 Cynthia Perez
Cynthia Perez Isabelle Gruber
Isabelle Gruber Caroline Arber
Caroline Arber- Department of Oncology UNIL CHUV, Ludwig Institute for Cancer Research Lausanne, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland
Chimeric antigen receptor (CAR) engineered T cell therapies individually prepared for each patient with autologous T cells have recently changed clinical practice in the management of B cell malignancies. Even though CARs used to redirect polyclonal T cells to the tumor are not HLA restricted, CAR T cells are also characterized by their endogenous T cell receptor (TCR) repertoire. Tumor-antigen targeted TCR-based T cell therapies in clinical trials are thus far using “conventional” αβ-TCRs that recognize antigens presented as peptides in the context of the major histocompatibility complex. Thus, both CAR- and TCR-based adoptive T cell therapies (ACTs) are dictated by compatibility of the highly polymorphic HLA molecules between donors and recipients in order to avoid graft-versus-host disease and rejection. The development of third-party healthy donor derived well-characterized off-the-shelf cell therapy products that are readily available and broadly applicable is an intensive area of research. While genome engineering provides the tools to generate “universal” donor cells that can be redirected to cancers, we will focus our attention on third-party off-the-shelf strategies with T cells that are characterized by unique natural features and do not require genome editing for safe administration. Specifically, we will discuss the use of virus-specific T cells, lipid-restricted (CD1) T cells, MR1-restricted T cells, and γδ-TCR T cells. CD1- and MR1-restricted T cells are not HLA-restricted and have the potential to serve as a unique source of universal TCR sequences to be broadly applicable in TCR-based ACT as their targets are presented by the monomorphic CD1 or MR1 molecules on a wide variety of tumor types. For each cell type, we will summarize the stage of preclinical and clinical development and discuss opportunities and challenges to deliver off-the-shelf targeted cellular therapies against cancer.
Introduction
Engineered T cell therapies using chimeric antigen receptors (CARs) against CD19+ B cell malignancies have been commercialized and have changed clinical practice. Current commercial products are manufactured in a highly personalized way for each individual patient with autologous peripheral blood αβ-TCR T cells (1, 2). Challenges with the use of autologous products include aspects related to previous chemotherapies or allogeneic hematopoietic cell transplantation (HCT) that can impact on the quantity and quality of the starting material, uncontrollable interpatient variability, and (too) long waiting times for the patients due to global manufacturing chains (3–5). Thus, the development of readily available off-the-shelf allogeneic immune effector cell (IEC) therapy products is an attractive alternative approach. Cell banks can be generated in advance; donors can be well-characterized according to the desired biological parameters of the final product. Major challenges to allogenic IEC therapies include the possibility of dual rejection: infused cells may produce graft-versus-host disease (GVHD), or the host immune system may reject the infused cells (4–6).
In this review, we will seek, evaluate and discuss challenges and opportunities for T cell-based IEC therapies, using naturally occurring “universal” donor T cells. These cells are either characterized by the recognition of well-defined HLA-restricted conventional αβ-TCR antigens, or are HLA-independent and recognize lipids, metabolites or phosphoantigens presented in the context of non-polymorphic receptors on target cells. By definition, these “universal” donor T cells do not produce GVHD, do not require genome editing for safe application as a therapeutic product, and have the capacity to potentially target a wide variety of cancers. We will focus our review on human preclinical and clinical developments including αβ-TCR T cells [virus specific T cells (VSTs), CD1-, and MR1-restricted T cells] as well as γδ-TCR T cells. Finally, we also discuss the potential use of universal TCRs that can be inserted as transgenes into IECs. Engineering of these “universal” donor T cells aims to combine and simultaneously exploit the endogenous natural properties of the cells with engineered properties that enhance the anti-tumor potential of the final product (e.g., recognition of tumor-derived lipids or metabolites by endogenous TCR and cell surface antigen by the introduced CAR).
Naturally Occurring “Universal” Donor T Cells as Platforms for T Cell Engineering
αβ-TCR T Cell Subsets
Conventional αβ-TCR T cells express HLA-restricted TCRs composed of an α- and a β-chain and recognize peptides presented by HLA molecules on the cell surface of target cells. Selected in the thymus, these T cells constitute the majority of the circulating T cells in the human body (7). Under physiologic conditions αβ-TCR T cells are polyclonal and express an extremely diverse TCR repertoire to cover a wide range of potential target antigens. This TCR diversity is reduced in memory T cell pools that form upon antigen specific expansion and clearance of a pathogen. VSTs directed against cytomegalovirus (CMV) for example are characterized by their oligoclonality, with a limited number of high avidity TCRs dominating the pool of memory VSTs that can re-expand upon repeated viral challenge (8–10). Unconventional αβ-TCR T cells are non-HLA-restricted and recognize non-peptide targets that are presented in the context of non-polymorphic molecules. In fact, several types of unconventional αβ-TCR T cells express semi-invariant TCRs, such as, for example, invariant natural killer T (iNKT) cells that recognize targets in the context of the monomorphic antigen-presenting molecule CD1, or mucosal-associated invariant T (MAIT) cells that recognize targets in the context of MR1 (Figure 1). Several of these αβ-TCR T cell subsets therefore harbor unique features that could potentially qualify them as universal donor cells for adoptive T cell therapy (ACT) (Table 1).
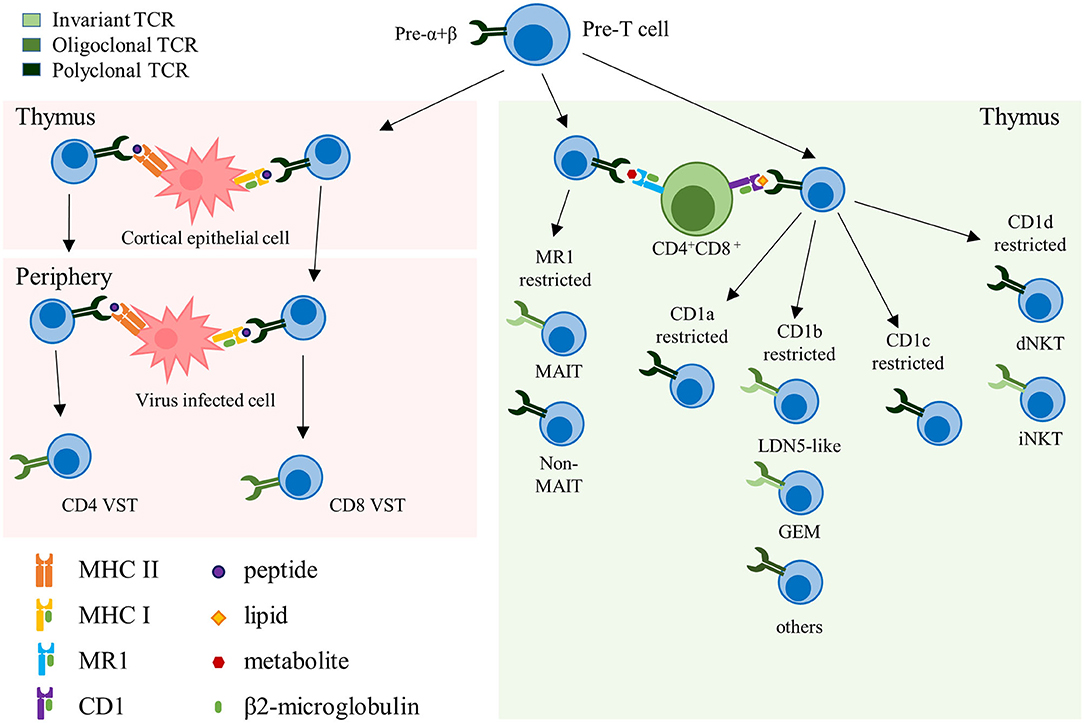
Figure 1. Representation of the different αβ-TCR subsets. Schematic representation summarizing the different αβ-TCR subsets, their development and restricting elements. The color grading of the TCR represents its clonality, from light to dark green (monoclonal to polyclonal, respectively). TCR, T cell receptor; VST, virus-specific T cell; MHC, major histocompatibility complex; MAIT, mucosal-associated invariant T cell; GEM, germline-encoded mycolyl lipid-reactive; dNKT, diverse natural killer T cell; iNKT, invariant natural killer T cell.
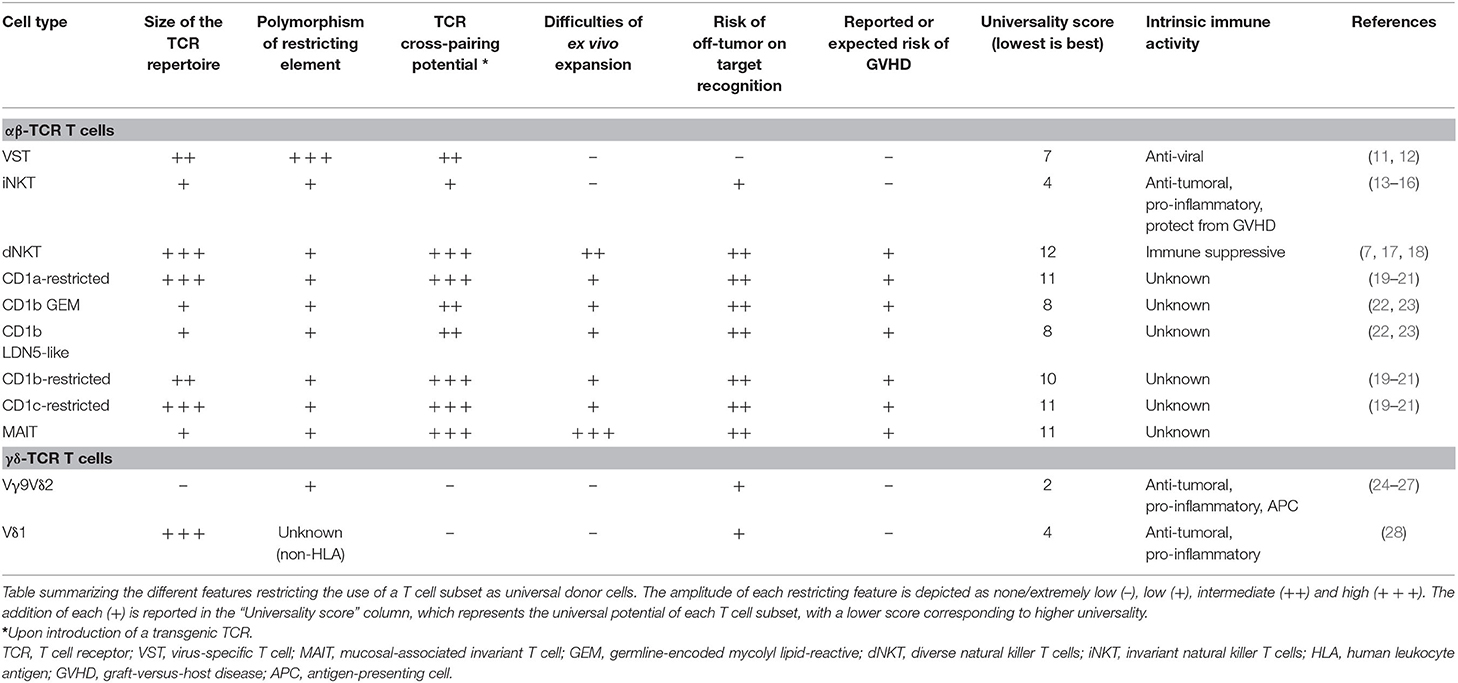
Table 1. Features of universal donor cells.
Virus-Specific T Cells (VSTs)
Adoptive transfer of VSTs to prevent or treat infections and/or EBV-associated post-transplant lymphoproliferation
Viral reactivations and infections after allogeneic HCT remain a major cause of morbidity and mortality (29). Current anti-viral drugs are associated with dose-limiting end organ toxicities or lack of efficacy due to primary or secondary resistance, and only virus-specific immune reconstitution can resolve the issue of recurrent infections. Thus, in patients that do not concomitantly present with GVHD, adoptive transfer of VSTs is a safe and efficient therapy to accelerate immune reconstitution. The various approaches of VST manufacturing for adoptive transfer as well as clinical trial results have recently been reviewed (30–32). Allogeneic HCT donor derived VSTs have shown significant clinical activity against Epstein-Barr Virus (EBV), CMV, adenovirus, BK virus, and Human Herpes Virus 6. Importantly, impressive anti-viral responses have been reported across studies, and significant alloreactivity or GVHD has only been described in a very limited number of patients. HCT donor derived Epstein-Barr Virus specific T cells (EBVSTs) are also active against post-transplant EBV+ lymphoproliferative disorders in 65–85% of treated patients (11, 33, 34), and their long-term persistence was demonstrated in gene marking studies (11).
With the goal to facilitate rapid access to VSTs including for patients with seronegative donors, allogeneic third-party VST banks have been developed by several groups and institutions (12, 30, 35–37) and have entered commercialization. Safety and anti-viral activity of adoptively transferred allogeneic third-party donor VSTs are excellent, and the overall response rate (ORR) when treating viral infections can reach up to 92% (36, 38). When targeting EBV+ lymphoproliferation, ORRs were 68% in HCT and 54% in solid organ transplant (SOT) patients (12). Because persistence is a key parameter for tumor targeted T cell therapies but has not been summarized in the recent review articles on VSTs, we selected clinical trials with information on persistence on the infused third-party VSTs and summarized the results in Table 2. Available information is quite scarce. Each group used different tracking methods, no gene-marking was available to easily detect the in vivo fate of the infused cells, and long-term follow-up is mostly lacking. Available data suggests that third-party VSTs do not engraft and persist as well as HLA-matched HCT donor-derived VSTs. The variability between patients and trials was broad. Gallot et al. for example were not able to detect significant levels of cells derived from the infused VST lines (41). But others found a correlation between detection of anti-viral activity (12, 38, 39, 42) or VST line derived TCR sequences (37, 40) and viral clearance. The limited persistence is most likely due to rejection of the infused third party VSTs by the host immune system. Thus, in order to overcome this problem, novel types of chimeric alloimmune defense receptors (ADRs) have been developed. One strategy consists of a chimeric receptor using the extracellular part of human β2-microglobulin and signaling through CD3ζ and was shown to protect VSTs from alloreactive T cells in vitro, but this approach cannot protect from NK cell mediated rejection (43). More recently, a receptor recognizing 4-1BB (CD137)—temporarily upregulated on both activated T and NK cells—and signaling through CD3ζ was developed. 4-1BBζ ADR-engineered T cells were protected from T and NK cell mediated rejection in vitro and in vivo in a mouse xenograft model, and CARs retained their antitumor function when co-expressed with the 4-1BBζ ADR (44). Thus, ADRs have the potential to further enhance the persistence, efficacy, universality and safety of third-party engineered VSTs, and can be co-engineered with CARs. In the post-transplant setting, endogenous immune reconstitution also plays an important role in the establishment of long-term viral control.
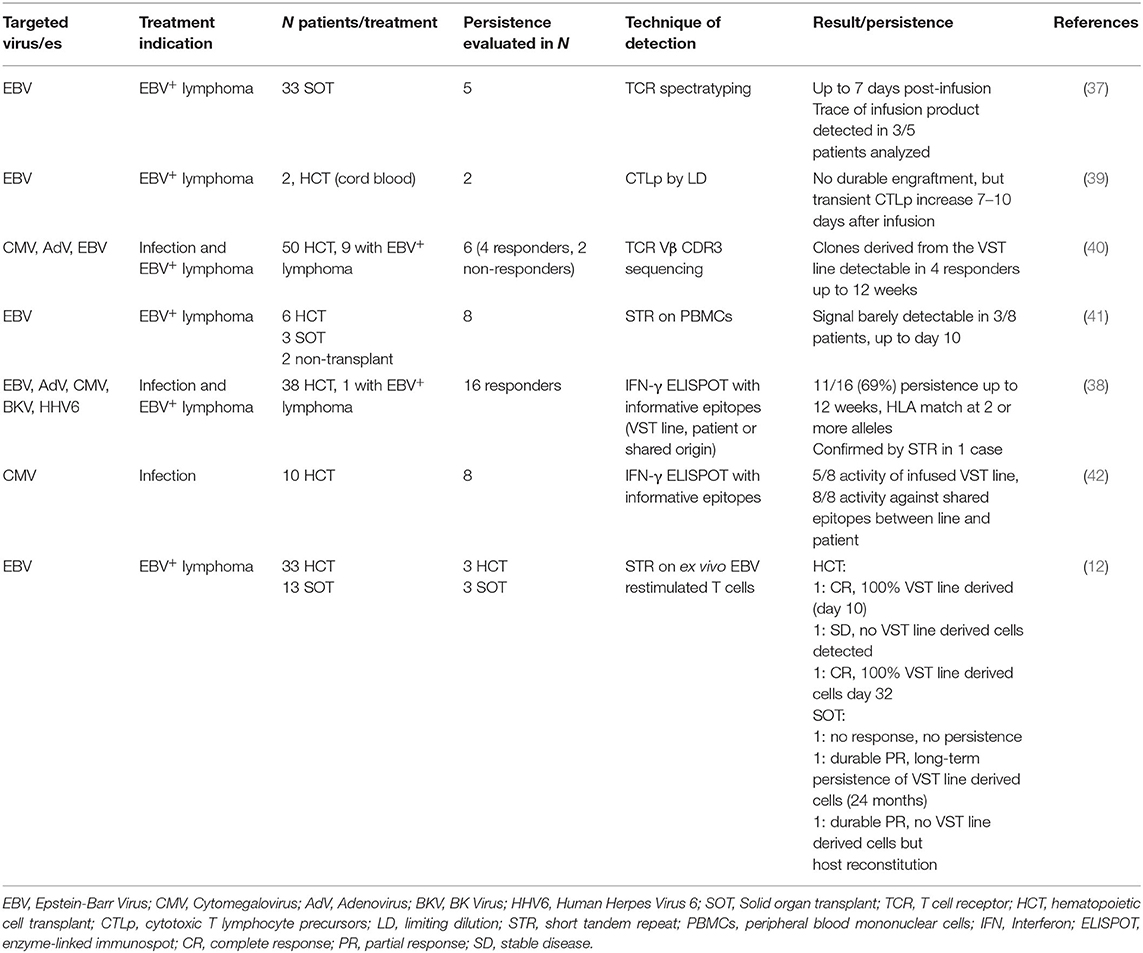
Table 2. In vivo persistence of third-party off-the-shelf VSTs.
We still need to learn more about the contributions of infused cells and endogenously reconstituted anti-viral immunity upon third-party VST adoptive transfer when targeting viral infections or EBV-associated malignancies. Systematic assessment of VST persistence with standardized methods across clinical trials would facilitate this understanding.
Engineered VSTs in clinical trials
VSTs have been clinically validated as a cellular therapy platform to genetically redirect antigen specificity against tumor-associated antigens (Table 3). With this approach, endogenous anti-viral TCR specificities can be exploited for the in vivo expansion and stimulation of the transgenic VSTs or targeting of viral-associated malignancies. Indeed, autologous CAR-engineered VSTs have been evaluated in clinical trials targeting GD2 for neuroblastoma (45, 46), and allogeneic HCT donor-derived CAR+ VSTs targeting CD19 in B-acute lymphoblastic leukemia (47–49). Reactivation of CMV in patients after allogeneic HCT and CD19 CAR+ CMVST infusion for example led to significant in vivo re-expansion of the infused cells and CD19 directed cytotoxic activity with elimination of B cells (49). To assess whether vaccination could be used to in vivo re-expand CAR+ VSTs, a clinical trial is underway to assess autologous GD2-CAR+ Varizella Zoster VST (VZVSTs) cell infusions in combination with vaccination (NCT01953900). Preclinical investigations had shown that anti-tumor function of GD2-CAR+ VZVSTs could be rescued in vitro upon stimulation with VZV peptide-pulsed dendritic cells (DC) (50). A clinical trial with gene-modified third-party partially HLA-matched healthy donor-derived banked CAR+ EBVSTs targeting CD30 in patients with EBV-associated CD30+ lymphomas is in preparation at Baylor College of Medicine (NCT04288726).
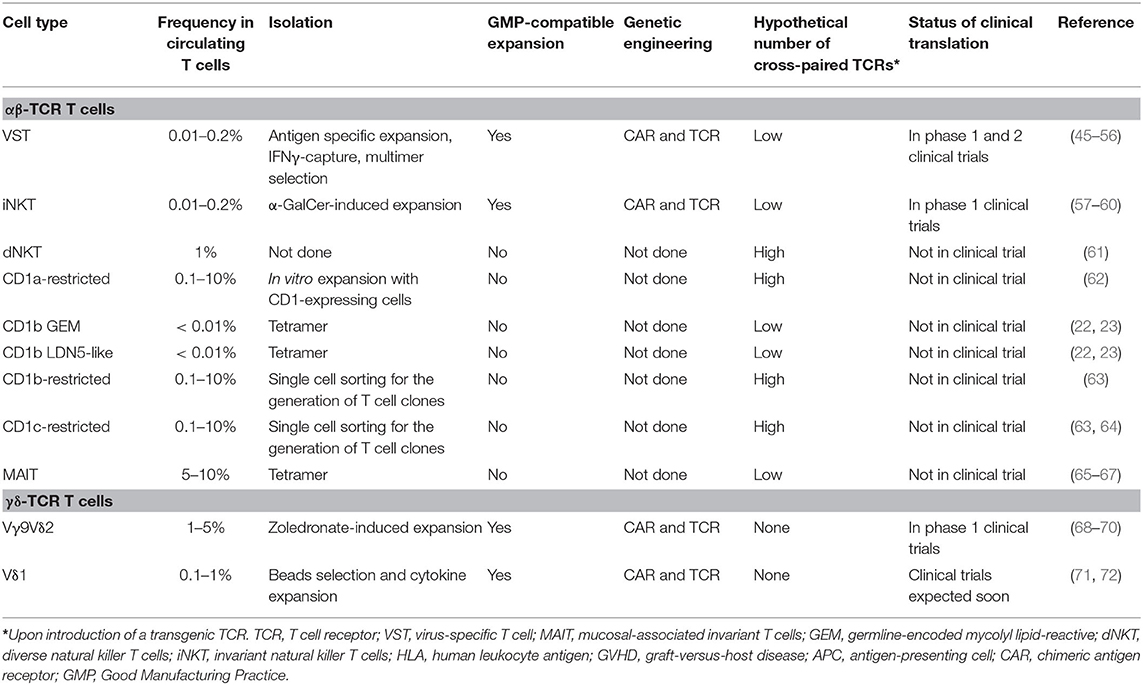
Table 3. Overview of clinical development status.
Engineering VSTs with a tumor-targeted transgenic TCR has been more challenging than with CARs, as forced expression of a transgenic TCR leads to downregulation of the endogenous TCRs (51). Indeed, these findings were confirmed in TCR transgenic VSTs, where reduction of antiviral activity was reported in several preclinical studies (52–55) and in one clinical trial (56). Interestingly, one report showed that TCR+ VSTs can shift their antigenic predominance depending on the type of antigenic exposure given to the cells (viral or tumor antigen) (55). The oligoclonal features of VSTs minimize the risk of cross-pairing between transgenic and endogenous TCR chains, and thus the use of VSTs to express a transgenic TCR is thought to reduce the risk of both off-target toxicities and GVHD. In the only clinical trial reported to date, a Wilms tumor antigen-1 (WT-1) specific TCR was expressed in single epitope specific EBVSTs generated from the HLA-matched HCT donor. The clinical responses in high-risk AML patients who received allogeneic HCT followed by prophylactic WT-1 TCR+ EBVST infusions were impressive (56). Unfortunately, no viral reactivation occurred in the cohort of 12 treated patients, so the question whether the level of anti-viral specificity is sufficient to mediate in vivo re-expansion of TCR+ VSTs upon viral reactivation and to protect against viral disease remains elusive. TCR transgenic third-party VSTs have not yet been evaluated clinically.
VSTs as platform for engineered ACT: how universal can they be?
Third-party banked VSTs have been established as a safe and efficient ACT to treat infections and EBV+ lymphoproliferation after allogeneic HCT or SOT. The oligoclonal nature of VSTs limits their capacity to induce GVHD in this patient population. Despite the polymorphic nature of HLA, VST banks can be built with a limited number of well-chosen and characterized donors to cover a highly diverse patient population (42). Furthermore, VSTs can efficiently be redirected to tumors with both CARs and TCRs for clinical use. Drawbacks of third-party VSTs are that (i) their use has so far been limited to HCT and SOT patients, (ii) the in vivo re-stimulation through the endogenous TCR depends on unpredictable endogenous viral reactivations or scheduled vaccinations, and (iii) their long-term persistence has not yet been conclusively assessed. The clinical development of engineering strategies such as incorporation of ADRs may overcome these limitations in the future and make VSTs safer and more universal.
CD1-Restricted T Cells
Background on CD1 molecules
The monomorphic CD1 family is constituted of five members, four extracellular (CD1a-CD1d) and one intracellular molecule (CD1e). CD1a, CD1b, and CD1c belong to the group 1 CD1, while group 2 is solely constituted of CD1d (7, 19). Circulating within the different secretory and endosomal compartments of the cells, CD1 molecules present a large array of lipid antigens to T cells (7, 73–75). So far, only a limited number of CD1-restricted antigens are known, consisting of lipids shared by multiple microorganisms and of self-lipids that accumulate during cellular stress, for example in cancer (19, 20, 76). CD1d expression is constitutive and present on all antigen-presenting cells (APCs), while the expression of CD1a/b/c is inducible and limited to a subset of APCs. For example, B cells express CD1c, Langerhans cells express CD1a and CD1e, while myeloid cells can express all five CD1 molecules (19, 20).
CD1d-Restricted T cells: their implication in tumor immunity
CD1d-restricted T cells, also called NKT cells, are selected in the thymus after recognition of CD1d molecules expressed by CD4+CD8+ double positive thymocytes (13, 19, 77). CD1d-restricted T cells are classified into two distinct groups based on their ability to recognize α-galactosylceramide (α-GalCer), a glycosphingolipid originally derived from marine sponge. Type I NKT, or iNKT, express a semi-invariant TCR consisting of an invariant TCRα chain (TRAV10-TRAJ18 in human) paired with a limited number of TCRβ chains and recognize α-GalCer. Type II NKT, or dNKT, express a more diverse polyclonal TCR repertoire, and are unresponsive to α-GalCer [(7); Figure 1].
α-GalCer is produced by the gut flora and many mammalian tissues and acts as a potent iNKT cell stimulator (14, 78–80). Dysregulated lipid production in tumors is also a source of antigenic lipids capable of stimulating iNKT cells [e.g., GD3 and GM3 in melanoma (81, 82), or α-fucosylceramides in colorectal and pancreatic adenocarcinomas (83)]. Upon TCR engagement, iNKTs rapidly secrete high levels of cytokines (e.g., IFNγ, TNFα, IL4, IL13, and IL17) and lytic granules (granzymes and perforin), and upregulate killing receptors such as Fas ligand and TRAIL (17, 84). Thus, iNKTs are rapidly cytotoxic and strongly modulate the tumor microenvironment by direct targeting of CD1d+ tumors, tumor-associated macrophages and myeloid-derived suppressor cells (15, 85). Modulation of the immune response occurs by transactivating NK cells, licensing DCs and activating γδ-TCR T cells (16, 19, 86). This crosstalk leads to a strong activation of the endogenous adaptive immune system (87). That iNKTs play an important role in anti-tumor immunity is inferred from the facts that low frequency of iNKT cells in patients with hematologic or head and neck cancers correlated with poor prognosis (88–90), while higher iNKT cell infiltration in colorectal cancer correlated with longer survival (91). After allogeneic HCT, higher doses of iNKT cells contained in the graft were associated with protection from acute GVHD (92), and early donor-derived iNKT cell reconstitution post-transplant correlated with reduced acute GVHD and lower non-relapse mortality while maintaining graft-versus-leukemia effects (93).
dNKTs are less well characterized and thought to have a more immune regulatory role (17, 18, 94). In multiple myeloma for example, dNKTs have been involved in suppression of anti-tumor immunity in an IL-13-dependent manner (94). Potential immunotherapeutic applications for dNKT cells have been reviewed elsewhere (61).
Ex vivo expanded iNKT cells in clinical trials
Due to their biology, iNKT cells are an attractive cell type to investigate for cancer immunotherapy (Table 3). However, establishing GMP compatible ex vivo expansion protocols for iNKT cells has been a hurdle to broader development. To date, results from two clinical trials assessing the safety of adoptively transferred ex vivo expanded autologous iNKT cells in cancer patients were reported (95, 96). In a lung cancer trial, autologous iNKT cells from 6 patients were expanded in the presence of α-GalCer and IL2, reaching 0.1–25% iNKT cells in the final products. Infusions were safe but no significant clinical responses were seen (96). In a melanoma trial, autologous iNKT cells were sorted from PBMCs and expanded ex vivo for 6–8 weeks with anti-CD3 and IL2. Purity post-expansion ranged from 13 to 87%. The nine treated patients had only minimal or no evidence of disease at time of infusion and were not lymphodepleted. A clear correlation between iNKT cell infusions, immune parameters and outcome could not be established (95).
Meanwhile, ex vivo expansion methods have been refined and now allow genetic engineering of iNKT cells (57, 58). Dual targeting by harnessing endogenous and engineered properties of iNKT cells produced very promising pre-clinical results in neuroblastoma with GD2-CAR iNKT cells also incorporating transgenic IL15 (57, 59) and in lymphoma with CD19-CAR iNKT cells expanded in media containing IL21 (58, 60). Both approaches have started phase I clinical evaluation at Baylor College of Medicine. Safety of autologous GD2-CAR.IL15 engineered iNKT cells is evaluated in patients with neuroblastoma (NCT03294954). Since iNKT cells are not alloreactive and clinical studies suggest that iNKTs can suppress GVHD (92, 93, 97), a clinical trial with third-party allogeneic off-the-shelf iNKT cells genetically engineered to express a CD19-CAR and IL15 is underway to assess safety, in vivo expansion and persistence, and responses in patients with B-cell malignancies (NCT03774654).
CD1d-Restricted T cells as platform for ACT
iNKT cells have the ability to kill CD1d+ tumor cells and immune suppressive cells in the tumor microenvironment through direct cytotoxicity, but also modulate the immune response of NK cells and DCs through cytokine secretion, producing enhanced anti-tumor responses of conventional endogenous T cells (16, 19, 86). In addition to its anti-tumor activity, iNKT cells can protect the patient from developing GVHD after allogeneic HCT, as better iNKT cell recovery correlated with a reduced risk of GVHD (92, 93, 97). With their lack of HLA-restriction, semi-invariant TCR and protective potential against GVHD, iNKT cells possess several unique features required for universal donor cells (Table 1). Their in vivo persistence will need to be analyzed. The field is currently moving toward evaluating the safe use of iNKT cells from allogeneic third-party universal donors in engineered ACT (NCT03774654).
CD1a/b/c-Restricted T cells and their implication in tumor immunity
The current knowledge on T cells restricted to group 1 CD1 is limited, and mostly results from studies performed in vitro on human T cell clones (62). Recognizing diverse microbial and self-lipid antigens, group 1 CD1-restricted T cells are relatively abundant among circulating lymphocytes in healthy individuals (63), and the majority has a polyclonal TCR repertoire (19–21). Two subsets of CD1b-restricted T cells, the germline-encoded, mycolyl lipid–reactive (GEM), and the LDN5-like T cells, which recognize glucose monomycolate, a lipid antigen derived from Mycobacterium tuberculosis, express an invariant TCR (TRAV1-TRAJ9, and TRAV17-TRBV4-1, respectively) (22, 23).
Group 1 CD1-restricted T cells are thought to participate in immune surveillance of hematologic malignancies. Analysis of a limited number of patient samples revealed positivity for CD1c in 51% and CD1b in 54% of AML patients (n = 33), 71% of B-ALL samples expressed CD1c (n = 7), and 75% of pediatric T-ALL samples expressed CD1a and CD1b (n = 8) (64). Methyl-lysophosphatidic acid (mLPA) is a self-lipid antigen presented in the context of CD1c on hematological malignancies (64). T cell clones recognizing mLPA in the context of CD1c produced higher levels of IFNγ when stimulated with malignant than with normal hematopoietic cells. Intracellular accumulation of mLPA in tumor cells is thought to increase CD1c-restricted presentation of mLPA on the cell surface compared to normal cells (64), and therefore leading to differential recognition of malignant cells with mLPA-restricted T cell clones.
Group 1 CD1-Restricted T cells as platform for ACT
Despite expression of CD1c on APC, tumor-reactive T cells differentially recognized CD1c-restricted mLPA presented by tumor cells, suggesting that CD1c-restricted lipid antigens specifically accumulate in malignant cells but not normal APCs (64). If CD1c-restricted T cells are made amenable to genetic engineering, they could be an interesting population to investigate for ACT. Similar prospects apply to CD1a- and CD1b-restricted T cells, as both CD1a and CD1b expression is restricted to APCs (19, 20). Due to their polyclonal TCR repertoire, CAR engineering could be more straightforward than TCR engineering due to potential cross-pairing of transgenic and endogenous TCR chains and higher risk to produce off-target toxicities.
CD1b-restricted invariant TCR T cells (GEM and LDN5-like T cells) theoretically are top candidates as universal donor cells for both CAR and TCR-based ACT (Table 1). However, their frequency is extremely low in M. tuberculosis positive patients [<0.01% of peripheral blood T cells (22)], and has not yet been described in healthy donors.
MR1-Restricted T Cells
General definition
The MR1 molecule is an evolutionary conserved, monomorphic protein (98, 99). Ubiquitously expressed, MR1 cell surface expression is however modulated by antigen abundance. Under physiological conditions, MR1 is almost undetectable at the cell surface. Bound antigen is needed for its trafficking to the cell surface, and cell surface expression is further enhanced by exogenous antigen loading (19, 98, 100). Known to be involved in the immunity against bacterial and yeast infections (101, 102), MR1 presents small metabolites derived from the metabolic pathways of vitamin B9 (folate) or B2 (riboflavin) (103). Only a few antigens have been identified so far, but the list is growing, and includes small cyclic molecules utilized as pharmacological agents, such as the aspirin analog 3-FSA (3-formylsalicylic acid) or the non-steroidal anti-inflammatory drug diclofenac, which suggests that MR1 may be involved in drug hypersensitivity (104, 105). Even if self-derived MR1-restricted antigens have not been identified yet, several studies suggest that such antigens exist (106). For several years, mucosal-associated invariant T cells (MAIT) were the only known T cells with MR1 restriction. Now there is growing evidence that MR1-restricted non-MAIT cells exist (107–109), but much more needs to be learned.
MR1-Restricted MAIT cells
MAIT cells develop upon interaction with CD4+CD8+ double positive cortical thymocytes, and continue to mature and expand after leaving the thymus [(105, 110, 111); Figure 1]. In humans, their numbers continuously increase during the first 25 years of life, and then slowly decrease with age (112, 113). Their expansion is thought to be dependent on stimulation with microbial antigens, as germ-free mice do not have any MAIT cells in the periphery, despite positive selection in the thymus (106).
Originally identified in the gut, MAIT cells are characterized by the expression of a semi-invariant TCR, constituted of an invariant TCR Vα chain (Vα7.2-Jα3.3) paired with a limited number of Vβ chains. While byproducts of the microbial riboflavin biosynthesis, such as 5-OP-RU (5-[2-oxopropylideneamino]-6-D-ribitylaminouracil), are known to strongly activate MAIT cells, the folate biosynthesis pathway generates molecules, such as 6-formyl-pterin, that exert inhibitory effects on MAIT cells (103, 114). Although MAIT cells constitute only 5% of the total T cell pool in humans, their frequency can greatly vary in different organs (105). MAIT cells are abundant in the liver, lung and gastro-intestinal tract, as well as in the blood. In the liver, for example, 45% of resident T cells are MAIT (115). In the periphery, MAIT cells constitute up to 10% of the circulating T cells (112). Similar to conventional T cells, MAIT activities can be modulated by the antigen recognized, the cytokines present in the microenvironment and the tissue to which they naturally home. In the colon, for example, MAIT cells preferentially display a Th1-type of cytokine secretion, and reside in the lamina propria and the intraepithelial compartment of the mucosa, while in lung and liver MAIT cells resemble tissue-resident memory T cells (116).
TCR engagement together with co-stimulation leads to rapid MAIT cell activation, in a memory-like manner (117, 118). Activated MAIT cells display both direct and indirect cytotoxic functions, through the secretion of granzyme B, perforin, and a large range of Th1 and Th17 type of cytokines (105). Together with their ability to home to infected sites, MAIT cells constitute an important player in anti-microbial defense.
MAIT cells in tumor immunity
MAIT cells are part of the tumor infiltrating lymphocyte population in colorectal cancer (116, 119, 120), hepatocellular carcinoma (121, 122), or kidney and brain tumors (123). In some tumors, an inverse correlation between circulating and tumor infiltrating MAIT cells was observed, but it is not clear yet if tumor-infiltrating MAIT cells are pro- or anti-tumorigenic. In brain and kidney tumors, for example, MAIT cell infiltration was associated with higher levels of pro-inflammatory cytokines (123). On the other hand, MAIT cell infiltration in colorectal cancer and hepatocellular carcinoma was associated with unfavorable clinical outcome. Failing to produce pro-inflammatory cytokines such as IFNγ upon ex vivo stimulation, these tumor-infiltrating MAIT cells were functionally impaired (119, 124). One study even showed by transcriptome sequencing analyses that MAIT cells infiltrating hepatocellular carcinoma acquired a pro-tumorigenic phenotype (122). Surprisingly, peripheral MAIT cells seem to be unaffected and retain their ability to respond to bacterial antigens and even to tumor cells (116, 120, 124).
In patients after allogeneic HCT, robust peripheral blood MAIT cell reconstitution has been associated with a lower risk for the development of subsequent severe acute GVHD (118, 125), and activated MAIT cells suppressed proliferation of CD4+ T cells in vitro. Correlations between gut microbiota composition, the related riboflavin pathway, and MAIT reconstitution exist (118, 126). Further investigations are necessary to decipher the precise role of MAIT cells in human GVHD and whether adoptively transferred ex vivo expanded MAIT cells could be immune suppressive.
MAIT cells as platform for ACT
MAIT cells possess unique features that would make them interesting candidates as universal donor cells for ACT (Table 1), and methods for their isolation and ex vivo expansion are being established (65–67). However, their precise role in anti-tumor immunity remains to be defined in more detail. Some of the burning questions include (i) if it is possible to reprogram them in vitro to express a stable Th1 profile, (ii) if adoptively transferred MAIT cells efficiently migrate to the tumor site and retain their anti-tumor function, and (iii) if they can be genetically engineered during ex vivo expansion. We are convinced that these and more questions will be answered soon, and MAIT cells will be investigated as universal donor cells.
γδ-TCR T Cell Subsets
γδ-TCR T cells constitute 1–5% of total circulating T cells (14, 127). Selected in the thymus, the rearrangement process of γδ-TCR is highly complex and not fully understood yet. Constituted of a far smaller number of gene segments than αβ-TCRs, only 4–6 functional Vγ and 8 Vδ vs. 46 Vα and 48 Vβ chains, the potential of γδ-TCR diversity is however thought to surpass the diversity of αβ-TCRs (127, 128). Vδ4-Vδ7 gene segments rearrange with segments of the TCR α-chain, and have alternative TRAV names. Vδ1-Vδ2 preferentially rearrange with Dδ, Jδ, and Cδ to create TCR δ-chains, though few reports showed that Vδ1 and Vδ3 could also rearrange with segments of the TCRα locus, thus generating a δ/αβ-TCR T cell (129–132).
Vδ usage pre-determines γδ-TCR T cells function and localization: the majority of peripheral γδ-TCR T cells expresses Vδ2, while tissue-resident γδ-TCR T cells favor Vδ1 and Vδ3 (127). In humans, the majority of γδ-TCR T cells consists of Vδ1 and Vδ2 T cells (129). Their ligands are not well-characterized. The Vγ9Vδ2 T cell subset recognizes phosphorylated antigens, presented by the butyrophilin (BTN) molecules. Vδ1 γδ-TCR T cells can recognize antigens presented on CD1c, CD1d, and MR1 molecules. Both Vδ1 and Vδ2 γδ-TCR T cells are able to recognize stress-related molecules such as MIC A/B either via their TCR or via NK receptors such as NKG2D (129, 133, 134).
BTN-Restricted Vγ9Vδ2 T Cells
General background
The Vγ9Vδ2 T cell subset is relatively abundant in circulating lymphocytes, and represents 1–5% of all T cells in healthy individuals, and 50–95% of γδ-TCR T cells (127, 135, 136). This T cell subset expresses an invariant TCR that recognizes phosphorylated isoprenoid metabolites, also called phosphoantigens, derived from the mevalonate pathway. These metabolites, such as the isopentenyl pyrophosphate (IPP), can accumulate in transformed and infected cells because of their dysregulated metabolism (127, 135, 137). IPP accumulates when the activity of the IPP-metabolizing enzyme farnesyl-diphosphate-synthase (FPPS) is blocked (135). The use of aminobiphosphonates, such as zoledronate, inhibits FFPS, which leads to an increase in intracellular level of IPP and the activation of Vγ9Vδ2 T cells (135, 138–140).
IPPs are presented to T cells by butyrophilin molecules. Belonging to the immunoglobulin superfamily, these glycoproteins are divided into three subfamilies (BTN1, BTN2, BTN3) (141). Only BTN3A (CD277) presents phosphoantigens to Vγ9Vδ2 T cells. Constituted of three isoforms, BTN3A molecules are expressed by the majority of human immune cells, including γδ-TCR T cells (142). Phosphoantigens bind the intracellular domain B30.2 of BTN3A, which induces conformational changes to the receptor and increases binding force of Vγ9Vδ2 TCR to BTN3A (129, 143, 144). Therefore, Vγ9Vδ2 T cells are able to recognize altered metabolites present in infected or cancer cells.
Vγ9Vδ2 T cells in tumor immunity
Once activated, Vγ9Vδ2 T cells acquire similar effector functions as conventional αβ-TCR T cells: they directly kill tumor cells upon engagement of death receptors (e.g., FAS, TRAIL, NKG2D) or by secreting granzymes and perforins (24). In addition, Vγ9Vδ2 T cells produce various pro-inflammatory cytokines such as TNFα or IFNγ, and can modulate the immune response. For example, Vδ9Vδ2 T cells can license and accelerate DC maturation (145, 146) and provide help to B cells (147).
Human γδ-TCR T cells can be expanded in vitro to clinically relevant numbers, are able to migrate to and kill tumors, and are amenable to genetic engineering (68–70, 148, 149). Both HLA class I- and class II-restricted αβ-TCRs have been successfully introduced into Vγ9Vδ2 T cells and recognized the cognate peptide when co-transduced with CD4 or CD8 co-receptors (69, 70). TCR mispairing between α-/β-chains and γ-/δ-chains cannot occur. Thus, γδ-TCR T cells are optimal recipients for transgenic αβ-TCRs. αβ-TCR-transgenic γδ-TCR T cells express both αβ- and γδ-TCRs and mediate tumor cytotoxicity through both TCRs (69). Vγ9Vδ2 T cells were also engineered to transiently express a TCR or a CAR and exerted both endogenous and engineered properties (68).
One unique feature of Vγ9Vδ2 T cells is their capacity to differentiate into professional APCs upon IPP stimulation. Vγ9Vδ2 T cells can phagocytose cells and crosspresent antigens, leading to the proliferation of both CD4+ and CD8+ T cells (25, 26). This interesting feature is maintained in genetically engineered Vγ9Vδ2 T cells. For example, GD2-CAR-transduced Vγ9Vδ2 T cells killed GD2+ neuroblastoma, while retaining their ability to endocytose long peptides derived from the melanoma antigen MART-1 and inducing the proliferation of autologous T cells transduced with a MART-1-specific TCR (27).
Non-Vδ2 γδ-TCR T Cells
General background
Non-Vδ2 γδ-TCR T cells consist of Vδ1 and Vδ3 γδ-TCR T cells, and are mostly tissue-resident T cells present in barrier epithelium, though some of these cells are also circulating in blood (127). Between these two subsets, Vδ1 are the most abundant. From the original diverse repertoire present in neonates, only few Vδ1 γδ-TCR T cell clones will expand and ultimately dominate the adult Vδ1 repertoire (150). Even though the antigens recognized by Vδ1 γδ-TCR T cells are mostly unknown, they were shown to recognize ligands presented by CD1a, CD1c, CD1d, and MR1 molecules as well as various stress-induced ligands (133, 151, 152).
CD1-Restricted Vδ1 γδ-TCR T cells
CD1 molecules were among the first ligands identified for γδ-TCR T cells (153), but only few CD1-restricted lipids recognized by Vδ1 γδ-TCR T cells have been identified so far. Exogenous antigens comprise pollen-derived phospholipids and bacterial lipids, while known self-lipids consist of glycolipid sulfatides, which are present in tissues where Vδ1 γδ-TCR T cells reside (154–157). γδ-TCR T cells have been reported to be involved in tissue repair and homeostasis (158, 159), and predominate among γδ-TCR T cells that infiltrate various tumors (see below). However, the presence of CD1-resticted γδ-TCR T cells in tumors, and their involvement in tumor immunity, have not been investigated yet.
Crystal structure of CD1d-Vδ1 binding reveals a distinct TCR recognition: CD1d recognition by Vδ1 γδ-TCR T cells is solely mediated by the germline-encoded CDR1 loop, independently of the bound antigen. The antigen is in contact with the CDR3 region, which determines the antigen specificity (156, 160). Similarly, CD1c recognition was also shown to be dictated solely by Vδ1. Bound antigens modulate TCR recognition: some self-lipids were shown to permit TCR binding, while other self-lipids blocked it (154).
MR1-Restricted Vδ1 γδ-TCR T cells
γδ-TCR T cells recognizing antigens presented by MR1 have only been recently identified (152). This novel γδ-TCR T cell subset is rare with a frequency between <0.001 and 0.1% of total CD3+ T cells, or <0.1–5% of γδ-TCR T cells in blood of healthy donors, but has also been found in TILs of a Merkel cell carcinoma patient. MR1-restricted γδ-TCR T cells preferentially expressed Vδ1 (72% of the 76 TCRs analyzed). Structural studies revealed that TCR recognition occurred by binding to the MR1 α3 domain situated underneath the antigen-binding site and independently of the bound antigen (152) suggesting inherent autoreactivity of these cells.
Vδ1 γδ-TCR T cells in tumor immunity
Consistent with their preferential localization in epithelial tissues, Vδ1 γδ-TCR T cells are the predominant γδ-TCR T cell population in the majority of solid tumors (28, 161–163). Vδ1 γδ-TCR T cell infiltration correlated with favorable prognosis in several cancer types, such as triple negative breast cancer (TNBC) or CLL (162, 164). Upon in vitro expansion, Vδ1 γδ-TCR T cells isolated from PBMCs of cancer patients displayed strong IFNγ secretion and cytotoxic responses against several autologous tumors including melanoma (165), TNBC (162), colon cancer (166), AML (71), CLL (164, 167), diffuse large B cell lymphoma (168), and multiple myeloma (169). In vitro expanded Vδ1 γδ-TCR T cells were able to kill autologous CLL in vitro and spare healthy B cells isolated from the same patient, thus showing their ability to distinguish transformed cells from healthy cells (164). Moreover, these Vδ1 γδ-TCR T cells restrained tumor growth and prolonged the survival of immunodeficient mice engrafted with either human colon cancer or AML (71, 166). In other tumors, such as breast cancer (161, 170), colorectal cancer (163), melanoma (171), or squamous cell carcinoma (172), Vδ1 γδ-TCR T cell infiltration was associated with poor prognosis, as these cells displayed an immunosuppressive phenotype promoting tumor growth (170, 171). Vδ1 γδ-TCR T cells were shown to differentiate into Th17-like T cells, producing elevated level of IL-17, and other immunosuppressive factors, such as IL-10, IL-18, and adenosine (161, 170). Cancer cells were directly responsible for the skewing of Vδ1 γδ-TCR T cells toward Th17 regulatory profile. Breast cancer, for example, secretes exosomes containing the IncRNA SNHG16, a long non-coding RNA inducing the expression of SMAD5, and therefore TGF-β1, in Vδ1 γδ-TCR T cells (161). In colorectal cancer patients, cancer stem cells directly promoted IL-17 production by Vδ1 γδ-TCR T cells by secreting immunomodulatory molecules. By multiplex analyses on 50 different cytokines, the authors identified IL-18 and VEGF as the two most promising candidates responsible for the skewing of Vδ1 γδ-TCR T cells toward IL-17-producing immunosuppressive cells (163).
So far, no clinical trials have investigated the safety and efficacy of Vδ1 γδ-TCR T cells as anti-tumor therapy (Table 3). However, several groups established GMP-compatible protocols to expand and genetically engineer Vδ1 γδ-TCR T cells in vitro with the goal to translate this approach to the clinic (27, 28, 71, 72). Vδ1 γδ-TCR T cells can be expanded from healthy donor or patient PBMCs using a cocktail of different cytokines and anti-CD3 antibody stimulation. Over 3 weeks of culture, cells expanded more than 3-log fold and differentiated into cytotoxic Th1-like T cells, capable of controlling tumor growth in xenograft mouse models (28, 71). Moreover, Vδ1 γδ-TCR T cells expanded from PBMCs could be transduced with an anti-GD2 CAR and killed GD2-positive neuroblastoma cells lines that were not naturally recognized by Vδ1 γδ-TCR T cells (27).
γδ-TCR T Cells in Clinical Trials
Published and ongoing clinical trials investigating the safety and antitumor function of γδ-TCR T cells have recently been reviewed (173). Overall, the results of published trials outside the setting of allogeneic HCT have been disappointing, demonstrating safety but no efficacy. A major limitation lies in the lack of understanding of γδ-TCR diversity and their potential target antigens (Table 3). Some of the current and future clinical trials are trying to address these issues. We will focus our discussion on efforts in developing allogeneic γδ-TCR T cell therapies, with the overall goal to move their application to third-party banked off-the-shelf therapies. We identified five registered clinical trials, but more are expected to emerge. Three trials explore the safety of allogeneic ex vivo expanded adoptively transferred γδ-TCR T cells in hematologic malignancies. In one trial, γδ-TCR T cells are expanded from the haploidentical stem cell donor and infused to the patient post-transplant in combination with post-transplant cyclophosphamide treatment (NCT03533816, Incysus Therapeutics). Safety and effects on post-transplant GVHD are investigated. Another trial assesses the adoptive transfer of ex vivo expanded γδ-TCR T cells derived from related haploidentical or HLA-matched donors in patients with relapsed/refractory AML after lymphodepleting chemotherapy (NCT03790072, TC Biopharm). Safety and efficacy are analyzed. Future prospects are to move to allogeneic third-party banked γδ-TCR T cell products, and to include genetic engineering with CARs. A third active trial is also investigating ex vivo expanded γδ-TCR T cells from allogeneic related donors in patients with relapsed/refractory AML (NCT04008381, Wuhan Hospital). For solid tumors, a randomized clinical trial investigates safety and efficacy of tumor reducing surgery alone or in combination with adoptive transfer of ex vivo expanded γδ-TCR T cells in patients with locally advanced pancreatic cancer. The source of the γδ-TCR T cells is not entirely clear (NCT03180437, Fuda Cancer Hospital). Finally, haploidentical NKG2DL-CAR engineered γδ-TCR T cells for a variety of relapsed or refractory solid tumors will be investigated in a phase I dose escalation trial (NCT04107142, Cytomed Therapeutics).
γδ-TCR T Cells as Platform for ACT
γδ-TCR T cell expansion protocols allow the incorporation of genetic engineering to redirect γδ-TCR T cells to tumor-associated antigens recognized by CARs or αβ-TCRs (28, 68, 71, 72). Preclinical data suggest that γδ-TCR T cells are particularly suitable for αβ-TCR-based engineering, as the risk of TCR mispairing is inexistent. Both TCRs remain well-expressed, and the redirected T cells can exert anti-tumor functions through both TCRs. As γδ-TCR T cells are not HLA restricted (137), there is theoretically no risk of causing GVHD in the recipients, but formal demonstration in a clinical trial with third-party donor derived γδ-TCR T cells is currently lacking. γδ-TCR T cells meet many features required for universal donor T cell therapies (Table 1), and the interest in the field of exploiting this cell type is high.
Universal TCR
TCRs derived from both αβ- and γδ-TCR repertoires that allow the targeting of a broad range of tumors in an HLA independent manner have been identified. These TCRs can be considered “universal,” as they redirect immune cells to broadly shared tumor-specific antigens. TCRs recognizing targets derived from altered cell metabolism are of particular interest, as these TCRs reliably distinguish between cancer and healthy cells. Three examples from the literature include Vγ9Vδ2 TCRs (174, 175), mLPA-specific CD1c-restricted αβ-TCRs (64), and an MR1-restricted αβ-TCR with unknown specificity (108). These TCRs have been successfully introduced into polyclonal αβ-TCR T cells and were able to redirect the engineered cells to a variety of cancers in an HLA-independent manner in preclinical studies. Autologous Vγ9Vδ2-TCR engineered αβ-TCR T cells are currently under phase I clinical evaluation in patients with hematologic malignancies (NTR6541, UMC Utrecht). More TCRs with similar features are likely to be identified in the future. TCRs targeting cancer-specific ligands in the context of non-polymorphic molecules are likely to become interesting candidates for engineered ACTs.
Conclusions and Future Outlook
VSTs and unconventional T cells possess several features that would enable their universal use without the need of genome editing to avoid unacceptable alloreactivity. Among these different T cell subsets, γδ-TCR T cells, especially Vγ9Vδ2 T cells, and iNKT cells show the highest universal potential (Table 1). However, whether or not host-mediated rejection of the infused cells will affect their engraftment and long-term persistence remains to be addressed in the upcoming clinical trials. The only data available to date on persistence, safety, and efficacy come from third-party banked VSTs where, despite excellent clinical activity, persistence seems to be reduced compared to other trials that used HLA-matched products. Characterized by a memory-like status, VSTs and unconventional T cells respond rapidly to antigen exposure, leading to strong cytolytic activity, and cytokine production (19). Several of these T cell types have successfully been redirected to tumors by genetic engineering with a CAR or a TCR and mostly retain their intrinsic characteristics. For example, CAR+ VSTs maintain their anti-viral responses (48, 49), γδ-TCR T cells express both endogenous and introduced αβ-TCR (69), and CAR+ iNKT cells continue to respond to α-GalCer (57, 59, 176). The retention of these cell-specific features can also promote their in vivo re-expansion after ACT through re-exposure to their natural cognate antigen (49). Another advantage is the relatively restricted pattern of target antigens recognized by their endogenous TCRs. CD1-restricted and γδ-TCR T cells, for example, recognize ligands derived from altered tumor metabolism, and thus spare the corresponding healthy cells (36, 43). Despite still limited understanding of the biology of certain unconventional T cell subsets, the developing clinical translational pipelines outlined in this review demonstrate that the future potential for some of these experimental therapies as off-the-shelf products is high (Table 3). We expect that some of these cell types or universal receptors will become important players in the field cancer immunotherapy.
Author Contributions
CP and CA: concept and writing of the manuscript. IG: preparation of the figure. All authors have read and agreed with the final version of the manuscript.
Funding
CA was supported by a Swiss Cancer Research grant KFS-4542-08-2018-R, the Helmut Horten Foundation and the Department of oncology UNIL CHUV, Ludwig Institute for Cancer Research, Lausanne University Hospital, and University of Lausanne. CP was supported by a John Hansen Research Grant DKMS-SLS-JHRG-2020-03.
Conflict of Interest
All authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chow VA, Shadman M, Gopal AK. Translating anti-CD19 CAR T-cell therapy into clinical practice for relapsed/refractory diffuse large B-cell lymphoma. Blood. (2018) 132:777–81. doi: 10.1182/blood-2018–04-839217
2. Cerrano M, Ruella M, Perales MA, Vitale C, Faraci DG, Giaccone L, et al. The advent of CAR T-cell therapy for lymphoproliferative neoplasms: integrating research into clinical practice. Front Immunol. (2020) 11:888. doi: 10.3389/fimmu.2020.00888
3. Papathanasiou MM, Stamatis C, Lakelin M, Farid S, Titchener-Hooker N, Shah N. Autologous CAR T-cell therapies supply chain: challenges and opportunities? Cancer Gene Ther. (2020). doi: 10.1038/s41417–019-0157-z
4. McCreedy BJ, Senyukov VV, Nguyen KT. Off the shelf T cell therapies for hematologic malignancies. Best Pract Res Clin Haematol. (2018) 31:166–75. doi: 10.1016/j.beha.2018.03.001
5. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-shelf' allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. (2020) 19:185–99. doi: 10.1038/s41573–019-0051–2
6. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. (2009) 373:1550–61. doi: 10.1016/S0140–6736(09)60237–3
7. Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB. The burgeoning family of unconventional T cells. Nat Immunol. (2015) 16:1114–23. doi: 10.1038/ni.3298
8. Peggs KS, Verfuerth S, Pizzey A, Khan N, Guiver M, Moss PA, et al. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet. (2003) 362:1375–7. doi: 10.1016/S0140–6736(03)14634-X
9. Wallace DL, Masters JE, de Lara CM, Henson SM, Worth A, Zhang Y, et al. Human cytomegalovirus-specific CD8(+) T-cell expansions contain long-lived cells that retain functional capacity in both young and elderly subjects. Immunology. (2011) 132:27–38. doi: 10.1111/j.1365–2567.2010.03334.x
10. Schober K, Buchholz VR, Busch DH. TCR repertoire evolution during maintenance of CMV-specific T-cell populations. Immunol Rev. (2018) 283:113–28. doi: 10.1111/imr.12654
11. Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. (2010) 115:925–35. doi: 10.1182/blood-2009–08-239186
12. Prockop S, Doubrovina E, Suser S, Heller G, Barker J, Dahi P, et al. Off-the-shelf EBV-specific T cell immunotherapy for rituximab-refractory EBV-associated lymphoma following transplantation. J Clin Invest. (2020) 130:733–47. doi: 10.1172/JCI121127
13. Tilloy F, Treiner E, Park SH, Garcia C, Lemonnier F, de la Salle H, et al. An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J Exp Med. (1999) 189:1907–21. doi: 10.1084/jem.189.12.1907
14. Godfrey DI, Le Nours J, Andrews DM, Uldrich AP, Rossjohn J. Unconventional T cell targets for cancer immunotherapy. Immunity. (2018) 48:453–73. doi: 10.1016/j.immuni.2018.03.009
15. Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest. (2009) 119:1524–36. doi: 10.1172/JCI37869
16. Paget C, Chow MT, Duret H, Mattarollo SR, Smyth MJ. Role of gammadelta T cells in alpha-galactosylceramide-mediated immunity. J Immunol. (2012) 188:3928–39. doi: 10.4049/jimmunol.1103582
17. Nair S, Dhodapkar MV. Natural killer T cells in cancer immunotherapy. Front Immunol. (2017) 8:1178. doi: 10.3389/fimmu.2017.01178
18. Gomes AQ, Correia DV, Silva-Santos B. Non-classical major histocompatibility complex proteins as determinants of tumour immunosurveillance. EMBO Rep. (2007) 8:1024–30. doi: 10.1038/sj.embor.7401090
19. Mori L, Lepore M, de Libero G. The immunology of CD1- and MR1-restricted T cells. Annu Rev Immunol. (2016) 34:479–510. doi: 10.1146/annurev-immunol-032414–112008
20. Consonni M, Dellabona P, Casorati G. Potential advantages of CD1-restricted T cell immunotherapy in cancer. Mol Immunol. (2018) 103:200–8. doi: 10.1016/j.molimm.2018.09.025
21. Consonni M, de Lalla C, Bigi A, Dellabona P, Casorati G. Harnessing the CD1 restricted T cell response for leukemia adoptive immunotherapy. Cytokine Growth Factor Rev. (2017) 36:117–23. doi: 10.1016/j.cytogfr.2017.06.007
22. van Rhijn I, Gherardin NA, Kasmar A, de Jager W, Pellicci DG, Kostenko L, et al. TCR bias and affinity define two compartments of the CD1b-glycolipid-specific T Cell repertoire. J Immunol. (2014) 192:4054–60. doi: 10.4049/jimmunol.1400158
23. van Rhijn I, Kasmar A, de Jong A, Gras S, Bhati M, Doorenspleet ME, et al. A conserved human T cell population targets mycobacterial antigens presented by CD1b. Nat Immunol. (2013) 14:706–13. doi: 10.1038/ni.2630
24. Gomes AQ, Martins DS, Silva-Santos B. Targeting gammadelta T lymphocytes for cancer immunotherapy: from novel mechanistic insight to clinical application. Cancer Res. (2010) 70:10024–7. doi: 10.1158/0008–5472.CAN-10–3236
25. Brandes M, Willimann K, Bioley G, Levy N, Eberl M, Luo M, et al. Cross-presenting human gammadelta T cells induce robust CD8+ alphabeta T cell responses. Proc Natl Acad Sci USA. (2009) 106:2307–12. doi: 10.1073/pnas.0810059106
26. Wu Y, Wu W, Wong WM, Ward E, Thrasher AJ, Goldblatt D, et al. Human gamma delta T cells: a lymphoid lineage cell capable of professional phagocytosis. J Immunol. (2009) 183:5622–9. doi: 10.4049/jimmunol.0901772
27. Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, et al. Chimeric antigen receptor-engineered human gamma delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol Ther. (2018) 26:354–65. doi: 10.1016/j.ymthe.2017.12.001
28. Almeida AR, Correia DV, Fernandes-Platzgummer A, da Silva CL, da Silva MG, Anjos DR, et al. Delta one T cells for immunotherapy of chronic lymphocytic leukemia: clinical-grade expansion/differentiation and preclinical proof of concept. Clin Cancer Res. (2016) 22:5795–804. doi: 10.1158/1078–0432.CCR-16–0597
29. Hill JA, Mayer BT, Xie H, Leisenring WM, Huang ML, Stevens-Ayers T, et al. The cumulative burden of double-stranded DNA virus detection after allogeneic HCT is associated with increased mortality. Blood. (2017) 129:2316–25. doi: 10.1182/blood-2016–10-748426
30. Bollard CM, Heslop HE. T cells for viral infections after allogeneic hematopoietic stem cell transplant. Blood. (2016) 127:3331–40. doi: 10.1182/blood-2016–01-628982
31. Sutrave G, Gottlieb DJ. Adoptive cell therapies for posttransplant infections. Curr Opin Oncol. (2019) 31:574–90. doi: 10.1097/CCO.0000000000000580
32. Keller MD, Bollard CM. Virus-specific T-cell therapies for patients with primary immune deficiency. Blood. (2020) 135:620–8. doi: 10.1182/blood.2019000924
33. Icheva V, Kayser S, Wolff D, Tuve S, Kyzirakos C, Bethge W, et al. Adoptive transfer of epstein-barr virus (EBV) nuclear antigen 1-specific t cells as treatment for EBV reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J Clin Oncol. (2013) 31:39–48. doi: 10.1200/JCO.2011.39.8495
34. Doubrovina E, Oflaz-Sozmen B, Prockop SE, Kernan NA, Abramson S, Teruya-Feldstein J, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood. (2012) 119:2644–56. doi: 10.1182/blood-2011–08-371971
35. Tischer S, Priesner C, Heuft HG, Goudeva L, Mende W, Barthold M, et al. Rapid generation of clinical-grade antiviral T cells: selection of suitable T-cell donors and GMP-compliant manufacturing of antiviral T cells. J Transl Med. (2014) 12:336. doi: 10.1186/s12967-014-0336-5
36. Withers B, Blyth E, Clancy LE, Yong A, Fraser C, Burgess J, et al. Long-term control of recurrent or refractory viral infections after allogeneic HSCT with third-party virus-specific T cells. Blood Adv. (2017) 1:2193–205. doi: 10.1182/bloodadvances.2017010223
37. Haque T, Wilkie GM, Jones MM, Higgins CD, Urquhart G, Wingate P, et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood. (2007) 110:1123–31. doi: 10.1182/blood-2006-12-063008
38. Tzannou I, Papadopoulou A, Naik S, Leung K, Martinez CA, Ramos CA, et al. Off-the-shelf virus-specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, epstein-barr virus, and adenovirus infections after allogeneic hematopoietic stem-cell transplantation. J Clin Oncol. (2017) 35:3547–57. doi: 10.1200/JCO.2017.73.0655
39. Barker JN, Doubrovina E, Sauter C, Jaroscak JJ, Perales MA, Doubrovin M, et al. Successful treatment of EBV-associated posttransplantation lymphoma after cord blood transplantation using third-party EBV-specific cytotoxic T lymphocytes. Blood. (2010) 116:5045–9. doi: 10.1182/blood-2010-04-281873
40. Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, Antin JH, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. (2013) 121:5113–23. doi: 10.1182/blood-2013–02-486324
41. Gallot G, Vollant S, Saiagh S, Clemenceau B, Vivien R, Cerato E, et al. T-cell therapy using a bank of EBV-specific cytotoxic T cells: lessons from a phase I/II feasibility and safety study. J Immunother. (2014) 37:170–9. doi: 10.1097/CJI.0000000000000031
42. Tzannou I, Watanabe A, Naik S, Daum R, Kuvalekar M, Leung KS, et al. “Mini” bank of only 8 donors supplies CMV-directed T cells to diverse recipients. Blood Adv. (2019) 3:2571–80. doi: 10.1182/bloodadvances.2019000371
43. Quach DH, Becerra-Dominguez L, Rouce RH, Rooney CM. A strategy to protect off-the-shelf cell therapy products using virus-specific T-cells engineered to eliminate alloreactive T-cells. J Transl Med. (2019) 17:240. doi: 10.1186/s12967–019-1988-y
44. Mo F, Watanabe N, McKenna MK, Hicks MJ, Srinivasan M, Gomes-Silva D, et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat Biotechnol. (2020). doi: 10.1038/s41587-020-0601-5
45. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. (2008) 14:1264–70. doi: 10.1038/nm.1882
46. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. (2011) 118:6050–6. doi: 10.1182/blood-2011–05-354449
47. Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. (2013) 122:2965–73. doi: 10.1182/blood-2013-06-506741
48. Rossig C, Pule M, Altvater B, Saiagh S, Wright G, Ghorashian S, et al. Vaccination to improve the persistence of CD19CAR gene-modified T cells in relapsed pediatric acute lymphoblastic leukemia. Leukemia. (2017) 31:1087–95. doi: 10.1038/leu.2017.39
49. Lapteva N, Gilbert M, Diaconu I, Rollins LA, Al-Sabbagh M, Naik S, et al. T-cell receptor stimulation enhances the expansion and function of CD19 chimeric antigen receptor-expressing T cells. Clin Cancer Res. (2019) 25:7340–50. doi: 10.1158/1078–0432.CCR-18–3199
50. Tanaka M, Tashiro H, Omer B, Lapteva N, Ando J, Ngo M, et al. Vaccination targeting native receptors to enhance the function and proliferation of chimeric antigen receptor (CAR)-modified T cells. Clin Cancer Res. (2017) 23:3499–509. doi: 10.1158/1078–0432.CCR-16–2138
51. Hart DP, Xue SA, Thomas S, Cesco-Gaspere M, Tranter A, Willcox B, et al. Retroviral transfer of a dominant TCR prevents surface expression of a large proportion of the endogenous TCR repertoire in human T cells. Gene Ther. (2008) 15:625–31. doi: 10.1038/sj.gt.3303078
52. Heemskerk MH, Hoogeboom M, Hagedoorn R, Kester MG, Willemze R, Falkenburg JH. Reprogramming of virus-specific T cells into leukemia-reactive T cells using T cell receptor gene transfer. J Exp Med. (2004) 199:885–94. doi: 10.1084/jem.20031110
53. Griffioen M, van Egmond HM, Barnby-Porritt H, van der Hoorn MA, Hagedoorn RS, Kester MG, et al. Genetic engineering of virus-specific T cells with T-cell receptors recognizing minor histocompatibility antigens for clinical application. Haematologica. (2008) 93:1535–43. doi: 10.3324/haematol.13067
54. van Loenen MM, de Boer R, van Liempt E, Meij P, Jedema I, Falkenburg JH, et al. A good manufacturing practice procedure to engineer donor virus-specific T cells into potent anti-leukemic effector cells. Haematologica. (2014) 99:759–68. doi: 10.3324/haematol.2013.093690
55. van Loenen MM, Hagedoorn RS, Kester MG, Hoogeboom M, Willemze R, Falkenburg JH, et al. Kinetic preservation of dual specificity of coprogrammed minor histocompatibility antigen-reactive virus-specific T cells. Cancer Res. (2009) 69:2034–41. doi: 10.1158/0008–5472.CAN-08–2523
56. Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med. (2019) 25:1064–72. doi: 10.1038/s41591–019-0472–9
57. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood. (2014) 124:2824–33. doi: 10.1182/blood-2013–11-541235
58. Ngai H, Tian G, Courtney AN, Ravari SB, Guo L, Liu B, et al. IL-21 selectively protects CD62L(+) NKT cells and enhances their effector functions for adoptive immunotherapy. J Immunol. (2018) 201:2141–53. doi: 10.4049/jimmunol.1800429
59. Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, et al. NKT cells coexpressing a GD2-specific chimeric antigen receptor and IL15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin Cancer Res. (2019) 25:7126–38. doi: 10.1158/1078–0432.CCR-19–0421
60. Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J Clin Invest. (2016) 126:2341–55. doi: 10.1172/JCI83476
61. Kato S, Berzofsky JA, Terabe M. Possible therapeutic application of targeting type II natural killer T cell-mediated suppression of tumor immunity. Front Immunol. (2018) 9:314. doi: 10.3389/fimmu.2018.00314
62. de Jong A, Pena-Cruz V, Cheng TY, Clark RA, Van Rhijn I, Moody DB. CD1a-autoreactive T cells are a normal component of the human alphabeta T cell repertoire. Nat Immunol. (2010) 11:1102–9. doi: 10.1038/ni.1956
63. de Lalla C, Lepore M, Piccolo FM, Rinaldi A, Scelfo A, Garavaglia C, et al. High-frequency and adaptive-like dynamics of human CD1 self-reactive T cells. Eur J Immunol. (2011) 41:602–10. doi: 10.1002/eji.201041211
64. Lepore M, de Lalla C, Gundimeda SR, Gsellinger H, Consonni M, Garavaglia C, et al. A novel self-lipid antigen targets human T cells against CD1c(+) leukemias. J Exp Med. (2014) 211:1363–77. doi: 10.1084/jem.20140410
65. Di Blasi D, Vacchini A, de Libero G, Mori L. Isolation and characterization of MAIT cells from human tissue biopsies. Methods Mol Biol. (2020) 2098:23–38. doi: 10.1007/978–1-0716–0207-2_2
66. Hagel JP, Garner LC, Bilton M, Mehta H, Leng T, Hackstein CP, et al. Human MAIT cell activation in vitro. Methods Mol Biol. (2020) 2098:97–124. doi: 10.1007/978–1-0716–0207-2_7
67. Wakao H. Reprogramming of MAIT cells to pluripotency and redifferentiation. Methods Mol Biol. (2020) 2098:237–57. doi: 10.1007/978–1-0716–0207-2_16
68. Harrer DC, Simon B, Fujii SI, Shimizu K, Uslu U, Schuler G, et al. RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an alpha/beta T-cell receptor: a safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer. (2017) 17:551. doi: 10.1186/s12885–017-3539–3
69. Hiasa A, Nishikawa H, Hirayama M, Kitano S, Okamoto S, Chono H, et al. Rapid alphabeta TCR-mediated responses in gammadelta T cells transduced with cancer-specific TCR genes. Gene Ther. (2009) 16:620–8. doi: 10.1038/gt.2009.6
70. van der Veken LT, Hagedoorn RS, van Loenen MM, Willemze R, Falkenburg JH, Heemskerk MH. Alphabeta T-cell receptor engineered gammadelta T cells mediate effective antileukemic reactivity. Cancer Res. (2006) 66:3331–7. doi: 10.1158/0008–5472.CAN-05–4190
71. Di Lorenzo B, Simoes AE, Caiado F, Tieppo P, Correia DV, Carvalho T, et al. Broad cytotoxic targeting of acute myeloid leukemia by polyclonal delta one T cells. Cancer Immunol Res. (2019) 7:552–8. doi: 10.1158/2326–6066.CIR-18–0647
72. Polito VA, Cristantielli R, Weber G, Del Bufalo F, Belardinilli T, Arnone CM, et al. Universal ready-to-use immunotherapeutic approach for the treatment of cancer: expanded and activated polyclonal gammadelta memory T cells. Front Immunol. (2019) 10:2717. doi: 10.3389/fimmu.2019.02717
73. Moody DB, Cotton RN. Four pathways of CD1 antigen presentation to T cells. Curr Opin Immunol. (2017) 46:127–33. doi: 10.1016/j.coi.2017.07.013
74. de Libero G, Mori L. Novel insights into lipid antigen presentation. Trends Immunol. (2012) 33:103–11. doi: 10.1016/j.it.2012.01.005
75. Sugita M, Cernadas M, Brenner MB. New insights into pathways for CD1-mediated antigen presentation. Curr Opin Immunol. (2004) 16:90–5. doi: 10.1016/j.coi.2003.11.014
76. Hakomori S. Glycosphingolipids in cellular interaction, differentiation, and oncogenesis. Annu Rev Biochem. (1981) 50:733–64. doi: 10.1146/annurev.bi.50.070181.003505
77. Tilloy F, Di Santo JP, Bendelac A, Lantz O. Thymic dependence of invariant V alpha 14+ natural killer-T cell development. Eur J Immunol. (1999) 29:3313–83. doi: 10.1002/(SICI)15214141(199910)29:10<3313::AIDIMMU3313>3.0.CO;28
78. Wieland Brown LC, Penaranda C, Kashyap PC, Williams BB, Clardy J, Kronenberg M, et al. Production of alpha-galactosylceramide by a prominent member of the human gut microbiota. PLoS Biol. (2013) 11:e1001610. doi: 10.1371/journal.pbio.1001610
79. Kain L, Webb B, Anderson BL, Deng S, Holt M, Costanzo A, et al. The identification of the endogenous ligands of natural killer T cells reveals the presence of mammalian alpha-linked glycosylceramides. Immunity. (2014) 41:543–54. doi: 10.1016/j.immuni.2014.08.017
80. Brennan PJ, Tatituri RV, Heiss C, Watts GF, Hsu FF, Veerapen N, et al. Activation of iNKT cells by a distinct constituent of the endogenous glucosylceramide fraction. Proc Natl Acad Sci USA. (2014) 111:13433–8. doi: 10.1073/pnas.1415357111
81. Wu DY, Segal NH, Sidobre S, Kronenberg M, Chapman PB. Cross-presentation of disialoganglioside GD3 to natural killer T cells. J Exp Med. (2003) 198:173–81. doi: 10.1084/jem.20030446
82. Park JE, Wu DY, Prendes M, Lu SX, Ragupathi G, Schrantz N, et al. Fine specificity of natural killer T cells against GD3 ganglioside and identification of GM3 as an inhibitory natural killer T-cell ligand. Immunology. (2008) 123:145–55. doi: 10.1111/j.1365–2567.2007.02760.x
83. Veerapen N, Reddington F, Bricard G, Porcelli SA, Besra GS. Synthesis and biological activity of alpha-L-fucosyl ceramides, analogues of the potent agonist, alpha-D-galactosyl ceramide KRN7000. Bioorg Med Chem Lett. (2010) 20:3223–6. doi: 10.1016/j.bmcl.2010.04.079
84. Salio M, Silk JD, Jones EY, Cerundolo V. Biology of CD1- and MR1-restricted T cells. Annu Rev Immunol. (2014) 32:323–66. doi: 10.1146/annurev-immunol-032713–120243
85. de Santo C, Salio M, Masri SH, Lee LY, Dong T, Speak AO, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest. (2008) 118:4036–48. doi: 10.1172/JCI36264
86. Metelitsa LS, Naidenko OV, Kant A, Wu HW, Loza MJ, Perussia B, et al. Human NKT cells mediate antitumor cytotoxicity directly by recognizing target cell CD1d with bound ligand or indirectly by producing IL-2 to activate NK cells. J Immunol. (2001) 167:3114–22. doi: 10.4049/jimmunol.167.6.3114
87. Fujii SI, Shimizu K. Exploiting antitumor immunotherapeutic novel strategies by deciphering the cross talk between invariant NKT cells and dendritic cells. Front Immunol. (2017) 8:886. doi: 10.3389/fimmu.2017.00886
88. Molling JW, Langius JA, Langendijk JA, Leemans CR, Bontkes HJ, van der Vliet HJ, et al. Low levels of circulating invariant natural killer T cells predict poor clinical outcome in patients with head and neck squamous cell carcinoma. J Clin Oncol. (2007) 25:862–8. doi: 10.1200/JCO.2006.08.5787
89. Najera Chuc AE, Cervantes LA, Retiguin FP, Ojeda JV, Maldonado ER. Low number of invariant NKT cells is associated with poor survival in acute myeloid leukemia. J Cancer Res Clin Oncol. (2012) 138:1427–32. doi: 10.1007/s00432–012-1251-x
90. Gorini F, Azzimonti L, Delfanti G, Scarfo L, Scielzo C, Bertilaccio MT, et al. Invariant NKT cells contribute to chronic lymphocytic leukemia surveillance and prognosis. Blood. (2017) 129:3440–51. doi: 10.1182/blood-2016–11-751065
91. Tachibana T, Onodera H, Tsuruyama T, Mori A, Nagayama S, Hiai H, et al. Increased intratumor Valpha24-positive natural killer T cells: a prognostic factor for primary colorectal carcinomas. Clin Cancer Res. (2005) 11:7322–7. doi: 10.1158/1078–0432.CCR-05–0877
92. Chaidos A, Patterson S, Szydlo R, Chaudhry MS, Dazzi F, Kanfer E, et al. Graft invariant natural killer T-cell dose predicts risk of acute graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Blood. (2012) 119:5030–6. doi: 10.1182/blood-2011–11-389304
93. Rubio MT, Moreira-Teixeira L, Bachy E, Bouillie M, Milpied P, Coman T, et al. Early posttransplantation donor-derived invariant natural killer T-cell recovery predicts the occurrence of acute graft-versus-host disease and overall survival. Blood. (2012) 120:2144–54. doi: 10.1182/blood-2012–01-404673
94. Chang DH, Deng H, Matthews P, Krasovsky J, Ragupathi G, Spisek R, et al. Inflammation-associated lysophospholipids as ligands for CD1d-restricted T cells in human cancer. Blood. (2008) 112:1308–16. doi: 10.1182/blood-2008–04-149831
95. Exley MA, Friedlander P, Alatrakchi N, Vriend L, Yue S, Sasada T, et al. Adoptive transfer of invariant NKT cells as immunotherapy for advanced melanoma: a phase i clinical trial. Clin Cancer Res. (2017) 23:3510–9. doi: 10.1158/1078–0432.CCR-16–0600
96. Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, et al. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. (2006) 12:6079–86. doi: 10.1158/1078–0432.CCR-06–0114
97. Mavers M, Maas-Bauer K, Negrin RS. Invariant natural killer T cells as suppressors of graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Front Immunol. (2017) 8:900. doi: 10.3389/fimmu.2017.00900
98. Miley MJ, Truscott SM, Yu YY, Gilfillan S, Fremont DH, Hansen TH, et al. Biochemical features of the MHC-related protein 1 consistent with an immunological function. J Immunol. (2003) 170:6090–8. doi: 10.4049/jimmunol.170.12.6090
99. Huang S, Martin E, Kim S, Yu L, Soudais C, Fremont DH, et al. MR1 antigen presentation to mucosal-associated invariant T cells was highly conserved in evolution. Proc Natl Acad Sci USA. (2009) 106:8290–5. doi: 10.1073/pnas.0903196106
100. Salio M, Awad W, Veerapen N, Gonzalez-Lopez C, Kulicke C, Waithe D, et al. Ligand-dependent downregulation of MR1 cell surface expression. Proc Natl Acad Sci USA. (2020) 117:10465–75. doi: 10.1073/pnas.2003136117
101. Le Bourhis L, Martin E, Peguillet I, Guihot A, Froux N, Core M, et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol. (2010) 11:701–8. doi: 10.1038/ni.1890
102. Gold MC, Cerri S, Smyk-Pearson S, Cansler ME, Vogt TM, Delepine J, et al. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol. (2010) 8:e1000407. doi: 10.1371/journal.pbio.1000407
103. Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature. (2012) 491:717–23. doi: 10.1038/nature11605
104. Keller AN, Eckle SB, Xu W, Liu L, Hughes VA, Mak JY, et al. Drugs and drug-like molecules can modulate the function of mucosal-associated invariant T cells. Nat Immunol. (2017) 18:402–11. doi: 10.1038/ni.3679
105. Godfrey DI, Koay HF, McCluskey J, Gherardin NA. The biology and functional importance of MAIT cells. Nat Immunol. (2019) 20:1110–28. doi: 10.1038/s41590–019-0444–8
106. Koay HF, Gherardin NA, Enders A, Loh L, Mackay LK, Almeida CF, et al. A three-stage intrathymic development pathway for the mucosal-associated invariant T cell lineage. Nat Immunol. (2016) 17:1300–11. doi: 10.1038/ni.3565
107. Lepore M, Kalinichenko A, Calogero S, Kumar P, Paleja B, Schmaler M, et al. Functionally diverse human T cells recognize non-microbial antigens presented by MR1. Elife. (2017) 6:e24476. doi: 10.7554/eLife.24476
108. Crowther MD, Dolton G, Legut M, Caillaud ME, Lloyd A, Attaf M, et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat Immunol. (2020) 21:178–85. doi: 10.1038/s41590–019-0578–8
109. Gherardin NA, Keller AN, Woolley RE, Le Nours J, Ritchie DS, Neeson PJ, et al. Diversity of T cells restricted by the MHC class i-related molecule MR1 facilitates differential antigen recognition. Immunity. (2016) 44:32–45. doi: 10.1016/j.immuni.2015.12.005
110. Martin E, Treiner E, Duban L, Guerri L, Laude H, Toly C, et al. Stepwise development of MAIT cells in mouse and human. PLoS Biol. (2009) 7:e54. doi: 10.1371/journal.pbio.1000054
111. Seach N, Guerri L, Le Bourhis L, Mburu Y, Cui Y, Bessoles S, et al. Double-positive thymocytes select mucosal-associated invariant T cells. J Immunol. (2013) 191:6002–9. doi: 10.4049/jimmunol.1301212
112. Gherardin NA, Souter MN, Koay HF, Mangas KM, Seemann T, Stinear TP, et al. Human blood MAIT cell subsets defined using MR1 tetramers. Immunol Cell Biol. (2018) 96:507–25. doi: 10.1111/imcb.12021
113. Novak J, Dobrovolny J, Novakova L, Kozak T. The decrease in number and change in phenotype of mucosal-associated invariant T cells in the elderly and differences in men and women of reproductive age. Scand J Immunol. (2014) 80:271–5. doi: 10.1111/sji.12193
114. Corbett AJ, Eckle SB, Birkinshaw RW, Liu L, Patel O, Mahony J, et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature. (2014) 509:361–5. doi: 10.1038/nature13160
115. Dusseaux M, Martin E, Serriari N, Peguillet I, Premel V, Louis D, et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood. (2011) 117:1250–9. doi: 10.1182/blood-2010–08-303339
116. Sundstrom P, Szeponik L, Ahlmanner F, Sundquist M, Wong JSB, Lindskog EB, et al. Tumor-infiltrating mucosal-associated invariant T (MAIT) cells retain expression of cytotoxic effector molecules. Oncotarget. (2019) 10:2810–23. doi: 10.18632/oncotarget.26866
117. Slichter CK, McDavid A, Miller HW, Finak G, Seymour BJ, McNevin JP, et al. Distinct activation thresholds of human conventional and innate-like memory T cells. JCI Insight. (2016) 1:e86292. doi: 10.1172/jci.insight.86292
118. Bhattacharyya A, Hanafi LA, Sheih A, Golob JL, Srinivasan S, Boeckh MJ, et al. Graft-derived reconstitution of mucosal-associated invariant T cells after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. (2018) 24:242–51. doi: 10.1016/j.bbmt.2017.10.003
119. Zabijak L, Attencourt C, Guignant C, Chatelain D, Marcelo P, Marolleau JP, et al. Increased tumor infiltration by mucosal-associated invariant T cells correlates with poor survival in colorectal cancer patients. Cancer Immunol Immunother. (2015) 64:1601–8. doi: 10.1007/s00262–015-1764–7
120. Ling L, Lin Y, Zheng W, Hong S, Tang X, Zhao P, et al. Circulating and tumor-infiltrating mucosal associated invariant T (MAIT) cells in colorectal cancer patients. Sci Rep. (2016) 6:20358. doi: 10.1038/srep20358
121. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. (2017) 169:1342–56.e16. doi: 10.1016/j.cell.2017.05.035
122. Duan M, Goswami S, Shi JY, Wu LJ, Wang XY, Ma JQ, et al. Activated and exhausted MAIT cells foster disease progression and indicate poor outcome in hepatocellular carcinoma. Clin Cancer Res. (2019) 25:3304–16. doi: 10.1158/1078–0432.CCR-18–3040
123. Peterfalvi A, Gomori E, Magyarlaki T, Pal J, Banati M, Javorhazy A, et al. Invariant Valpha7.2-Jalpha33 TCR is expressed in human kidney and brain tumors indicating infiltration by mucosal-associated invariant T (MAIT) cells. Int Immunol. (2008) 20:1517–25. doi: 10.1093/intimm/dxn111
124. Shaler CR, Tun-Abraham ME, Skaro AI, Khazaie K, Corbett AJ, Mele T, et al. Mucosa-associated invariant T cells infiltrate hepatic metastases in patients with colorectal carcinoma but are rendered dysfunctional within and adjacent to tumor microenvironment. Cancer Immunol Immunother. (2017) 66:1563–75. doi: 10.1007/s00262–017-2050–7
125. Kawaguchi K, Umeda K, Hiejima E, Iwai A, Mikami M, Nodomi S, et al. Influence of post-transplant mucosal-associated invariant T cell recovery on the development of acute graft-versus-host disease in allogeneic bone marrow transplantation. Int J Hematol. (2018) 108:66–75. doi: 10.1007/s12185–018-2442–2
126. Konuma T, Kohara C, Watanabe E, Takahashi S, Ozawa G, Suzuki K, et al. Reconstitution of circulating mucosal-associated invariant T cells after allogeneic hematopoietic cell transplantation: its association with the riboflavin synthetic pathway of gut microbiota in cord blood transplant recipients. J Immunol. (2020) 204:1462–73. doi: 10.4049/jimmunol.1900681
127. Legut M, Cole DK, Sewell AK. The promise of gammadelta T cells and the gammadelta T cell receptor for cancer immunotherapy. Cell Mol Immunol. (2015) 12:656–68. doi: 10.1038/cmi.2015.28
128. Davis MM. The evolutionary and structural 'logic' of antigen receptor diversity. Semin Immunol. (2004) 16:239–43. doi: 10.1016/j.smim.2004.08.003
129. Lee HW, Chung YS, Kim TJ. Heterogeneity of human gammadelta T cells and their role in cancer immunity. Immune Netw. (2020) 20:e5. doi: 10.4110/in.2020.20.e5
130. Pellicci DG, Uldrich AP, Le Nours J, Ross F, Chabrol E, Eckle SB, et al. The molecular bases of delta/alphabeta T cell-mediated antigen recognition. J Exp Med. (2014) 211:2599–615. doi: 10.1084/jem.20141764
131. Miossec C, Faure F, Ferradini L, Roman-Roman S, Jitsukawa S, Ferrini S, et al. Further analysis of the T cell receptor gamma/delta+ peripheral lymphocyte subset. The V delta 1 gene segment is expressed with either C alpha or C delta. J Exp Med. (1990) 171:1171–88. doi: 10.1084/jem.171.4.1171
132. Peyrat MA, Davodeau F, Houde I, Romagne F, Necker A, Leget C, et al. Repertoire analysis of human peripheral blood lymphocytes using a human V delta 3 region-specific monoclonal antibody. Characterization of dual T cell receptor (TCR) delta-chain expressors and alpha beta T cells expressing V delta 3J alpha C alpha-encoded TCR chains. J Immunol. (1995) 155:3060–7.
133. Das H, Sugita M, Brenner MB. Mechanisms of Vdelta1 gammadelta T cell activation by microbial components. J Immunol. (2004) 172:6578–86. doi: 10.4049/jimmunol.172.11.6578
134. Corvaisier M, Moreau-Aubry A, Diez E, Bennouna J, Mosnier JF, Scotet E, et al. V gamma 9V delta 2 T cell response to colon carcinoma cells. J Immunol. (2005) 175:5481–8. doi: 10.4049/jimmunol.175.8.5481
135. Herrmann T, Fichtner AS, Karunakaran MM. An update on the molecular basis of phosphoantigen recognition byVγ9Vδ2 T cells. Cells. (2020) 9:1433. doi: 10.3390/cells9061433
136. Blazquez JL, Benyamine A, Pasero C, Olive D. New insights into the regulation of gammadelta T cells by BTN3A and other BTN/BTNL in tumor immunity. Front Immunol. (2018) 9:1601. doi: 10.3389/fimmu.2018.01601
137. Tanaka Y, Morita CT, Tanaka Y, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature. (1995) 375:155–8. doi: 10.1038/375155a0
138. Kunzmann V, Bauer E, Feurle J, Weissinger F, Tony HP, Wilhelm M. Stimulation of gammadelta T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood. (2000) 96:384–92. doi: 10.1182/blood.V96.2.384
139. Li J, Herold MJ, Kimmel B, Muller I, Rincon-Orozco B, Kunzmann V, et al. Reduced expression of the mevalonate pathway enzyme farnesyl pyrophosphate synthase unveils recognition of tumor cells by Vgamma9Vdelta2 T cells. J Immunol. (2009) 182:8118–24. doi: 10.4049/jimmunol.0900101
140. Gober HJ, Kistowska M, Angman L, Jeno P, Mori L, De Libero G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J Exp Med. (2003) 197:163–8. doi: 10.1084/jem.20021500
141. Malinowska M, Tokarz-Deptula B, Deptula W. Butyrophilins: an important new element of resistance. Cent Eur J Immunol. (2017) 42:399–403. doi: 10.5114/ceji.2017.72806
142. Arnett HA, Viney JL. Immune modulation by butyrophilins. Nat Rev Immunol. (2014) 14:559–69. doi: 10.1038/nri3715
143. Sandstrom A, Peigne CM, Leger A, Crooks JE, Konczak F, Gesnel MC, et al. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vgamma9Vdelta2 T cells. Immunity. (2014) 40:490–500. doi: 10.1016/j.immuni.2014.03.003
144. Yang Y, Li L, Yuan L, Zhou X, Duan J, Xiao H, et al. A structural change in butyrophilin upon phosphoantigen binding underlies phosphoantigen-mediated Vgamma9Vdelta2 T cell activation. Immunity. (2019) 50:1043–53.e5. doi: 10.1016/j.immuni.2019.02.016
145. Devilder MC, Maillet S, Bouyge-Moreau I, Donnadieu E, Bonneville M, Scotet E. Potentiation of antigen-stimulated V gamma 9V delta 2 T cell cytokine production by immature dendritic cells (DC) and reciprocal effect on DC maturation. J Immunol. (2006) 176:1386–93. doi: 10.4049/jimmunol.176.3.1386
146. Conti L, Casetti R, Cardone M, Varano B, Martino A, Belardelli F, et al. Reciprocal activating interaction between dendritic cells and pamidronate-stimulated gammadelta T cells: role of CD86 and inflammatory cytokines. J Immunol. (2005) 174:252–60. doi: 10.4049/jimmunol.174.1.252
147. Bansal RR, Mackay CR, Moser B, Eberl M. IL-21 enhances the potential of human gammadelta T cells to provide B-cell help. Eur J Immunol. (2012) 42:110–9. doi: 10.1002/eji.201142017
148. Alnaggar M, Xu Y, Li J, He J, Chen J, Li M, et al. Allogenic Vγ9Vδ2 T cell as new potential immunotherapy drug for solid tumor: a case study for cholangiocarcinoma. J Immunother Cancer. (2019) 7:36. doi: 10.1186/s40425–019-0501–8
149. Kobayashi H, Tanaka Y, Yagi J, Osaka Y, Nakazawa H, Uchiyama T, et al. Safety profile and anti-tumor effects of adoptive immunotherapy using gamma-delta T cells against advanced renal cell carcinoma: a pilot study. Cancer Immunol Immunother. (2007) 56:469–76. doi: 10.1007/s00262–006-0199–6
150. Davey MS, Willcox CR, Joyce SP, Ladell K, Kasatskaya SA, McLaren JE, et al. Clonal selection in the human Vdelta1 T cell repertoire indicates gammadelta TCR-dependent adaptive immune surveillance. Nat Commun. (2017) 8:14760. doi: 10.1038/ncomms14760
151. Russano AM, Bassotti G, Agea E, Bistoni O, Mazzocchi A, Morelli A, et al. CD1-restricted recognition of exogenous and self-lipid antigens by duodenal gammadelta+ T lymphocytes. J Immunol. (2007) 178:3620–6. doi: 10.4049/jimmunol.178.6.3620
152. Le Nours J, Gherardin NA, Ramarathinam SH, Awad W, Wiede F, Gully BS, et al. A class of gammadelta T cell receptors recognize the underside of the antigen-presenting molecule MR1. Science. (2019) 366:1522–7. doi: 10.1126/science.aav3900
153. Porcelli S, Brenner MB, Greenstein JL, Balk SP, Terhorst C, Bleicher PA. Recognition of cluster of differentiation 1 antigens by human CD4-CD8-cytolytic T lymphocytes. Nature. (1989) 341:447–50. doi: 10.1038/341447a0
154. Roy S, Ly D, Castro CD, Li NS, Hawk AJ, Altman JD, et al. Molecular analysis of lipid-reactive Vdelta1 gammadelta T cells identified by CD1c tetramers. J Immunol. (2016) 196:1933–42. doi: 10.4049/jimmunol.1502202
155. Bai L, Picard D, Anderson B, Chaudhary V, Luoma A, Jabri B, et al. The majority of CD1d-sulfatide-specific T cells in human blood use a semiinvariant Vdelta1 TCR. Eur J Immunol. (2012) 42:2505–10. doi: 10.1002/eji.201242531
156. Luoma AM, Castro CD, Mayassi T, Bembinster LA, Bai L, Picard D, et al. Crystal structure of Vdelta1 T cell receptor in complex with CD1d-sulfatide shows MHC-like recognition of a self-lipid by human gammadelta T cells. Immunity. (2013) 39:1032–42. doi: 10.1016/j.immuni.2013.11.001
157. Takahashi T, Suzuki T. Role of sulfatide in normal and pathological cells and tissues. J Lipid Res. (2012) 53:1437–50. doi: 10.1194/jlr.R026682
158. Komano H, Fujiura Y, Kawaguchi M, Matsumoto S, Hashimoto Y, Obana S, et al. Homeostatic regulation of intestinal epithelia by intraepithelial gamma delta T cells. Proc Natl Acad Sci USA. (1995) 92:6147–51. doi: 10.1073/pnas.92.13.6147
159. Toulon A, Breton L, Taylor KR, Tenenhaus M, Bhavsar D, Lanigan C, et al. A role for human skin-resident T cells in wound healing. J Exp Med. (2009) 206:743–50. doi: 10.1084/jem.20081787
160. Uldrich AP, Le Nours J, Pellicci DG, Gherardin NA, McPherson KG, Lim RT, et al. CD1d-lipid antigen recognition by the gammadelta TCR. Nat Immunol. (2013) 14:1137–45. doi: 10.1038/ni.2713
161. Ni C, Fang QQ, Chen WZ, Jiang JX, Jiang Z, Ye J, et al. Breast cancer-derived exosomes transmit lncRNA SNHG16 to induce CD73+gammadelta1 Treg cells. Signal Transduct Target Ther. (2020) 5:41. doi: 10.1038/s41392–020-0129–7
162. Wu Y, Kyle-Cezar F, Woolf RT, Naceur-Lombardelli C, Owen J, Biswas D, et al. An innate-like Vdelta1(+) gammadelta T cell compartment in the human breast is associated with remission in triple-negative breast cancer. Sci Transl Med. (2019) 11:eaax9364. doi: 10.1126/scitranslmed.c
163. Meraviglia S, Lo Presti E, Tosolini M, La Mendola C, Orlando V, Todaro M, et al. Distinctive features of tumor-infiltrating gammadelta T lymphocytes in human colorectal cancer. Oncoimmunology. (2017) 6:e1347742. doi: 10.1080/2162402X.2017.1347742
164. Poggi A, Venturino C, Catellani S, Clavio M, Miglino M, Gobbi M, et al. Vdelta1 T lymphocytes from B-CLL patients recognize ULBP3 expressed on leukemic B cells and up-regulated by trans-retinoic acid. Cancer Res. (2004) 64:9172–9. doi: 10.1158/0008–5472.CAN-04–2417
165. Donia M, Ellebaek E, Andersen MH, Straten PT, Svane IM. Analysis of Vdelta1 T cells in clinical grade melanoma-infiltrating lymphocytes. Oncoimmunology. (2012) 1:1297–304. doi: 10.4161/onci.21659
166. Wu D, Wu P, Wu X, Ye J, Wang Z, Zhao S, et al. Ex vivo expanded human circulating Vdelta1 gammadeltaT cells exhibit favorable therapeutic potential for colon cancer. Oncoimmunology. (2015) 4:e992749. doi: 10.4161/2162402X.2014.992749
167. Simoes C, Silva I, Carvalho A, Silva S, Santos S, Marques G, et al. Quantification and phenotypic characterization of peripheral blood Vdelta1 + T cells in chronic lymphocytic leukemia and monoclonal B cell lymphocytosis. Cytometry B Clin Cytom. (2019) 96:164–8. doi: 10.1002/cyto.b.21645
168. Reboursiere E, Gac AC, Garnier A, Salaun V, Reman O, Pham AD, et al. Increased frequencies of circulating and tumor-resident Vdelta1(+) T cells in patients with diffuse large B-cell lymphoma. Leuk Lymphoma. (2018) 59:187–95. doi: 10.1080/10428194.2017.1321751
169. Knight A, Mackinnon S, Lowdell MW. Human Vdelta1 gamma-delta T cells exert potent specific cytotoxicity against primary multiple myeloma cells. Cytotherapy. (2012) 14:1110–8. doi: 10.3109/14653249.2012.700766
170. Chabab G, Barjon C, Abdellaoui N, Salvador-Prince L, Dejou C, Michaud HA, et al. Identification of a regulatory Vdelta1 gamma delta T cell subpopulation expressing CD73 in human breast cancer. J Leukoc Biol. (2020) 107:1057–67. doi: 10.1002/JLB.3MA0420–278RR
171. Wistuba-Hamprecht K, Di Benedetto S, Schilling B, Sucker A, Schadendorf D, Garbe C, et al. Phenotypic characterization and prognostic impact of circulating gammadelta and alphabeta T-cells in metastatic malignant melanoma. Int J Cancer. (2016) 138:698–704. doi: 10.1002/ijc.29818
172. Lo Presti E, Toia F, Oieni S, Buccheri S, Turdo A, Mangiapane LR, et al. Squamous cell tumors recruit gammadelta T cells producing either IL17 or IFNgamma depending on the tumor stage. Cancer Immunol Res. (2017) 5:397–407. doi: 10.1158/2326–6066.CIR-16–0348
173. Sebestyen Z, Prinz I, Dechanet-Merville J, Silva-Santos B, Kuball J. Translating gammadelta (gammadelta) T cells and their receptors into cancer cell therapies. Nat Rev Drug Discov. (2020) 19:169–84. doi: 10.1038/s41573–019-0038-z
174. Scotet E, Martinez LO, Grant E, Barbaras R, Jeno P, Guiraud M, et al. Tumor recognition following Vgamma9Vdelta2 T cell receptor interactions with a surface F1-ATPase-related structure and apolipoprotein A-I. Immunity. (2005) 22:71–80. doi: 10.1016/j.immuni.2004.11.012
175. Marcu-Malina V, Heijhuurs S, van Buuren M, Hartkamp L, Strand S, Sebestyen Z, et al. Redirecting alphabeta T cells against cancer cells by transfer of a broadly tumor-reactive gammadeltaT-cell receptor. Blood. (2011) 118:50–9. doi: 10.1182/blood-2010–12-325993
Keywords: allogeneic off-the-shelf T cells, virus-specific T cells, unconventional T cells, engineered, CD1, MR1, GVHD, rejection
Citation: Perez C, Gruber I and Arber C (2020) Off-the-Shelf Allogeneic T Cell Therapies for Cancer: Opportunities and Challenges Using Naturally Occurring “Universal” Donor T Cells. Front. Immunol. 11:583716. doi: 10.3389/fimmu.2020.583716
Received: 15 July 2020; Accepted: 07 September 2020;
Published: 11 November 2020.
Edited by:
Alice Bertaina, Stanford University, United StatesReviewed by:
Nicole Andrea Mifsud, Monash University, AustraliaSergio Querol, Banc de Sang i Teixits, Spain
Copyright © 2020 Perez, Gruber and Arber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Arber, Y2Fyb2xpbmUuYXJiZXJAdW5pbC5jaA==