 Ju-Yang Jung
Ju-Yang Jung Ji-Won Kim
Ji-Won Kim Chang-Hee Suh
Chang-Hee Suh Hyoun-Ah Kim
Hyoun-Ah Kim
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 05 November 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.583513
This article is part of the Research Topic DAMPs across the Tree of Life, Volume 3: Human Diseases View all 10 articles
Systemic juvenile idiopathic arthritis (JIA) and adult-onset Still’s disease (AOSD) are systemic inflammatory disorders that manifest as high-spiking fever, joint pain, evanescent skin rash, and organomegaly. Their pathogenesis is unclear, but inflammation is triggered by activation of the innate immune system with aberrant production of proinflammatory cytokines. Along with extrinsic factors, intrinsic pathways can trigger an unexpected immune response. Damage-associated molecular patterns (DAMPs) induce the activation of innate immune cells, leading to sterile inflammation in systemic JIA and AOSD. These endogenous proteins interact with Toll-like receptors (TLRs), which are pattern recognition receptors, and mediate immune signaling following stimulation by pathogen-associated molecular patterns and DAMPs. Several DAMPs, such as S100 proteins, play a role in the development or severity of systemic JIA and AOSD, in which their interactions with TLRs are altered. Also, the expression levels of genes encoding DAMPs contribute to the susceptibility to systemic JIA and AOSD. Herein, we review reports that TLR and DAMP signaling initiates and/or maintains the inflammatory response in systemic JIA and AOSD, and their correlations with the clinical characteristics of those diseases. In addition, we assess their utility as biomarkers or therapeutics for systemic JIA and AOSD.
Systemic juvenile idiopathic arthritis (JIA) and adult-onset Still’s disease (AOSD) are systemic autoinflammatory diseases characterized by spiking fever, skin rash, polyarticular arthralgia, hepatosplenomegaly, and leukocytosis (1–4). Although the role of the adaptive immune response is limited, activation of the innate immune system plays a pivotal role in both diseases. This has been demonstrated by the activation of innate immune cells and overproduction of proinflammatory cytokines including interleukin (IL)-1β, IL-18, IL-6, and tumor necrosis factor-α (TNF-α) in systemic JIA and AOSD (5, 6). Human leukocyte antigen (HLA) and IL-18, IL-6, and macrophage inhibitory factor (MIF) polymorphisms are associated with the occurrence of systemic JIA and AOSD (7–11). Several viruses, such as rubella, measles, echovirus 7, coxsackievirus, cytomegalovirus, Epstein–Barr virus, parainfluenza, influenza, adenovirus, hepatitis B and C, and parvovirus B19, are reported to trigger both diseases (12–16).
These factors activate the innate immune system; macrophages and neutrophils increase in number, and the levels of markers indicating their activation increase abnormally. Patients with systemic JIA or AOSD had a high serum level of the neutrophil activation marker, CD64 (FcɣRI), the level of which was correlated with disease severity (17, 18). The level of macrophage-colony stimulating factor, which is involved in macrophage differentiation and survival, was increased in the serum of patients with AOSD (19). The serum levels of calprotectin, a calcium-binding protein released during activation of neutrophils and macrophages, and soluble CD163, which is released by activated macrophages, were higher in patients with systemic JIA or AOSD; moreover, their levels were correlated with disease activity (17, 20, 21).
During an innate immune response, pattern-recognition receptors (PRRs) on immune cells interact with pathogen-associated molecular patterns (PAMPs) to induce an immune response. PAMPs include microbial components such as lipopolysaccharides (LPS) from Gram-negative bacteria and viral single-stranded RNA (22). Also, after infectious or non-infectious tissue injury, the release and binding of intracellular or extracellular factors to PRRs on immune cells leads to sterile inflammation. Damage-associated molecular patterns [DAMPs; e.g., high mobility group box 1 (HMGB1), histones, cell-free DNA, and S100 proteins] are potent activators of the immune system.
Several DAMPs play a role in the pathogenesis of systemic JIA and AOSD (23). The level of HMGB1 in serum was elevated in patients with systemic JIA or AOSD, and then downregulated after disease resolution (24, 25). The serum levels of S100A8 (calgranulin A or myeloid-related protein 8, MRP8) and S100A9 (calgranulin B or MRP14) were increased in patients with systemic JIA and AOSD, and these factors were deposited in the skin or lymph nodes of the latter patients (21, 26, 27). Such DAMPs are recognized by members of the PRR family, including Toll-like receptors (TLRs), nucleotide-binding oligomerization domain receptors (NOD-like receptors; NLRs), C-type lectin receptors, and retinoic-acid-inducible gene 1-like receptors, in several inflammatory disorders.
Changes in the adaptive immune system in systemic JIA and AOSD, and the effects of IL-1 and IL-18 on T cell differentiation and activity have been established, suggesting that innate and adaptive immune responses are linked in both disorders (28). The frequencies of circulating Th17 cells were elevated and correlated with disease activity in patients with AOSD, and the proportions of IFN-γ- and IL-17-producing CD4+T cells and IL-17-producing CD3+CD4- T cells were increased in systemic JIA (29, 30). Moreover, an increased population of activated regulatory T cell expressing IL-17 and a prominent Th17 gene expression signature were observed in acute systemic JIA (31). The proportion of CD8+naïve T cells was elevated in AOSD patients and correlated with disease activity (32).
Systemic JIA and AOSD are diagnosed based on clinical manifestations and laboratory results, because there is no reliable biomarker. Although criteria based on combinations of typical clinical and laboratory findings have been established, there is no way to differentiate systemic JIA and AOSD from other conditions (e.g., infections or neoplasms) (33). High levels of inflammatory markers, such as ferritin, have poor diagnostic specificity for systemic JIA and AOSD (34).
Here, we review TLRs and their ligands, and the mechanisms by which they induce an innate inflammatory response. We also summarize the roles of TLRs and their ligands in rheumatic diseases, focusing on systemic JIA and AOSD. An understanding of their pathogenesis will enable the identification and development of reliable biomarkers of both diseases.
As a link between infection/tissue damage and inflammation, TLRs sense and transfer danger signals to intracellular signaling pathways (35). TLRs interact with different PAMPs and transmit signals via specific sets of adaptors and transcription factors in various immune and non-immune cells (Table 1). For example, TLR4 recognizes LPS, TLR9 senses unmethylated CpG from bacteria, viruses, or parasites; and TLR7 and 8 recognize viral or parasitic ssRNAs.

Table 1 Toll like receptors and their ligands with involved adaptors.
TLRs are expressed on macrophages, neutrophils, dendritic cells (DCs), natural killer (NK) cells, mast cells, T- and B-cells, and some types of nonimmune cells (e.g., epithelial and endothelial cells) (36). Most TLRs are type I transmembrane proteins located in the plasma membrane, intracellular endosomes, or both. TLR2 and TLR4 are extracellular receptors, and TLR3, TLR7, TLR8, and TLR9 are located in the endosomal compartment (37). TLRs comprise extracellular domain-containing leucine-rich repeats (LRRs) and a cytoplasmic Toll/IL-1 receptor (TIR) domain (35, 36). Upon ligand interaction, the homodimerization or heterodimerization of TLRs, with the exception of TLR3, triggers the production of the adaptor molecule myeloid differentiation primary response protein 88 (MYD88), which interacts with IL-1R-activating kinase (IRAK)-4 and IRAK-2. TLR3 and TLR4 interact with TIR domain-containing adaptor molecule 1 (TICAM-1 or TRIF) via stimulation by dsRNA viruses (38). The dimerization of IRAK-4 and -2 activates TNF-receptor-associated factor 6 (TRAF6); the final protein complex (Myddosome) induces nuclear transcriptional activity and the transcription of nuclear factor kappa B (NF-κB), interferon regulatory factor (IRF), and AP-1 (39, 40). The various TLR signaling pathways involve different transcription factors, and trigger various cellular responses, resulting in the expression of genes encoding inflammatory cytokines such as type I IFN and IFN-inducible genes. The production of IL-1, IL-6, TNF-α, IL-12, IFNs, chemokines, adhesion molecules, costimulatory molecules, and tissue-degrading enzymes is induced by interactions between ligands and TLRs (41). Diverse factors, ranging from microbial agents to self-DNAs, trigger multiple TLR pathways in a variety of cell types and induce the expression of distinct subsets of genes.
TLR-mediated stimulation is implicated in diverse diseases, including infections, sepsis, autoimmune diseases, and malignancies. DAMPs interact with TLRs as endogenous ligands and initiate signal transduction, inducing inflammatory responses. Most DAMPs, which include nucleic acids, intracellular proteins, and extracellular matrix components, are released by damaged tissues or dying cells. Their interactions are implicated in the pathogenesis of sterile inflammatory conditions, including rheumatic diseases, cancer, and wound healing.
S100A8 and S100A9 are released during the apoptosis or necrosis of neutrophils and monocytes or the formation of neutrophil extracellular traps (NETs) (42). They interact with TLR4 and receptor for advanced glycation end products (RAGE), and S100A8/S100A9-TLR4 signaling in human monocytes has effects similar to LPS-TLR4 signaling. HMGB1 is a proinflammatory mediator that binds several PRRs, including RAGE, TLR4, TLR9, C-X-C chemokine receptor type 4 (CXCR4), and T cell immunoglobulin mucin-3 (TIM-3) (43). HMGB1 is released during apoptosis or necrosis, acting as a DAMP, and inflammatory cytokines including TNF-α and IFN-ɣ enhance the release of HMGB1 (44, 45). Extracellular HMGB1 binds to TLR4 adaptor myeloid differentiation factor-2 (MD-2), triggering the activation of NF-κB and the transcription of proinflammatory cytokines (46). In addition, the HMGB1–CpG-ODN complex binds to TLR9, promoting cytokine production (37). As an acute-phase reactant and DAMP, serum amyloid A (SAA) can induce proinflammatory cytokines during injury, sterile inflammation, and infection, and SAA promotes the production of IL-1β via activation of the NLRP3 inflammasome (47). The action of SAA is dependent on TLR signaling. SAA activity decreases in TLR4 deficiency and SAA induces G-CSF and IL-8/CXCL8 via TLR2 (48–50).
TLR3, TLR7, and TLR9 on endosomes are activated by self-nucleic acids such as self-DNA and RNA-protein particles (41). Single-stranded RNA, double-stranded RNA, and unmethylated CpG DNA stimulate the TLR7, TLR3, and TLR9 signaling pathways, respectively. Internalization of self-nucleic acids activates the TLR7 and TLR9 signaling pathways and stimulates transcription of IFN-α in patients with autoimmune diseases, such as SLE.
TLR signaling promotes activation of the NLR family pyrin domain-containing 3 (NLRP3) inflammasome, which is a cytoplasmic protein complex that modulates the innate immune response (51). TLR signaling via MyD88 or TRIF stimulates transcription of Nlrp3 and synthesis of pro-IL-1β, resulting in inflammasome assembly and activity (52). TLR signaling and inflammasome activity also promote pyroptosis and inflammatory caspase-dependent lytic cell death, inducing the release of IL-1β, IL-18, and HMGB1 in sterile inflammation (53, 54).
Rheumatoid arthritis (RA), which is the most common autoimmune disease, is characterized by joint inflammation and destruction. A variety of genetic and environmental factors are related to the chronic inflammatory response in RA. TLR signaling is implicated in the development and maintenance of RA, and potential strategies targeting TLR signaling are currently under investigation (55). TLR2, TLR3, TLR4, and TLR7 were highly expressed on synovial tissue, and synovial tissue macrophages, and the levels of TLR2 and TLR4 expression were associated with IL-12 and IL-18 levels in synovial tissue in RA (56, 57). Endogenous DAMPs, such as heat shock proteins, fibronectin, HMGB1, and S100 proteins, activate synovial macrophages or DCs via TLR signaling pathways (58, 59). TLR2 signaling induces the release of chemokines by synovial fibroblasts, and the MYD88-dependent pathway is involved in joint inflammation in RA (60). Synthetic dsRNA and necrotic synovial fluid stimulated TLR3 on synovial fibroblasts, inducing the production of IFN-β, CXCL10, CCL5, and IL-6. Also, TLR3 and TLR4 signaling in synovial fluid macrophages resulted in overproduction of TNF-α and IL-6 (61, 62). TLR-3 and TLR-7 were highly expressed in the RA synovium, and TLR2 and TLR4 signaling in DCs from patients with RA triggered the production of inflammatory mediators (63). TLR7 expression in RA monocytes induced TNF-α production and correlated with the disease activity of RA (64).
TLR2 signaling promotes joint inflammation during the acute phase, and TLR4 promotes matrix metalloproteinase (MMP)-mediated cartilage destruction, osteoclast formation, and IL-17 production in the chronic phase (65). A study using a serotonin receptor antagonist showed that TLR8 signaling induced TNF production in RA (66). The expression of TLR3 and TLR7 on DCs and synovial fibroblasts was controlled by type I IFN, but not by IL-1β, IL-18, or TNF-α (67). TLR5 is strongly expressed in the synovium of RA patients, and ligation of TLR5 induces TNF-α and promotes monocyte migration to synovial tissue and osteoclastic development of myeloid cells (68). Some TLR4, TLR5, TLR7, and TLR9 polymorphisms are associated with susceptibility to RA (69). TLR4 polymorphisms are also associated with shared epitope and disease activity (70).
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease involving multiple organs, an autoantibody response, and immune complex deposition. The TLR7/MyD88 signaling pathway in plasmacytoid DCs and B cells plays a role in the pathogenesis of SLE (71). After internalization, immune complexes bound to DNA or RNA interact with endosomal TLR7 and TLR8, promoting a type I IFN response (72). TLR7 was overexpressed in models of severe lupus, and in patients with SLE, and the inhibition or attenuation of TLR7 signaling ameliorated the inflammatory response in the lung and kidney of lupus-prone mice (73, 74). TLR9 expression induced the generation of autoantibodies (e.g., anti-dsDNA antibodies) and B-cell activation via immune complexes (75). Although TLR9 signaling contributes to the development of lupus nephritis, enhanced TLR9 signaling prevents severe manifestations and defective TLR9 expression promotes inflammation in murine lupus (76–78). The role of TLR9 in SLE is controversial. TLR8 signaling modulates TLR7 activation, and deletion of TLR8 promotes autoantibody production and inflammation (79).
In addition, blockade of TLR signaling in major immune cells attenuates the inflammatory response in SLE, confirming a role for TLR in its pathogenesis. The inhibition of IRAK-4, which involves the TLR7 signaling pathway, on plasmacytoid DCs not only reduces the expression of IFN-responsive genes but also ameliorates inflammation in lupus nephritis (80). A TLR inhibitor prevented tissue damage caused by immune complex deposition and cellular infiltration, and alleviated collagen-induced arthritis and the manifestations of SLE (81). Mutations in TLR-trafficking chaperone, which suppresses TLR7 signaling, ameliorated systemic inflammation (82). TLR7 and TLR9 polymorphisms are associated with SLE susceptibility, particularly in Asians (83).
Sjögren syndrome is an autoimmune disease characterized by lymphocytic infiltration and inflammation of the salivary and lacrimal glands. There is aberrant expression of TLRs on PBMCs, minor salivary gland (MSG) biopsy tissue and salivary gland epithelial cells (SGECs) in Sjögren syndrome. TLR2, TLR3, and TLR4 were strongly expressed on the SGECs of patients with Sjögren syndrome, and TLR2 signaling induces the production of IL-23/IL-17 via IL-6 and signal transducer and activator of transcription (STAT) in the NF-κB pathway in Sjögren syndrome (84, 85). TLR7, TL9, and TLR7/8 signaling is also associated with a salivary inflammatory response involving antigen presentation and the secretion of proinflammatory cytokines (86).
Systemic sclerosis (SSc) is an autoimmune disease characterized by fibrosis of the skin and/or internal organs and vasculopathy (87). Autoantibody-producing or autoreactive cells induce endothelial activation and progressive fibrosis in SSc. TLR signaling pathways play roles in the pathogenesis of SSc, and their pharmacological inhibition ameliorates disease progression. The level of fibronectin, an endogenous TLR4 ligand, is increased in the serum and skin tissue of patients with SSc, and disruption of TLR4 signaling abrogated collagen production and myofibroblast differentiation (88). Also, the level of the extracellular matrix glycoprotein tenascin-C is elevated in the serum, fibroblasts, and skin lesions of patients with SSc, inducing collagen gene expression and myofibroblast transformation via TLR4 signaling (89). Mitochondrial DNA and CpG oligonucleotides trigger TLR9 signaling, leading to TGFβ production and fibroblast activation in patients with SSc (90). Liquid crystalline complexes composed of CXCL4 and self-DNA or microbial DNA amplify the activation of plasmacytoid DCs and IFN-α production via TLR9 signaling in SSc (91).
Antiphospholipid antibody syndrome is characterized by recurrent thrombosis and complications of pregnancy associated with an autoimmune-mediated inflammatory response. Antiphospholipid antibodies (aPLs) include anti-cardiolipin antibodies (aCLs), lupus anticoagulant (LAC), and anti-β2 glycoprotein I (GPI) antibodies. aPLs induce an inflammatory response by interacting with TLR2, TLR4, TLR7, TLR8, and TLR9 (92–95). In placental inflammation or thrombosis, anti-β2 GPI activates TLR4, impairing autophagy and activating the inflammasome in endothelial cells (96, 97).
Gout, which is the most common rheumatic disease, is characterized by an acute inflammatory response against monosodium urate monohydrate (MSU) crystals in the joints. TLR2 and TLR4 signaling via MyD88 is triggered by MSU crystals, and induces the production of proinflammatory cytokines (including IL-1β and TNF-α) by activating the NALP3 inflammasome (98–100). S100A8 and S100A9 were released by MSU crystal-activated phagocytes, and stimulated IL-1β secretion in a TLR4-dependent manner. The S100A8 and S100/A9 levels were elevated in patients with gout and were correlated with disease activity (101). An inhibitor of the NALP3 inflammasome and TLR2 suppressed MSU crystal uptake by macrophages, and alleviated swelling and pain in MSU-injected joints (102).
Table 2 summarizes the putative pathological roles of TLR and its ligands in several rheumatic diseases.
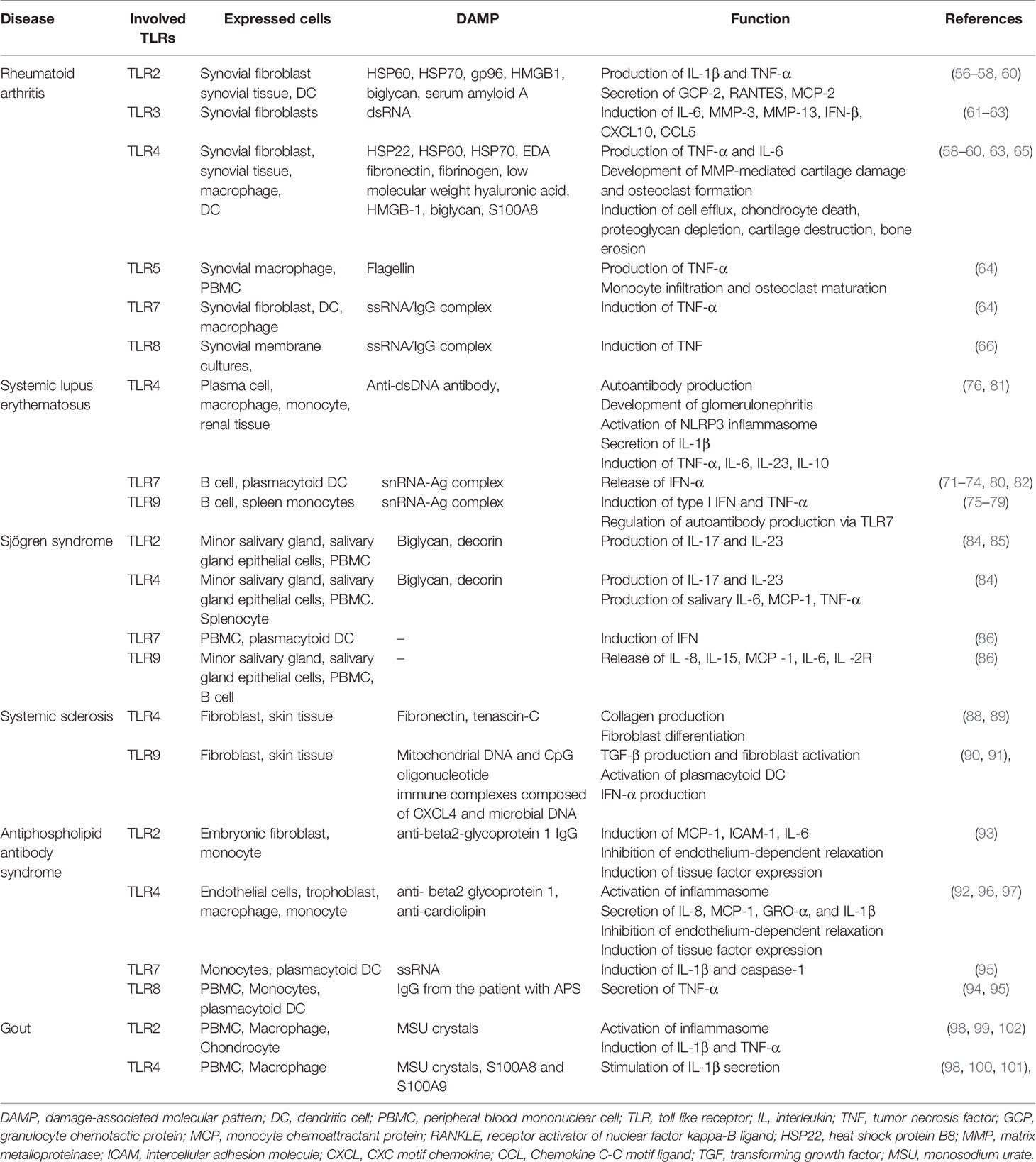
Table 2 Toll like receptors in rheumatic diseases.
Macrophages and monocytes are highly activated in systemic JIA and AOSD. Altered TLR signaling and its ligands have been implicated in the pathogenesis of systemic JIA and AOSD (Figure 1). We review the role of TLR and its DAMP molecules in the pathogenesis of systemic JIA and AOSD.
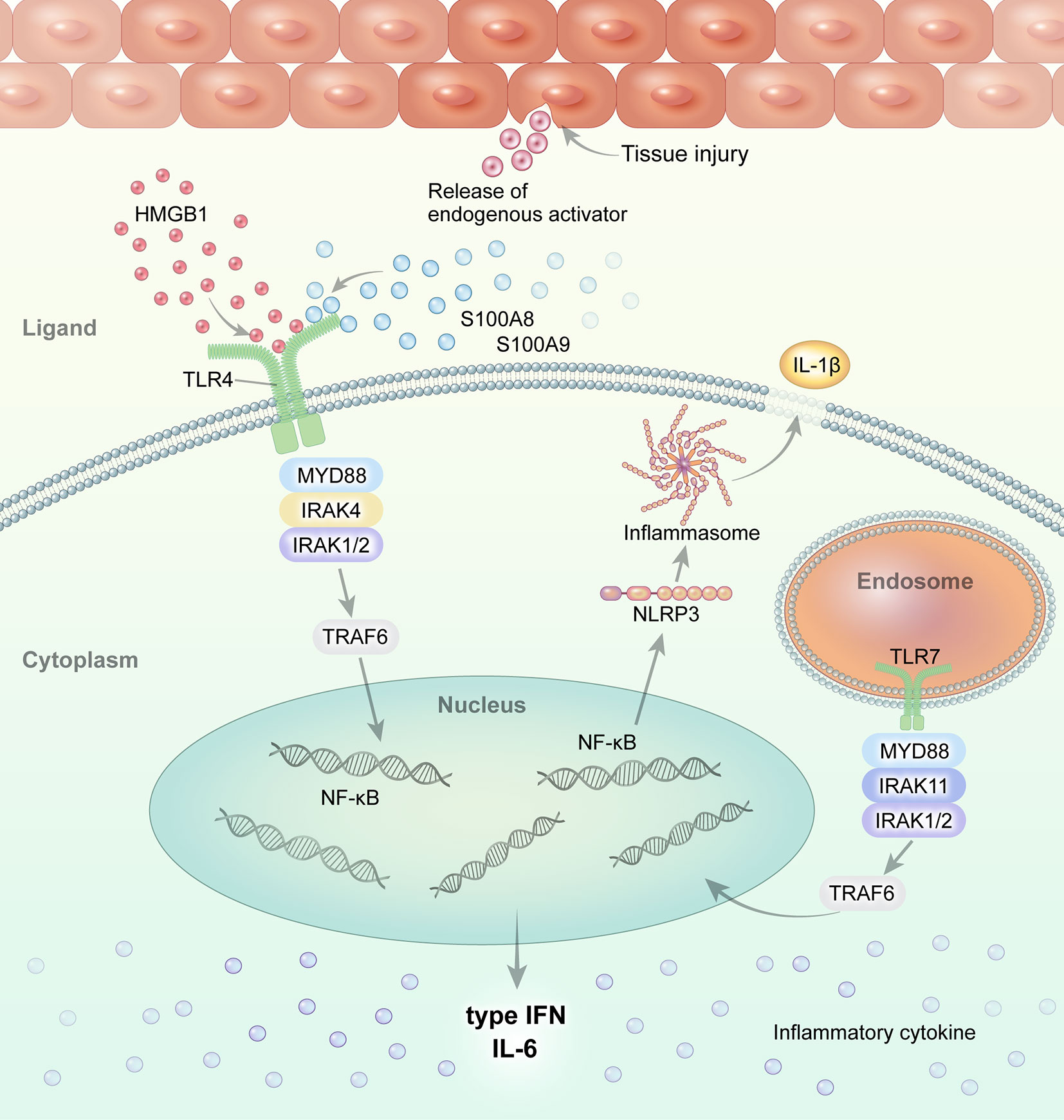
Figure 1 Overview of TLR signaling and the endogenous DAMP pathway in systemic JIA and AOSD. Regarding the role of TLR and its ligands in the pathogenesis of systemic JIA and AOSD, current evidence suggests that endogenous ligands, such as S100A8, S100A9, and S100A8/A9, and HMGB1, interact with and stimulate the TLR4 pathway. Activated TLR4 and TLR7 induce NLRP3 inflammasome activation and the secretion of IL-1β in systemic JIA and AOSD.
S100A8, S100A9, S100A8/A9 (calprotectin), and S100A12 are calcium-binding proteins released from activated phagocytic myeloid cells that act as proinflammatory endogenous TLR4 ligands during sterile inflammation (42). The interaction of S100 proteins and TLR4 in the context of sterile inflammation or tissue injury is independent of the presence of PAMP. S100A8, S100A9, and S100A12 induce proinflammatory cytokines via TLR4 (103–106). S100A8 interacts with the TLR4/MD2 complex, and S100A8/S100A9 activities are locally restricted through hiding the TLR4/MD2-binding site by (S100A8/S100A9)2 tetramer formation (107).
S100A8/S100A9 binding to TLR4 also induces the transcription of inflammatory cytokine genes, such as IFN regulatory factor 3 (IRF3) (108). Injection of S100A8 enhanced the expression of the Fcɣ receptor (FcɣR) on macrophages in the synovium of a chronic experimental arthritis mice model, while up-regulated expression of FcɣR was abrogated in the synovium of TLR4 knockout mice (109). In an inflammatory autoimmune disease model, S100A8 and S100A9 expression was up-regulated and contributed to the development of IL-17-expressing CD8+ T cells (110). The interaction of S100A8/A9 and TLR4 upregulates IL-17 expression in CD8+ T cells.
Serum S100A8 and S100A9 levels were highly elevated in patients with systemic JIA or AOSD (26, 65). Serum levels of S100A8/A9 were significantly higher in patients with systemic JIA compared to those with systemic infection or other types of JIA, and were correlated with IL-1β expression on phagocytes (111). Serum S100A8/A9 levels were elevated in patients with AOSD, including those with lymphadenopathy and skin rash (21, 26, 112). The serum S100A12 levels were also increased in systemic JIA and AOSD, suggesting pathological roles as DAMPs (113, 114). As endogenous ligands, S100A8/A9 and S100A12 activate TLR4 and RAGE signaling, promoting the production of proinflammatory cytokines such as IL-1β, IL-6, and TNF-α (103, 115, 116). Elevated levels of these cytokines activate inflammatory cells, including neutrophils and monocytes, to produce the TLR4 ligand S100, leading to disease progression. Furthermore, a recent study identified neutrophil activation in both active and clinically inactive systemic JIA patients, characterized by the expression of proinflammatory genes, such as S100A8, and inflammasome components, reflecting persistent innate immune activation (117). They showed that neutrophils in patients with both active disease and longstanding clinically inactive disease had significantly increased capacity to release S100A8/A9 upon activation. However, there was no association between two functional single nucleotide polymorphisms in TLR4 and susceptibility to JIA (118). Furthermore, TLR4 expression is reportedly significantly decreased on the monocytes of patients with SLE and JIA, including systemic JIA. Reduced TLR4 expression was suggested to be a cause of chronic arthritis or the result of a feedback loop (119).
HMGB1, which is a DNA-binding protein released by necrotic or damaged cells, is an extracellular DAMP that links tissue injury to the innate immune response (43). HMGB1 binds to RAGE and TLR4, propagating inflammatory signals. HMGB1 is implicated in systemic inflammation in sepsis, liver injury, arthritis, and SLE. The serum HMGB1 level is elevated in patients with AOSD and systemic JIA compared to healthy controls (HCs) (24, 25), and is correlated with the systemic AOSD score (24). HMGB1 is released from NETs, which are important in the pathophysiology of AOSD (120). The serum levels of NET molecules (including cell-free DNA, myeloperoxidase (MPO)-DNA complex, and α-defensin) were increased in patients with AOSD (121). The serum from active AOSD patients induced NETosis in neutrophils from HCs. NET molecules induced IL-1β production by monocytes, representing a novel pathogenic mechanism of AOSD. Therefore, NET molecules, as ligands of TLR4, might be associated with activation of the TLR signaling pathway in systemic JIA and AOSD.
TLR7 signaling, which is triggered by nucleic acids from damaged host cells, contributes to chronic inflammatory disorders. The expression of TLR7 on circulating precursors of myeloid DCs (pre-mDCs) and mDCs was markedly elevated in patients with AOSD compared to HCs (51). The transcript levels of TLR7, Myd88, IRAK4, TRAF6, and IFN-α were correlated with the serum IL-1β and IFN-α levels in patients with AOSD. After remission, the expression of TLR7 on circulating pre-mDCs, and the transcript levels of TRAF6 and IRAK4, were significantly decreased. NLRP3 inflammasome activation via the TLR7-MyD88 pathway promotes secretion of IL-1β and IL-18, leading to chronic inflammation. The levels of the NLRP3 inflammasome and its byproducts were significantly elevated in patients with AOSD and correlated with disease activity (122). A TLR7 agonist upregulated the levels of NLRP3 inflammasome pathway components (caspase-1, IL-1β, and IL-18) in patients with AOSD, but not in HCs.
IL-6, as one of the proinflammatory cytokines upregulated in systemic JIA and AOSD, plays an important role in systemic inflammation. IL-6 enhances the TLR-induced inflammatory response in vivo and in vitro (123). The administration of IL-6 increased the levels of IL-1β, and some chemokines, in peripheral blood monocytes and synovial fluid mononuclear cells (RA synoviocytes), suggesting amplification of the TLR signaling-mediated cytokine and chemokine response (124). Therefore, an elevated IL-6 level may upregulate TLR signaling in systemic JIA and AOSD.
SAA is synthesized in response to inflammatory stimuli, and induces IL-β production by activating the NLRP3 inflammasome; this is involved in the pathogenesis of systemic JIA and AOSD. Circulating SAA levels were higher in JIA patients, and correlated with the disease activity of JIA (125). A SAA gene polymorphism study revealed that one T allele of rs12218 is associated with disease susceptibility in AOSD patients (126). SAA plays a role in sterile inflammation through activation of TLR2 or TLR4 and their signal pathways. The activation of TLR2 or TLR4 signaling by SAA might contribute to the inflammation seen in systemic JIA and AOSD, although further research on this is needed.
Few studies have evaluated the role of TLRs as biomarkers of systemic JIA and AOSD. Moreover, only a few studies have evaluated the utility of serum levels of their ligands as biomarkers, because cell surface TLR levels are difficult to evaluate. TLR4 ligands, such as S100A8/A9, S100A12, HMGB1, and NET molecules, have become reliable biomarkers for diagnosing and evaluating disease activity in systemic JIA and AOSD. The serum level of the TLR4 ligand S100A8/A9 was correlated with disease activity (based on acute-phase reactants and subjective assessments), and an elevated level during clinical remission was predictive of further disease flares in systemic JIA patients (20). S100A8/A9 was superior to C-reactive protein (CRP) for differentiating systemic JIA from other autoinflammatory syndromes and systemic undifferentiated recurring fever syndrome (127). The serum S100A8/A9 level was correlated with the drug response, suggesting its utility for monitoring disease activity in subclinical systemic JIA (20). Similarly, the serum levels of S100A8 and S100A9 were elevated in patients with AOSD, and were correlated with markers of disease activity, including the systemic disease score (21). Furthermore, the sensitivity and specificity of S100A8/A9 for differentiation of AOSD were 63 and 80.1%, respectively (128). Data on the role of S100A12 as a biomarker are similar to those on S100A9/A9 in systemic JIA and AOSD patients. Serum S100A12 levels were elevated in active AOSD patients relative to HCs and correlated with systemic inflammatory markers, such as ESR, CRP, and ferritin (114). The S100A12 levels were also elevated in patients with systemic JIA compared to patients with infection and HCs (113). The sensitivity and specificity of S100A12 for distinguishing between infection and systemic JIA were 66 and 94%, respectively. One study investigated whether patients with systemic JIA at risk of relapse could be identified using biomarkers, and found that the best single biomarker for predicting flare was S100A12 (129). These data suggest that serum S100A8/A9 and S100A12 levels are good biomarkers for diagnosing systemic JIA and AOSD, predicting relapse, and evaluating disease activity. The utility of serum HMGB1 as a biomarker of systemic JIA and AOSD is limited. The HMGB1 level was higher in patients with AOSD than in HC, but was weakly correlated with the CRP level and systemic score (24). The HMGB1 level was also higher in patients with systemic JIA than in HC, and was associated with serositis and hepatosplenomegaly (25). Another study found positive correlations of the serum level of HMGB1 with ESR, CRP, and α2 globulin in patients with JIA, including systemic JIA. In a NET study, patients with AOSD had higher levels of cell-free DNA and NET-DNA complexes, and their neutrophils released more NETs, compared to HCs (130). The serum levels of cell-free DNA, MPO-DNA, and α-defensin were significantly increased in patients with AOSD compared to HCs (121). Furthermore, these levels were correlated with the levels of several disease-activity markers, and neutrophil elastase and MPO-positive inflammatory cells were detected in the lymph nodes and skin of patients with active AOSD.
One study evaluated the frequencies of TLR7-expressing pre-mDCs and mDCs by flow cytometry in patients with AOSD and SLE, as well as in HC (51). The levels of TLR7 signaling molecules were elevated and positively correlated with disease activity in patients with AOSD.
We have reviewed the roles of TLRs and their ligands, as PRRs and DAMPs, in aggravating inflammation, including sterile inflammation, in two rheumatic diseases (systemic JIA and AOSD). TLRs and their ligands contribute to inflammation in patients with systemic JIA and AOSD. The levels of DAMPs, such as S100 proteins, HMGB1, and MPO-DNA complex, are elevated in the blood of patients with active AOSD or systemic JIA, suggesting that they aggravate inflammation by activating TLR4 in inflammatory cells. Furthermore, the TLR4 ligands S100A8/A9 and MPO-DNA complex have potential as biomarkers for diagnosis and disease activity in patients with systemic JIA and AOSD.
Although the triggers of inflammation are unknown, viral or bacterial infection, as well as environmental factors, could act as danger signals promoting TLR activation. This could in turn lead to the activation of neutrophils and macrophages, and to the production of proinflammatory cytokines such as IL-6 and DAMPs, via activated neutrophils and macrophages. This would sustain sterile inflammation in systemic JIA and AOSD. However, some questions remain to be resolved. The first concerns whether TLR signaling is triggered, or occurs due to the loss of inhibitory signals, during initial inflammation. The second concerns which subset of patients is more likely to develop systemic JIA or AOSD even after being exposed to similar environmental factors or viral/bacterial infections. Therefore, further studies should determine the genetic factors associated with onset or exacerbation of the initial TLR responses. The third question concerns whether the TLR–TLR ligand response is associated with chronic features of systemic JIA and AOSD; this is currently unclear, despite the known link between initial acute inflammation and TLR responses. Further basic and clinical research, including large, multicenter, prospective studies, is needed to confirm the role of TLRs and their ligands in systemic JIA and AOSD. The current findings enhance our understanding of the pathogenesis of systemic JIA and AOSD, and will facilitate the development of diagnostic and prognostic biomarkers, as well as novel therapeutics targeting TLR signaling in systemic JIA and AOSD.
All authors listed have made a substantial, direct and, intellectual contribution to the work and approved it for publication.
This work was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology [Grand Number. 2019R1A2C1005988].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Pardeo M, Bracaglia C, De Benedetti F. Systemic juvenile idiopathic arthritis: New insights into pathogenesis and cytokine directed therapies. Best Pract Res Clin Rheumatol (2017) 31:505–16. doi: 10.1016/j.berh.2018.02.002
2. Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still’s disease. J Autoimmun (2018) 93:24–36. doi: 10.1016/j.jaut.2018.07.018
3. Kadavath S, Efthimiou P. Adult-onset Still’s disease-pathogenesis, clinical manifestations, and new treatment options. Ann Med (2015) 47:6–14. doi: 10.3109/07853890.2014.971052
4. Swart JF, de Roock S, Prakken BJ. Understanding inflammation in juvenile idiopathic arthritis: How immune biomarkers guide clinical strategies in the systemic onset subtype. Eur J Immunol (2016) 46:2068–77. doi: 10.1002/eji.201546092
5. Choi JH, Suh CH, Lee YM, Suh YJ, Lee SK, Kim SS, et al. Serum cytokine profiles in patients with adult onset Still’s disease. J Rheumatol (2003) 30:2422–7.
6. Kessel C, Lippitz K, Weinhage T. Proinflammatory Cytokine Environments Can Drive Interleukin-17 Overexpression by γ/δ T Cells in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol (2017) 69:1480–94. doi: 10.1002/art.40099
7. Youm JY, Woo JH, Kim TH, Bae SC, Yoo DH. Interleukin-1beta and interleukin-1 receptor antagonist gene polymorphisms in Korean patients with adult-onset Still’s disease. Scand J Rheumatol (2007) 36:390–3. doi: 10.1080/03009740701340081
8. Ombrello MJ, Remmers EF, Tachmazidou I, Grom A, Foell D, Haas JP, et al. HLA-DRB1*11 and variants of the MHC class II locus are strong risk factors for systemic juvenile idiopathic arthritis. Proc Natl Acad Sci U S A (2015) 112:15970–5. doi: 10.1073/pnas.1520779112
9. Modesto C, Patiño-García A, Sotillo-Piñeiro E, Merino J, García-Consuegra J, Merino R, et al. TNF-alpha promoter gene polymorphisms in Spanish children with persistent oligoarticular and systemic-onset juvenile idiopathic arthritis. Scand J Rheumatol (2005) 34:451–4. doi: 10.1080/03009740510026652
10. Asano T, Furukawa H, Sato S, Yashiro M, Kobayashi H, Watanabe H, et al. Effects of HLA-DRB1 alleles on susceptibility and clinical manifestations in Japanese patients with adult onset Still’s disease. Arthritis Res Ther (2017) 19:199. doi: 10.1186/s13075-017-1406-x
11. Wang FF, Huang XF, Shen N, Leng L, Bucala R, Chen SL, et al. A genetic role for macrophage migration inhibitory factor (MIF) in adult-onset Still’s disease. Arthritis Res Ther (2013) 15:R65. doi: 10.1186/ar4239
12. Chen DY, Chen YM, Lan JL, Tzang BS, Lin CC, Hsu TC. Significant association of past parvovirus B19 infection with cytopenia in both adult-onset Still’s disease and systemic lupus erythematosus patients. Clin Chim Acta (2012) 413:855–60. doi: 10.1016/j.cca.2012.01.027
13. Bento DP, Tavares R, Baptista Leite R, Miranda A, Ramos S, Ventura F, et al. Adult-Onset Still’s Disease and cytomegalovirus infection. Acta Rheumatol Port (2010) 35:259–63.
14. Gambichler T, Paech V, Rotterdam S, Stücker M, Boms S, Altmeyer P. Hepatitis B-assocciated adult-onset Still’s disease presenting with neutrophilic urticaria. Eur J Med Res (2003) 8:527–30.
15. Valtonen JM, Kosunen TU, Karjalainen J, Valtonen M, Leirisalo-Repo M, Valtonen VV. Serological findings in patients with acute syndromes fulfilling the proposed criteria of adult onset Still’s disease. Scan J Rheumatol (1997) 26:342–5. doi: 10.3109/03009749709065695
16. Luder AS, Naphtali V, Ben Porat E, Lahat N. Still’s disease associated with adenovirus infection and defect in adenovirus directed natural killing. Ann Rheum Dis (1989) 48:81–786. doi: 10.1136/ard.48.9.781
17. Thornton S, Tan R, Sproles A, Do T, Schick J, Grom AA, Schulert GS, et al. A Multiparameter Flow Cytometry Analysis Panel to Assess CD163 mRNA and Protein in Monocyte and Macrophage Populations in Hyperinflammatory Diseases. J Immunol (2019) 202:1635–43. doi: 10.4049/jimmunol.1800765
18. Komiya A, Matsui T, Nogi S, Iwata K, Futami H, Takaoka H, et al. Neutrophil CD64 is upregulated in patients with active adult-onset Still’s disease. Scand J Rheumatol (2012) 41:156–8. doi: 10.3109/03009742.2011.644325
19. Matsui K, Tsuchida T, Hiroishi K, Tominaga K, Hayashi N, Hada T, et al. High serum level of macrophage-colony stimulating factor (M-CSF) in adult-onset Still’s disease. Rheumatol (Oxford) (1999) 38:477–8. doi: 10.1093/rheumatology/38.5.477
20. Holzinger D, Frosch M, Kastrup A, Prince FH, Otten MH, Van Suijlekom-Smit LW, et al. The Toll-like receptor 4 agonist MRP8/14 protein complex is a sensitive indicator for disease activity and predicts relapses in systemic-onset juvenile idiopathic arthritis. Ann Rheum Dis (2012) 71:974–80. doi: 10.1136/annrheumdis-2011-200598
21. Kim HA, An JM, Nam JY, Jeon JY, Suh CH. Serum S100A8/A9, but not follistatin-like protein 1 and interleukin 18, may be a useful biomarker of disease activity in adult-onset Still’s disease. J Rheumatol (2012) 39:1399–406. doi: 10.3899/jrheum.120079
22. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu Rev Pathol (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
23. Jung JY, Suh CH, Kim HA. The role of damage-associated molecular pattern for pathogenesis and biomarkers in adult-onset Still’s disease. Expert Rev Mol Diagn (2019) 19:459–68. doi: 10.1080/14737159.2019.1615449
24. Jung JY, Suh CH, Sohn S, Nam JY, Kim HA. Elevated high-mobility group B1 levels in active adult-onset Still’s disease associated with systemic score and skin rash. Clin Rheumatol (2016) 35:1937–42. doi: 10.1007/s10067-016-3314-x
25. Bobek D, Grčević D, Kovačić N, Lukić IK, Jelušić M. The presence of high mobility group box-1 and soluble receptor for advanced glycation end-products in juvenile idiopathic arthritis and juvenile systemic lupus erythematosus. Pediatr Rheumatol Online J (2014) 12:50. doi: 10.1186/1546-0096-12-50
26. Kim HA, Han JH, Kim WJ, Noh HJ, An JM, Yim H, et al. TLR4 Endogenous Ligand S100A8/A9 Levels in Adult-Onset Still’s Disease and Their Association with Disease Activity and Clinical Manifestations. Int J Mol Sci (2016) 17:1342. doi: 10.3390/ijms17081342
27. Kim HA, Kim YH, Jeon YK, Yang WI, Kwon JE, Han JH. Histopathology and expression of the chemokines CXCL10, CXCL13, and CXCR3 and the endogenous TLR-4 ligand S100A8/A9 in lymph nodes of patients with adult-onset Still’s disease. Sci Rep (2019) 9:7517. doi: 10.1038/s41598-019-44032-6
28. Kessel C, Hedrich CM, Foell D. Innately Adaptive or Truly Autoimmune: Is There Something Unique About Systemic Juvenile Idiopathic Arthritis? Arthritis Rheumatol (2020) 72:210–9. doi: 10.1002/art.41107
29. Omoyinmi E, Hamaoui R, Pesenacker A, Nistala K, Moncrieffe H, Ursu S, et al. Th1 and Th17 cell subpopulations are enriched in the peripheral blood of patients with systemic juvenile idiopathic arthritis. Rheumatol (Oxford) (2012) 51:1881–6. doi: 10.1093/rheumatology/kes162
30. Chen DY, Chen YM, Lan JL, Lin CC, Chen HH, Hsieh CW. Potential role of Th17 cells in the pathogenesis of adult-onset Still’s disease. Rheumatol (Oxford) (2010) 49:2305–12. doi: 10.1093/rheumatology/keq284
31. Henderson LA, Hoyt KJ, Lee PY, Rao DA, Jonsson AH, Nguyen JP, et al. Th17 reprogramming of T cells in systemic juvenile idiopathic arthritis. JCI Insight (2020) 5:e132508. doi: 10.1172/jci.insight.132508
32. Jung JY, Choi B, Sayeed HM, Suh CH, Kim YW, Kim HA, et al. Characteristic patterns of HLA presentation and T cell differentiation in adult-onset Still’s disease. Int J Immunopathol Pharmacol (2018) 32:2058738418791284. doi: 10.1177/2058738418791284
33. Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, Kashiwagi H, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol (1992) 19:424–30.
34. Fautrel B, Le Moël G, Saint-Marcoux B, Taupin P, Vignes S, Rozenberg S, et al. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J Rheumatol (2001) 28:322–9.
35. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm (2010) 2010:672395. doi: 10.1155/2010/672395
36. Fitzgerald KA, Kagan JC. Toll-like Receptors and the Control of Immunity. Cell (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
37. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol (2007) 8:487–96. doi: 10.1038/ni1457
38. Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, et al. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med (2003) 198:1043–55. doi: 10.1084/jem.20031023
39. Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A. The human toll signaling pathway: divergence of nuclear factor kappaB and JNK/SAPK activation upstream of tumor necrosis factor receptor-associated factor 6 (TRAF6). J Exp Med (1998) 187:2097–101. doi: 10.1084/jem.187.12.2097
40. Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell (1998) 2:253–8. doi: 10.1016/s1097-2765(00)80136-7
41. Chen JQ, Szodoray P, Zeher M. Toll-Like Receptor Pathways in Autoimmune Diseases. Clin Rev Allergy Immunol (2016) 50:1–17. doi: 10.1007/s12016-015-8473-z
42. Holzinger D, Tenbrock K, Roth J. Alarmins of the S100-Family in Juvenile Autoimmune and Auto-Inflammatory Diseases. Front Immunol (2019) 10:182. doi: 10.3389/fimmu.2019.00182
43. Paudel YN, Angelopoulou E, Piperi C, Balasubramaniam V, Othman I, Shaikh MF. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur J Pharmacol (2019) 858:172487. doi: 10.1016/j.ejphar.2019.172487
44. Jiang W, Pisetsky DS. The role of IFN-alpha and nitric oxide in the release of HMGB1 by RAW 264.7 cells stimulated with polyinosinic-polycytidylic acid or lipopolysaccharide. J Immunol (2006) 177:3337–43. doi: 10.4049/jimmunol.177.5.3337
45. Bell CW, Jiang W, Reich CF,3, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol (2006) 291:C1318–25. doi: 10.1152/ajpcell.00616.2005
46. Yang H, Wang H, Ju Z, Ragab AA, Lundbäck P, Long W, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med (2015) 212:5–14. doi: 10.1084/jem.20141318
47. Niemi K, Teirilä L, Lappalainen J, Rajamäki K, Baumann MH, Öörni K, et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol (2011) 186:6119–28. doi: 10.4049/jimmunol.1002843
48. He RL, Zhou J, Hanson CZ, Chen J, Cheng N, Ye RD. Serum amyloid A induces G-CSF expression and neutrophilia via Toll-like receptor 2. Blood (2009) 113:429–37. doi: 10.1182/blood-2008-03-139923
49. Sandri S, Rodriguez D, Gomes E, Monteiro HP, Russo M, Campa A. Is serum amyloid A an endogenous TLR4 agonist? J Leukoc Biol (2008) 83:1174–80. doi: 10.1189/jlb.0407203
50. De Buck M, Berghmans N, Pörtner N, Vanbrabant L, Cockx M, Struyf S, et al. Serum amyloid A1α induces paracrine IL-8/CXCL8 via TLR2 and directly synergizes with this chemokine via CXCR2 and formyl peptide receptor 2 to recruit neutrophils. J Leukoc Biol (2015) 98:1049–60. doi: 10.1189/jlb.3A0315-085R
51. Chen DY, Lin CC, Chen YM, Lan JL, Hung WT, Chen HH, et al. Involvement of TLR7 MyD88-dependent signaling pathway in the pathogenesis of adult-onset Still’s disease. Arthritis Res Ther (2013) 15:R39. doi: 10.1186/ar4193
52. Shenderov K, Riteau N, Yip R, Mayer-Barber KD, Oland S, Hieny S, et al. Cutting edge: Endoplasmic reticulum stress licenses macrophages to produce mature IL-1β in response to TLR4 stimulation through a caspase-8- and TRIF-dependent pathway. J Immunol (2014) 192:2029–33. doi: 10.4049/jimmunol.1302549
53. Nyström S, Antoine DJ, Lundbäck P, Lock JG, Nita AF, Högstrand K, et al. TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J (2013) 32:86–99. doi: 10.1038/emboj.2012.328
54. Gurung P, Malireddi RK, Anand PK, Demon D, Vande Walle L, Liu Z, et al. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-β (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem (2012) 287:34474–83. doi: 10.1074/jbc.M112.401406
55. Joosten LA, Abdollahi-Roodsaz S, Dinarello CA, O’Neill L, Netea MG. Toll-like receptors and chronic inflammation in rheumatic diseases: new developments. Nat Rev Rheumatol (2016) 12:344–57. doi: 10.1038/nrrheum.2016.61
56. Seibl R, Birchler T, Loeliger S, Hossle JP, Gay RE, Saurenmann T, et al. Expression and regulation of Toll-like receptor 2 in rheumatoid arthritis synovium. Am J Pathol (2003) 162:1221–7. doi: 10.1016/s0002-9440(10)63918-1
57. Radstake TR, Roelofs MF, Jenniskens YM, Oppers-Walgreen B, van Riel PL, Barrera P, et al. Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma. Arthritis Rheumatol (2004) 50:3856–65. doi: 10.1002/art.20678
58. Huang QQ, Sobkoviak R, Jockheck-Clark AR, Shi B, Mandelin AM,2, Tak PP, et al. Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J Immunol (2009) 182:4965–73. doi: 10.4049/jimmunol.0801563
59. Grevers LC, de Vries TJ, Vogl T, Abdollahi-Roodsaz S, Sloetjes AW, Leenen PJ, et al. S100A8 enhances osteoclastic bone resorption in vitro through activation of Toll-like receptor 4: implications for bone destruction in murine antigen-induced arthritis. Arthritis Rheumatol (2011) 63:1365–75. doi: 10.1002/art.30290
60. Pierer M, Rethage J, Seibl R, Lauener R, Brentano F, Wagner U, et al. Chemokine secretion of rheumatoid arthritis synovial fibroblasts stimulated by Toll-like receptor 2 ligands. J Immunol (2004) 172:1256–65. doi: 10.4049/jimmunol.172.2.1256
61. Ospelt C, Brentano F, Rengel Y, Stanczyk J, Kolling C, Tak PP, et al. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheumatol (2008) 58:3684–92. doi: 10.1002/art.24140
62. Brentano F, Schorr O, Gay RE, Gay S, Kyburz D. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3. Arthritis Rheumatol (2005) 52:2656–65. doi: 10.1002/art.21273
63. Roelofs MF, Joosten LA, Abdollahi-Roodsaz S, van Lieshout AW, Sprong T, van den Hoogen FH, et al. The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheumatol (2005) 52:2313–22. doi: 10.1002/art.21278
64. Chamberlain ND, Kim SJ, Vila OM, Volin MV, Volkov S, Pope RM, et al. Ligation of TLR7 by rheumatoid arthritis synovial fluid single strand RNA induces transcription of TNFα in monocytes. Ann Rheum Dis (2013) 72:418–26. doi: 10.1136/annrheumdis-2011-201203
65. Abdollahi-Roodsaz S, Joosten LA, Roelofs MF, Radstake TR, Matera G, Popa C, et al. Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheumatol (2007) 56:2957–67. doi: 10.1002/art.22848
66. Sacre SM, Lo A, Gregory B, Simmonds RE, Williams L, Feldmann M, et al. Inhibitors of TLR8 reduce TNF production from human rheumatoid synovial membrane cultures. J Immunol (2008) 181:8002–9. doi: 10.4049/jimmunol.181.11.8002
67. Roelofs MF, Wenink MH, Brentano F, Abdollahi-Roodsaz S, Oppers-Walgreen B, Barrera P, et al. Type I interferons might form the link between Toll-like receptor (TLR) 3/7 and TLR4-mediated synovial inflammation in rheumatoid arthritis (RA). Ann Rheum Dis (2009) 68:1486–93. doi: 10.1136/ard.2007.086421
68. Kim SJ, Chen Z, Chamberlain ND, Essani AB, Volin MV, Amin MA, et al. Ligation of TLR5 promotes myeloid cell infiltration and differentiation into mature osteoclasts in rheumatoid arthritis and experimental arthritis. J Immunol (2014) 193:3902–13. doi: 10.4049/jimmunol.1302998
69. Lee YH, Bae SC, Kim JH, Song GG. Toll-like receptor polymorphisms and rheumatoid arthritis: a systematic review. Rheumatol Int (2014) 34:111–6. doi: 10.1007/s00296-013-2666-7
70. Sánchez E, Orozco G, López-Nevot MA, Jiménez-Alonso J, Martín J. Polymorphisms of toll-like receptor 2 and 4 genes in rheumatoid arthritis and systemic lupus erythematosus. Tissue Antigens (2004) 63:54–7. doi: 10.1111/j.1399-0039.2004.00162.x
71. Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity (2018) 49:725–739.e726. doi: 10.1016/j.immuni.2018.08.015
72. Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med (2005) 202:1575–85. doi: 10.1084/jem.20051696
73. Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders HJ. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol (2007) 18:1721–31. doi: 10.1681/asn.2006101162
74. Pacheco GV, Novelo Noh IB, Velasco Cárdenas RM, Angulo Ramírez AV, López Villanueva RF, Quintal Ortiz IG, et al. Expression of TLR-7, MyD88, NF-kB, and INF-α in B Lymphocytes of Mayan Women with Systemic Lupus Erythematosus in Mexico. Front Immunol (2016) 7:22. doi: 10.3389/fimmu.2016.00022
75. Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med (2005) 202:321–31. doi: 10.1084/jem.20050338
76. Summers SA, Hoi A, Steinmetz OM, O’Sullivan KM, Ooi JD, Odobasic D, et al. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J Autoimmun (2010) 35:291–8. doi: 10.1016/j.jaut.2010.05.004
77. Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol (2010) 184:1840–8. doi: 10.4049/jimmunol.0902592
78. Tilstra JS, John S, Gordon RA, Leibler C, Kashgarian M, Bastacky S, et al. B cell-intrinsic TLR9 expression is protective in murine lupus. J Clin Invest (2020) 130:3172–87. doi: 10.1172/jci132328
79. Desnues B, Macedo AB, Roussel-Queval A, Bonnardel J, Henri S, Demaria O, et al. TLR8 on dendritic cells and TLR9 on B cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice. Proc Natl Acad Sci U S A (2014) 111:1497–502. doi: 10.1073/pnas.1314121111
80. Corzo CA, Varfolomeev E, Setiadi AF, Francis R, Klabunde S, Senger K, et al. The kinase IRAK4 promotes endosomal TLR and immune complex signaling in B cells and plasmacytoid dendritic cells. Sci Signal (2020) eaaz1053:13. doi: 10.1126/scisignal.aaz1053
81. Shah M, Kim GY, Achek A, Cho EY, Baek WY, Choi YS, et al. The αC helix of TIRAP holds therapeutic potential in TLR-mediated autoimmune diseases. Biomaterials (2020) 245:119974. doi: 10.1016/j.biomaterials.2020.119974
82. Majer O, Liu B, Kreuk LSM, Krogan N, Barton GM. UNC93B1 recruits syntenin-1 to dampen TLR7 signalling and prevent autoimmunity. Nature (2019) 575:366–70. doi: 10.1038/s41586-019-1612-6
83. Lee YH, Lee HS, Choi SJ, Ji JD, Song GG. Associations between TLR polymorphisms and systemic lupus erythematosus: a systematic review and meta-analysis. Clin Exp Rheumatol (2012) 30:262–5.
84. Spachidou MP, Bourazopoulou E, Maratheftis CI, Kapsogeorgou EK, Moutsopoulos HM, Tzioufas AG, et al. Expression of functional Toll-like receptors by salivary gland epithelial cells: increased mRNA expression in cells derived from patients with primary Sjögren’s syndrome. Clin Exp Immunol (2007) 147:497–503. doi: 10.1111/j.1365-2249.2006.03311.x
85. Kwok SK, Cho ML, Her YM, Oh HJ, Park MK, Lee SY, et al. TLR2 ligation induces the production of IL-23/IL-17 via IL-6, STAT3 and NF-kB pathway in patients with primary Sjogren’s syndrome. Arthritis Res Ther (2012) 14:R64. doi: 10.1186/ar3780
86. Karlsen M, Jonsson R, Brun JG, Appel S, Hansen T. TLR-7 and -9 Stimulation of Peripheral Blood B Cells Indicate Altered TLR Signalling in Primary Sjögren’s Syndrome Patients by Increased Secretion of Cytokines. Scand J Immunol (2015) 82:523–31. doi: 10.1111/sji.12368
87. Frasca L, Lande R. Toll-like receptors in mediating pathogenesis in systemic sclerosis. Clin Exp Immunol (2020) 201:14–24. doi: 10.1111/cei.13426
88. Bhattacharyya S, Tamaki Z, Wang W, Hinchcliff M, Hoover P, Getsios S, et al. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci Transl Med (2014) 6:232ra250. doi: 10.1126/scitranslmed.3008264
89. Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, et al. Tenascin-C drives persistence of organ fibrosis. Nat Commun (2016) 7:11703. doi: 10.1038/ncomms11703
90. Fang F, Marangoni RG, Zhou X, Yang Y, Ye B, Shangguang A, et al. Toll-like Receptor 9 Signaling Is Augmented in Systemic Sclerosis and Elicits Transforming Growth Factor β-Dependent Fibroblast Activation. Arthritis Rheumatol (2016) 68:1989–2002. doi: 10.1002/art.39655
91. Lande R, Lee EY, Palazzo R, Marinari B, Pietraforte I, Santos GS, et al. CXCL4 assembles DNA into liquid crystalline complexes to amplify TLR9-mediated interferon-α production in systemic sclerosis. Nat Commun (2019) 10:1731. doi: 10.1038/s41467-019-09683-z
92. Schenkein HA, Thomas RR. Anticardiolipin (aCL) in sera from periodontitis subjects activate Toll-like receptor 4 (TLR4). PLoS One (2018) 13:e0203494. doi: 10.1371/journal.pone.0203494
93. Satta N, Dunoyer-Geindre S, Reber G, Fish RJ, Boehlen F, Kruithof EK, et al. The role of TLR2 in the inflammatory activation of mouse fibroblasts by human antiphospholipid antibodies. Blood (2007) 109:1507–14. doi: 10.1182/blood-2005-03-024463
94. Döring Y, Hurst J, Lorenz M, Prinz N, Clemens N, Drechsler MD, et al. Human antiphospholipid antibodies induce TNFalpha in monocytes via Toll-like receptor 8. Immunobiology (2010) 215:230–41. doi: 10.1016/j.imbio.2009.03.002
95. Hurst J, Prinz N, Lorenz M, Bauer S, Chapman J, Lackner KJ, et al. TLR7 and TLR8 ligands and antiphospholipid antibodies show synergistic effects on the induction of IL-1beta and caspase-1 in monocytes and dendritic cells. Immunobiology (2009) 214:683–91. doi: 10.1016/j.imbio.2008.12.003
96. Mulla MJ, Weel IC, Potter JA, Gysler SM, Salmon JE, Peraçoli MTS, et al. Antiphospholipid Antibodies Inhibit Trophoblast Toll-Like Receptor and Inflammasome Negative Regulators. Arthritis Rheumatol (2018) 70:891–902. doi: 10.1002/art.40416
97. Raschi E, Chighizola CB, Grossi C, Ronda N, Gatti R, Meroni PL, et al. β2-glycoprotein I, lipopolysaccharide and endothelial TLR4: three players in the two hit theory for anti-phospholipid-mediated thrombosis. J Autoimmun (2014) 55:42–50. doi: 10.1016/j.jaut.2014.03.001
98. Liu-Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R. Innate immunity conferred by Toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheumatol (2005) 52:2936–46. doi: 10.1002/art.21238
99. Mylona EE, Mouktaroudi M, Crisan TO, Makri S, Pistiki A, Georgitsi M, et al. Enhanced interleukin-1β production of PBMCs from patients with gout after stimulation with Toll-like receptor-2 ligands and urate crystals. Arthritis Res Ther (2012) 14:R158. doi: 10.1186/ar3898
100. Rossato MF, Hoffmeister C, Trevisan G, Bezerra F, Cunha TM, Ferreira J, et al. Monosodium urate crystal interleukin-1β release is dependent on Toll-like receptor 4 and transient receptor potential V1 activation. Rheumatol (Oxford) (2020) 59:233–42. doi: 10.1093/rheumatology/kez259
101. Holzinger D, Nippe N, Vogl T, Marketon K, Mysore V, Weinhage T, et al. Myeloid-related proteins 8 and 14 contribute to monosodium urate monohydrate crystal-induced inflammation in gout. Arthritis Rheumatol (2014) 66:1327–39. doi: 10.1002/art.38369
102. Han Q, Bing W, Di Y, Hua L, Shi-He L, Yu-Hua Z, et al. Kinsenoside screening with a microfluidic chip attenuates gouty arthritis through inactivating NF-κB signaling in macrophages and protecting endothelial cells. Cell Death Dis (2016) 7:e2350. doi: 10.1038/cddis.2016.255
103. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med (2007) 13:1042–9. doi: 10.1038/nm1638
104. Riva M, Källberg E, Björk P, Hancz D, Vogl T, Roth J, et al. Induction of nuclear factor-κB responses by the S100A9 protein is Toll-like receptor-4-dependent. Immunology (2012) 137:172–82. doi: 10.1111/j.1365-2567
105. Foell D, Wittkowski H, Kessel C, Lüken A, Weinhage T, Varga G, et al. Proinflammatory S100A12 can activate human monocytes via Toll-like receptor 4. Am J Respir Crit Care Med (2013) 187:1324–34. doi: 10.1164/rccm.201209-1602OC
106. Kessel C, Fuehner S, Zell J, Zimmermann B, Drewianka S, Brockmeyer S, et al. Calcium and zinc tune autoinflammatory Toll-like receptor 4 signaling by S100A12. J Allergy Clin Immunol (2018) 142:1370–3.e8. doi: 10.1016/j.jaci.2018.06.027
107. Vogl T, Stratis A, Wixler V, Völler T, Thurainayagam S, Jorch SK, et al. Autoinhibitory regulation of S100A8/S100A9 alarmin activity locally restricts sterile inflammation. J Clin Invest (2018) 128:1852–66. doi: 10.1172/JCI89867
108. Wang J, Vodovotz Y, Fan L, Li Y, Liu Z, Namas R, et al. Injury-induced MRP8/MRP14 stimulates IP-10/CXCL10 in monocytes/macrophages. FASEB J (2015) 29:250–62. doi: 10.1096/fj.14-255992
109. van Lent PL, Grevers LC, Schelbergen R, Blom A, Geurts J, Sloetjes A, et al. S100A8 causes a shift toward expression of activatory Fcγ receptors on macrophages via toll-like receptor 4 and regulates Fcγ receptor expression in synovium during chronic experimental arthritis. Arthritis Rheumatol (2010) 62:3353–64. doi: 10.1002/art.27654
110. Loser K, Vogl T, Voskort M, Lueken A, Kupas V, Nacken W, et al. The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat Med (2010) 16:713–7. doi: 10.1038/nm.2150
111. Frosch M, Ahlmann M, Vogl T, Wittkowski H, Wulffraat N, Foell D, et al. The myeloid-related proteins 8 and 14 complex, a novel ligand of toll-like receptor 4, and interleukin-1beta form a positive feedback mechanism in systemic-onset juvenile idiopathic arthritis. Arthritis Rheumatol (2009) 60:883–91. doi: 10.1002/art.24349
112. Jung SY, Park YB, Ha YJ, Lee KH, Lee SK. Serum calprotectin as a marker for disease activity and severity in adult-onset Still’s disease. J Rheumatol (2010) 37:1029–34. doi: 10.3899/jrheum.091120
113. Wittkowski H, Frosch M, Wulffraat N, Goldbach-Mansky R, Kallinich T, Kuemmerle-Deschner J, et al. S100A12 is a novel molecular marker differentiating systemic-onset juvenile idiopathic arthritis from other causes of fever of unknown origin. Arthritis Rheumatol (2008) 58:3924–31. doi: 10.1002/art.24137
114. Bae CB, Suh CH, An JM, Jung JY, Jeon JY, Nam JY, et al. Serum S100A12 may be a useful biomarker of disease activity in adult-onset Still’s disease. J Rheumatol (2014) 41:2403–8. doi: 10.3899/jrheum.140651
115. Chen B, Miller AL, Rebelatto M, Brewah Y, Rowe DC, Clarke L, et al. S100A9 induced inflammatory responses are mediated by distinct damage associated molecular patterns (DAMP) receptors in vitro and in vivo. PLoS One (2015) 10:e0115828. doi: 10.1371/journal.pone.0115828
116. Ma L, Sun P, Zhang JC, Zhang Q, Yao SL. Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int J Mol Med (2017) 40:31–8. doi: 10.3892/ijmm.2017.2987
117. Brown RA, Henderlight M, Do T, Yasin S, Grom AA, DeLay M, et al. Neutrophils From Children With Systemic Juvenile Idiopathic Arthritis Exhibit Persistent Proinflammatory Activation Despite Long-Standing Clinically Inactive Disease. Front Immunol (2018) 9:2995. doi: 10.3389/fimmu.2018.02995
118. Lamb R, Zeggini E, Thomson W, Donn R. Toll-like receptor 4 gene polymorphisms and susceptibility to juvenile idiopathic arthritis. Ann Rheum Dis (2005) 64:767–9. doi: 10.1136/ard.2004.026930
119. Kirchner M, Sonnenschein A, Schoofs S, Schmidtke P, Umlauf VN, Mannhardt-Laakmann W. Surface expression and genotypes of Toll-like receptors 2 and 4 in patients with juvenile idiopathic arthritis and systemic lupus erythematosus. Pediatr Rheumatol Online J (2013) 11:9. doi: 10.1186/1546-0096-11-9
120. Torres-Ruiz J, Carrillo-Vázquez DA, Tapia-Rodríguez M, Garcia-Galicia JA, Alcocer-Varela J, Gómez-Martín D. The role of low density granulocytes and NETosis in the pathogenesis of adult-onset Still’s Disease. Clin Exp Rheumatol (2019) 37(Suppl 121):74–82.
121. Ahn MH, Han JH, Chwae YJ, Jung JY, Suh CH, Kwon JE, et al. Neutrophil Extracellular Traps May Contribute to the Pathogenesis in Adult-onset Still Disease. J Rheumatol (2019) 46:1560–9. doi: 10.3899/jrheum.181058
122. Hsieh CW, Chen YM, Lin CC, Tang KT, Chen HH, Hung WT, et al. Elevated Expression of the NLRP3 Inflammasome and Its Correlation with Disease Activity in Adult-onset Still Disease. J Rheumatol (2017) 44:1142–50. doi: 10.3899/jrheum.161354
123. Strippoli R, Carvello F, Scianaro R, De Pasquale L, Vivarelli M, Petrini S, et al. Amplification of the response to Toll-like receptor ligands by prolonged exposure to interleukin-6 in mice: implication for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol (2012) 64:1680–8. doi: 10.1002/art.33496
124. Caiello I, Minnone G, Holzinger D, Vogl T, Prencipe G, Manzo A, et al. IL-6 amplifies TLR mediated cytokine and chemokine production: implications for the pathogenesis of rheumatic inflammatory diseases. PLoS One (2014) 9:e107886. doi: 10.1371/journal.pone.0107886
125. Cantarini L, Giani T, Fioravanti A, Iacoponi F, Simonini G, Pagnini I, et al. Serum amyloid A circulating levels and disease activity in patients with juvenile idiopathic arthritis. Yonsei Med J (2012) 53:1045–8. doi: 10.3349/ymj.2012.53.5.1045
126. Yashiro M, Furukawa H, Asano T, Sato S, Kobayashi H, Watanabe H, et al. Serum amyloid A1 (SAA1) gene polymorphisms in Japanese patients with adult-onset Still’s disease. Med (Baltimore) (2018) 97:e13394. doi: 10.1097/MD.0000000000013394
127. Aljaberi N, Tronconi E, Schulert G, Grom AA, Lovell DJ, Huggins JL, et al. The use of S100 proteins testing in juvenile idiopathic arthritis and autoinflammatory diseases in a pediatric clinical setting: a retrospective analysis. Pediatr Rheumatol Online J (2020) 18:7. doi: 10.1186/s12969-020-0398-2
128. Guo Q, Zha X, Li C, Jia Y, Zhu L, Guo J, et al. Serum calprotectin–a promising diagnostic marker for adult-onset Still’s disease. Clin Rheumatol (2016) 35:73–9. doi: 10.1007/s10067-015-3108-6
129. Gerss J, Roth J, Holzinger D, Ruperto N, Wittkowski H, Frosch M, et al. Paediatric Rheumatology International Trials Organization (PRINTO). Phagocyte-specific S100 proteins and high-sensitivity C reactive protein as biomarkers for a risk-adapted treatment to maintain remission in juvenile idiopathic arthritis: a comparative study. Ann Rheum Dis (2012) 71:1991–7. doi: 10.1136/annrheumdis-2012-201329
Keywords: damage-associated molecular pattern, toll-like receptor, systemic juvenile idiopathic arthritis, adult onse still disease, inflammation
Citation: Jung J-Y, Kim J-W, Suh C-H and Kim H-A (2020) Roles of Interactions Between Toll-Like Receptors and Their Endogenous Ligands in the Pathogenesis of Systemic Juvenile Idiopathic Arthritis and Adult-Onset Still’s Disease. Front. Immunol. 11:583513. doi: 10.3389/fimmu.2020.583513
Received: 15 July 2020; Accepted: 12 October 2020;
Published: 05 November 2020.
Edited by:
Sy Seong, Seoul National University, South KoreaReviewed by:
Christoph Kessel, University Hospital Muenster, GermanyCopyright © 2020 Jung, Kim, Suh and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyoun-Ah Kim, bmFraGFkYUBuYXZlci5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.