 Yang Wang
Yang Wang Jilong Shen
Jilong Shen Li Yu
Li Yu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Immunol. , 16 December 2020
Sec. Molecular Innate Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.583193
Inflammasomes are multimeric protein complexes regulating the innate immune response to invading pathogens or stress stimuli. Recent studies have reported that nucleotide-binding leucine-rich repeat-containing (NLRs) proteins and DNA sensor absent in melanoma 2 (AIM2) serve as inflammasome sentinels, whose stimulation leads to the proteolytic activation of caspase-1, proinflammatory cytokine secretion, and pyroptotic cell death. Toxoplasma gondii, an obligate intracellular parasite of phylum Apicomplexans, is reportedly involved in NLRP1, NLRP3 and AIM2 inflammasomes activation; however, mechanistic evidence regarding the activation of these complexes is preliminary. This review describes the current understanding of inflammasome signaling in rodent and human models of T. gondii infection.
Toxoplasma gondii is an obligate intracellular protozoan parasite potentially invading all nucleated cells of warm-blooded vertebrates, infecting approximately one-third of the human population worldwide. In immunocompetent individuals, a generally asymptomatic acute infection is followed by the onset of a quiescent bradyzoite parasite stage, leading to long-term chronic infections. However, in immunosuppressed individuals, the bradyzoites in the cyst become proliferating tachyzoites and can induce marked tissue damage, causing severe toxoplasmosis and distant dissemination. Previous studies have reported that multiple genetic types of T. gondii are distributed worldwide, of which types I, II and III are the classical strains in North America and Europe (1). Furthermore, T. gondii strains are much more genetically diverse in South America; however, most parasites in East Asia fall within one nonclassical lineage: Chinese 1 (2). Among laboratory mice, excluding wild mice or other intermediate hosts (3), type I parasites kill the host in the acute phase of the infection, possibly due to hyperinflammation-inducible cytokine storm and uncontrolled parasite dissemination. Type II and III strains typically exhibit less virulence and may even be avirulent.
Pioneer studies have reported that, at the early stage of T. gondii infections, dendritic cells (DCs), monocytes, and macrophages are the initial host responders. Innate immune cells use pattern recognition receptors (PRRs) to sense molecules associated with invaders, called pathogen-associated molecular patterns (PAMPs) (Figure 1). Host PRRs are involved in pathogen recognition, including membrane-bound Toll-like receptors (TLRs), cytosolic nucleotide-binding leucine-rich repeat-containing (NLRs) proteins, and the cytosolic DNA sensor absent in melanoma 2 (AIM2), which play vital physiological roles including antigen presentation, cell death, and cytokine secretion (4, 5). Myeloid differentiation primary response 88 (MyD88), a primary adapter protein, influences the function of multiple TLRs and regulates IL-12 secretion from DCs, macrophages, and neutrophils in response to a T. gondii infection, implying that TLRs are involved in microbial recognition. Thus far, numerous studies have reported that specific TLRs participate in T. gondii recognition, including TLR2, TLR4, TLR7, TLR9, TLR11, and TLR12. Among these, TLR2 and TLR4 participate in recognizing parasites by macrophages via interaction with glycosylphosphatidylinositol (GPI) anchors protein of T. gondii, whereas TLR7 and TLR9 detect the parasite’s nucleic acids (6). In contrast to GPI, profilin (PRF), another T. gondii PAMP, is mediated through recognition by TLR11 and TLR12 in DCs, since it is highly susceptible to T. gondii infections in mice deficient in UNC93B1, a chaperon for these two endosomal nucleic acid-sensing TLRs (7, 8). Concurrently, both TLR11–/– and TLR12–/– mice are highly susceptible to infection, displaying severe deficits in IL-12 and IFN-γ production. Therefore, TLR11 and TLR12 have been considered major players in the recognition of T. gondii by DCs. Furthermore, recent studies have reported that the interaction between T. gondii surface adhesins microneme protein (MIC) 1/4 with TLR2/4 N-glycans triggers IL-12 secretion in both DCs and macrophages (9, 10). Hence, further studies are required to investigate the function of TLR signaling in proinflammatory cytokine production and host protection after T. gondii infection in innate immune cells.
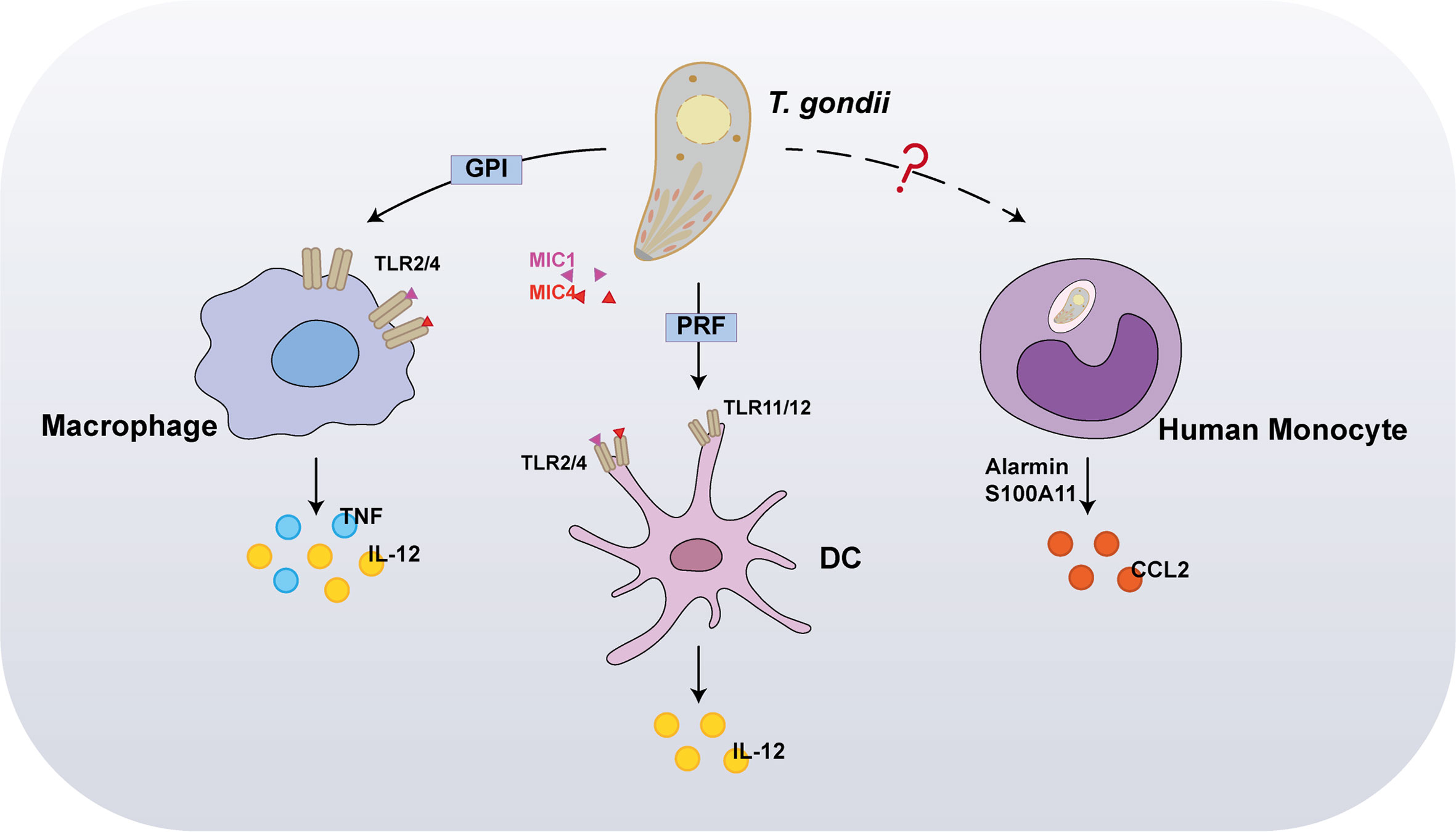
Figure 1 Recognition of Toxoplasma gondii by innate immunity. In the early stages of infection, the first host cells to respond are dendritic cells (DCs), monocytes, and macrophages. Interaction between T. gondii profilin and Toll-like receptor 11 (TLR11) is important for host production of interleukin-12 (IL-12). In addition to stimulating IL-12 production, macrophages also induce tumor necrosis factor (TNF) in response to TLR2 and TLR4-mediated detection of glycosylphosphatidylinositol (GPI). Furthermore, the interaction between T. gondii microneme protein (MIC) 1/4 with TLR2/4 N-glycans triggers IL-12 secretion in both DCs and macrophages. Alarmin S100A11, a damage-associated molecule (DAM) released from infected cells, was recently reported to stimulate chemokine CCL2 secretion in a RAGE-dependent manner against T. gondii infection on human monocytes.
In marked contrast to murine cells, in human cells, TLR11/TLR12 was reportedly do not function in T. gondii PAMPs recognition, owing to a lack of functional TLR11 and TLR12 genes. Recently, alarmin S100A11, a damage-associated molecule (DAM) released from infected cells, was recently reported to stimulate chemokine CCL2 secretion in a RAGE-dependent manner against T. gondii infection on monocytes (11); however, it remains unknown whether the innate immune sensor directly interacts with T. gondii PAMPs. Furthermore, inflammasome activation is partly required for T. gondii recognition in both murine and human cells. NLRPs are cytosolic proteins, which are poised to respond specifically to protozoan parasites. Indeed, NLRP1, NLRP3, and AIM2 form inflammasome complexes with the effector caspase in response to a T. gondii infection.
Inflammasomes are cytosolic multimeric protein complexes responding to microbial molecules or stress signals, which defend the host against pathogens. In general, sensor proteins, including NLRPs or AIM2, detect PAMPs or danger-associated molecular patterns (DAMPs) in the cytoplasm to initiate oligomerization, facilitating the recruitment of the adapter apoptosis-associated speck-like protein containing a caspase-activation and recruitment domain (CARD) (ASC) (12). The CARD of ASC or the CARD of the sensor protein recruits inflammatory caspase-1 monomers via homotypic protein domain interactions. The inflammasome platform leads to the dimerization of caspase-1, thus activating protease function, and caspase-1 subsequently triggers the release of mature interleukin-1β (IL-1β) and IL-18 (13). Moreover, activated caspase-1 cleaves gasdermin D (GSDMD) into its N-terminal form, yielding a pore-forming fragment that targets the plasma membrane, leading to pyroptosis (14). All proteins of the NLR family contain a central nucleotide-binding domain (NACHT), which comprises an NBD, helical domain 1 (HD1), winged-helix domain (WHD) and helical domain 2 (HD2); a short C-terminal leucine-rich repeat (LRR) domain; and most have a variable N-terminal domain. NLRP3, the most studied and characterized inflammasome, contains an N-terminal effector domain pyrin domain (PYD). The PYD of NLRP3 interacts with the PYD of ASC, and the CARD domain of ASC interacts with the CARD domain of effector caspases (15). The mitotic kinase NEK7 was recently reported to be required for the stimulus-dependent NLRP3 activation by building oligomerization (16). Human NLRP1 also contains a domain termed function to find domain (FIIND), which is an autolytic cleavage domain, and a C-terminal CARD domain. In contrast, mouse NLRP1 (mouse genome encodes for three NLRP1 paralogs, a-c), which does not contain a PYD domain, directly interacts with a caspase (17). Therefore, the adapter protein ASC is necessary for activating the NLRP3 inflammasome to induce caspase cleavage and is not necessary for NLRP1. Furthermore, ASC contributes to NLRP-independent inflammasome activation. AIM2 contains a nuclear localization (HIN) 200 domain responsible for cytosolic double-strands DNA (dsDNA) binding and a PYD domain that interacts with ASC and subsequently activates the AIM2 inflammasome (18) (Figure 2A).
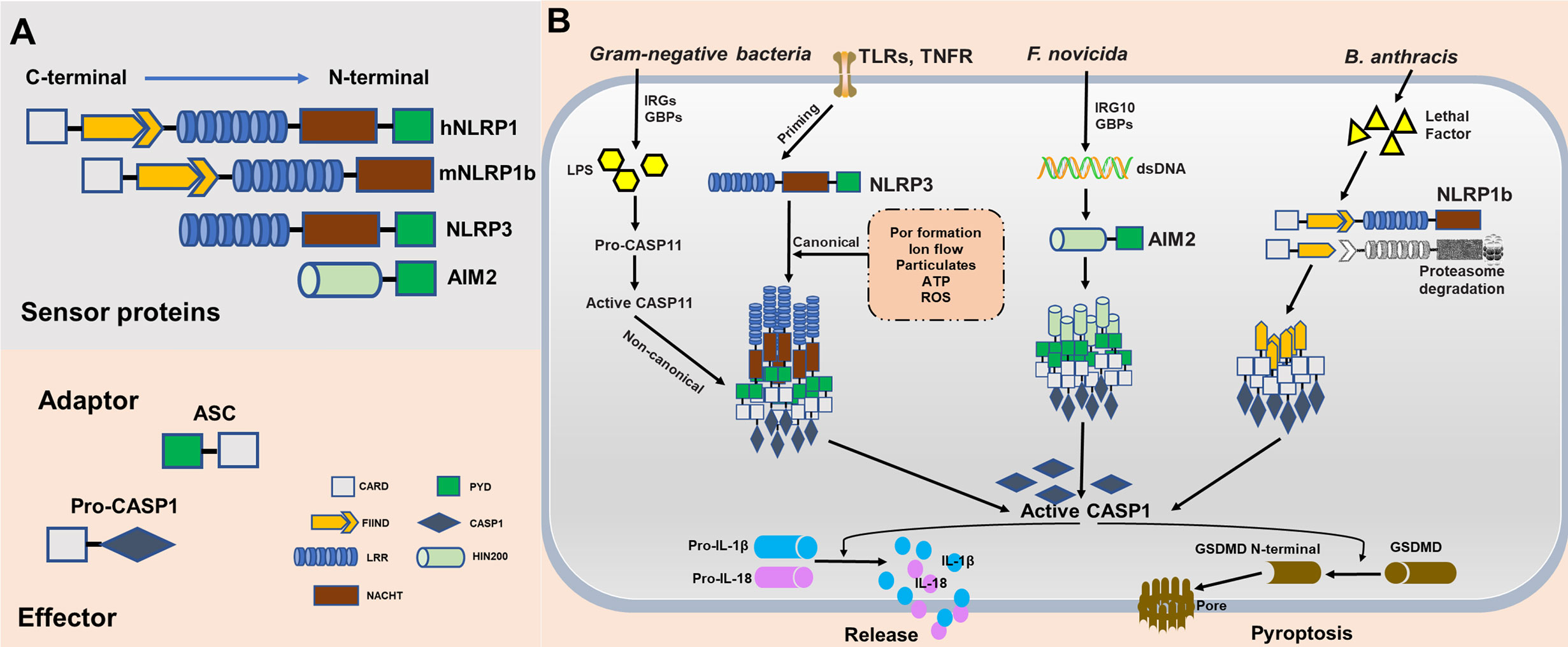
Figure 2 Molecular composition and activation of the NLRP1, NLRP3, and absent in melanoma 2 (AIM2), inflammasomes. (A) Inflammasome sensor proteins containing a leucine-rich-repeat domain (LRR), a nucleotide-binding domain (NACHT), a HIN-200 domain, a caspase-activation and recruitment domain (CARD) and/or a pyrin domain (PYD). PYD of hNLRP1, NLRP3, and AIM2 potentially recruit adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) to mediate CARD–CARD interactions with the effector caspase. As opposed to humans, mouse NLRP1b does not encode PYD. However, the CARD of mNLRP1b can interact directly with caspase without the ASC. (B) NLRP1b, NLRP3, and AIM2 are activated in response to various stimuli and/or pathogens. Activated NLRP1b, NLRP3, and AIM2 recruits and activates caspase-1 to initiate pyroptosis through gasdermin D (GSDMD) cleavage. Caspase-1 also releases IL-1β and IL-18 through pro-interleukin (IL)-1β and pro-IL-18 cleavage.
Although NLRP1, NLRP3, and AIM2 together contribute to host defense, they are distinct in terms of ligand binding, complex composition, and activation mechanisms. NLRP1 was the first reported NLR to form an inflammasome complex; however, its activation mechanism remains unclear (19). mNLRP1b is activated through direct cleavage by the lethal factor (LF) protease secreted by Bacillus anthracis. An underlying activation mechanism, termed “functional degradation”, has been proposed. The self-cleavage of NLRP1b within its FIIND(ZU5+UPA) domain is required for subsequent inflammasome activation. Pathogenic trigger, such as LF cleaves the N-terminal fragment of NLRP1b, leading to ubiquitination and degradation of the unstable fragment by the E3 ubiquitin ligase UBR2. This functional degradation event releases autoinhibition of the CARD-containing C-terminal fragment of the protein, permitting it to oligomerize and form an activated inflammasome platforms (20–23). Consistent with this model, another pathogenic enzyme named IpaH7.8, a Shigella flexneri ubiquitin ligase secreted effector, was found to induce NLRP1b degradation and activation. However, it remains unclear whether this functional degradation event triggers NLRP1b activation in T. gondii. Currently, the mechanism underlying the activation of the NLRP3 inflammasome remains unclear. Initial priming and activation are required for complete activation of NLRP3. In both human and murine cells, the initial priming step requires recognition of PAMPs by PRRs to activate NLRP3 expression, ensuring that the cell is ready to respond to NLRP3 triggers. Numerous triggers, including ion flow (potassium efflux and calcium influx), ATP, lysosomal disruption by crystalline particulates, and reactive oxygen species (24–26), have been proposed to activate canonical NLRP3 signaling, implying numerous potential mechanism of action for this inflammasome. For instance, Leishmania induces the generation of reactive oxygen species (ROS) to activate the NLRP3 inflammasome, and the major glycoconjugate present on the surface of this parasite triggers caspase-11 dependent NLRP3 inflammasome activation (27, 28). Similarly, a Neospora caninum-induced potassium efflux helps activate the NLRP3 inflammasome (29). Furthermore, NLRP3 activation by gram-negative bacteria or cytosolic LPS has been referred to as non-canonical signaling. This activation mechanism dependents on murine caspase-11, human caspase-4, and caspase-5. Stimulation by LPS and caspase-4/5/11 facilitates the cleavage of GSDMD, leading to pyroptosis (30, 31). The mechanisms of action of the AIM2 inflammasome are less clear than those of the NLRP1 and NLRP3 inflammasome. Several groups independently identify guanylate-binding proteins (GBPs) and IRGB10 as essential mediators of AIM2 activation in Francisella novicida infections (32). Mechanistically, these IFN-inducible GTPases target cytosolic pathogens and promote bacteriolysis, liberating bacterial dsDNA into the cytosol, which is then recognized by the DNA-binding protein AIM2 (33, 34) (Figure 2B).
T. gondii infects nearly all nucleated cell types, exhibiting an extremely broad host range. Compared with mice, humans and rats are markedly resistant to T. gondii infection, generally having a subclinical chronic infection. Lewis rats, however, can clear the T. gondii completely, thus inhibiting cysts formation in the host brain and muscle tissues. T. gondii resistance is partially mediated by Toxo1, a major locus of control on chromosome 10 containing Nlrp1 (35). Accordingly, SNP, whole transcriptome and functional studies have identified T. gondii as an activator of the NLRP1 inflammasome in rat macrophages (36, 37). Bone-marrow-derived macrophages (BMDMs) extract from Lewis and spontaneously hypertensive (SHR) rats, both expressing Nlrp1 allele 5, displayed pyroptosis upon stimulation by T. gondii. In contrast, T. gondii induced markedly lesser pyroptosis in Brown Norway (BN), Sprague Dawley (SD), and Fischer (CDF) rat BMDMs expressing Nlrp1 allele 1 or allele 2 (36–38). These T. gondii-resistant rats have T. gondii-sensitive macrophages, indicating a protective role of macrophage pyroptosis upon parasite infection. It should be emphasized that rat Nlrp1 alleles that confer susceptibility to T gondii are the opposite of those conferring LT susceptibility (38, 39).
The NLRP3 inflammasome is dispensable for T. gondii infection in rats, in contrast, both NLRP1 and NLRP3 are important for controlling T. gondii in mice (40, 41). Mice are more susceptible to T. gondii infection than rats. T. gondii induces undetectable or far less cell pyroptosis in mouse BMDMs than in rat BMDMs. Previous investigation of caspase-11 function focused on its LPS sensing abilities, which proved to be a powerful trigger for signal transduction in non-canonical inflammasome (42). One study demonstrated that caspase-11 functions to protect the host by enhancing inflammation during the early phase of infection (43). In mouse macrophages, activation of caspase-11 requires the lysis of vacuole-bearing pathogens by GBPs during infections with gram-negative bacteria (44). When considering type I T. gondii in mice, lethal overproduction of Th1 cytokines, including IL-1β and IL-18, may be implicated in inflammasome activation (45). Moreover, the deletion of mitogen-activated protein kinase 1 (MAPK1) and MAPK2 in type I parasites suppresses NLRP1/3 inflammasome activation, as opposed to wide-type parasites, but the virulence in mice decreased (46). The possible reason causes this discrepancy about the functions of NLRP3 inflammasome in the host against T. gondii is that ΔMAPK1 and ΔMAPK2 parasites caused high levels of IFN-β, resulting in the inhibition of inflammasome activation (47), and decreased virulence of parasite may partly attribute to the importance of type I IFN in control of toxoplasmosis as recently reported (48). Nonetheless, the mechanism underlying NLRP1 activation in response to T. gondii infection remains unclear in mice and rats. Recent studies have investigated the mechanism underlying NLRP3 activation in T. gondii-infected mouse macrophages. P2X7R, a proinflammatory receptor, was found to be involved in canonical NLRP3 inflammasome activation depending on extracellular ATP (eATP) and thus mediate T. gondii control (49) (Figure 3A). In addition to NLRP1/3, NOD2 plays a crucial role in the host adaptive response to T. gondii, regulating T cell immune responses. One of the premiere studies on Toxoplasma and NLRs reported that NOD2 provides an intrinsic signal in T cells, contributing to the generation of immunity via protective T helper cells (Th) 1 against T. gondii infection (50). Nod2-/- mice reportedly failed to mount an effective adaptive response, with impaired production of IL-2 and nuclear accumulation of the transcriptional factor c-Rel. Furthermore, it should be mentioned here that NOD2 may also bind to NLRP1 or NLRP3, thereby participating in IL-1β secretion (51–54). Accordingly, further studies are required to investigate the role of NLRP1, NLRP3, and other NLR proteins in T. gondii infection.

Figure 3 Role of the inflammasome complex in modulating immune responses in a Toxoplasma gondii infection. (A) T. gondii-mediated NLRP1 inflammasome activation in Lewis rat bone-marrow-derived macrophages (BMDMs), subsequently leads to the generation of the biologically active IL-1β and IL-18 cytokines and cell pyroptosis. T. gondii dense granule proteins GRA35, GRA42, and GRA43 are involved in NLRP1 inflammasome activation. In murine cells, multiple mechanisms including potassium K+ efflux, ATP, reactive oxygen species (ROS), and CASP11 are involved in NLRP3 activation in a T. gondii infection. Furthermore, NLRP1b is activated in parasitic infections, albeit through unclear mechanisms. (B) T. gondii infection activates both NLRP1 and NLRP3 inflammasomes in human monocytic cells. T. gondii induces a Syk-CARD9/MALT-1-NF-κB signaling pathway and activates the NLRP3 inflammasome for IL-1β release, and GRA15 directly activates NF-κB. Recent studies have reported that in human macrophages, IFN-γ impels guanylate binding protein 1 (GBP1) to mediate cell apoptosis via the absent in melanoma (AIM2) inflammasome, ASC, and caspase-8 in a T. gondii infection.
IFN-γ is the major mediator of resistance to T. gondii infection and is essential for generating various antimicrobial activities in numerous cell types, especially in murine cells. These activities involve either direct destruction of the parasitophorous vacuole membranes (PVMs, targeted by IFN-γ inducible GTPases, IRGs, and GBPs), production of NO and ROS, acidification of the intravacuolar environment, or the activation of cell death upon infection (55–57). IL-18 and IL-1β are both required for IFN-γ-mediated immune responses. Therefore, inflammasome activation is expected to potentially affect IFN-γ production and host resistance to T. gondii. Concurrently, a previous study recently reported that in the absence of TLR11, inflammasome activation is critical for maintaining CD4+ T cell-derived IFN-γ production and mouse resistance to T. gondii strain ME49 (58). However, the role of the inflammasome is dispensable and limited when TLR11 remains functional, due to TLR11-dependent IL-12 production is sufficient to generate Th1 immunity to the parasite without inflammasome dependent release of IL-18. Furthermore, Angel K. Kongsomboonvech et al. recently reported that Naïve CD8 T cell IFN-γ responses to T. gondii require the inflammasome-independent NLRP3 pathway and rhoptry proteins (ROP) 5 (59). This paper was identified that the NLRP3 regulates T cell function and underscore the need for NLRP3-activating adjuvants in vaccines targeted at inducing CD8 T cell-derived IFN-γ responses to T. gondii. Therefore, further experiments will have to investigate the crosstalk between T cell function with the inflammasome pathway.
Because TLR11 is a pseudogene and TLR12 is not expressed in humans, it remains unclear whether inflammasome activation has more pronounced effects in human cells than in murine cells. Moreover, T. gondii-induced inflammasome activation varies among different human cell types (Figure 3B). A previous study on hNLRP1 activation in toxoplasmosis initially suggested that human susceptibility to congenital toxoplasmosis was mapped to an allele in the NLRP1 gene locus, suggesting a role of the hNLRP1 inflammasome in the human immune response to T. gondii (60). In T. gondii-infected human monocytic cells, siRNA-mediated silencing of hNLRP1 directly influences cell death, parasite replication, and inflammatory cytokine production. Another study reported that active T. gondii invasion stimulated IL-1β secretion in a manner dependent on ASC and caspase-1, and on strain-specific virulence factor GRA15II (an NF−κB activator) (61). Moreover, this study shows that NLRP3 is an inflammasome sensor for T. gondii-infected primary human monocytes, resulting in rapid IL-1β release from cells (61). Potassium efflux contributed to this phenomenon, since IL-1β production was significantly reduced in monocytes after extracellular potassium treatment. Furthermore, syk, a spleen tyrosine kinase, participates in NLRP3 activation during fungal and viral infection and is critical IL-1β release from infected primary monocytes (62). Syk phosphorylation was also detected during T. gondii infection, activating the downstream PKCδ-CARD9/MALT-1 pathway and subsequently leading to intranuclear NF-κB translocation, which in turn promotes the expression of IL1B and NLRP3 (63). Notably, IL-1β production was not altered in GSDMD-knockout THP-1 cells, indicating that IL-1β, which is released from T. gondii-infected human monocytes, is independent of GSDMD cleavage and cell pyroptosis in THP-1 cells. Interestingly, unlike in T. gondii-infected primary human monocytes, T. gondii-infected human macrophages did not secrete IL-1β or harbor activated NLRP3. Fisch et al. recently provided novel insights into the mechanisms underlying inflammasome activation by type I and type II T. gondii strains in human macrophages. They found T. gondii induces an atypical apoptosis pathway involving the AIM2 and promotes apoptosis via ASC and Caspase-8. Interestingly, this phenomenon depends on IFN-γ-stimulated human GBP1 (64). In human small intestinal epithelial cells, P2X7R/NLRP3 pathway was also found to be activated and play important roles in IL-1β secretion and inhibition of T. gondii proliferation (65). Furthermore, T. gondii evades neutrophil-mediated defense by impairing the production of IL-1β and inhibiting the activation of the NLRP3 inflammasome, while facilitating the propagation of the parasite (66). It remains unknown whether the inflammasome is activated in DCs in human and murine cells.
A successful T. gondii infection requires a delicate balance between host immune responses and the parasite immune evasion, leading to both the host and parasite survival. From another perspective, T. gondii takes advantage of the NLRP3 inflammasome activation, potentially compromising host mechanisms of parasite control. A previous study reported that, T. gondii-infected human monocytes induce IL-1β production in a manner dependent on T. gondii effector protein GRA15 and NLRP3 inflammasome (61). IL-1β combined with IFN-γ robustly stimulates inducible nitric oxide synthase (iNOS) expression. Consequently, indole 2,3-dioxygenase 1 (IDO1) production is severely impaired and facilitates parasite growth in human monocytes (67). GRA7 of T. gondii was previously found to induce expression of pro-inflammatory cytokine genes including IL-1β in macrophages (68). Moreover, it was recently found that it serves as a substrate of PKCα and the recombinant N-terminal -GRA7 interacted with the PYD domain of ASC to promote anti-mycobacterial host defense (69). Whether this effector binds to ASC and induce the expression of pro-inflammatory cytokine in host cells infected with T. gondii needs further confirmed. Three other dense granule proteins (GRA35, GRA42, and GRA43) were found to be required for inducing pyroptosis and IL-1β secretion in Lewis rat BMDMs (70). It should be emphasized that T. gondii GRAs are involved in NLRP1 inflammasome activation, albeit the mechanism underlying has not been extensively studied. Furthermore, the T. gondii redox enzyme peroxiredoxin (rTgPrx) was reported to promote alternatively activated macrophage polarization, increasing IL-10 release, while simultaneously impairing caspase-1 activation and IL-1β secretion (71).
Inflammasome activation can be triggered as a protective host response in the process of pathogen infection. NLRP1, NLRP3 and AIM2 inflammasomes have been identified to participate in the immune response during T. gondii infection; however, whether other inflammasomes, e.g., pyrin or newly emerged CARD8 are involved in T. gondii infection is required further investigation. As one of the most successful intracellular parasites, Toxoplasma develops a series of strategies to fight against host defense. How the parasite fights against inflammasome pathway is still a mystery. Overall, studies on the roles of inflammasomes during T. gondii infection are still in their infancy, and their exact roles and activation mechanisms among different cell types remain unknown. Further studies are required to investigate how T. gondii is resistant to pyroptosis, and whether the GSDMD plays a role in toxoplasmosis? Although numerous factors including eATP, ROS and potassium efflux are reportedly involved in inflammasome activation, the precise T. gondii -derived molecules initiating this process remain unknown. Moreover, AIM2 function as a DNA sensor has been extensively described in virus infection. However, T. gondii is a unique intracellular pathogen different from virus, and it resides within a non-fusogenic parasitophorous vacuole (PV), providing a physical niche for the parasite to shield from intracellular cytoplasmic defense mechanisms. The IFN-γ-inducible GTPases including IRGs and GBPs are essential to disrupt the PV membrane, thereby releasing parasite DNA into the cytoplasm. Are these GTPases involved in AIM2 inflammasome pathway? How inflammasomes crosstalk with interferon-inducible GTPase-mediated host defense? To address these questions, further researches are needed in future. A comprehensive understanding of the mechanisms underlying inflammasome regulation in a T. gondii infection will help us strengthen the knowledge that how the host immune responses to this parasite.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
LY and YW designed the work. YW and JZ drafted the article and diagramming; YC, JS, and LY did critical revision of the article. All authors contributed to the article and approved the submitted version.
This work was supported in part by the National Key R&D Program of China (2017YFD0500400 to LY), National Natural Science Foundation of China (82072304, 81871671 and 81572022 to LY, 81802003 to YC), Outstanding Young Scholars Financial Support of Anhui Medical University (0113014104 to LY), and promotion plan of basic and clinical cooperative research in Anhui Medical University (2019xkjT023 to LY).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Howe DK, Sibley LD. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J Infect Dis (1995) 172(6):1561–6. doi: 10.1093/infdis/172.6.1561
2. Wang L, Cheng HW, Huang KQ, Xu YH, Li YN, Du J, et al. Toxoplasma gondii prevalence in food animals and rodents in different regions of China: isolation, genotyping and mouse pathogenicity. Parasit Vectors (2013) 6:273. doi: 10.1186/1756-3305-6-273
3. Murillo-Leon M, Muller UB, Zimmermann I, Singh S, Widdershooven P, Campos C, et al. Molecular mechanism for the control of virulent Toxoplasma gondii infections in wild-derived mice. Nat Commun (2019) 10(1):1233. doi: 10.1038/s41467-019-09200-2
4. Hayward JA, Mathur A, Ngo C, Man SM. Cytosolic Recognition of Microbes and Pathogens: Inflammasomes in Action. Microbiol Mol Biol Rev (2018) 82(4):e00015–18. doi: 10.1128/MMBR.00015-18
5. O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol (2013) 13(6):453–60. doi: 10.1038/nri3446
6. Debierre-Grockiego F, Campos MA, Azzouz N, Schmidt J, Bieker U, Resende MG, et al. Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii. J Immunol (2007) 179(2):1129–37. doi: 10.4049/jimmunol.179.2.1129
7. Koblansky AA, Jankovic D, Oh H, Hieny S, Sungnak W, Mathur R, et al. Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity (2013) 38(1):119–30. doi: 10.1016/j.immuni.2012.09.016
8. Andrade WA, Souza Mdo C, Ramos-Martinez E, Nagpal K, Dutra MS, Melo MB, et al. Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe (2013) 13(1):42–53. doi: 10.1016/j.chom.2012.12.003
9. Costa Mendonca-Natividade F, Duque Lopes C, Ricci-Azevedo R, Sardinha-Silva A, Figueiredo Pinzan C, Paiva Alegre-Maller AC, et al. Receptor Heterodimerization and Co-Receptor Engagement in TLR2 Activation Induced by MIC1 and MIC4 from Toxoplasma gondii. Int J Mol Sci (2019) 20(20):5001. doi: 10.3390/ijms20205001
10. Sardinha-Silva A, Mendonca-Natividade FC, Pinzan CF, Lopes CD, Costa DL, Jacot D, et al. The lectin-specific activity of Toxoplasma gondii microneme proteins 1 and 4 binds Toll-like receptor 2 and 4 N-glycans to regulate innate immune priming. PloS Pathog (2019) 15(6):e1007871. doi: 10.1371/journal.ppat.1007871
11. Safronova A, Araujo A, Camanzo ET, Moon TJ, Elliott MR, Beiting DP, et al. Alarmin S100A11 initiates a chemokine response to the human pathogen Toxoplasma gondii. Nat Immunol (2019) 20(1):64–72. doi: 10.1038/s41590-018-0250-8
12. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3
13. Rathinam VA, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell (2016) 165(4):792–800. doi: 10.1016/j.cell.2016.03.046
14. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
15. Clay GM, Sutterwala FS, Wilson ME. NLR proteins and parasitic disease. Immunol Res (2014) 59(1-3):142–52. doi: 10.1007/s12026-014-8544-x
16. Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature (2019) 570(7761):338–43. doi: 10.1038/s41586-019-1295-z
17. Chavarria-Smith J, Vance RE. The NLRP1 inflammasomes. Immunol Rev (2015) 265(1):22–34. doi: 10.1111/imr.12283
18. Marim FM, Franco MMC, Gomes MTR, Miraglia MC, Giambartolomei GH, Oliveira SC. The role of NLRP3 and AIM2 in inflammasome activation during Brucella abortus infection. Semin Immunopathol (2017) 39(2):215–23. doi: 10.1007/s00281-016-0581-1
19. Christgen S, Place DE, Kanneganti TD. Toward targeting inflammasomes: insights into their regulation and activation. Cell Res (2020) 30(4):315–27. doi: 10.1038/s41422-020-0295-8
20. Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science (2019) 364(6435):eaau1330. doi: 10.1126/science.aau1330
21. Xu H, Shi J, Gao H, Liu Y, Yang Z, Shao F, et al. The N-end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. EMBO J (2019) 38(13):e101996. doi: 10.15252/embj.2019101996
22. Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, et al. N-terminal degradation activates the NLRP1B inflammasome. Science (2019) 364(6435):82–5. doi: 10.1126/science.aau1208
23. Mitchell PS, Sandstrom A, Vance RE. The NLRP1 inflammasome: new mechanistic insights and unresolved mysteries. Curr Opin Immunol (2019) 60:37–45. doi: 10.1016/j.coi.2019.04.015
24. Wang L, Negro R, Wu H. TRPM2, linking oxidative stress and Ca(2+) permeation to NLRP3 inflammasome activation. Curr Opin Immunol (2020) 62:131–5. doi: 10.1016/j.coi.2020.01.005
25. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science (2008) 320(5876):674–7. doi: 10.1126/science.1156995
26. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature (2016) 530(7590):354–7. doi: 10.1038/nature16959
27. Zamboni DS, Sacks DL. Inflammasomes and Leishmania: in good times or bad, in sickness or in health. Curr Opin Microbiol (2019) 52:70–6. doi: 10.1016/j.mib.2019.05.005
28. de Carvalho RVH, Andrade WA, Lima-Junior DS, Dilucca M, de Oliveira CV, Wang K, et al. Leishmania Lipophosphoglycan Triggers Caspase-11 and the Non-canonical Activation of the NLRP3 Inflammasome. Cell Rep (2019) 26(2):429–37 e5. doi: 10.1016/j.celrep.2018.12.047
29. Wang X, Gong P, Zhang X, Li S, Lu X, Zhao C, et al. NLRP3 Inflammasome Participates in Host Response to Neospora caninum Infection. Front Immunol (2018) 9:1791:1791. doi: 10.3389/fimmu.2018.01791
30. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
31. Yi YS. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology (2017) 152(2):207–17. doi: 10.1111/imm.12787
32. Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, et al. IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell (2016) 167(2):382–96 e17. doi: 10.1016/j.cell.2016.09.012
33. Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Ruhl S, et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol (2015) 16(5):476–84. doi: 10.1038/ni.3119
34. Man SM, Place DE, Kuriakose T, Kanneganti TD. Interferon-inducible guanylate-binding proteins at the interface of cell-autonomous immunity and inflammasome activation. J Leukoc Biol (2017) 101(1):143–50. doi: 10.1189/jlb.4MR0516-223R
35. Cavailles P, Sergent V, Bisanz C, Papapietro O, Colacios C, Mas M, et al. The rat Toxo1 locus directs toxoplasmosis outcome and controls parasite proliferation and spreading by macrophage-dependent mechanisms. Proc Natl Acad Sci U S A (2006) 103(3):744–9. doi: 10.1073/pnas.0506643103
36. Cavailles P, Flori P, Papapietro O, Bisanz C, Lagrange D, Pilloux L, et al. A highly conserved Toxo1 haplotype directs resistance to toxoplasmosis and its associated caspase-1 dependent killing of parasite and host macrophage. PloS Pathog (2014) 10(4):e1004005. doi: 10.1371/journal.ppat.1004005
37. Cirelli KM, Gorfu G, Hassan MA, Printz M, Crown D, Leppla SH, et al. Inflammasome sensor NLRP1 controls rat macrophage susceptibility to Toxoplasma gondii. PloS Pathog (2014) 10(3):e1003927. doi: 10.1371/journal.ppat.1003927
38. Newman ZL, Printz MP, Liu S, Crown D, Breen L, Miller-Randolph S, et al. Susceptibility to anthrax lethal toxin-induced rat death is controlled by a single chromosome 10 locus that includes rNlrp1. PloS Pathog (2010) 6(5):e1000906. doi: 10.1371/journal.ppat.1000906
39. Taabazuing CY, Griswold AR, Bachovchin DA. The NLRP1 and CARD8 inflammasomes. Immunol Rev (2020) 297(1):13–25. doi: 10.1111/imr.12884
40. Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, Koller BH, et al. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. mBio (2014) 5(1):e01117–13. doi: 10.1128/mBio.01117-13
41. Ewald SE, Chavarria-Smith J, Boothroyd JC. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect Immun (2014) 82(1):460–8. doi: 10.1128/IAI.01170-13
42. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
43. Coutermarsh-Ott SL, Doran JT, Campbell C, Williams TM, Lindsay DS, Allen IC. Caspase-11 Modulates Inflammation and Attenuates Toxoplasma gondii Pathogenesis. Mediators Inflammation (2016) 2016:9848263. doi: 10.1155/2016/9848263
44. Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature (2014) 509(7500):366–70. doi: 10.1038/nature13157
45. Mordue DG, Monroy F, La Regina M, Dinarello CA, Sibley LD. Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J Immunol (2001) 167(8):4574–84. doi: 10.4049/jimmunol.167.8.4574
46. Wang S, Wang Z, Gu Y, Li Z, Li Z, Wei F, et al. Toxoplasma gondii mitogen-activated protein kinases are associated with inflammasome activation in infected mice. Microbes Infect (2016) 18(11):696–700. doi: 10.1016/j.micinf.2016.07.004
47. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity (2011) 34(2):213–23. doi: 10.1016/j.immuni.2011.02.006
48. Matta SK, Olias P, Huang Z, Wang Q, Park E, Yokoyama WM, et al. Toxoplasma gondii effector TgIST blocks type I interferon signaling to promote infection. Proc Natl Acad Sci U S A (2019) 116(35):17480–91. doi: 10.1073/pnas.1904637116
49. Lees MP, Fuller SJ, McLeod R, Boulter NR, Miller CM, Zakrzewski AM, et al. P2X7 receptor-mediated killing of an intracellular parasite, Toxoplasma gondii, by human and murine macrophages. J Immunol (2010) 184(12):7040–6. doi: 10.4049/jimmunol.1000012
50. Shaw MH, Reimer T, Sanchez-Valdepenas C, Warner N, Kim YG, Fresno M, et al. T cell-intrinsic role of Nod2 in promoting type 1 immunity to Toxoplasma gondii. Nat Immunol (2009) 10(12):1267–74. doi: 10.1038/ni.1816
51. Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci U S A (2008) 105(22):7803–8. doi: 10.1073/pnas.0802726105
52. Kim YG, Park JH, Shaw MH, Franchi L, Inohara N, Nunez G. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity (2008) 28(2):246–57. doi: 10.1016/j.immuni.2007.12.012
53. Pan Q, Mathison J, Fearns C, Kravchenko VV, Da Silva Correia J, Hoffman HM, et al. MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. J Leukoc Biol (2007) 82(1):177–83. doi: 10.1189/jlb.1006627
54. Strober W, Watanabe T. NOD2, an intracellular innate immune sensor involved in host defense and Crohn’s disease. Mucosal Immunol (2011) 4(5):484–95. doi: 10.1038/mi.2011.29
55. Hakimi MA, Olias P, Sibley LD. Toxoplasma Effectors Targeting Host Signaling and Transcription. Clin Microbiol Rev (2017) 30(3):615–45. doi: 10.1128/CMR.00005-17
56. Yamamoto M, Okuyama M, Ma JS, Kimura T, Kamiyama N, Saiga H, et al. A cluster of interferon-gamma-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity (2012) 37(2):302–13. doi: 10.1016/j.immuni.2012.06.009
57. Sturge CR, Benson A, Raetz M, Wilhelm CL, Mirpuri J, Vitetta ES, et al. TLR-independent neutrophil-derived IFN-gamma is important for host resistance to intracellular pathogens. Proc Natl Acad Sci U S A (2013) 110(26):10711–6. doi: 10.1073/pnas.1307868110
58. Lopez-Yglesias AH, Camanzo E, Martin AT, Araujo AM, Yarovinsky F. TLR11-independent inflammasome activation is critical for CD4+ T cell-derived IFN-gamma production and host resistance to Toxoplasma gondii. PloS Pathog (2019) 15(6):e1007872. doi: 10.1371/journal.ppat.1007872
59. Kongsomboonvech AK, Rodriguez F, Diep AL, Justice BM, Castallanos BE, Camejo A, et al. Naive CD8 T cell IFNgamma responses to a vacuolar antigen are regulated by an inflammasome-independent NLRP3 pathway and Toxoplasma gondii ROP5. PloS Pathog (2020) 16(8):e1008327. doi: 10.1371/journal.ppat.1008327
60. Witola WH, Mui E, Hargrave A, Liu S, Hypolite M, Montpetit A, et al. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect Immun (2011) 79(2):756–66. doi: 10.1128/IAI.00898-10
61. Gov L, Karimzadeh A, Ueno N, Lodoen MB. Human innate immunity to Toxoplasma gondii is mediated by host caspase-1 and ASC and parasite GRA15. mBio (2013) 4(4):e00255–13. doi: 10.1128/mBio.00255-13
62. Callaway JB, Smith SA, McKinnon KP, de Silva AM, Crowe JE Jr., Ting JP. Spleen Tyrosine Kinase (Syk) Mediates IL-1beta Induction by Primary Human Monocytes during Antibody-enhanced Dengue Virus Infection. J Biol Chem (2015) 290(28):17306–20. doi: 10.1074/jbc.M115.664136
63. Pandori WJ, Lima TS, Mallya S, Kao TH, Gov L, Lodoen MB. Toxoplasma gondii activates a Syk-CARD9-NF-kappaB signaling axis and gasdermin D-independent release of IL-1beta during infection of primary human monocytes. PloS Pathog (2019) 15(8):e1007923. doi: 10.1371/journal.ppat.1007923
64. Fisch D, Bando H, Clough B, Hornung V, Yamamoto M, Shenoy AR, et al. Human GBP1 is a microbe-specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J (2019) 38(13):e100926. doi: 10.15252/embj.2018100926
65. Quan JH, Huang R, Wang Z, Huang S, Choi IW, Zhou Y, et al. P2X7 receptor mediates NLRP3-dependent IL-1beta secretion and parasite proliferation in Toxoplasma gondii-infected human small intestinal epithelial cells. Parasit Vectors (2018) 11(1):1. doi: 10.1186/s13071-017-2573-y
66. Lima TS, Gov L, Lodoen MB. Evasion of Human Neutrophil-Mediated Host Defense during Toxoplasma gondii Infection. mBio (2018) 9(1):e02027–17. doi: 10.1128/mBio.02027-17
67. Bando H, Lee Y, Sakaguchi N, Pradipta A, Ma JS, Tanaka S, et al. Inducible Nitric Oxide Synthase Is a Key Host Factor for Toxoplasma GRA15-Dependent Disruption of the Gamma Interferon-Induced Antiparasitic Human Response. mBio (2018) 9(5):e01738–18. doi: 10.1128/mBio.01738-18
68. Yang CS, Yuk JM, Lee YH, Jo EK. Toxoplasma gondii GRA7-Induced TRAF6 Activation Contributes to Host Protective Immunity. Infect Immun (2016) 84(1):339–50. doi: 10.1128/IAI.00734-15
69. Koh HJ, Kim YR, Kim JS, Yun JS, Jang K, Yang CS. Toxoplasma gondii GRA7-Targeted ASC and PLD1 Promote Antibacterial Host Defense via PKCalpha. PloS Pathog (2017) 13(1):e1006126. doi: 10.1371/journal.ppat.1006126
70. Wang Y, Cirelli KM, Barros PDC, Sangare LO, Butty V, Hassan MA, et al. Three Toxoplasma gondii Dense Granule Proteins Are Required for Induction of Lewis Rat Macrophage Pyroptosis. mBio (2019) 10(1):e02388–18. doi: 10.1128/mBio.02388-18
Keywords: Toxoplasma gondii, NLRP1, NLRP3, AIM2, inflammasome, infection
Citation: Wang Y, Zhu J, Cao Y, Shen J and Yu L (2020) Insight Into Inflammasome Signaling: Implications for Toxoplasma gondii Infection. Front. Immunol. 11:583193. doi: 10.3389/fimmu.2020.583193
Received: 04 August 2020; Accepted: 17 November 2020;
Published: 16 December 2020.
Edited by:
Uday Kishore, Brunel University London, United KingdomReviewed by:
Li-Chung Hsu, National Taiwan University, TaiwanCopyright © 2020 Wang, Zhu, Cao, Shen and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Yu, bGlseXl1MzNAMTI2LmNvbQ==; Jilong Shen, c2hlbmppbG9uZzUzQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.