 Jungmin Yang
Jungmin Yang Inhwa Hwang1
Inhwa Hwang1 Sung Jae Shin
Sung Jae Shin Je-Wook Yu
Je-Wook Yu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 18 November 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.581165
Bacteria-released components can modulate host innate immune response in the absence of direct host cell–bacteria interaction. In particular, bacteria-derived outer membrane vesicles (OMVs) were recently shown to activate host caspase-11-mediated non-canonical inflammasome pathway via deliverance of OMV-bound lipopolysaccharide. However, further precise understanding of innate immune-modulation by bacterial OMVs remains elusive. Here, we present evidence that flagellated bacteria-released OMVs can trigger NLRC4 canonical inflammasome activation via flagellin delivery to the cytoplasm of host cells. Salmonella typhimurium-derived OMVs caused a robust NLRC4-mediated caspase-1 activation and interleukin-1β secretion in macrophages in an endocytosis-dependent, but guanylate-binding protein-independent manner. Notably, OMV-associated flagellin is crucial for Salmonella OMV-induced inflammasome response. Flagellated Pseudomonas aeruginosa-released OMVs consistently promoted robust NLRC4 inflammasome activation, while non-flagellated Escherichia coli-released OMVs induced NLRC4-independent non-canonical inflammasome activation leading to NLRP3-mediated interleukin-1β secretion. Flagellin-deficient Salmonella OMVs caused a weak interleukin-1β production in a NLRP3-dependent manner. These findings indicate that Salmonella OMV triggers NLRC4 inflammasome activation via OMV-associated flagellin in addition to a mild induction of non-canonical inflammasome signaling via OMV-bound lipopolysaccharide. Intriguingly, flagellated Salmonella-derived OMVs induced more rapid inflammasome response than flagellin-deficient Salmonella OMV and non-flagellated Escherichia coli-derived OMVs. Supporting these in vitro results, Nlrc4-deficient mice showed significantly reduced interleukin-1β production after intraperitoneal challenge with Salmonella-released OMVs. Taken together, our results here propose that NLRC4 inflammasome machinery is a rapid sensor of bacterial OMV-bound flagellin as a host defense mechanism against bacterial pathogen infection.
Host immune system attempts to detect invasion of bacterial pathogens via pattern-recognition receptors on tissue-resident sentinel cells, such as macrophages (1). Recognition of bacterial ligand by these receptors promptly results in the production of proinflammatory cytokines, which then trigger host inflammatory responses by recruiting more leukocytes from the bloodstream to clear the invading pathogens (2). Among the proinflammatory cytokines produced during the initial stage of bacterial infection, interleukin (IL)-1β has a crucial role in initiation of the inflammatory process (3). The maturation and secretion of IL-1β requires formation of inflammasome multi-protein complex (4). Upon bacterial infection, bacterial ligands drive inflammasome complex assembly. This complex consists mostly of sensor protein, adaptor protein, and procaspase-1, leading to the generation of active caspase-1 (5). Caspase-1 then processes inactive pro-IL-1β into an active form and facilitates leaderless IL-1β secretion into the extracellular space.
Several cytosolic inflammasome sensor molecules detect intracellular bacteria-specific ligands in direct or indirect ways (2, 5). NOD-like receptor family, CARD domain-containing 4 (NLRC4) is the most professional sensor molecule that responds to bacterial infections (6). NLRC4 monomer is assembled into active oligomeric complex upon engagement by bacterial flagellin-bound neuronal apoptosis inhibitory protein 5 (NAIP5), which is the actual sensor for bacterial flagellin (7). Similarly, both NAIP1 and NAIP2 recognize type 3 secretion system (T3SS) needle and rod proteins in Gram-negative bacteria (8, 9), and this NAIPs–T3SS protein complex promotes the assembly of NLRC4 inflammasome. Nlrc4-deficient mice showed increased susceptibility to infection with bacterial pathogens such as Salmonella typhimurium (10, 11), which indicates that NLRC4 inflammasome confers innate immune protection against pathogenic bacteria.
In addition to NLRC4, absent in melanoma 2 (AIM2) directly detects cytosolic bacterial double-stranded DNA to form AIM2 inflammasome (12). Caspase-11 recognizes intracellular lipopolysaccharide (LPS), derived from invasive Gram-negative bacteria, leading to the activation of non-canonical inflammasome pathways (13, 14). In addition to this direct sensing of bacterial ligands by inflammasome sensor molecules, some bacterial toxins can indirectly drive inflammasome activation. For example, pore-forming toxins from Staphylococcus aureus or Streptococcus pneumoniae induce NLR family, pyrin domain-containing 3 (NLRP3)-mediated inflammasome activation (5). But some bacterial toxins promote pyrin inflammasome signaling via Rho GTPase inhibition (15).
Outer membrane vesicles (OMVs) are produced mostly from Gram-negative bacteria by vesiculation process (16). The physiological function of bacterial OMVs remains to be further determined, but the generated OMVs eliminate toxic compounds (e.g., misfolded proteins) to promote bacterial survival under stress conditions (16). In addition, OMVs can increase bacterial pathogenicity by delivering antibiotic resistance genes to other bacteria and virulence factors to host cells (17). Hence, host immune recognition of OMVs is beneficial for detection of bacterial invasion and inhibition of the spread of bacterial pathogenicity. Intriguingly, recent studies revealed that bacterial OMVs can induce caspase-11-mediated non-canonical inflammasome activation via deliverance of OMV-bound LPS into the cytosol (18, 19). In this study, we present evidence that OMVs from flagellated bacteria are able to trigger NLRC4-mediated canonical inflammasome activation in a mainly flagellin-dependent manner. This bacterial OMV-driven NLRC4 inflammasome signaling may provide an efficient host defense mechanism as a rapid sensing machinery against bacterial infection.
C57BL/6 and Nlrp3−/− mice were obtained from The Jackson Laboratory. Nlrc4−/− mice were obtained from Genentech through Dr. Tatsuya Saitoh (Osaka University). Gbp2−/− mice were provided by Korea Mouse Phenotyping Center. All mice were bred at Yonsei University College of Medicine and maintained under specific pathogen-free conditions. 9- to 12-week old mice were used for the experiments. For some experiments, mice were injected with Salmonella OMVs (50 µg) or phosphate-buffered saline (PBS) via the intraperitoneal route. Protocols for the animal experiments were approved by the Institutional Ethical Committee, Yonsei University College of Medicine. All experiments were performed in accordance with the approved guidelines of the Institutional Ethical Committee.
LPS, ATP, nigericin, DOTAP liposomal transfection reagent, proteinase K, cytochalasin D, MCC950, and OptiPrep density gradient were purchased from Sigma-Aldrich. Pitstop2 was obtained from Abcam. Flagellin purified from S. typhimurium CDC 6516-60 (ATCC 14028) was purchased from InvivoGen. CytoTox96 non-radioactive cytotoxicity assay kit was obtained from Promega. The following antibodies were used for detecting caspase-1 (Adipogen), NLRP3 (Adipogen), IL-1β (R&D Systems), ASC (Santa Cruz Biotechnology), flagellin (InvivoGen), OmpA (Biorbyt), and gasdermin D (Novus Biologicals). Anti-NLRC4 antibody was prepared using mouse NLRC4 peptide (AbFrontier).
Mouse bone marrow-derived macrophages (BMDMs) were prepared from mouse femurs as previously described (12). Immortalized Asc−/− BMDMs were provided by Dr. E.S. Alnemri (Thomas Jefferson University, Philadelphia, USA). All BMDMs were maintained in L929-conditioned DMEM supplemented with 10% fetal bovine serum and antibiotics. Cell death was determined by extracellular release of lactate dehydrogenase (LDH) using a CytoTox96 non-radioactive cytotoxicity assay kit. The LDH release was calculated as [extracellular LDH/(intracellular LDH + extracellular LDH) × 100].
Salmonella enterica serovar typhimurium strains, SL1344 and 14028s, were used in this study. Flagellin- or prgJ deletion Salmonella mutants (fliC–fljB, prgJ, and fliC–fljB–prgJ) were generated by homologous recombination and kindly provided by Dr. F. Shao (National Institute of Biological Sciences, Beijing, China). Pseudomonas aeruginosa strain PAO1 was kindly provided by Dr. S.S. Yoon (Yonsei University College of Medicine). Escherichia coli B (BL21) and DH5α strains were obtained from RBC bioscience. Bacterial strains were grown overnight in LB medium with shaking and aeration. Overnight-cultured bacteria were normalized by bacterial number, diluted 1:50 with fresh LB medium and grown for an additional 6 or 1–12 h. Then, the bacterial suspension was centrifuged to pellet the bacteria and further filtered using 0.2-µm syringe filter to remove bacteria. The final filtrate was used for bacteria-free CS. The prepared bacterial CS was added onto LB agar plate and incubated overnight to confirm the absence of bacterial contamination. To inactivate protein moiety in the bacterial CS, CSs were heated at 98°C for 30 min (heat treatment) or treated with proteinase K (10 µg/ml) at 37°C for 30 min and subsequently boiled at 98°C for 30 min to eliminate proteinase K activity.
To prepare outer membrane vesicles, bacteria-free CSs (1L) were concentrated to 25–50 ml by using the protein concentrator PES 100K MWCO (Thermo Fisher Sci) and ultracentrifuged at 150,000 × g at 4°C for 3 h. The pelleted fraction was washed with PBS and re-ultracentrifuged. The final pellet was resuspended with PBS (1 ml). The concentration of isolated OMVs was determined using a BCA protein assay kit. Isolated OMVs were analyzed using a Nanoparticle Tracking Analysis system (NS300, Malvern Panalytical Ltd). The analyses were performed with purified OMVs from at least three-independent isolation. To further fractionate OMVs, OptiPrep gradients were layered as: 1.7 ml 45%, 1.7 ml 40%, 1.7 ml 35%, 1.7 ml 30%, 1.7 ml 20%, and 1.7 ml 10%. Isolated OMVs (500 μl) were loaded on top of the OptiPrep gradients. The gradients were ultracentrifuged at 150,000 × g for 20 h at 4°C, and 1 ml of each fraction was obtained. Obtained fractions were precipitated for immunoblotting by methanol/chloroform extraction method. In several experiments, isolated OMVs were heated at 98°C for 30 min (heat treatment) or treated with proteinase K (10 µg/ml) at 37°C for 30 min and subsequently added with PMSF (1 mM) to eliminate proteinase K activity.
Cells were lysed in buffer containing 20 mM HEPES (pH 7.5), 0.5% Nonidet P-40, 50 mM KCl, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, and protease inhibitors. Soluble lysates were fractionated by SDS-PAGE and then transferred to PVDF membranes. In some experiments, cell culture supernatants were precipitated by methanol/chloroform as described previously (20) and then immunoblotted. All the blots shown are representative image of at least three-independent experiments.
To induce a conventional NLRP3 inflammasome activation, BMDMs were primed with LPS (0.25 µg/ml, 3 h), followed with ATP treatment (2–2.5 mM, 30–45 min) (21). To induce a non-canonical inflammasome activation, cells were treated with Pam3CSK4 (1 µg/ml, 4 h), followed by the transfection of LPS (2 µg/ml, 5 h) using a DOTAP (DT). Inflammasome activation was determined by the presence of active caspase-1 p20 and active IL-1β from culture supernatants in immunoblots and by the extracellular IL-1β quantification using ELISA. To measure extracellular levels of IL-1β or IL-6, culture supernatants were assayed using an IL-1β- or IL-6-specific ELISA kit (BioLegend) according to the manufacturer’s instructions.
Isolated OMVs were labeled by incubating with 1% (v/v) Vybrant Dil (Thermo Fisher Scientific) at 37°C for 30 min. Unbound dyes were removed by ultracentrifugation and PBS washing three times. PBS was also incubated with dye in the same way as the OMVs and used for negative control. After treating with labeled OMVs, cells were fixed with 4% para-formaldehyde. Fixed cells on coverslip were mounted using ProLong Gold reagent with DAPI. The cells were then examined using confocal microscopy (LSM700, Carl Zeiss, Oberkochen, Germany).
All values were expressed as the mean ± SEM of individual samples. All the data were obtained from at least three-independent experiments. Data were analyzed using Student’s t-test or one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test for comparison of all groups with control group as indicated in the figure legend. The level of statistical significance was set at p ≤ 0.05. Analyses were performed using GraphPad Prism 5.
Salmonella typhimurium is the most common enteric pathogen causing gastroenteritis. To examine whether S. typhimurium-secreted molecules can trigger host inflammasome signaling pathways regardless of bacterial contact with host macrophages, we used a transwell culture system (Figure 1A). As flagellin is the major ligand for inflammasome activation during S. typhimurium infection (22, 23), we tested flagellin-deficient (ΔfliC–fljB) S. typhimurium as well as wild-type bacteria. Culture media from wild-type and flagellin-deficient (ΔfliC–fljB) S. typhimurium in the upper wells clearly drove a robust inflammasome activation of mouse bone marrow-derived macrophages (BMDMs) in the lower wells, as determined by the presence of cleaved caspase-1 (p20) and matured IL-1β in the supernatant (Figure 1B). To further confirm the inflammasome-stimulating capacity of S. typhimurium-secreted molecules, we carefully collected bacteria-free culture supernatants (CS) from S. typhimurium and treated them into BMDMs. Notably, CS from wild-type or flagellin mutant S. typhimurium caused a marked caspase-1 activation (Figure 1C). This Salmonella CS-induced inflammasome activation was dependent on NLRC4 and apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), which is a crucial adaptor protein of the inflammasome complex (Figure 1D and Figure S1A). However, activation was not dependent on NLRP3 (Figure 1E). These findings indicated that Salmonella-secreted components are able to promote NLRC4-mediated inflammasome activation in the absence of direct bacterial contact with host macrophages.
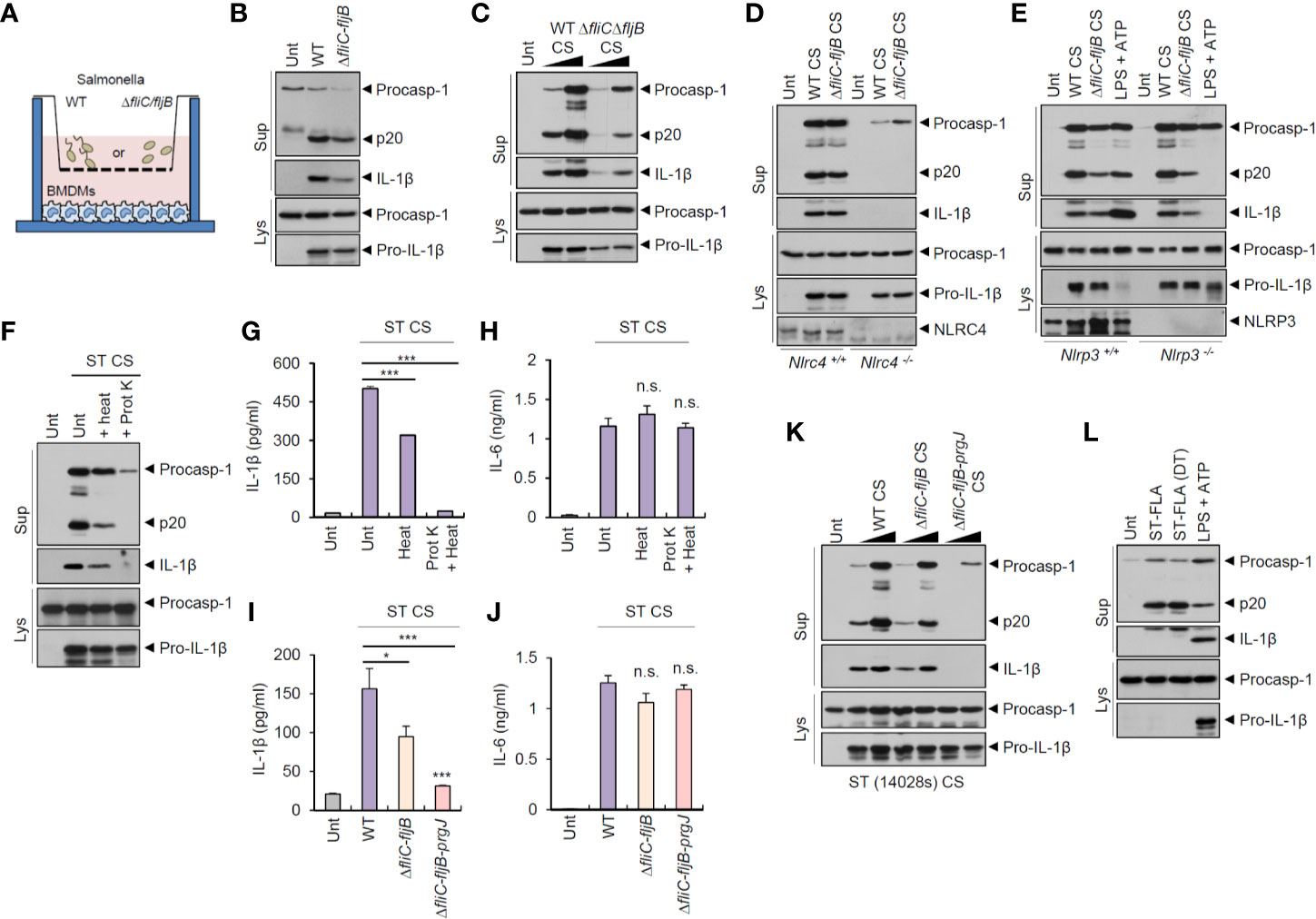
Figure 1 Culture supernatants from Salmonella typhimurium promotes NLRC4-dependent inflammasome activation. (A) Illustration of experimental scheme to determine inflammasome activation in BMDMs by bacteria-secreted molecules using transwell plate. (B) Immunoblots from mouse BMDMs in the lower well (A) incubated with wild-type (WT) or ΔfliC-fljB S. typhimurium SL1344 in the upper well (A, MOI 30) for 7 h. (C) Immunoblots from mouse BMDMs untreated or treated with culture supernatant (CS, 1/33 or 1/10 volume of culture medium), derived from 6 h culture of WT or ΔfliC–fljB S. typhimurium, for 6 h. (D) Immunoblots from Nlrc4+/+ or Nlrc4−/− mice BMDMs treated with WT or ΔfliC–fljB S. typhimurium CS (1/10) for 6 h. (E) Immunoblots from Nlrp3+/+ or Nlrp3−/− mice BMDMs treated with WT or ΔfliC–fljB S. typhimurium CS (1/10) for 6 h, or primed with LPS (0.25 µg/ml, 3 h), followed by treatment with ATP (2.5 mM, 40 min). (F) Immunoblots from mouse BMDMs incubated with S. typhimurium CS (1/20) with or without heat treatment (97°C, 30 min) or proteinase K treatment (10 µg/ml, 30 min), for 6 h. (G, H) Quantification of IL-1β (G) or IL-6 (H) in the supernatant of mouse BMDMs treated with S. typhimurium CS (1/20) as same as in (F). (n = 3, one-way ANOVA) (I, J) Quantification of IL-1β (I) or IL-6 (J) in the culture supernatants of mouse BMDMs treated with WT, ΔfliC–fljB, or ΔfliC–fljB–prgJ S. typhimurium CS (1/100) for 6 h. (n = 4, one-way ANOVA). (K) Immunoblots from mouse BMDMs treated with culture supernatant (CS, 1/100 or 1/20), derived from 6 h culture of WT, ΔfliC–fljB, ΔfliC–fljB–prgJ S. typhimurium 14028s, for 6 h. (L) Immunoblots from mouse BMDMs treated with S. typhimurium flagellin (ST-FLA, 250 ng/ml) with or without premixing with DOTAP (DT) liposomal transfection reagent for 6 h or treated with LPS (0.25 µg/ml, 3 h), followed by ATP treatment (2.5 mM, 40 min). Culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies. Data were expressed as the mean ± SEM. Asterisks indicate significant differences compared with S. typhimurium CS-treated group. (*P < 0.05, ***P < 0.001, n.s. not significant).
To determine the bacterial components responsible for Salmonella CS-induced NLRC4 inflammasome activation, we first examined a potential role of protein present in the CS. Inactivation of protein by heat treatment applied to S. typhimurium CS significantly reduced caspase-1 activation and active IL-1β secretion (Figures 1F, G). Additionally, protein degradation via proteinase treatment completely abrogated S. typhimurium CS-induced inflammasome activation (Figures 1F, G). Indeed, proteinase treatment, but not heat treatment, caused a complete degradation of bacterial proteins in the CS and lysates (Figures S1B, C). However, neither heat inactivation nor proteinase treatment impaired IL-6 production by Salmonella CS (Figure 1H).
Flagellin and T3SS protein are the main stimulants for NAIP-dependent NLRC4 inflammasome activation during S. typhimurium infection (22, 23). We thus examined a putative role of flagellin or PrgJ, a T3SS rod protein, in S. typhimurium CS-induced inflammasome activation. CS from flagellin-deficient (ΔfliC–fljB) Salmonella caused significantly less IL-1β production than wild-type Salmonella CS (Figure 1I). Furthermore, S. typhimurium CS from flagellin/prgJ-deficient mutants (ΔfliC–fljB–prgJ) induced remarkably attenuated IL-1β secretion from BMDMs (Figure 1I). However, both proteins were not required for CS-induced IL-6 production from BMDMs (Figure 1J). Consistent with these findings, flagellin/prgJ-deficient Salmonella-derived CS failed to induce a robust activation of caspase-1 and cell death in BMDMs (Figure 1K and Figure S2).
To further confirm the role of flagellin or prgJ in the CS, we directly treated a purified Salmonella flagellin protein into BMDMs with or without DOTAP (N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate) liposomal transfection agent. Consistent with the results of previous study (22), cytosolic delivery of flagellin using DOTAP caused caspase-1 activation in BMDMs (Figure 1L). Notably, 6 h treatment with S. typhimurium flagellin alone also drove caspase-1 processing (Figure 1L). This unexpected inflammasome activation by recombinant flagellin required the expression of NLRC4 and ASC but not of NLRP3 (Figures S3A–S3C). This result suggested that Salmonella-released flagellin and T3SS rod protein PrgJ are capable of inducing host NLRC4 inflammasome activation.
To examine whether bacteria-secreted vesicle fractions mediated inflammasome activation, we isolated pelleted fractions after 150,000 × g centrifugation of Salmonella CS. Intriguingly, pelleted-vesicles from S. typhimurium CS still induced caspase-1 activation and active IL-1β secretion (Figure 2A). We then further isolated bacterial OMVs from Salmonella CS (Figure S4A). Outer membrane protein A (OmpA) was present in Salmonella OMV fractions (Figure 2B). The results of the nanoparticle tracking analysis (Figure 2C) and transmission electron microscopy (Figure 2D and Figure S4B) indicated that S. typhimurium-released OMV size ranged from 50 to 200 nm. In addition, we confirmed the absence of bacterial contamination in the isolated OMV fractions or the CS fraction (Figure S4C) by overnight culture in the LB agar plate to exclude a possibility that free bacteria can affect the results.
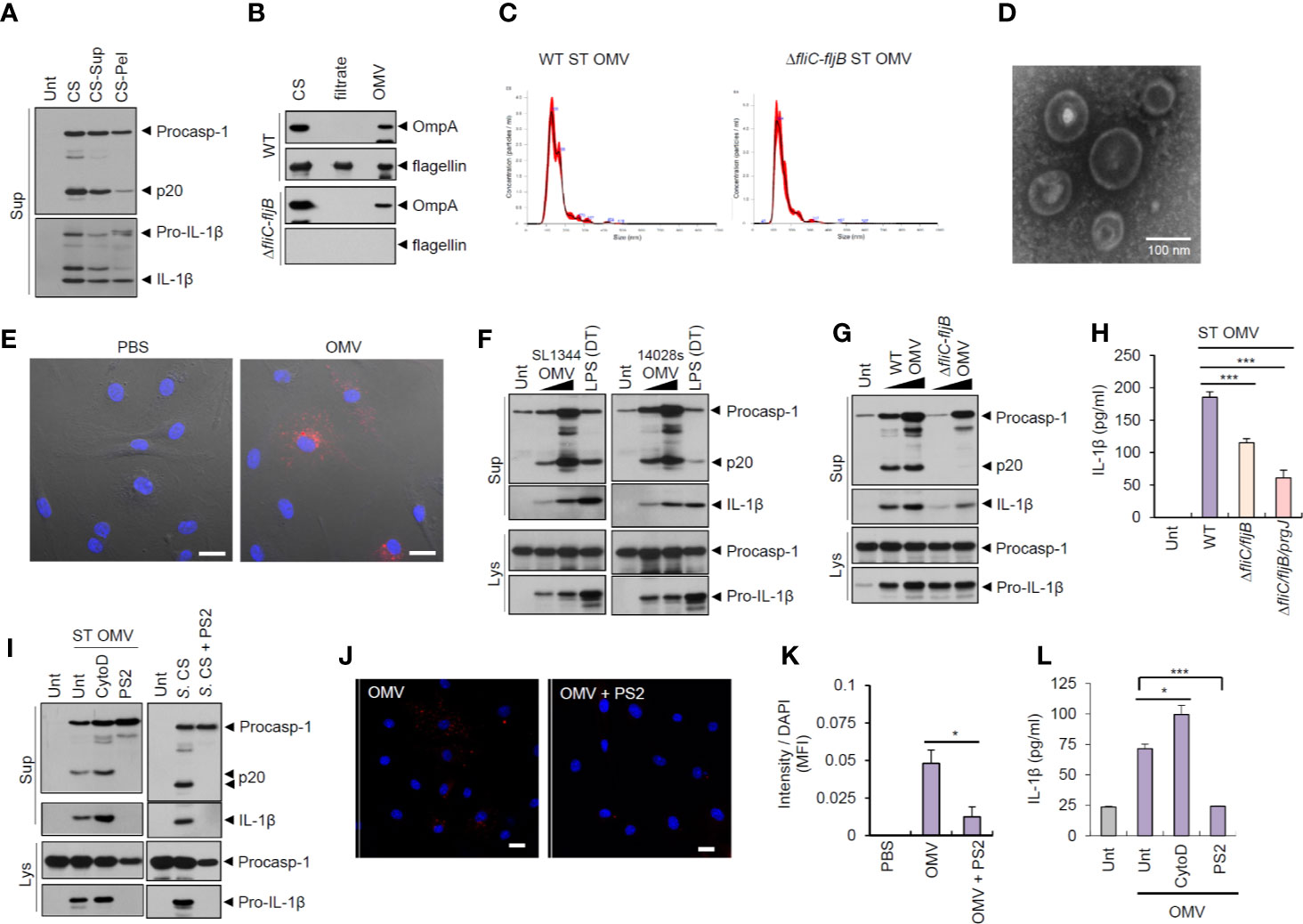
Figure 2 Outer membrane vesicles from Salmonella typhimurium activate inflammasome signaling via receptor-mediated endocytosis. (A) Immunoblots from the culture supernatants of mouse BMDMs treated with S. typhimurium CS, or supernatant (CS-Sup) or pellet (CS-Pel) from ultracentrifugation (150,000 × g, 18 h) of S. typhimurium CS. (B) Immunoblots from wild-type (WT) or ΔfliC–fljB S. typhimurium CS, filtrates from Centricon concentration (Supplementary Figure S3A) and the extract of OMVs. (C) Nanoparticle Tracking Analysis of OMVs isolated from WT or ΔfliC–fljB S. typhimurium. (D) Transmission electron microscopy of OMVs isolated from wild-type S. typhimurium. Scale bar, 100 nm. (E) Representative confocal microscopy images of BMDMs treated with Vybrant Dil-labeled PBS or S. typhimurium OMVs. Scale bar, 20 µm. (F) Immunoblots from mouse BMDMs treated with OMVs isolated from S. typhimurium SL1344 or 14028s (0.5 or 5 µg/ml) for 8 h or treated with Pam3CSK4 (1 µg/ml, 4 h), followed by the transfection of LPS (2 µg/ml, 5 h) using a DOTAP (DT). (G) Immunoblots from mouse BMDMs treated with WT or ΔfliC–fljB S. typhimurium (SL1344) OMV (1 or 5 µg/ml, 8 h). (H) Quantification of IL-1β in the culture supernatants of mouse BMDMs treated with WT, ΔfliC–fljB, or ΔfliC–fljB–prgJ S. typhimurium (SL1344) OMVs (5 µg/ml, 8 h) (n = 7, one-way ANOVA). (I) Immunoblots from mouse BMDMs treated with S. typhimurium OMV (5 µg/ml, 6 h) in the presence of cytochalasin D (5 µM) or Pitstop 2 (PS2, 10 µM), or treated with S. typhimurium CS (1/20) in the presence of PS2 (10 µM) pretreatment (10 min before Salmonella CS treatment). (J) Representative confocal microscopy images of BMDMs treated with Vybrant Dil-labeled S. typhimurium OMVs (5 µg/ml, 6 h) in the presence of Pitstop 2 treatment (10 µM, 30 min pretreat). Scale bar, 20 µm. (K) Relative Dil fluorescence intensity per DAPI signals of BMDMs as treated in (J). (n = 6, one-way ANOVA). (L) Quantification of IL-1β in the culture supernatants of mouse BMDMs treated as in (I, left panel). (n = 3, one-way ANOVA). Culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies. Data were expressed as the mean ± SEM. Asterisks indicate significant differences compared with S. typhimurium CS-treated group. (*P < 0.05, ***P < 0.001).
Next, Salmonella-derived OMVs were treated into BMDMs to examine the innate immune responses against bacterial OMVs. Vybrant Dil-labeled OMVs were clearly detected in the cytosol of BMDMs (Figure 2E), suggesting that bacteria-derived OMVs can deliver bacterial components into the cytoplasm of host cells. Of note, treatment with OMVs isolated from S. typhimurium (SL1344 or 14028s) caused a robust caspase-1 activation and IL-1β secretion in BMDMs (Figure 2F). S. typhimurium SL1344 OMV-triggered caspase-1 activation was markedly impaired by flagellin deficiency (Figure 2G). Consistently, OMVs from flagellin-deficient Salmonella SL1344 induced significantly less IL-1β secretion than wild-type Salmonella OMVs in BMDMs (Figure 2H). OMVs from flagellin-deficient S. typhimurium strain 14028s also failed to trigger robust caspase-1 activation and IL-1β production (Figures S5A, B). In contrast, flagellin deficiency did not affect the Salmonella OMV-induced IL-6 production (Figure S5C).
To examine whether Salmonella OMVs did act to transport bacterial proteins to the cytosol, we blocked phagocytosis using cytochalasin D and clathrin-mediated endocytosis using Pitstop 2 (PS2), respectively. PS2 treatment, but not cytochalasin D treatment, markedly inhibited inflammasome activation in BMDMs in response to S. typhimurium-released OMVs and CS (Figure 2I). PS2 treatment also significantly inhibited cytosolic entry of labeled-OMVs (Figures 2J, K), indicating that entry of S. typhimurium OMVs was facilitated by clathrin-mediated endocytosis. Consistently, PS2 significantly reduced Salmonella OMV-triggered IL-1β secretion (Figure 2L). These findings support the hypothesis that Salmonella OMVs function as a cargo carrier for cytosolic delivery of bacterial proteins through clathrin-mediated endocytosis.
To elucidate the mechanisms underlying the inflammasome activation by S. typhimurium-derived OMVs, we examined which sensor molecule was associated with OMV-mediated inflammasome activation. Previous studies found that OMVs from Escherichia coli trigger caspase-11-mediated non-canonical inflammasome activation via LPS delivery into the cytoplasm, leading to NLRP3-dependent IL-1β secretion (18, 19). Unexpectedly, the results indicated that S. typhimurium OMVs were capable of inducing caspase-1 activation, even in Nlrp3-deficient BMDMs (Figure 3A). As a control for caspase-11-mediated non-canonical inflammasome signaling, cytosolic delivery of LPS using DOTAP caused NLRP3-dependent caspase-1 activation (Figure 3A). In contrast, S. typhimurium OMV-induced caspase-1 activation was remarkably abolished in Nlrc4- and Asc-deficient BMDMs (Figures 3B, C). This result indicate that NLRC4 was a major sensor molecule for the S. typhimurium OMVs. Flagellin-deficient Salmonella-released OMVs caused very faint caspase-1 activation in macrophages (Figure 3D). This attenuated caspase-1 activation by flagellin-lacking OMVs apparently depended on the expression of NLRP3 in BMDMs (Figure 3D). Consistently, flagellin-deficient S. typhimurium OMVs induced a weak IL-1β production in a NLRP3-dependent manner (Figure S5D).
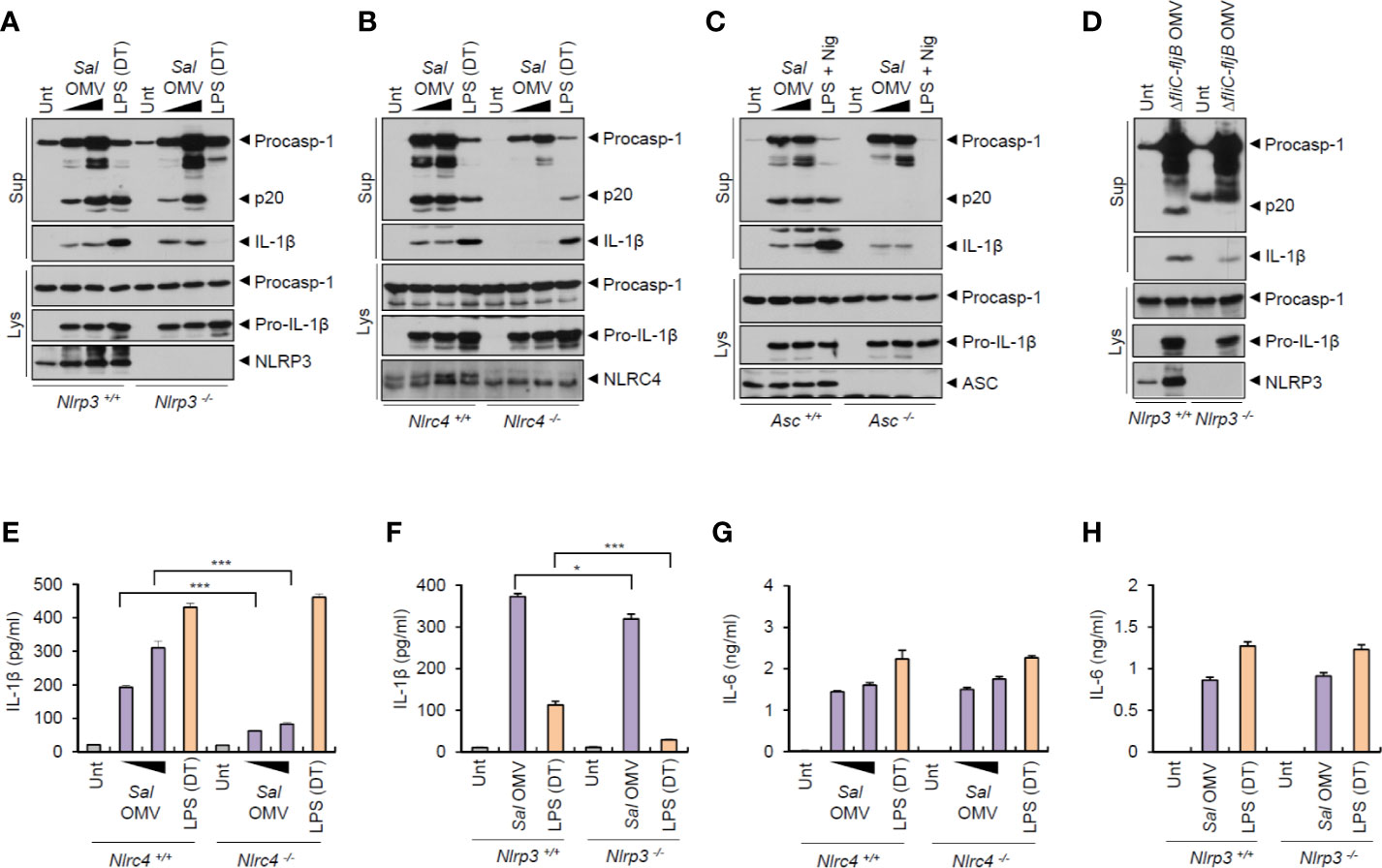
Figure 3 Salmonella-released outer membrane vesicles promotes NLRC4-dependent inflammasome activation. (A, B) Immunoblots from Nlrp3+/+ or Nlrp3−/− (A) or Nlrc4+/+ or Nlrc4−/− (B) mice BMDMs treated with S. typhimurium OMVs (3 or 10 µg/ml, 6 h, A; 5 or 12.5 µg/ml, 6 h, B), or treated with Pam3CSK4 (1 µg/ml, 4 h), followed by the transfection of LPS (1 µg/ml, 6 h) using a DOTAP (DT). (C) Immunoblots from Asc+/+ or Asc −/− immortalized BMDMs treated with S. typhimurium OMVs (5 or 12.5 µg/ml, 6 h), or primed with LPS (1 µg/ml, 6 h), followed by the treatment with nigericin (5 µM, 40 min). (D) Immunoblots from Nlrp3+/+ or Nlrp3−/− mice BMDMs treated with ΔfliC–fljB S. typhimurium OMVs (10 µg/ml) for 6 h. (E, F) Quantification of IL-1β in the culture supernatants of Nlrc4+/+ or Nlrc4−/− (E) or Nlrp3+/+ or Nlrp3−/− (F) mice BMDMs treated with S. typhimurium OMVs (1 or 7 µg/ml, E; 10 µg/ml, F) for 8 h, or treated with Pam3CSK4 (1 µg/ml, 3 h), followed by the transfection of LPS (2 µg/ml, E; 1 µg/ml, F) for 6 h. (n = 3) (G, H) Quantification of IL-1β in the culture supernatants of Nlrc4 +/+ or Nlrc4−/− (G) or Nlrp3+/+ or Nlrp3−/− (H) mice BMDMs treated with as same as in (E, F). (n = 3) Culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies. Data were expressed as the mean ± SEM. Asterisks indicate significant differences compared with the group in the Nlrp3+/+ cells. (*P < 0.05, ***P < 0.001).
In support of these findings, S. typhimurium OMV-induced IL-1β production was significantly impaired in Nlrc4-knockout macrophages (Figure 3E). It was only very slightly reduced in NLRP3-knockout macrophages (Figure 3F). But Salmonella OMV treatment resulted in robust NLRC4- and NLRP3-independent IL-6 production (Figures 3G, H). Taken together, these findings indicate that Salmonella-released OMVs can trigger the activation of NLRC4/ASC-mediated inflammasome signaling pathways in host macrophages.
To examine whether OMVs derived from bacteria other than Salmonella can induce NLRC4-mediated inflammasome activation, we tested the effects of the other Gram-negative bacterial pathogen, Pseudomonas aeruginosa PAO1. Consistent with the S. typhimurium results, P. aeruginosa-released OMVs also caused a robust caspase-1 activation and active IL-1β secretion (Figure 4A). This P. aeruginosa OMV-driven inflammasome activation depended on NLRC4, but not on NLRP3 (Figures 4B, C). Consistent with these findings, P. aeruginosa OMV treatment resulted in robust secretion of IL-1β in a NLRC4-dependent manner (Figure 4D). Instead, NLRP3 deficiency caused a slight reduction in IL-1β production by Pseudomonas OMVs (Figure 4E).
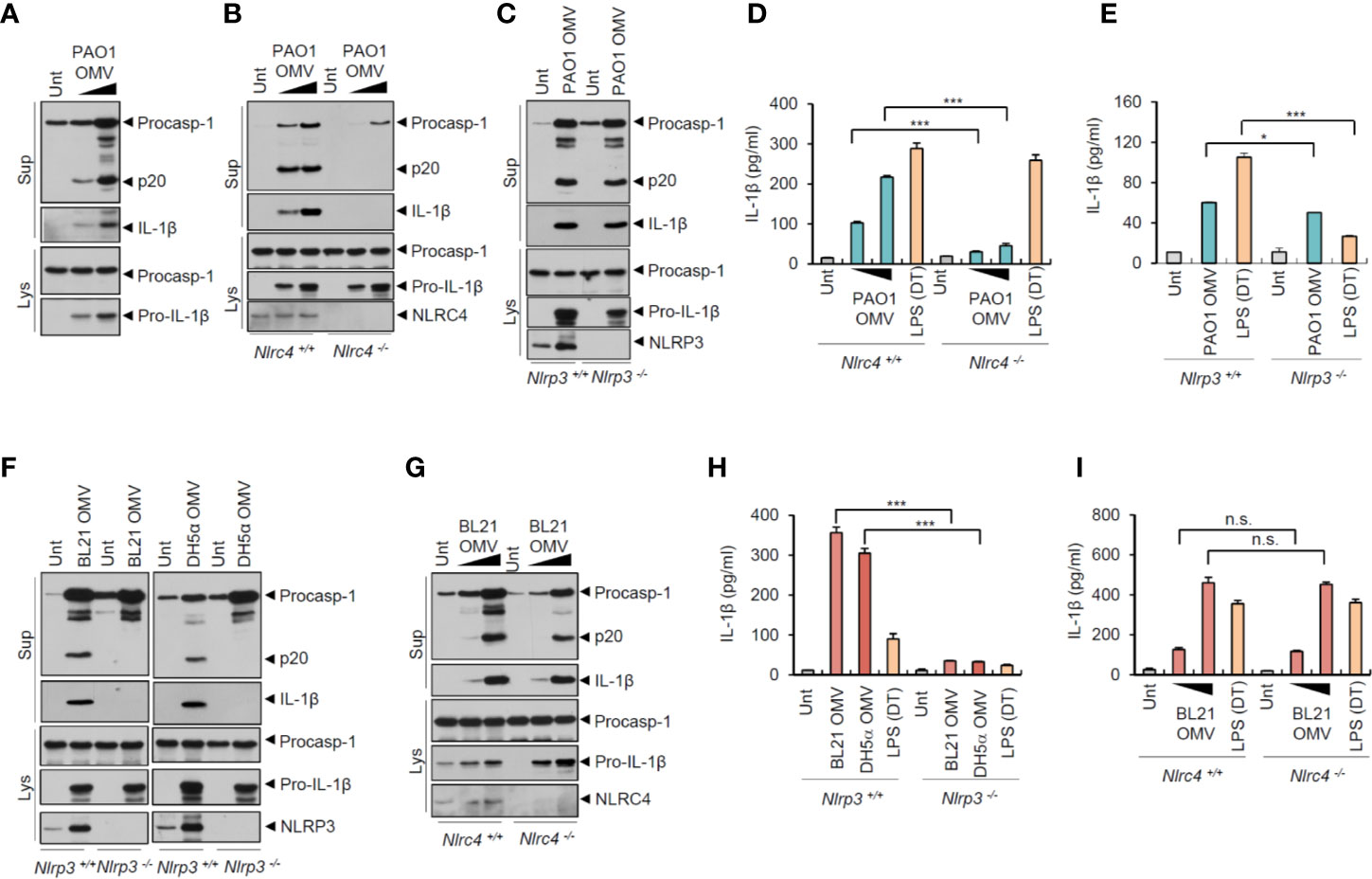
Figure 4 Flagellated bacteria-released outer membrane vesicles trigger NLRC4-dependent inflammasome activation. (A) Immunoblots from mouse BMDMs treated with OMV (0.5 or 3 µg/ml) isolated from Pseudomonas aeruginosa PAO1 for 8 h. (B) Immunoblots from Nlrc4+/+ or Nlrc4−/− mice BMDMs treated with P. aeruginosa OMV (1 or 7 µg/ml) for 8 h. (C) Immunoblots from Nlrp3 +/+ or Nlrp3−/− mice BMDMs treated with P. aeruginosa OMV (5 µg/ml) for 8 h. (D, E) Quantification of IL-1β in the culture supernatants of Nlrc4+/+ or Nlrc4−/− (D) or Nlrp3+/+ or Nlrp3−/− (E) mice BMDMs treated with P. aeruginosa OMV (1 or 7 µg/ml, D; 5 µg/ml, E) for 8 h, or treated with Pam3CSK4 (1 µg/ml, 3 h), followed by the transfection of LPS (2 µg/ml, D; 1 µg/ml, E) for 6 h. (n = 3) (F) Immunoblots from Nlrp3+/+ or Nlrp3−/− mice BMDMs treated with E. coli BL21 or DH5α OMVs (5 µg/ml) for 8 h. (G) Immunoblots from Nlrc4+/+ or Nlrc4−/− mice BMDMs treated with E. coli BL21 OMVs (1 or 5 µg/ml) for 8 h. (H, I) Quantification of IL-1β in the culture supernatants of Nlrp3+/+ or Nlrp3−/− (H) or Nlrc4+/+ or Nlrc4−/− (I) mice BMDMs treated with E. coli BL21-derived OMVs (5 µg/ml, H; 1 or 5 µg/ml, I) or DH5α-derived OMVs (5 µg/ml, H) for 8 h, or treated with Pam3CSK4 (1 µg/ml, 3 h), followed by the transfection of LPS (1 µg/ml, H; 2 µg/ml, I) for 6 h. (n = 3) Culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies. Data were expressed as the mean ± SEM. Asterisks indicate significant differences compared with the group in the Nlrp3+/+ cells. (*P < 0.05, ***P < 0.001, n.s. not significant).
Given that flagellin may be the main stimulus for Salmonella-released OMV-induced NLRC4 inflammasome activation, we examined the roles of OMVs from non-flagellated bacteria, such as E. coli strains BL21 and DH5α. Unlike Salmonella or Pseudomonas OMVs, E. coli (BL21 and DH5α)-released OMVs induced robust caspase-1 activation only in NLRP3-expressing macrophages (Figure 4F). In contrast, NLRC4 deficiency did not affect E. coli OMV-driven caspase-1 activation (Figure 4G). Consistent with these results, non-flagellated E. coli OMVs promoted NLRP3-dependent, but NLRC4-independent, IL-1β secretion (Figures 4H, I). Furthermore, E. coli OMV-induced inflammasome activation was blocked by NLRP3-selective inhibitor MCC950 (Figure S6). These findings indicate that non-flagellated bacteria-released OMVs induce NLRP3 inflammasome activation probably via triggering caspase-11-mediated non-canonical inflammasome signaling. In contrast, flagellated bacteria-released OMVs can facilitate canonical NLRC4 inflammasome pathways.
As the results indicated, flagellin in the OMVs had a pivotal role in Salmonella OMV-induced caspase-1 activation. We thus examined whether flagellin was indeed located in the OMVs derived from Salmonella culture. Isolated OMVs were further fractionated using an OptiPrep density gradient medium. Flagellin was detected in the same fraction as in OmpA, an unique OMV marker (Figure 5A), indicating that flagellin was present in the S. typhimurium-released OMVs. Moreover, flagellin was observed in the low density fraction of bacterial CS, but not of OMVs (Figure 5A), suggesting that isolated OMVs contain no or very little free flagellin outside OMVs. Nevertheless, to further confirm whether flagellin is present as inserted forms in the OMV membrane or inside OMVs, isolated OMVs were subjected to heat and proteinase K treatment. Flagellin in the OMVs was almost intact against heat- and proteinase treatment (Figure 5B), while flagellin in the bacterial CS was completely degraded by proteinase treatment (Figure S1B). Indeed, heat treatment significantly impaired bacterial CS-induced inflammasome activation (Figures 1F, G). Similarly, heat-inactivation of recombinant Salmonella flagellin protein greatly impaired inflammasome activation (Figure 5C). However, heat treatment did not inhibit rather increase S. typhimurium OMV-triggered caspase-1 activation and IL-1β secretion (Figures 5D, E). These results demonstrate that OMVs may function as a cargo for cytosolic delivery of flagellin against heat- or proteinase-induced damages.
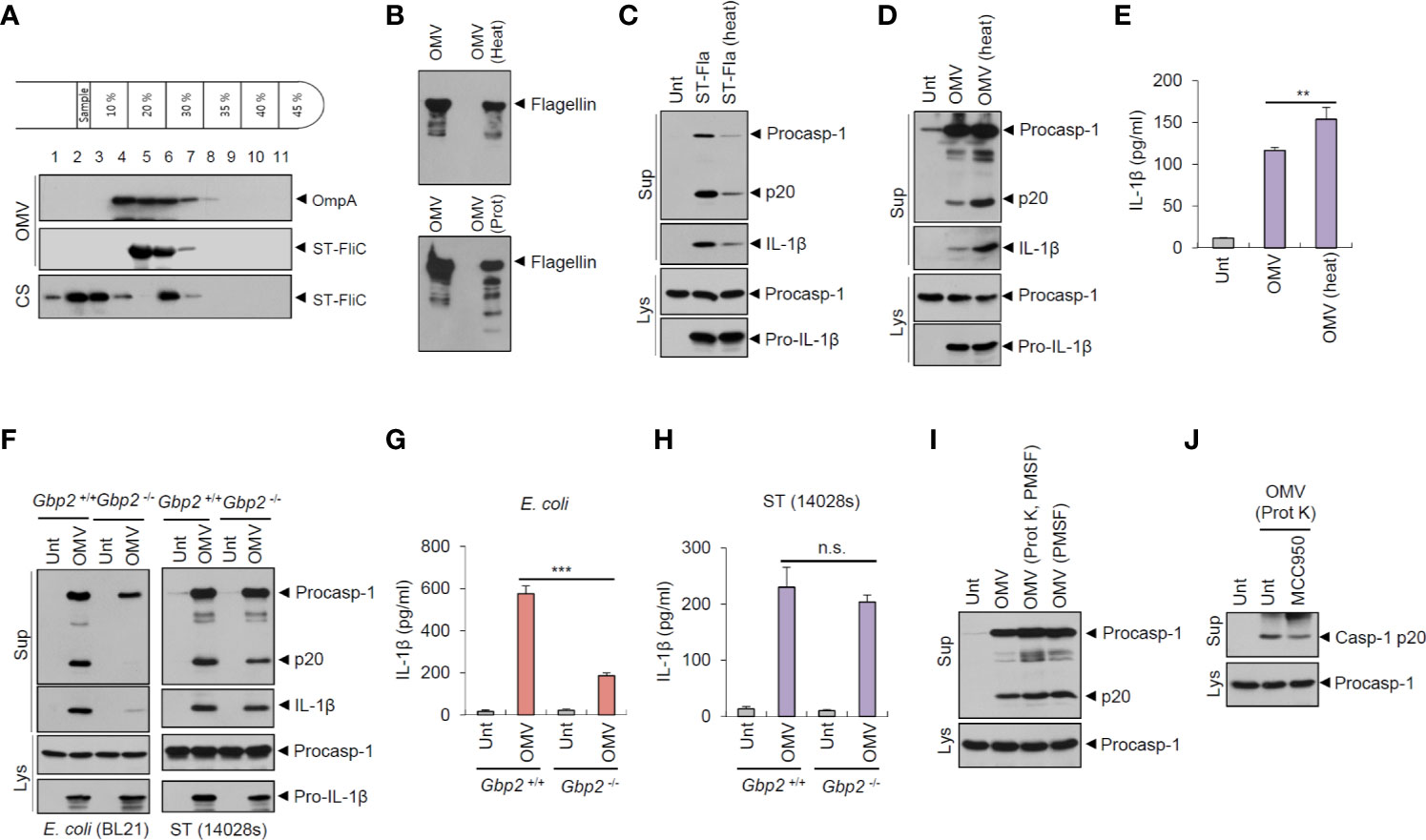
Figure 5 Salmonella-released outer membrane vesicles exhibit heat-resistant and GBP2-independent inflammasome activation potential. (A) Immunoblots of OptiPrep density gradient fractions of S. typhimurium OMVs. (B) Immunoblots of the extracts of OMVs in the presence of heat treatment (97°C, 30 min) or proteinase K treatment (10 µg/ml, 30 min) by anti-ST-flagellin antibody. (C) Immunoblots from mouse BMDMs primed with LPS (0.25 µg/ml, 3 h), followed by the treatment with intact or heat-treated S. typhimurium flagellin (250 ng/ml) for 6 h. (D) Immunoblots of mouse BMDMs treated with intact or heat-treated S. typhimurium OMVs (5 µg/ml) for 6 h. (E) Quantification of IL-1β in the culture supernatants of mouse BMDMs treated with intact or heat-treated S. typhimurium OMVs for 6 h. (n = 5). (F) Immunoblots from Gbp2+/+ or Gbp2−/− mice BMDMs treated with E. coli BL21 OMVs (5 µg/ml) or S. typhimurium (14028s) OMVs (5 µg/ml) or for 8 h. (G, H) Quantification of IL-1β in the culture supernatants of Gbp2+/+ or Gbp2−/− mice BMDMs treated as same as (F) (n = 3). (I, J) Immunoblots of mouse BMDMs treated with intact or proteinase-treated S. typhimurium OMVs (10 µg/ml) in the presence of MCC 950 (100 nM) as indicated for 8 h. After proteinase treatment, PMSF was added to eliminate proteinase K activity. Culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies. Data were expressed as the mean ± SEM. Asterisks indicate significant differences. (**P < 0.01, ***P < 0.001, n.s. not significant).
Guanylate-binding proteins (GBPs) are required for bacterial OMV-mediated non-canonical inflammasome activation in the previous studies (19, 24). The results suggest that GBP2 and GBP5 facilitate the release of LPS from OMVs, which enables the interaction between LPS and caspase-11. In this context, we investigated the potential involvement of GBP2 on S. typhimurium OMV-induced inflammasome activation. GBP2 deficiency clearly abrogated caspase-1 activation and IL-1β secretion by OMVs isolated from E. coli culture (Figures 5F, G). In contrast to E. coli-derived OMVs, wild-type Salmonella-derived OMVs caused similar levels of caspase-1 processing and IL-1β production from Gbp2-deficient macrophages (Figures 5F, H). However, flagellin-deficient (ΔfliC–fljB) S. typhimurium OMVs led to a weak GBP2-dependent IL-1β production (Figure S7), indicating that S. typhimurium OMVs are also able to mediate non-canonical inflammasome activation to a lesser degree. These results demonstrate that E. coli OMVs and flagellin-deficient S. typhimurium OMVs can trigger GBP2-NLRP3-mediated non-canonical inflammasome activation.
In addition, we further examined inflammasome-activating capacity of proteinase-treated S. typhimurium OMVs to exclude a possibility that free flagellin contamination in the isolated OMVs may affect our results. Proteinase-treated OMVs caused a similar extent of caspase-1 activation to intact OMVs (Figure 5I). Furthermore, proteinase-treated OMVs induced a robust caspase-1 activation regardless of MCC950 treatment (Figure 5J). These observations further support that S. typhimurium OMVs can trigger NLRC4 inflammasome signaling via OMV-associated flagellin.
To further evaluate the role of flagellin for inflammasome-stimulating potential of Salmonella OMVs, we examined time-dependent inflammasome activity induced by wild-type and flagellin-deficient S. typhimurium OMVs. We found that flagellin-deficient Salmonella-released OMVs caused a delayed and weaker inflammasome activation than wild-type S. typhimurium OMVs in BMDMs (Figures 6A, B). Interestingly, flagellin-deficient Salmonella OMVs induced a significant weaker cell death of macrophages, as measured by LDH release, until 8 h post OMV treatment compared to wild-type Salmonella OMVs (Figure 6C). However, prolonged (16 h) treatment with Salmonella OMVs caused similar IL-1β production and pyroptotic cell death irrespective of flagellin expression (Figures 6B, C).
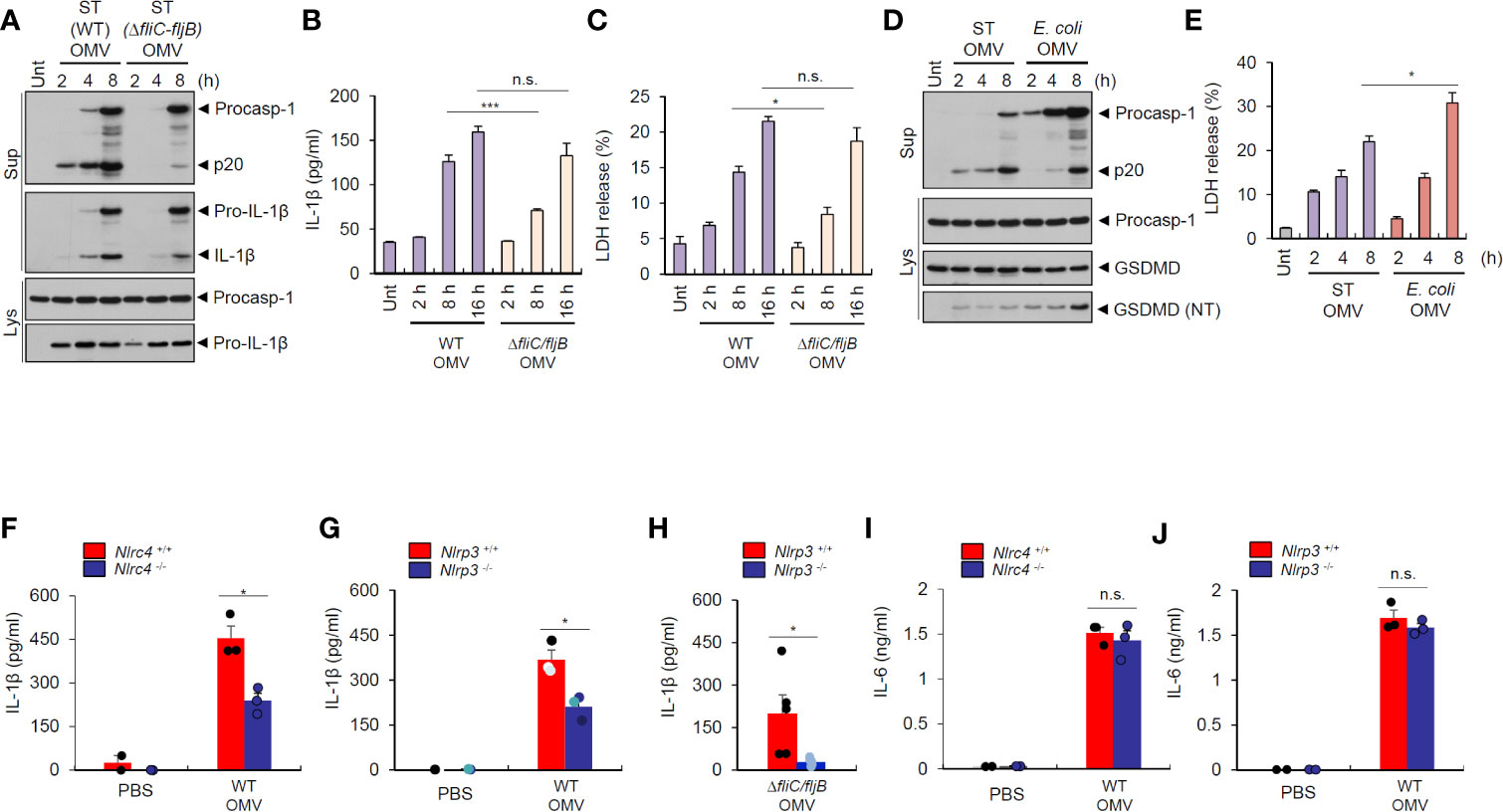
Figure 6 Salmonella-released outer membrane vesicles trigger in vivo NLRC4-dependent interleukin-1β secretion. (A) Immunoblots from mouse BMDMs treated with wild-type (WT) or ΔfliC–fljB S. typhimurium (SL1344) OMV (5 µg/ml) for 2, 4, 8 h. (B) Quantification of IL-1β in the culture supernatants of mouse BMDMs treated with WT or ΔfliC–fljB S. typhimurium (SL1344) OMVs (5 µg/ml) for 2, 8, 16 h. (n = 3) (C) Quantification of LDH release into culture supernatants of mouse BMDMs treated with WT or ΔfliC–fljB S. typhimurium (SL1344) OMVs (5 µg/ml) for 2, 8, 16 h. (n = 3) (D) Immunoblots from mouse BMDMs treated with S. typhimurium (SL1344) or E. coli (BL21) OMVs (5 µg/ml) for 2, 4, 8 h as indicated. GSDMD, gasdermin D (E) Quantification of LDH release into culture supernatants of mouse BMDMs treated with S. typhimurium (SL1344) or E. coli (BL21) OMVs (5 µg/ml) for 2, 4, 8 h as indicated. (n = 3). (F, G) Quantification of IL-1β in the peritoneal lavage fluid of Nlrc4+/+ or Nlrc4−/− (F), or Nlrp3+/+ or Nlrp3−/− (G) mice 6 h after intraperitoneal injection of PBS or S. typhimurium OMVs (50 µg/mice). (n = 2, PBS; n = 3, OMV) (H) Quantification of IL-1β in the peritoneal lavage fluid of Nlrp3+/+ or Nlrp3−/− mice 6 h after intraperitoneal injection of ΔfliC–fljB S. typhimurium OMVs (50 µg/mice). (n = 5, Nlrp3+/+; n = 6, Nlrp3−/−) (I, J) Quantification of IL-6 in the peritoneal lavage fluid of Nlrc4+/+ or Nlrc4−/− (I), or Nlrp3+/+ or Nlrp3−/− (J) mice 6 h after intraperitoneal injection of PBS or S. typhimurium OMVs (50 µg/mice). (n = 2, PBS; n = 3, OMV) Culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies. Data were expressed as the mean ± SEM. Asterisks indicate significant differences. (*P < 0.05, ***P < 0.001, n.s. not significant).
Then, we checked time-dependent inflammasome response from macrophages upon treatment with Salmonella- or E.coli-derived OMVs. S. typhimurium OMVs promoted rapid caspase-1 activation as early as 2 h post treatment, whereas E. coli OMVs did not induce robust caspase-1 activation until 8 h-treatment (Figure 6D). These findings suggest that S. typhimurium OMV-associated flagellin can trigger more rapid host inflammasome activation than E. coli OMV-bound LPS. Instead, E. coli OMVs promoted stronger gasdermin D cleavage (Figure 6D), as determined by the presence of cleaved GSDMD (NT) in the cytosol, and pyroptotic cell death (Figure 6E) than S. typhimurium OMVs at 8 h-treatment to macrophages. These results propose that NLRC4 inflammasome machinery is a critical sensor for rapid detection of flagellated bacteria-released OMVs as a host defense mechanism, whereas non-flagellated bacteria-induced caspase-11 non-canonical inflammasome signaling rather contributes to pyroptosis in response to bacterial OMV-delivered LPS.
To determine in vivo role of NLRC4 against S. typhimurium–released OMVs, we intraperitoneally injected bacterial OMVs into mice. Analysis of peritoneal lavage contents from mice revealed that intraperitoneal challenge with S. typhimurium-released OMVs caused robust IL-1β production (Figure 6F). Nlrc4-deficient mice showed significantly reduced IL-1β production (Figure 6F) after S. typhimurium OMV challenge. Interestingly, NLRP3 deficiency significantly reduced IL-1β production in the peritoneal lavage of mice after intraperitoneal injection of Salmonella-derived OMVs (Figure 6G). Flagellin-deficient S. typhimurium OMVs also caused NLRP3-dependent IL-1β production from mice (Figure 6H), indicating that Salmonella-derived OMVs can facilitate both NLRC4- and NLRP3-mediated inflammasome activation in mice. However, IL-6 production was promoted regardless of NLRC4 and NLRP3 presence in the peritoneal lavage of mice upon S. typhimurium OMV challenge (Figures 6I, J). These findings further support the hypothesis that Salmonella-derived OMVs can induce in vivo NLRC4-mediated canonical inflammasome activation in addition to caspase-11 non-canonical inflammasome activation.
Gram-negative bacteria-released OMVs can facilitate the spread of bacterial pathogenicity through delivery of virulence factors and antibiotic resistance genes (16). Bacterial OMVs can also trigger host innate sensing machinery (e.g., inflammasome pathways), which helps to increase bacterial clearance (17). In particular, bacteria-released OMVs promote caspase-11-mediated non-canonical inflammasome responses via OMV-bound LPS (18, 24, 25). OMV-mediated intracellular delivery of LPS results in the activation of caspase-11, a cytosolic LPS sensor, with the help of GBPs (19). Active caspase-11 then induces gasdermin D pore formation in the plasma membrane, which leads to potassium efflux and subsequent NLRP3 inflammasome activation (26). In this context, bacterial OMVs can induce IL-1β secretion in a caspase-11-mediated indirect manner, as described above. However, our study is the first to find that bacterial OMVs can directly trigger host NLRC4-mediated canonical inflammasome activation by delivering bacterial flagellin into the cytoplasm of host cells.
Our results indicate that inflammasome activation by OMVs from flagellated bacteria such as S. typhimurium and P. aeruginosa was markedly abolished in Nlrc4-deficient macrophages, but slightly decreased in Nlrp3-deficent cells. Consistently, OMVs released from S. typhimurium lacking flagellin and T3SS protein prgJ failed to induce strong inflammasome activation. These findings further support the potential role of bacterial OMV-associated flagellin for the NLRC4 inflammasome activation. Bacterial flagellin is essential for bacterial motility and invasion, and thus is considered a significant virulence factor. Additionally, flagellin is the most abundant protein in the secreted fraction of S. typhimurium cultures (27). Previous studies initially suggested that Salmonella flagellin is delivered to the cytoplasm of host cells through T3SS-dependent direct secretion and bacterial phagocytosis (22, 28). However, recent studies found that flagellin is also detected in OMVs from S. typhimurium and P. aeruginosa (29–31). In this context, OMV cargo is capable of delivering bacterial flagellin into the cytoplasm leading to intracellular NLRC4 inflammasome responses.
Our data demonstrate that heat treatment significantly reduced inflammasome-stimulating potential of Salmonella CS, but not of Salmonella OMVs. Considering that heat treatment did not induce flagellin degradation in the bacterial CS (Figure S1B), we speculate that heat treatment may disrupt the conformation of flagellin or prgJ, resulting in the attenuation of the ability to activate the inflammasomes. In contrast, heat treatment slightly increased Salmonella OMV-driven inflammasome activation. It is possible that heat may cause a weak disruption of OMV structure, facilitating more efficient delivery to the cytoplasm. These findings suggest that OMVs can protect flagellin from heat-induced damage during the delivery to the target cells.
Somewhat different from the in vitro results in macrophages, in vivo IL-1β production by S. typhimurium OMV was dependent on both NLRC4 and NLRP3 after intraperitoneal challenge with OMVs (Figure 6). These in vivo results suggest that Salmonella OMV-bound LPS was also important for inflammasome activation in the peritoneal lavage contents of mice. Both NLRC4 and NLRP3 are likely to be redundant for detecting Salmonella-derived OMVs to trigger inflammasome responses, at least in peritoneal lavage contents. Flagellin was the main stimulant for S. typhimurium OMV-driven inflammasome activation in the BMDMs, but flagellin-deficient S. typhimurium OMVs also triggered a slight inflammasome activation in a NLRP3-dependent manner (Figure 3D). However, we found that flagellin-deficient S. typhimurium OMVs caused a delayed inflammasome response in BMDMs compared with wild-type S. typhimurium OMVs (Figure 6A). Furthermore, flagellated S. typhimurium OMVs induced more rapid inflammasome activation than non-flagellated E. coli OMVs in BMDMs (Figure 6D). These results indicate that at least in macrophages, NLRC4 inflammasome machinery is a more rapid sensor for detecting Salmonella-released OMVs than caspase-11. Particularly, NLRC4-mediated pathogen sensing is crucial for host defense against enteric infection, but not against intraperitoneal infection (10). In this context, NLRC4 inflammasome signaling for flagellated bacteria-released OMVs might have a major role in host defense, especially in the gut.
GBPs, interferon-inducible GTPases, are required for clearance of intracellular pathogens by inducing lysis of pathogen-containing vacuoles (32–34). GBPs also facilitates the intracellular interaction of OMV-bound LPS with caspase-11 by exposing lipid A (19). Thus, GBPs have a pivotal role in non-canonical inflammasome activation by OMVs. However, GBP2 seemed not to be critical for inflammasome activation by S. typhimurium-released OMVs (Figure 5H). These results indicated that GBP2 was not involved in activation of NLRC4 inflammasome signaling by OMV-associated flagellin. The precise role of other GBPs for OMV-mediated NLRC4 inflammasome requires further investigation.
A physiological significance of NLRC4 inflammasome activation by bacterial OMVs remains to be further determined. Based on previous studies and our present results, both caspase-11 non-canonical and NLRC4 canonical inflammasomes promote caspase-1-dependent IL-1β production in response to bacteria-released OMVs (Figure 7). Interestingly, both caspase-11-deficient and Gbp-deficient mice showed increased survival after poly I:C and the subsequent E. coli OMV injection (19). This finding indicates that caspase-11 non-canonical inflammasome activation by E. coli OMV-associated LPS significantly contributes to endotoxic shock via inducing a robust pyroptosis. Instead, NLRC4 inflammasome signaling by flagellated bacteria-released OMVs can contribute to rapid detection of bacterial infection leading to the inhibition of bacterial dissemination and virulence as a host defense mechanism (Figure 7). Taken together, our findings suggest that NLRC4 is a bona-fide sensor for detecting flagellated bacteria-released OMVs to mediate inflammasome signaling. These results provide molecular insights into host defense mechanisms against flagellin-containing bacterial pathogens such as S. typhimurium or P. aeruginoasa.
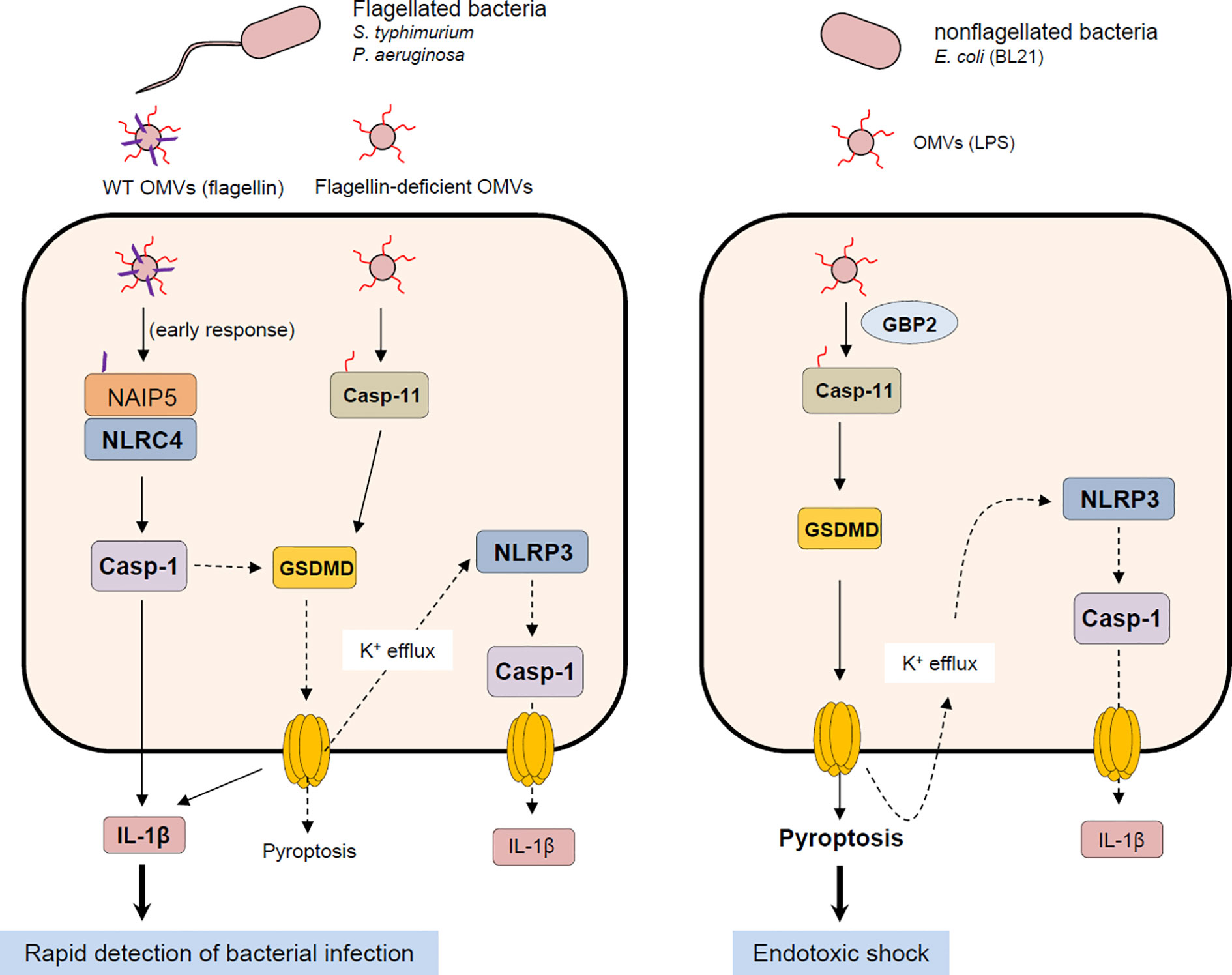
Figure 7 Proposed mechanism of host inflammasome activation by flagellated or non-flagellated bacteria-released outer membrane vesicles. Flagellated bacteria such as S. typhimurium or P. aeruginosa facilitate the delivery of bacterial flagellin and LPS into the cytoplasm of host cells via OMV cargo. In the initial phase of OMV exposure, OMV-delivered flagellin causes the assembly of NLRC4 inflammasome leading to caspase-1 activation and the subsequent IL-1β secretion. Besides flagellin, intracellular OMV-associated LPS can activate caspase-11 non-canonical inflammasome signaling, which includes GSDMD-dependent pyroptosis and NLRP3-mediated caspase-1 activation and IL-1β secretion. On the contrary, OMVs derived from non-flagellated bacteria such as E. coli mediates the cytosolic delivery of bacterial LPS. Non-flagellated bacterial -delivered LPS mainly causes the activation of caspase-11 and GSDMD-mediated pyroptosis, contributing to potential endotoxic shock. NAIP5, NLR family, apoptosis inhibitory protein 5; NLRC4, NLR family, CARD domain-containing protein 4; NLRP3, NLR family, pyrin domain-containing protein 3; GSDMD, gasdermin D.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Institutional Ethical Committee, Yonsei University College of Medicine.
JY designed and conducted most of the experiments in this study. IH and EL conducted the experiments. SS, E-JL, and JR provided critical materials and technical advice. J-WY supervised the entire project and wrote the manuscript with JY. All authors contributed to the article and approved the submitted version.
This work was supported by the National Research Foundation of Korea Grant funded by the Korean Government (2015M3A9B6073856, 2017R1A2B2007467, 2020R1A2B5B02001823).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank F. Shao (National Institute of Biological Sciences, China) for providing S. typhimurium mutants, and S.J. Oh and O.S. Shin (Korea University) for the help of Nanoparticle Tracking Analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.581165/full#supplementary-material
1. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
2. Storek KM, Monack DM. Bacterial recognition pathways that lead to inflammasome activation. Immunol Rev (2015) 265(1):112–29. doi: 10.1111/imr.12289
3. Sahoo M, Ceballos-Olvera I, del Barrio L, Re F. Role of the inflammasome, IL-1beta, and IL-18 in bacterial infections. ScientificWorldJournal (2011) 11:2037–50. doi: 10.1100/2011/212680
4. Yu JW, Lee MS. Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance. Arch Pharm Res (2016) 39(11):1503–18. doi: 10.1007/s12272-016-0827-4
5. Broz P. Recognition of Intracellular Bacteria by Inflammasomes. Microbiol Spectr (2019) 7(2):BAI-0003-2019. doi: 10.1128/microbiolspec.BAI-0003-2019
6. Duncan JA, Canna SW. The NLRC4 Inflammasome. Immunol Rev (2018) 281(1):115–23. doi: 10.1111/imr.12607
7. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477(7366):596–600. doi: 10.1038/nature10510
8. Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U S A (2013) 110(35):14408–13. doi: 10.1073/pnas.1306376110
9. Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science (2015) 350(6259):404–9. doi: 10.1126/science.aac5789
10. Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, Suzuki S, et al. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol (2012) 13(5):449–56. doi: 10.1038/ni.2263
11. Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A (2014) 111(20):7403–8. doi: 10.1073/pnas.1402911111
12. Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol (2010) 11(5):385–93. doi: 10.1038/ni.1859
13. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science (2013) 341(6151):1250–3. doi: 10.1126/science.1240988
14. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341(6151):1246–9. doi: 10.1126/science.1240248
15. Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol (2016) 17(8):914–21. doi: 10.1038/ni.3457
16. Schwechheimer C, Kuehn MJ. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat Rev Microbiol (2015) 13(10):605–19. doi: 10.1038/nrmicro3525
17. Pathirana RD, Kaparakis-Liaskos M. Bacterial membrane vesicles: Biogenesis, immune regulation and pathogenesis. Cell Microbiol (2016) 18(11):1518–24. doi: 10.1111/cmi.12658
18. Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, et al. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell (2016) 165(5):1106–19. doi: 10.1016/j.cell.2016.04.015
19. Santos JC, Dick MS, Lagrange B, Degrandi D, Pfeffer K, Yamamoto M, et al. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. EMBO J (2018) 37(6):e98089. doi: 10.15252/embj.201798089
20. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458(7237):509–13. doi: 10.1038/nature07710
21. Gim E, Shim DW, Hwang I, Shin OS, Yu JW. Zika Virus Impairs Host NLRP3-mediated Inflammasome Activation in an NS3-dependent Manner. Immune Netw (2019) 19(6):e40. doi: 10.4110/in.2019.19.e40
22. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol (2006) 7(6):576–82. doi: 10.1038/ni1346
23. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477(7366):592–5. doi: 10.1038/nature10394
24. Finethy R, Luoma S, Orench-Rivera N, Feeley EM, Haldar AK, Yamamoto M, et al. Inflammasome Activation by Bacterial Outer Membrane Vesicles Requires Guanylate Binding Proteins. MBio (2017) 8(5):e01188-17. doi: 10.1128/mBio.01188-17
25. Chen S, Yang D, Wen Y, Jiang Z, Zhang L, Jiang J, et al. Dysregulated hemolysin liberates bacterial outer membrane vesicles for cytosolic lipopolysaccharide sensing. PLoS Pathog (2018) 14(8):e1007240. doi: 10.1371/journal.ppat.1007240
26. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
27. Matsui H, Eguchi M, Ohsumi K, Nakamura A, Isshiki Y, Sekiya K, et al. Azithromycin inhibits the formation of flagellar filaments without suppressing flagellin synthesis in Salmonella enterica serovar typhimurium. Antimicrob Agents Chemother (2005) 49(8):3396–403. doi: 10.1128/AAC.49.8.3396-3403.2005
28. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature (2004) 430(6996):213–8. doi: 10.1038/nature02664
29. Liu Q, Liu Q, Yi J, Liang K, Hu B, Zhang X, et al. Outer membrane vesicles from flagellin-deficient Salmonella enterica serovar Typhimurium induce cross-reactive immunity and provide cross-protection against heterologous Salmonella challenge. Sci Rep (2016) 6:34776. doi: 10.1038/srep34776
30. Bai J, Kim SI, Ryu S, Yoon H. Identification and characterization of outer membrane vesicle-associated proteins in Salmonella enterica serovar Typhimurium. Infect Immun (2014) 82(10):4001–10. doi: 10.1128/IAI.01416-13
31. Bauman SJ, Kuehn MJ. Purification of outer membrane vesicles from Pseudomonas aeruginosa and their activation of an IL-8 response. Microbes Infect (2006) 8(9-10):2400–8. doi: 10.1016/j.micinf.2006.05.001
32. Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature (2014) 509(7500):366–70. doi: 10.1038/nature13157
33. Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Ruhl S, et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol (2015) 16(5):476–84. doi: 10.1038/ni.3119
Keywords: outer membrane vesicles, NLRC4, inflammasome, interleukin-1, caspase-1, flagellin, host defense
Citation: Yang J, Hwang I, Lee E, Shin SJ, Lee E-J, Rhee JH and Yu J-W (2020) Bacterial Outer Membrane Vesicle-Mediated Cytosolic Delivery of Flagellin Triggers Host NLRC4 Canonical Inflammasome Signaling. Front. Immunol. 11:581165. doi: 10.3389/fimmu.2020.581165
Received: 08 July 2020; Accepted: 12 October 2020;
Published: 18 November 2020.
Edited by:
Soohyun Kim, Konkuk University, South KoreaReviewed by:
Richard Louis Ferrero, Hudson Institute of Medical Research, AustraliaCopyright © 2020 Yang, Hwang, Lee, Shin, Lee, Rhee and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Je-Wook Yu, amV3b29reXVAeXVocy5hYw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.