 11-mer peptide and a 15-mer peptide matching the reference wild-type sequence, with an arginine replacing the variant leucine (Figure 2B). In intracellular cytokine staining (ICS) assays, a bulk T cell line generated against A3C-A and A3C-B strongly recognized the 11-mer peptide LW11, which matched the intersecting region of the overlapping 15-mer peptides (Figure 3). A similar response was observed with the 10-mer peptide SW10. In contrast, weaker responses were observed with all other peptides, including LT10.
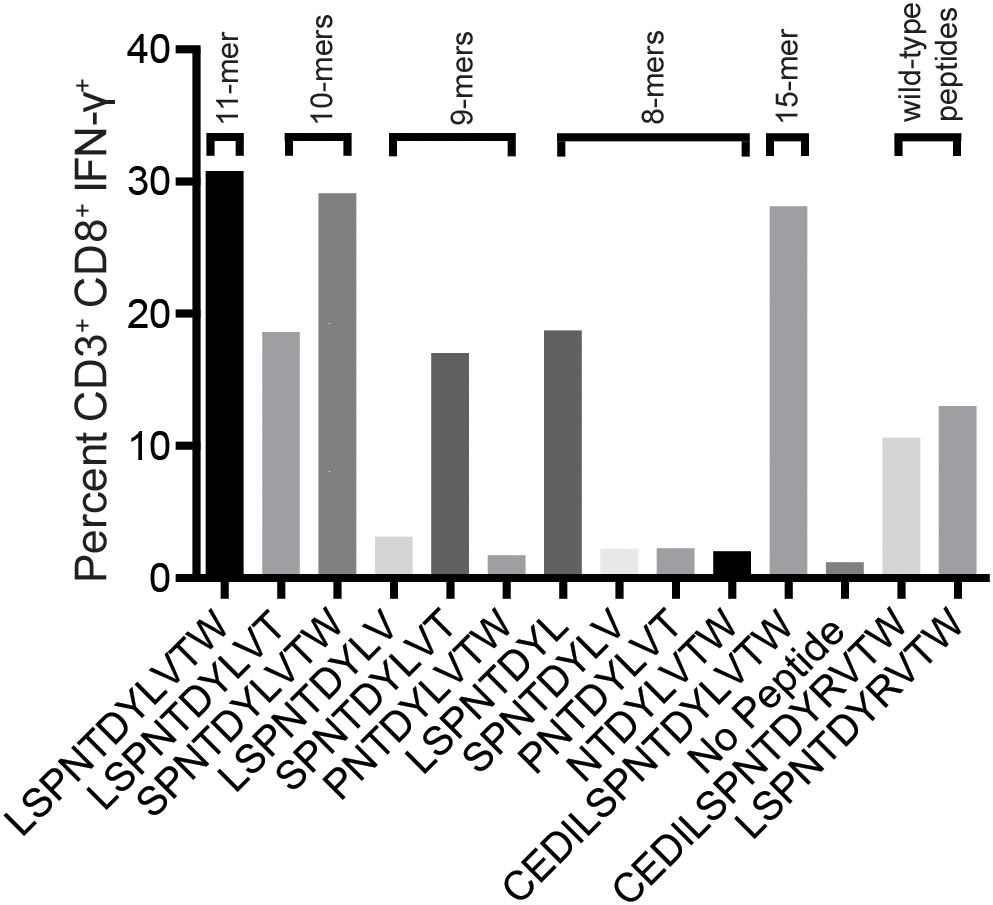
Figure 3. Select peptides corresponding to the APOBEC3C variant stimulate an mHAg-specific T cell line. An APOBEC3C mHAg-specific T cell line was incubated with an mHAgneg BLC line pulsed with peptides, including the 11-mer peptide corresponding to the shared region between the 15-mer peptides A3C-A and A3C-B (Figure 2B) and the corresponding wild-type 11-mer and 15-mer peptides. Unpulsed autologous BLCs were included as a negative control, and A3C-A peptide-pulsed autologous BLCs were included as a positive control. Displayed is the percentage of CD3+CD8+IFN-γ+ cells determined by ICS.
To confirm these results, we conducted similar ICS assays using serial dilutions of each peptide that elicited a response at a concentration of 1 μM, namely LL8, ST9, LT10, SW10, and LW11. Sharp reductions in IFN-γ production were observed with decreasing concentrations of LL8, ST9, and LT10 (Figure 4A). In contrast, SW10 and LT11 elicited similar levels of IFN-γ production throughout the dilution series, which extended down to 1 nM. In further titration experiments, SW10 and LT11 exhibited largely equivalent dose-response curves down to a concentration of 1 fM, but SW10 elicited IFN-γ production more potently at concentrations of 1 nM and 100 pM, suggesting this peptide was the minimal optimal mHAg epitope in APOBEC3C (Figure 4B).
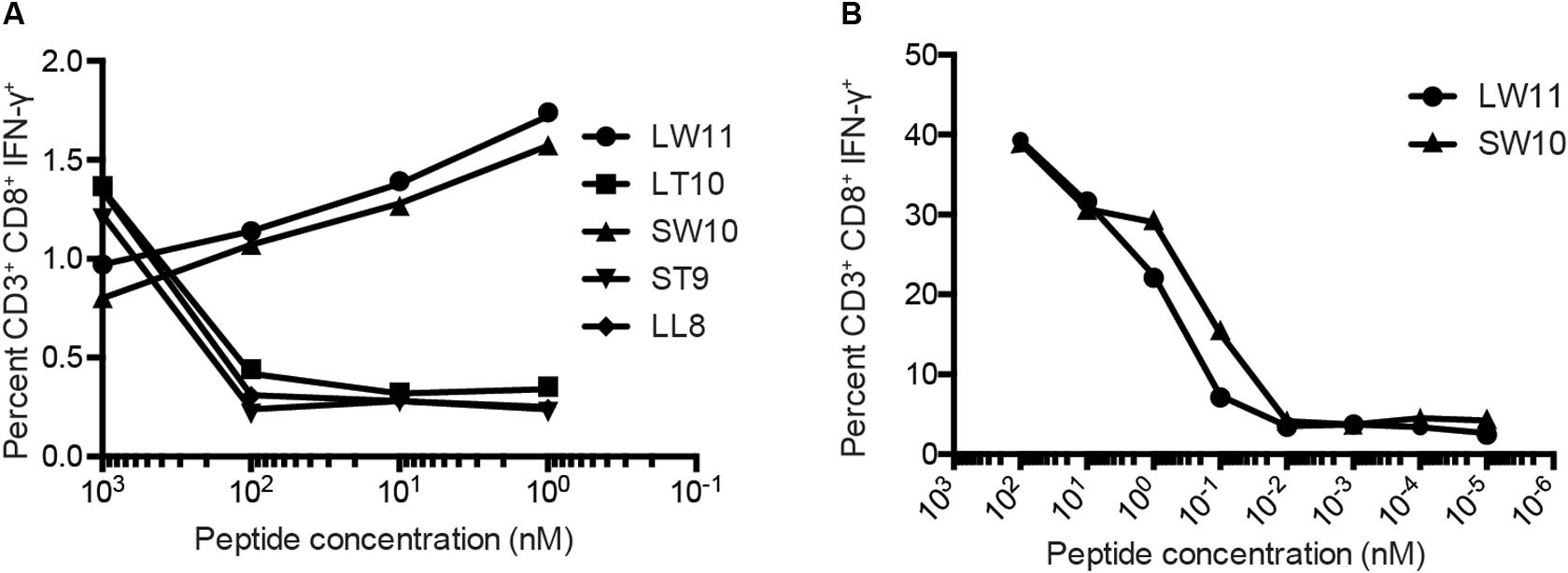
Figure 4. Fine mapping of the mHAg epitope in APOBEC3C. (A) An APOBEC3C mHAg-specific T cell line was incubated with mHAgneg BLCs pulsed with serial dilutions of the indicated peptides from 1 μM to 1 nM. (B) As in panel (A) with serial dilutions of the peptides SW10 and LW11 from 100 nM to 1 fM. Displayed is the percentage of CD3+CD8+IFN-γ+ cells determined by ICS.
MHC Class I Restriction of the mHAg Epitope in APOBEC3C
To determine the restricting MHC-I molecule for APOBEC3C SW10, we used three immortalized human MHC-I-null cell lines individually expressing Mafa-A or Mafa-B allomorphs from the MCM M2 MHC haplotype (34). Each of these MHC-I “transferents” was pulsed with the SW10 peptide and incubated with the APOBEC3C-specific T cell line. In ICS assays, only T cells incubated with the peptide-pulsed Mafa-A1∗063 transferent induced IFN-γ production above background levels, indicating the formation of stable complexes with SW10 (Figure 5).
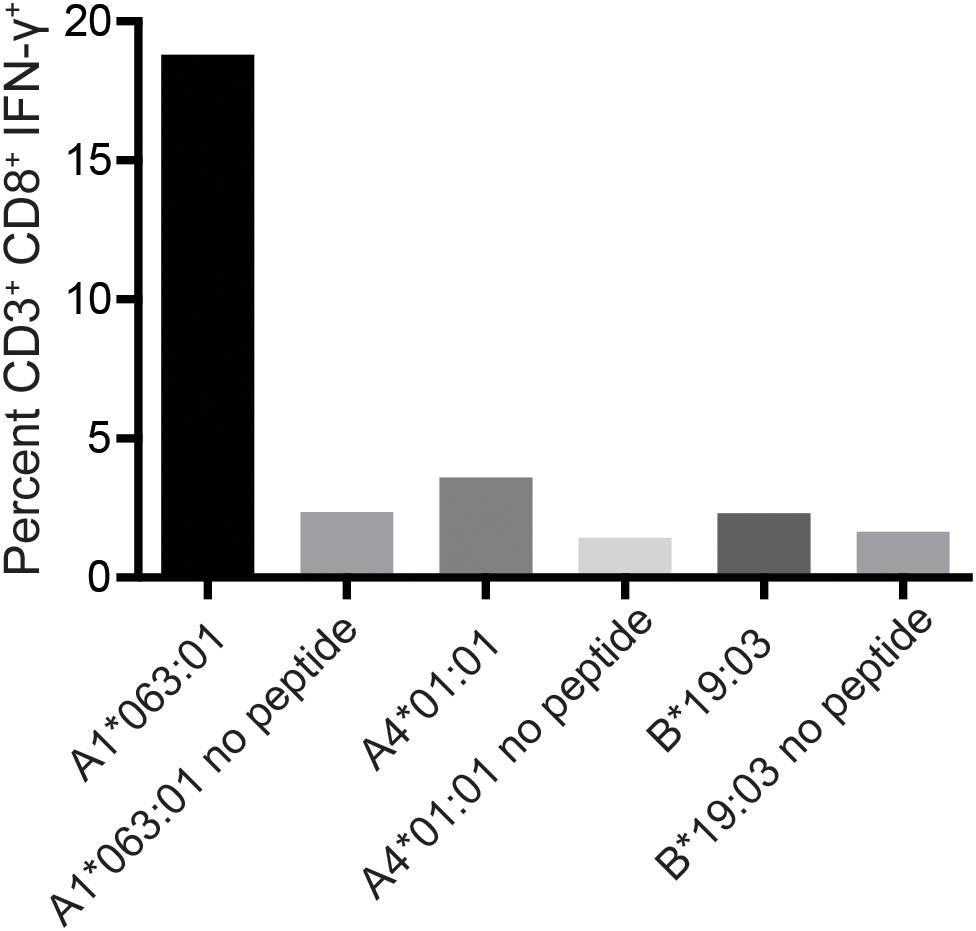
Figure 5. Restriction analysis of the mHAg epitope in APOBEC3C. An APOBEC3C mHAg-specific T cell line was incubated with unpulsed or SW10 peptide-pulsed MHC-I transferents expressing Mafa-A1*063:01, Mafa-A4*01:01, or Mafa-B*19:03. Displayed is the percentage of CD3+CD8+IFN-γ+ cells determined by ICS.
The SW10 epitope incorporates the previously described Mafa-A1∗063 peptide-binding motif, with a serine at position one, a proline at position two, an asparagine at position three, and a tryptophan at the carboxy terminus of the peptide (25, 35). We were therefore able to generate tetrameric complexes of Mafa-A1∗063/SW10. In line with the functional data, these recombinant antigen complexes robustly stained APOBEC3C-specific T cells, confirming Mafa-A1∗063 as the restriction element for SW10 (Figure 6).
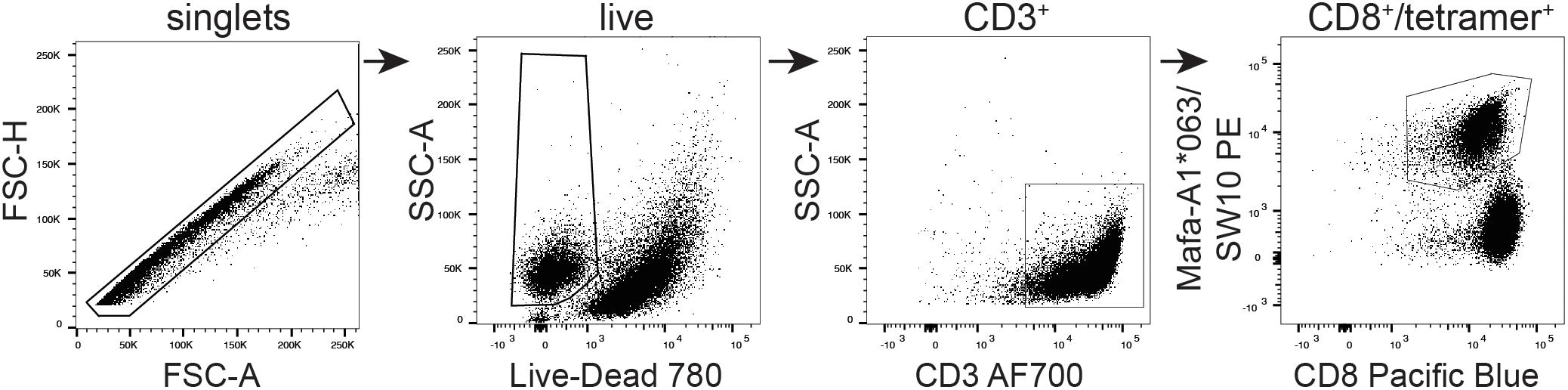
Figure 6. Tetrameric complexes of Mafa-A1*063/SW10 stain APOBEC3C SW10-specific CD8+ T cells. An APOBEC3C mHAg-specific T cell line was stained with PE-conjugated tetrameric complexes of Mafa-A1*063/SW10. Displayed is the flow cytometric gating strategy for data analysis, progressively selecting singlets, live cells, CD3+, and CD8+/tetramer+ cells.
Tissue Distribution of the APOBEC3C mHAg Epitope
The tissue distribution of mHAgs can be exploited for therapeutic gain (36). This strategy is epitomized by the infusion of allogeneic T cells to treat hematologic cancers, which operates via the selective targeting of mHAgs expressed by recipient leukocytes, including malignant cells, to eliminate tumors without causing severe GvHD. We therefore sought to determine whether immune recognition of the SW10 epitope is similarly limited to the hematopoietic system, given that CD4+ T cells are known to express high levels of APOBEC3C (37).
To test non-hematopoietic cells, we collected skin explants and isolated fibroblasts from the APOBEC3C SW10pos MCM, cy0743. In IFN-γ ELISpot assays, SW10-specific T cell lines isolated from two alloimmunized MCMs displayed greater reactivity against the mHAgpos BLCs compared with either the corresponding autologous BLCs or mHAgpos fibroblasts, suggesting limited expression of SW10 outside the hematopoietic system (Figure 7). The APOBEC3C SW10 epitope may therefore serve as a model hematopoietic tissue-restricted antigen in MCMs. Additional testing with inflammatory cytokine-treated fibroblasts and PBMCs will be required to confirm the precise expression profile of APOBEC3C SW10.
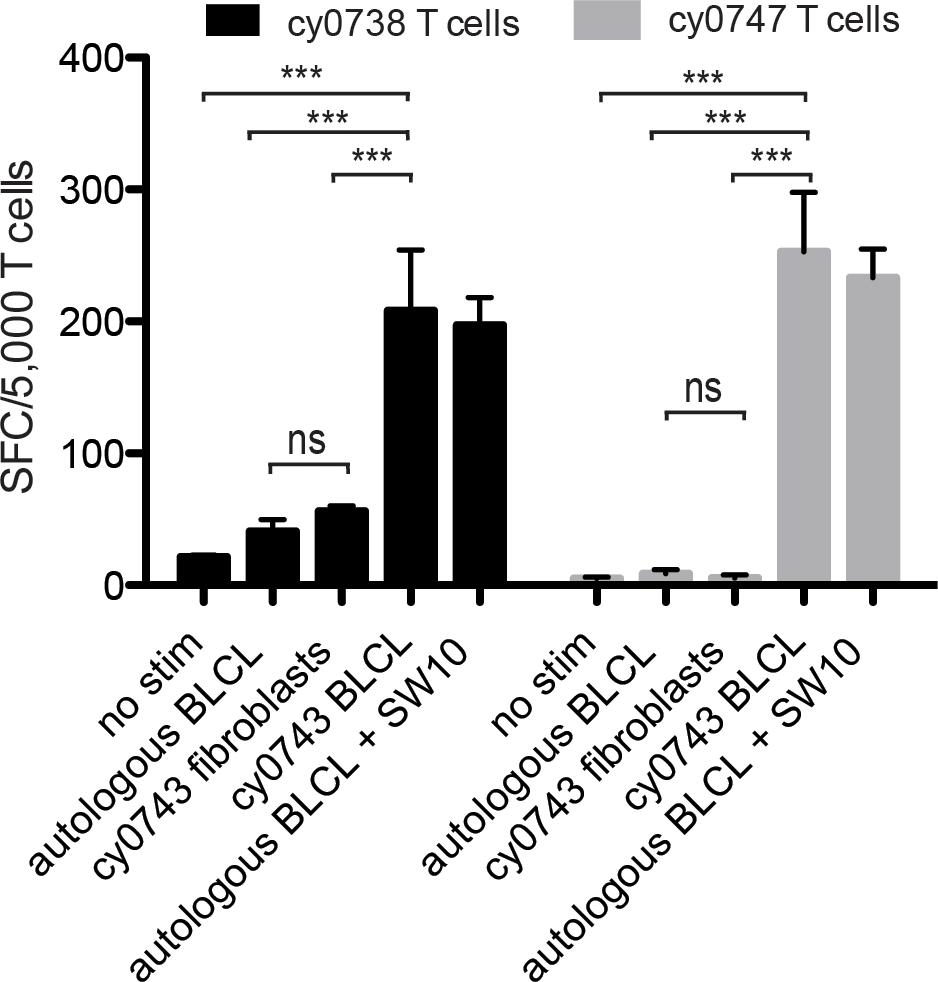
Figure 7. Tissue restriction of the mHAg epitope in APOBEC3C. APOBEC3C SW10-specific T cell lines from MCMs cy0738 and cy0747 were incubated with equal numbers of fibroblasts or BLCs from the mHAgpos animal cy0743 in an IFN-γ ELISpot assay. T cells cultured alone or incubated with equal numbers of the corresponding unpulsed autologous BLCs were included as negative controls, and T cells incubated with equal numbers of the corresponding SW10 peptide-pulsed autologous BLCs were included as a positive control. Data are shown as a mean ± SD for each condition. Differences between groups were assessed using a one-way ANOVA with Tukey’s test for multiple comparisons. ***p < 0.0001; ns = not significant. SFC, spot-forming cell.
Frequency of the SW10-Encoding SNP in 68 MCMs
To determine the prevalence of the SW10-encoding SNP, irrespective of MHC haplotype, we expanded our analysis to include an additional 47 MCMs. Across the entire WES dataset, five animals had a SNP at position NC_027902.1:80990325 in APOBEC3C, representing a population frequency of 7.35% among 68 MCMs. Four animals had the G > T polymorphism at this position, encoding the leucine variant found in SW10. The other animal had a G > A polymorphism at this position, encoding glutamine, which although not tested in this study, may similarly induce alloreactive T cell responses in Mafa-A1∗063pos MCMs.
Discussion
In this study, we used a combination of alloimmunization, WES, and bioinformatics to identify an NHP mHAg. Although a previous study described the conservation of human HA-1, HA-2, and H-Y mHAgs in chimpanzees and rhesus macaques (38), the MHC restriction and immunogenicity of these mHAgs were not confirmed in vivo. Accordingly, the SW10 epitope in APOBEC represents the first clearly defined mHAg in NHPs.
Our investigation was made possible by the unique genetics of MCMs. In contrast to most outbred populations, MCMs passed through an artificial bottleneck approximately 400 years ago (39, 40), which resulted in a 23% reduction in the number of SNPs (40) and limited MHC diversity compared with mainland cynomolgus macaques (27, 28). These genetic peculiarities made it possible to assemble 21 MHC-identical animals, subdivide them into discrete groups based on mHAg expression, and map the SW10 epitope following the identification of a SNP in APOBEC3C.
Fortuitously, the MHC class I allomorph Mafa-A1∗063 restricts APOBEC3C SW10. Mafa-A1∗063 is a “universal” A locus MHC-I allele expressed by the three most common MCM MHC haplotypes (M1, M2, and M3) (28). As a result, approximately 88% of MCMs have the potential to present APOBEC3C SW10. However, the SNP encoding this mHAg is present at a much lower frequency, estimated at only ∼7% based on our analysis of 68 MCMs. Ideally, this SNP would be present at a higher frequency for ease of study. It should be noted that similar frequency limitations apply to MHC-homozygous MCMs (27). Although potentially limiting for future interventional studies, breeding programs could be established to increase the frequency of APOBEC3C SW10 mHAgpos MCMs.
APOBEC3C belongs to a family of well-known restriction factors that limit the replication of HIV and SIV. Apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3 proteins are cytidine deaminases that protect against endogenous retroelements, retroviruses, and lentiviruses by inducing the hypermutation of viral genomes during reverse transcription (41). APOBEC3 genes are expressed widely in immune cells (37, 42, 43). In line with these gene expression studies, we found that APOBEC3C SW10-specific T cells recognized BLCs but not fibroblasts isolated from an mHAgpos MCM. However, APOBEC3C is also expressed in a variety of human tissues, including the gut and the lungs (37, 43). The corresponding tissues may therefore be susceptible to attack by APOBEC3C SW10-specific T cells. Our analysis mirrors the screening process for selecting tissue-restricted mHAg-specific T cell clones to infuse into patients with refractory leukemia (44). Of the seven patients receiving mHAg-specific T cell infusions in this clinical trial, two developed severe GvHD. These results suggest that APOBEC3C SW10 is sufficiently tissue-restricted for further study, but MCMs will need to be monitored for the development of GvHD.
T cell immunotherapy is a useful and often curative strategy for treating hematologic cancers (44–53). A similar approach could be used to target latent reservoirs of HIV (54, 55). Such reservoirs are limited to immune cells and are thought to be reduced or even eliminated by GvH responses after allo-HSCT (10–12). However, it is difficult to test this hypothesis in humans, because very few HIV+ patients undergo allo-HSCT. Moreover, different conditioning regimens are typically used in these rare cases, and efficacy likely depends to some extent on the receipt of HSCs homozygous for the CCR5Δ32 mutation. Our results may therefore facilitate the development of standardized NHP models to investigate the antiviral effects of GvH responses.
Our immunization strategy was adapted from human clinical trials investigating paternal lymphocyte immunotherapy (LIT) as a treatment for recurrent miscarriages (56, 57). In these studies, LIT was well tolerated and did not increase the risk of autoimmunity or GvHD (58, 59). In MHC-matched individuals, this alloimmunization strategy likely enriches for T cells targeting leukocyte-expressed mHAgs and could feasibly be extended to map mHAgs in other species, like outbred macaques or humans. Such in vivo enrichment protocols may offer advantages over traditional in vitro approaches for identifying tissue-restricted immunodominant mHAgs (60–63).
Further refinements to the methods described here could expedite the identification of mHAgs. A key limitation in our study was the inefficient generation of BLCs. Although we generated WES data from 21 MHC-identical MCMs, we were only able to immortalize B cells from 10 animals, which constrained our ability to sieve candidate SNPs. The inclusion of a statistical component in the WES segregation analysis would also have likely enhanced the identification and prioritization of candidate SNPs (23, 62). In addition, we focused our analysis on coding regions, which precluded the capture of intron-located or splice variant-generated SNPs (64). Whole-genome sequencing and/or mass spectrometric approaches would circumvent this particular limitation and allow the identification of such “cryptic” mHAgs.
Conclusion
In conclusion, we identified an mHAg epitope in APOBEC3C restricted by the common MCM MHC-I allomorph Mafa-A1∗063. Importantly, allorecognition of this epitope was focused on cells of hematopoietic origin but not dermal fibroblasts, suggesting potential utility as a model antigen in NHPs for testing mHAg-targeted immunotherapies. In particular, we anticipate that our discovery will enable the systematic evaluation of GvH responses as a therapeutic strategy to combat latent immunodeficiency viruses, which are readily modeled in MCMs.
Author’s Note
This manuscript was previously released on the preprint server bioRxiv as Weinfurter et al. (65).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/sra, SAMN12325046–SAMN12325066.
Ethics Statement
The animal study was reviewed and approved by University of Wisconsin Institutional Animal Care and Use Committee.
Author Contributions
JW performed experiments, acquired and analyzed data, and wrote the manuscript. MG and AE performed the whole exome segregation analysis. LM acquired and analyzed data. SL-L and DP generated the Mafa-A1∗063/SW10 tetramer. RW contributed to the sample preparation and study methodology. MR acquired funding, conceived and directed the project, and wrote the manuscript. All authors edited the manuscript and approved the final version.
Funding
This study was funded by the National Institutes of Health (NIH) via grants R01 AI118495 and R24 OD021322 awarded to MR. Additional support was provided by the Office of Research Infrastructure Programs/OD via grant P51OD011106 awarded to the WNPRC at the University of Wisconsin-Madison. This research was conducted in part at a facility constructed with support from the Research Facilities Improvement Program via grants RR15459-01 and RR020141-01. DP was supported by a Wellcome Trust Senior Investigator Award (100326/Z/12/Z). The funders had no role in study design, collection, analysis, interpretation of the data, or manuscript preparation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the veterinary staff at the Wisconsin National Primate Research Center (WNPRC) for their assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.586251/full#supplementary-material
Supplementary Table 1 | ELISpot responses for 69 T cell clones incubated with 10 (n = 47) or fewer (n = 22) BLC lines. Numbers indicate spot-forming cells (SFCs) per well. Clones that were selected for further analysis are in boxes, with positive responses highlighted in bold.
Supplementary Table 2 | Segregation analysis of APOBEC3C, IGHM, OR4K3 and IL20RA or MAP3K5. Displayed are the segregation scores for the selected alloreactive T cell clones. MCMs were placed in response or non-response groups based on the recognition of their BLCs by the respective T cell clones. Haplotype indicates the nucleotide SNP present at the chromosome and nucleotide position relative to the Mmu-8.0.1 reference genome.
References
1. Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci USA. (1997) 94:13193–7. doi: 10.1073/pnas.94.24.13193
2. Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. (1999) 5:512–7. doi: 10.1038/8394
3. Sengupta S, Siliciano RF. Targeting the latent reservoir for HIV-1. Immunity. (2018) 48:872–95. doi: 10.1016/j.immuni.2018.04.030
4. Wong JK, Hezareh M, Günthard HF, Havlir DV, Ignacio CC, Spina CA, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. (1997) 278:1291–5.
5. Chun TW, Davey RTJ, Engel D, Lane HC, Fauci AS. Re-emergence of HIV after stopping therapy. Nature. (1999) 401:874–5. doi: 10.1038/44755
6. Davey RT, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci USA. (1999) 96:15109–14. doi: 10.1073/pnas.96.26.15109
7. Crooks AM, Bateson R, Cope AB, Dahl NP, Griggs MK, Kuruc JD, et al. Precise quantitation of the latent HIV-1 reservoir: implications for eradication strategies. J Infect Dis. (2015) 212:1361–5. doi: 10.1093/infdis/jiv218
8. Deeks SG, Autran B, Berkhout B, Benkirane M, Cairns S, Chomont N, et al. Towards an HIV cure: a global scientific strategy. Nat Rev Immunol. (2012) 12:607–14. doi: 10.1038/nri3262
9. Kim Y, Anderson JL, Lewin SR. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe. (2018) 23:14–26. doi: 10.1016/j.chom.2017.12.004
10. Gupta RK, Abdul-Jawad S, McCoy LE, Mok HP, Peppa D, Salgado M, et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature. (2019) 568:244–8. doi: 10.1038/s41586-019-1027-4
11. Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. (2009) 360:692–8. doi: 10.1056/NEJMoa0802905
12. Hutter G. Stem cell transplantation in strategies for curing HIV/AIDS. AIDS Res Ther. (2016) 13:31. doi: 10.1186/s12981-016-0114-y
13. Gupta RK, Peppa D, Hill AL, Gálvez C, Salgado M, Pace M, et al. Evidence for HIV-1 cure after CCR5Δ32/Δ32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: a case report. Lancet HIV. (2020) 7:30069. doi: 10.1016/S2352-3018(20)30069-2
14. Henrich TJ, Hu Z, Li JZ, Sciaranghella G, Busch MP, Keating SM, et al. Long-term reduction in peripheral blood HIV type 1 reservoirs following reduced-intensity conditioning allogeneic stem cell transplantation. J Infect Dis. (2013) 207:1694–702. doi: 10.1093/infdis/jit086
15. Koelsch KK, Rasmussen TA, Hey-Nguyen WJ, Pearson C, Xu Y, Bailey M, et al. Impact of allogeneic hematopoietic stem cell transplantation on the HIV reservoir and immune response in 3 HIV-infected individuals. J Acquir Immune Defic Syndr. (2017) 75:328–37. doi: 10.1097/QAI.0000000000001381
16. Salgado M, Kwon M, Gálvez C, Badiola J, Nijhuis M, Bandera A, et al. Mechanisms that contribute to a profound reduction of the HIV-1 reservoir after allogeneic stem cell transplant. Ann Intern Med. (2018) 169:674–83. doi: 10.7326/M18-0759
17. Vincent K, Roy DC, Perreault C. Next-generation leukemia immunotherapy. Blood. (2011) 118:2951–9. doi: 10.1182/blood-2011-04-350868
18. Griffioen M, van Bergen CA, Falkenburg JH. Autosomal minor histocompatibility antigens: how genetic variants create diversity in immune targets. Front Immunol. (2016) 7:100. doi: 10.3389/fimmu.2016.00100
19. Warren EH, Deeg HJ. Dissecting graft-versus-leukemia from graft-versus-host-disease using novel strategies. Tissue Antigens. (2013) 81:183–93. doi: 10.1111/tan.12090
20. Pitman MC, Lau JSY, McMahon JH, Lewin SR. Barriers and strategies to achieve a cure for HIV. Lancet HIV. (2018) 5:e317–28. doi: 10.1016/S2352-3018(18)30039-0
21. Spierings E, Kim YH, Hendriks M, Borst E, Sergeant R, Canossi A, et al. Multicenter analyses demonstrate significant clinical effects of minor histocompatibility antigens on GvHD and GvL after HLA-matched related and unrelated hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. (2013) 19:1244–53. doi: 10.1016/j.bbmt.2013.06.001
22. Granados DP, Rodenbrock A, Laverdure JP, Côté C, Caron-Lizotte O, Carli C, et al. Proteogenomic-based discovery of minor histocompatibility antigens with suitable features for immunotherapy of hematologic cancers. Leukemia. (2016) 30:1344–54. doi: 10.1038/leu.2016.22
23. Fuchs KJ, Honders MW, van der Meijden ED, Adriaans AE, van der Lee DI, Pont MJ, et al. Optimized whole genome association scanning for discovery of HLA class I-restricted minor histocompatibility antigens. Front Immunol. (2020) 11:659. doi: 10.3389/fimmu.2020.00659
24. Lawler SH, Sussman RW, Taylor LL. Mitochondrial DNA of the Mauritian macaques (Macaca fascicularis): an example of the founder effect. Am J Phys Anthropol. (1995) 96:133–41. doi: 10.1002/ajpa.1330960203
25. Budde ML, Wiseman RW, Karl JA, Hanczaruk B, Simen BB, O’Connor DH. Characterization of Mauritian cynomolgus macaque major histocompatibility complex class I haplotypes by high-resolution pyrosequencing. Immunogenetics. (2010) 62:773–80. doi: 10.1007/s00251-010-0481-9
26. O’Connor SL, Blasky AJ, Pendley CJ, Becker EA, Wiseman RW, Karl JA, et al. Comprehensive characterization of MHC class II haplotypes in Mauritian cynomolgus macaques. Immunogenetics. (2007) 59:449–62. doi: 10.1007/s00251-007-0209-7
27. Wiseman RW, Wojcechowskyj JA, Greene JM, Blasky AJ, Gopon T, Soma T, et al. Simian immunodeficiency virus SIVmac239 infection of major histocompatibility complex-identical cynomolgus macaques from Mauritius. J Virol. (2007) 81:349–61. doi: 10.1128/JVI.01841-06
28. Wiseman RW, Karl JA, Bohn PS, Nimityongskul FA, Starrett GJ, O’Connor DH. Haplessly hoping: macaque major histocompatibility complex made easy. ILAR J. (2013) 54:196–210. doi: 10.1093/ilar/ilt036
29. Holman N, Weinfurter JT, Harsla TR, Wiseman RW, Belli AJ, Michaels AJ, et al. Isolation of a monoclonal antibody from a phage display library binding the rhesus macaque MHC class I allomorph Mamu-A1∗001. PLoS One. (2017) 12:e0179039. doi: 10.1371/journal.pone.0179039
30. Reynolds MR, Weiler AM, Piaskowski SM, Piatak MJ, Robertson HT, Allison DB, et al. A trivalent recombinant Ad5 gag/pol/nef vaccine fails to protect rhesus macaques from infection or control virus replication after a limiting-dose heterologous SIV challenge. Vaccine. (2012) 30:4465–75. doi: 10.1016/j.vaccine.2012.04.082
31. Caskey JR, Wiseman RW, Karl JA, Baker DA, Lee T, Maddox RJ, et al. MHC genotyping from rhesus macaque exome sequences. Immunogenetics. (2019) 71:531–44. doi: 10.1007/s00251-019-01125-w
32. Prall TM, Graham ME, Karl JA, Wiseman RW, Ericsen AJ, Raveendran M, et al. Improved full-length killer cell immunoglobulin-like receptor transcript discovery in Mauritian cynomolgus macaques. Immunogenetics. (2017) 69:325–39. doi: 10.1007/s00251-017-0977-7
33. Lupski JR, Gonzaga-Jauregui C, Yang Y, Bainbridge MN, Jhangiani S, Buhay CJ, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome Med. (2013) 5:57. doi: 10.1186/gm461
34. Shimizu Y, DeMars R. Production of human cells expressing individual transferred HLA-A,-B,-C genes using an HLA-A,-B,-C null human cell line. J Immunol. (1989) 142:3320–8.
35. Burwitz BJ, Pendley CJ, Greene JM, Detmer AM, Lhost JJ, Karl JA, et al. Mauritian cynomolgus macaques share two exceptionally common major histocompatibility complex class I alleles that restrict simian immunodeficiency virus-specific CD8+ T cells. J Virol. (2009) 83:6011–9. doi: 10.1128/JVI.00199-09
36. Bleakley M, Riddell SR. Exploiting T cells specific for human minor histocompatibility antigens for therapy of leukemia. Immunol Cell Biol. (2011) 89:396–407. doi: 10.1038/icb.2010.124
37. Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. (2010) 38:4274–84. doi: 10.1093/nar/gkq174
38. den Haan JM, Bontrop RE, Pool J, Sherman N, Blokland E, Engelhard VH, et al. Conservation of minor histocompatibility antigens between human and non-human primates. Eur J Immunol. (1996) 26:2680–5. doi: 10.1002/eji.1830261120
39. Bonhomme M, Blancher A, Cuartero S, Chikhi L, Crouau-Roy B. Origin and number of founders in an introduced insular primate: estimation from nuclear genetic data. Mol Ecol. (2008) 17:1009–19. doi: 10.1111/j.1365-294X.2007.03645.x
40. Osada N, Hettiarachchi N, Adeyemi Babarinde I, Saitou N, Blancher A. Whole-genome sequencing of six Mauritian cynomolgus macaques (Macaca fascicularis) reveals a genome-wide pattern of polymorphisms under extreme population bottleneck. Genome Biol Evol. (2015) 7:821–30. doi: 10.1093/gbe/evv033
41. Olson ME, Harris RS, Harki DA. APOBEC Enzymes as targets for virus and cancer therapy. Cell Chem Biol. (2018) 25:36–49. doi: 10.1016/j.chembiol.2017.10.007
42. Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, et al. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol. (2011) 85:11220–34. doi: 10.1128/JVI.05238-11
43. Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol. (2009) 83:9474–85. doi: 10.1128/JVI.01089-09
44. Warren EH, Fujii N, Akatsuka Y, Chaney CN, Mito JK, Loeb KR, et al. Therapy of relapsed leukemia after allogeneic hematopoietic cell transplantation with T cells specific for minor histocompatibility antigens. Blood. (2010) 115:3869–78. doi: 10.1182/blood-2009-10-248997
45. Allers K, Hutter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, et al. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood. (2011) 117:2791–9. doi: 10.1182/blood-2010-09-309591
46. Dickinson AM, Norden J, Li S, Hromadnikova I, Schmid C, Schmetzer H, et al. Graft-versus-leukemia effect following hematopoietic stem cell transplantation for leukemia. Front Immunol. (2017) 8:496. doi: 10.3389/fimmu.2017.00496
47. Falkenburg JH, Warren EH. Graft versus leukemia reactivity after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. (2011) 17:S33–8. doi: 10.1016/j.bbmt.2010.11.009
48. Fontaine P, Roy-Proulx G, Knafo L, Baron C, Roy DC, Perreault C. Adoptive transfer of minor histocompatibility antigen-specific T lymphocytes eradicates leukemia cells without causing graft-versus-host disease. Nat Med. (2001) 7:789–94. doi: 10.1038/89907
49. Goulmy E, Gratama JW, Blokland E, Zwaan FE, van Rood JJA. minor transplantation antigen detected by MHC-restricted cytotoxic T lymphocytes during graft-versus-host disease. Nature. (1983) 302:159–61. doi: 10.1038/302159a0
50. Janelle V, Rulleau C, Del Testa S, Carli C, Delisle JS. T-cell immunotherapies targeting histocompatibility and tumor antigens in hematological malignancies. Front Immunol. (2020) 11:276. doi: 10.3389/fimmu.2020.00276
51. Li N, Matte-Martone C, Zheng H, Cui W, Venkatesan S, Tan HS, et al. Memory T cells from minor histocompatibility antigen-vaccinated and virus-immune donors improve GVL and immune reconstitution. Blood. (2011) 118:5965–76. doi: 10.1182/blood-2011-07-367011
52. Van Der Harst D, Goulmy E, Falkenburg JH, Kooij-Winkelaar YM, van Luxemburg-Heijs SA, Goselink HM, et al. Recognition of minor histocompatibility antigens on lymphocytic and myeloid leukemic cells by cytotoxic T-cell clones. Blood. (1994) 83:1060–6. doi: 10.1182/blood.v83.4.1060.1060
53. Vincent K, Hardy MP, Trofimov A, Laumont CM, Sriranganadane D, Hadj-Mimoune S, et al. Rejection of leukemic cells requires antigen-specific T cells with high functional avidity. Biol Blood Marrow Transplant. (2014) 20:37–45. doi: 10.1016/j.bbmt.2013.10.020
54. Margolis DM, Archin NM, Cohen MS, Eron JJ, Ferrari G, Garcia JV, et al. Curing HIV: seeking to target and clear persistent infection. Cell. (2020) 181:189–206. doi: 10.1016/j.cell.2020.03.005
55. Mitsuyasu R. Curing HIV: lessons from cancer therapy. Curr Opin HIV AIDS. (2013) 8:224–9. doi: 10.1097/COH.0b013e32835ef0a1
56. Leith JG, Clark DA, Matthews TJ, Rosenthal KL, Luscher MA, Barber BH, et al. Assessing human alloimmunization as a strategy for inducing HIV type 1 neutralizing anti-HLA responses. AIDS Res Hum Retroviruses. (2003) 19:957–65. doi: 10.1089/088922203322588305
57. Mowbray JF, Underwood JL, Michel M, Forbes PB, Beard RW. Immunisation with paternal lymphocytes in women with recurrent miscarriage. Lancet. (1987) 2:679–80. doi: 10.1016/s0140-6736(87)92457-3
58. Cavalcante MB, Sarno M, Araujo Júnior E, Da Silva Costa F, Barini R. Lymphocyte immunotherapy in the treatment of recurrent miscarriage: systematic review and meta-analysis. Arch Gynecol Obstet. (2017) 295:511–8. doi: 10.1007/s00404-016-4270-z
59. Kling C, Steinmann J, Westphal E, Magez J, Kabelitz D. Adverse effects of intradermal allogeneic lymphocyte immunotherapy: acute reactions and role of autoimmunity. Hum Reprod. (2006) 21:429–35. doi: 10.1093/humrep/dei316
60. Griffioen M, Honders MW, van der Meijden ED, van Luxemburg-Heijs SA, Lurvink EG, Kester MG, et al. Identification of 4 novel HLA-B∗40:01 restricted minor histocompatibility antigens and their potential as targets for graft-versus-leukemia reactivity. Haematologica. (2012) 97:1196–204. doi: 10.3324/haematol.2011.049478
61. Oostvogels R, Lokhorst HM, Minnema MC, van Elk M, van den Oudenalder K, Spierings E, et al. Identification of minor histocompatibility antigens based on the 1000 genomes project. Haematologica. (2014) 99:1854–9. doi: 10.3324/haematol.2014.109801
62. Van Bergen CA, Rutten CE, Van Der Meijden, Van Luxemburg-Heijs SA, Lurvink EG, Houwing-Duistermaat JJ, et al. High-throughput characterization of 10 new minor histocompatibility antigens by whole genome association scanning. Cancer Res. (2010) 70:9073–83. doi: 10.1158/0008-5472.CAN-10-1832
63. Warren EH, Greenberg PD, Riddell SR. Cytotoxic T-lymphocyte-defined human minor histocompatibility antigens with a restricted tissue distribution. Blood. (1998) 91:2197–207. doi: 10.1182/blood.v91.6.2197
64. Laumont CM, Vincent K, Hesnard L, Audemard É, Bonneil É, Laverdure JP, et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med. (2018) 10:eaau5516. doi: 10.1126/scitranslmed.aau5516
Keywords: minor histocompatibility antigen (mHAg), APOBEC, cynomolgus macaque, non-human primate (NHP), CD8 T cell, epitope, HIV, cellular immunotherapy
Citation: Weinfurter JT, Graham ME, Ericsen AJ, Matschke LM, Llewellyn-Lacey S, Price DA, Wiseman RW and Reynolds MR (2020) Identifying a Minor Histocompatibility Antigen in Mauritian Cynomolgus Macaques Encoded by APOBEC3C. Front. Immunol. 11:586251. doi: 10.3389/fimmu.2020.586251
Received: 22 July 2020; Accepted: 08 September 2020;
Published: 26 October 2020.
Edited by:
Ralf Dressel, University Medical Center Göttingen, GermanyReviewed by:
Lutz Walter, Deutsches Primatenzentrum, GermanyMarieke Griffioen, Leiden University Medical Center, Netherlands
Copyright © 2020 Weinfurter, Graham, Ericsen, Matschke, Llewellyn-Lacey, Price, Wiseman and Reynolds. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew R. Reynolds, bWF0dC5yZXlub2xkc0B3aXNjLmVkdQ==