 Morgan Brisse
Morgan Brisse Sophia M. Vrba
Sophia M. Vrba Natalie Kirk
Natalie Kirk Yuying Liang
Yuying Liang Hinh Ly
Hinh Ly- 1Biochemistry, Molecular Biology, and Biophysics Graduate Program, University of Minnesota Twin Cities, St. Paul, MN, United States
- 2Department of Veterinary & Biomedical Sciences, University of Minnesota Twin Cities, St. Paul, MN, United States
- 3Comparative Molecular Biosciences Graduate Program, University of Minnesota Twin Cities, St. Paul, MN, United States
Despite the success of vaccination to greatly mitigate or eliminate threat of diseases caused by pathogens, there are still known diseases and emerging pathogens for which the development of successful vaccines against them is inherently difficult. In addition, vaccine development for people with compromised immunity and other pre-existing medical conditions has remained a major challenge. Besides the traditional inactivated or live attenuated, virus-vectored and subunit vaccines, emerging non-viral vaccine technologies, such as viral-like particle and nanoparticle vaccines, DNA/RNA vaccines, and rational vaccine design, offer innovative approaches to address existing challenges of vaccine development. They have also significantly advanced our understanding of vaccine immunology and can guide future vaccine development for many diseases, including rapidly emerging infectious diseases, such as COVID-19, and diseases that have not traditionally been addressed by vaccination, such as cancers and substance abuse. This review provides an integrative discussion of new non-viral vaccine development technologies and their use to address the most fundamental and ongoing challenges of vaccine development.
Introduction
Beginning with the discovery of cowpox inoculation that can protect humans against smallpox infection by Edward Jenner in the late 18th century, vaccination has become an important means to prevent disease. Despite the success of vaccination in the eradication or control of some major pathogens, several challenges remain in vaccine development and administration. Several widespread infectious diseases such as HIV, tuberculosis, and influenza continue to pose great challenges for fully protective vaccination. Emerging and reemerging pathogens present a pressing need for expediting vaccine development and approval as a rapid response to epidemics, such as the current COVID-19 global pandemic caused by the SARS-CoV-2 virus. The advantages and disadvantages of the various vaccine platforms can make the choice for preferred platform(s) to use for vaccine development during a pandemic complicated. The traditional methods to produce a vaccine, such as live attenuated and inactivated vaccines or protein subunit vaccines have their advantages and disadvantages, which have been extensively reviewed elsewhere (1–3). Briefly, live attenuated vaccines present the risk of reversion to a highly pathogenic form while inactivated vaccines may not be sufficiently immunogenic or in some cases can lead to an enhanced disease pathology (3). Additionally, most pandemic vaccines have to be clinically tested during an active outbreak in order to obtain sufficient safety and efficacy data, thereby limiting the number of vaccine candidates that can be deployed to save life during an emergency situation.
Less conventional approaches to vaccinology include the non-viral vaccine technologies that are the topic of this review, as well as viral vector platforms. Viral vector vaccines rely on antigen delivered on an unrelated, non-pathogenic viral backbone. This technology was developed almost forty years ago using a vaccinia virus vector expressing the hepatitis B surface antigen (HBsAg), which provided protective immunity to chimpanzees exposed to hepatitis B (4, 5). Since then, viral vectors have been used successfully in many veterinary species (6–12), although only a single viral vector has been licensed for human vaccination (rVSV-ZEBOV for Ebola virus) (13). A number of viruses have been developed as vectors for vaccine development, including poxviruses, adenoviruses, herpesviruses, arenaviruses, retroviruses, paramyxoviruses, and flaviviruses, among others (14–16). The main advantage of viral vectors over traditional vaccines is their ability to evoke a robust adaptive immune response in the absence of an adjuvant (17). However, the tradeoff for enhanced immunogenicity is the concern for potential reversion of the attenuated viral vector to virulence, especially when using a replication-competent vector (18). Replication-defective and single-cycle viral vectors are attractive alternatives that have an increased safety profile and, in some cases, are still able to elicit a strong immune response (19, 20). More details about the known viral vectors and their recent advances in vaccine development will be discussed in our forthcoming review article (Vrba, S.M., et al., in preparation).
Other fundamental challenges toward successful vaccination include the ever-changing and highly divergent nature of some viruses that allow for the potential to escape vaccine coverage, pre-existing immunity of the vaccinated populations, and pre-existing medical conditions that can prevent vaccines from being fully effective and safe. Vaccination could also provide an enticing alternative therapy against diseases such as cancers and substance abuse. However, the efficacy of these vaccines is limited by the disease complexity and the lack of a more complete understanding of protective immunity in these medical conditions. The relative contribution and balance of the different arms of host immunity, i.e., antibodies and cell-mediated responses, toward protection without adverse effects remains a challenging issue that needs to be addressed for individual disease (21). Furthermore, the immune response to vaccination can be influenced by numerous factors such as gender, age, co-existing medical conditions, genetic variations, and lifestyle (22). While vaccines have traditionally been delivered as inactivated or attenuated preparations, recent developments of non-viral vaccine systems offer potential additional solutions to meet the new challenges of vaccine development, especially during epidemic or pandemic situations. This article focuses on new non-viral vaccine development technologies and their implications for combating on-going and emerging communicable and non-communicable diseases.
Emerging Technologies in Non-Viral Vaccine Development
Virus-Like Particle and Nanoparticle Subunit Vaccines
Subunit vaccines deliver antigens as purified proteins, which confer the advantage of enhanced safety and scalability compared to whole-pathogen vaccines due to the lack of the requirement for the expression of all viral components and the ability to express and purify any particular antigens of interest in large quantity. A disadvantage of subunit vaccines is that they are generally less immunogenic in nature and therefore require adjuvants and repeated vaccination doses (2). Several approaches have been used to increase the immunogenicity and stability of subunit vaccines, such as virus-like particle (VLP) vaccines and nanoparticle (NP) vaccines.
VLP vaccines use platforms capable of producing particles that mimic the structure of authentic viruses. VLP vaccines can be produced by expressing antigenic proteins in a eukaryotic or prokaryotic system, resulting in the formation of particles with an inherent ability of the antigenic proteins to self-assemble (23) (Figure 1). Alternatively, VLP vaccines can also be made by producing blank VLP templates and then chemically linking antigenic peptides onto the pre-formed particles (23). Because these VLPs do not contain a viral genome, they are unable to replicate in cells and therefore have an improved safety profile compared to live viral vaccines (24). Yet, VLP vaccines can often fully activate immune systems of the vaccinated individual. VLPs are taken up by dendritic cells, where they are processed and presented on MHC class I and II molecules to activate the adaptive immune response. Subsequent stimulation of CD8+ T cells and CD4+ T helper cells leads to activation of cell mediated responses and B cells (and antibody production), respectively (23, 25–29). As a result, VLP vaccines are considered highly immunogenic and can stimulate robust cellular and humoral immune responses due to their highly repetitive display of antigenic epitopes (30, 31). A number of VLP vaccine candidates are now clinically applicable with some notable examples including the hepatitis B vaccine (HBV) Engerix (32), the human papillomavirus vaccine (HPV) Cervarix (33) from GlaxoSmithKline (GSK), the HBV vaccine Recombivax® (34), and the human papillomavirus (HPV) vaccine Gardasil® (35) from Merck & Co, Inc. VLP vaccines that are currently in clinical trials include vaccines for malaria (36, 37), influenza (38), rotavirus (39, 40), tuberculosis (41), Zika virus (42), and HIV (43, 44). Efforts to further increase the immunogenicity of VLP vaccines include optimizing antigen design and production platforms (of primarily bacterial origin) (45).
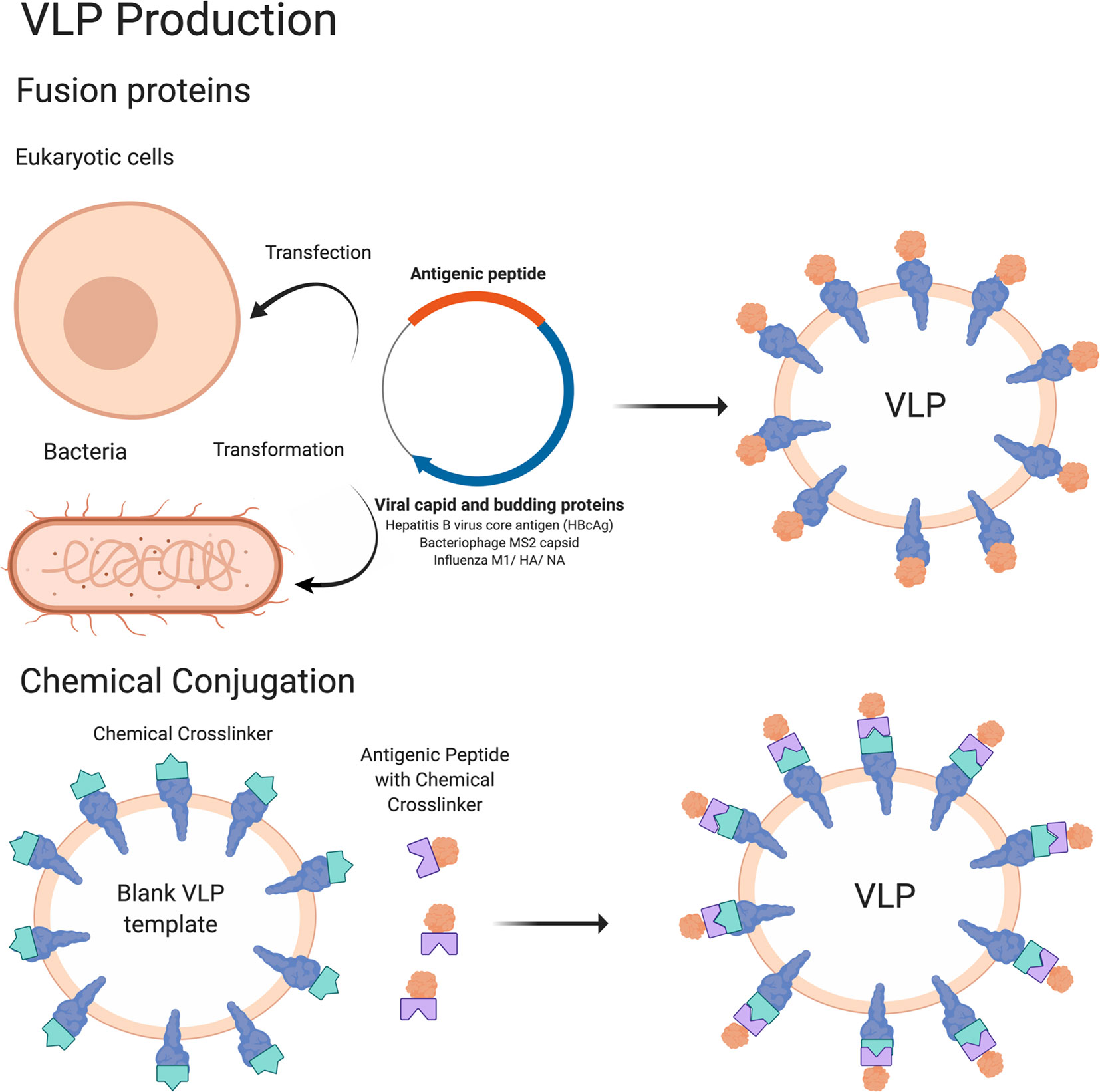
Figure 1 Schematic of VLP vaccine production. The methodology to produce VLP vaccines is summarized in this cartoon. In brief, VLP vaccines are produced by transfecting eukaryotic cells or transforming bacterial cells with a DNA plasmid encoding an antigenic peptide attached to a viral capsid and/or other protein that is sufficient to form VLPs. The antigenic peptide is present on the outside of the VLP which becomes available for interaction with the immune system. Antigens conjugated with a chemical crosslinker can also be attached to VLPs containing external proteins conjugated to a complementary chemical crosslinker, which will result in antigens being linked to the VLP and being presented on the outside edges. Figure created using BioRender software.
NP vaccines are produced by chemically crosslinking protein antigens and carrier molecules to increase immunogenicity and decrease degradation of the antigens (45). These carriers can be organic (primarily lipid-based) or in-organic (primarily polymeric or metal based) (Figure 2) (46–49). More recently, self-assembling protein NPs, which consist of oligomers of monomeric protein, have been found in some cases to also provide the benefit normally afforded by a carrier (47, 50). NPs have similarly high rates of stability as VLPs, but they do not stimulate the innate immune response to the same extent as VLPs. However, NPs are simpler in design than VLPs by lacking the multiple protein components of VLPs, which further decrease their cost of production and increase their reproducibility and safety. The challenges associated with decreased immunogenicity of NP vaccines as compared to VLP vaccines can be partly addressed by adjusting the carrier to the desired antigen, based on factors such as size, surface charge, shape and hydrophobicity (46, 47, 50). Additionally, carriers can be used to directly target NPs to immune cells and to increase cross-presentation by antigen-presenting cells (APCs) (46, 47).
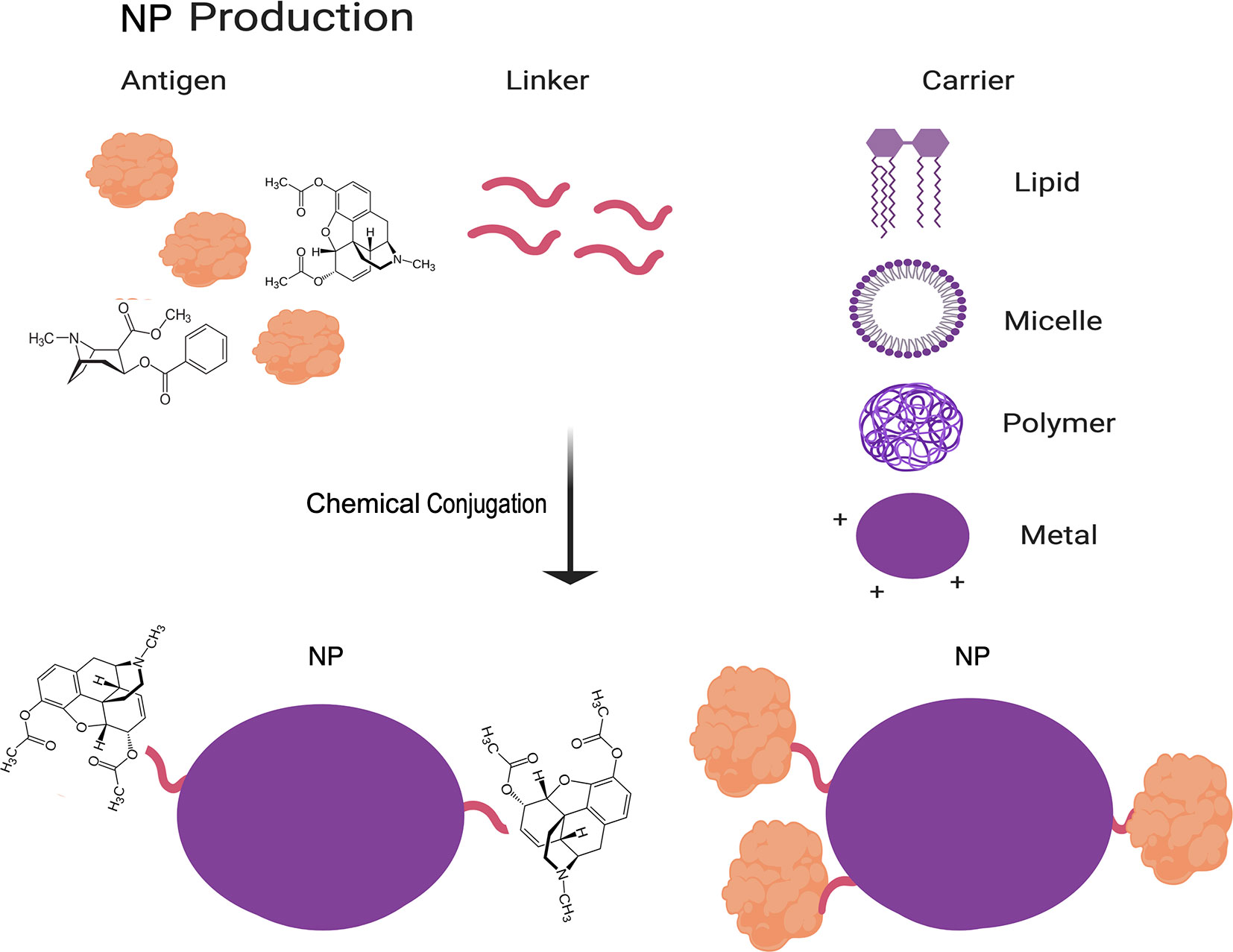
Figure 2 Schematic of NP vaccine production. The methodology to produce NP vaccines is summarized in this cartoon. In brief, NP vaccines are produced by assembling a complex of antigens, a linker molecule, and a carrier molecule by chemical conjugation. Figure created using BioRender software.
DNA and RNA Vaccines
Another promising area of vaccine development includes vaccines that are based on nucleic acids: DNA or RNA vaccines. These vaccines have gained popularity due to their cost-effectiveness, ease of design and production, attractive biosafety profile, and, in the case of DNA, stability. Nucleic acid vaccines have gained particular attention for their potential to rapidly produce vaccines against emerging infectious diseases such as those currently being tested against SARS-CoV-2, the causative agent of COVID-19, which will be discussed in some detail below.
The immunogenic and protective efficacy of DNA vaccines have been demonstrated repeatedly in vitro and in small animal models, and a limited number of DNA vaccines have been approved for veterinary use (51). However, DNA vaccines tend to induce poor immune responses in humans and other large animal models (52). One possible explanation may be that intramuscular injection, which has been the most studied route of DNA vaccine administration in humans, tends to elicit mostly cell-mediated immune responses (53), which is likely due to significantly lower APC populations residing in muscles and antigen presentation dominated by MHC I (51). Alternatively, DNA vaccines can be coated with gold NPs and administered intradermally by a gene gun (Figure 3). While preliminary data suggest that this method may increase humoral responses to DNA-based vaccines (51), it is limited by its low dose per administration (54). In vivo electroporation (permeabilization of the skin by an electric current to allow plasmid DNA uptake) has thus far been shown to have the highest immunogenicity in multiple small animal models (51, 54) and has been tested in two phase I clinical trials for HIV vaccination with some promising results. In the first clinical trial, the immune system was primed with a DNA vaccine encoding the IL12 gene followed by a boost dose with the recombinant VSV-based HIV vaccine (55). The second trial evaluated the cellular immunity induced by HIV DNA vaccines through intramuscular injection administered by electroporation (56). Other efforts are being undertaken to increase the immunogenicity of DNA vaccines such as codon optimization, optimal promoter usage and epigenetic design, generating nanocarrier plasmids to increase stability and plasmids fused to proteins that specifically target APCs, adjuvant use (which will be discussed in some detail below) and short hairpin RNA (shRNA) targeting of host cells that decrease immunogenicity to DNA vaccines. These approaches have been extensively reviewed elsewhere (51, 54).
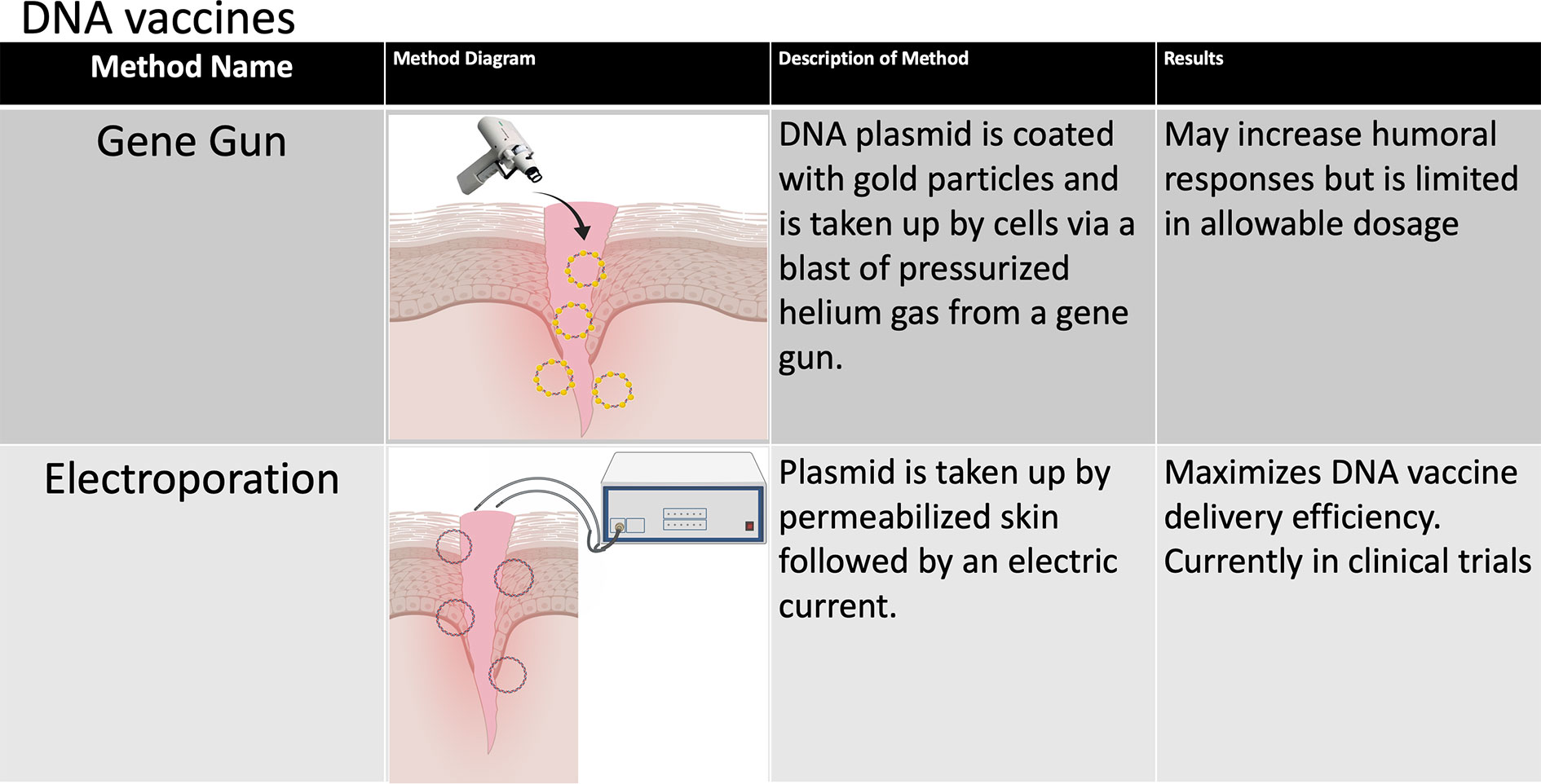
Figure 3 Methods of improving DNA vaccines. The various methods that have been developed to improve the stability and immunogenicity of DNA vaccines are summarized in this chart. A number of design and delivery mechanisms have contributed to improving the performance of nucleic acid vaccines, such as methods of clinical delivery, genetic engineering, and linking nucleic acid vaccines to cells or biomolecules. Figure created using BioRender software.
A recent development involves the successful use of mRNA as a protective vaccine. While mRNA was originally found to be viable for in vivo gene transfer in the early 1990’s, the development of mRNA vaccines was initiated much later due to the inherent instability of mRNA compared to DNA (57). The efficacy of mRNA vaccines can be increased by several factors, such as ensuring mRNA purity, adding 5’ Kozak and cap sequences, 3’ poly-A sequences and modified nucleosides to increase mRNA stability and decrease detection by the receptors of innate immune cells, codon optimization, introduction by intramuscular, and intradermal injection to reduce RNA degradation, and by generating thermostable mRNA (57–59) (Figures 4, 5). Methods to encapsulate RNA have also been explored to increase the stability and immunogenicity of RNA vaccines, as has been used with exosome encapsulated RNA (60) and RNA-transfected dendritic cells (61, 62). When fully optimized, RNA vaccines may have an immunogenic advantage over DNA vaccines due to the presence of multiple cellular pathways that activate innate immunity in response to foreign RNA such as the toll-like receptors (TLRs) and RIG-I-like receptors (RLRs) (63, 64).
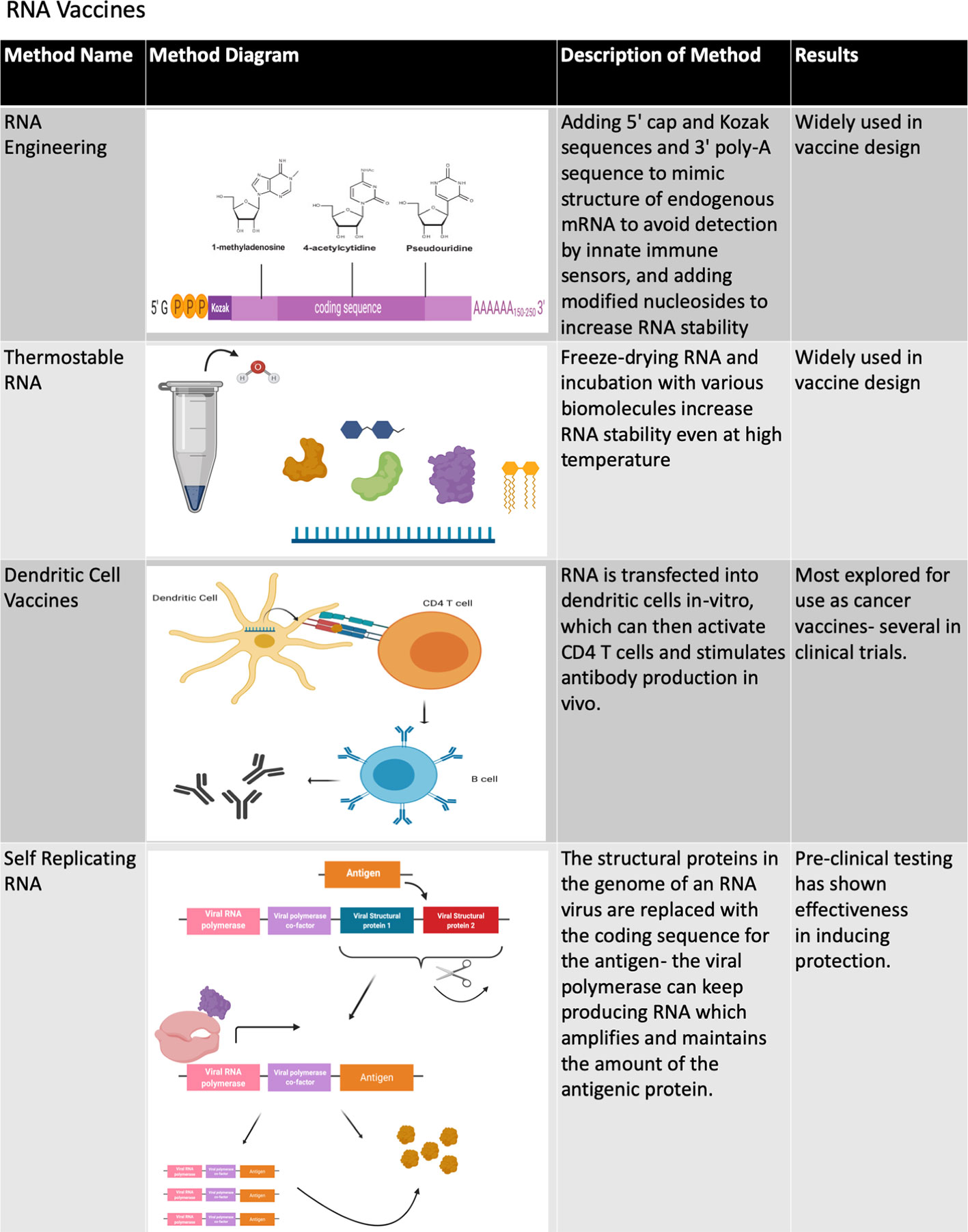
Figure 4 Methods of improving RNA vaccines. The various methods that have been developed to improve the stability and immunogenicity of RNA vaccines are summarized in this chart. A number of design and delivery mechanisms have contributed to improving the performance of nucleic acid vaccines, such as methods of clinical delivery, genetic engineering, and linking nucleic acid vaccines to cells or biomolecules. Figure created using BioRender software.
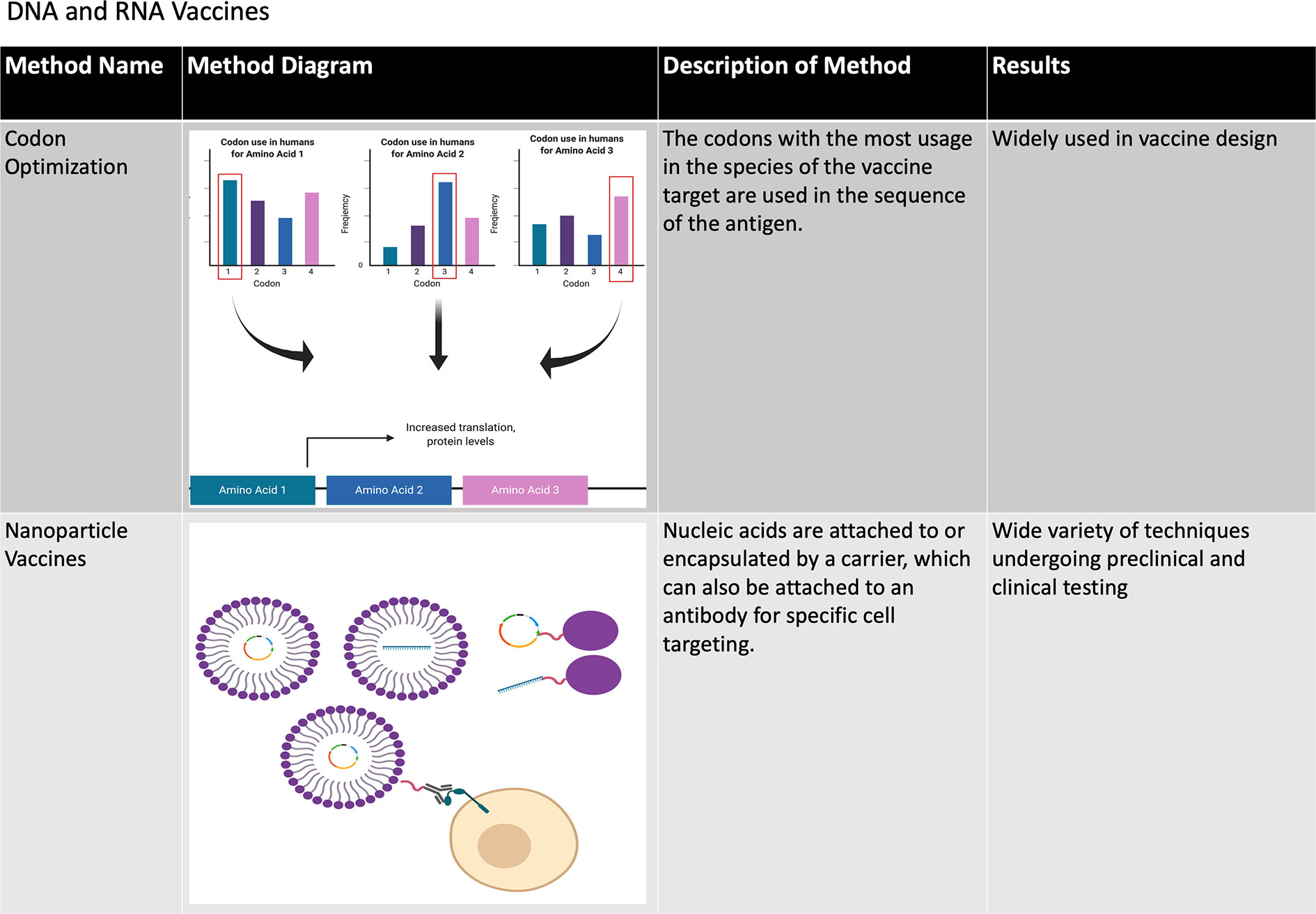
Figure 5 Methods of improving DNA and RNA vaccines. The various methods that have been developed to improve the stability and immunogenicity of both DNA and RNA vaccines are summarized in this chart. A number of design and delivery mechanisms have contributed to improving the performance of nucleic acid vaccines, such as methods of clinical delivery, genetic engineering, and linking nucleic acid vaccines to cells or biomolecules. Figure created using BioRender software.
In addition to the aforementioned non-replicating RNA, RNA vaccines can include self-replicating or self-amplifying RNA molecules that are normally based on positive-strand RNA viruses of which the structural genes are replaced by antigens (57, 58) (Figure 4). One study comparing the efficacy of conventional mRNA versus self-amplifying RNA found that both were effective in protecting mice against influenza infection, but that self-amplifying RNA elicited protection at a much lower RNA dose and induced a delayed yet longer-lasting antigen expression (65). Self-replicating RNA transfected into dendritic cells (66) has been shown to induce an immune response in vivo (67). RNA vaccines have been used in a number of studies in animal models (68) and have recently completed phase 1 clinical trials for rabies (57, 69) and influenza (68, 70). Both trials had similar safety profiles with most patients experiencing mild to moderate reactions to administration and a few patients experiencing more severe reactions. Both vaccines also demonstrated immunogenicity through neutralizing antibody levels, though antibody levels with the rabies vaccine were more highly dependent on dose and route, with needle-free intradermal dose able to sustain neutralizing antibody levels in half of the number of vaccinated individuals one year after injection but not with those receiving intramuscular or intradermal injections (57, 69). Additionally, phase 1 clinical trials are currently underway to test the self-replicating RNA vaccines for HIV and Zika virus (68).
Several challenges face the development of DNA/RNA vaccines. First, while DNA and RNA vaccines may avoid the safety concerns due to microorganism-based vaccine formulations, they have safety concerns of their own. While an early study suggested that DNA vaccination might result in some instances of random chromosomal integration, it was determined that this occurred with a significantly lower frequency than random genetic mutations (71). However, a subsequent study did not observe chromosomal integration to occur following DNA vaccination (51). The possibility of introducing unwanted bacterial DNA elements (such as antibiotic resistance genes to the gut microbiome) has been raised as a safety concern for DNA vaccination, but as of yet it has not been proven (51). As such, regulatory guidelines have been put in place for new DNA vaccine clinical trials in the United States and Europe (72). Vaccine formulation based on mRNA has the advantage of being produced in cell-free systems that can eliminate the concern of bacterial contamination and also lack the potential for chromosomal integration and long-lasting expression (57). While the World Health Organization (WHO) has recently classified mRNA vaccines as its own therapeutic class (73), similar regulations have not yet been developed due to the more limited testing of mRNA vaccines in humans.
It has been found that DNA vaccination primarily induces antigen expression at the site of administration with significantly lower levels being observed elsewhere (51), which may partly explain its poor immunogenicity. While less is known about the levels of on- and/or off-target expressions seen with RNA vaccination, they are presumed to be generally lower than DNA vaccine due to the decreased stability of RNA. However, off-target antigen expression may be relatively minor in shaping the immune response, as the route and mode of DNA/RNA vaccine delivery can markedly alter vaccine immunity, but the mechanism is yet to be fully understood. Generally, intramuscular or intradermal injection is used in animal models and human volunteers to elicit protection against infectious disease to maximize delivery to APCs, while intraperitoneal or intravenous injection has been used in selected animal models to induce systemic expression in therapeutic models, such as cancer vaccination (57). These findings implicating localized dosage routes as most effective for eliciting immunity from nucleic acid vaccines may help explain why gene gun and electroporation have been found to be the most effective routes for DNA vaccine administration. The most effective dosage routes may also be similar for RNA vaccines, as intradermal and intramuscular injection have repeatedly been found to be the most effective delivery routes for RNA vaccines, and needle-free delivery systems may also be more effective than injections (57). Interestingly, immunity can still result from injection of naked RNA in certain models, but it has been found in particular that IV administration of naked RNA results in rapid RNA degradation (57).
Rationally Designed Vaccines
A key aspect of non-viral vaccine development involves the selection of antigens that can effectively elicit a protective immune response. Whereas traditional vaccines are generally developed through attenuation or inactivation of pathogens and through the incorporation of few selected antigens as vaccine components, new technologies have recently been applied toward antigen discovery and design. For example, “reverse vaccinology” refers to the ability to screen the complete antigen sets based on whole-genome sequencing of pathogens for the ability to induce protective antibody responses. Combinations of reverse vaccinology and traditional vaccine approaches allow for an efficient development of immunogenic vaccine candidates (74, 75).
Bioinformatic tools have contributed greatly to vaccine design and evaluation in recent years. Computational approaches are continually improving in their ability to predict T and B-cell epitopes from the complete antigen pools and to rationally design antigens with potential long-lasting protective immunity. Such algorithms calculate antigen-antibody interaction energies and structures (76) that increasingly bridge modeling based on existing templates and free-modeling based on heterologous structures and database consensus design (77). Deep sequencing combined with computational analysis allows for thoroughly characterizing the B cell repertoire in survivors of disease to identify the protective immunity (78).
A computationally designed antigen found to be protective in animal models was first reported for the respiratory syncytial virus (RSV) F antigen in 2013 (79). Many challenging vaccine targets have since been developed rationally for HIV (80–82), hepatitis C (83, 84), herpes (85), Zika (86, 87), RSV (82), HPV (88), as well as for bacteria (82, 89), fungi (90), and cancers (91). Rational vaccine design has also been utilized to improve VLP and NP vaccines by selecting a repetitive and predictive protein backbone structure for enhanced antigen presentation (92). Finally, rational antigen design is being explored for activation of dendritic cells (93) such as targeting C-type lectin receptors to activate antigen presentation in the context of the pathogens (e.g., Ebola and HIV) (94). The first rationally-designed vaccine to undergo human clinical trials is the anti-malarial vaccine Mosquirix, which was approved for use by the European Medicines Agency in 2015 (95). Human clinical trials have not yet begun for other rationally designed vaccines, however. A key point to note is that rationally designed vaccines require a comprehensive knowledge of the biology and immune response to a pathogen (95), and rational design is therefore more difficult to implement for rapidly emerging diseases.
A major challenge to rational vaccine antigen design is the lack of knowledge of T cell epitopes compared to B cell epitopes. Most successful antigens are expected to elicit both B and T cell responses. Quantitative databases have been developed more for predicting B cell receptor (BCR) epitopes than T cell receptor (TCR) epitopes. BCR epitopes can be predicted in part by the structural and chemical properties of the epitopes due to BCRs recognizing primary and tertiary antigen structures, while TCR epitopes have to be predicted based on known TCR epitopes because they only recognize the primary structures. Increasing capacity for identification of TCR epitopes by machine learning from known epitopes will likely help to mitigate this inequity (74, 75, 96).
Non-Viral Vaccine Systems to Address Ongoing Challenges of Vaccine Development
Vaccines for Immunosuppressed Individuals
A fundamental challenge to vaccination is the limited immunogenic response to vaccines seen in immunosuppressed individuals, namely, young children, the elderly, and those who are immunocompromised for medical reasons. The underlying causes of immunosuppression in each of these populations vary, and their underlying mechanisms should be taken into account when creating the best vaccine approach.
Young children, in particular infants and neonates, are considered to be immunosuppressed due to age-specific immune system developments that result in children being particularly susceptible to infection (97). The specific mechanisms for the immunosuppression in this population vary, but one prominent example is the decreased expression of Th17 supporting cytokines by TLR receptors and increased expression of anti-inflammatory cytokines in neonates and particularly in premature newborns (98). On the other hand, the immunosuppressive phenomenon observed in the elderly has been referred to as immunosenescence (99) and it is caused by a number of complex changes resulting in impaired innate and adaptive immune responses (100–104), degradation of lymphoid architecture (105), and increasing proinflammatory cytokines and chemokines (106, 107). To highlight a few important changes, dendritic cells have reduced uptake of antigens (108, 109), macrophages are unable to phagocytose apoptotic cells (110), the number of naïve T cells decreases (111, 112), and B cell repertoire decreases (113). These age-related changes in the ability of the immune system to respond to infection differ from the challenges to vaccination presented by conditions or medications that result in immunosuppression. One such example of a medication that results in immunosuppression is steroids, which have been reviewed extensively elsewhere (114). Steroids exert many effects on immune cells, such as the reprogramming of dendritic cells to tolerogenic dendritic cells (115–117). These cells induce the formation of regulatory T cells (118).
The development of vaccines that can potently activate the innate immune response without using live attenuated vaccines is a central focus of vaccine development for immunocompromised individuals. This is particularly relevant for subunit vaccines, as they do not contain potential viral genomic elements that can act as pattern-associated molecular patterns (PAMPs) to activate innate immune responses. A major approach toward increasing the immunogenic response to a subunit vaccine is through the use of adjuvants. Originally discovered by including food products in equine vaccines and inducing sites of localized sterile inflammation and abscesses, the adjuvant repertoire has since been greatly expanded (119). The so-called “first generation adjuvants”, which remain the most common adjuvants in clinical use, include aluminum salts (alum) and mineral oil-in-water emulsions, which function by promoting the migration of APCs to the sites of intramuscular injection (120). However, the use of these adjuvants is limited greatly by the recruitment of only a comparatively small population of immune cells that are made up of APCs (120) and a markedly Th2 response with little to no cellular immune response (120). This has become the aim of current research to design new adjuvants that can increase the breadth of the innate immune responses to the vaccine.
Much effort has been focused on enhancing the usable adjuvant repertoire to further customize the immune response and to avoid the Th2-dominated immune response seen with some adjuvants (e.g., alum) and instead support a Th1 response in certain circumstances. Specifically, a response skewed toward Th2 response is desired for antibody production and antiparasitic immune response, while a Th1 response is desired for intracellular or viral pathogens. Skewing toward Th1 or Th2 is thought to occur after APCs stimulate certain cytokine gene expression profiles (121). For example, LPS-derivative–based AS04 is being used in hepatitis B and HPV vaccines (122) and has been found to increase cell-mediated immune responses in patients with end-stage renal disease (123). Other adjuvants that have been developed to induce a Th1 immune response include IC31® (124, 125), GLA-SE (125, 126), and CAF01 (125, 127). In addition to the Th1 skewed immune response that these adjuvants displayed, GLA-SE induced antibodies and CAF01 showed a Th1/Th17 response (125). Increasing the breadth of the immune response to vaccines thus can enhance the safety and effectiveness of vaccines for both immunosuppressed populations as well as the general population.
There has been an increasing effort toward developing new vaccines that may induce a safe and immunogenic response in immunocompromised individuals. As an example, DNA vaccines could be used to encode for antibodies that could be safely and temporarily expressed in immunocompromised patients, such as throughout the course of an influenza season. Recent studies that tested the efficacy of influenza neutralizing antibodies found that protection against lethal disease could be conferred by plasmids expressing antibodies given intramuscularly by electroporation (128). However, several major considerations need to be fully addressed before these techniques can be developed for human use. Specifically, the duration and the stability of plasmid vaccination have not yet been fully characterized in humans. Additionally, it has been shown that anti-dsDNA antibodies can be produced by primary B cells isolated from mice treated with plasmid DNA (129), which appear similar to anti-dsDNA antibodies that have been shown to be expressed during systemic lupus erythematosus (130, 131). The anti-dsDNA antibodies may prefer to bind to certain CpG-rich sequences on bacterial DNA of the plasmid (129), which might serve as a means for DNA vaccine optimization. Finally, the purity of plasmid DNA stocks needs to be thoroughly confirmed in order to avoid possible stimulation of unwanted immune reactions.
Other novel adjuvant approaches include the surfactant and emulsifier-based AS03 that are currently being used in influenza-pandemic vaccines (132). LPS-derivative–based AS04 is being used in hepatitis B and HPV vaccines (122) and has been found to increase cell-mediated immune responses in patients with end-stage renal disease (123). Lipid products that form micelles in solution and act as solid particle carriers are another form of adjuvants that can activate innate immunity, as seen with CAF01 (133, 134) and AS01B/E formulation from GlaxoSmithKline (135), which are used in the only currently available vaccine for malaria (136, 137). Several other adjuvants currently in use primarily function as TLR agonists (138–140). TLR agonists have shown promise in aged and young mice (141) as various TLR agonists [e.g., CpG (TLR9), poly(I:C) (TLR3), and pam3CSK4 (TLR1 and TLR2)] can induce expression of co-stimulatory molecules on APCs (141). Another adjuvant approach taken to overcome the immune challenges presented by young children is the use of ß-glucan. These sugars, found in the cell walls of some pathogens, activate dendritic cells through the CLEC71-SYK-CARD9 pathway, and it was shown to provide protection against tuberculosis infection (142). Recently, defective-interfering (DI) viral particle vaccines have also been explored for use as adjuvants to increase the innate immune response (143–152). These are VLPs with aberrant and defective genomes, which have been found in some cases to increase the innate immune response when compared to the replicating virion. Taken together, several innovative strategies are currently being developed to increase the immunogenicity and safety of vaccines for immunocompromised populations.
Vaccines With Non-Traditional Antigens
Because of their increased safety and versatility, such as the ability to deliver a diverse range of molecules as antigens, VLP and NP vaccines have the potential for use to provide immunity against non-protein antigens. A prime example is the development of NP vaccines to treat substance abuse disorders by attaching a drug molecule to a hapten carrier (Figure 2). Vaccines against drugs of abuse aim to elicit a humoral (antibody) response that can neutralize the drug target before it crosses the blood-brain barrier to induce psychotropic effects, thereby decreasing positive associations with and hopefully dependency on the addictive drug. Such vaccines have an advantage for long-term therapeutic use over pharmaceuticals targeting neural receptors by eliminating the medical complications and safety concerns of directly modulating neural signaling network. They also differ from other vaccines in that they are given to active users of drugs of abuse to prevent escalation of use or relapse and do not depend on herd immunity for effectiveness, so their efficacy is determined by individual responses to the vaccines (153).
A hapten carrier, which is a potent B cell antigen, is used to stimulate B cell responses and thereby also activates B cell responses to the attached drug. Therefore, hapten and linker design are of particular interest to ensure structural integrity and to maximize B cell responses (153–170). The vaccine can also be linked to a protein carrier designed to activate T cell responses (and particularly CD4 T cell response to then activate B cells) (153, 160, 171–181), though there has not been a clear determination of whether an increased CD4 Th2:Th1 ratio correlates with efficacy for vaccines against drugs of abuse (153). Finally, an additional consideration in designing vaccines against drugs of abuse is determining whether to target the drug itself or its possibly more psychotropic metabolites that can provide a greater level of protection. A prime example of this is heroin vaccines seemingly being most effective when they can structurally mimic the psychotropic heroin metabolite 6-acetylmorphine (6AM) (166, 172, 182). It should be noted that clinical trial results have only been reported for vaccines against nicotine and cocaine addictions, with the vaccines demonstrating efficacy only in a subset of patients that were able to achieve high neutralizing antibody titers (153, 183–186). The recent vaccine developments to ameliorate drug abuse have been reviewed more extensively elsewhere (153, 160, 187).
Additionally, VLP and NP vaccines are being used in toxoid vaccine formulations, which provide quick neutralization against a cytotoxic molecule (primarily bacterial toxins) that cannot be expressed in its full and activated form. The most well-known example is the diphtheria, tetanus and pertussis (DTaP) inactivated subunit vaccine which has been in use for decades and can elicit effective immune responses against the toxin produced by any of these bacteria if/when the vaccinated individual happens to be exposed to them. The pertussis component of DTaP has a demonstrated high level of safety that it is one of only two known vaccines (besides influenza) that is given to pregnant women in several countries (188). Current clinical trials are focusing on testing potentially more effective toxoid vaccines for pertussis (189) as well as Haemophilius influenzae type b, polio virus, and hepatitis B virus (190). It has also been found that using bacterial membrane or red blood cell (RBC) membrane micelles as a carrier can significantly increase the immunogenicity of the vaccines, and pre-clinical testing is currently underway to use these carriers in vaccine development against the multi-drug resistant bacteria Staphylococcus aureus (MRSA) (191).
Therapeutic Vaccines for Noncommunicable Diseases
As our capabilities for vaccine development and production have expanded, a paradigm shift has recently taken place to use vaccines for disease treatment in addition to disease prevention. These therapeutic vaccine designs rely on the identification of protein markers unique to a disease phenotype which may evade the development of an immune response due to the markers not being recognized by the APCs. For example, cancer vaccines to elicit immune responses against cancer-specific antigens are one of the most widely studied therapeutic vaccines to date due to the inherent challenges of developing effective cancer therapeutics and a need for targeted treatment. The immunosuppressive environment present in cancers has made developing cancer vaccines a significant challenge, especially for vaccines that rely on viral vectors. Therefore, the improved safety profile of non-viral vaccines offers an attractive potential for cancer vaccine development. Non-viral vaccines also confer an additional advantage for developing cancer vaccines in that cancer vaccines may be most effective when an antigen specific to the mutational profile of the individual cancer is used (192–194), particularly in combination to overcome immune tolerance (195). The time needed to make a vaccine against an individual antigen or against a combination of individual antigens is greatly reduced with non-viral vaccines due to the ease of encoding an antigen on a nucleic acid vaccine or purifying protein for a subunit vaccine in comparison to incorporating a personalized antigen into a viral vaccine, growing viral stocks and verifying its expression (196).
Nucleic acid vaccines have been a key area in recent developments for cancer treatment. While a number of DNA cancer vaccine candidates have entered into early phase clinical trials (197), RNA vaccines are thought to have particularly encouraging potential due to their increased immunogenicity compared to DNA vaccines (68, 198). Preliminary results indicate that intranodal injection of naked tumor antigen-encoding mRNA can control tumor growth in mouse cancer models (199–202). Additionally, naked mRNA was found to be immunogenic via intradermal injection in a phase I/II clinical trial for prostate cancer (203). However, a key challenge in developing RNA cancer vaccines has been the need to further ensure the stability of the RNA and to increase its targeting to APCs (204, 205) in order to overcome the immunosuppressive environment of cancers. Developing a delivery vehicle for RNA cancer vaccines has therefore been a central focus of research and development in this area.
Loading RNA into liposomes is one method that has shown some success in controlling cancer growth in mouse models (206–208) and has demonstrated some preliminary efficacy in early stage clinical trials for use as a delivery system for anti-cancer genes (209, 210) and siRNAs (211), with different liposome constructs targeting RNA localization to the spleen. RNA-loaded liposomes can also be targeted directly to T cells by using RNA that encodes for anti-CD3 along with the cancer antigens, bypassing the need for recognition by APCs. This concept has notably been tested in conjunction with chimeric antigen receptor (CAR) T cell therapy (198, 212, 213). Another method bypasses targeting RNA to APCs by directly transfecting dendritic cells (DCs) with RNA extracted from tumors or RNA encoding tumor antigens (214–217) (Figure 4) and then introducing the engineered DCs into patients with a combination of cytokines and/or checkpoint blockades (218, 219). However, DC-directed RNA vaccines are currently limited by the restrained immune environment present in cancers, which can limit the activity of DCs and increases the activity of regulatory T cells (216, 220, 221). It is thought that these challenges could be mitigated by optimizing the use of cytokines and other factors that would act as adjuvants in combination with cancer RNA vaccines (195, 222–225) and by optimizing DC isolation and culturing conditions (226). A few DC-directed RNA vaccine candidates are currently in clinical trials, including those in phase III (196, 221).
Subunit vaccines have also been developed for use as cancer vaccines (227), which are being tested with many of the same delivery systems as nucleic acid-based cancer vaccines to maximize vaccine targeting to immune cells (228). NP-based vaccines in particular have been developed and tested for use as cancer treatments (228–235), the most notable of which are several HPV vaccines for prevention of cervical cancer (236). While less development has been done on VLP-based cancer vaccines, one notable target that has been used is the widely expressed cancer antigen human epidermal growth factor receptor-2/neu (HER2), which has shown to be immunogenic in mouse models (237–244) and in early clinical testing in humans (245) and dogs (246), but as a whole these vaccines have had to undergo additional design in order to overcome B cell tolerance (227, 247) and to fully characterize their anti-tumor activity.
Because subunit vaccines require antigen presentation in order to elicit an immune response, a primary challenge in VLP- and NP-based cancer vaccine developments has been optimizing their uptake by APCs (228). Vaccine uptake by APCs can be optimized by engineering VLP- and NP-based cancer vaccines to resemble the structure of viral particles as closely as possible, such as by using certain types of carriers (liposomes, polymers and ferritin cages), sizes (20–45 nm) and a spherical shape (Figure 4). Subsequently, these vaccines may be most successful when combined with checkpoint blockade treatment by encouraging clonal expansion of lymphocytes (248). VLPs and NPs can also be used as immuno-enhancers, e.g., to deliver cytokines and TLR agonists to target sites, which has been found to boost localized immune responses while avoiding immunopathogenic and possibly systemic inflammation (46).
Vaccines for Rapidly Emerging Viral Diseases
Emerging and reemerging pathogens, such as West Nile virus, pandemic influenza virus, Ebola virus, dengue virus, Zika virus, and the on-going global pandemic SARS-CoV-2 pose great challenges to the public health system. Rapid development and deployment of vaccines are critical to quickly build up resistance against these and other disease “X”, which is a term used by the WHO to refer to future unknown disease pandemics (249). The ideal vaccine platform in a pandemic situation must be cost-effective and can be rapidly developed and produced on a large scale to meet global demands. Temperature sensitivity is also a consideration, as cold chain storage can be particularly difficult to maintain in developing countries. Development of heat stable vaccines like the oral bovine rotavirus pentavalent vaccine (BRV-PV, Rotasil® by the Serum Institute of India), which was prequalified by the WHO in 2018, can provide protection against serious diseases in regions where transportation and refrigeration are unreliable (250). In comparison, the only FDA approved Ebola virus vaccine (rVSV-ZEBOV, ERVEBO® by Merck and Co., Inc.) must be stored at −80°C or −60°C (251), which presents a major obstacle for affected countries. Rapid production of low cost, scalable, and temperature stable vaccines is an ongoing challenge in the face of emerged and emerging global disease pandemics.
Currently, rapid development of vaccines is greatly limited by the resources and regulatory policies needed to bring a vaccine from its conceptualization stage to the clinic, which has been estimated to cost between $200 and $500 million dollars and to take 5–18 years (252). Vaccines also tend to be manufactured in countries with larger economic and technical prowess and more robust disease surveillance systems than developing countries and therefore can unfairly influence the equity of vaccine distribution and usage. This was seen in the 2009–2010 influenza pandemic, where 80% of the vaccines were manufactured and used in seven industrialized regions (United States, Canada, Australia, western Europe, Russia, China, and Japan), while the majority of developing regions in the world did not receive any pandemic influenza vaccines until January 2010, 9 months after the WHO declared the influenza pandemic (253). In addition, as mentioned previously, most pandemic vaccines have to be clinically tested during an active outbreak in order to obtain sufficient safety and efficacy data, thereby limiting the number of vaccine candidates that can be deployed to save lives. This was seen during the Ebola outbreak of 2013–2015, when two vaccines were fully developed in advance of clinical trials but only one (the simian adenovirus-based Ebola vaccine ChAd3-EBO-Z) was tested early enough in the outbreak to obtain sufficient clinical data (254). Similar challenges are also seen in selecting vaccine candidates for the large sample sizes needed for phase III clinical trials for HIV vaccine candidates. Statistical ranking systems to prioritize candidates are being developed to aid in this selection process (255). Zoonotic diseases present additional considerations, as it is economical to vaccinate the multiple species that may act as reservoirs of the pathogen(s) in order to control the spread of the disease. The first vaccine to provide protection in multiple species is the simian adenovirus-based vaccine candidate ChAdOx1 RVF, which has been shown to provide effective protection against Rift Valley Fever virus in sheep, goats, and cattle and is currently undergoing testing in larger livestock field trials and in humans (254, 256).
Other measures have been undertaken to expedite the process of vaccine development and reduce the cost of vaccine production. International institutions allow for collaborative groups to rapidly co-operate on vaccine development and shorten the vaccine manufacturing process. The Coalition for Epidemic Preparedness and Innovations (CEPI) provides funds for clinical trial and stockpiling of vaccines that would not have market incentive in a traditional funding mechanism of vaccine development and manufacturing (257). Such international collaborations will help to bridge the differing vaccine development policies and investitures across countries and use these combined resources to develop vaccines to primarily benefit those living in either underdeveloped or developing nations (258). In a recent example of this, CEPI, Gavi, and the WHO have come together to form COVAX, the vaccines pillar of the Access to COVID-19 Tools (ACT) Accelerator, with the mission to expedite the production of a COVID-19 vaccine to be equitably distributed throughout the world (259).
Technical challenges in vaccine production process can be an impediment. For example, the use of fertilized chicken eggs in vaccine production can pose challenges such as the restricted capacity of egg production, egg allergies, and the emergence of viruses with egg-culture-adapted mutations that can reduce vaccine efficacy (260). The use of animal cells for certain vaccines can also present significant challenges of cost, slow production rates, and potential high risk of contaminations. Other vaccine production systems, such as VLP vaccines produced in yeasts, insect cells and bacterial systems, as well as DNA/RNA vaccines, can benefit from increased robustness of antigen production, decreased risk of contaminations, and quicker time of response (252). This may especially be the case for DNA vaccines, where the increasing capacity of next-generation DNA sequencing, for example, has lowered the time for development of a DNA vaccine from 20 months following the 2003 SARS outbreak to 3.25 months following the 2016 Zika outbreak (261).
COVID-19 Pandemic as a Case Study to Rapidly Develop Non-Viral Vaccines
The ongoing COVID-19 pandemic has presented unique opportunities as well as challenges for vaccine development. Unlike the influenza vaccines, no coronavirus vaccines existed prior to the COVID-19 pandemic. Such a rapid and widespread need for a completely novel vaccine for COVID-19 has resulted in a drive to significantly reduce the length of time required to produce a new vaccine. It has also highlighted the necessity to use non-viral vaccine platforms with overlapping stages of vaccine development, including preclinical and clinical testing and manufacturing that would otherwise be required to happen in a stepwise process for a traditional vaccine development effort (262–264). However, a rapid progression of clinical testing will need to be balanced by the need for obtaining quality data on vaccine safety and efficacy (265), especially considering previous reports of pathological antibody-dependent enhancement responses in some patients immunized with the 2003 SARS vaccine candidates (266, 267). As with previous pandemics, widespread global availability and resource management will be another key consideration for vaccine selection, especially considering the near ubiquitous presence of COVID-19 around the globe and its disproportionate impact on populations of low socio-economic status (268, 269). It is also likely that the approval of multiple vaccine candidates will be most optimal to controlling and ending the pandemic should more than one vaccine prove to be effective in preventing COVID-19 disease. Multiple COVID-19 vaccines would allow for more clinical and regulatory choices to accommodate differences in patient responses (particularly in more vulnerable patient populations) and manufacturing and distribution capabilities (270, 271). Perhaps, with these considerations in mind, non-viral COVID-19 vaccine platforms (e.g., DNA and mRNA) have been selected among the first candidates to enter clinical testing, partly for their aforementioned reasons of safety profiles and relative ease of manufacturing (Table 1). Some of the RNA-based COVID-19 vaccines (all of which are currently in various stages of clinical trials) include but are not necessarily limited to:
1. The mRNA-1273 vaccine developed by the U.S. biotech company Moderna (272).
2. The mRNA CVnCoV vaccine developed by the German company CureVac (273).
3. A group of 4 RNA vaccines under the name BNT162 developed by the German company Biontech that consists of two nucleoside-modified mRNAs, a uridine-containing mRNA and a self-amplifying mRNA (274), which in an early phase I/II trial, the nucleoside-modified mRNA BNT1621b has been shown to elicit neutralizing antibodies (275) and is better tolerated particularly in older adults than BNT1621a (276, 277).
4. The self-amplifying mRNA LNP-nCoVsaRNA (COVAC1) vaccine from the Imperial College London (278).
5. The mRNA vaccine LUNAR-COV19 (ARCT-021) from US company Arcturus Therapeutics (279).
6. An unnamed mRNA vaccine candidate from Chinese company Yunnan Walvax Biotechnology (280).
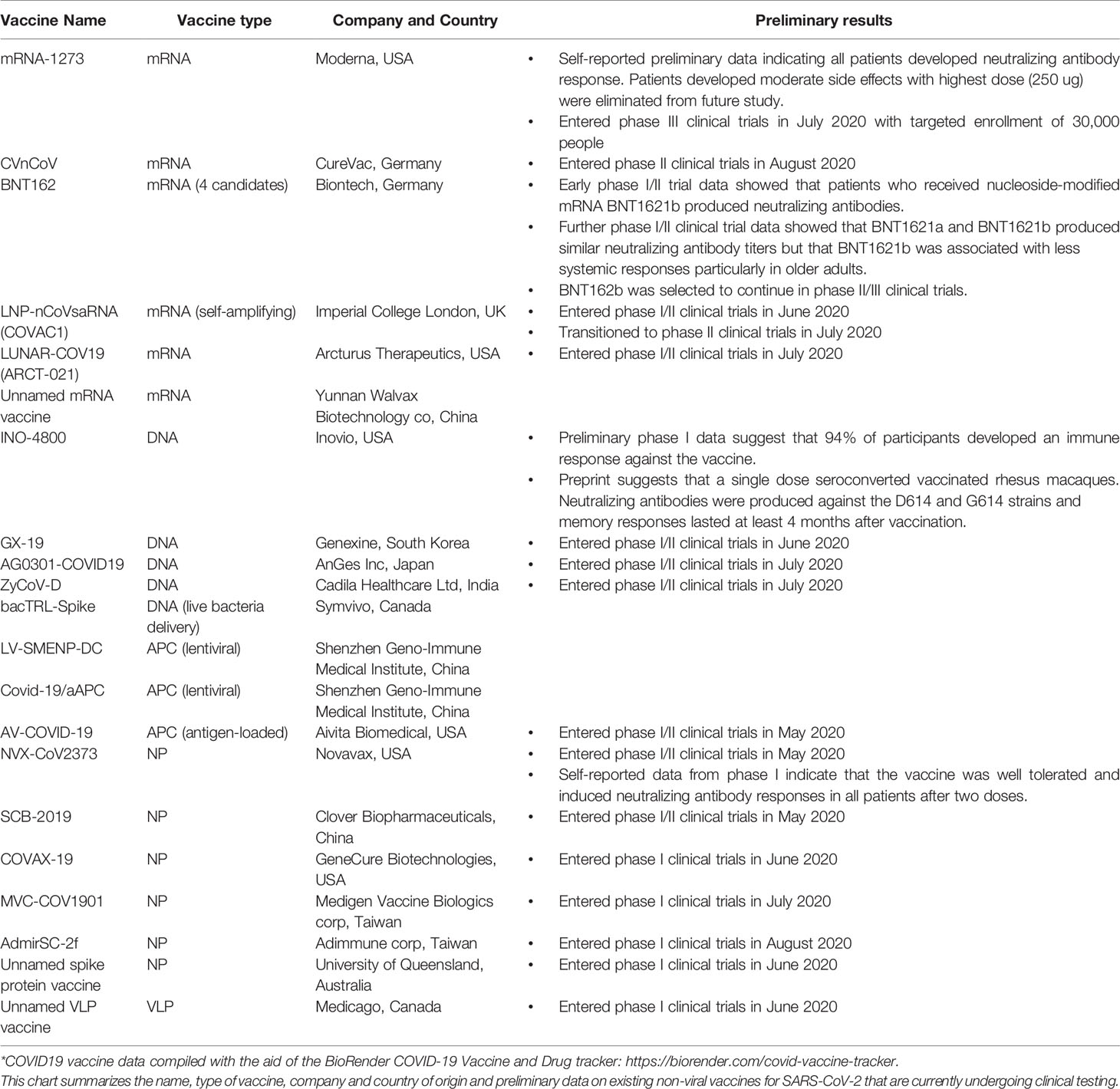
Table 1 Non-viral vaccines currently in development for SARS-CoV-2*.
Some of the COVID-19 DNA vaccines (all of which are also in various stages of clinical trials) include but are not necessarily limited to:
1. The INO-4800 vaccine developed by the U.S. pharmaceutical company Inovio (281) with preliminary phase I data suggesting that 94% of participants might have developed an immune response against it following vaccine administration by electroporation (282) and that vaccination in rhesus macaques elicited neutralizing antibodies against both the D614 and G614 SARS-CoV-2 strains (283).
2. The GX-19 vaccine developed by the South Korean company Genexine (284).
3. The AG0301-COVID19 vaccine developed by the Japanese company AnGes, Inc. (285, 286).
4. The ZyCoV-D vaccine developed by the Indian company Cadila Healthcare Ltd (287).
5. The live bacteria-mediated plasmid delivery system bacTRL-Spike developed by the Canadian company Symvivo (288).
Genetically engineered APCs are also being pursued as potential COVID-19 vaccine candidates, with DCs transfected with lentiviral vectors expressing COVID-19 antigens currently being tested in China (289, 290) and in the United States (291). Meanwhile, COVID-19 VLP- and NP-based vaccines have also advanced into clinical trials, including the NP NVX-CoV2373 vaccine from the U.S. company Novavax (292), the NP SCB-2019 vaccine from the Chinese company Clover Biopharmaceuticals (293), the NP COVAX-19 vaccine from the U.S. company GeneCure Biotechnologies (294), the NP vaccine from the Taiwanese company Medigen Vaccine Biologics (295), the NP vaccine AdmirSC-2f from Taiwanese company Adimmune corp (296), an unnamed NP vaccine from the University of Queensland (297, 298) and an unnamed VLP vaccine from the Canadian company Medicago (299). CEPI has collaborated in the development and testing of a selected number of these vaccine candidates (273, 281, 298).
It should also be noted that several viral vectored vaccine candidates for COVID-19 have also entered in clinical testing (Table 2). Several adenovirus vectored vaccines are currently the furthest along in clinical testing. One example is the vaccine candidate AZD1222 (formerly known as ChAdOx1 nCov-19), a replication-defective chimpanzee adenovirus developed by Oxford University which entered phase III clinical trials in August 2020. This viral vector was chosen due to its previous application as a vaccine vector for Middle East respiratory syndrome coronavirus (MERS-CoV). The ChAdOx1 vector encoding the spike (S) protein provided protection against six different strains of MERS-CoV in rhesus macaques (25), demonstrating its ability to be an effective vaccine for coronaviruses. Specific to COVID-19, AZ1222 was found to induce humoral and cell mediated immune repsonses in phase I/II cliical trial and did not result in any instances of severe side effects (300). It has recently found that AZD1222 could induce a robust humoral, CD8 and Th1 dominant CD4 response in mice and rhesus macaques and that both a prime and a prime-boost regimen protected rhesus macaques against COVID-19 related pneumonia. However, it should be noted that there was no difference in the amount of nasal virus shedding in vaccinated vs unvaccinated animals challenged with SARS-CoV-2 (301).
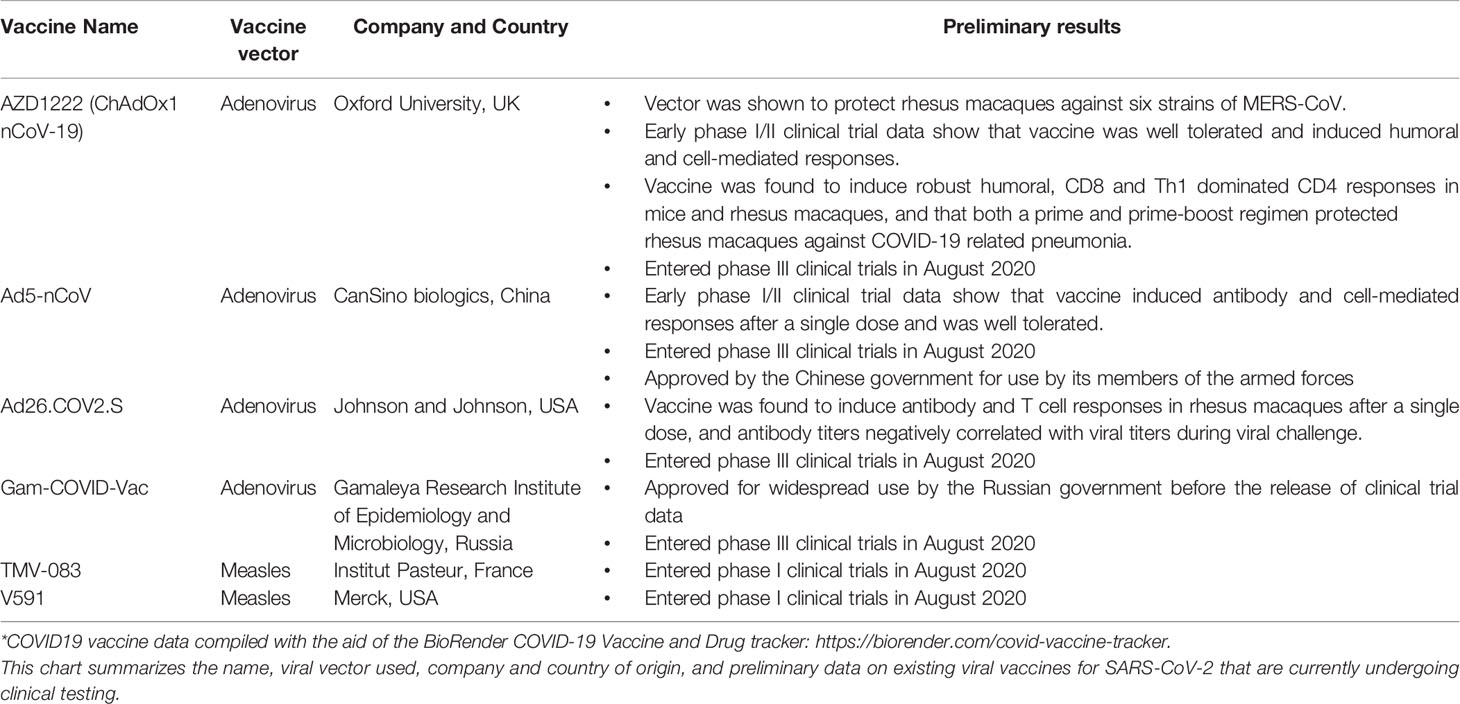
Table 2 Viral vaccines currently in development for SARS-CoV-2*.
Two other replication-incompetent adenoviral vectored vaccines for COVID-19 have also entered clinical trials. The Ad5-nCoV candidate from Chinese company CanSino biologics was shown in early clinical trial data to induce significant antibody and T cell responses after a single dose and to have only rare instances of severe side effects that were more prevalent among the higher dose groups (302, 303). The Chinese government has recently approved the vaccine for use among its members of the armed forces (304). Additionally, the Ad26.COV2.S from Johnson & Johnson induced antibody and T cell responses in rhesus macaques after a single dose, and antibody titers negatively correlated with viral titers during viral challenge (305). Finally, the Gam-COVID-Vac candidate from the Gamaleya Research Institute of Epidemiology and Microbiology in Russia is another adenovirus-based vaccine that is the first COVID-19 vaccine to gain government approval for widespread use after a phase I trial. Phase III trials for this vaccine began in August 2020 (306).
Finally, two COVID-19 vaccine candidates based on live-attenuated measles platforms have also entered into early clinical trials. The TMV-083 candidate from the Institut Pasteur and with collaboration with CEPI is a measles vectored vaccine expressing a modified SARS-CoV-2 surface glycoprotein that entered phase I clinical trials in August 2020 (307), while the V591 candidate from Merck also entered phase I clinical trials in August 2020 (308). Many other viral vectored vaccine candidates for COVID-19 are also in preclinical stages of development.
Summary
The emergence of new non-viral vaccine technologies has significantly advanced the scope and efficacy of traditional vaccine formulations that are generally based on single protein subunit vaccines or attenuated or killed vaccines. Non-viral vaccine technologies have allowed for new applications to address ongoing challenges of vaccination with customization in the areas of safety, immunogenicity, breadth of protection, scalability, and ease of production. These new technologies have also expanded the notion of what is possible with vaccination by extending their reach to once untenable areas, such as cancer treatment and neutralization of drugs of abuse. It is clear that continued development and optimization of vaccines will require multi-faceted approaches that can only be implemented with extensive cross-field collaboration and periodic review of the current state of vaccinology. These challenges as well as opportunities ensure that vaccine development will remain on the cutting edge of science for decades to come to combat new and emerging pathogens as well as other noncommunicable diseases.
Author Contributions
MB, SV, NK, and HL contributed to the literature review and writing of the manuscript. MB prepared all figures and tables. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in parts by NIH NIAID grant R01 AI131586, USDA-NIFA-Capacity Funds (Hatch and Animal Health), and the University of Minnesota School of Medicine Academic Investment Research Program (AIRP) and COVID-19 Rapid Response Funds to HL and YL, USDA-NIFA AFRI grant #2019-05384 and Minnesota Agricultural Experiment Station Rapid Agricultural Response Fund to HL, and by a pre-doctoral NIH fellowship T32 DA007097 to MB. NIH T32 training grant in Comparative Medicine and Pathology (5T32 OD010993-17) for NK.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Liu MA. Immunologic basis of vaccine vectors. Immunity (2010) 33:504–15. doi: 10.1016/j.immuni.2010.10.004
2. Plotkin S. History of vaccination. Proc Natl Acad Sci U S A (2014) 111:12283–7. doi: 10.1073/pnas.1400472111
3. Rauch S, Jasny E, Schmidt KE, Petsch B. New vaccine technologies to combat outbreaksituations. Front Immunol (2018) 9(1963):1–24. doi: 10.3389/fimmu.2018.01963
4. Smith GL, MacKett M, Moss B. Infectious vaccinia virus recombinants that express hepatitis B virus surface antigen. Nature (1983) 302:490–5. doi: 10.1038/302490a0
5. Moss B, Smith GL, Gerin JL, Purcell RH. Live recombinant vaccinia virus protects chimpanzees against hepatitis B. Nature (1984) 311:67–9. doi: 10.1038/311067a0
6. Tartaglia J, Jarrett O, Neil JC, Desmettre P, Paoletti E. Protection of cats against feline leukemia virus by vaccination witha canarypox virus recombinant, ALVAC-FL. J Virol(1993) 67:2370–5. doi: 10.1128/JVI.67.4.2370-2375.1993
7. Darteil R, Bublot M, Laplace E, Bouquet JF, Audonnet JC, Riviè M. Herpesvirus of turkey recombinant viruses expressing infectious bursal disease virus (IBDV) VP2 immunogen induce protection against an IBDV virulent challenge in chickens. Virology (1995) 211:481–90. doi: 10.1006/viro.1995.1430
8. Pastoret PP, Brochier B. The development and use of a vaccinia-rabies recombinant oral vaccine for the control of wildlife rabies; A link between Jenner and Pasteur. Epidemiol Infect (1996) 116:235–40. doi: 10.1017/s0950268800052535
9. Welter J, Taylor J, Tartaglia J, Paoletti E, Stephensen CB. Vaccination against Canine Distemper Virus Infection in Infant Ferrets with and without Maternal Antibody Protection, Using Recombinant Attenuated Poxvirus Vaccines. J Virol (2000) 74:6358–67. doi: 10.1128/jvi.74.14.6358-6367.2000
10. Pardo MC, Tanner P, Bauman J, Silver K, Fischer L. Immunization of Puppies in the Presence of Maternally Derived Antibodies Against Canine Distemper Virus. J Comp Pathol (2007) 137:S72. doi: 10.1016/j.jcpa.2007.04.015
11. Jas D, Coupier C, Toulemonde CE, Guigal PM, Poulet H. Three-year duration of immunity in cats vaccinated with a canarypox-vectored recombinant rabies virus vaccine. Vaccine (2012) 30:6991–6. doi: 10.1016/j.vaccine.2012.09.068
12. Esaki M, Godoy A, Rosenberger JK, Rosenberger SC, Gardin Y, Yasuda A, et al. Protection and antibody response caused by turkey herpesvirus vector newcastle disease vaccine. Avian Dis (2013) 57:750–5. doi: 10.1637/10540-032613-Reg.1
13. First FDA-approved vaccine for the prevention of Ebola virus disease, marking a critical milestone in public health preparedness and response | FDA . Available at: https://www.fda.gov/news-events/press-announcements/first-fda-approved-vaccine-prevention-ebola-virus-disease-marking-critical-milestone-public-health (Accessed September 1, 2020).
14. Draper SJ, Heeney JL. Viruses as vaccine vectors for infectious diseases and cancer. Nat Rev Microbiol (2010) 8:62–73. doi: 10.1038/nrmicro2240
15. Dhanwani R, Zhou Y, Huang Q, Verma V, Dileepan M, Ly H, et al. A Novel Live Pichinde Virus-Based Vaccine Vector Induces Enhanced Humoral and Cellular Immunity after a Booster Dose. J Virol (2015) 90:2551–60. doi: 10.1128/JVI.02705-15
16. Dhanwani R, Ly H, Liang Y. Recombinant Tri-Segmented Pichinde Virus as a Novel Live Viral Vaccine Platform. In: Clifton NJ, editor. Methods in molecular biology. New York, NY: Humana Press (2017). p. 169–79. doi: 10.1007/978-1-4939-6869-5_10
17. Peng B, Wang LR, Gomez-Roman VR, Davis-Warren A, Montefiori DC, Kalyanaraman VS, et al. Replicating Rather than Nonreplicating Adenovirus-HumanImmunodeficiency Virus Recombinant Vaccines Are Better at Eliciting Potent Cellular Immunity andPriming High-Titer Antibodies. J Virol (2017) 79:10200–9. doi: 10.1128/jvi.79.16.10200-10209.2005
18. Condit RC, Williamson AL, Sheets R, Seligman SJ, Monath TP, Excler JL, et al. Unique safety issues associated with virus-vectored vaccines: Potential for and theoretical consequences of recombination with wild type virus strains. Vaccine (2016) 34:6610–6. doi: 10.1016/j.vaccine.2016.04.060
19. Dudek T, Knipe DM. Replication-defective viruses as vaccines and vaccine vectors. Virology (2006) 344:230–9. doi: 10.1016/j.virol.2005.09.020
20. Barry M. Single-cycle adenovirus vectors in the current vaccine landscape. Expert Rev Vaccines (2018) 17:163–73. doi: 10.1080/14760584.2018.1419067
21. Clem AS. Fundamentals of vaccine immunology. J GlobalInfect Dis (2011) 3:373–8. doi: 10.4103/0974-777X.77299
22. Zimmermann P, Curtis N. Factors that influence the immune response tovaccination. Clin Microbiol Rev (2019) 32:e00084–18. doi: 10.1128/CMR.00084-18
23. Kushnir N, Streatfield SJ, Yusibov V. Virus-like particles as a highly efficient vaccine platform: Diversity of targets and production systems and advances in clinical development. Vaccine (2012) 31:58–83. doi: 10.1016/j.vaccine.2012.10.083
24. Mohsen MO, Zha L, Cabral-Miranda G, Bachmann MF. Major findings and recent advances in virus–like particle (VLP)-based vaccines. Semin Immunol (2017) 34:123–32. doi: 10.1016/j.smim.2017.08.014
25. Deml L, Speth C, Dierich MP, Wolf H, Wagner R. Recombinant HIV-1 Pr55gag virus-like particles: Potent stimulators of innate and acquired immune responses. Mol Immunol (2005) 42:259–77. doi: 10.1016/j.molimm.2004.06.028
26. Paliard X, Liu Y, Wagner R, Wolf H, Baenziger J, Walker CM. Priming of strong, broad, and long-lived HIV type 1 p55(gag)-specific CD8+ cytotoxic T cells after administration of a virus-like particle vaccine in rhesus macaques. AIDS Res Hum Retroviruses (2000) 16:273–82. doi: 10.1089/088922200309368
27. Murata K, Lechmann M, Qiao M, Gunji T, Altert HJ, Liang TJ. Immunization with hepatitis C virus-like particles protects mice from recombinant hepatitis C virus-vaccinia infection. Proc Natl Acad Sci U.S.A. (2003) 100:6753–8. doi: 10.1073/pnas.1131929100
28. Schirmbeck R, Böhm W, Reimann J. Virus-like Particles Induce MHC Class I-Restricted T-Cell Responses: Lessons learned from the hepatitis B small surface antigen. Intervirology (1996) 39:111–9. doi: 10.1159/000150482
29. Win SJ, Ward VK, Dunbar PR, Young SL, Baird MA. Cross-presentation of epitopes on virus-like particles via the MHC i receptor recycling pathway. Immunol Cell Biol (2011) 89:681–8. doi: 10.1038/icb.2010.161
30. Grgacic EVL, Anderson DA. Virus-like particles: Passport to immune recognition. Methods (2006) 40:60–5. doi: 10.1016/j.ymeth.2006.07.018
31. Chackerian B. Virus-like particles: Flexible platforms for vaccine development. Expert Rev Vaccines (2007) 6:381–90. doi: 10.1586/14760584.6.3.381
32. glaxosmithkline. ENGERIX-B [Hepatitis B Vaccine (Recombinant)] injectable suspension, for intramuscular use. Available at: www.vaers.hhs.gov.
33. glaxosmithkline. Cervarix, Suspension for Intramuscular Injection. Available at: www.vaers.hhs.gov.
34. RECOMBIVAX HB®. Hepatitis B Vaccine (Recombinant) Suspension for intramuscular injection. Available at: www.vaers.hhs.gov.
35. Merck. GARDASIL® [Human Papillomavirus Quadrivalent (Types 6, 11, 16, and 18) Vaccine, Recombinant] Suspension for intramuscular injection. Available at: www.vaers.hhs.gov.
36. Agnandji ST, Lell B, Fernandes JF, Abossolo BP, Methogo BGNO, Kabwende AL, et al. A phase 3 trial of RTS,S/AS01 malaria vaccine in African infants. N Engl J Med (2012) 367:2284–95. doi: 10.1056/NEJMoa1208394
37. Agnandji ST, Lell B, Soulanoudjingar SS, Fernandes JF, Abossolo BP, Conzelmann C, et al. First results of phase 3 trial of RTS,S/AS01 malaria vaccine in African children. N Engl J Med (2011) 365:1863–75. doi: 10.1056/NEJMoa1102287
38. Song JM, Wang BZ, Park KM, van Rooijen N, Quan FS, Kim MC, et al. Influenza virus-like particles containing M2 induce broadly crossprotective immunity. PloS One (2011) 6:e14538. doi: 10.1371/journal.pone.0014538
39. El-Attar L, Oliver SL, Mackie A, Charpilienne A, Poncet D, Cohen J, et al. Comparison of the efficacy of rotavirus VLP vaccines to a live homologous rotavirus vaccine in a pig model of rotavirus disease. Vaccine (2009) 27:3201–8. doi: 10.1016/j.vaccine.2009.03.043
40. Zhou H, Guo L, Wang M, Qu J, Zhao Z, Wang J, et al. Prime immunization with rotavirus VLP 2/6 followed by boosting withan adenovirus expressing VP6 induces protective immunization against rotavirus inmice. Virol J (2011) 8(3):1–8. doi: 10.1186/1743-422X-8-3
41. Dhanasooraj D, Kumar RA, Mundayoor S. Subunit protein vaccine delivery system for tuberculosis based on hepatitis B virus core VLP (HBc-VLP) particles. In: Methods in Molecular Biology. Totowa, NJ: Humana Press Inc. (2016). p. 377–92. doi: 10.1007/978-1-4939-3389-1_26
42. Boigard H, Alimova A, Martin GR, Katz A, Gottlieb P, Galarza JM. Zika virus-like particle (VLP) based vaccine. PloS Negl Trop Dis (2017) 11:e0005608. doi: 10.1371/journal.pntd.0005608
43. Franco D, Liu W, Gardiner DF, Hahn BH, Ho DD. CD40L-containing virus-like particle as a candidate HIV-1 vaccine targeting dendritic cells. J Acquir Immune Defic Syndr (2011) 56:393–400. doi: 10.1097/QAI.0b013e31820b844e
44. Pillay S, Shephard EG, Meyers AE, Williamson AL, Rybicki EP. HIV-1 sub-type C chimaeric VLPs boost cellular immune responses inmice. J Immune Based Ther Vaccines (2010) 8(7):1–6. doi: 10.1186/1476-8518-8-7
45. Frietze KM, Peabody DS, Chackerian B. Engineering virus-like particles as vaccine platforms. Curr Opin Virol (2016) 18:44–9. doi: 10.1016/j.coviro.2016.03.001
46. Pati R, Shevtsov M, Sonawane A. Nanoparticle vaccines against infectious diseases.Front Immunol (2018) 9(2224):1–16. doi: 10.3389/fimmu.2018.02224
47. Zhao L, Seth A, Wibowo N, Zhao CX, Mitter N, Yu C, et al. Nanoparticle vaccines. Vaccine (2014) 32:327–37. doi: 10.1016/j.vaccine.2013.11.069
48. Han J, Zhao D, Li D, Wang X, Jin Z, Zhao K. Polymer-based nanomaterials and applications for vaccines anddrugs. Polym (Basel) (2018) 10(31):1–14. doi: 10.3390/polym10010031
49. Li H, Li Y, Wang X, Hou Y, Hong X, Gong T, et al. Rational design of polymeric hybrid micelles to overcome lymphaticand intracellular delivery barriers in cancer immunotherapy. Theranostics (2017) 7:4383–98. doi: 10.7150/thno.20745
50. Al-Halifa S, Gauthier L, Arpin D, Bourgault S, Archambault D. Nanoparticle-Based Vaccines Against RespiratoryViruses. Front Immunol (2019) 10:22. doi: 10.3389/fimmu.2019.00022
51. Hobernik D, Bros M. DNA vaccines—How far from clinical use? Int J Mol (2018) 19:3605. doi: 10.3390/ijms19113605
52. Khan KH. DNA vaccines: Roles against diseases. GERMS (2013) 3:26–35. doi: 10.11599/germs.2013.1034
53. Kennedy NJ, Spithill TW, Tennent J, Wood PR, Piedrafita D. DNA vaccines in sheep: CTLA-4 mediated targeting and CpG motifs enhance immunogenicity in a DNA prime/protein boost strategy. Vaccine (2006) 24:970–9. doi: 10.1016/j.vaccine.2005.08.076
54. Li L, Saade F, Petrovsky N. The future of human DNA vaccines. J Biotechnol (2012) 162:171–82. doi: 10.1016/j.jbiotec.2012.08.012
55. Li SS, Kochar NK, Elizaga M, Hay CM, Wilson GJ, Cohen KW, et al. DNA priming increases frequency of T-cell responses to a vesicularstomatitis virus HIV vaccine with specific enhancement of CD8 T-cell responses by interleukin-12plasmid DNA. Clin Vaccine Immunol (2017) 24:1–14e00263-17. doi: 10.1128/CVI.00263-17
56. Haidari G, Cope A, Miller A, Venables S, Yan C, Ridgers H, et al. Combined skin and muscle vaccination differentially impact thequality of effector T cell functions: The CUTHIVAC-001 randomized trial. SciRep (2017) 7:13011. doi: 10.1038/s41598-017-13331-1
57. Zhang C, Maruggi G, Shan H, Li J. Advances in mRNA vaccines for infectious diseases. Front Immunol (2019) 10:594. doi: 10.3389/fimmu.2019.00594
58. Lundstrom K. Latest development on RNA-based drugs and vaccines.Futur Sci OA (2018) 4(5):FSO300. doi: 10.4155/fsoa-2017-0151
59. Uchida S, Yoshinaga N, Yanagihara K, Yuba E, Kataoka K, Itaka K. Designing immunostimulatory double stranded messenger RNA with maintained translational activity through hybridization with poly A sequences for effective vaccination. Biomaterials (2018) 150:162–70. doi: 10.1016/J.BIOMATERIALS.2017.09.033
60. Hood JL. Post isolation modification of exosomes for nanomedicine applications. Nanomedicine (2016) 11:1745–56. doi: 10.2217/nnm-2016-0102
61. Benteyn D, Heirman C, Bonehill A, Thielemans K, Breckpot K. mRNA-based dendritic cell vaccines. Expert Rev Vaccines (2014) 14:161–76. doi: 10.1586/14760584.2014.957684
62. Diken M, Kreiter S, Selmi A, Britten CM, Huber C, Türeci Ö, et al. Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation. Gene Ther (2011) 18:702–8. doi: 10.1038/gt.2011.17
63. Van den Boorn JG, Hartmann G. Turning Tumors into Vaccines: Co-opting the Innate Immune System. Immunity (2013) 39:27–37. doi: 10.1016/j.immuni.2013.07.011
64. Elion DL, Cook RS, Elion DL, Cook RS. Harnessing RIG-I and intrinsic immunity in the tumor microenvironment for therapeutic cancer treatment. Oncotarget (2018) 9:29007–17. doi: 10.18632/oncotarget.25626
65. Vogel AB, Lambert L, Kinnear E, Busse D, Erbar S, Reuter KC, et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol Ther (2018) 26:446–55. doi: 10.1016/j.ymthe.2017.11.017
66. Démoulins T, Englezou PC, Milona P, Ruggli N, Tirelli N, Pichon C, et al. Self-replicating RNA vaccine delivery to dendriticcells. Methods Mol Biol (2017) 1499:37–75. doi: 10.1007/978-1-4939-6481-9_3
67. Englezou PC, Sapet C, Démoulins T, Milona P, Ebensen T, Schulze K, et al. Self-Amplifying Replicon RNA Delivery to Dendritic Cells by Cationic Lipids. Mol Ther Nucleic Acids (2018) 12:118–34. doi: 10.1016/j.omtn.2018.04.019
68. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines-a new era in vaccinology. Nat Rev Drug Discovery (2018) 17:261–79. doi: 10.1038/nrd.2017.243
69. Alberer M, Gnad-Vogt U, Hong HS, Mehr KT, Backert L, Finak G, et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet (2017) 390:1511–20. doi: 10.1016/S0140-6736(17)31665-3
70. Bahl K, Senn JJ, Yuzhakov O, Bulychev A, Brito LA, Hassett KJ, et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol Ther (2017) 25:1316–27. doi: 10.1016/j.ymthe.2017.03.035
71. Wang Z, Troilo PJ, Wang X, Griffiths TG, Pacchione SJ, Barnum AB, et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther (2004) 11:711–21. doi: 10.1038/sj.gt.3302213
72. Myhr AI. DNA Vaccines: Regulatory Considerations and Safety Aspects. Curr Issues Mol Biol (2017) 22:79–88. doi: 10.21775/cimb.022.079
73. Kramps T, Elbers K. Introduction to RNA vaccines. Methods MolBiol (2017) 1499:1–11. doi: 10.1007/978-1-4939-6481-9_1
74. Rueckert C, Guzmán CA. Vaccines: From Empirical Development to RationalDesign. PloS Pathog (2012) 8:e1003001. doi: 10.1371/journal.ppat.1003001
75. Peri C, Gagni P, Combi F, Gori A, Chiari M, Longhi R, et al. Rational epitope design for protein targeting. ACS Chem Biol (2013) 8:397–404. doi: 10.1021/cb300487u
76. Yamashita T. Toward rational antibody design: Recent advancements in molecular dynamics simulations. Int Immunol (2018) 30:133–40. doi: 10.1093/intimm/dxx077
77. He L, Zhu J. Computational tools for epitope vaccine design and evaluation. Curr Opin Virol (2015) 11:103–12. doi: 10.1016/J.COVIRO.2015.03.013
78. Rappuoli R, Bottomley MJ, D’Oro U, Finco O, De Gregorio E. Reverse vaccinology 2.0: Human immunology instructs vaccine antigen design. J Exp Med (2016) 213:469–81. doi: 10.1084/JEM.20151960
79. McLellan JS, Chen M, Joyce MG, Sastry M, Stewart-Jones GBE, Yang Y, et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Sci (80 ) (2013) 342:592–8. doi: 10.1126/science.1243283
80. Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Sci (80) (2010) 329:856–61. doi: 10.1126/science.1187659
81. Zhou T, Xu K. Structural features of broadly neutralizing antibodies and rational design of vaccine. In: Advances in Experimental Medicine and Biology. New York, NY: Springer New York LLC (2018). p. 73–95. doi: 10.1007/978-981-13-0484-2_4
82. Ferlenghi I, Cozzi R, Scarselli M. Structural Vaccinology: A Three-dimensional View for Vaccine Development. Curr Top Med Chem (2013) 13:2629–37. doi: 10.2174/15680266113136660187
83. Keck M-L, Wrensch F, Pierce BG, Baumert TF, Foung SKH. Mapping Determinants of Virus Neutralization and Viral Escape forRational Design of a Hepatitis C Virus Vaccine. Front Immunol (2018) 9:1194. doi: 10.3389/fimmu.2018.01194
84. He L, Tzarum N, Lin X, Shapero B, Sou C, Mann CJ, et al. Proof of concept for rational design of hepatitis C virus E2 core nanoparticle vaccines. Sci Adv (2020) 6:eaaz6225. doi: 10.1126/sciadv.aaz6225
85. Kaufmann JK, Flechtner JB. Evolution of rational vaccine designs for genital herpes immunotherapy. Curr Opin Virol (2016) 17:80–6. doi: 10.1016/j.coviro.2016.01.021
86. López-Camacho C, Abbink P, Larocca RA, Dejnirattisai W, Boyd M, Badamchi-Zadeh A, et al. Rational Zika vaccine design via the modulation of antigen membraneanchors in chimpanzee adenoviral vectors. Nat Commun (2018) 9(1):2441. doi: 10.1038/s41467-018-04859-5
87. Tai W, Chen J, Zhao G, Geng Q, He L, Chen Y, et al. Rational Design of Zika Virus Subunit Vaccine with EnhancedEfficacy. J Virol (2019) 93(17):e02187-18. doi: 10.1128/jvi.02187-18
88. Li Z, Song S, He M, Wang D, Shi J, Liu X, et al. Rational design of a triple-type human papillomavirus vaccine bycompromising viral-type specificity. Nat Commun (2018) 9(1):5360. doi: 10.1038/s41467-018-07199-6
89. Ong E, Wong MU, He Y. Identification of new features from known bacterial protectivevaccine antigens enhances rational vaccine design. Front Immunol (2017) 8:1382. doi: 10.3389/fimmu.2017.01382
90. Hurtgen BJ, Hung CY. Rational design to T lymphocyte epitope-based vaccines against Coccidioides infection. Methods Mol Biol (2017) 1625:45–64. doi: 10.1007/978-1-4939-7104-6_4
91. Martínez-Sáez N, Peregrina JM, Corzana F. Principles of mucin structure: Implications for the rational design of cancer vaccines derived from MUC1-glycopeptides. Chem Soc Rev (2017) 46:7154–75. doi: 10.1039/c6cs00858e
92. Karch CP, Burkhard P. Vaccine technologies: From whole organisms to rationally designed protein assemblies. Biochem Pharmacol (2016) 120:1–14. doi: 10.1016/j.bcp.2016.05.001
93. Zupančič E, Curato C, Paisana M, Rodrigues C, Porat Z, Viana AS, et al. Rational design of nanoparticles towards targeting antigen-presenting cells and improved T cell priming. J Control Release (2017) 258:182–95. doi: 10.1016/j.jconrel.2017.05.014
94. Porkolab V, Chabrol E, Varga N, Ordanini S, Sutkevičiute I, Thépaut M, et al. Rational-Differential Design of Highly Specific Glycomimetic Ligands: Targeting DC-SIGN and Excluding Langerin Recognition. ACS Chem Biol (2018) 13:600–8. doi: 10.1021/acschembio.7b00958
95. Oyarzún P, Kobe B. Recombinant and epitope-based vaccines on the road to the market and implications for vaccine design and production. Hum Vaccines Immunother (2016) 12:763–7. doi: 10.1080/21645515.2015.1094595
96. Liljeroos L, Malito E, Ferlenghi I, Bottomley MJ. Structural and Computational Biology in the Design of ImmunogenicVaccine Antigens. J Immunol Res (2015) 2015:156241. doi: 10.1155/2015/156241
97. Zhang X, Zhivaki D, Lo-Man R. Unique aspects of the perinatal immune system. Nat Rev Immunol (2017) 17:495–507. doi: 10.1038/nri.2017.54
98. Kollmann TR, Levy O, Montgomery RR, Goriely S. Innate Immune Function by Toll-like Receptors: Distinct Responses in Newborns and the Elderly. Immunity (2012) 37:771–83. doi: 10.1016/j.immuni.2012.10.014
99. Aiello A, Farzaneh F, Candore G, Caruso C, Davinelli S, Gambino CM, et al. Immunosenescence and its hallmarks: How to oppose aging strategically? A review of potential options for therapeutic intervention. Front Immunol (2019) 10:2247:2247. doi: 10.3389/fimmu.2019.02247
100. Hazeldine J, Lord JM. Innate immunesenescence: underlying mechanisms and clinical relevance. Biogerontology (2015) 16:187–201. doi: 10.1007/s10522-014-9514-3
101. Montgomery RR, Shaw AC. Paradoxical changes in innate immunity in aging: recent progress and new directions. J Leukoc Biol (2015) 98:937–43. doi: 10.1189/jlb.5mr0315-104r
102. Chinn IK, Blackburn CC, Manley NR, Sempowski GD. Changes in primary lymphoid organs with aging. Semin Immunol (2012) 24:309–20. doi: 10.1016/j.smim.2012.04.005
103. Kline GH, Hayden TA, Klinman NR. B cell maintenance in aged mice reflects both increased B cell longevity and decreased B cell generation. J Immunol (1999) 162:3342–9.
104. Stephan RP, Lill-Elghanian DA, Witte PL. Development of B cells in aged mice: decline in the ability of pro-Bcells to respond to IL-7 but not to other growth factors. J Immunol (1997) 158:1598–609.
105. Becklund BR, Purton JF, Ramsey C, Favre S, Vogt TK, Martin CE, et al. The aged lymphoid tissue environment fails to support naive T cellhomeostasis. Sci Rep (2016) 6:30842. doi: 10.1038/srep30842
106. Ferrucci L, Corsi A, Lauretani F, Bandinelli S, Bartali B, Taub DD, et al. The origins of age-related proinflammatory state. Blood (2005) 105:2294–9. doi: 10.1182/blood-2004-07-2599
107. De Martinis M, Franceschi C, Monti D, Ginaldi L. Inflamm-ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett (2005) 579:2035–9. doi: 10.1016/j.febslet.2005.02.055
108. Cumberbatch M, Dearman RJ, Kimber I. Influence of ageing on Langerhans cell migration in mice: Identification of a putative deficiency of epidermal interleukin-1α. Immunology (2002) 105:466–77. doi: 10.1046/j.1365-2567.2002.01381.x
109. Desai A, Grolleau-Julius A, Yung R. Leukocyte function in the aging immune system. J Leukoc Biol (2010) 87:1001–9. doi: 10.1189/jlb.0809542
110. Aprahamian T, Takemura Y, Goukassian D, Walsh K. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin Exp Immunol (2008) 152:448–55. doi: 10.1111/j.1365-2249.2008.03658.x
111. Cambier J. Immunosenescence: a problem of lymphopoiesis, homeostasis, microenvironment, and signaling. John Cambier. Immunol Rev (2005) 205:5–6. doi: 10.1111/j.0105-2896.2005.00276.x
112. Wertheimer AM, Bennett MS, Park B, Uhrlaub JL, Martinez C, Pulko V, et al. Aging and Cytomegalovirus Infection Differentially and Jointly Affect Distinct Circulating T Cell Subsets in Humans. J Immunol (2014) 192:2143–55. doi: 10.4049/jimmunol.1301721
113. Kogut I, Scholz JL, Cancro MP, Cambier JC. B cell maintenance and function in aging. Semin Immunol (2012) 24:342–9. doi: 10.1016/j.smim.2012.04.004
114. Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol (2011) 335:2–13. doi: 10.1016/j.mce.2010.04.005
115. Rea D, Van Kooten C, Van Meijgaarden KE, Ottenhoff THM, Melief CJM, Offringa R. Glucocorticoids transform CD40-triggering of dendritic cells into an alternative activation pathway resulting in antigen-presenting cells that secrete IL-10. Blood (2000) 95:3162–7. doi: 10.1182/blood.v95.10.3162
116. Rutella S, Lemoli RM. Regulatory T cells and tolerogenic dendritic cells: From basic biology to clinical applications. Immunol Lett (2004) 94:11–26. doi: 10.1016/j.imlet.2004.04.015
117. Luther C, Adamopoulou E, Stoeckle C, Brucklacher-Waldert V, Rosenkranz D, Stoltze L, et al. Prednisolone Treatment Induces Tolerogenic Dendritic Cells and a Regulatory Milieu in Myasthenia Gravis Patients. J Immunol (2009) 183:841–8. doi: 10.4049/jimmunol.0802046
118. Chamorro S, García-Vallejo JJ, Unger WWJ, Fernandes RJ, Bruijns SCM, Laban S, et al. TLR Triggering on Tolerogenic Dendritic Cells Results in TLR2 Up-Regulation and a Reduced Proinflammatory Immune Program. J Immunol (2009) 183:2984–94. doi: 10.4049/jimmunol.0801155
119. Pasquale A, Preiss S, Silva F, Garçon N. Vaccine Adjuvants: from 1920 to 2015 and Beyond.Vaccines (2015) 3:320–43. doi: 10.3390/vaccines3020320
120. Moyer TJ, Zmolek AC, Irvine DJ. Beyond antigens and adjuvants: Formulating futurevaccines. J Clin Invest (2016) 126:799–808. doi: 10.1172/JCI81083
121. Grogan JL, Mohrs M, Harmon B, Lacy DA, Sedat JW, Locksley RM. Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity (2001) 14:205–15. doi: 10.1016/S1074-7613(01)00103-0
122. Didierlaurent AM, Morel S, Lockman L, Giannini SL, Bisteau M, Carlsen H, et al. AS04, an Aluminum Salt- and TLR4 Agonist-Based Adjuvant System, Induces a Transient Localized Innate Immune Response Leading to Enhanced Adaptive Immunity. J Immunol (2009) 183:6186–97. doi: 10.4049/jimmunol.0901474
123. Kundi M. New hepatitis B vaccine formulated with an improved adjuvant system. Expert Rev Vaccines (2007) 6:133–40. doi: 10.1586/14760584.6.2.133
124. Olafsdottir TA, Lingnau K, Nagy E, Jonsdottir I. IC31 ® , a Two-Component Novel Adjuvant Mixed with a Conjugate Vaccine Enhances Protective Immunity against Pneumococcal Disease in Neonatal Mice. Scand J Immunol (2009) 69:194–202. doi: 10.1111/j.1365-3083.2008.02225.x
125. Knudsen NPH, Olsen A, Buonsanti C, Follmann F, Zhang Y, Coler RN, et al. Different human vaccine adjuvants promote distinct antigen-independent immunological signatures tailored to different pathogens. Sci Rep (2016) 6:1–13. doi: 10.1038/srep19570
126. Behzad H, Huckriede ALW, Haynes L, Gentleman B, Coyle K, Wilschut JC, et al. GLA-SE, a synthetic toll-like receptor 4 agonist, enhances T-cell responses to influenza vaccine in older adults. J Infect Dis (2012) 205:466–73. doi: 10.1093/infdis/jir769
127. Agger EM, Rosenkrands I, Hansen J, Brahimi K, Vandahl BS, Aagaard C, et al. Cationic Liposomes Formulated with Synthetic Mycobacterial Cordfactor (CAF01): A Versatile Adjuvant for Vaccines with Different Immunological Requirements. PloS One (2008) 3:e3116. doi: 10.1371/journal.pone.0003116
128. Elliott STC, Kallewaard NL, Benjamin E, Wachter-Rosati L, McAuliffe JM, Patel A, et al. DMAb inoculation of synthetic cross reactive antibodies protectsagainst lethal influenza A and B infections. NPJ Vaccines (2017) 2(18):1–18. doi: 10.1038/s41541-017-0020-x
129. Uccellini MB, Busto P, Debatis M, Marshak-Rothstein A, Viglianti GA. Selective binding of anti-DNA antibodies to native dsDNA fragmentsof differing sequence. Immunol Lett (2012) 143:85–91. doi: 10.1016/j.imlet.2012.01.003
130. Vallin H, Perers A, Alm GV, Ronnblom L. Anti-Double-Stranded DNA Antibodies and Immunostimulatory Plasmid DNA in Combination Mimic the Endogenous IFN-α Inducer in Systemic Lupus Erythematosus. J Immunol (1999) 163:6306–13.
131. Wellmann U, Letz M, Herrmann M, Angermuller S, Kalden JR, Winkler TH. The evolution of human anti-double-stranded DNAautoantibodies. Proc Natl Acad Sci (2005) 102:9258–63. doi: 10.1073/pnas.0500132102
132. Morel S, Didierlaurent A, Bourguignon P, Delhaye S, Baras B, Jacob V, et al. Adjuvant System AS03 containing α-tocopherol modulates innate immune response and leads to improved adaptive immunity. Vaccine (2011) 29:2461–73. doi: 10.1016/j.vaccine.2011.01.011
133. Henriksen-Lacey M, Christensen D, Bramwell VW, Lindenstrøm T, Agger EM, Andersen P, et al. Liposomal cationic charge and antigen adsorption are important properties for the efficient deposition of antigen at the injection site and ability of the vaccine to induce a CMI response. J Control Release (2010) 145:102–8. doi: 10.1016/j.jconrel.2010.03.027
134. van Dissel JT, Joosten SA, Hoff ST, Soonawala D, Prins C, Hokey DA, et al. A novel liposomal adjuvant system, CAF01, promotes long-lived Mycobacterium tuberculosis-specific T-cell responses in human. Vaccine (2014) 32:7098–107. doi: 10.1016/j.vaccine.2014.10.036
135. Garçon N, Chomez P, Van Mechelen M. GlaxoSmithKline Adjuvant Systems in vaccines: Concepts, achievements and perspectives. Expert Rev Vaccines (2007) 6:723–39. doi: 10.1586/14760584.6.5.723
136. Bejon P, Lusingu J, Olotu A, Leach A, Lievens M, Vekemans J, et al. Efficacy of RTS,S/AS01E vaccine against malaria in children 5 to 17 months of age. N Engl J Med (2008) 359:2521–32. doi: 10.1056/NEJMoa0807381
137. Olotu A, Lusingu J, Leach A, Lievens M, Vekemans J, Msham S, et al. Efficacy of RTS,S/AS01E malaria vaccine and exploratory analysis on anti-circumsporozoite antibody titres and protection in children aged 5-17 months in Kenya and Tanzania: A randomised controlled trial. Lancet Infect Dis (2011) 11:102–9. doi: 10.1016/S1473-3099(10)70262-0
138. Stanberry L, Strugnell R. Vaccines of the future. Perspect Vaccinol (2011) 1:151–99. doi: 10.1016/j.pervac.2011.05.006
139. Duthie MS, Windish HP, Fox CB, Reed SG. Use of defined TLR ligands as adjuvants within humanvaccines. Immunol Rev (2011) 239:178–96. doi: 10.1111/j.1600-065X.2010.00978.x
140. Gnjatic S, Sawhney NB, Bhardwaj N. Toll-like receptor agonists are they good adjuvants? Cancer J (2010) 16:382–91. doi: 10.1097/PPO.0b013e3181eaca65
141. Jones SC, Brahmakshatriya V, Huston G, Dibble J, Swain SL. TLR-Activated Dendritic Cells Enhance the Response of Aged Naive CD4 T Cells via an IL-6–Dependent Mechanism. J Immunol (2010) 185:6783–94. doi: 10.4049/jimmunol.0901296
142. Gill MA, Palucka AK, Barton T, Ghaffar F, Jafri H, Banchereau J, et al. Mobilization of plasmacytoid and myeloid dendritic cells to mucosal sites in children with respiratory syncytial virus and other viral respiratory infections. J Infect Dis (2005) 191:1105–15. doi: 10.1086/428589
143. Dimmock NJ, Easton AJ. Defective Interfering Influenza Virus RNAs: Time To Reevaluate Their Clinical Potential as Broad-Spectrum Antivirals? J Virol (2014) 88:5217–27. doi: 10.1128/JVI.03193-13
144. Dimmock NJ, Rainsford EW, Scott PD, Marriott AC. Influenza Virus Protecting RNA: an Effective Prophylactic and Therapeutic Antiviral. J Virol (2008) 82:8570–8. doi: 10.1128/jvi.00743-08
145. Duhaut SD, Dimmock NJ. Defective influenza A virus generated entirely from plasmids: Its RNA is expressed in infected mouse lung and modulates disease. J Virol Methods (2003) 108:75–82. doi: 10.1016/S0166-0934(02)00260-4
146. Scott PD, Meng B, Marriott AC, Easton AJ, Dimmock NJ. Defective interfering virus protects elderly mice from influenza. Virol J (2011) 8:212. doi: 10.1186/1743-422X-8-212
147. Dimmock NJ, Dove BK, Scott PD, Meng B, Taylor I, Cheung L, et al. Cloned Defective Interfering Influenza Virus Protects Ferrets from Pandemic 2009 Influenza A Virus and Allows Protective Immunity to Be Established. PloS One (2012) 7:49394. doi: 10.1371/journal.pone.0049394
148. Dimmock NJ, Dove BK, Meng B, Scott PD, Taylor I, Cheung L, et al. Comparison of the protection of ferrets against pandemic 2009 influenza A virus (H1N1) by 244 DI influenza virus and oseltamivir. Antiviral Res (2012) 96:376–85. doi: 10.1016/j.antiviral.2012.09.017
149. Scott PD, Meng B, Marriott AC, Easton AJ, Dimmock NJ. Defective interfering influenza virus confers only short-lived protection against influenza virus disease: Evidence for a role for adaptive immunity in DI virus-mediated protection in vivo. Vaccine (2011) 29:6584–91. doi: 10.1016/j.vaccine.2011.06.114
150. Scott PD, Meng B, Marriott AC, Easton AJ, Dimmock NJ. Defective interfering influenza A virus protects in vivo against disease caused by a heterologous influenza B virus. J Gen Virol (2011) 92:2122–32. doi: 10.1099/vir.0.034132-0
151. Easton AJ, Scott PD, Edworthy NL, Meng B, Marriott AC, Dimmock NJ. A novel broad-spectrum treatment for respiratory virus infections: Influenza-based defective interfering virus provides protection against pneumovirus infection in vivo. Vaccine (2011) 29:2777–84. doi: 10.1016/j.vaccine.2011.01.102
152. Yang Y, Lyu T, Zhou R, He X, Ye K, Xie Q, et al. The antiviral and antitumor effects of defective interfering particles/genomes and their mechanisms. Front Microbiol (2019) 10:1852:1852. doi: 10.3389/fmicb.2019.01852
153. Pravetoni M. Biologics to treat substance use disorders: Current status and new directions. Hum Vaccines Immunother (2016) 12:3005–19. doi: 10.1080/21645515.2016.1212785
154. Carrera MRA, Ashley JA, Parsons LH, Wirsching P, Koob GF, Janda KD. Suppression of psychoactive effects of cocaine by active immunization. Nature (1995) 378:727–30. doi: 10.1038/378727a0
155. Jalah R, Torres OB, Mayorov AV, Li F, Antoline JFG, Jacobson AE, et al. Efficacy, but Not Antibody Titer or Affinity, of a Heroin Hapten Conjugate Vaccine Correlates with Increasing Hapten Densities on Tetanus Toxoid, but Not on CRM197 Carriers. Bioconjug Chem (2015) 26:1041–53. doi: 10.1021/acs.bioconjchem.5b00085
156. McCluskie MJ, Thorn J, Mehelic PR, Kolhe P, Bhattacharya K, Finneman JI, et al. Molecular attributes of conjugate antigen influence function of antibodies induced by anti-nicotine vaccine in mice and non-human primates. Int Immunopharmacol (2015) 25:518–27. doi: 10.1016/j.intimp.2015.02.030
157. Kimishima A, Wenthur CJ, Zhou B, Janda KD. An advance in prescription opioid vaccines: Overdose mortality reduction and extraordinary alteration of drug half-life. ACS Chem Biol (2017) 12:36–40. doi: 10.1021/acschembio.6b00977
158. Bremer PT, Kimishima A, Schlosburg JE, Zhou B, Collins KC, Janda KD. Combatting Synthetic Designer Opioids: A Conjugate Vaccine Ablates Lethal Doses of Fentanyl Class Drugs. Angew Chemie Int Ed (2016) 55:3772–5. doi: 10.1002/anie.201511654
159. Collins KC, Schlosburg JE, Bremer PT, Janda KD. Methamphetamine Vaccines: Improvement through Hapten Design. J Med Chem (2016) 59:3878–85. doi: 10.1021/acs.jmedchem.6b00084
160. Bremer PT, Janda KD. Conjugate Vaccine Immunotherapy for Substance Use Disorder. Pharmacol Rev (2017) 69:298–315. doi: 10.1124/pr.117.013904
161. Sulima A, Jalah R, Antoline JFG, Torres OB, Imler GH, Deschamps JR, et al. A Stable Heroin Analogue That Can Serve as a Vaccine Hapten to Induce Antibodies That Block the Effects of Heroin and Its Metabolites in Rodents and That Cross-React Immunologically with Related Drugs of Abuse. J Med Chem (2018) 61:329–43. doi: 10.1021/acs.jmedchem.7b01427
162. Nguyen JD, Bremer PT, Ducime A, Creehan KM, Kisby BR, Taffe MA, et al. Active vaccination attenuates the psychostimulant effects of α-PVP and MDPV in rats. Neuropharmacology (2017) 116:1–8. doi: 10.1016/j.neuropharm.2016.12.005
163. Isomura S, Wirsching P, Janda KD. An immunotherapeutic program for the treatment of nicotine addiction: Hapten design and synthesis. J Org Chem (2001) 66:4115–21. doi: 10.1021/jo001442w
164. Peterson EC, Gunnell M, Che Y, Goforth RL, Carroll FI, Henry R, et al. Using hapten design to discover therapeutic monoclonal antibodies for treating methamphetamine abuse. J Pharmacol Exp Ther (2007) 322:30–9. doi: 10.1124/jpet.106.117150
165. Moreno AY, Mayorov AV, Janda KD. Impact of distinct chemical structures for the development of a methamphetamine vaccine. J Am Chem Soc (2011) 133:6587–95. doi: 10.1021/ja108807j
166. Stowe GN, Vendruscolo LF, Edwards S, Schlosburg JE, Misra KK, Schulteis G, et al. A vaccine strategy that induces protective immunity against heroin. J Med Chem (2011) 54:5195–204. doi: 10.1021/jm200461m
167. Pravetoni M, Le Naour M, Harmon TM, Tucker AM, Portoghese PS, Pentel PR. An oxycodone conjugate vaccine elicits drug-specific antibodies that reduce oxycodone distribution to brain and hot-plate analgesia. J Pharmacol Exp Ther (2012) 341:225–32. doi: 10.1124/jpet.111.189506
168. Pryde DC, Jones LH, Gervais DP, Stead DR, Blakemore DC, Selby MD, et al. Selection of a Novel Anti-Nicotine Vaccine: Influence of Antigen Design on Antibody Function in Mice. PloS One (2013) 8:e76557. doi: 10.1371/journal.pone.0076557
169. Pravetoni M, Le Naour M, Tucker AM, Harmon TM, Hawley TM, Portoghese PS, et al. Reduced antinociception of opioids in rats and mice by vaccination with immunogens containing oxycodone and hydrocodone haptens. J Med Chem (2013) 56:915–23. doi: 10.1021/jm3013745
170. Collins KC, Janda KD. Investigating hapten clustering as a strategy to enhance vaccines against drugs of abuse. Bioconjug Chem (2014) 25:593–600. doi: 10.1021/bc500016k
171. Stevens MW, Gunnell MG, Tawney R, Owens SM. Optimization of a methamphetamine conjugate vaccine for antibody production in mice. Int Immunopharmacol (2016) 35:137–41. doi: 10.1016/j.intimp.2016.03.028
172. Bremer PT, Schlosburg JE, Banks ML, Steele FF, Zhou B, Poklis JL, et al. Development of a Clinically Viable Heroin Vaccine. J Am Chem Soc (2017) 139:8601–11. doi: 10.1021/jacs.7b03334
173. Pravetoni M, Pentel PR, Potter DN, Chartoff EH, Tally L, LeSage MG. Effects of an oxycodone conjugate vaccine on oxycodone self-administration and oxycodone-induced brain gene expression in rats. PloS One (2014) 9:101807. doi: 10.1371/journal.pone.0101807
174. Kosten TR, Rosen M, Bond J, Settles M, Roberts JSC, Shields J, et al. Human therapeutic cocaine vaccine: Safety and immunogenicity. Vaccine (2002) 20:1196–204. doi: 10.1016/S0264-410X(01)00425-X
175. Martell BA, Mitchell E, Poling J, Gonsai K, Kosten TR. Vaccine pharmacotherapy for the treatment of cocaine dependence. Biol Psychiatry (2005) 58:158–64. doi: 10.1016/j.biopsych.2005.04.032
176. Moreno AY, Azar MR, Warren NA, Dickerson TJ, Koob GF, Janda KD. A critical evaluation of a nicotine vaccine within a self-administration behavioral model. Mol Pharm (2010) 7:431–41. doi: 10.1021/mp900213u
177. Rüedi-Bettschen D, Wood SL, Gunnell MG, West CM, Pidaparthi RR, Carroll FI, et al. Vaccination protects rats from methamphetamine-induced impairment of behavioral responding for food. Vaccine (2013) 31:4596–602. doi: 10.1016/j.vaccine.2013.07.038
178. Cornish KE, De Villiers SHL, Pravetoni M, Pentel PR. Immunogenicity of individual vaccine components in a bivalent nicotine vaccine differ according to vaccine formulation and administration conditions. PloS One (2013) 8:82557. doi: 10.1371/journal.pone.0082557
179. Kosten TR, Domingo CB, Shorter D, Orson F, Green C, Somoza E, et al. Vaccine for cocaine dependence: A randomized double-blind placebo-controlled efficacy trial. Drug Alcohol Depend (2014) 140:42–7. doi: 10.1016/j.drugalcdep.2014.04.003
180. Pravetoni M, Vervacke JS, Distefano MD, Tucker AM, Laudenbach M, Pentel PR. Effect of Currently Approved Carriers and Adjuvants on the Pre-Clinical Efficacy of a Conjugate Vaccine against Oxycodone in Mice and Rats. PloS One (2014) 9:e96547. doi: 10.1371/journal.pone.0096547
181. McCluskie MJ, Thorn J, Gervais DP, Stead DR, Zhang N, Benoit M, et al. Anti-nicotine vaccines: Comparison of adjuvanted CRM197 and Qb-VLP conjugate formulations for immunogenicity and function in non-human primates. Int Immunopharmacol (2015) 29:663–71. doi: 10.1016/j.intimp.2015.09.012
182. Schlosburg JE, Vendruscolo LF, Bremer PT, Lockner JW, Wade CL, Nunes AAK, et al. Dynamic vaccine blocks relapse to compulsive intake of heroin. Proc Natl Acad Sci U S A (2013) 110:9036–41. doi: 10.1073/pnas.1219159110
183. Cornuz J, Zwahlen S, Jungi WF, Osterwalder J, Klingler K, van Melle G, et al. A vaccine against nicotine for smoking cessation: A randomizedcontrolled trial. PloS One (2008) 3(6):e2547. doi: 10.1371/journal.pone.0002547
184. Hatsukami DK, Jorenby DE, Gonzales D, Rigotti NA, Glover ED, Oncken CA, et al. Immunogenicity and smoking-cessation outcomes for a novel nicotine immunotherapeutic. Clin Pharmacol Ther (2011) 89:392–9. doi: 10.1038/clpt.2010.317
185. Fahim RE, Kessler PD, Kalnik MW. Therapeutic vaccines against tobacco addiction. Expert Rev Vaccines (2013) 12:333–42. doi: 10.1586/erv.13.13
186. Martell BA, Orson FM, Poling J, Mitchell E, Rossen RD, Gardner T, et al. Cocaine vaccine for the treatment of cocaine dependence in methadone-maintained patients: A randomized, double-blind, placebo-controlled efficacy trial. Arch Gen Psychiatry (2009) 66:1116–23. doi: 10.1001/archgenpsychiatry.2009.128
187. Townsend EA, Banks ML. Preclinical Evaluation of Vaccines to Treat Opioid Use Disorders: How Close are We to a Clinically Viable Therapeutic? CNS Drugs (2020) 34:449–61. doi: 10.1007/s40263-020-00722-8
188. Raya BA, Edward KM, Scheifele DW, Halperin SA. Pertussis and influenza immunisation during pregnancy: a landscape review. Lancet Infect Dis (2017) 17:209–22. doi: 10.1016/S1473-3099(17)30190-1
189. Hozbor D. New Pertussis Vaccines: A Need and aChallenge. Pertussis Infection and Vaccines (book), New Pertussis Vaccines: A Need and a Challenge (book section) (2019). pp. 115–26. doi: 10.1007/5584_2019_407
190. Kitchin NRE. Review of diphtheria, tetanus and pertussis vaccines in clinical development. Expert Rev Vaccines (2011) 10:605–15. doi: 10.1586/erv.11.60
191. Angsantikul P, Fang RH, Zhang L. Toxoid Vaccination against Bacterial Infection Using Cell Membrane-Coated Nanoparticles. Bioconjug Chem (2018) 29:604–12. doi: 10.1021/acs.bioconjchem.7b00692
192. Li L, Goedegebuure SP, Gillanders WE. Preclinical and clinical development of neoantigen vaccines. Ann Oncol (2017) 28:xii11–7. doi: 10.1093/annonc/mdx681
193. Aurisicchio L, Pallocca M, Ciliberto G, Palombo F. The perfect personalized cancer therapy: Cancer vaccines against neoantigens. J Exp Clin Cancer Res (2018) 37:86. doi: 10.1186/s13046-018-0751-1
194. Maeng HM, Berzofsky JA. Strategies for developing and optimizing cancervaccines. F1000Research (2019) 8:654. doi: 10.12688/f1000research.18693.1
195. Tagliamonte M, Petrizzo A, Tornesello ML, Buonaguro FM, Buonaguro L. Antigen-specific vaccines for cancer treatment. Hum Vaccines Immunother (2014) 10:3332–46. doi: 10.4161/21645515.2014.973317
196. Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Sci (80) (2018) 359:1355–60. doi: 10.1126/science.aar7112
197. Lopes A, Vandermeulen G, Préat V. Cancer DNA vaccines: current preclinical and clinical developmentsand future perspectives. J Exp Clin Cancer Res (2019) 38:146. doi: 10.1186/s13046-019-1154-7
198. Sayour EJ, Mendez-Gomez HR, Mitchell DA. Cancer vaccine immunotherapy with RNA-loadedliposomes. Int J Mol Sci (2018) 19:2890. doi: 10.3390/ijms19102890
199. Granstein RD, Ding W, Ozawa H. Induction of anti-tumor immunity with epidermal cells pulsed with tumor- derived RNA or intradermal administration of RNA. J Invest Dermatol (2000) 114:632–6. doi: 10.1046/j.1523-1747.2000.00929.x
200. Scheel B, Aulwurm S, Probst J, Stitz L, Hoerr I, Rammensee H-G, et al. Therapeutic anti-tumor immunity triggered by injections of immunostimulating single-stranded RNA. Eur J Immunol (2006) 36:2807–16. doi: 10.1002/eji.200635910
201. Van Lint S, Goyvaerts C, Maenhout S, Goethals L, Disy A, Benteyn D, et al. Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res (2012) 72:1661–71. doi: 10.1158/0008-5472.CAN-11-2957
202. Bialkowski L, Van Weijnen A, Van Der Jeught K, Renmans D, Daszkiewicz L, Heirman C, et al. Intralymphatic mRNA vaccine induces CD8 T-cell responses that inhibit the growth of mucosally located tumours. Sci Rep (2016) 6:1–15. doi: 10.1038/srep22509
203. Kübler H, Scheel B, Gnad-Vogt U, Miller K, Schultze-Seemann W, vom Dorp F, et al. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: a first-in-man phase I/IIa study. J Immunother Cancer (2015) 3:26. doi: 10.1186/s40425-015-0068-y
204. Audouy SAL, De Leij LFMH, Hoekstra D, Molema G. In vivo characteristics of cationic liposomes as delivery vectors for gene therapy. Pharm Res (2002) 19:1599–605. doi: 10.1023/A:1020989709019
205. Lorenzer C, Dirin M, Winkler AM, Baumann V, Winkler J. Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J Control Release (2015) 203:1–15. doi: 10.1016/j.jconrel.2015.02.003
206. Hess PR, Boczkowski D, Nair SK, Snyder D, Gilboa E. Vaccination with mRNAs encoding tumor-associated antigens and granulocyte-macrophage colony-stimulating factor efficiently primes CTL responses, but is insufficient to overcome tolerance to a model tumor/self antigen. Cancer Immunol Immunother (2006) 55:672–83. doi: 10.1007/s00262-005-0064-z
207. Phua KKL, Staats HF, Leong KW, Nair SK. Intranasal mRNA nanoparticle vaccination induces prophylactic andtherapeutic anti-tumor immunity. Sci Rep (2014) 4:5128. doi: 10.1038/srep05128
208. Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A, et al. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett (2017) 17:1326–35. doi: 10.1021/acs.nanolett.6b03329
209. Lu C, Stewart DJ, Lee JJ, Ji L, Ramesh R, Jayachandran G, et al. Phase I clinical trial of systemically administeredTUSC2(FUS1)-nanoparticles mediating functional gene transfer in humans. PloS One (2012) 7(4):e34833. doi: 10.1371/journal.pone.0034833
210. Cho EA, Moloney FJ, Cai H, Au-Yeung A, China C, Scolyer RA, et al. Safety and tolerability of an intratumorally injected DNAzyme, Dz13, in patients with nodular basal-cell carcinoma: A phase 1 first-in-human trial (DISCOVER). Lancet (2013) 381:1835–43. doi: 10.1016/S0140-6736(12)62166-7
211. Wagner MJ, Mitra R, Mcarthur MJ, Baze W, Barnhart K, Wu SY, et al. Preclinical mammalian safety studies of EPHARNA (DOPC Nanoliposomal EphA2-Targeted siRNA). Mol Cancer Ther (2017) 16:1114–23. doi: 10.1158/1535-7163.MCT-16-0541
212. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N Engl J Med (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
213. Stadler CR, Bähr-Mahmud H, Celik L, Hebich B, Roth AS, Roth RP, et al. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat Med (2017) 23:815–7. doi: 10.1038/nm.4356
214. Wilgenhof S, Van Nuffel AMT, Benteyn D, Corthals J, Aerts C, Heirman C, et al. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann Oncol (2013) 24:2686–93. doi: 10.1093/annonc/mdt245
215. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature (2016) 534:396–401. doi: 10.1038/nature18300
216. McNamara MA, Nair SK, Holl EK. RNA-Based Vaccines in Cancer Immunotherapy. J Immunol Res (2015) 2015:794528. doi: 10.1155/2015/794528
217. Sayour EJ, De Leon G, Pham C, Grippin A, Kemeny H, Chua J, et al. Systemic activation of antigen-presenting cells via RNA-Loadednanoparticles. Oncoimmunology (2017) 6(1):e1256527. doi: 10.1080/2162402X.2016.1256527
218. Wilgenhof S, Corthals J, Heirman C, Van Baren N, Lucas S, Kvistborg P, et al. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patientswith pretreated advanced melanoma. J Clin Oncol (2016) 34:1330–8. doi: 10.1200/JCO.2015.63.4121
219. Wang Y, Zhang L, Xu Z, Miao L, Huang L. mRNA Vaccine with Antigen-Specific Checkpoint Blockade Induces an Enhanced Immune Response against Established Melanoma. Mol Ther (2018) 26:420–34. doi: 10.1016/j.ymthe.2017.11.009
220. Tan YS, Sansanaphongpricha K, Prince MEP, Sun D, Wolf GT, Lei YL. Engineering Vaccines to Reprogram Immunity against Head and Neck Cancer. J Dent Res (2018) 97:627–34. doi: 10.1177/0022034518764416
221. Mastelic-Gavillet B, Balint K, Boudousquie C, Gannon PO, Kandalaft LE. Personalized dendritic cell vaccines-recent breakthroughs andencouraging clinical results. Front Immunol (2019) 10:766. doi: 10.3389/fimmu.2019.00766
222. Pollard C, De Koker S, Saelens X, Vanham G, Grooten J. Challenges and advances towards the rational design of mRNA vaccines. Trends Mol Med (2013) 19:705–13. doi: 10.1016/j.molmed.2013.09.002
223. Becker PD, Guzmán CA. Rational design of vaccination strategies to promote antigen entryinto the MHC class I-restricted presentation pathway. Transfus MedHemother (2017) 77:398–411. doi: 10.1159/000082484
224. Gatti-Mays ME, Redman JM, Collins JM, Bilusic M. Cancer vaccines: Enhanced immunogenic modulation through therapeutic combinations. Hum Vaccines Immunother (2017) 13:2561–74. doi: 10.1080/21645515.2017.1364322
225. Vermaelen K. Vaccine strategies to improve anticancer cellular immuneresponses. Front Immunol (2019) 10:8. doi: 10.3389/fimmu.2019.00008
226. Perez CR, De Palma M. Engineering dendritic cell vaccines to improve cancerimmunotherapy. Nat Commun (2019) 10:5408. doi: 10.1038/s41467-019-13368-y
227. Sander AF, Lollini P-L. Virus-like antigen display for cancer vaccine development, what is the potential? Expert Rev Vaccines (2018) 17:285–8. doi: 10.1080/14760584.2018.1455505
228. Wen R, Umeano AC, Kou Y, Xu J, Farooqi AA. Nanoparticle systems for cancer vaccine. Nanomedicine (2019) 8:627–48. doi: 10.2217/nnm-2018-0147
229. Butts C, Socinski MA, Mitchell PL, Thatcher N, Havel L, Krzakowski M, et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): A randomised, double-blind, phase 3 trial. Lancet Oncol (2014) 15:59–68. doi: 10.1016/S1470-2045(13)70510-2
230. Rosalia RA, Cruz LJ, van Duikeren S, Tromp AT, Silva AL, Jiskoot W, et al. CD40-targeted dendritic cell delivery of PLGA-nanoparticle vaccines induce potent anti-tumor responses. Biomaterials (2015) 40:88–97. doi: 10.1016/j.biomaterials.2014.10.053
231. Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro Oncol (2015) 17:854–61. doi: 10.1093/neuonc/nou348
232. Riabov V, Tretyakova I, Alexander RB, Pushko P, Klyushnenkova EN. Anti-tumor effect of the alphavirus-based virus-like particle vector expressing prostate-specific antigen in a HLA-DR transgenic mouse model of prostate cancer. Vaccine (2015) 33:5386–95. doi: 10.1016/j.vaccine.2015.08.062
233. Singer C, Pfeiler G, Hubalek M, Bartsch R, Stoeger H, Pichler A, et al. Efficacy and safety of the therapeutic cancervaccine tecemotide (L-BLP25) in early breast cancer: Results from a prospective, randomized,neoadjuvant phase-II study (ABCSG-34). Eur J Cancer (2020) 132:43–52. doi: doi:10.1016/j.ejca.2020.03.018
234. Varypataki EM, Benne N, Bouwstra J, Jiskoot W, Ossendorp F. Efficient eradication of established tumors in mice with cationic liposome-based synthetic long-peptide vaccines. Cancer Immunol Res (2017) 5:222–33. doi: 10.1158/2326-6066.CIR-16-0283
235. Zupančič E, Curato C, Kim JS, Yeini E, Porat Z, Viana AS, et al. Nanoparticulate vaccine inhibits tumor growth via improved T cell recruitment into melanoma and huHER2 breast cancer. Nanomed Nanotechnol Biol Med (2018) 14:835–47. doi: 10.1016/j.nano.2017.12.011
236. Stanley M. Tumour virus vaccines: Hepatitis B virus and humanpapillomavirus. Philos Trans R Soc B Biol Sci (2017) 372:20160268. doi: 10.1098/rstb.2016.0268
237. Tegerstedt K, Lindencrona JA, Curcio C, Andreasson K, Tullus C, Forni G, et al. A single vaccination with polyomavirus VP1/VP2Her2 virus-like particles prevents outgrowth of HER-2/neu-expressing tumors. Cancer Res (2005) 65:5953–7. doi: 10.1158/0008-5472.CAN-05-0335
238. Tegerstedt K, Franzén A, Ramqvist T, Dalianis T. Dendritic cells loaded with polyomavirus VP1/VP2Her2 virus-like particles efficiently prevent outgrowth of a Her2/neu expressing tumor. Cancer Immunol Immunother (2007) 56:1335–44. doi: 10.1007/s00262-007-0281-8
239. Andreasson K, Tegerstedt K, Eriksson M, Curcio C, Cavallo F, Forni G, et al. Murine pneumotropic virus chimeric Her2/ neu virus-like particles as prophylactic and therapeutic vaccines against Her2/ neu expressing tumors. Int J Cancer (2009) 124:150–6. doi: 10.1002/ijc.23920
240. Jalali SA, Sankian M, Tavakkol-Afshari J, Jaafari MR. Induction of tumor-specific immunity by multi-epitope rat HER2/neu-derived peptides encapsulated in LPD Nanoparticles. Nanomed Nanotechnol Biol Med (2012) 8:692–701. doi: 10.1016/j.nano.2011.09.010
241. Singer J, Manzano-Szalai K, Fazekas J, Thell K, Bentley-Lukschal A, Stremnitzer C, et al. Proof of concept study with an HER-2 mimotope anticancer vaccinededuced from a novel AAV-mimotope library platform. Oncoimmunology (2016) 5(7):e1171446. doi: 10.1080/2162402X.2016.1171446
242. Shukla S, Myers JT, Woods SE, Gong X, Czapar AE, Commandeur U, et al. Plant viral nanoparticles-based HER2 vaccine: Immune response influenced by differential transport, localization and cellular interactions of particulate carriers. Biomaterials (2017) 121:15–27. doi: 10.1016/j.biomaterials.2016.12.030
243. Arab A, Nicastro J, Slavcev R, Razazan A, Barati N, Nikpoor AR, et al. Lambda phage nanoparticles displaying HER2-derived E75 peptide induce effective E75-CD8+ T response. Immunol Res (2018) 66:200–6. doi: 10.1007/s12026-017-8969-0
244. Palladini A, Thrane S, Janitzek CM, Pihl J, Clemmensen SB, de Jongh WA, et al. Virus-like particle display of HER2 induces potent anti-cancerresponses. Oncoimmunology (2018) 7(3):e1408749. doi: 10.1080/2162402X.2017.1408749
245. Wiedermann U, Wiltschke C, Jasinska J, Kundi M, Zurbriggen R, Garner-Spitzer E, et al. A virosomal formulated Her-2/neu multi-peptide vaccine induces Her-2/neu-specific immune responses in patients with metastatic breast cancer: A phase i study. Breast Cancer Res Treat (2010) 119:673–83. doi: 10.1007/s10549-009-0666-9
246. Mason NJ, Gnanandarajah JS, Engiles JB, Gray F, Laughlin D, Gaurnier-Hausser A, et al. Immunotherapy with a HER2-Targeting listeria induces HER2-Specific immunity and demonstrates potential therapeutic effects in a phase I trial in canine osteosarcoma. Clin Cancer Res (2016) 22:4380–90. doi: 10.1158/1078-0432.CCR-16-0088
247. Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, et al. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat Nanotechnol (2016) 11:295–303. doi: 10.1038/nnano.2015.292
248. Neek M, Il KT, Wang SW. Protein-based nanoparticles in cancer vaccine development. Nanomed Nanotechnol Biol Med (2019) 15:164–74. doi: 10.1016/j.nano.2018.09.004
249. World Health Organization. List of Blueprint priority diseases. Available at: http://www.emro.who.int/pandemic-epidemic-diseases/news/list-of-blueprint-priority-diseases.html. (2018).
250. Isanaka S, Guindo O, Langendorf C, Matar Seck A, Plikaytis BD, Sayinzoga-Makombe N, et al. Efficacy of a Low-Cost, Heat-Stable Oral Rotavirus Vaccine in Niger. N Engl J Med (2017) 376:1121–30. doi: 10.1056/NEJMoa1609462
251. Fda, Cber. . Available at: https://www.fda.gov/media/133748/download (Accessed September 1, 2020).
252. Kis Z, Shattock R, Shah N, Kontoravdi C. Emerging Technologies for Low-Cost, Rapid VaccineManufacture. Biotechnol J (2019) 14:1–14. doi: 10.1002/biot.201800376
253. Berlanda Scorza F, Tsvetnitsky V, Donnelly JJ. Universal influenza vaccines: Shifting to better vaccines. Vaccine (2016) 34:2926–33. doi: 10.1016/J.VACCINE.2016.03.085
254. Gilbert SC, Warimwe GM. Rapid development of vaccines against emerging pathogens: The replication-deficient simian adenovirus platform technology. Vaccine (2017) 35:4461–4. doi: 10.1016/J.VACCINE.2017.04.085
255. Huang Y, Diazgranados C, Janes H, Huang Y, DeCamp AC, Metch B, et al. Selection of HIV vaccine candidates for concurrent testing in an efficacy trial. Curr Opin Virol (2016) 17:57–65. doi: 10.1016/j.coviro.2016.01.007
256. Warimwe GM, Gesharisha J, Carr BV, Otieno S, Otingah K, Wright D, et al. Chimpanzee adenovirus vaccine provides multispecies protectionagainst rift valley fever. Sci Rep (2016) 6:20617. doi: 10.1038/srep20617
257. Plotkin SA. Vaccines for epidemic infections and the role of CEPI. Hum Vaccin Immunother (2017) 13:2755–62. doi: 10.1080/21645515.2017.1306615
258. Seib K, Pollard AJ, de Wals P, Andrews RM, Zhou F, Hatchett RJ, et al. Policy making for vaccine use as a driver of vaccine innovation and development in the developed world. Vaccine (2017) 35:1380–9. doi: 10.1016/J.VACCINE.2016.10.080
259. 172 countries & multiple candidate vaccines engaged in COVID-19 Vaccine Global Access Facility – CEPI . Available at: https://cepi.net/news_cepi/172-countries-multiple-candidate-vaccines-engaged-in-covid-19-vaccine-global-access-facility/ (Accessed September 1, 2020).
260. Wu NC, Lv H, Thompson AJ, Paulson JC, Mok CKP, Wilson IA. Preventing an Antigenically Disruptive Mutation in Egg-Based H3N2 Seasonal Influenza Vaccines by Mutational Incompatibility. Cell Host Microbe (2019) 25:836–44. doi: 10.1016/j.chom.2019.04.013
261. Graham BS, Mascola JR, Fauci AS. Novel vaccine technologies: Essential Components of an AdequateResponse to Emerging Viral Diseases. JAMA (2018) 319:1431. doi: 10.1001/jama.2018.0345
262. Lurie N, Saville M, Hatchett R, Halton J. Developing Covid-19 Vaccines at Pandemic Speed. N Engl J Med (2020) 382:1969–73. doi: 10.1056/NEJMp2005630
263. Chen WH, Strych U, Hotez PJ, Bottazzi ME. The SARS-CoV-2 Vaccine Pipeline: an Overview. Curr Trop Med Rep (2020) 7:61–4. doi: 10.1007/s40475-020-00201-6
264. Amanat F, Krammer F. SARS-CoV-2 Vaccines: Status Report. Immunity (2020) 52:583–9. doi: 10.1016/j.immuni.2020.03.007
265. Jiang S. Don’t rush to deploy COVID-19 vaccines and drugs without sufficient safety guarantees. Nature (2020) 579:321. doi: 10.1038/d41586-020-00751-9
266. Lv H, Wu NC, Mok CKP. COVID-19 vaccines: knowing the unknown. EurJ Immunol (2020) 50:939–43. doi: 10.1002/eji.202048663
267. Iwasaki A, Yang Y. The potential danger of suboptimal antibody responses in COVID-19. Nat Rev Immunol (2020) 20:339–41. doi: 10.1038/s41577-020-0321-6
268. Usher AD. COVID-19 vaccines for all? Lancet (2020) 395:1822–3. doi: 10.1016/s0140-6736(20)31354-4
269. Yamey G, Schäferhoff M, Hatchett R, Pate M, Zhao F, McDade KK. Ensuring global access to COVID-19 vaccines. Lancet (2020) 395:1405–6. doi: 10.1016/S0140-6736(20)30763-7
270. Survival of the Most: Why Multiple Vaccines May Be the Best Way to Stop the COVID-19 Pandemic . Available at: https://www.massgeneral.org/news/coronavirus/research/vaccines/multiple-vaccines-best-way-stop-covid-19-pandemic (Accessed August 30, 2020).
271. Multiple vaccines could be needed to fully stop COVID-19 pandemic, UW vaccinologist says - GeekWire . Available at: https://www.geekwire.com/2020/multiple-vaccines-needed-fully-stop-covid-19-pandemic-uw-vaccinologist-says/ (Accessed August 30, 2020).
272. Safety and Immunogenicity Study of 2019-nCoV Vaccine (mRNA-1273) for Prophylaxis of SARS-CoV-2 Infection (COVID-19) - Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT04283461 (Accessed July 4, 2020).
273. A Study to Evaluate the Safety, Reactogenicity and Immunogenicity of Vaccine CVnCoV in Healthy Adults - Full Text View - ClinicalTrials.gov . Available at: https://www.clinicaltrials.gov/ct2/show/NCT04449276 (Accessed July 4, 2020).
274. A Trial Investigating the Safety and Effects of Four BNT162 Vaccines Against COVID-2019 in Healthy Adults - Full Text View - ClinicalTrials.gov . Available at: https://clinicaltrials.gov/ct2/show/NCT04380701 (Accessed July 4, 2020).
275. Mulligan MJ, Lyke KE, Kitchin N, Absalon J, Gurtman A, Lockhart SP, et al. Phase 1/2 Study to Describe the Safety and Immunogenicity of aCOVID-19 RNA Vaccine Candidate (BNT162b1) in Adults 18 to 55 Years of Age: Interim Report. medRxiv (2020) . doi: 10.1101/2020.06.30.20142570
276. Walsh EE, Frenck R, Falsey AR, Kitchin N, Absalon J, Gurtman A, et al. RNA-Based COVID-19 Vaccine BNT162b2 Selected for a Pivotal Efficacy Study. medRxiv Prepr Serv Heal Sci (2020). doi: 10.1101/2020.08.17.20176651. 2020.08.17.20176651.
277. Mulligan MJ, Lyke KE, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Phase 1/2 study of COVID-19 RNA vaccine BNT162b1 in adults. Nature (2020), 1–8. doi: 10.1038/s41586-020-2639-4
278. ISRCTN. ISRCTN17072692: Clinical trial to assess the safety of a coronavirus vaccine in healthy men and women . Available at: http://www.isrctn.com/ISRCTN17072692 (Accessed July 4, 2020).
279. Ascending Dose Study of Investigational SARS-CoV-2 Vaccine ARCT-021 in Healthy Adult Subjects - Full Text View - ClinicalTrials.gov . Available at: https://clinicaltrials.gov/ct2/show/NCT04480957 (Accessed September 6, 2020).
280. A Phase I clinical trial to evaluate the safety, tolerance and preliminary immunogenicity of different doses of a SARS-CoV-2 mRNA vaccine in population aged 18-59 years and 60 years and above . Available at: http://www.chictr.org.cn/showprojen.aspx?proj=55524 (Accessed September 6, 2020).
281. Safety, Tolerability and Immunogenicity of INO-4800 for COVID-19 in Healthy Volunteers - Full Text View - ClinicalTrials.gov . Available at: https://clinicaltrials.gov/ct2/show/NCT04336410 (Accessed July 4, 2020).
282. INOVIO. Reports Positive Interim Phase I Data for COVID-19 DNA Vaccine, Joins “Warp Speed” Primate Study . Available at: https://www.genengnews.com/news/inovio-reports-positive-interim-phase-i-data-for-covid-19-dna-vaccine-joins-warp-speed-primate-study/?utm_medium=newsletter&utm_source=GEN+Daily+News+Highlights&utm_content=01&utm_campaign=GEN+Daily+News+Highlights_20200701 (Accessed July 4, 2020).
283. Patel A, Walters J, Reuschel EL, Schultheis K, Parzych E, Gary EN, et al. Intradermal-delivered DNA vaccine provides anamnestic protection in a rhesus macaque SARS-CoV-2 challenge model. bioRxiv (2020). doi: 10.1101/2020.07.28.225649. 2020.07.28.225649.
284. Safety and Immunogenicity Study of GX-19, a COVID-19 Preventive DNA Vaccine in Healthy Adults - Full Text View - ClinicalTrials.gov . Available at: https://clinicaltrials.gov/ct2/show/NCT04445389 (Accessed July 4, 2020).
285. Study of COVID-19 DNA Vaccine (AG0301-COVID19) - Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT04463472 (Accessed September 6, 2020).
286. Study of COVID-19 DNA Vaccine (AG0302-COVID19) - Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT04527081 (Accessed September 6, 2020).
287. A prospective, randomized, adaptive, phase I/II clinical study to evaluate the safety andimmunogenicity of Novel Corona Virus -2019-nCov vaccine candidate of M/s Cadila Healthcare Limitedby intradermal route in healthy subjects. Available at: http://ctri.nic.in/Clinicaltrials/showallp.php?mid1=45306&EncHid=&userName=Zydus.
288. Evaluating the Safety, Tolerability and Immunogenicity of bacTRL-Spike Vaccine for Prevention of COVID-19 - Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT04334980 (Accessed July 4, 2020).
289. Immunity and Safety of Covid-19 Synthetic Minigene Vaccine - Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT04276896 (Accessed July 4, 2020).
290. Safety and Immunity of Covid-19 aAPC Vaccine - Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT04299724 (Accessed July 4, 2020).
291. Phase Ib-II Trial of Dendritic Cell Vaccine to Prevent COVID-19 in Adults - Full Text View - ClinicalTrials.gov . Available at: https://clinicaltrials.gov/ct2/show/NCT04386252 (Accessed July 4, 2020).
292. Evaluation of the Safety and Immunogenicity of a SARS-CoV-2 rS (COVID-19) Nanoparticle Vaccine With/Without Matrix-M Adjuvant - Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT04368988?term=novavax&recrs=ab&draw=2&rank=1 (Accessed July 4, 2020).
293. SCB-2019 as COVID-19 Vaccine - Full Text View - ClinicalTrials.gov . Available at: https://clinicaltrials.gov/ct2/show/NCT04405908 (Accessed July 4, 2020).
294. Therapeutic Vaccine Trial of COVID-19 for Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection - Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT04428073 (Accessed July 4, 2020).
295. A Phase I, Prospective, Open-Labeled Study to Evaluate the Safety and Immunogenicity of MVC-COV1901 - Full Text View - ClinicalTrials.gov . Available at: https://clinicaltrials.gov/ct2/show/NCT04487210 (Accessed September 6, 2020).
296. A Study to Evaluate the Safety and Immunogenicity of COVID-19 (AdimrSC-2f) Vaccine - Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT04522089 (Accessed September 6, 2020).
297. An interventional study to evaluate the safety and immune response of a vaccine against Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV-2, the virus that causes COVID-19 infection) when given to healthy adult participants . Available at: https://www.australianclinicaltrials.gov.au/anzctr/trial/ACTRN12620000674932 (Accessed September 6, 2020).
298. A Study on the Safety, Tolerability and Immune Response of SARS-CoV-2 Sclamp (COVID-19) Vaccine in Healthy Adults - Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT04495933 (Accessed September 6, 2020).
299. Safety, Tolerability and Immunogenicinity of a Coronavirus-Like Particle COVID-19 Vaccine in Adults Aged 18-55 Years. - Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT04450004 (Accessed September 6, 2020).
300. Folegatti PM, Ewer KJ, Aley PK, Angus B, Becker S, Belij-Rammerstorfer S, et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet (2020) 396:467–78. doi: 10.1016/S0140-6736(20)31604-4
301. van Doremalen N, Lambe T, Spencer A, Belij-Rammerstorfer S, Purushotham JN, Port JR, et al. ChAdOx1 nCoV-19 vaccine prevents SARS-CoV-2 pneumonia in rhesus macaques. Nature (2020) 1–8. doi: 10.1038/s41586-020-2608-y
302. Zhu FC, Li YH, Guan XH, Hou LH, Wang WJ, Li JX, et al. Safety, tolerability, and immunogenicity of a recombinant adenovirustype-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-humantrial. Lancet (2020) 395:1845–54. doi: 10.1016/S0140-6736(20)31208-3
303. Zhu FC, Guan XH, Li YH, Huang JY, Jiang T, Hou LH, et al. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet (2020) 396:479–88. doi: 10.1016/S0140-6736(20)31605-6
304. Mahase E. Covid-19: Where are we on immunity and vaccines? BMJ (2020) 370:m3096. doi: 10.1136/bmj.m3096
305. Mercado NB, Zahn R, Wegmann F, Loos C, Chandrashekar A, Yu J, et al. Single-shot Ad26 vaccine protects against SARS-CoV-2 in rhesus macaques. Nature (2020), 1–11. doi: 10.1038/s41586-020-2607-z
306. Mahase E. Covid-19: Russia approves vaccine without large scale testing or published results. BMJ (2020) 370:m3205. doi: 10.1136/bmj.m3205
307. Clinical Trial to Evaluate the Safety and Immunogenicitiy of the COVID-19 Vaccine - Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT04497298 (Accessed September 6, 2020).
308. A Study to Assess Safety, Tolerability, and Immunogenicity of V591 (COVID-19 Vaccine) in Healthy Participants (V591-001) - Full Text View - ClinicalTrials.gov. Available at: https://www.clinicaltrials.gov/ct2/show/NCT04498247 (Accessed [Accessed September 6, 2020]).
Keywords: non-viral DNA-RNA vaccines, nanoparticle vaccines, virus-like particle vaccines, cancer vaccines, substance abuse, noncommunicable disease, infectious disease, COVID19
Citation: Brisse M, Vrba SM, Kirk N, Liang Y and Ly H (2020) Emerging Concepts and Technologies in Vaccine Development. Front. Immunol. 11:583077. doi: 10.3389/fimmu.2020.583077
Received: 14 July 2020; Accepted: 14 September 2020;
Published: 30 September 2020.
Edited by:
Katie Ewer, University of Oxford, United KingdomReviewed by:
Arun Kumar, Coalition for Epidemic Preparedness Innovations (CEPI), NorwayHannah Sharpe, University of Oxford, United Kingdom
Copyright © 2020 Brisse, Vrba, Kirk, Liang and Ly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hinh Ly, aGx5QHVtbi5lZHU=