
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 18 January 2021
Sec. Alloimmunity and Transplantation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.579151
 Chloé Spilleboudt1*†
Chloé Spilleboudt1*† Virginie De Wilde1,2†Philippe Lewalle3Ludovic Cabanne4,5
Virginie De Wilde1,2†Philippe Lewalle3Ludovic Cabanne4,5 Mathieu Leclerc4,5Florence Beckerich4,5
Mathieu Leclerc4,5Florence Beckerich4,5 Dominique Bories4,5Silvia Cardoso6
Dominique Bories4,5Silvia Cardoso6 Miguel P. Soares6Benoît Vokaer1Jean-Michel Hougardy7
Miguel P. Soares6Benoît Vokaer1Jean-Michel Hougardy7 Véronique Flamand1Judith Racapé7,8Marc Abramowicz9
Véronique Flamand1Judith Racapé7,8Marc Abramowicz9 Sébastien Maury4,5‡
Sébastien Maury4,5‡ Alain Le Moine1,7*‡
Alain Le Moine1,7*‡Graft-versus-host disease (GVHD) remains a major clinical drawback of allogeneic hematopoietic stem cell transplantation (HSCT). Here, we investigated how the stress responsive heme catabolizing enzyme heme oxygenase-1 (HO-1, encoded by HMOX1) regulates GVHD in response to allogeneic hematopoietic stem cell transplantation in mice and humans. We found that deletion of the Hmox1 allele, specifically in the myeloid compartment of mouse donor bone marrow, promotes the development of aggressive GVHD after allogeneic transplantation. The mechanism driving GVHD in mice transplanted with allogeneic bone marrow lacking HO-1 expression in the myeloid compartment involves enhanced T cell alloreactivity. The clinical relevance of these observations was validated in two independent cohorts of HSCT patients. Individuals transplanted with hematopoietic stem cells from donors carrying a long homozygous (GT)n repeat polymorphism (L/L) in the HMOX1 promoter, which is associated with lower HO-1 expression, were at higher risk of developing severe acute GVHD as compared to donors carrying a short (GT)n repeat (S/L or S/S) polymorphism associated with higher HO-1 expression. In this study, we showed the unique importance of donor-derived myeloid HO-1 in the prevention of lethal experimental GVHD and we corroborated this observation by demonstrating the association between human HMOX1 (GT)n microsatellite polymorphisms and the incidence of severe acute GVHD in two independent HSCT patient cohorts. Donor-derived myeloid HO-1 constitutes a potential therapeutic target for HSCT patients and large-scale prospective studies in HSCT patients are necessary to validate the HO-1 L/L genotype as an independent risk factor for developing severe acute GVHD.
Acute graft-versus-host disease (GVHD) remains a major cause of mortality after allogeneic HSCT (1, 2). This pathologic process is counter-regulated by regulatory T cells (TREGS) (3, 4) as well as by myeloid-derived suppressor cells (MDSCs) (5, 6). The latter are a heterogeneous population of immature myeloid cells harboring monocytic or polymorphonuclear markers (7–10), that suppress T cell responses via diverse and not fully understood mechanisms. In an experimental mouse model, MDSCs suppress acute GVHD via an IL-13- and arginase-1-dependent mechanism that regulates graft-versus-host alloreactivity (11). In other experimental settings, MDSCs have also been show to exert T cell suppression through indoleamine 2,3-dioxygenase (IDO) production or through nitric oxide synthase (NOS)-dependent mechanisms which are associated or not with the expression of heme oxygenase-1 (HO-1) (12–14). Of note, IDO and iNOS are heme proteins that are tightly regulated by mechanisms that control heme-iron metabolism in monocyte cells (15). Moreover, heme catabolism by HO-1 counter-regulates the rejection of transplanted cells, tissues, or organs in a variety of experimental conditions (16). In keeping with this notion, we have previously shown that MDSCs block T cell-mediated allograft rejection via a mechanism that is dependent on the expression of HO-1 (17). It has also been reported that non-specific pharmacologic HO-1 overexpression regulates T cell alloreactivity in a manner that prevents the onset of experimental GVHD (18–21). However, whether natural endogenous levels of HO-1 limit GVHD and its sources, and the contribution of donor versus recipient HO-1 remain critical open questions.
The human HMOX1 promoter has a microsatellite polymorphism in which the number of (GT) repeats (GT)n is inversely correlated with HMOX1 transcriptional activity and, ultimately, HO-1 expression (22). This (GT)n polymorphism has been associated with the clinical outcome of a variety of inflammatory diseases (23, 24). Specifically, longer (GT)n repeats are associated with low levels of HO-1 expression (21) and correlate with higher disease susceptibility, while shorter (GT)n repeats are associated with higher levels of HO-1 expression and correlate with lower disease susceptibility. This has been illustrated for cardiovascular disease (25–28), chronic kidney disease (29, 30), HIV-induced central nervous system neuroinflammation (31, 32), rheumatoid arthritis (33), chronic obstructive pulmonary disease (34), and organ transplantation (35, 36). In sharp contrast, however, shorter (GT)n repeats are associated with higher levels of HO-1 expression and promote metaflammation associated with the onset of experimental metabolic disease (37).
Herein, we demonstrated the unique importance of donor-derived myeloid HO-1 in the prevention of lethal experimental GVHD and we corroborated this observation with the association between the human HMOX1 (GT)n microsatellite polymorphisms and the incidence of severe acute GVHD in two independent HSCT patient cohorts.
Hmox1−/− mice (38) were generated by Hmox1+/− mating and genotyped, as previously described (38, 39). C57BL/6 LysMCre/wtHmox1Δ/Δ mice were generated by mating of C57BL/6 Hmox1lox/lox mice (40) with C57BL/6 LysMCre/wt mice (kind gift from S.A. Nedospasov) (41). Mice received humane care in compliance with the Principles of Laboratory Animal Care formulated by the National Institutes of Health (Bethesda, MA), and protocols were approved by the local committee for animal welfare (agreement # LA2500519).
Recipients were lethally irradiated with 750cGy (BALB/c) or 980cGy (C57BL/6) from a 137Cs source, one day before bone marrow (BM) and whole spleen cell infusions (2.5 × 106 BM and 2 × 106 spleen C57BL/6 cells into BALB/c recipients and 5 × 106 BM with 15 × 106 spleen BALB/c cells into C57BL/6 recipients into the tail vein in 0.2 ml of RPMI solution). GVHD was monitored as previously described (42). For both CD8+ and CD4+ T cell depletion, 1 mg of a pair of monoclonal antibodies (mAbs) were injected i.p. at day 1 posttransplant (clone YTS 169 plus YTS 156 for CD8+ T cells and clones YTS191 plus YTA 3.1.2 for CD4+ T cells (43)) or YCATE mAb (control isotype), all kindly provided by Dr. S. Cobbold, Dunn School of Pathology, Oxford University, UK.
For MLR, responder cells (2.5 × 106 cells/ml) isolated from host spleens were stimulated with irradiated adequate splenocytes (2000 Rad; 2.5 × 106 cells/ml), in 48-well, flat-bottom plates. IFN-γ, IL-17A, and IL-10 production was measured in the culture supernatants after 48 or 72 h using commercially available ELISA kits (Duoset, R&D Systems, Minneapolis, MN). The detection threshold was 10 pg/ml for IFN-γ, IL-17A, and IL-10. For HO-1 plasma quantification, the HO-1 ELISA kit (ADI-960-0F1, Enzo Life Sciences) was used following manufacturer instructions.
Total RNA was extracted from liver and spleen using the MagNaPure LC RNA Isolation Kit III for tissue (Roche Diagnostics 03330591001) or from bone marrow-derived macrophages (BMDMs) with the MagNaPure LC RNA Isolation Kit-High Performance (Roche Diagnostics 03542394001). Reverse transcription and real-time PCR were performed using LightCycler-RNA Master Hybridization Probes (one-step procedure) on a LightCycler apparatus (Roche Diagnostics). The number of mRNA copies was evaluated using a standard curve for each gene of interest and was normalized to β-actin as a housekeeping gene. Primer and probe sequences are detailed in Table S1.
Commercially available fluorescent antibodies were used following manufacturer’s instructions. FITC-conjugated anti-mouse F4/80 (clone BM8), APC-conjugated anti-mouse H2kb (clone AF6-88.5.5.3), eFluor450-conjugated anti-mouse Ly-6G (clone RC57BL/6-8C5), eFluor450-conjugated anti-mouse H2kd (clone SF1-1.1.1), PerCP-Cy5.5-conjugated anti-mouse Ly-6C (clone HK1.4), and anti-mouse CD16/CD32 (Fc block, clone 93) were purchased from eBioscience. FITC-conjugated anti-mouse IA/IE (clone 2G9), PE-conjugated anti-mouse CD11c (cloneHL3), PE-conjugated anti-mouse CD49d (clone 9C10), PB-conjugated anti-mouse CD3 (clone 500A2), APC-conjugated anti-mouse CD8 (clone 53–6.7), and APC-conjugated anti-mouse CD11b (clone M1/70) were purchased from BD Biosciences.
HO-1 intracytoplasmic staining was performed using the Intracellular Fixation and Permeabilization Buffer Set (88–8824, eBioscience) and unconjugated anti-mouse HO-1 (clone HO-1-1, Abcam), followed by FITC-conjugated anti-mouse IgG1 or V450-conjugated anti-mouse IgG1(clone A85-1, BD Biosciences). The isotype clone NCG01 was used as a control antibody. Flow cytometry analysis was performed on a BD LSRFortessa cell analyzer using BD FACSDiva software (BD Biosciences).
Hemoglobin levels, blood leukocyte and platelet counts were performed using the automated system ADVIA 120 (Siemens, Munich, Germany).
Serum aspartate aminotransferase (AST) levels were measured by specific absorbance (Cobas 501, Roche Diagnostics).
1 × 106 BM cells/ml isolated from femurs and tibias were cultured in DMEM medium supplemented with L-Glutamine, 4.5 g/L glucose (BE12-604F, Lonza), 10% heat-inactivated fetal calf serum (FCS), amino acids, sodium pyruvate, penicillin/streptomycin, β-mercaptoethanol, and 20% supernatant from an M-CSF-transfected L929 cell culture. Three days later, 5 ml of complete medium containing 60% of L929 cell supernatant was added. At day 7, BM-derived macrophages (BMDMs) were stimulated with 100 ng/ml of LPS (L2630, Sigma-Aldrich) for 24 h in fresh medium with 2% L929 supernatant.
After a wash with phosphate-buffered saline (PBS), target spleen cells were resuspended at 1 × 106 cells/ml and labeled with 0.2 (BALB/C) or 2 µM (C57BL/6) of CFSE (Cell trace CFSE Cell Proliferation Kit) in pre-warmed PBS-BSA 0.1% for 10 min at 37°C. The reaction was stopped by the addition of an equal volume of cold PBS, followed by 5 min of incubation on ice. Then, 100 µl of CFSE-labeled cell solution (200 × 106 cells/ml of BALB/C and C57BL/6 mice) was injected IV into mice with GVHD. Two hours later, spleen cells were analyzed by FACS.
Data were collected from two independent cohorts of 120 and 160 patients. The first cohort included 120 patients at the Henri Mondor hospital in Paris (France) who underwent HSCT between January 2007 and December 2012. The second cohort included 160 patients who underwent HSCT at the Jules Bordet Institute (Brussels, Belgium) between January 2001 and December 2011. In each cohort, all HSCT patients were retrospectively included during the fixed period without exclusion criteria. All grafts were from HLA-matched related donors (MRD) or HLA-matched unrelated donors (MUD). Patient and donor characteristics at the time of transplantation are shown in Tables 1 and 2. Patient characteristics in the two cohorts were compared using Pearson’s chi-square test for categorical data and Student’s t test for continuous variables distributed normally. Myeloablative regimens are detailed in the supplementary data. Early disease stage was defined as being either the first complete remission (CR) of acute leukemia or a chronic phase of chronic myeloid leukemia, whereas late disease stage included patients who did not achieve CR or were beyond the first CR.
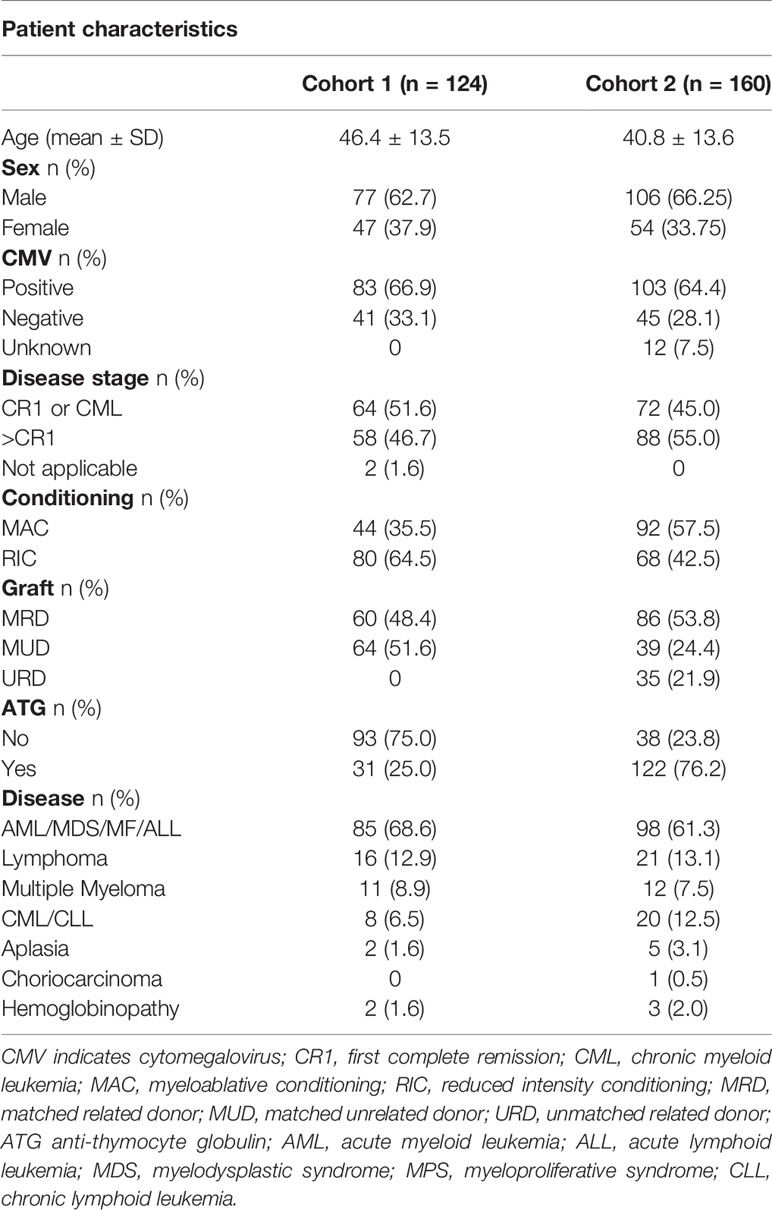
Table 1 Patient characteristics.
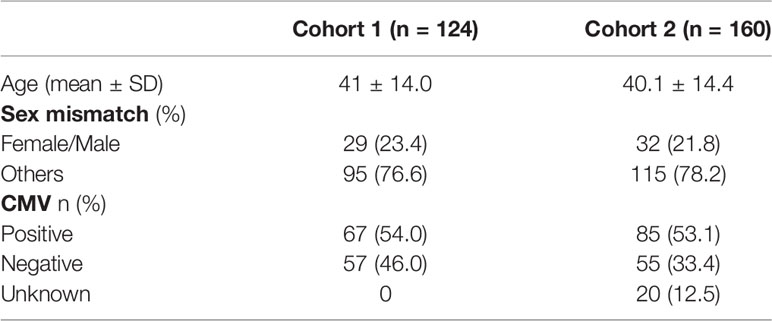
Table 2 HSC donor characteristics.
Acute GVHD was defined according to the National Institutes of Health consensus criteria based upon the timing of presentation, the typical clinical features, and the absence of diagnostic or distinctive features of chronic GVHD (44) and was graded according to the Glucksberg criteria (45). DNA samples from healthy volunteers were used for the control group.
DNA samples were obtained from residual material. In controls, DNA samples were prepared from peripheral blood stem cells (PBSCs) using standard procedures and commercially available kits. Genetic analyses were retrospectively performed with the approval of the Ethics Committees of Erasme Hospital and the Jules Bordet Institute (numbers P2011/255 and 2012/193, respectively). The Henri Mondor University Hospital (Créteil, France) has been accredited according to the international JACIE program since 2005. All patients and donors signed informed consent for (1) registration into the Promise database, and (2) cryopreservation of their biological material for research purposes.
The 5′-flanking region containing a poly (GT)n repeat of the HO-1 gene was amplified by PCR. A FAM-labelled sense primer, 5′-AGA-GCC-TGC-AGC-TTC-TCA-GA-3′, and an antisense primer, 5′-ACA AAG-TCT-GGC-CAT-AGG-ACC-3′, were used to amplify 50 ng of genomic DNA. Primers were purchased from Eurogentec. PCR was performed over 20 touchdown cycles of denaturation at 94°C for 20 s, annealing at 62°C to 52°C (temperature decreasing by 0.5°C at each cycle) and extension at 72°C for 30 s. To determine fragment lengths, the PCR product was loaded onto a POP-6 matrix (Applied BioSystems), and calculations of fragment lengths were laser based through an automated fragment analyzer (model 3130XL, Applied BioSystems) and comparisons with sequenced alleles of different lengths (GS-500 ROX Applied BioSystems).
In relation to functional data on GT repeats described by others (22, 27), alleles were classified into two groups: short (S) alleles with less than 30 GT repeats, and long (L) alleles with more than 30 GT repeats. This classification entails three possible genotypes within the donor and host populations: S/S, S/L, and L/L.
To compare categorical data for allelic distribution, genotype distribution, Hardy-Weinberg equilibrium, and GVHD frequency, either Fisher’s exact test or Pearson’s chi-square test was used, as appropriate. The association between GVHD and risk factors was determined by univariate and multivariate analyses. When estimating cumulative incidence of GVHD, we took into account death without GVHD as a competing risk and used the Fine and Gray model to estimate its subdistribution hazard ratio (SHR). Since graft rejection was an exclusion criterion for the study, it was not considered as a competing event for GVHD occurrence. Multivariate analysis included variables significantly associated with GVHD severity, if shown by univariate analysis. Due to the number of cases and the collinearity between the donor and the recipient genotypes, only three variables were included in our model: graft, radiotherapy, and donor genotype. The adjusted SHRs (aSHRs) are reported. All statistical analyses were performed using STATA 12.0 software. A p-value <0.05 was considered statistically significant. For mouse experiments, differences between groups were estimated using the two-tailed Mann-Whitney nonparametric test. Mouse survival curves were compared using the log-rank test. A p-value <0.05 was considered statistically significant.
We asked whether the expression of HO-1 was induced after experimental allogeneic HSCT and tested this hypothesis initially in BALB/c to C57BL/6 HSCT. Expression of HO-1 was induced in CD11b+Ly6ChighCD49d+MHC-IIhigh monocytic MDSCs and in CD11b+Ly6ClowMHC-IIlow monocytes (Figure 1A). Both monocytic cell populations were F4/80+, CD11c- and expressed intermediate levels of Ly6G (Figure 1A). This demonstrates that HSCT is associated with the induction of HO-1 in donor-derived hematopoietic cells.
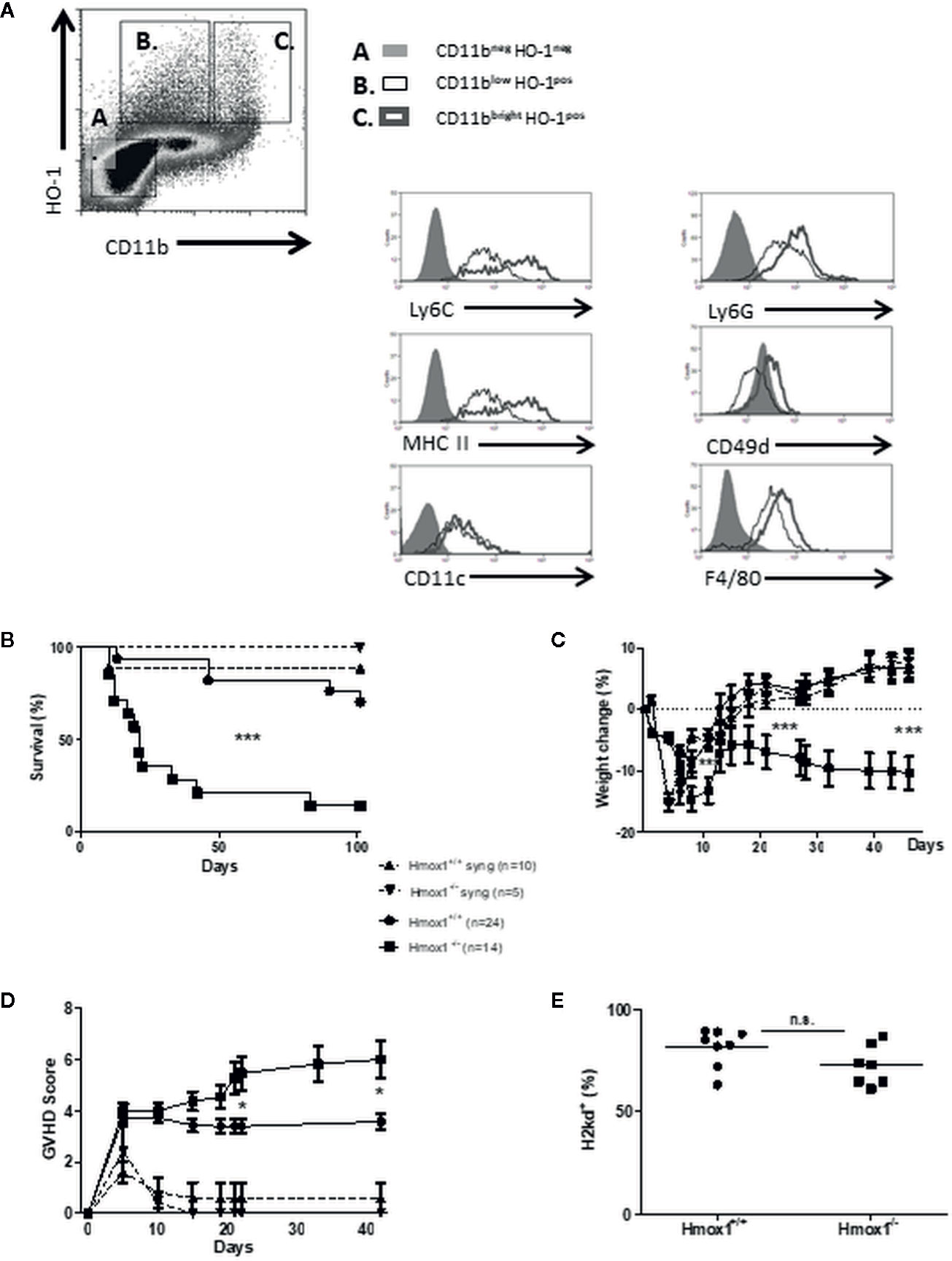
Figure 1 Donor-derived HO-1 regulates GVHD and survival. (A) BM cells from HSCT recipients were harvested on day 7 posttransplantation and characterized by FACS analysis. Two cell populations (B, C) expressing intracytoplasmic HO-1 were identified, based on CD11b, Ly6C, Ly6G, CD49d, F4/80, and MHC-II expression. Results are representative of five mice, seven days after transplantation. (B–D) GVHD characteristics were compared after syngeneic (BALB/c to BALB/c) and allogeneic (BALB/c to C57BL/6) BM transplantation. In lethally irradiated hosts, 5 × 106 BM cells and 15 × 106 spleen cells from either wild type (WT) or Hmox1−/− donors were injected. Survival (B), % weight loss (C) and GVHD scores (D) were compared between groups. (E) Host reconstitutions by allogeneic (H2kd) BM and spleen cells are shown in peripheral blood monocytes (PBMCs). Three or four independent experiments involving 5–6 animals per group were pooled. n.s., no significant difference; *P <.05; **P <.01; ***P <.001.
Survival of C57BL/6 recipients transplanted with allogeneic BM and spleen cells from BALB/c mice was compromised when the donor cells lacked HO-1 expression (BALB/c Hmox1−/− donors), as compared to C57BL/6 recipients transplanted with donor cells expressing HO-1 (BALB/c Hmox1+/+ donors) (Figure 1B). Survival in C57BL/6 recipients transplanted with allogeneic BALB/c Hmox1−/− BM and spleen cells was correlated with progressive body weight loss (Figure 1C) and clinical signs of GVHD (Figure 1D). This was not associated, however, with overall changes in hematopoietic chimerism (Figure 1E). Expression of HO-1 had no impact on the survival of syngeneic BALB/C recipients transplanted with BM and spleen cells from BALB/c Hmox1−/− or Hmox1+/+ mice (Figures 1B, D). This demonstrates that donor-derived HO-1 expression is essential to prevent the development of allogeneic GVHD after HSCT.
We then assessed whether the protective effect exerted by HO-1 depends on the myeloid BM cells or the alloreactive spleen cells within the HSCT. Survival of C57BL/6 recipients was compromised only when the transplanted BM, but not spleen cells, were isolated from allogeneic BALB/c Hmox1−/− mice, as compared to control C57BL/6 recipients transplanted with allogeneic BM and/or spleen cells from BALB/c Hmox1+/+ mice (Figures 2A, C). This suggests that expression of HO-1 by donor myeloid cells is required to suppress GVHD and all further experiments were performed using wild type Hmox1+/+ spleen cells.
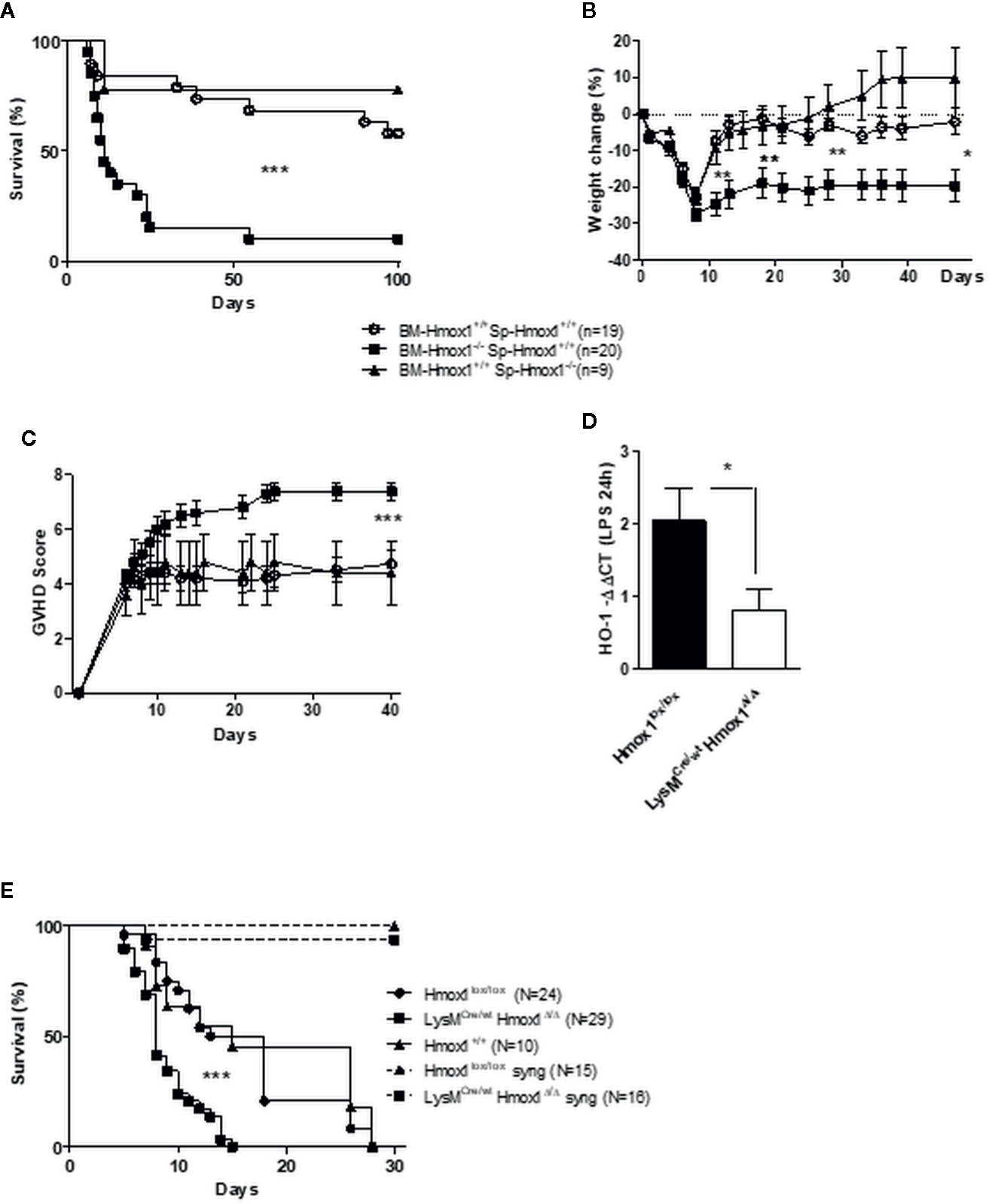
Figure 2 Myeloid HO-1 but not T cell-derived HO-1 regulates GVHD. Survival (A), weight change (B) and GVHD score (C) were compared between C57BL/6 irradiated hosts transplanted with a mix of BM and T cells from wild type (Hmox1+/+) BALB/c donors, with a mix of Hmox1−/− BM and Hmox1+/+ BALB/c T cells, or with a mix of Hmox1+/+ BM and Hmox1−/− BALB/c T cells, respectively. Results were pooled from three independent experiments including 6–7 mice per group. Donor chimerism (H-2Kd positive cells) was similar in both groups (data not shown). (D–E) A conditional HO-1 ablation in myeloid cells mimics the picture we observed with Hmox1−/− BM donors. The specific myeloid HO-1 deficiency was confirmed by comparing HO-1 transcription by lipopolysaccharide (LPS)-stimulated bone marrow-derived macrophages (BMDM) from control littermates Hmox1lox/lox or LysMCre/wtHmox1Δ/Δ mice (D). BMDM were generated as described in supplementary methods. Lethally-irradiated BALB/c mice were grafted with 2 × 106 spleen cells from Hmox1lox/lox littermates and 2.5 × 106 BM cells from either Hmox1lox/lox or LysMCre/wtHmox1Δ/Δ donors. Three experiments each including 8–10 mice were pooled. (E) Transplantation of Hmox1lox/lox or LysMCre/wtHmox1Δ/Δ BM cells into syngeneic C57BL/6 hosts did not trigger GVHD, in two separate experiments each with 7–8 mice. n.s. indicates no significant difference, *P <.05, **P <.01, ***P <.001.
To prove unequivocally that expression of HO-1 by donor myeloid cells is required to suppress GVHD, we used C57BL/6 LysMCre/wtHmox1Δ/Δ mice in which the Hmox1 allele is deleted specifically in the myeloid compartment, as confirmed in LPS-stimulated BM-derived monocytic cells by qRT-PCR (Figure 2D). Incidence of lethality (Figure 2E) and GVHD severity (data not shown) were higher in BALB/c recipients transplanted with BM from C57BL/6 LysMCre/wtHmox1Δ/Δ mice, as compared to control BALB/c recipients transplanted with BM from wild type C57BL/6 Hmox1+/+ or with C57BL/6 Hmox1flox/flox mice (Figure 2E). This was not the case for syngeneic C57BL/6 recipients transplanted with BM from C57BL/6 LysMCre/wtHmox1Δ/Δ mice, as compared to control C57BL/6 recipients receiving wild type Hmox1+/+ or Hmox1flox/flox BM (Figure 2E).
We then investigated the mechanism by which the expression of HO-1 in donor myeloid cells suppresses the development of GVHD. The number of donor-derived CD4+ and CD8+ T cells did not differ between recipients of either Hmox1−/− or Hmox1+/+ BM donors (Figure 3A). To address whether HO-1 expression modulates T cell alloreactive responses underlying GVHD, we performed standard mixed lymphocyte reactions (MLR) using spleen cells harvested seven days after HSCT and syngeneic, allogeneic, or third-party stimulatory cells. Leukocytes isolated from C57BL/6 recipients transplanted with BM from BALB/c Hmox1−/− mice produced higher levels of IL-17 and IFN-γ while the production of IL-10 was reduced, as compared to leukocytes isolated from C57BL/6 recipients transplanted with BM from BALB/c Hmox1+/+ mice (Figures 3B, D). Surprisingly, in the syngeneic cultures (left columns), IFN-γ production by leukocytes isolated from C57BL/6 recipients transplanted with BM from BALB/c Hmox1−/− mice was higher than that in leukocytes isolated from C57BL/6 recipients transplanted with BM from BALB/c Hmox1+/+ mice (Figure 3B). It is possible that spleen cells that have been seeded in culture still contain allogeneic and alloreactive cells, allowing ex-vivo IFN-γ production independently of the MLR stimulatory condition. In line with this, the highest amounts of IFN-γ are produced by B6 hosts transplanted with Hmox1−/− donors. This effect is observed with IFN-γ but not IL-17 or IL-10 due to the larger amounts of cytokine production.
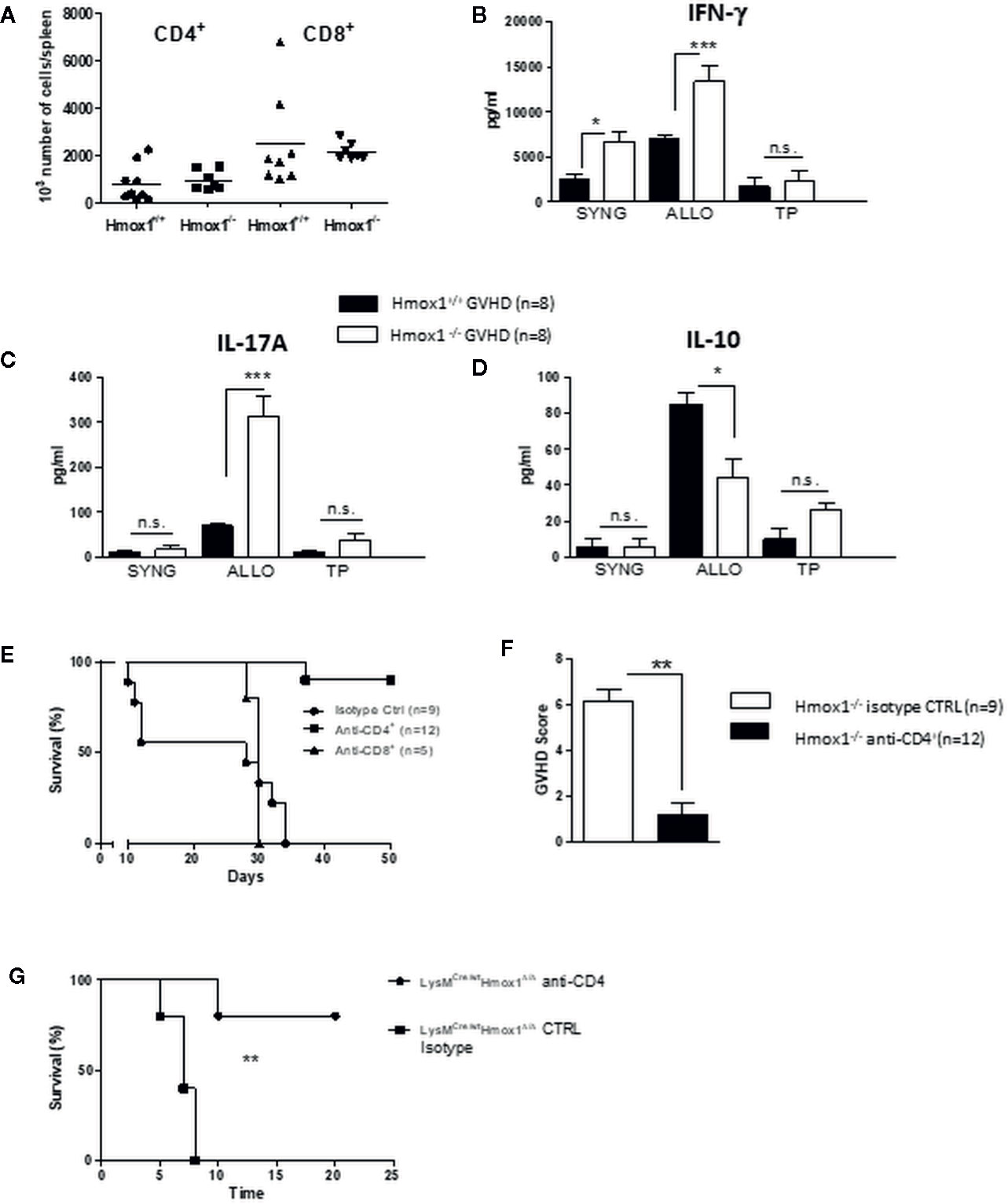
Figure 3 Donor-derived myeloid HO-1 controls T cell alloreactivity. (A) The number of donor-derived CD4+ and CD8+ T cells was quantified by flow cytometry at day 7 post-transplantation in C57BL/6 hosts grafted with Hmox1−/− BM and WT BALB/c T cells (Hmox1−/− GVHD) and compared with controls (WT GVHD). (B–D) Spleen cells from Hmox1−/− versus Hmox1+/+ BM reconstituted hosts were cultured with donor type (SYNG: syngeneic BALB/c), host type (ALLO: allogeneic C57BL/6), or third party (TP) (CBA/Ca) irradiated stimulatory cells. IFN-γ, IL-17A, and IL-10 were measured in MLR supernatants. Two independent experiments involving 8 animals per group were pooled. (E, F) The effects of either CD4+ or CD8+ T cell depletion (see materials and methods) on host survival and GVHD score are shown in the BALB/c (Hmox1−/− versus Hmox1+/+ BM) to C57BL/6 graft versus host combination. (G) The effect of CD4+ depletion on host survival in the BALB/c recipients transplanted with BM from C57BL/6 LysMCre/wtHmox1Δ/Δ mice. n.s., no significant difference; *P <.05; **P <.01; ***P <.001.
Finally, the onset of acute GVHD aggravation in C57BL/6 recipients transplanted with BM from BALB/c Hmox1−/− mice was also reflected by the increase in IFN-γ serum levels, IL-17A or IFN-γ mRNA in the liver, serum aspartate aminotransferase (AST) levels, and oxidative stress-induced molecules (Figures S1A, G). Of note, this was not observed in the syngeneic conditions (data not shown).
The enhanced mortality in C57BL/6 hosts transplanted with BM from BALB/c Hmox1−/− mice was T cell-mediated since CD4+ T cell depletion dramatically prevented GVHD and mortality (Figures 3E, F). CD8+ T cell depletion also modulated GVHD, but to a lesser extent (Figure 3E). However, in the CD8+ T cell depletion experiments, the results should be interpreted cautiously due to the small number of animals included. A comparable effect of CD4 T cell depletion was observed in the BALB/c recipients transplanted with BM from C57BL/6 LysMCre/wtHmox1Δ/Δ mice (Figure 3G).
We then asked whether expression of HO-1 could modulate alloreactive cytotoxic CD8+ T cell (CTL) responses, a critical component in the pathogenesis of GVHD. CTL activity was compared in vivo in BALB/c recipients transplanted with BM from C57BL/6 LysMCre/wtHmox1Δ/Δ versus BM from wild type C57BL/6 Hmox1+/+ or C57BL/6 Hmox1flox/flox mice. CTL activity was similar in all allogeneic BM transplant combinations, as assessed in vivo by using CFSE-labeled splenocytes corresponding to the transplanted BM genotype (Figure S2).
In order to investigate the clinical relevance of the previous experimental data, we assessed the impact of donor and recipient HO-1 polymorphisms in the outcome of HSCT in patients.
A total of 120 and 160 HSCT procedures in two independent cohorts were investigated. The minimum follow-up was 1 year. Tables 1 and 2 summarize patient and donor characteristics and treatments. In both cohorts, underlying diseases are listed according to their frequency. Types of transplantation have been divided into three categories: matched related donor (MRD), matched unrelated donor (MUD), and unmatched related donor (URD). Conditioning treatments were separated in two categories: myeloablative conditioning (MAC) and reduced intensity conditioning (RIC). The two cohorts differed in the following characteristics: (1) there was no transplantation from URD in the first cohort, versus 22% in the second one; (2) there was a higher proportion of RIC in the first cohort; (3) there was a lower rate of radiotherapy used in the first cohort; (4) GVHD prophylaxis with ATG was used in 25% of patients in the first cohort versus 76.2% in the second.
The bimodal distribution of (GT)n polymorphisms in the HMOX1 promoter in donor and recipients from cohorts 1 and 2 and healthy controls (Figures 4A, B), ranged from 12 to 42 and peaked at 23 and 30 (GT)n, similar to that observed in previous studies (27–29, 35, 46, 47). The (GT)n alleles were segregated into short (S), i.e. (GT)n < 30, and long (L), i.e. (GT)n > 30, and individuals were classified as L/L, L/S, or S/S genotypes, as described before (23). The distributions of S or L alleles and genotype frequencies were similar in recipients and donors, in both cohorts and in the control group (Table 3): Hardy-Weinberg equilibrium was met.
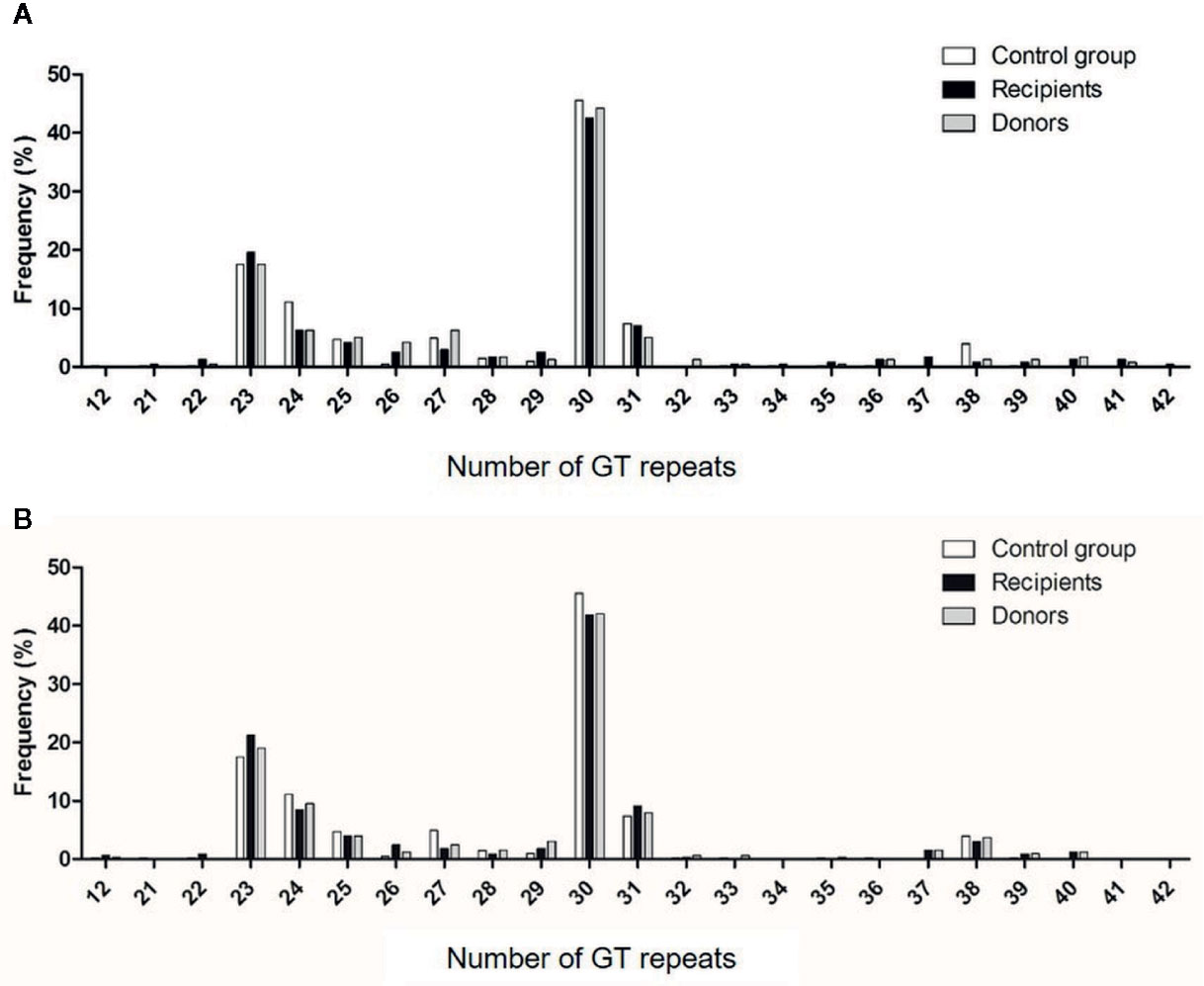
Figure 4 Distribution of the number of (GT)n repeats in the microsatellite region of the HO-1 gene promoter. Numbers of (GT) repeats are shown in HSC donors (gray bars) and recipients (black bars) of cohorts 1 (A) and 2 (B) and in healthy volunteers (white bars).
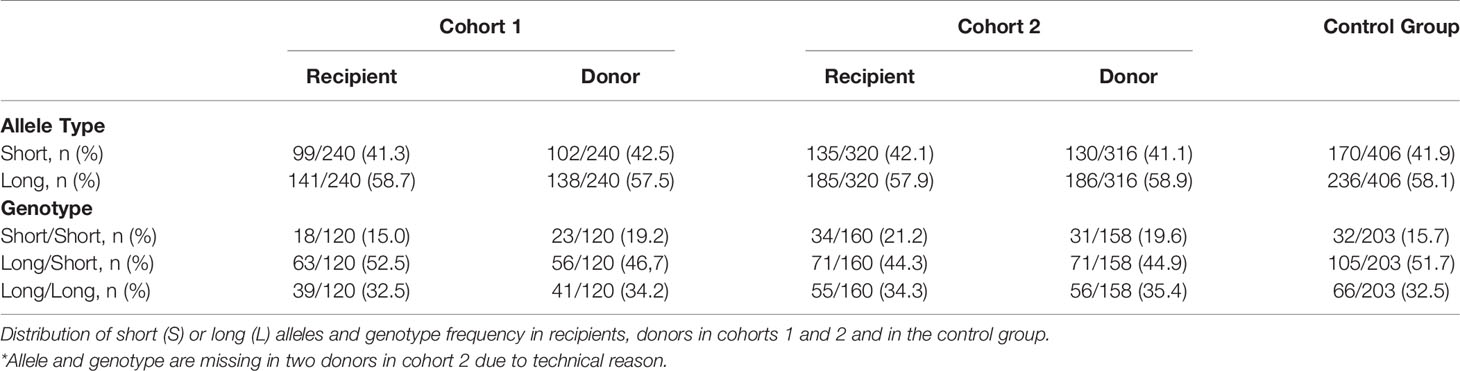
Table 3 Allele and genotype frequencies in the two cohorts and the control group.
As shown in Table 4, the proportion of L alleles in the recipient was not associated with GVHD severity, whereas a trend for a higher proportion of L alleles in the donor was observed in patients with grade III/IV acute GVHD (S vs L allele: 28.6% vs 71.4%, p=0.05 and 33.8 vs 66.2%, p=ns in cohorts 1 and 2, respectively). The L/L genotype in the donor was associated with a higher proportion of grade III/IV acute GVHD in both cohorts (S/S or S/L vs L/L: 46.4% vs 53.5%, p<0.05 and 47.0% vs 52.9% p<0.05 in cohorts 1 and 2, respectively), whereas an L/L genotype in the recipient did not influence GVHD severity (Table 5).
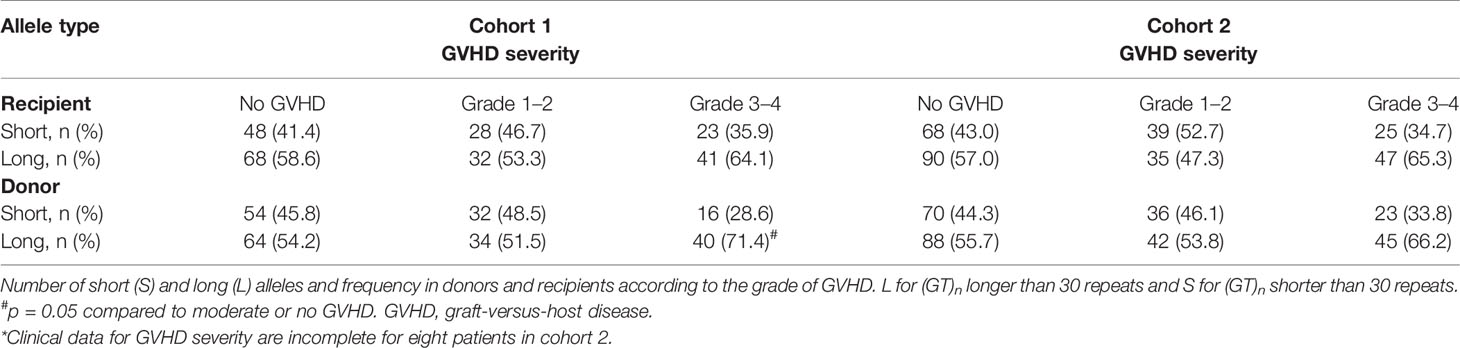
Table 4 Allele distribution according to GVHD severity.
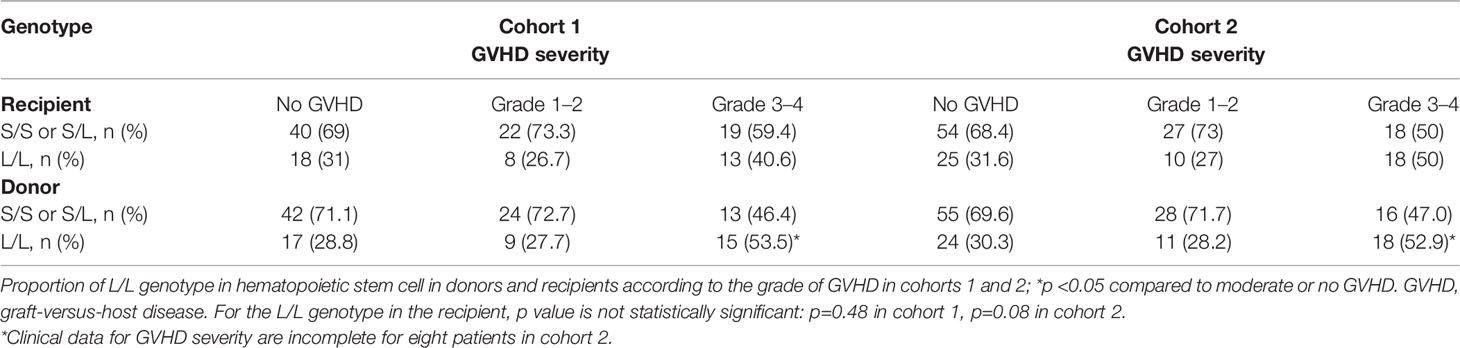
Table 5 Genotype distribution according to GVHD severity.
The association between GVHD and risk factors was analyzed by univariate and multivariate analyses, considering L/L donor genotype, age, cytomegalovirus (CMV) status, gender (donor and recipient), type of transplantation, intensity of conditioning, and use of radiotherapy (Tables 6 and 7). Subdistribution hazard ratio (SHR) for grade III/IV acute GVHD was significantly higher in cases involving an L/L genotype in the donor (SHR: 2.3; CI: 1.2–4.4, p=0.01 and SHR: 2.0; CI: 1.1–3.6, p=0.02, in cohorts 1 and 2, respectively) and of an L/L genotype in the recipient only in cohort 2 (SHR: 2.0; CI: 1.1–3.6; p=0.02). Other acknowledged risk factors for developing GVHD (48) remained significant after multivariate analysis, such as radiotherapy or the type of transplantation (Table 7). Radiotherapy was a risk factor for GVHD in cohort 2 but not in cohort 1, in which radiotherapy was much less frequently used. Graft origin (MUD) was identified as a risk factor for GVHD in cohort 1 but not in cohort 2, probably related to the systematic use of ATG (Tables 6 and 7). Regarding ATG use, multivariate analysis was not performed because univariate analysis did not find statistical correlation when performed in the cohort with the most abundant use of ATG (data not shown). Importantly, multivariate analysis revealed that the donor’s L/L genotype remained a significant risk factor for GVHD, independently of radiotherapy and of the graft source (aSHR: 2.6; CI: 1.3–4.9, p=0.005 and aSHR: 1.9; CI: 1.0–3.5, p=0.04 for cohorts 1 and 2, respectively). In summary, HSCT from a donor harboring the L/L genotype increases the risk for developing severe acute GVHD. This corroborates our experimental data in mice, strongly suggesting that the relative level of HO-1 expression in donor BM plays a critical role in the onset of GVHD.
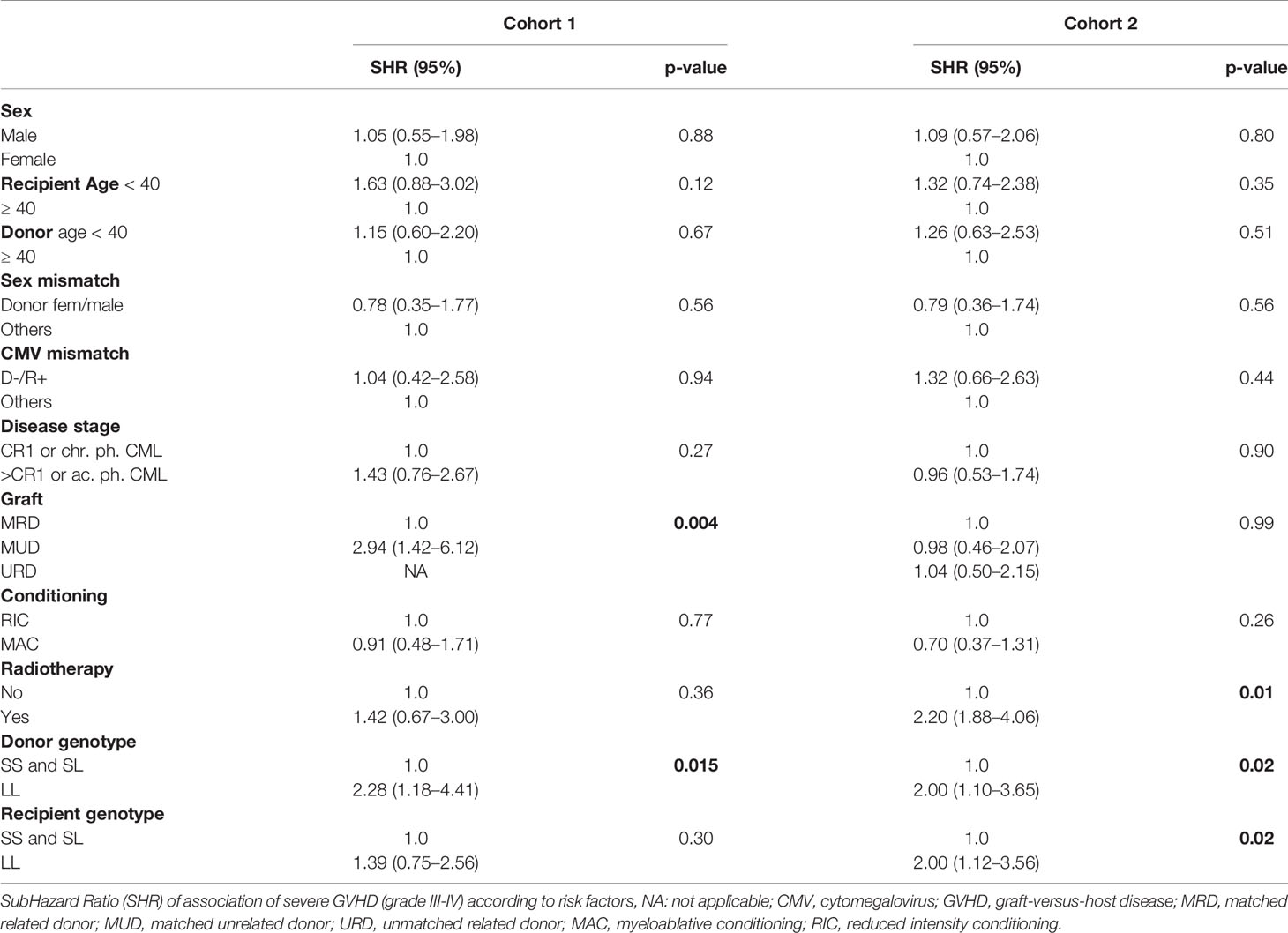
Table 6 Univariate analysis of GVHD risk factors.
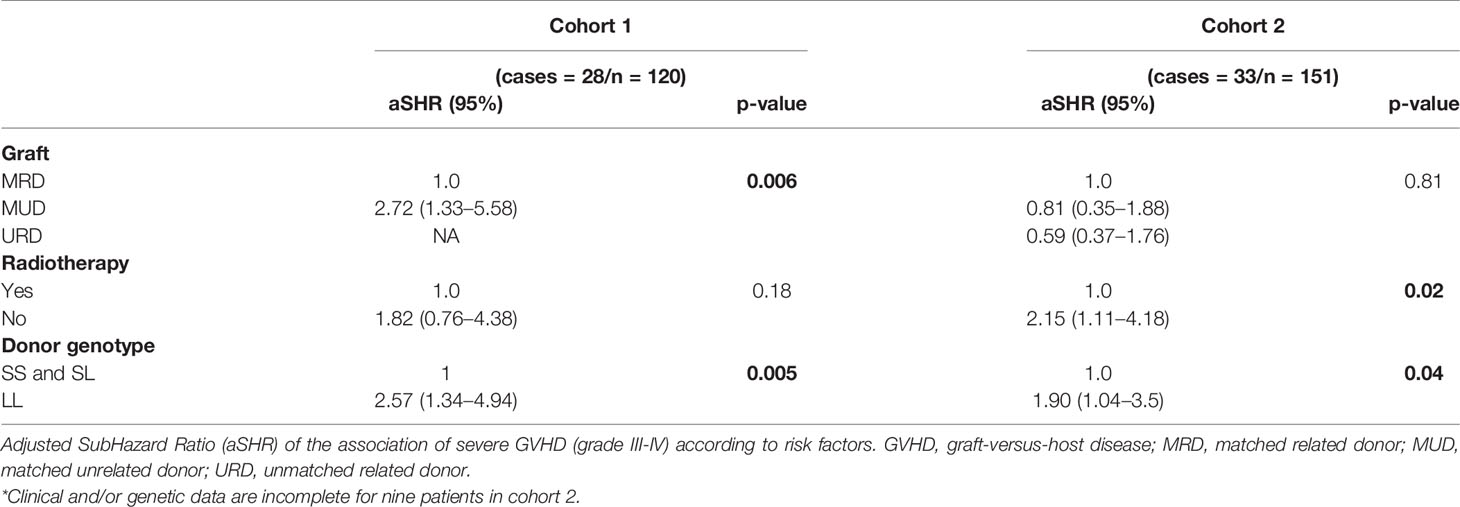
Table 7 Multivariate analysis of GVHD risk factors.
In this study, we provide evidence that donor-derived myeloid HO-1 prevents the onset of acute GVHD after HSCT. Importantly, the protective effect against GVHD associated with the expression of HO-1 was determined via a genetic loss-of-function approach, as compared to previous studies in which non-specific pharmacological induction of HO-1 was shown to be associated with suppression of GHVD (18–20). It should be noted, however, that these studies have not provided irrefutable evidence that the beneficial effects associated with these pharmacological approaches are indeed driven by the upregulation of HO-1 expression (18–20).
HO-1 can be stress-induced in almost all cell types, including epithelial or endothelial cells (49). In our mouse models, host animals are always in a wild type background and can upregulate HO-1 normally in response to many stress types, including GVHD. We have measured plasma HO-1 levels by ELISA and detected large amounts of plasma HO-1, whether the BMT donor was HO-1 sufficient or deficient (data not shown). Nevertheless, that HO-1 production did not afford any protection against GVHD, in contrast to the HO-1 from the BM donor.
Another study that enhanced expression of HO-1 in mesenchymal stem cells (MSC) by lentiviral gene transfer in a mouse GVHD model, showed a significant diminution of clinical and pathological GVHD scores in mice, but not on survival (50). As such, the current study is the first demonstration that expression of HO-1 in myeloid cells suppresses GVHD after HSCT in mice, and likely in humans as well, as suggested by the association between a (GT)n L/L genotype associated with low HO-1 expression and GVHD severity.
Another study in HSCT patients that investigated the association between HMOX1 (GT)n promoter polymorphisms and transplantation-related mortality reported results that are in apparent contrast to ours (51). Gerbitz et al. (2008) investigated 92 patient-donor pairs that were classified as either high or low HO-1 expressors based on the presence or the absence of a single long allele (≥ 30 GT repeats). This “dominant” approach differs from the “recessive” approach we adopted (to be considered a low expressor requires the L/L genotype). In their study, Gerbitz et al. (2008) showed that HSCT from matched unrelated donors defined as high HO-1 expressors negatively influenced overall survival and transplantation-related mortality. Regarding the risk of acute GVHD, there was only a trend but no statistical difference, with a higher rate of grade III/IV acute GVHD when the unrelated donor was a low HO-1 expressor. This apparent discrepancy may result from cohort characteristics (only matched donor/recipient pairs), genotype definition (L allele dominance assumed), and selected outcomes. Indeed, transplantation-related mortality and overall survival are composite outcomes and the higher mortality observed by Gerbitz et al. (2008) might be due to increased susceptibility to infections and disease relapse rather than GVHD (23, 27–29, 52). Another study observed a higher incidence of grade III/IV acute GVHD in the donor L/L genotype group in the non MAC regimens cohort but not in the MAC regimens cohort (53). In the MAC cohort, they also found a significantly higher relapse incidence in the donor S/S genotype group, but no difference in overall survival. In this study, they did not observe the same effect of HMOX1 (GT)n promoter polymorphisms in their two cohorts and this may be due to the small size of the cohorts. A recent study that investigated the influence of the single nucleotide polymorphism (SNP) rs2071746 (-413A>T) in the HO-1 promoter on HSCT outcomes in 593 patients with hematological malignancies showed that the donor A/A or A/T genotype was associated with better 5-year overall survival and disease-free survival compared to the donor T/T genotype. The A allele has been associated with higher HO-1 expression levels than the T allele. The donor HO-1 genotype showed no effects on GVHD (54). This apparent discrepancy might be explained by the use of different genotyping methodologies (SNP versus (GT)n polymorphism) and we did not assess the incidence of overall survival, non-relapse mortality, and GVHD relapse-free survival.
There are potentially multiple overlapping mechanisms by which myeloid HO-1 can prevent the onset of severe GVHD (55). In line with previous observations (17), we demonstrate that expression of HO-1 by myeloid cells regulates alloreactive T cell responses in a manner that suppresses the onset of GVHD, suggesting, therefore, that pharmacological induction of HO-1 in this cell compartment may be used as a therapeutic approach in the treatment of GVHD. This finding is also in keeping with previous observations indicating that HO-1 activity is essential for and promotes tolerance to transplanted allogeneic organs (56). In accordance with this notion, we found that when expressed by myeloid cells, HO-1 suppresses alloreactive Th1 and Th17 cell responses involved in the onset of GVHD. Notably, when expressed in a non-myeloid compartment, HO-1 was unable to control these alloreactive responses.
The control of anti-host alloreactive T cells via HO-1 is in agreement with our previous observation that MDSCs can suppress alloreactive T cell responses and skin allograft rejection through an HO-1-dependent mechanism. This suggests that physiologic processes by which MDSCs control adaptive immunity can be regulated via HO-1-dependent mechanisms (17). The exact mechanism by which HO-1 exerts these effects is not clear and is likely to be multifactorial.
One possibility could be that HO-1 generates one or several metabolic products that act in MDSCs and/or directly on alloreactive T cells to suppress their activation and proliferation. In keeping with this notion, pharmacologic blockade of HO-1 activity is associated with the accumulation of reactive oxygen species (ROS) levels in dendritic cells (DCs) and, through a p38 mitogen-activated protein kinase (p38 MAPK)-dependent pathway, induces DC maturation and CD8+ T cell activation (57, 58).
Another possibility could be that under stress conditions, such as those associated with transplantation, HO-1 becomes the rate-limiting factor in the catabolism of heme, generating equimolar amounts of biliverdin, carbon monoxide (CO), and labile iron (59). Heme can be released from hemoproteins, under stress conditions, such as to generate labile heme (60). Labile heme is recognized by pattern recognition receptors, including toll-like receptor 4 and NACHT, LRR, and Pyrin Domain Containing 3 (NLRP3) (60) acting as a bona fide alarmin that activates monocyte/macrophages. Whether these effects of labile heme promote T cell alloreactivity and, hence, the rejection mediated by transplanted cells, tissues, or organs has not been established. What is clear, however, is that heme catabolism by HO-1 counters the pro-oxidant effects of labile heme in a manner that inhibits its pro-inflammatory and cytotoxic effects. Moreover, heme catabolism by HO-1 generates CO, which limits the production of labile heme while regulating monocyte/macrophage activation in a manner that inhibits the production of cytotoxic cytokines, e.g. TNF, while promoting that of immunoregulatory cytokines, e.g. IL-10 (58, 59). Macrophages are considered principal regulators of immune homeostasis and have been categorized into two subtypes: M1, which are proinflammatory via a Th1 response, and M2, which harbor an anti-inflammatory phenotype via a Th2 response (61, 62). Upregulation of HO-1 promotes M2 polarization (63–65). In line with this, Zhang et al. (2018) recently reported that M2 polarization through myeloid HO-1 partially prevents ischemia-reperfusion injury (66). In our GVHD model, it is not excluded that donor myeloid HO-1 controls T cell alloreactivity through M2 polarization. Another group has shown that monocytes co-cultured with expanded TREGs downregulate costimulatory and MHC-class II molecules and upregulate M2 markers, including HO-1 and IL-10 (67).
We very recently observed in a mouse tumor model that the deletion of HO-1 in the myeloid compartment enhances the beneficial effects of a therapeutic antitumor vaccine by boosting CD8 T cell proliferation and cytotoxicity (68). This underlines the possible modulation of adaptive immune response through myeloid HO-1. In addition, myeloid HO-1 deficiency induced a modulation of tumor-associated macrophage transcriptional and epigenetic programs. Comparable mechanisms might be involved in our GVHD model.
An intriguing possibility is that MHC class I-restricted presentation of HO-1-derived peptides exerts immunoregulatory effects on alloreactive T cells, much like that demonstrated in cancer patients, where HLA-A2–restricted CD8+ TREGs specific for HO-1-derived peptides suppress tumor specific T cell responses (69). Nevertheless, our data suggests that CD8+ T cell depletion does not protect against GVHD in mice receiving BM allogeneic transplants in which the expression of HO-1 is deleted in myeloid cells, which does not fully support that an immunoregulatory mechanism based on CD8+ TREG to be operational here.
It is also possible that the protective effect of HO-1 could act via additional mechanisms such as the CO generated by heme catabolism which promotes myeloid cell expansion and differentiation (70). In addition, CO exerts potent cytoprotective (71) and anti-inflammatory effects (72, 73) that should restrain the severity of GVHD as well.
Expression of HO-1 in myeloid cells may also play a critical role in restoring iron metabolism after transplantation. In support of this notion, Kovtunovych et al. reported that subablative BMT cured Hmox1−/− mice by repopulating tissue macrophages in a syngeneic context, restoring iron metabolism parameters, and by normalizing oxidative stress-induced molecules in the liver and the kidney (74). Nevertheless, in our experiments, the lack of HO-1 in syngeneic BMT only marginally affected outcomes, which underlines the synergy of alloantigens in the pathogenicity of myeloid-restricted HO-1 deficiency. Based on the results presented here and those of others, it might be reasonable to consider HO-1 induction, either to prevent GVHD or as adjuvant therapy to treat GVHD. Indeed, a broad range of HO-1 inducers that have been recently described (75, 76) might be used in the clinic.
We conclude that donor-derived myeloid HO-1 is a key regulator of T cell alloreactivity and acute GVHD severity and constitutes a potential therapeutic target for HSCT patients. Large-scale prospective studies in HSCT patients are necessary to validate the HO-1 L/L genotype as an independent risk factor to developing severe acute GVHD. Additional mechanistic approaches should allow a better understanding of how HO-1 regulates GVHD and T cell functions. If the role of HO-1 is confirmed, the HO-1 (GT)n polymorphism of the donor might be considered as a biomarker before BMT in a personalized medicine strategy combined or not with therapeutic use of HO-1 inducers.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by Erasme Hospital and the Jules Bordet Institute (numbers P2011/255 and 2012/193, respectively). The Henri Mondor University Hospital (Créteil, France) has been accredited according to the international JACIE program since 2005. All patients and donors signed informed consent for (1) registration into the Promise database, and (2) cryopreservation of their biological material for research purposes. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements. The animal study was reviewed and approved by local committee for animal welfare (agreement # LA2500519).
CS, VW designed and performed the research, analyzed the data, and wrote the paper. BV, J-MH, and VF helped to perform the research. PL, LC, CR, FB, and DB provided the materials and performed the research. JR performed the statistical analysis and wrote the paper. MA critically reviewed the manuscript and analyzed the genetic data. MS provided the material and critically reviewed and wrote the manuscript. SM and AL designed the research, analyzed the data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
CS was also funded by the Société Francophone de Transplantation (France), the Alice and David Van Buuren Foundation, and the Fonds Carine Vyghen pour le don d’organes. MS is supported by Fundação Calouste Gulbenkian and by competitive grants from the Fundação para a Ciência e Tecnologia (PTDC/SAU TOX/116627/2010, HMSP-ICT/0022/2010) and by the European Community 7th Framework Grant ERC-2011-AdG. 294709.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank Louis-Marie Charbonnier, Maxime Rossi and Michel Goldman for helpful discussions and technical assistance. The authors acknowledge the contribution of a medical writer, Sandy Field, PhD, for editing the manuscript, Dr. Philippe Horlait, Laurent Depret, Gregory Waterlot, Christophe Notte for animal care, Frédéric Paulart for technical assistance, and The Plateforme de Resources Biologiques, Cellulothèque, GH Mondor, Créteil, France (Dr Flavia Castellano) for providing human samples. CS, VW, and BV are research fellows of the Fonds National de la Recherche Scientifique (FNRS)-Belgium. J-MH is a research fellow of the Fonds Erasme (Erasme Hospital, Brussels, Belgium). CS received a grant from the Société Francophone de Transplantation. AL is an honorary research associate of the FNRS.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.579151/full#supplementary-material
1. Zeiser R, Blazar BR. Acute Graft-versus-Host Disease - Biologic Process, Prevention, and Therapy. N Engl J Med (2017) 377(22):2167–79. doi: 10.1056/NEJMra1609337
2. Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol (2012) 12(6):443–58. doi: 10.1038/nri3212
3. Bluestone JA, Tang Q. Treg cells-the next frontier of cell therapy. Science (2018) 362(6411):154–5. doi: 10.1126/science.aau2688
4. Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med (2002) 196(3):389–99. doi: 10.1084/jem.20020399
5. Koehn BH, Blazar BR. Role of myeloid-derived suppressor cells in allogeneic hematopoietic cell transplantation. J Leukoc Biol (2017) 102(2):335–41. doi: 10.1189/jlb.5MR1116-464R
6. Morecki S, Gelfand Y, Yacovlev E, Eizik O, Shabat Y, Slavin S. CpG-induced myeloid CD11b+Gr-1+ cells efficiently suppress T cell-mediated immunoreactivity and graft-versus-host disease in a murine model of allogeneic cell therapy. Biol Blood Marrow Transplant (2008) 14(9):973–84. doi: 10.1016/j.bbmt.2008.06.018
7. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
8. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest (2015) 125(9):3365–76. doi: 10.1172/JCI80006
9. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
10. Budhwar S, Verma P, Verma R, Rai S, Singh K. The Yin and Yang of Myeloid Derived Suppressor Cells. Front Immunol (2018) 9:2776. doi: 10.3389/fimmu.2018.02776
11. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood (2010) 116(25):5738–47. doi: 10.1182/blood-2010-06-287839
12. Rosborough BR, Castellaneta A, Natarajan S, Thomson AW, Turnquist HR. Histone deacetylase inhibition facilitates GM-CSF-mediated expansion of myeloid-derived suppressor cells in vitro and in vivo. J Leukoc Biol (2012) 91(5):701–9. doi: 10.1189/jlb.0311119
13. Dilek N, van Rompaey N, Le Moine A, Vanhove B. Myeloid-derived suppressor cells in transplantation. Curr Opin Organ Transplant (2010) 15(6):765–8. doi: 10.1097/MOT.0b013e3283401742
14. Mougiakakos D, Jitschin R, von Bahr L, Poschke I, Gary R, Sundberg B, et al. Immunosuppressive CD14+HLA-DRlow/neg IDO+ myeloid cells in patients following allogeneic hematopoietic stem cell transplantation. Leukemia (2013) 27(2):377–88. doi: 10.1038/leu.2012.215
15. Soares MP, Hamza I. Macrophages and Iron Metabolism. Immunity (2016) 44(3):492–504. doi: 10.1016/j.immuni.2016.02.016
16. Soares MP, Brouard S, Smith RN, Bach FH. Heme oxygenase-1, a protective gene that prevents the rejection of transplanted organs. Immunol Rev (2001) 184:275–85. doi: 10.1034/j.1600-065x.2001.1840124.x
17. De Wilde V, Van Rompaey N, Hill M, Lebrun JF, Lemaitre P, Lhomme F, et al. Endotoxin-induced myeloid-derived suppressor cells inhibit alloimmune responses via heme oxygenase-1. Am J Transplant (2009) 9(9):2034–47. doi: 10.1111/j.1600-6143.2009.02757.x
18. Gerbitz A, Ewing P, Wilke A, Schubert T, Eissner G, Dietl B, et al. Induction of heme oxygenase-1 before conditioning results in improved survival and reduced graft-versus-host disease after experimental allogeneic bone marrow transplantation. Biol Blood Marrow Transplant (2004) 10(7):461–72. doi: 10.1016/j.bbmt.2004.04.001
19. Ewing P, Hildebrandt GC, Planke S, Andreesen R, Holler E, Gerbitz A. Cobalt protoporphyrine IX-mediated heme oxygenase-I induction alters the inflammatory cytokine response, but not antigen presentation after experimental allogeneic bone marrow transplantation. Int J Mol Med (2007) 20(3):301–8. doi: 10.3892/ijmm.20.3.301
20. Woo J, Iyer S, Mori N, Buelow R. Alleviation of graft-versus-host disease after conditioning with cobalt-protoporphyrin, an inducer of heme oxygenase-1. Transplantation (2000) 69(4):623–33. doi: 10.1097/00007890-200002270-00026
21. McDaid J, Yamashita K, Chora A, Ollinger R, Strom TB, Li XC, et al. Heme oxygenase-1 modulates the allo-immune response by promoting activation-induced cell death of T cells. FASEB J (2005) 19(3):458–60. doi: 10.1096/fj.04-2217fje
22. Hirai H, Kubo H, Yamaya M, Nakayama K, Numasaki M, Kobayashi S, et al. Microsatellite polymorphism in heme oxygenase-1 gene promoter is associated with susceptibility to oxidant-induced apoptosis in lymphoblastoid cell lines. Blood (2003) 102(5):1619–21. doi: 10.1182/blood-2002-12-3733
23. Exner M, Minar E, Wagner O, Schillinger M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic Biol Med (2004) 37(8):1097–104. doi: 10.1016/j.freeradbiomed.2004.07.008
24. Garcia-Santos D, Chies JA. HO-1 polymorphism as a genetic determinant behind the malaria resistance afforded by haemolytic disorders. Med Hypotheses (2010) 74(5):807–13. doi: 10.1016/j.mehy.2009.12.010
25. Hsu LA, Yeh YH, Kuo CT, Chen YH, Chang GJ, Tsai FC, et al. Microsatellite polymorphism in the heme oxygenase-1 gene promoter and the risk of atrial fibrillation in Taiwanese. PloS One (2014) 9(9):e108773. doi: 10.1371/journal.pone.0108773
26. Pechlaner R, Willeit P, Summerer M, Santer P, Egger G, Kronenberg F, et al. Heme oxygenase-1 gene promoter microsatellite polymorphism is associated with progressive atherosclerosis and incident cardiovascular disease. Arterioscler Thromb Vasc Biol (2015) 35(1):229–36. doi: 10.1161/ATVBAHA.114.304729
27. Chen YH, Lin SJ, Lin MW, Tsai HL, Kuo SS, Chen JW, et al. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Hum Genet (2002) 111(1):1–8. doi: 10.1007/s00439-002-0769-4
28. Chen YH, Chau LY, Lin MW, Chen LC, Yo MH, Chen JW, et al. Heme oxygenase-1 gene promotor microsatellite polymorphism is associated with angiographic restenosis after coronary stenting. Eur Heart J (2004) 25(1):39–47. doi: 10.1016/j.ehj.2003.10.009
29. Chen YH, Kuo KL, Hung SC, Hsu CC, Chen YH, Tarng DC. Length polymorphism in heme oxygenase-1 and risk of CKD among patients with coronary artery disease. J Am Soc Nephrol (2014) 25(11):2669–77. doi: 10.1681/ASN.2013111205
30. Leaf DE, Body SC, Muehlschlegel JD, McMahon GM, Lichtner P, Collard CD, et al. Length Polymorphisms in Heme Oxygenase-1 and AKI after Cardiac Surgery. J Am Soc Nephrol (2016) 27(11):3291–7. doi: 10.1681/ASN.2016010038
31. Gill AJ, Garza R, Ambegaokar SS, Gelman BB, Kolson DL. Heme oxygenase-1 promoter region (GT)n polymorphism associates with increased neuroimmune activation and risk for encephalitis in HIV infection. J Neuroinflam (2018) 15(1):70. doi: 10.1186/s12974-018-1102-z
32. Garza R, Gill AJ, Bastien BL, Garcia-Mesa Y, Gruenewald AL, Gelman BB, et al. Heme oxygenase-1 promoter (GT) n polymorphism associates with HIV neurocognitive impairment. Neurol Neuroimmunol Neuroinflamm (2020) 7(3):e710. doi: 10.1212/NXI.0000000000000710
33. Rueda B, Oliver J, Robledo G, Lopez-Nevot MA, Balsa A, Pascual-Salcedo D, et al. HO-1 promoter polymorphism associated with rheumatoid arthritis. Arthritis Rheumatol (2007) 56(12):3953–8. doi: 10.1002/art.23048
34. Zhou H, Ying X, Liu Y, Ye S, Yan J, Li Y. Genetic polymorphism of heme oxygenase 1 promoter in the occurrence and severity of chronic obstructive pulmonary disease: a meta-analysis. J Cell Mol Med (2017) 21(5):894–903. doi: 10.1111/jcmm.13028
35. Baan C, Peeters A, Lemos F, Uitterlinden A, Doxiadis I, Claas F, et al. Fundamental role for HO-1 in the self-protection of renal allografts. Am J Transplant (2004) 4(5):811–8. doi: 10.1111/j.1600-6143.2004.00420.x
36. Ozaki KS, Marques GM, Nogueira E, Feitoza RQ, Cenedeze MA, Franco MF, et al. Improved renal function after kidney transplantation is associated with heme oxygenase-1 polymorphism. Clin Transplant (2008) 22(5):609–16. doi: 10.1111/j.1399-0012.2008.00832.x
37. Jais A, Einwallner E, Sharif O, Gossens K, Lu TT, Soyal SM, et al. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell (2014) 158(1):25–40. doi: 10.1016/j.cell.2014.04.043
38. Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, et al. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clin Invest (1999) 103(8):R23–9. doi: 10.1172/JCI6163
39. Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, et al. A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med (2010) 2(51):51ra71. doi: 10.1126/scitranslmed.3001118
40. Mamiya T, Katsuoka F, Hirayama A, Nakajima O, Kobayashi A, Maher JM, et al. Hepatocyte-specific deletion of heme oxygenase-1 disrupts redox homeostasis in basal and oxidative environments. Tohoku J Exp Med (2008) 216(4):331–9. doi: 10.1620/tjem.216.331
41. Allie N, Grivennikov SI, Keeton R, Hsu NJ, Bourigault ML, Court N, et al. Prominent role for T cell-derived tumour necrosis factor for sustained control of Mycobacterium tuberculosis infection. Sci Rep (2013) 3:1809. doi: 10.1038/srep01809
42. Colson YL, Christopher K, Glickman J, Taylor KN, Wright R, Perkins DL. Absence of clinical GVHD and the in vivo induction of regulatory T cells after transplantation of facilitating cells. Blood (2004) 104(12):3829–35. doi: 10.1182/blood-2004-01-0393
43. Graca L, Le Moine A, Lin CY, Fairchild PJ, Cobbold SP, Waldmann H. Donor-specific transplantation tolerance: the paradoxical behavior of CD4+CD25+ T cells. Proc Natl Acad Sci U S A (2004) 101(27):10122–6. doi: 10.1073/pnas.0400084101
44. Vigorito AC, Campregher PV, Storer BE, Carpenter PA, Moravec CK, Kiem HP, et al. Evaluation of NIH consensus criteria for classification of late acute and chronic GVHD. Blood (2009) 114(3):702–8. doi: 10.1182/blood-2009-03-208983
45. Thomas ED, Storb R, Clift RA, Fefer A, Johnson L, Neiman PE, et al. Bone-marrow transplantation (second of two parts). N Engl J Med (1975) 292(17):895–902. doi: 10.1056/NEJM197504242921706
46. Yamada N, Yamaya M, Okinaga S, Nakayama K, Sekizawa K, Shibahara S, et al. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am J Hum Genet (2000) 66(1):187–95. doi: 10.1086/302729
47. Guenegou A, Leynaert B, Benessiano J, Pin I, Demoly P, Neukirch F, et al. Association of lung function decline with the heme oxygenase-1 gene promoter microsatellite polymorphism in a general population sample. Results from the European Community Respiratory Health Survey (ECRHS), France. J Med Genet (2006) 43(8):e43. doi: 10.1136/jmg.2005.039743
48. Jagasia M, Arora M, Flowers ME, Chao NJ, McCarthy PL, Cutler CS, et al. Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood (2012) 119(1):296–307. doi: 10.1182/blood-2011-06-364265
49. Terry CM, Clikeman JA, Hoidal JR, Callahan KS. TNF-alpha and IL-1alpha induce heme oxygenase-1 via protein kinase C, Ca2+, and phospholipase A2 in endothelial cells. Am J Physiol (1999) 276(5):H1493–501. doi: 10.1152/ajpheart.1999.276.5.H1493
50. Yu M, Wang J, Fang Q, Liu P, Chen S, Zhe N, et al. High expression of heme oxygenase-1 in target organs may attenuate acute graft-versus-host disease through regulation of immune balance of TH17/Treg. Transpl Immunol (2016) 37:10–7. doi: 10.1016/j.trim.2016.05.002
51. Gerbitz A, Hillemanns P, Schmid C, Wilke A, Jayaraman R, Kolb HJ, et al. Influence of polymorphism within the heme oxygenase-I promoter on overall survival and transplantation-related mortality after allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2008) 14(10):1180–9. doi: 10.1016/j.bbmt.2008.08.002
52. Walther M, De Caul A, Aka P, Njie M, Amambua-Ngwa A, Walther B, et al. HMOX1 gene promoter alleles and high HO-1 levels are associated with severe malaria in Gambian children. PloS Pathog (2012) 8(3):e1002579. doi: 10.1371/journal.ppat.1002579
53. Kollgaard T, Kornblit B, Petersen J, Klausen TW, Mortensen BK, Braendstrup P, et al. (GT)n Repeat Polymorphism in Heme Oxygenase-1 (HO-1) Correlates with Clinical Outcome after Myeloablative or Nonmyeloablative Allogeneic Hematopoietic Cell Transplantation. PloS One (2016) 11(12):e0168210. doi: 10.1371/journal.pone.0168210
54. Horio T, Morishita E, Mizuno S, Uchino K, Hanamura I, Espinoza JL, et al. Donor Heme Oxygenase-1 Promoter Gene Polymorphism Predicts Survival after Unrelated Bone Marrow Transplantation for High-Risk Patients. Cancers (Basel) (2020) 12(2):424. doi: 10.3390/cancers12020424
55. Soares MP, Marguti I, Cunha A, Larsen R. Immunoregulatory effects of HO-1: how does it work? Curr Opin Pharmacol (2009) 9(4):482–9. doi: 10.1016/j.coph.2009.05.008
56. Yamashita K, Ollinger R, McDaid J, Sakahama H, Wang H, Tyagi S, et al. Heme oxygenase-1 is essential for and promotes tolerance to transplanted organs. FASEB J (2006) 20(6):776–8. doi: 10.1096/fj.05-4791fje
57. Al-Huseini LM, Aw Yeang HX, Hamdam JM, Sethu S, Alhumeed N, Wong W, et al. Heme oxygenase-1 regulates dendritic cell function through modulation of p38 MAPK-CREB/ATF1 signaling. J Biol Chem (2014) 289(23):16442–51. doi: 10.1074/jbc.M113.532069
58. Ryter SW, Choi AM. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl Res (2016) 167(1):7–34. doi: 10.1016/j.trsl.2015.06.011
59. Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol (2010) 50:323–54. doi: 10.1146/annurev.pharmtox.010909.105600
60. Soares MP, Bozza MT. Red alert: labile heme is an alarmin. Curr Opin Immunol (2016) 38:94–100. doi: 10.1016/j.coi.2015.11.006
61. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122(3):787–95. doi: 10.1172/JCI59643
62. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity (2010) 32(5):593–604. doi: 10.1016/j.immuni.2010.05.007
63. Weis N, Weigert A, von Knethen A, Brune B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol Biol Cell (2009) 20(5):1280–8. doi: 10.1091/mbc.e08-10-1005
64. Naito Y, Takagi T, Higashimura Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch Biochem Biophys (2014) 564:83–8. doi: 10.1016/j.abb.2014.09.005
65. Vijayan V, Wagener F, Immenschuh S. The macrophage heme-heme oxygenase-1 system and its role in inflammation. Biochem Pharmacol (2018) 153:159–67. doi: 10.1016/j.bcp.2018.02.010
66. Zhang M, Nakamura K, Kageyama S, Lawal AO, Gong KW, Bhetraratana M, et al. Myeloid HO-1 modulates macrophage polarization and protects against ischemia-reperfusion injury. JCI Insight (2018) 3(19):e120596. doi: 10.1172/jci.insight.120596
67. Romano M, Fanelli G, Tan N, Nova-Lamperti E, McGregor R, Lechler RI, et al. Expanded Regulatory T Cells Induce Alternatively Activated Monocytes With a Reduced Capacity to Expand T Helper-17 Cells. Front Immunol (2018) 9:1625. doi: 10.3389/fimmu.2018.01625
68. Alaluf E, Vokaer B, Detavernier A, Azouz A, Splittgerber M, Carrette A, et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI Insight (2020) 5(11):e133929. doi: 10.1172/jci.insight.133929
69. Andersen MH, Sorensen RB, Brimnes MK, Svane IM, Becker JC, thor Straten P. Identification of heme oxygenase-1-specific regulatory CD8+ T cells in cancer patients. J Clin Invest (2009) 119(8):2245–56. doi: 10.1172/JCI38739
70. Wegiel B, Hedblom A, Li M, Gallo D, Csizmadia E, Harris C, et al. Heme oxygenase-1 derived carbon monoxide permits maturation of myeloid cells. Cell Death Dis (2014) 5:e1139. doi: 10.1038/cddis.2014.97
71. Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, et al. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med (2000) 192(7):1015–26. doi: 10.1084/jem.192.7.1015
72. Brouard S, Berberat PO, Tobiasch E, Seldon MP, Bach FH, Soares MP. Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-kappa B to protect endothelial cells from tumor necrosis factor-alpha-mediated apoptosis. J Biol Chem (2002) 277(20):17950–61. doi: 10.1074/jbc.M108317200
73. Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med (2000) 6(4):422–8. doi: 10.1038/74680
74. Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood (2010) 116(26):6054–62. doi: 10.1182/blood-2010-03-272138
75. Funes SC, Rios M, Fernandez-Fierro A, Covian C, Bueno SM, Riedel CA, et al. Naturally Derived Heme-Oxygenase 1 Inducers and Their Therapeutic Application to Immune-Mediated Diseases. Front Immunol (2020) 11:1467. doi: 10.3389/fimmu.2020.01467
Keywords: Heme oxygenase-1, graft-versus-host disease, transplantation, hematopoeietic stem cell transplantation, polymorphism, myeloid-derived suppressor cells
Citation: Spilleboudt C, De Wilde V, Lewalle P, Cabanne L, Leclerc M, Beckerich F, Bories D, Cardoso S, Soares MP, Vokaer B, Hougardy J-M, Flamand V, Racapé J, Abramowicz M, Maury S and Le Moine A (2021) Donor-Derived Myeloid Heme Oxygenase-1 Controls the Development of Graft-Versus-Host Disease. Front. Immunol. 11:579151. doi: 10.3389/fimmu.2020.579151
Received: 01 July 2020; Accepted: 30 November 2020;
Published: 18 January 2021.
Edited by:
Hans-Jochem Kolb, Kolb Consulting UG, GermanyReviewed by:
Hung Nguyen, University of Central Florida, United StatesCopyright © 2021 Spilleboudt, De Wilde, Lewalle, Cabanne, Leclerc, Beckerich, Bories, Cardoso, Soares, Vokaer, Hougardy, Flamand, Racapé, Abramowicz, Maury and Le Moine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chloé Spilleboudt, Y2hsb2Uuc3BpbGxlYm91ZHRAYm9yZGV0LmJl; Alain Le Moine, YWxlbW9pbmVAdWxiLmFjLmJl
†These authors have contributed equally to this work
‡These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.