 Davide Quaresmini
Davide Quaresmini Michele Guida
Michele Guida
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 29 October 2020
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.584903
This article is part of the Research Topic Angiogenesis and Immune System: Two Players in Cancer Field in the Era of Immunotherapy View all 5 articles
Neoangiogenesis is a recognized hallmark of cancer, granting tumor cells to dispose of metabolic substrates through a newly created vascular supply. Neoangiogenesis was also confirmed in melanoma, where vascular proliferation is associated with increased aggressiveness and poorer prognosis. Furthermore, melanoma cells show the so-called vascular mimicry, consisting in the assumption of endothelial-like features inducing the expression of pro-angiogenic receptors and ligands, which take part in the interplay with extracellular matrix (ECM) components and are potentiated by the ECM remodeling and the barrier molecule junction alterations that characterize the metastatic phase. Although neoangiogenesis was biologically proven and clinically associated with worse outcomes in melanoma patients, in the past anti-angiogenic therapies were employed with poor improvement of the already unsatisfactory results associated with chemotherapic agents. Among the novel therapies of melanoma, immunotherapy has led to previously unexpected outcomes of treatment, yet there is a still strong need for potentiating the results, possibly by new regimens of combination therapies. Molecular models in many cancer types showed mutual influences between immune responses and vascular normalization. Recently, clinical trials are investigating the efficacy of the association between anti-angiogenetic agents and immune-checkpoint inhibitors to treat advanced stage melanoma. This paper reviews the biological bases of angiogenesis in melanoma and summarizes the currently available clinical data on the use of anti-angiogenetic compounds in melanoma.
Melanoma is an aggressive cancer arising from melanocytic precursors, with high potential for locoregional and metastatic spread. As a common hallmark of cancers, when the dimensions of the primary tumoral mass of melanoma reach the threshold for nutrient diffusion, a cancer-specific net of blood vessels is fundamental to provide substrates for cancer cell survival and growth (1). The newborn neoplastic vasculature is aberrant and incomplete with distorted, dilated, and leaky vessels, insufficient pericyte coverage, abnormal endothelial cell proliferation, an uneven distribution within cancer tissues, and wide fenestrations that ultimately contribute to tumor cell sprouting through the vascular flow (2–5). The process of activation of quiescent vasculature by cancer cells is called “angiogenic switch” and is sustained by a disequilibrium between molecular activators and inhibitors of angiogenesis, in favor of angiogenesis (6). Angiogenesis is essential in growth and progression of cancers, including melanoma (7). Warren first described angiogenesis in a melanoma graft animal model, observing remarkable angiogenic capacity of melanoma tissue (8). Since then, angiogenesis in melanoma has been furtherly investigated to identify a possibly more complex and heterogeneous behavior in the handling of blood vessels by this neoplasm (9). First, melanoma cells induce the spread of new blood vessels by pre-existing ones; secondly, melanoma cells recruit bone marrow progenitors that reach hypoxic areas within the tumor microenvironment, where they can induce vascular formation; finally, melanoma cells themselves can acquire an endothelial-like phenotype in the so-called vascular mimicry phenomenon, directly taking part into the structure of blood vessels (9). So far, inhibitors of angiogenesis have been used in oncology in other cancers including renal, colorectal, and ovarian neoplasms, but data from clinical melanoma research are lacking. The current therapies for advanced stage disease in melanoma are based on the inhibition of either the aberrantly activated BRAF/MEK pathway, or the immune checkpoints PD1 and CTLA4. Despite obtaining previously unexpected outcomes in patients with advanced disease, leading to improved survival rates, these therapies still have potential for further improvements. This paper summarizes the biological bases of angiogenesis in melanoma matching to the most recent development of treatments, and reviews the currently available preclinical and clinical data on the use of anti-angiogenetic compounds in patients with melanoma.
The recent evolution of melanoma treatment has led to a primary role of targeted and immune therapy both in the adjuvant and the metastatic setting. Interestingly, the molecular architecture of melanoma microenvironment is far more complex than previously thought, and a strong cross-talk has been clearly demonstrated between the angiogenic and the immune components of cancer stroma, with mutual influences in the molecular compartments that are involved in response to systemic treatments. Angiogenic factors are produced by tumor cells, stromal cells including cancer-associated fibroblasts (CAFs), and inflammatory cells like lymphocytes and macrophages. The hyperproduction of pro-angiogenetic factors is induced by several mechanisms, primarily the activation of the hypoxia-induced HIF1α pathway, the oncogene-induced transcription of Vascular Endothelial Growth Factor (VEGF), and the loss of oncosuppressor genes including p53, which both stimulates the production of antiangiogenic factors like thrombospondin-1 and inhibits the expression of proangiogenic factors (10, 11).
VEGF-A is the archetype and most biologically relevant among proangiogenic factors, with a strong effect on endothelial survival and migration (12), and on vasculogenic mimicking properties in melanoma cells (13). VEGF-A is upregulated by HIF-1α and oncogene signaling pathways (14–16). Remarkably, VEGF-A has also shown immunosuppressive capacity. In particular, the increased production of VEGF-A in cancer leads to inhibition of T cells in several ways: 1) reducing the activity of functional T cells both directly (17) and indirectly through the endothelial PGE2-mediated suppressive action on T cells (18); 2) decreasing neoantigen presentation to lymphocytes by inhibiting the maturation of dendritic cells (DCs) (19), mainly interfering with NF-kB activation (20); 3) recruiting immunosuppressive T regulatory cells (Tregs) into the tumor microenvironment (21, 22); 4) limiting endothelial cytokine response and adhesion molecule expression, hence affecting vascular functional permeability to leukocytes and their peripheral recruitment to cancer microenvironment (23, 24). Relevantly for immunotherapy, VEGF pathway activation also enhances T cell exhaustion mediated by immune checkpoints like PDL1, CTLA4, TIM3, and LAG3 (25). While lowering the immunogenic compartment of immunity, VEGF potentiates the counteracting immunopermissive microenvironment (26) both by recruiting immune-suppressive Tregs (27) and myeloid derived stromal cells (MDSCs) (28), and activating tumor-associated macrophages (TAMs) at the tumor site (29). To confirm this role of VEGF, the anti-VEGF antibody Bevacizumab induces DC maturation and a reduction in Tregs and MDSCs recruitment to cancer sites (22, 30, 31). VEGF can be produced by cancer cells and immune cells from tumor microenvironment, mostly from Tregs and in smaller proportions from TAMs, MDSCs and DCs (32), creating cellular communications that either directly or indirectly convey on the inhibition of cytotoxic T lymphocytes (33).
ANG-2 is an antagonist cytokine of the Angiopoietin-1/Tie2 pathway that acts as a facilitator of VEGF-dependent angiogenesis (34). ANG-2 has long been considered an exclusive product of endothelium, but more recently Pari and colleagues demonstrated that it can also be produced by melanoma (35). ANG-2 levels in sera are increased in stage III and IV melanoma patients, but not in stages I and II (36). Consistently with this evidence, ANG-2 is produced by melanoma cells themselves, especially by metastatic sites (35). Differently from the stromal-derived ANG-2, melanoma-derived ANG-2 was not shown to increase the microvessel density of melanoma microenvironment but rather showed a protection of tumor cells from oxidative stress and a role in reactive oxygen species associated metastatization to the lungs in a mouse model (35). High serum ANG-2 levels were correlated with worse overall response rate to immunotherapy in melanoma (37). ANG-2 contributes to immune microenvironment composition, by acting on the Tie-2 expressing subpopulation of circulating monocytes, that are recruited by ANG-2 and converted to M2-like macrophages (38) and secrete IL10, which is a known promoter of Treg expansion and inhibitor of effector T cell activity (39).
TLRs are a family of pattern recognition receptors involved in antimicrobial immunity, apoptotic cell clearance, and cancer. Among all family members, TLR-4 is expressed in 90% of primary and 93% of metastatic melanomas, where it plays a role in the aggressive behavior of cancer cells (40). TLR-4 signaling involves the activation of signal transducer and activator of transcription 3 (STAT3), which on turn promotes melanoma growth and aggressiveness associated features including angiogenesis and epithelial to mesenchymal transition (41). During melanomagenesis, ultraviolet radiation recruits and activates neutrophils in a TLR4-mediated mechanism, inducing an inflammation that facilitates angiogenesis and favors melanoma angiotropism (40). Moreover, STAT3 has also been associated to immunosuppression in melanoma (42).
In cancer microenvironment, there is a constant dynamic cross-talk between all resident cells which is far beyond the mere activity of cancer cells alone: the dynamic interaction between all cell components is responsible for the biological behavior of cancer. Accordingly, angiogenesis in cancer is not only induced by cancer cells themselves: the immune cells in tumor microenvironment can sustain angiogenesis in cancer (43). Globally, tumor cells can influence immune infiltrates towards an immune permissive phenotype. VEGF, for example, is produced by TAMs, tumor-associated neutrophils, regulatory DCs, myeloid derived suppressor cells, NK cells, and γδT17 cells (43). VEGF-R1 and -2 are expressed on DCs, which can promote angiogenesis (44). Neutrophils and TAMs secrete proangiogenic factors including VEGF, TNFα, IL8, and chemokines (45), together with matrix metalloproteases, which are essential to remodel the extracellular matrix during angiogenesis and metastatization (46). In an analysis on mouse models of breast cancers, extended to some of TCGA databases excluding melanoma, Tian et al. (47) demonstrated that the activation of vessel normalization (pericyte coverage, reduced vascular leakage, improved blood perfusion) is one of the effects of immune checkpoint inhibitors (ICIs) mediated by the activation of CD4+ T lymphocytes, in particular IFNγ producing Th1 cells (47). In particular, the authors postulated the existence of a positive feedback loop, according to which Th1 cells localize proximal to tumoral vessels and change the local tumor microenvironment via CKs like IFNγ, which induces a reduction in VEGF-A production and an increase in Th1- and pericyte-recruiting chemokines CXCL9, CXCL10, CXCL11 (47). Such finding would confirm the evidence that inhibition of IFNγ signaling is associated to secondary resistance to immune checkpoint inhibitors (48). Furthermore, after interacting with DCs, T cells can acquire neurophilin-1 which is a ligand of VEGF-A that promotes angiogenesis (49). Endothelial cells, formerly considered passive lining cells of blood vessels, are actively responsible of the intense reciprocal cellular interaction consisting in the so-called angiocrine signaling that is essential in normal organ development (50) and can be exacerbated in cancer. Tumor cells can then induce endothelial activation mediated by CKs including Angiopoietin-2, which are responsible for the autocrine induction of STAT3 signaling in the endothelium, followed by the expression of chemokines (CCL2) and adhesion molecules (ICAM1) that recruit CCR2+ macrophages to the cancer site (37). Endothelial cells take part in granulocyte differentiations in physiology and pathology, given the common developmental origin between endothelial cells and hematopoietic cells (37): the endothelium secretes CKs (SCF, CXCL12) that contribute to the quiescence of hematopoietic cells in the bone marrow, but can also promote granulopoiesis in case of inflammation (51). The hypoxia-regulated Endothelin B receptor on tumor endothelium acts as an obstacle to T cell adhesion and has been identified in some cases of resistance to immune therapy (52).
Antiangiogenics are a class of kinase inhibitors that bind either angiogenic factors or their receptors. The first antiangiogenic agent to be developed was Bevacizumab, an anti-VEGF monoclonal antibody, which is still indicated in the treatment of cancers including colorectal, ovarian, or uterine carcinomas. Many other agents were synthesized furtherly, presenting a wider spectrum of pharmacodynamic targeting, including Sunitinib, Pazopanib, Ramucirumab, Regorafenib, Sorafenib, Aflibercept, being so far approved in daily practice either alone or in combination. Antiangiogenic agents model the irregular and leaky vessels of cancer to create an almost normalized intratumoral vascular network, at least transiently. Such improved vascular efficiency of cancer microcirculation is thus the main responsible for a more efficient transport of chemotherapic agents to cancer cells, and also the molecular background to the association of antiangiogenic therapies with traditional chemotherapies (53). Part of the effects of antiangiogenics in cancer may also be attributed to the modulation of immune cell composition in tumor microenvironment, triggered by the reduction in the tissue hypoxia that is associated with the immature cancer vasculature. The response to hypoxia favors the polarization of the tumor microenvironment towards an immune-suppressive phenotype in terms of increase in Tregs and M2-TAMs, reduction of DC activity, and increase in PDL1 expression on endothelial cells, TAMs, DCs, and cytotoxic lymphocytes (33). Antiangiogenics can then interfere with both CD8+ T cells trafficking and TAMs repolarization, inducing an immunostimulatory milieu (37). Anti-VEGF-A agents also improve immune responses (54). In a preclinical model of antiangiogenic-driven vascular normalization in melanoma and other primary cancers, Schmittnaegel et al. demonstrated that the administration of a bispecific anti-VEGF-ANG2 antibody was associated with increased recruitment and activation of CD4+ and CD8+ T cells at the areas with normalized vessels (55). In the cross-talk between adaptive and innate immunity, IFNγ from CD4+ and CD8+ T cells then stimulates M1-TAMs to angiostatic activity and antagonize endothelial cell proliferation (56). In a preclinic mouse model with multiple cancer cell lines including melanoma, DeAlmeida and Colleagues demonstrated that therapy with anti-VEGFA targeting agent induces the HIF1α-mediated activation of intratumoral CD8+ cells resulting in an increase in IFNγ and TNFα production (57). Unfortunately, the response is usually transient (58) and short in duration (59), due to mechanisms of escape that include the activation of metabolic stress responses in cancer cells; the activation of alternative angiogenic pathways like the ANG-2 pathway; the participation to new vessel growth according to the vascular co-option mechanism; the normalization of cancer vessels (56). In renal cell carcinoma, two different molecular subsets have been identified: the angiogenic subset, characterized by upregulation of angiogenesis-associated genes and responsiveness to antiangiogenic therapy, and the inflammatory subset, presenting an upregulation in immune-related genes and refractoriness to antiangiogenic therapy (60). In contrast, no similar dichotomy has been identified in melanoma, for which resistance to antiangiogenic therapies may be intrinsically associated to its biology: as evidenced by Donnem, melanoma primary and metastatic lesions strongly rely on vessel co-option for their vascular supply, as an alternative to angiogenesis (61). In melanoma, the mutation of the BRAF gene has primary importance, since mutations at the codon 600 are druggable and clinically associated with significant responses. The constitutive activation of BRAF kinase is not only a driver mutation in melanoma, but also has effects on melanoma microenvironment composition (62). In particular, BRAF mutations are associated with an increased density of Treg infiltrate (63), and a reduction in T cell activity by the indirect secretion of IL-1α and IL-1β from CAFs (64). PDL1 expression in immune infiltrate is also more prevalent in BRAF mutated tumor specimens (65). Finally, BRAF mutated cells also have angiogenic capacity (66), since they can induce other cancer cells and microenvironmental cells to secrete CXCL8 and CCL2, two pro-tumorigenic chemokines, leading to cancer cell proliferation and macrophage-mediated angiogenesis (62). Furthermore, the pathway of VEGFR-1 has recently been identified as an escape mechanism to the BRAF-inhibitor Vemurafenib (67). As evidenced by Aztori and colleagues, melanoma cell lines express higher VEGF-receptors when transformed into Vemurafenib-resistant, and silencing of such receptors can prolong the maintenance of sensitivity to Vemurafenib (67). Together with targeted therapy for BRAF mutated patients, the backbone of treatment in advanced melanoma is represented by the immune therapy with either anti-CTLA4 or anti-PD1 agents, that remove immune checkpoint inhibition to potentiate the immune response to melanoma. Despite an overall improvement in the outcomes of treatment, still immune therapy is often associated to secondary resistance and progression and, yet more rarely, to early resistance. Among the possible mechanisms underpinning these resistances, recent evidence also identified some vascular-related mechanisms. In particular, high serum ANG-2 levels correlated with worse overall response rate to ICI therapy in melanoma, possibly because ANG-2 can recruit monocytes and induce PDL1 expression in M2-macrophages (37). Wu and colleagues identified a subset of melanoma patients characterized by a significant tumor infiltration of CD68+ macrophages that particularly responded to treatment with Ipilimumab and Bevacizumab with a neat decrease in ANG-2 expression (68). Allen and colleagues demonstrated that the combination therapy of antiangiogenics and anti-PD1 agents induced an increase in intratumoral high endothelial venules responsible for selective leukocyte infiltration and for the switch of microenvironment towards immunosensitive features (69). A bispecific anti-VEGFA and anti-ANG2 was also shown to potentiate the efficacy of an anti-PD1 treatment (55).
The first trials involving antiangiogenetic drugs in melanoma date back to early experiences two decades ago, when chemotherapy was the only available treatment for advanced stage disease, with palliative intent and dramatically poor outcomes. Most of these studies are phase 1 or 2 trials for stage III unresectable or stage IV melanoma, either in single or double arm of treatment, with small cohorts of recruited patients, usually 20–30 (Table 1). Despite preclinical data suggesting the advantage of a more regular vascular network in the distribution of chemotherapics to cancer cells, these trials did not provide satisfying results from the association of antiangiogenics with common chemotherapics, showing no statistically significant improvement in the outcomes of traditional chemotherapy schedules, therefore they have never been investigated in wider phase 3 clinical trials and have never been adopted in everyday practice. More recently, immune therapy became the new gold standard for systemic therapy, together with anti-BRAF and anti-MEK targeted therapy for BRAF mutated patients, and was tested in the association with antiangiogenic agents, given the evidence of efficacy from the combination of antiangiogenics with ICIs not only in preclinical, but also in the clinical settings for other cancers including renal clear cell or non-small cell lung cancer. Hodi and colleagues performed an investigational phase I trial in 46 patients with advanced melanoma without brain metastases receiving a first (37%) or second (63%) line treatment with Ipilimumab and Bevacizumab: the best overall response rate (ORR) was 19.6% with a disease control rate of 67.4% and a median time to progression of 9 months (78). The immunohistochemical analysis of serial biopsies of target lesions revealed changes in melanoma-associated endothelium with increased expression of E-selectin (78). In a more recent phase IB/II trial, Taylor and colleagues treated patients with advanced solid tumors including melanoma with the association of Pembrolizumab and Lenvatinib, an inhibitor of multiple kinases including VEGFR. Among the 21 patients of the melanoma subcohort, the ORR was 33% and also included 1 complete response, while the disease control rate was 81% (79). Recently, Arance and colleagues presented the preliminary results of the phase 2 LEAP004 trial investigating the association of Lenvatinib and Pembrolizumab in 103 patients with advanced melanoma progressing on immunotherapy in second or further line of treatment. The median progression-free survival was 4.2 months with a median overall survival of 13.9 months and a 21.4% response rate, supporting a possible role in overcoming resistance to immunotherapy by Lenvatinib (80). These encouraging data are now furtherly being investigated in a randomized phase III trial specifically dedicated to advanced melanoma. Moreover, other studies are currently ongoing, mostly still in the recruitment phase, for treatment associations of antiangiogenics with anti-PD1 (Nivolumab or Pembrolizumab) or anti-PDL1 (Avelumab) inhibitors (Table 2). As previously described, angiogenesis plays a major role in the natural history of melanoma, from its intrinsic aggressiveness to some forms of resistance to systemic therapy. Despite widely intertwined mechanisms between angiogenesis and immunity, the efficacy of antiangiogenic therapies is currently insufficient. Hence, much interest is addressed to the ongoing clinical trials of combined antiangiogenic and immune therapies, to pursue better outcomes in the therapy of advanced melanoma.
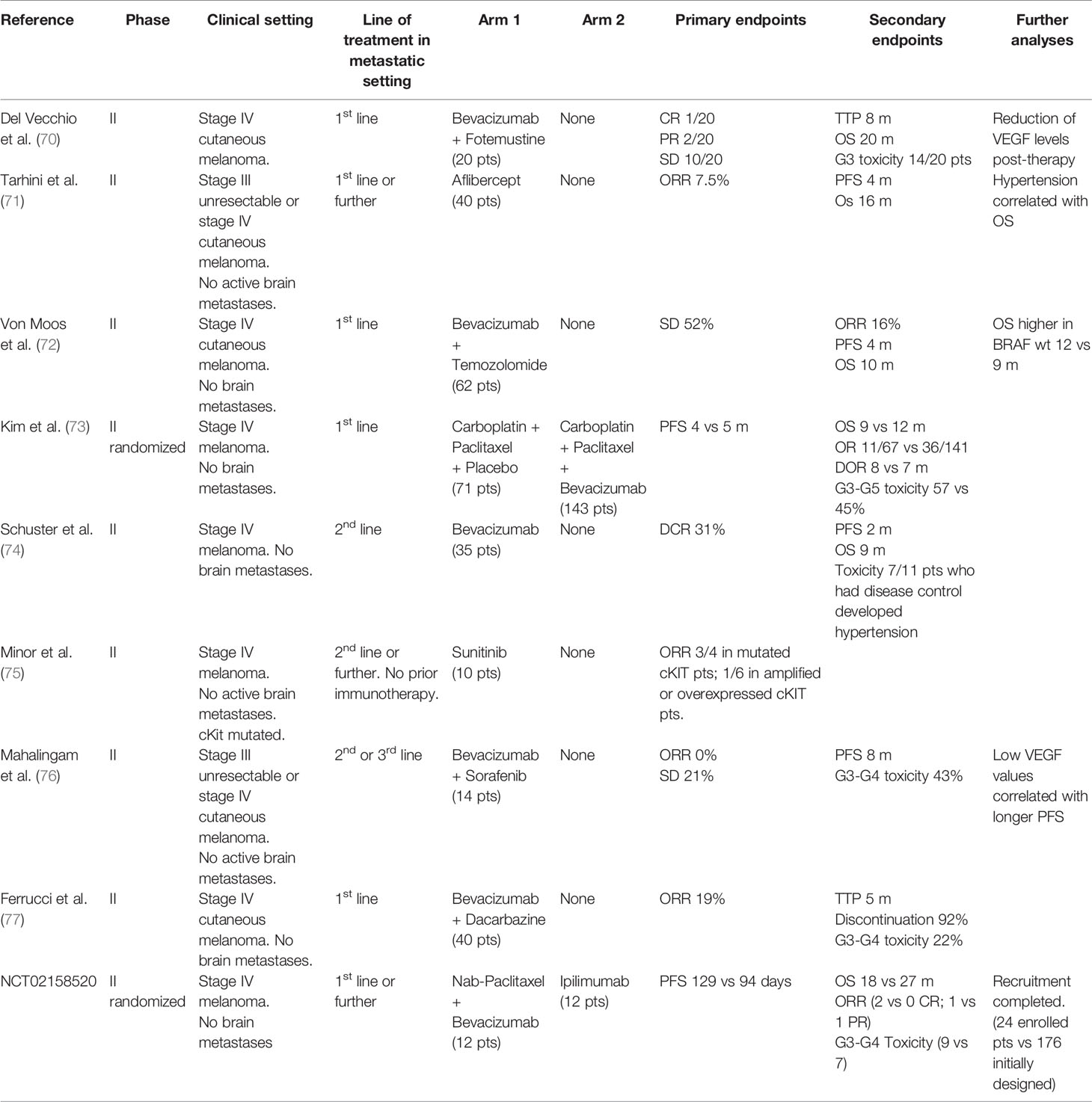
Table 1 Clinical trials of the association of antiangiogenic treatments with old drugs in advanced melanoma.
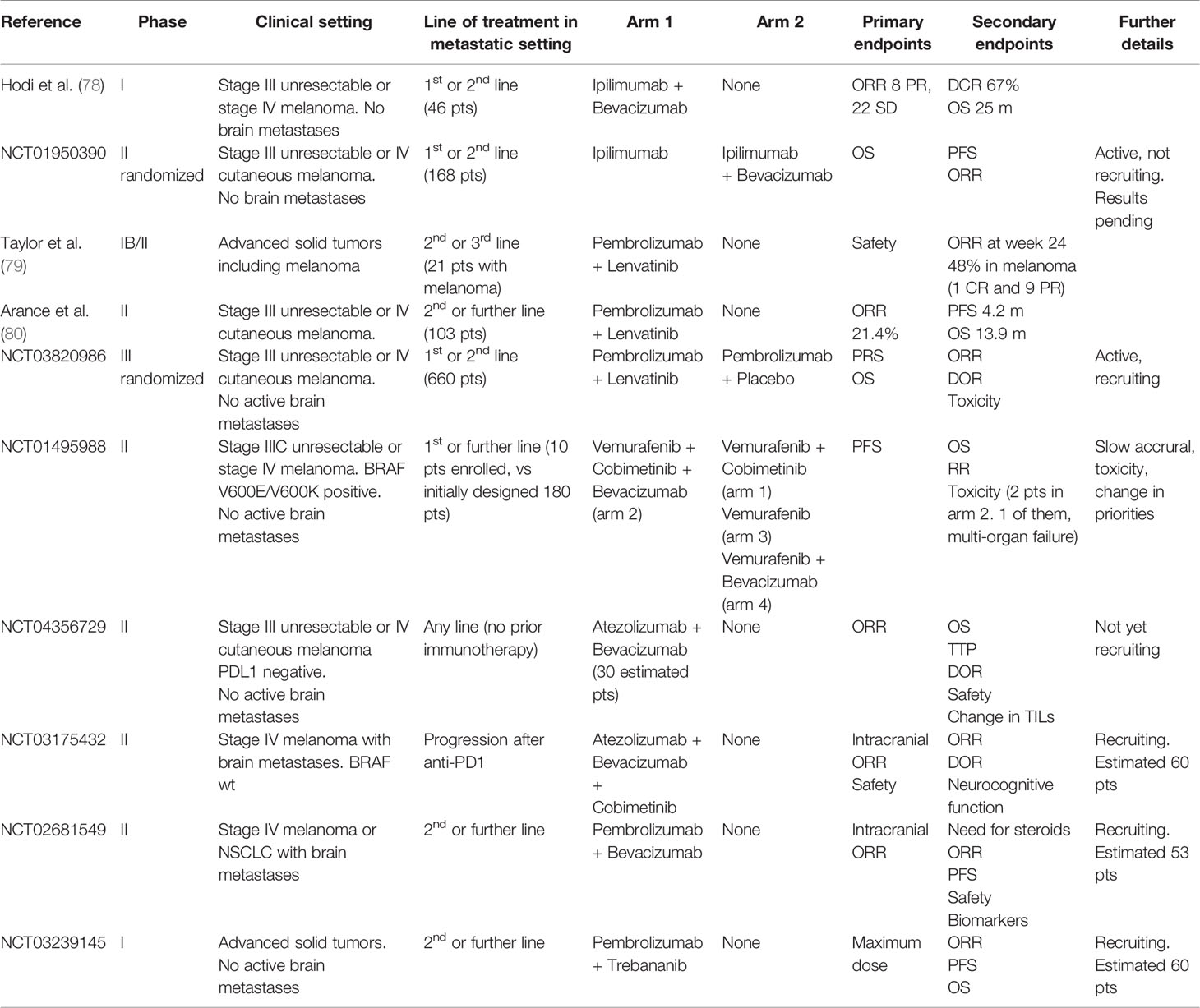
Table 2 Clinical trials on the association of antiangiogenic with immunotherapy or anti-BRAF/anti-MEK targeted therapy in advanced melanoma.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MM declared a shared affiliation with one of the authors, DQ, MG, to the handling editor at time of review.
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
2. Cooke VG, LeBleu VS, Keskin D, Khan Z, O’Connell JT, Teng Y, et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell (2012) 21(1):66–81. doi: 10.1016/j.ccr.2011.11.024
3. Nagy JA, Chang SH, Shih SC, Dvorak AM, Dvorak HF. Heterogeneity of the tumor vasculature. Semin Thromb Hemost (2010) 36(3):321–31. doi: 10.1055/s-0030-1253454
4. Baluk P, Hashizume H, McDonald DM. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev (2005) 15(1):102–11. doi: 10.1016/j.gde.2004.12.005
5. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell (2014) 26(5):605–22. doi: 10.1016/j.ccell.2014.10.006
6. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell (1996) 86(3):353–64. doi: 10.1016/s0092-8674(00)80108-7
7. Cho WC, Jour G, Aung PP. Role of angiogenesis in melanoma progression: update on key angiogenic mechanisms and other associated components. Semin Cancer Biol (2019) 59:175–86. doi: 10.1016/j.semcancer.2019.06.015
8. Warren BA, Shubik P. The growth of the blood supply to melanoma transplants in the hamster cheek pouch. Lab Invest (1966) 15(2):464–78
9. Jour G, Ivan D, Aung PP. Angiogenesis in melanoma: an update with a focus on current targeted therapies. J Clin Pathol (2016) 69(6):472–83. doi: 10.1136/jclinpath-2015-203482
10. Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol (2009) 19(5):329–37. doi: 10.1016/j.semcancer.2009.05.003
11. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer (2003) 3(6):401–10. doi: 10.1038/nrc1093
12. Apte RS, Chen DS, Ferrara N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell (2019) 176(6):1248–64. doi: 10.1016/j.cell.2019.01.021
13. Vartanian A, Stepanova E, Grigorieva I, Solomko E, Baryshnikov A, Lichinitser M. VEGFR1 and PKCα signaling control melanoma vasculogenic mimicry in a VEGFR2 kinase-independent manner. Melanoma Res (2011) 21(2):91–8. doi: 10.1097/CMR.0b013e328343a237
14. Ferrara N. Vascular endothelial growth factor. Arterioscler Thromb Vasc Biol (2009) 29(6):789–91. doi: 10.1161/ATVBAHA.108.179663
15. Mac Gabhann F, Popel AS. Systems biology of vascular endothelial growth factors. Microcirculation (2008) 15(8):715–38. doi: 10.1080/10739680802095964
16. Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology (2005) 69 Suppl 3:4–10. doi: 10.1159/000088478
17. Ziogas AC, Gavalas NG, Tsiatas M, Tsitsilonis O, Politi E, Terpos E, et al. VEGF directly suppresses activation of T cells from ovarian cancer patients and healthy individuals via VEGF receptor Type 2. Int J Cancer (2012) 130(4):857–64. doi: 10.1002/ijc.26094
18. Mulligan JK, Rosenzweig SA, Young MR. Tumor secretion of VEGF induces endothelial cells to suppress T cell functions through the production of PGE2. J Immunother (2010) 33(2):126–35. doi: 10.1097/CJI.0b013e3181b91c9c
19. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med (1996) 2(10):1096–103. doi: 10.1038/nm1096-1096
20. Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol (1998) 160(3):1224–32.
21. Li B, Lalani AS, Harding TC, Luan B, Koprivnikar K, Huan Tu G, et al. Vascular endothelial growth factor blockade reduces intratumoral regulatory T cells and enhances the efficacy of a GM-CSF-secreting cancer immunotherapy. Clin Cancer Res (2006) 12(22):6808–16. doi: 10.1158/1078-0432.CCR-06-1558
22. Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res (2013) 73(2):539–49. doi: 10.1158/0008-5472.CAN-12-2325
23. Griffioen AW, Damen CA, Blijham GH, Groenewegen G. Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood (1996) 88(2):667–73. doi: 10.1002/(sici)1097-0215(19990118)80:2<315::aid-ijc23>3.0.co;2-I
24. Dirkx AE, Oude Egbrink MG, Kuijpers MJ, van der Niet ST, Heijnen VV, Bouma-ter Steege JC, et al. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res (2003) 63(9):2322–9.
25. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med (2015) 212(2):139–48. doi: 10.1084/jem.20140559
26. Rahma OE, Hodi FS. The Intersection between Tumor Angiogenesis and Immune Suppression. Clin Cancer Res (2019) 25(18):5449–57. doi: 10.1158/1078-0432.CCR-18-1543
27. Wada J, Suzuki H, Fuchino R, Yamasaki A, Nagai S, Yanai K, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res (2009) 29(3):881–8.
28. Huang Y, Chen X, Dikov MM, Novitskiy SV, Mosse CA, Yang L, et al. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood (2007) 110(2):624–31. doi: 10.1182/blood-2007-01-065714
29. Varney ML, Johansson SL, Singh RK. Tumour-associated macrophage infiltration, neovascularization and aggressiveness in malignant melanoma: role of monocyte chemotactic protein-1 and vascular endothelial growth factor-A. Melanoma Res (2005) 15(5):417–25. doi: 10.1097/00008390-200510000-00010
30. Osada T, Chong G, Tansik R, Hong T, Spector N, Kumar R, et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol Immunother (2008) 57(8):1115–24. doi: 10.1007/s00262-007-0441-x
31. Kusmartsev S, Eruslanov E, Kübler H, Tseng T, Sakai Y, Su Z, et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J Immunol (2008) 181(1):346–53. doi: 10.4049/jimmunol.181.1.346
32. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature (2011) 475(7355):226–30. doi: 10.1038/nature10169
33. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol (2018) 15(5):325–40. doi: 10.1038/nrclinonc.2018.29
34. Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol (2009) 10(3):165–77. doi: 10.1038/nrm2639
35. Abdul Pari AA, Singhal M, Hübers C, Mogler C, Schieb B, Gampp A, et al. Tumor Cell-Derived Angiopoietin-2 Promotes Metastasis in Melanoma. Cancer Res (2020) 80(12):2586–98. doi: 10.1158/0008-5472.CAN-19-2660
36. Helfrich I, Edler L, Sucker A, Thomas M, Christian S, Schadendorf D. Augustin HG.Angiopoietin-2 levels are associated with disease progression in metastatic malignant melanoma. Clin Cancer Res (2009) 15(4):1384–92. doi: 10.1158/1078-0432.CCR-08-1615
37. Singhal M, Augustin HG. Beyond Angiogenesis: Exploiting Angiocrine Factors to Restrict Tumor Progression and Metastasis. Cancer Res (2020) 80(4):659–62. doi: 10.1158/0008-5472.CAN-19-3351
38. Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res (2010) 70(13):5270–80. doi: 10.1158/0008-5472.CAN-10-0012
39. Coffelt SB, Chen YY, Muthana M, Welford AF, Tal AO, Scholz A, et al. Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J Immunol (2011) 186(7):4183–90. doi: 10.4049/jimmunol.1002802
40. Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez-Ramos D, et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature (2014) 507(7490):109–13. doi: 10.1038/nature13111
41. Fu XQ, Liu B, Wang YP, Li JK, Zhu PL, Li T, et al. Activation of STAT3 is a key event in TLR4 signaling-mediated melanoma progression. Cell Death Dis (2020) 11(4):246. doi: 10.1038/s41419-020-2440-1
42. Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev (2005) 24(2):315–27. doi: 10.1007/s10555-005-1580-1
43. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front Immunol (2018) 9:527. doi: 10.3389/fimmu.2018.00527
44. Riboldi E, Musso T, Moroni E, Urbinati C, Bernasconi S, Rusnati M, et al. Cutting edge: proangiogenic properties of alternatively activated dendritic cells. J Immunol (2005) 175(5):2788–92. doi: 10.4049/jimmunol.175.5.2788
45. Owen JL, Mohamadzadeh M. Macrophages and chemokines as mediators of angiogenesis. Front Physiol (2013) 4:159. doi: 10.3389/fphys.2013.00159
46. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer (2008) 8(8):618–31. doi: 10.1038/nrc2444
47. Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature (2017) 544(7649):250–4. doi: 10.1038/nature21724
48. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell (2016) 167(2):397–404.e9. doi: 10.1016/j.cell.2016.08.069
49. Bourbié-Vaudaine S, Blanchard N, Hivroz C, Roméo PH. Dendritic cells can turn CD4+ T lymphocytes into vascular endothelial growth factor-carrying cells by intercellular neuropilin-1 transfer. J Immunol (2006) 177(3):1460–9. doi: 10.4049/jimmunol.177.3.1460
50. Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature (2016) 529(7586):316–25. doi: 10.1038/nature17040
51. Boettcher S, Gerosa RC, Radpour R, Bauer J, Ampenberger F, Heikenwalder M, et al. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood (2014) 124(9):1393–403. doi: 10.1182/blood-2014-04-570762
52. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med (2008) 14(1):28–36. doi: 10.1038/nm1699
53. Dewhirst MW, Secomb TW. Transport of drugs from blood vessels to tumour tissue. Nat Rev Cancer (2017) 17(12):738–50. doi: 10.1038/nrc.2017.93
54. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front Immunol (2018) 9:978. doi: 10.3389/fimmu.2018.00978
55. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassará A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med (2017) 9(385):eaak9670. doi: 10.1126/scitranslmed.aak9670
56. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer (2017) 17(8):457–74. doi: 10.1038/nrc.2017.51
57. De Almeida PE, Mak J, Hernandez G, Jesudason R, Herault A, Javinal V, et al. Anti-VEGF Treatment Enhances CD8+ T-cell Antitumor Activity by Amplifying Hypoxia. Cancer Immunol Res (2020) 8(6):806–18. doi: 10.1158/2326-6066.CIR-19-0360
58. Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell (2004) 6(6):553–63. doi: 10.1016/j.ccr.2004.10.011
59. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A (2012) 109(43):17561–6. doi: 10.1073/pnas.1215397109
60. Şenbabaoğlu Y, Gejman RS, Winer AG, Liu M, Van Allen EM, de Velasco G, et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol (2016) 17(1):231. doi: 10.1186/s13059-016-1092-z
61. Donnem T, Reynolds AR, Kuczynski EA, Gatter K, Vermeulen PB, Kerbel RS, et al. Non-angiogenic tumours and their influence on cancer biology. Nat Rev Cancer (2018) 18(5):323–36. doi: 10.1038/nrc.2018.14
62. Croce L, Coperchini F, Magri F, Chiovato L, Rotondi M. The multifaceted anti-cancer effects of BRAF-inhibitors. Oncotarget (2019) 10(61):6623–40. doi: 10.18632/oncotarget.27304
63. Leslie C, Bowyer SE, White A, Grieu-Iacopetta F, Trevenen M, Iacopetta B, et al. FOXP3+ T regulatory lymphocytes in primary melanoma are associated with BRAF mutation but not with response to BRAF inhibitor. Pathology (2015) 47(6):557–63. doi: 10.1097/PAT.0000000000000314
64. Khalili JS, Liu S, Rodríguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res (2012) 18(19):5329–40. doi: 10.1158/1078-0432.CCR-12-1632
65. Kim K, Jeon S, Kim TM, Jung CK. Immune Gene Signature Delineates a Subclass of Papillary Thyroid Cancer with Unfavorable Clinical Outcomes. Cancers (Basel) (2018) 10(12):494. doi: 10.3390/cancers10120494
66. Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res (2005) 65(6):2412–21. doi: 10.1158/0008-5472.CAN-04-2423
67. Atzori MG, Ceci C, Ruffini F, Trapani M, Barbaccia ML, Tentori L, et al. Role of VEGFR-1 in melanoma acquired resistance to the BRAF inhibitor vemurafenib. J Cell Mol Med (2020) 24(1):465–75. doi: 10.1111/jcmm.14755
68. Wu X, Giobbie-Hurder A, Liao X, Connelly C, Connolly EM, Li J, et al. Angiopoietin-2 as a Biomarker and Target for Immune Checkpoint Therapy. Cancer Immunol Res (2017) 5(1):17–28. doi: 10.1158/2326-6066.CIR-16-0206
69. Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med (2017) 9(385):eaak9679. doi: 10.1126/scitranslmed.aak9679
70. Del Vecchio M, Mortarini R, Canova S, Di Guardo L, Pimpinelli N, Sertoli MR, et al. Bevacizumab plus fotemustine as first-line treatment in metastatic melanoma patients: clinical activity and modulation of angiogenesis and lymphangiogenesis factors. Clin Cancer Res (2010) 16(23):5862–72. doi: 10.1158/1078-0432.CCR-10-2363
71. Tarhini AA, Frankel P, Margolin KA, Christensen S, Ruel C, Shipe-Spotloe J, et al. Aflibercept (VEGF Trap) in inoperable stage III or stage iv melanoma of cutaneous or uveal origin. Clin Cancer Res (2011) 17(20):6574–81. doi: 10.1158/1078-0432.CCR-11-1463
72. von Moos R, Seifert B, Simcock M, Goldinger SM, Gillessen S, Ochsenbein A, et al. Swiss Group for Clinical Cancer Research (SAKK). First-line temozolomide combined with bevacizumab in metastatic melanoma: a multicentre phase II trial (SAKK 50/07). Ann Oncol (2012) 23(2):531–6. doi: 10.1093/annonc/mdr126
73. Kim KB, Sosman JA, Fruehauf JP, Linette GP, Markovic SN, McDermott DF, et al. BEAM: a randomized phase II study evaluating the activity of bevacizumab in combination with carboplatin plus paclitaxel in patients with previously untreated advanced melanoma. J Clin Oncol (2012) 30(1):34–41. doi: 10.1200/JCO.2011.34.6270
74. Schuster C, Eikesdal HP, Puntervoll H, Geisler J, Geisler S, Heinrich D, et al. Clinical efficacy and safety of bevacizumab monotherapy in patients with metastatic melanoma: predictive importance of induced early hypertension. PloS One (2012) 7(6):e38364. doi: 10.1371/journal.pone.0038364
75. Minor DR, Kashani-Sabet M, Garrido M, O’Day SJ, Hamid O, Bastian BC. Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res (2012) 18(5):1457–63. doi: 10.1158/1078-0432.CCR-11-1987
76. Mahalingam D, Malik L, Beeram M, Rodon J, Sankhala K, Mita A, et al. Phase II study evaluating the efficacy, safety, and pharmacodynamic correlative study of dual antiangiogenic inhibition using bevacizumab in combination with sorafenib in patients with advanced malignant melanoma. Cancer Chemother Pharmacol (2014) 74(1):77–84. doi: 10.1007/s00280-014-2479-8
77. Ferrucci PF, Minchella I, Mosconi M, Gandini S, Verrecchia F, Cocorocchio E, et al. Dacarbazine in combination with bevacizumab for the treatment of unresectable/metastatic melanoma: a phase II study. Melanoma Res (2015) 25(3):239–45. doi: 10.1097/CMR.0000000000000146
78. Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res (2014) 2(7):632–42. doi: 10.1158/2326-6066.CIR-14-0053
79. Taylor MH, Lee CH, Makker V, Rasco D, Dutcus CE, Wu J, et al. Phase IB/II Trial of Lenvatinib Plus Pembrolizumab in Patients With Advanced Renal Cell Carcinoma, Endometrial Cancer, and Other Selected Advanced Solid Tumors. J Clin Oncol (2020) 38(11):1154–63. doi: 10.1200/JCO.19.01598
80. Arance Fernandez AM, O’Day SJ, De la Cruz Merino L, Petrella T, Jamar R, Ny L, et al. Lenvatinib (len) plus pembrolizumab (pembro) for advanced melanoma (MEL) that progressed on PD1 or PDL1 inhibitor: initial results of LEAP-004. Ann Oncol (2020) 31) suppl_4:S1142–215. doi: 10.1016/annonc/annonc325
Keywords: neoangiogenesis, melanoma, combination strategy, antiangiogenics, immunotherapy
Citation: Quaresmini D and Guida M (2020) Neoangiogenesis in Melanoma: An Issue in Biology and Systemic Treatment. Front. Immunol. 11:584903. doi: 10.3389/fimmu.2020.584903
Received: 18 July 2020; Accepted: 12 October 2020;
Published: 29 October 2020.
Edited by:
Ronca Roberto, University of Brescia, ItalyReviewed by:
Rodabe N. Amaria, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2020 Quaresmini and Guida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michele Guida, bWljZ3VpZGE1N0BnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.