
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 October 2020
Sec. Alloimmunity and Transplantation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.578314
This article is part of the Research Topic Managing Chronic GvHD in the Era of Personalized Medicine View all 8 articles
 Nathaniel Edward Bennett Saidu1,2*
Nathaniel Edward Bennett Saidu1,2* Chiara Bonini3
Chiara Bonini3 Anne Dickinson4
Anne Dickinson4 Magdalena Grce1
Magdalena Grce1 Marit Inngjerdingen2
Marit Inngjerdingen2 Ulrike Koehl5
Ulrike Koehl5 Antoine Toubert6,7
Antoine Toubert6,7 Robert Zeiser8
Robert Zeiser8 Sara Galimberti9*
Sara Galimberti9*Chronic graft-versus-host disease (cGvHD) is a severe complication of allogeneic hematopoietic stem cell transplantation that affects various organs leading to a reduced quality of life. The condition often requires enduring immunosuppressive therapy, which can also lead to the development of severe side effects. Several approaches including small molecule inhibitors, antibodies, cytokines, and cellular therapies are now being developed for the treatment of cGvHD, and some of these therapies have been or are currently tested in clinical trials. In this review, we discuss these emerging therapies with particular emphasis on tyrosine kinase inhibitors (TKIs). TKIs are a class of compounds that inhibits tyrosine kinases, thereby preventing the dissemination of growth signals and activation of key cellular proteins that are involved in cell growth and division. Because they have been shown to inhibit key kinases in both B cells and T cells that are involved in the pathophysiology of cGvHD, TKIs present new promising therapeutic approaches. Ibrutinib, a Bruton tyrosine kinase (Btk) inhibitor, has recently been approved by the Food and Drug Administration (FDA) in the United States for the treatment of adult patients with cGvHD after failure of first-line of systemic therapy. Also, Janus Associated Kinases (JAK1 and JAK2) inhibitors, such as itacitinib (JAK1) and ruxolitinib (JAK1 and 2), are promising in the treatment of cGvHD. Herein, we present the current status and future directions of the use of these new drugs with particular spotlight on their targeting of specific intracellular signal transduction cascades important for cGvHD, in order to shed some light on their possible mode of actions.
GvHD is a complication that often occurs after allogeneic hematopoietic stem cell transplantation (allo-HSCT) in up to 50% of cases, where donor T- and B cells derived from the graft recognize and attack host antigens (1). This is particularly important because HSCT is the treatment of choice for many types of malignant and non-malignant hematological and autoimmune diseases. According to the onset, GvHD can be acute (aGvHD) or chronic (cGvHD): aGvHD typically occurs in less than 100 days after transplantation and it is associated with inflammation in several organs or sites, such as skin, gastrointestinal tract, mouth, genital tract or liver, while cGvHD may present at any time after HSCT, even if it typically occurs after 100 days from graft infusion (2).
Thus, from the clinical point of view, cGvHD resembles an “autoimmune syndrome”, characterized by immune dysregulation and absence of functional tolerance. As in systemic rheumatologic diseases, its clinical manifestations are variable, varying from lichen planus-like lesions to full sclerosis, muscle pain or joint fasciitis, vulvo-vaginitis, bronchiolitis obliterans (BO), in addition to damage of gastrointestinal tract and liver (3).
The National Institute of Health (NIH) consensus established diagnostic criteria for cGvHD in 2005 (4), which were revised in 2014 (5). The authors defined cGvHD-specific organ manifestations and elaborated a scoring system by considering the severity of involvement of skin, mouth, eyes, gastrointestinal tract, liver, lungs, joint fascia, and genital tract differently from other hematological diseases, and from models for testing conditioning or immunosuppressive regimens. Unfortunately, the existing murine models of cGvHD fail to fully include the whole spectrum of human cGvHD symptoms, which makes it difficult to treat and to draw general conclusions about the efficacy of new therapeutic approaches based on animal models (6).
In the pathogenesis of cGvHD, the disease develops as a result of complex molecular networks, from thymus damage to unusual antigen presentation and aberrant T- and B cell interactions (Figures 1A, B). This consequently leads to an enhanced Th17 differentiation, macrophage sequestration in tissues, alloantibody formation, and fibrosis, with the latter highly dependent on nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation and inflammatory cytokines production (7). Usually, the initial phase starts with release of inflammatory cytokines from the host tissues damaged by the pre-transplant conditioning regimens. These cytokines [interleukin-1 (IL-1), tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), platelet-derived growth factor (PDGF)] in combination with antigen presenting cells, stimulate the activation of donor allo-reactive T cells, with expansion of donor helper T cells, cytotoxic T cells and natural killer (NK) cells that cause cytotoxicity against host cells. In addition, both colony-stimulating factor-1 (CSF-1) and granulocyte macrophage-colony stimulating factor (GM-CSF), two key cytokines that modulate the differentiation, proliferation and survival of macrophages, have been implicated in cGvHD (8, 9). The role of GM-CSF activated myeloid cells in mediating GvHD by favoring IL-1β and Reactive Oxygen Species (ROS) production by phagocytes has recently been highlighted (10). This tissue damage is not counterbalanced by the physiologic control exerted by the host thymus and of T regulatory lymphocytes (Tregs) that usually are fundamental for immune tolerance (11). Donor macrophages may also infiltrate organs like the skin and lungs of a HSCT recipient through CSF-1 receptor signaling; and studies have shown that these donor CSF-1R-dependent macrophages may contribute to both sclerodermatous (Scl)-cGvHD and lung cGvHD, otherwise known as bronchiolitis obliterans syndrome (BOS), via the expression of Transforming Growth Factor beta (TGF-β) (8). This is important because it is now well established that TGF-β is a fundamental pathogenic cytokine in fibrosis, and elevated levels of this cytokine has been found in cGvHD patients; though the mechanism through which it contributes to the pathogenesis of the disease remains elusive (3). It is, however, clear that in certain organ-specific cGvHDs, such as skin cGvHD, both the TGF-β and PDGF pathways appear to be up-regulated leading to the activation and differentiation of fibroblasts into alpha-smooth muscle actin (α-SMA)-expressing myofibroblasts. These α-SMA-expressing myofibroblasts then proliferate and mediate fibrosis in Scl-cGvHD (12, 13).
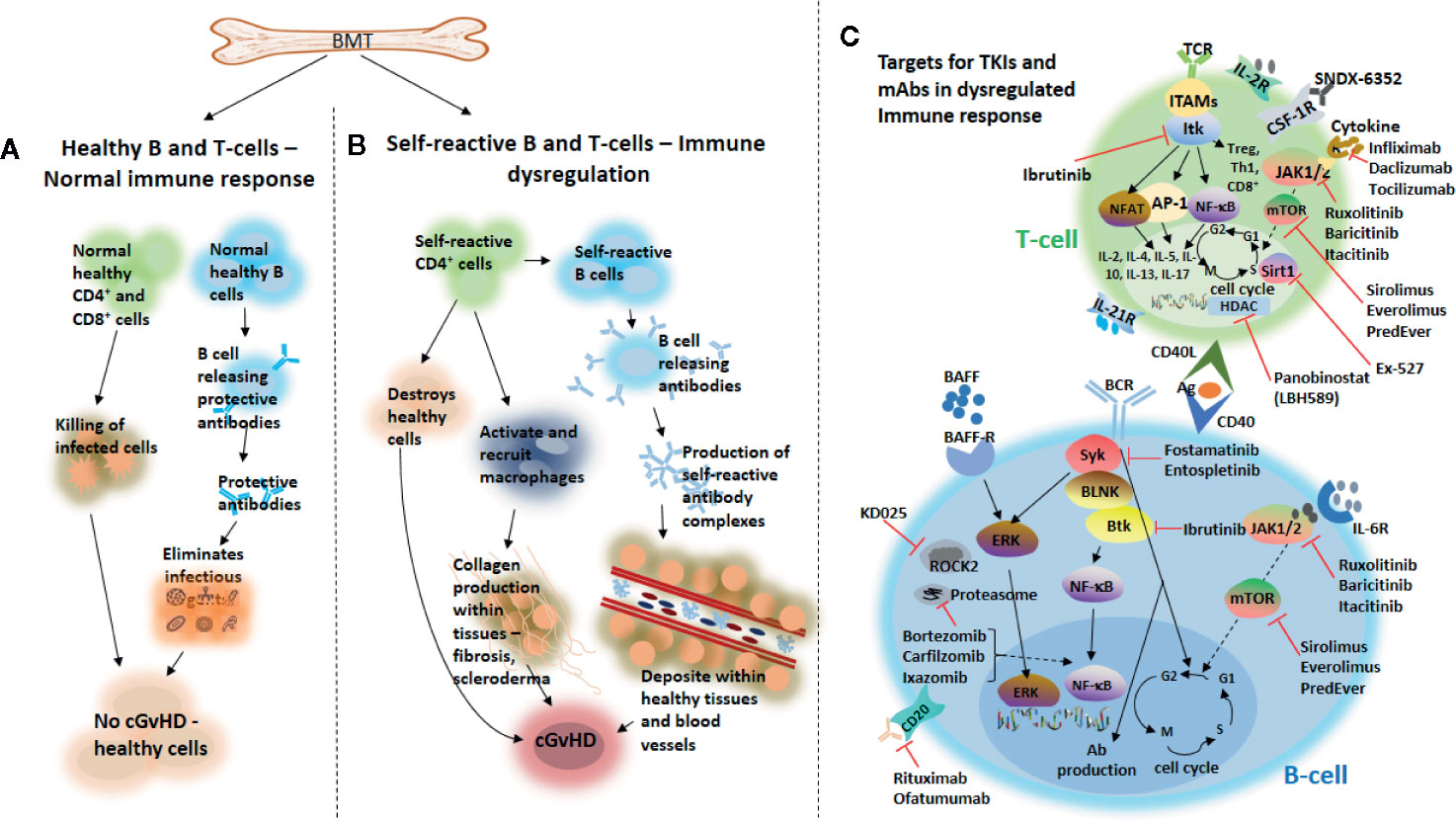
Figure 1 Chronic GvHD development and novel agents targeting B and T cells that are under investigation for the treatment of the disease. Following bone marrow transplantation, healthy production of effector B and T cells from the bone marrow may trigger a normal healthy immune response leading to a healthy immune homeostasis (A). Overproduction of self-reactive B and T cells from donor-derived bone marrow grafts may cause immune dysregulation, which on the one hand may lead to the destruction of healthy tissues, activate and recruit macrophages important for the production of collagen within tissues, thereby, causing fibrosis and scleroderma and subsequently, development of cGvHD. On the other hand, production of self-reactive antibody complexes may be triggered by self-reactive B cells from donor-derived bone marrow grafts, which may be deposited into healthy tissues and blood vessels and subsequently leading to the development of cGvHD (B). Novel agents targeting either B- or T cells that are under investigation for the treatment of cGvHD (C). TCR, T cell receptor; TKIs, tyrosine kinase inhibitors; IL-2R, interleukin-2 receptor; ITK, IL-2–inducible kinase; JAK1/2, Janus kinase 1/2; mTOR, mammalian target of rapamycin; HDAC, histone deacetylase; AP-1, activator protein 1; Sirt1, sirtuin 1; Tregs, regulatory T cells; Ab, antibody; Th1, Type 1 T-helper; ROCK2, Rho-associated coiled-coil kinase 2; BLNK, B cell linker; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NFAT, nuclear factor of activated T cells; ITAMS, immunereceptor tyrosine-based activation motifs; CSF-1R, colony-stimulating factor 1 receptor; BCR, B cell receptor; Btk, Bruton tyrosine kinase; Syk, splenic tyrosine kinase; BAFF, B cell activating factor; BAFF-R, BAFF receptor; ERK, extracellular signal-regulated kinase; CD20, cluster of differentiation 20; CD40L, cluster of differentiation 40 ligand; Ag, antigen; IL-6R, interleukin-6 receptor.
Fibroblasts are also fundamental in the pathogenesis of cGvHD involving lacrimal glands, in a different way in respect of Sjogren syndrome. Indeed, fibroblasts act as antigen presenting cells and communicate with various inflammatory cells leading to the invasion of ductal epithelium with its destruction and ductal ectasia of lacrimal glands (14). The involvement of the eyes is so clinically relevant because worsening of cGvHD score in the eyes, joints/fascia, or oral mucosa, when assessed at 6 months, are more likely to predict subsequent treatment failure; with 74% patients free from failure at 36 months when no impairment of symptoms and signs were observed at 6 months vs 26% of those that presented a higher eye, mouth or joints involvement and inflammation (15).
The “hyperinflammatory” status that characterizes cGvHD has been known for many years; in 2012, a group from Bethesda elaborated on a simple but effective score for predicting the severity of cGvHD using some parameters typical of autoimmune diseases, such as C reactive protein (CRP), complement (C3 and C4), platelets and albumin levels. When CRP is >0.7 mg/dl, C3 >140 mg/dl, C4 >28 mg/dl, platelets >250 K/μl and albumin <3.6 g/dl were all present, the chances of active cGvHD was 80% vs 69% when 1–3 parameters were present and 0% when all variables were lower than the cut off (16).
Moreover, an interesting association between the “inflammatory phenotype” of patients with cGvHD and microbiome composition is emerging. Indeed, gut microbiota of healthy patients has been compared with subjects who underwent allo-HSCT but did not develop cGvHD. The first cohort was enriched with Bacteroides, Lactobacillus, Clostridium, and Veillonella, whereas, in the group without cGvHD, two were enriched with Ruminococcus, Blautia, and Faecalibacterium. The latter is a well-known anti-inflammatory commensal bacterium, which was previously reported in patients with intestinal inflammatory diseases including active ulcerative colitis, and its presence correlated with higher amounts of IL-10 (17).
This is the prerequisite for employing anti-inflammatory agents (such as Btk inhibitors, JAK1/2 inhibitors or hydroxychloroquine) in the therapy of cGvHD as described below.
The psychological and economic impact of cGvHD is still enormous, as many complications can emerge from both the disease and its treatment (18). Chronic GvHD impairs the quality of life of the affected patients who require a continued medical follow-up, with higher risk of infection and death. Even the administration of corticosteroids is problematic: in a cohort of 162 patients, it was clearly demonstrated, that cGvHD and steroid treatment significantly prolonged time to recover normal body mass index (BMI) and muscle strength after HSCT (19). Acute GvHD occurs in 40%–50% of patients receiving allo-HSCT, which makes aGvHD a major risk factor for developing cGvHD (20). Other known risk factors include specific diseases, such as chronic myeloid leukemia (CML), age (older patients are more prone to cGvHD), use of mismatched or unrelated donors compared to matched sibling transplants, use of peripheral blood stem cells instead of bone marrow stem cells as a graft source, and sex mismatch between recipients (especially male) and donor (especially female) (21, 22). This review therefore, provides the current knowledge on the emerging therapies for cGvHD, with particular emphasis on TKIs, JAK1/JAK2, and proteasome inhibitors that are now ready for entering into clinical practice.
Standard of care in the treatment of cGvHD depends on the particular organ(s) or site(s) that is/are affected and adopted treatments can be topical or systemic. Nevertheless, about 50%–60% of patients with cGvHD will require a second-line treatment within 2 years, but at the moment there is no consensus on the optimal choice of agents for second or further lines of therapy (23). The National Comprehensive Cancer Network (NCCN) guidelines (https://www.nccn.org/professionals/physician_gls/pdf/hct.pdf) and the European Society for Blood and Marrow Transplantation (EBMT) consensus (24), both edited in 2020, agree in the use of steroids as first-line treatment, and sustain the use of ibrutinib, the only compound approved by the FDA for second- or further line treatment of cGvHD. Nevertheless, both guidelines clearly state that there are still no standard therapies for steroid-resistant (SR) patients, limiting the patients to the listed available drugs (see Table 1), and advising clinicians to possibly enroll these patients into clinical trials.
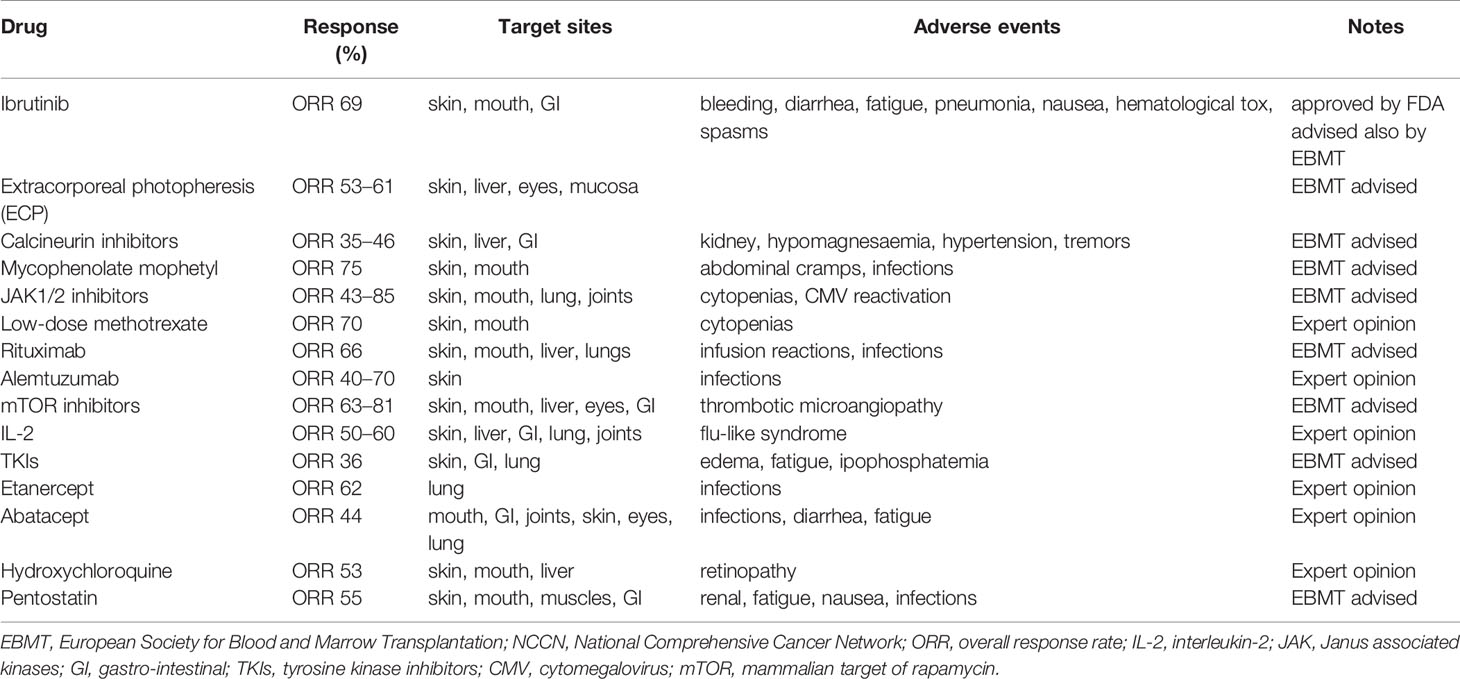
Table 1 Possible therapeutic approaches for steroid-resistant/refractory cGvHD patients according to the NCCN and EBMT 2020 guidelines (24).
It also must be noted that cGvHD severely impairs the quality of life. Hence, it is imperative that new treatment options be firstly aimed at improving the quality of life by reducing symptoms, preventing immune-mediated damage and disability, while at the same time avoiding toxicities that are associated to the treatment itself (25, 26). The long-term goal of cGvHD treatment on the other hand, is to institute an immunologic tolerance that will allow the patient to successfully withdraw from immunosuppressive treatments without relapse or clinically significant manifestations of disease activity while preserving the graft-versus-leukemia (GVL) effect (25, 26).
The current available treatment options for cGvHD firstly involve the use of corticosteroids. Due to their lymphopenic and anti-inflammatory properties, and generally based on controlled clinical trials, corticosteroids (such as prednisone) have been the mainstay of first-line treatment of cGvHD for the past three decades (27, 28). These corticosteroids can be administered alone or in combination with other immuno-suppressants such as calcineurin inhibitors, which suppress the immune system by preventing the production of IL-2 by T cells (26–29). This treatment option is sometimes however problematic as it is often inadequate or toxic, and can cause immunosuppression that may lead to an increase in malignancy relapse. Side effects, such as infections, myopathy, cataracts, hyperglycemia, decline in bone mass, and avascular necrosis have all been associated with prolonged corticosteroid use. Combination therapy with immuno-suppressants to reduce these side effects has shown little or no benefit (27, 28). Furthermore, deteriorating signs of cGvHD in previously affected organs along with the development of signs and symptoms of the disease in previously unaffected organs have both also been associated to corticosteroid treatment (26); all of which warrants a second line of treatment in nearly 50%–60% of patients who experience reoccurrence of cGvHD (16). Second-line cGvHD treatment is based on retrospective analyses, and in some cases, single-arm phase II trials (23).
Whatever line of therapy, the treatment of cGvHD has three different goals:
i. to reduce the activated status of B- and T cells: in this context, Btk inhibitors can reduce the pre-germinal B cells and T follicular helper (Tfh) lymphocytes; JAK1/2 inhibitors can decrease Th1, Th2, and Th17 activity, and anti-B and anti-T cells monoclonal antibodies (mAbs) seem to be also effective. Among the latter drugs, anti-CD20 and anti-CD52 antibodies have already entered into the clinical practice and the anti-CD26 mAb Begelomab seems to be very promising (30).
ii. to play an anti-inflammatory effect, in particular by reducing secretion of IL-6, TNF-α and IL-17. In this category, the most frequently adopted antibodies are Infliximab and Etanercept along with the JAK1 inhibitor Itacitinib.
iii. to slow down the development of fibrosis: in addition to macrophages and pathogenic TGF-β, recent studies have also suggested that fibroblasts isolated from cGvHD patients can promote fibrosis through the upregulation of collagen genes (COL1α1 and COL1α2), a mechanisms which was induced by hyperactive TGF-β signaling (31). In this context, inhibitors of CSF-1, the TGF-β pathway, PDGF, spleen tyrosine kinase (Syk), Rho-associated coiled-coil kinase 2 (ROCK2) and Hedgehog seem promising approaches [see in the following references (3, 32, 33)].
Table 2 highlights some of the agents that are currently being evaluated in patients with cGvHD. The use of some of these agents to treat cGvHD is based on their current usage in the clinic to treat certain autoimmune diseases, where their tolerability and efficacy profiles are known. In addition, patients with cGvHD often display symptoms resembling autoimmune diseases, making these agents appealing for cGvHD treatment. The mechanism of action of these agents on both B and T cells in cGvHD is illustrated in Figure 1C.
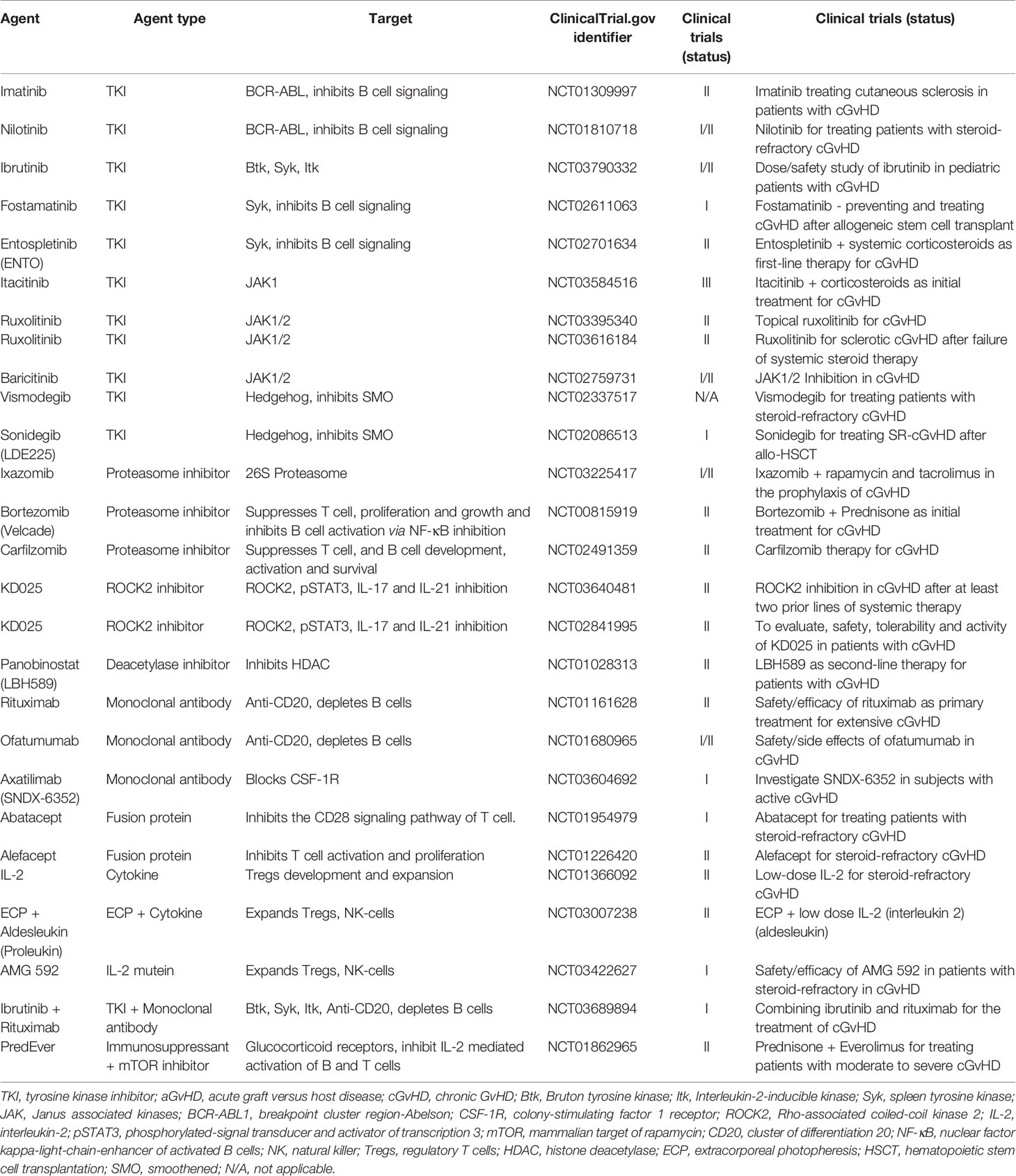
Table 2 Selected ongoing studies evaluating novel drug agents in patients with cGvHD.
It is now quite clear that in addition to alloreactive T cells, allo- and auto-reactive B cells along with several cytokines, play a fundamental role in the development of cGvHD (1, 3, 34–39). Undoubtedly, a central event in the pathogenesis of cGvHD is the recognition of antigen (Ag) via the B cell receptor (BCR). A major difference to the physiological recognition of antigen by the BCR has been demonstrated in the murine model, where B cells have a BCR-hyper-responsiveness in mice developing cGvHD (36–39). When pathogenic B cells are activated in cGvHD, they expand in a process that is guided by soluble factors, including IL-4, IL-17, IL-21, and B cell activating factor (BAFF), which is connected to the formation of germinal centers (GCs) (37, 40, 41). Donor Tfh cells cooperate with GC B cells that undergo somatic hypermutation by amassing B cells capable of producing antibodies to antigens, thereby, activating the BCR and thus, supporting cGvHD [as reviewed in (36)]. The increased number of circulating Tfh cells may thus serve as a surrogate marker of the presence of these Tfh cells in GCs: in a cohort of 70 patients, the number of circulating Tfh cells increased in patients with cGvHD, especially in those with higher severity of disease, where the expansion of Tfh was highly correlated with the generation of IgG1 antibodies (36).
Furthermore, clinical studies have demonstrated a striking increase of BAFF in the plasma of patients with cGvHD, and also of the BCR intercellular signaling molecules, such as Syk and B cell linker (BLNK) (42), which are relevant for cGvHD development. In preclinical studies, mice lacking Btk failed to develop cGvHD (39). It must be noted that when Syk is phosphorylated upon BCR engagement, it sustains B cell survival and proliferation, and promotes monocyte migration and fibrosis in cGvHD murine models (43). Moreover, after linking to its receptor, BAFF also activates extracellular regulated kinase (ERK), Protein kinase B (Akt), and NF-κB; hence, proteasome inhibitors, such as bortezomib and ixazomib, which are able to block the NF-κB signaling, can be good candidates for the treatment for cGvHD (6).
Chronic GvHD, as mentioned earlier, is an immunologic attack of host organs or tissues by donor T cells and B cells following HSCT. Donor T-helper (Th) cells are crucial in GvHD initiation, owing to their ability to differentiate into Th1 (secreting IL-2 and IFN-γ), Th2 (secreting IL-4, IL-5, IL-10 and IL-13), Th17 (secreting IL-17), and Tfh cells, which facilitate organ-specific GvHD (44). Previously published studies have shown that cGvHD results from the selective activation of allo-reactive donor CD4+ T cells that act as helpers to host B cells, thereby triggering B cell activation and auto-antibody production (45). In addition, CD4+ Foxp3+ regulatory T cells are important mediators of immune tolerance, and their impairment has been associated with cGvHD (46).
Some of the new therapeutic approaches for cGvHD are therefore aimed at targeting T cell responses or T cell signaling pathways. For example, the critical homeostatic cytokine IL-2, at low doses, can be used to target Treg development and expansion as shown in a phase I clinical trial, where 1.5 × 106 IU/m2/day of IL-2 was able to selectively target and enhance Tregs, with improvement of patients with cGvHD (47). Aldesleukin, a recombinant human IL-2, in combination with extracorporeal photopheresis (ECP) to treat SR-cGvHD, is also in a phase II clinical trial. Furthermore, the safety and tolerability of AMG 592, an IL-2 mutein, is been evaluated in a phase I/II clinical trial in subjects with SR-cGvHD (ClinicalTrials.gov identifier: NCT03007238, NCT03422627).
At the ASH meeting held in 2018, it was reported that artesunate (a drug used for treating malaria) improved the survival of mice with cGvHD by increasing the number of Tregs and decreasing that of Th17 both in peripheral blood (PB) and spleen, further sustaining the role of T cells in mediating cGvHD (48).
Other agents targeting T cells include abatacept and alefacept (both of which target and inhibits the activation and proliferation of T cells), the ROCK2 inhibitor KD025 (49), and the proteasome inhibitors ixazomib (50), bortezomib (51), carfilzomib (52), and ibrutinib (53). These agents are discussed in a more detail later in this review.
Syk, Btk and IL-2-inducible T cell kinase (Itk) are cytoplasmic TKs that are fundamental for the development of B- and T cells, playing multiple functions in the immune system. In general, Btk and Syk are crucial for the phosphorylation and activation of downstream effectors in BCR signaling, while Itk is crucial for the activation of downstream effectors in TCR signaling. Moreover, Syk is involved in immune receptor signaling, such as BCR signaling that promotes B cell proliferation and survival, but also in control of cell migration, cellular adhesion, innate immune recognition, platelet activation and vascular development, while Btk is responsible for B cell development, differentiation and signaling (54). Moreover, Btk is central in control of the Toll-like receptor (TLR)-induced IL-10 expression by B cells, and is an active part of the NLRP3 inflammasome, a multimeric protein complex that triggers the release of proinflammatory cytokines, such as IL-1β and IL-18, in many inflammatory conditions, including Alzheimer’s disease, diabetes, and infections (55), and lack of Btk can lead to a compromised BCR-induced proliferation and survival. Finally, Itk is responsible for T cell development, differentiation and signaling (56).
Recent studies demonstrated that Btk, Itk, and Syk, may all be required for the development of cGvHD (39, 43, 57). It is worth noting that upon engagement of the BCR and TCR with a signaling molecule, SRC family kinases, including Lck/Yes novel tyrosine kinase (Lyn) and lymphocyte-specific protein tyrosine kinase (Lck), are activated leading to the subsequent activation of Syk, Btk and Itk. These activated molecules then activate some transcription factors such as NF-κB in B cells and nuclear factor of activated T cells (NF-AT) in T cells, subsequently controlling cell survival, proliferation, and production of pro-inflammatory cytokines (54, 58) (see Figure 1B). This is important in the pathophysiology of cGvHD as Btk activation can drive the production of self-reactive antibody complexes which may be deposited within healthy tissues and blood vessels and hence, promote cGvHD development (Figure 1A). Furthermore, pro-inflammatory cytokines produced as a result of Syk, Btk, or Itk signaling can have a direct effect on cGvHD target tissues (58, 59).
Several agents are currently under investigation for the treatment of cGvHD, which act by either targeting B cells or T cells, or both, and/or macrophages. These agents include TKIs, anti-CD20 monoclonal antibodies and Syk inhibitors, which target B cells, proteasome inhibitors, which target T cells, and JAK1/2 and Btk inhibitors, which target both B and T cells (24). Some of these agents are discussed below according to their known mechanisms of action.
Tyrosine kinases (TKs) have been implicated in several cellular processes including differentiation, proliferation, anti-apoptosis, and B and T cell signaling (28, 53, 60–63). Consequently, the tyrosine kinase inhibitors (TKIs), by blocking B and T cell activation, could be appealing for the treatment of cGvHD. TKIs function by binding to ATP-binding catalytic sites of the tyrosine kinase domains on receptor (RTKs) and on non-receptor TKs. In the absence of TK activity, substrates necessary for RTK function cannot be phosphorylated and consequent cellular events are abrogated. Hence, TKIs can cause a blockade of downstream intracellular signaling and transcription of genes, including some pro-inflammatory cytokines (Figure 2).
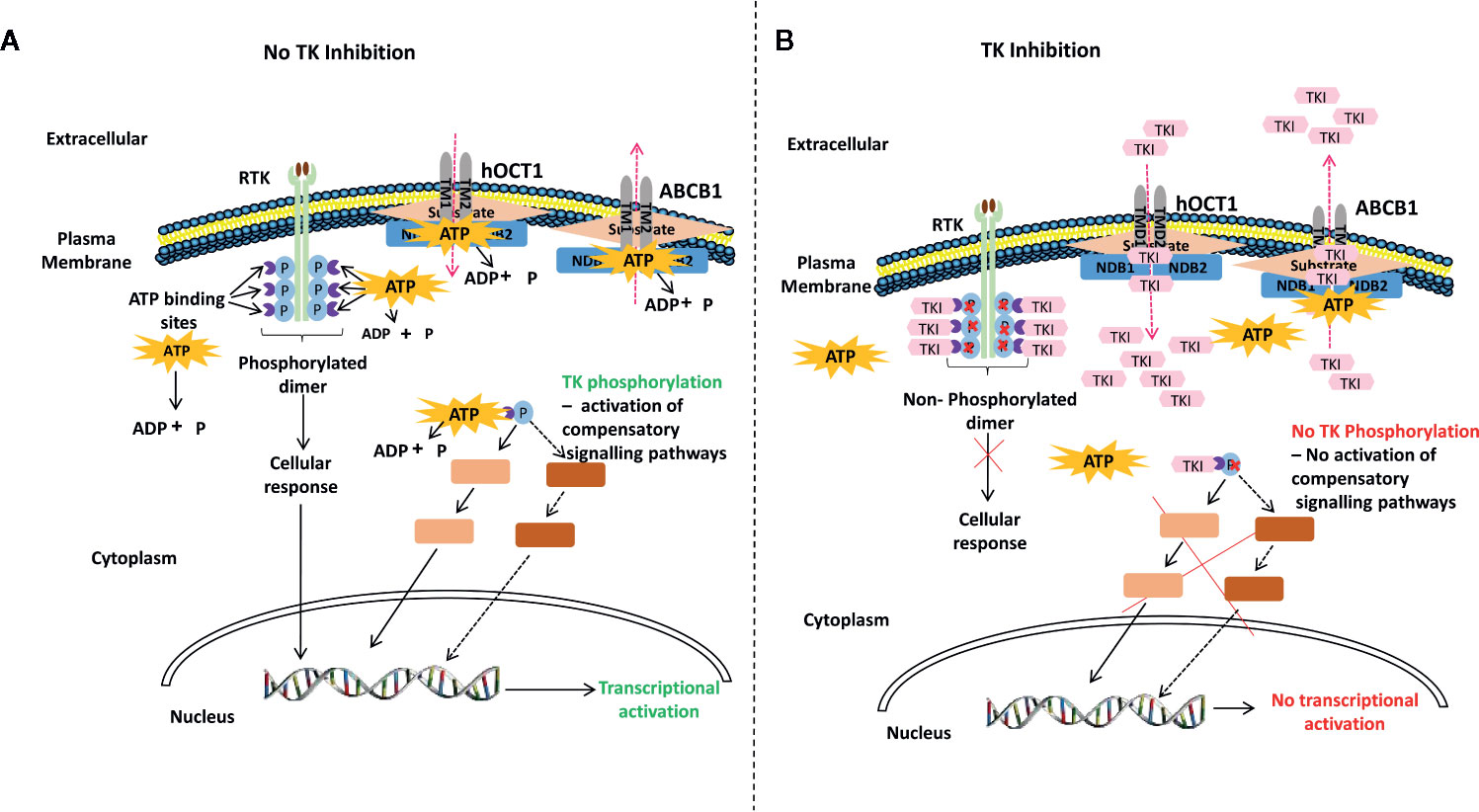
Figure 2 A generalized proposed mechanism of action of tyrosine kinase inhibitors (TKIs). (A) auto-phosphorylation of receptor TK (RTK) (light green) upon ligand (brown) binding activates TKs (purple) downstream leading to transcriptional activation. RTK functions by binding ATP and transferring phosphate from ATP to tyrosine residues on various substrates, which leads to their phosphorylation and subsequent cellular response; such as the excess proliferation of cells. Intracellular non-RTKs can also bind ATP and transfer phosphate from ATP to tyrosine residues on various substrates, which can also lead to their phosphorylation and subsequent cellular response. The plasma membrane also has influx and efflux transporters that are ATP-gated (left hand panel). (B) In the absence of TK activity, substrates necessary for RTK function cannot be phosphorylated and consequent cellular events are abrogated. Hence, TKIs function by binding to ATP-binding catalytic sites of the TK domains on RTKs or on non-RTKs, thereby, preventing TK phosphorylation and inhibiting down-stream intracellular signaling and transcription of genes such as pro-inflammatory cytokines. TKIs can also bind and occupy ATP- or substrate-binding sites on transmembrane transporters such as the hOTC1 influx protein and the ABC efflux pumps (64–66), thereby, affecting drug and cellular responses (right hand panel). RTK, receptor tyrosine kinase; TK, tyrosine kinase; hOCT1, human organic cation transport member 1; ABC, ATP-binding cassette; ABCB1, ABC sub-family B member 1 (P-glycoprotein 1); ATP, adenosine triphosphate; ADP, adenosine diphosphate; p, phosphate; NDB1, nucleotide binding domain 1; NDB2, nucleotide binding domain 2; TMD1, transmembrane domain 1; TMD2, transmembrane domain 2; TKI, tyrosine kinase inhibitor.
The efficacy profiles along with the mechanism of action of most of these TKIs are also known, as they have long been used to treat hematological diseases, such as acute leukemia (67), chronic lymphocytic (CLL) (68). In CML, for example, the rearrangement of BCR and ABL1 genes gives origin to a fusion gene where the control of the ABL1 kinase function is lost and so the new protein is constitutively active, with the consequent excess of proliferation of myeloid precursors and an abnormal interaction of the leukemic stem cells with the actin and the bone marrow stroma (69) (Figure 2, left hand panel).
TKIs can also bind and occupy ATP- or substrate-binding sites on transmembrane transporters, such as the ATP-binding cassette (ABC) efflux pumps and the human organic cation transport member 1 (hOTC1) influx protein (64–66) thereby affecting drug and cellular responses (Figure 2, right hand panel). In CML, TKIs such as imatinib and dasatinib, have been shown to not only target and inhibit BCR-ABL1, but also c-KIT, PDGF receptor (PDGFR), and Src family kinases, which results in the abrogation of the transcription of pro-inflammatory cytokines such as IL-1, IL-6, and TNF-α, and prevents the proliferation of myeloid cells (70).
In cGvHD, TKIs could be used to inhibit growth factor receptor pathways such as those of PDGFR, epidermal growth factor receptor (EGFR) and TGF-β pathways. Indeed, ABL1 is located downstream of TGF-β, and in preclinical models of systemic sclerosis, a disease which may resemble Scl-cGvHD, the production of proteins of the extra-cellular matrix by TGF-β was significantly decreased in ABL1-deficient cells (71). On the other hand, it has been clearly demonstrated that many TKs, such as PDGFR, vascular endothelial growth factor receptor (VEGFR) and EGFR are significantly increased during liver fibrosis development, transforming the hepatic stellate cells from resting to active (63, 72). In addition, other signaling pathways, such as MEK/ERK and phosphatidylinositol-3 kinase (PI3K)/Akt seem to be activated by TKs during hepatic stellate cells activation; this is the basis for employing TKIs in diseases such as liver fibrosis (72) Table 3.
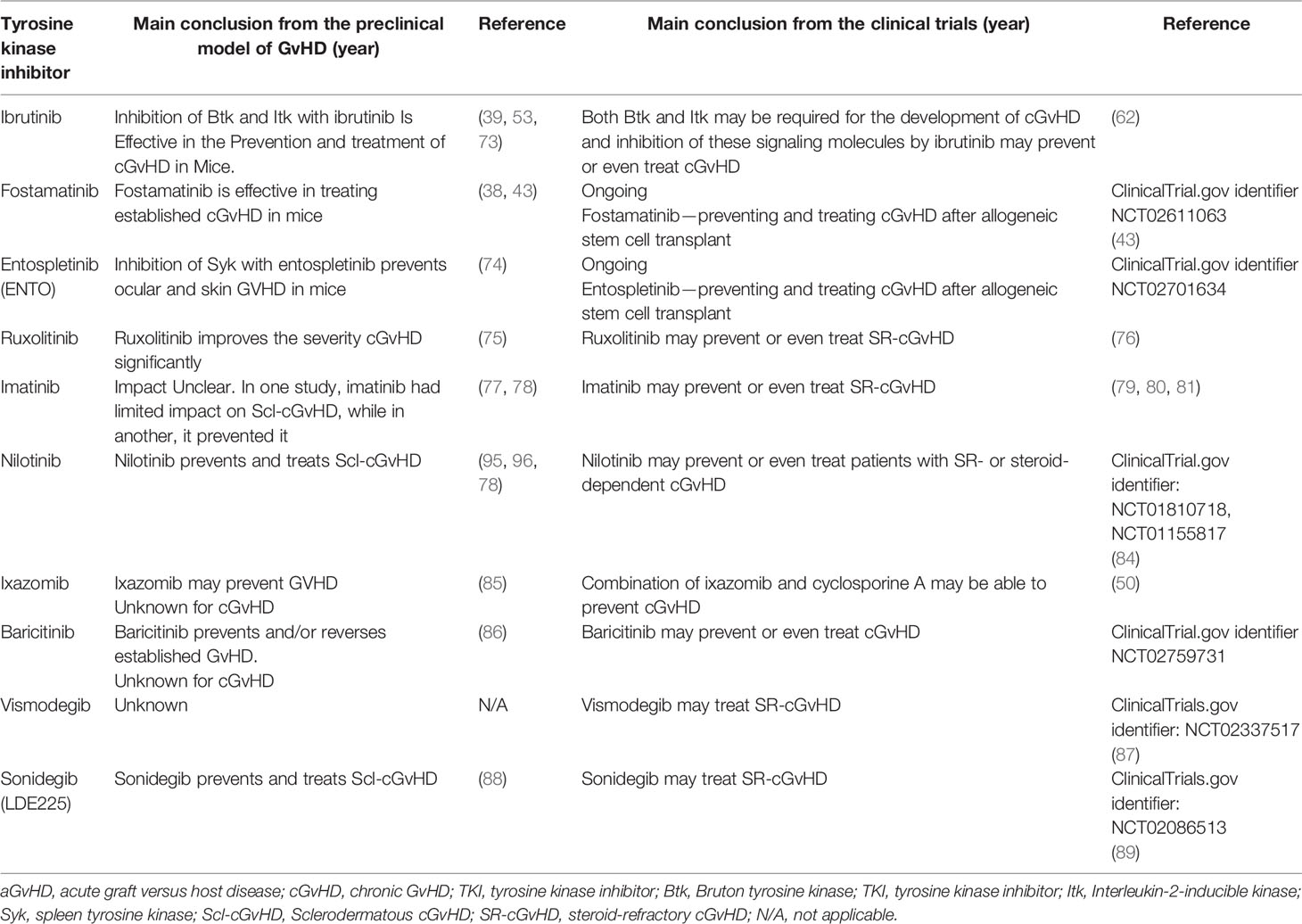
Table 3 Translation of selected tyrosine kinase inhibition strategies from animal models of cGvHD into clinical trials.
Imatinib is a first generation TKI approved for the treatment of CML and Philadelphia-positive acute lymphoblastic leukemia (ALL) and for myelodysplastic/myeloproliferative diseases carrying the PDGFR rearrangement. In addition to the BCR-ABL1 fusion protein, imatinib can also target and inhibit PDGFR and TGF-β pathways both of which play important roles in the fibrosing process occurring in Scl-cGvHD. Hence, targeting these growth factors could represent an important therapeutic strategy for treatment of cGvHD (90, 91).
In a murine model of bleomycin-induced fibrosis, imatinib prevented the differentiation of fibroblasts into myofibroblasts and reduced the synthesis and accumulation of proteins of the extra-cellular matrix in the skin, significantly ameliorating the mice phenotype (92, 93). Besides, imatinib was also effective against pulmonary, renal and liver fibrosis. In the same model, imatinib significantly prevented pulmonary fibrosis by inhibiting proliferation of mesenchymal cells (93).
In one preclinical study, Belle et al. used a murine model of Scl-cGvHD to assess the therapeutic impact of imatinib; published data indicated that imatinib is able to reduce the proliferation of both total T cells and T-regs in the spleen. Nevertheless, despite the inhibition of PDGFR, there was no significant clinical impact of imatinib on murine Scl-cGvHD (77). The impact of imatinib on liver fibrosis is more debated: indeed, it significantly decreased the expression of some fibrosis markers, such as α-SMA, Pcol1A1, and Hsp47 (94), but reduced only the early liver fibrogenesis without preventing the long-term fibrosis progression (95). Moreover, in a small series of CML patients, our group recently showed that imatinib is able to reduce the expression of several genes that usually are highly expressed in autoimmune diseases. Genes such as ANX4A, which is highly expressed in the Sjogren’s syndrome, CASP10, highly expressed in Crohn’s disease, CEACAM8 and CTSG, overexpressed in arthritis, ITGAM, elevated in psoriasis, and PGLYRP1, whose high levels have been documented in chronic gingivitis, were all reduced by imatinib; thus, supporting the “anti-inflammatory” power of this drug (96).
In 2002, it was reported for the first time that in a series of 23 CML patients with bone marrow fibrosis, treatment with imatinib for 3 months induced a marked regression of bone marrow fibrosis (97). Two years later, another group demonstrated that imatinib reduced fibrosis by at least 2 grades in 61% and by at least 1 grade in 85% of cases, independently on the cytogenetic response (98). Similar successes were reported in patients with treatment-resistant systemic sclerosis, where skin fibrosis generally was more responsive than lung fibrosis. In this context, the authors showed that imatinib decreased the phosphorylation both of PDFGR and of c-ABL1 in the skin of systemic sclerosis (SSc) patients (99). This finding is particularly interesting since SSc may resemble Scl-cGvHD, which paves the way for a similar imatinib treatment approach in Scl-cGvHD patients.
On this basis, in 2009, Olivieri et al. reported the efficacy of imatinib at 200 mg/day in a series of 19 patients with refractory cGvHD, where the organs mostly involved were skin, lungs and bowel. After 6 months of treatment, half of the patients showed a significant improvement of cGvHD signs, especially in skin fibrosis (79). The same conclusions were drawn by Magro et al. who reported that imatinib 400 mg/day was able to reduce skin sclerosis within 2 months of treatment. They investigated the safety and efficacy of imatinib in 14 patients with Scl-GVHD: with a median follow-up of 11.6 months, ORR was 50%, including 28% of CR and 71% of PR and a significant reduction of steroid dose. Imatinib was well tolerated in most patients but in 29% of them, adverse events (muscle cramps, nausea diarrhea, and fatigue) led to treatment discontinuation (100). In addition, there is some evidence to suggest that imatinib can improve the severity of cGvHD and at the same time allow for the withdrawal of systemic corticosteroids without further deteriorating the disease symptoms (79, 80, 99, 101, 102).
A possible explication of these different results could be probably found in some pharmacogenetic aspects. Imatinib is substrate of transmembrane influx and efflux pumps, as clearly showed in CML where the combination of polymorphisms of the human organic cation transporter type 1 (hOCT1) influx pump and of one of the most important efflux pumps (ATP-binding cassette sub-family B member 1; ABCB1) seem to influence either the plasma level or the drug intra-cellular concentrations, with a clear impact on toxicity and efficacy (103). Analogously, the impact of transporters in the intake of imatinib by dermal fibroblast has been recently assessed by a German group: OCT1, OCT2, organic cation transporter novel type 1 (OCTN1), OCTN2, and Multidrug and toxin extrusion 1 (MATE1) were expressed in dermal fibroblasts and the expression and activity of MATE1 were deemed responsible for imatinib uptake. On the other hand, MATE1 expression seemed to be correlated to cytokines, such as PDGF subunit B (PDGFB), and to the NOTCH signaling pathway. Indeed, in dermal fibroblasts, the NOTCH pathway is highly activated and blockade of this signaling network normalized the transporter function of MATE1 and increased the imatinib efficacy in fibroblasts from patients with SSc (104).
Nilotinib is a second-generation FDA approved TKI that could also be used as first-line treatment of CML. Like imatinib, it targets BCR-ABL1 and PDGFR with a higher affinity and lower IC50 compared with imatinib, so offering higher rates of early and deep molecular responses—conditions necessary also for a further interruption of treatment (105–108). In preclinical cGvHD models, nilotinib has been able to prevent Scl-cGvHD by targeting and inhibiting the aberrant activation of the pro-fibrotic c-ABL1 kinase and PDGFR (82, 83).
In another murine model of liver fibrosis, nilotinib decreased TNF-α, TGF-β, receptor for advance glycation endproducts (RAGE), and high-mobility group box 1 protein (HMGB1) mRNA expression, decreased nitric oxide levels and increased glutathione peroxidase activity, with significant reduction of fibrosis (83).
The anti-inflammatory and immunomodulatory effects of nilotinib on cGvHD were further highlighted when cultured CD3-positive peripheral blood mononuclear cells from healthy donors were exposed to varying concentrations of this TKI. Nilotinib suppressed the production of pro-inflammatory cytokines, including IL-2, IL-10, IL-17, IFN-γ and TNF-α; nevertheless, it had little or no effect on the frequency of Tregs, B and NK cells (109).
Elevated levels of TGF-β as mentioned earlier, has been found in cGvHD patients though the mechanism through which it contributes to the pathogenesis of the disease remains elusive. In their study, Busilacchi et al. demonstrated that nilotinib represses the growth of fibroblast isolated from cGvHD patients and causes earlier senescence in these cultured cells compared to normal fibroblasts. More importantly, nilotinib treatment led to the downregulation of both COL1α1 and COL1α2, along with TGF-β inhibition, which was associated with reduction of intracellular phospho-Smad2. Hence, suggesting that TGF-β inhibition at intracellular and systemic level denotes a crucial anti-fibrotic apparatus of nilotinib in a clinical setting (31).
Recently, Olivieri et al, conducted a phase I/II study in which they evaluated the safety and maximum tolerated dose (MTD) of nilotinib in 22 SR-cGvHD patients with multiorgan involvement. MTD could not be reached after dose increase of up to 600 mg/day, and according to the 2005 and 2014 NIH and GITMO [an exploratory approach, combining objective involvement (OI) without failure] response criteria, ORR at 6 months were 27.8%, 22.2%, and 55.6%, respectively. Overall survival (OS) at 48 months was 75%, whereas, the failure free survival (FFS), according to the NIH and GITMO criteria was 30% and 25%, respectively. Adverse events that were attributed to nilotinib treatment included itching, headache, nausea, cramps, mild anemia, and asthenia. Nonetheless, it was concluded that the safety profile of nilotinib and long-term outcome makes this TKI an attractive option in SR-cGvHD (110).
There are currently a number of ongoing phase I/II studies that are aimed at evaluating the safety and efficacy of nilotinib in patients with steroid-refractory or steroid-dependent cGvHD (ClinicalTrials.gov identifiers: NCT01810718, NCT01155817).
Ibrutinib is a first-generation TKI which in humans, targets and impedes the activation of B and T cells signaling by binding to and inhibiting Btk and Itk, respectively (31, 65). In a phase II trial, ibrutinib offered to SR cases resulted in 67% of rapid multiorgan responses (62). No major hemorrhages were reported and atrial fibrillation was reported only in 2% of cases. Overall, treatment discontinuation rate by 2 years of treatment occurred in one-third of cases. Ibrutinib is approved by the FDA for the treatment of SR-cGvHD, and recently, this drug showed a significant improvement of quality of life in a series of 42 patients affected by cGvHD (111).
Fostamatinib, like ibrutinib, is a small molecule first-generation TKI, but unlike ibrutinib, fostamatinib is able to ameliorate the severity of cGvHD by targeting Syk and blocking its down-stream signaling. It also prevents the production of chemokines and pro-inflammatory cytokines that are involved in the activation of CD4+ T cells and their subsequent differentiation into a Th1 or Th17 phenotype (36, 38, 43). Using mouse models of cGvHD with multiorgan system, Flynn et al. identified Syk-mediated BCR signaling in murine allogeneic B cells as a facilitator of cGvHD and they further validated targeting and inhibiting Syk with fostamatinib as an effective approach for the treatment of cGvHD (43). In a similar study, using a murine model of Scl-cGvHD, Huu et al. observed Syk activation and phosphorylation in B and T cells, as well as macrophages after allo-HSCT, and Syk inhibition using R788 (fostamatinib sodium) significantly reduced the severity and fibrosis of Scl-cGvHD. They further showed that R788 was not only effective at inhibiting the production of IL-6 by macrophages, but also production of IL-13, IL-17A, and IFN-γ by CD4+ T cells (38).
There is currently no reported clinical study highlighting the effects of fostamatinib in cGvHD. However, based on results from in vitro and in vivo preclinical data in cGvHD (38, 75), fostamatinib is currently being evaluated for the treatment and prevention of cGvHD in a phase I clinical trial. Enrolled patients will receive fostamatinib 100 mg qd, 150 mg qd, or 100 mg bid from day +90 days after transplant and up to 1 year (ClinicalTrials.gov identifier: NCT02611063). Results of this trial have not yet been presented.
Preclinical and clinical data suggest that JAK1/2 signaling may play an important role in the pathogenesis of B- and T cell-mediated GvHD (75, 113). This notion is supported by the requirement of a constant activation of cytokine receptors that are necessary for the production of a number of key cytokines which are implicated in the pathogenesis of GvHD by JAK1/2.
Ruxolitinib, a JAK1/2-inhibitor, has been used to suppress T cell activation via inhibition of cytokine receptor-mediated signaling. Inhibition of this pathway by ruxolitinib improves the outcome of subjects affected by cGvHD not only in murine models, but also in patients (75). JAK1/2 inhibition by ruxolitinib and the resulting attenuation of several murine cGvHD features, is possibly in part through the down-regulation of IFN-γ signaling (75). This drug has been reported to be effective also against the BOs, characterized by fixed airway obstruction and bronchiolar inflammation. In a series of five pediatric patients, two had a partial response (PR), and one an improvement in FEV1 rapidly after starting ruxolitinib (114). In addition, a recent meta-analysis, which included eight different studies that was focused on cGvHD, reported that ruxolitinib offered 62% of overall responses, with 27% of complete and 45% of partial resolution of GvHD signs and symptoms (115).
Itacitinib is a powerful anti JAK1 inhibitor that is effective in treating connective tissue diseases (116). In a phase-1 study on aGvHD, when administered at 200 or 300 mg, it induced 78.6% and 66.7% of overall response rate, respectively, with 70.6% of successes in steroid-refractory patients (117). On this basis, two studies trying itacitinib in cGvHD recently started the accrual (ClinicalTrials.gov identifier: NCT04200365, NCT03584516).
Bortezomib is a proteasome inhibitor, which in a phase II trial, administered at 1.3 mg/m2 i.v. on days 1, 8, 15, and 22 of each 35-day cycle for 3 cycles, was used in combination with prednisone for initial therapy of cGvHD. This combination was well tolerated, with limited occurrence of sensory peripheral neuropathy. The ORR was 80%, with only 10% of CRs, especially in skin (73%), gastrointestinal tract (75%), and liver (53%). One-third of cases showed a significant improvement of symptoms referred to joints, muscles, or fascia (118). More recently, it has been reported that bortezomib is well tolerated and effective in pulmonary cGvHD: in 17 patients, with a cGvHD lasting from more than 3 years, bortezomib induced a significant reduction of forced expiratory volume (FEV1) decline (from -1.06%/month before to -0.25%/month after treatment) (119).
Ixazomib. Interesting data are coming from the murine model where ixazomib, a second-generation proteasome inhibitor, has been used: it suppressed naïve human dendritic cells (DC) maturation, reducing their pro-inflammatory cytokine production, with a final improved survival of mice (85). Ixazomib was also able to induce apoptosis of activated T lymphocytes, reduce effector CD4+ T cells in bone marrow and spleen, increase Tregs in lymph nodes, Peyer patches and thymus, so suggesting the possibility of using this drug, usually employed in relapsed multiple myeloma (120), for treating cGvHD (50). When administered at 4 mg on days 1, 8, and 15 of a 28-day cycle for up to six total cycles, ixazomib showed 34% of ORR at 6 months; overall survival (OS) at 6 and 12 months was 92% and 90%, respectively. Interestingly, this therapy was well tolerated, because no subjects discontinued the treatment by 12 months (121). Moreover, after ixazomib exposure, serum BAFF levels decreased, so supporting a possible immune modulatory role for this new drug (122).
Like the TKIs, the efficacy profiles along with the mechanism of action of most of these mAbs and fusion proteins are known, as most of them are currently employed in the clinic to treat autoimmune diseases, such as rheumatoid arthritis (105). These compounds have been used to target various immune cells including B cells, T cells and macrophages in cGvHD settings. Rituximab, obinutuzumab, and ofatumumab are all anti-CD20 mAbs that have been shown to deplete B cells and thereby prevent B-cell clonal expansion in vivo cGvHD settings (106). The anti-CD52 mAb Alemtuzumab, which targets and deplete B cells, T cells, dendritic cells (DCs), and natural killer (NK) cells, is also promising for the treatment of cGvHD (107). In addition, a number of studies have reported the use of fusion proteins to treat cGvHD (108, 123). The use of some of these mAbs, antibody-drug conjugates, and fusion proteins for the treatment of cGvHD are discussed below.
Rituximab is a chimeric mAb that targets CD20, a cell-surface marker specifically found on pre-B and mature B-lymphocytes. Binding of rituximab to CD20 results in destruction of the lymphocytes by several mechanisms, including antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity and direct apoptosis (124, 125). It is usually employed at 375 mg/m2 weekly for four doses; it represents the mAb that is most frequently employed in cGvHD treatment (126–128), including Scl-cGvHD (129) in addition to other types of cGvHD (37, 130, 131). Recently, it has been reported that rituximab offered 41% of responses at 1 year, 69% at 2 years, and 77% at 3 years. Interestingly, 67% of responsive subjects tapered steroids. The probability of being alive and free from cGvHD was 36%, 55%, and 57% at 1, 2, and 3 years, respectively (132).
Finally, combining B cell depletion with rituximab (375 mg/m2, once per week, for 4 weeks) followed by TKI inhibition with nilotinib (200 mg, twice daily, for 6 months) have shown a more profound and long-lasting response (8% CR, 63% PR) with a higher survival rate (96.6% in 1 year) in patients with Scl-cGvHD (129).
Ofatumumab is another fully human anti-CD20 mAb, which, like Rituximab, targets, and depletes B lymphocytes (133). Administration of ofatumumab alone or in combination with glucocorticoids has demonstrated clinical improvement of cGvHD symptoms in different studies (134). In a small series, Ofatumumab was administered at 1000 mg on days 1 and 14 without dose-limiting toxicity. Fatigue, transaminitis, and infusional reactions were observed, but there were no cases of hepatitis B reactivation or progressive multifocal leukoencephalopathy. At 6 months, the ORR was 72%, with 36% of complete responders (CRs) and 90% of patients who reduced the steroid dose (135).
Obinutuzumab is a novel powerful humanized anti-CD20 mAb. A randomized study with this anti-CD20 antibody, infused at 3, 6, 9, and 12 months from transplantation, is now opened to enrollment (ClinicalTrials.gov identifier: NCT02867384).
Begelomab is a murine IgG2B mAb against CD26 that usually favors T cell migration. In a series of 28 patients, begelomab was administered at 2–4.5 mg/day for 5 days, with possible 6 additional doses in 3 weeks, and this cohort was compared with 82 matched controls who received anti-thymocyte globulin, ECP, mycophenolate, sirolimus, etanercept, high dose cyclophosphamide. Begelomab rendered a more effective result than conventional treatments, with an ORR of 75% vs 41%, with particular improvement in the gut (ORR 82% vs 34%). This translated into an advantage in terms of outcome, with 1-year OS of 50% vs 31% of the controls (136). In another recently published study, begelomab was shown to induce responses in over 60% of SR-aGvHD patients, including those with severe gut and liver GvHD, having failed one or more lines of treatment. Begelomab was administered to 69 patients with SR-aGvHD; of which, 8, 33, and 28 had severity of GvHD grades II, III, and IV, respectively. Notably, 28 of the 69 patients were from two prospective studies (EudraCT No. 2007-005809-21; 2012-001353-19) and 41 on a compassionate use study; the median age was 42 and 44, respectively. In both the prospective and compassionate groups, day 28 responses to begelomab were 75% and 61%, respectively, while the CRRs were 11% and 12%. Responses for grade III GvHD were recorded in 83% and 73% of patients, while responses in grade IV GvHD were recorded in 66% and 56% of patients in the two groups, respectively. Interestingly, non-relapse mortality at 6 months was 28% and 38%; and in general, 64%, 56%, 68% responses for skin, liver, and gut stage III–IV GvHD, respectively, were recorded. The 1-year OS was 50% for the prospective studies and 33% for the compassionate use patients. More importantly, of the 28 surviving patients, 12 developed cGvHD, where the severity was mild in five, moderate in five, and severe in two patients. In addition, no adverse events directly attributable to begelomab were reported (30). Begelomab was recently granted Orphan Drug Designations (ODD) by the FDA for the treatment of SR-aGvHD in the USA, EU and Switzerland. While the begelomab effect on aGvHD is an interesting and encouraging finding, there is to date, no clinical trials investigating the effect of begelomab in cGvHD patients. Nonetheless, studies have shown that CD26 may play a role in the development of pulmonary cGvHD, and that treating human umbilical cord blood (HuCB-NOG) transplant mice with the fusion protein caveolin-1-Ig (Cav-Ig), prevents the development of pulmonary cGvHD in these mice by binding to caveolin-1 on the surface of APCs and to CD26 on T cells, thereby, blocking the interaction between CD26 and caveolin-1 (137). Whether begelomab can have a similar effect in pulmonary cGvHD and/or other types of cGvHD remains an open question.
Infliximab is a chimeric monoclonal anti-TNF-α antibody, which has been successfully employed for treating aGvHD (29, 138). For cGvHD, it has been reported that two infusions at 10 mg/Kg i.v. resolved recurrent pericardial effusion related to cGvHD in a pediatric patient (139). Another anti-TNF-α agent, which is a fusion protein with promising signs of ameliorating cGvHD, is Etanercept. Etanercept was administered to 21 patients with steroid-refractory aGvHD (SR-aGvHD) and cGvHD at 25 mg twice weekly for 4 weeks, followed by 25 mg weekly for another month. Overall response rate (ORR) was 52%, with higher efficacy in gastro-intestinal aGvHD (140).
Alemtuzumab is an anti-CD52 mAb, which targets not only lymphocytes including B cells, T cells and NKs, but also DCs (141, 142). Alemtuzumab has already been employed for the treatment of chronic lymphocytic leukemia (107). In 2008, alemtuzumab was reported for the first time as an effective agent in a patient with extensive cutaneous cGvHD, who was already treated unsuccessfully with steroids, cyclosporine-A, sirolimus, tacrolimus, mychophenolate mofetil, infliximab, and rituximab. In this patient, all ulcers and pain were resolved by month 7 of therapy with alemtuzumab, administered at 10 mg/day subcutaneously for six consecutive days every 4 weeks (143). Importantly, not only is alemtuzumab effective in treating cGvHD, but is also able to prevent or lessen the incidence of the disease (144, 145), all of which highlights the significance of this agent in both prophylaxis and treatment of cGvHD.
Brentuximab Vedotin is an anti-CD30 antibody-drug conjugate already employed in Hodgkin’s (146) and in cutaneous lymphomas (147). When used to treat cGvHD, it offered 47% PRs, with 65% of patients able to decrease the steroid dose. Notwithstanding its good efficacy, the toxicity was judged too high, with 41% of patients developing grade 3 or 4 adverse events (in particular severe peripheral neuropathy), thus sustaining the need of larger studies in cGvHD with this drug (148).
Axatilimab (SNDX-6352) is a humanized IgG4 mAb that binds to the colony-stimulating factor 1 receptor (CSF-1R) and inhibits its function. CSF-1R is a receptor for the cytokine CSF-1, which is responsible for the production, differentiation and function of macrophages. By binding to CSF-1R and inhibiting it, axatilimab may be able to disrupt the activities of donor-derived pro-inflammatory macrophages that have been shown to promote cGvHD in the lung and skin (149). Axatilimab is currently being evaluated in a phase I study for its effects in patients with active cGvHD (ClinicalTrials.gov identifier: NCT03604692). Nevertheless, the role of anti-CSF-1R antibodies in cGvHD treatment needs further studies, because it has been reported that axatilimab is also able to reduce resident macrophages, while having no effect on inflammatory monocyte recruitment in models of aGvHD (149).
Alefacept is a full dimeric human leukocyte-function-associated antigen (LFA)-3/IgG1 fusion protein used for treating psoriasis; a disease which is now commonly classified as an autoimmune disease. Alefacept inhibits the activation of T cells by interfering with the CD2 receptor on T cells, and thus blocks T cell proliferation (150–152). Alefacept was demonstrated to be effective for also treating cGvHD, with eight out of 12 patients responding after 2 weeks of treatment. Unfortunately, this response was only temporary, and infections, pericarditis and a squamous cell carcinoma of the lip were observed (108).
Abatacept, a soluble fusion protein comprising the extracellular domain of human cytotoxic T-lymphocyte antigen (CTLA)-4 linked to the Fc portion of IgG1 and interferes with the CD28 signaling pathway of T cells by interfering with CD80 and CD86 expressed by antigen-presenting cells (APC). This compound is used for treating the autoimmune disease rheumatoid arthritis, and also seems promising for the treatment of Scl-cGvHD. When administered at 10 mg/kg, it has been reported to be well-tolerated and to offer 44% of PRs and reduction in prednisone usage to half of the responsive patients (123).
Other novel approaches to target cGvHD effector cells include:
Ⅰ Blocking cytokines, such as IL-17 and IL-22, which are responsible for Th17-mediated cGvHD (153, 154), or using low doses IL-2 to induce the expansion of Tregs, which will in turn suppress Tfh cells (155). Tfh cells can promote cGvHD through their support for B cells in GC development, allowing B cell differentiation into plasma cells, leading to the deposition of allo-antibodies into target organs (156).
The possibility of targeting ROCK2, which is able to bind phospho-STAT3 to form a ROCK2/STAT3/JAK2 complex and control the Th17 genesis, was recently reported (157). At the 2018 American society of hematology (ASH) meeting, it was demonstrated that the new ROCK2 inhibitor KD025 is able to reduce levels of IL-21 and IL-17 and to increase the number of Tregs. In a small series of 17 patients who already received three lines of therapy, KD025 induced within 2 months, 65% of responses across all affected organ systems except lung; 82% of them sustained for more than 20 weeks (158).
In addition, high levels of both IL-17 and IL-22 have been reported in the skin of patients with cutaneous cGvHD, which was also attributed to an IL-6-dependent increased secretion of these cytokines by donor CD4+ T and IL-22+-Th cells (153). The anti-IL-22 mAb could be a possible option for treating cGvHD, as already demonstrated in murine models for aGvHD, where it reduced TNFα and IFN-γ levels, but at the same time, increased Treg populations (159). Two clinical trials with the anti-IL-22 antibody in aGvHD are now in progress in the USA (ClinicalTrials.gov identifier: NCT02406651, NCT03763318), paving the way for a possible translation into a cGvHD setting.
ⅠⅠ Blocking CD40L and IL-21R to prevent Tfh and GC B cell response (37, 160).
ⅠⅠⅠ Inhibition of the NAD-dependent protein deacetylase sirtuin 1 (sirt1), which regulates different subsets of T cells and is required not only for maintenance of T cell tolerance, but also for promoting Th17 responses (161–164). The important role played by this enzyme in B- and T cell interaction during the development of GvHD was recently highlighted by Daenthanasanmak et al. (165). In a murine model, the authors demonstrated that donor T cells lacking sirt1 reduced B cell activation and differentiation but increased Tregs. Moreover, administration of the sirt1 inhibitor selisistat (EX 527) resulted in reduced Tfh responses along with preventing and treating cGvHD by downregulation of IFN-γ, IL-17, and IL-21, which have all been associated with cGvHD pathogenesis (165);
IV Inhibition of the Hedgehog (Hh) signaling pathway. This pathway is involved in the regulation of cellular differentiation during embryonic development (166) and in control of cell proliferation and carcinogenesis (167). The role of Hh signaling in cGvHD was investigated by Zerr et al. (88), where they clearly demonstrated how the Hh pathway plays an important role in the pathophysiology of cGvHD. Based on these preclinical results, Vismodegib, a potent inhibitor of Hh, and already employed for basal cell carcinoma, is currently being evaluated for the treatment of cGvHD (ClinicalTrials.gov identifier: NCT02337517). It has been reported in a very small series that Vismodegib at 150 mg/day induced partial responses in five out of eight patients; nevertheless, half of them stopped treatment due to toxicity (dysgeusia, fatigue, elevated lipase). The median time to response was 103 days and the duration of response was 7.8 months (87). Another Hh inhibitor, Sonidegib, offered 47% of partial response in skin in a small series of 8 patients with Scl-cGvHD; unfortunately, patients reported worsening of quality of life, especially in non-responders (89).
V Inhibition of NOTCH signaling. In a murine model of multi-organ cGvHD, it was recently reported that Delta-like ligand 4 (Dll4)-driven NOTCH signaling is essential for the development of cGvHD and that Ab-mediated (anti-Dll1/ anti-Dll4) blockade of this pathway prevented and treated cGvHD (168). Indeed, the activation of NOTCH signaling induces an increased production of pro-inflammatory cytokines, decreases Tregs and increases pathogenic GC B cell population through increase in Tfh cells, leading to an increased tissue damage and collage deposition. Inhibition of NOTCH signaling on the other hand, impedes the production of pro-inflammatory cytokines, increases Tregs and inhibits GC formation, hence, preventing target organ damage (168).
VI Inhibition of the mitogen activated extracellular signal regulated kinases 1 and 2 (MEK1 and MEK2), which are important components of the MAPK signaling pathway. In murine models, the RAS/MEK/ERK pathway is activated in naive but not in effector memory T cells (169). Consequently, the MEK inhibitor Trametinib suppressed GvHD-inducing T cells while sparing antitumor GVL and virus-specific T cells in murine models characterized by skin sclerosis and alopecia (170). Sclerosis and alopecia are both not cGvHD but are high frequency cutaneous manifestations that often occur in cGvHD (171, 172), which makes MEK inhibition relevant to cGvHD.
VII Inhibition of the PI3K signaling pathway. PI3Ks are a family of kinases of different classes involved in several cellular processes including metabolism, growth, differentiation, and proliferation. PI3K delta (PI3Kδ) is an isoform preferentially expressed in leukocytes; it seems to control IL-17 production, and its inhibition has been reported to decrease the symptoms of cGvHD in murine models (173, 174). It was shown in a bronchiolites obliterans cGvHD mouse model that PI3Kδ activity in donor T cells is required for the development of cGvHD. In the same study, the authors were able to interestingly demonstrate that the PI3Kδ inhibitor, GS‐649443, was able to treat either BO or Scl-cGvHD by limiting IL-17 production (175). The possibility of testing PI3Kδ inhibitors as a therapeutic strategy for cGvHD is sustained also by the observation that Idelalisib, which is also a PI3K inhibitor, is already employed in relapsed follicular lymphoma and chronic lymphocytic leukemia (176), where it was shown to not increase the onset of cGvHD when administered to patients relapsed after alloHSCT (177).
VIII Inhibition of Inositol Kinase B. Inositol triphosphate kinase B (ITPKB) is an enzyme involved in the regulation of calcium intracellular levels, which is fundamental for T cell activation and differentiation. It has been reported that ITPKB inhibition induces apoptosis of activated T cells, and can control T cell–mediated autoimmunity (178). Recently, it was shown in two murine models of cGvHD (Scl and OB) that the ITPKB inhibitor GNF362, significantly improved skin and lung GvHD scores either by reducing M2 macrophage infiltration, or by decreasing the number of IFN-γ and IL-22 producing CD4+ T cells, without impairing the graft-versus-tumor effect (179). There is however, to date, no clinical trials with ITPKB inhibitors for the treatment of cGvHD.
We have summarized the more recent advances regarding the preclinical and clinical activities of some newer immunotherapy-based strategies for SR-cGvHD treatment, that include monoclonal anti-B and T cell antibodies, along with TKIs, JAK-, MEK-, proteasome-, and PI3K-inhibitors. An algorithm showing possible second and third-line treatment options for cGvHD is given in Figure 3. All these new strategies seem to offer great opportunities for preventing or reversing cGvHD, as they are aimed at repairing/maintaining immune regulation by targeting B cell (responsible for the production of self-reactive antibody complexes) or T cell (responsible for pro-inflammatory and pro-fibrotic cytokine production) signaling and by reducing inflammation, which is fundamental in the pathogenesis of cGvHD (73). However, the biggest drawback for some of these emerging therapies for cGvHD is the available evidence for their tolerability and efficacies, which is frequently from preclinical or clinical trials that are trivial and often conflicting.
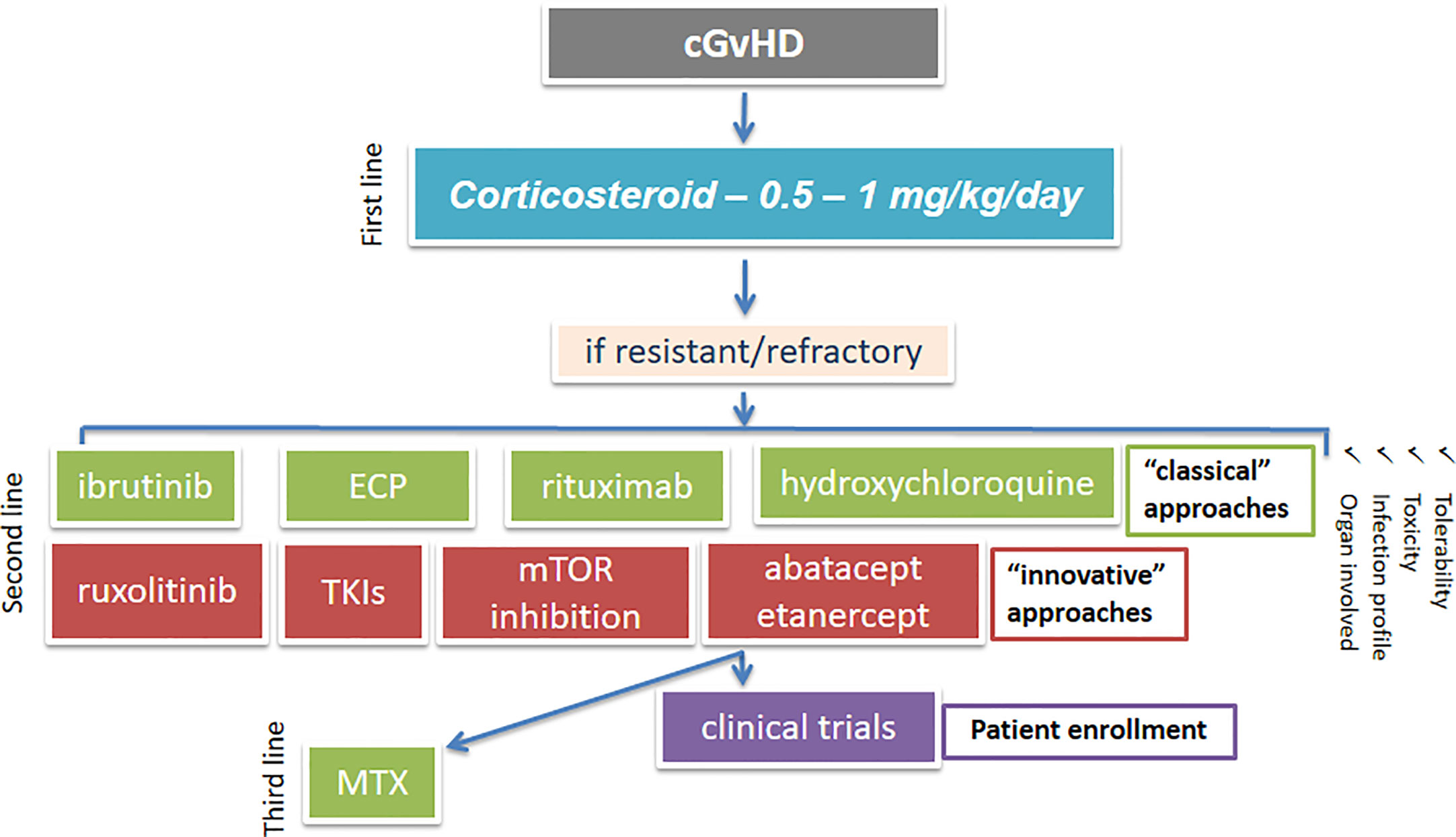
Figure 3 An algorithm showing possible treatment options for cGvHD. cGvHD, chronic graft-versus-host disease; mTOR, mammalian target of rapamycin; ECP, extracorporeal photopheresis; TKI, tyrosine kinase inhibitor; MTX, methotrexate.
Imatinib, dasatinib, nilotinib, ibrutinib, ixazomib, bortezomib, and mAbs (anti-CD20, -CD30, -CD52) are all today commonly employed in the clinical routine for treating acute and chronic leukemias, myeloma and lymphoma. Nevertheless, our knowledge on their efficacies and toxicities in the context of cGvHD is still quite incomplete and many questions remain unanswered; including: What is the tolerability of each of these promising approaches in the setting of cGvHD? What could be the less toxic and, at the same time, the most effective compound? Are the adverse events that are already observed in the treatment of cancers the same when these drugs are employed in patients already allo-transplanted and receiving many concomitant (also immunosuppressive) drugs? Is there an increased risk of secondary malignancies during these new treatments? Is it possible to design an algorithm that could help physicians to choose the most effective drug according to the organ-specific clinical features of cGvHD? Unfortunately, the majority of these questions are still unresolved and to date, the only FDA-approved drug for the treatment of adult patients with SR-cGvHD is ibrutinib, which, as mentioned above, seems to be effective and well tolerated (61). Importantly however, some mAbs, TKIs, and JAK1/2 inhibitors are now in their late stages of evaluation and some are even already employed in the clinics for “compassionate use”.
Perhaps some additional aspects, such as the pharmacogenetic aspects, could help us to design for each patient a “tailored” therapy. For example, it would be incorrect to use imatinib in patients without an effective influx pump or with a high efflux of the drug; in these cases, a TKI such nilotinib, which is superior to imatinib could be advantageous.
We did not summarize options for cGvHD treatment using cell-based therapies, such as mesenchymal stroma cells, regulatory T cells, exosomes, and others, which will be addressed in an additional paper.
Profound knowledge gaps in fully understanding the biology of cGvHD have until now mired the discovery and implementation of effective therapeutic strategies. Nonetheless, we are hopeful that murine models and clinical data, which are already available, will soon be able to help clinicians to control or eradicate cGvHD complications of HSCT, thus, ameliorating the quality of life of transplanted patients.
In this complex scenario, the COST ACTION 17138 “EUROGRAFT” project (see “gvhd.eu” website), in the context of whom this review has been realized, will lead to an improved understanding of the pathogenesis of cGvHD and its associated comorbidities, and will help us to develop a coordinated approach to therapy, via a strict international collaboration among experts in this field.
NS and SG planned and wrote the manuscript. NS and SG drew the figures. CB, AD, MG, MI, UK, AT, and RZ discussed the content and contributed to writing. All authors contributed to the article and approved the submitted version.
This work was supported by COST (European Cooperation in Science and Technology), www.cost.eu—CA17138 EUROGRAFT. AT was supported by the French Government’s Investissement d’Avenir Program, Laboratoire d’Excellence “Milieu Intérieur” Grant ANR-10-LABX-69-01 and by the by the Agence Nationale de la Recherche (Project RANKLthym ANR-19- CE18-0021-02).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Li X, Gao Q, Feng Y, Zhang X. Developing role of B cells in the pathogenesis and treatment of chronic GVHD. Br J Haematol (2019) 184:323–36. doi: 10.1111/bjh.15719
2. Nassereddine S, Rafei H, Elbahesh E, Tabbara I. Acute Graft Versus Host Disease: A Comprehensive Review. Anticancer Res (2017) 37:1547–55. doi: 10.21873/anticanres.11483
3. MacDonald KPA, Hill GR, Blazar BR. Chronic graft-versus-host disease: biological insights from preclinical and clinical studies. Blood (2017) 129:13–21. doi: 10.1182/blood-2016-06-686618
4. Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant (2005) 11:945–56. doi: 10.1016/j.bbmt.2005.09.004
5. Pavletic SZ, Vogelsang GB, Lee SJ. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: preface to the series. Biol Blood Marrow Transplant (2015) 21:387–8. doi: 10.1016/j.bbmt.2014.12.035
6. Nakasone H, Sahaf B, Miklos DB. Therapeutic benefits targeting B-cells in chronic graft-versus-host disease. Int J Hematol (2015) 101:438–51. doi: 10.1007/s12185-015-1782-4
7. Presland RB. Biology of chronic graft-vs-host disease: Immune mechanisms and progress in biomarker discovery. World J Transplant (2016) 6:608–19. doi: 10.5500/wjt.v6.i4.608
8. Alexander KA, Flynn R, Lineburg KE, Kuns RD, Teal BE, Olver SD, et al. CSF-1–dependant donor-derived macrophages mediate chronic graft-versus-host disease. J Clin Invest (2014) 124:4266–80. doi: 10.1172/JCI75935
9. Hotta M, Yoshimura H, Satake A, Tsubokura Y, Ito T, Nomura S. GM-CSF therapy inhibits chronic graft-versus-host disease via expansion of regulatory T cells. Eur J Immunol (2019) 49:179–91. doi: 10.1002/eji.201847684
10. Tugues S, Amorim A, Spath S, Martin-Blondel G, Schreiner B, De Feo D, et al. Graft-versus-host disease, but not graft-versus-leukemia immunity, is mediated by GM-CSF-licensed myeloid cells. Sci Transl Med (2018) 10(469):eaat8410. doi: 10.1126/scitranslmed.aat8410
11. 2019 Beyond Fundamentals of HCT . American Society for Transplantation and Cellular Therapy. Available at: https://www.asbmt.org/meetings-events/beyond-fundamentals77 (Accessed September 9, 2019).
12. Goussetis E, Spiropoulos A, Theodosaki M, Stefanaki K, Petrakou E, Graphakos S. Myofibroblasts generated in culture from sclerotic skin lesions of a patient with extensive chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation are of recipient origin. Stem Cells Dev (2010) 19:1285–7. doi: 10.1089/scd.2009.0401
13. Janin-Mercier A, Devergie A, Van Cauwenberge D, Saurat JH, Bourges M, Lapiere CM, et al. Immunohistologic and ultrastructural study of the sclerotic skin in chronic graft-versus-host disease in man. Am J Pathol (1984) 115:296–306.
14. Ogawa Y, Yamazaki K, Kuwana M, Mashima Y, Nakamura Y, Ishida S, et al. A significant role of stromal fibroblasts in rapidly progressive dry eye in patients with chronic GVHD. Invest Ophthalmol Vis Sci (2001) 42:111–9.
15. Pidala JA, Hamilton BK, Martin PJ, Onstad L, Storer BE, Palmer J, et al. The Chronic Graft-versus-Host Disease Failure-Free Survival (cGVHD-FFS) Index. Biol Blood Marrow Transplant (2019) 25:2468–73. doi: 10.1016/j.bbmt.2019.07.040
16. Grkovic L, Baird K, Steinberg SM, Williams KM, Pulanic D, Cowen EW, et al. Clinical laboratory markers of inflammation as determinants of chronic graft-versus-host disease activity and NIH global severity. Leukemia (2012) 26:633–43. doi: 10.1038/leu.2011.254
17. Farhadfar N, Gharaibeh RZ, Wingard JR, Lyon D, Jobin C, Wang GP, et al. Microbiota Phylogenic Analysis Revealed Decreased Abundance of Faecalibacterium Prausnitzii, an Anti-Inflammatory Commensal Bacterium, in Patients with Chronic Graft-Versus-Host Disease. Blood (2018) 132:2119–9. doi: 10.1182/blood-2018-99-113658
18. Andrews C, Smith S, Kennel M, Schilling S, Kalpakjian C. The Association of Performance Status and Disease Severity in Patients With Chronic Graft-vs-Host Disease. Arch Phys Med Rehabil (2019) 100:606–12. doi: 10.1016/j.apmr.2018.04.034
19. Hayakawa J, Miyamura D, Kimura S-I, Gomyo A, Tamaki M, Akahoshi Y, et al. Negative impact of chronic graft-versus-host disease and glucocorticoid on the recovery of physical function after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant (2019) 54:994–1003. doi: 10.1038/s41409-018-0365-4
20. Flowers MED, Inamoto Y, Carpenter PA, Lee SJ, Kiem H-P, Petersdorf EW, et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood (2011) 117:3214–9. doi: 10.1182/blood-2010-08-302109
21. Kumar AJ, Kim S, Hemmer MT, Arora M, Spellman SR, Pidala JA, et al. Graft-versus-host disease in recipients of male unrelated donor compared with parous female sibling donor transplants. Blood Adv (2018) 2:1022–31. doi: 10.1182/bloodadvances.2017013052
22. Randolph SSB, Gooley TA, Warren EH, Appelbaum FR, Riddell SR. Female donors contribute to a selective graft-versus-leukemia effect in male recipients of HLA-matched, related hematopoietic stem cell transplants. Blood (2004) 103:347–52. doi: 10.1182/blood-2003-07-2603
23. Wolff D, Schleuning M, von Harsdorf S, Bacher U, Gerbitz A, Stadler M, et al. Consensus Conference on Clinical Practice in Chronic GVHD: Second-Line Treatment of Chronic Graft-versus-Host Disease. Biol Blood Marrow Transplant (2011) 17:1–17. doi: 10.1016/j.bbmt.2010.05.011
24. Penack O, Marchetti M, Ruutu T, Aljurf M, Bacigalupo A, Bonifazi F, et al. Prophylaxis and management of graft versus host disease after stem-cell transplantation for haematological malignancies: updated consensus recommendations of the European Society for Blood and Marrow Transplantation. Lancet Haematol (2020) 7:e157–67. doi: 10.1016/S2352-3026(19)30256-X
25. Wolff D, Bertz H, Greinix H, Lawitschka A, Halter J, Holler E. The treatment of chronic graft-versus-host disease: consensus recommendations of experts from Germany, Austria, and Switzerland. Dtsch Arztebl Int (2011) 108:732–40. doi: 10.3238/arztebl.2011.0732
26. Flowers MED, Martin PJ. How we treat chronic graft-versus-host disease. Blood (2015) 125:606–15. doi: 10.1182/blood-2014-08-551994
27. Dignan FL, Amrolia P, Clark A, Cornish J, Jackson G, Mahendra P, et al. Diagnosis and management of chronic graft-versus-host disease. Br J Haematol (2012) 158:46–61. doi: 10.1111/j.1365-2141.2012.09128.x
28. Fowler DH, Pavletic SZ. Syk and tired of current chronic GVHD therapies. Blood (2015) 125:3974–5. doi: 10.1182/blood-2015-05-640672
29. Hamadani M, Hofmeister CC, Jansak B, Phillips G, Elder P, Blum W, et al. Addition of infliximab to standard acute graft-versus-host disease prophylaxis following allogeneic peripheral blood cell transplantation. Biol Blood Marrow Transplant (2008) 14:783–9. doi: 10.1016/j.bbmt.2008.04.006
30. Bacigalupo A, Angelucci E, Raiola AM, Varaldo R, Di Grazia C, Gualandi F, et al. Treatment of steroid resistant acute graft versus host disease with an anti-CD26 monoclonal antibody—Begelomab. Bone Marrow Transplant (2020) 55(8):1580–7 doi: 10.1038/s41409-020-0855-z
31. Marinelli Busilacchi E, Costantini A, Mancini G, Tossetta G, Olivieri J, Poloni A, et al. Nilotinib Treatment of Patients Affected by Chronic Graft-versus-Host Disease Reduces Collagen Production and Skin Fibrosis by Downmodulating the TGF-β and p-SMAD Pathway. Biol Blood Marrow Transplant (2020) 26:823–34. doi: 10.1016/j.bbmt.2020.01.014
32. MacDonald KP, Blazar BR, Hill GR. Cytokine mediators of chronic graft-versus-host disease. J Clin Invest (2017) 127:2452–63. doi: 10.1172/JCI90593
33. Hill L, Alousi A, Kebriaei P, Mehta R, Rezvani K, Shpall E. New and emerging therapies for acute and chronic graft versus host disease. Ther Adv Hematol (2018) 9:21–46. doi: 10.1177/2040620717741860
34. Cutler CS, Koreth J, Ritz J. Mechanistic approaches for the prevention and treatment of chronic GVHD. Blood (2017) 129:22–9. doi: 10.1182/blood-2016-08-686659
35. Poe JC, Jia W, Su H, Anand S, Rose JJ, Tata PV, et al. An aberrant NOTCH2-BCR signaling axis in B cells from patients with chronic GVHD. Blood (2017) 130:2131–45. doi: 10.1182/blood-2017-05-782466
36. Zeiser R, Sarantopoulos S. Blazar BR. B-cell targeting in chronic graft-versus-host disease. Blood (2018) 131:1399–405. doi: 10.1182/blood-2017-11-784017
37. Flynn R, Du J, Veenstra RG, Reichenbach DK, Panoskaltsis-Mortari A, Taylor PA, et al. Increased T follicular helper cells and germinal center B cells are required for cGVHD and bronchiolitis obliterans. Blood (2014) 123:3988–98. doi: 10.1182/blood-2014-03-562231
38. Le Huu D, Kimura H, Date M, Hamaguchi Y, Hasegawa M, Hau KT, et al. Blockade of Syk ameliorates the development of murine sclerodermatous chronic graft-versus-host disease. J Dermatol Sci (2014) 74:214–21. doi: 10.1016/j.jdermsci.2014.02.008
39. Dubovsky JA, Flynn R, Du J, Harrington BK, Zhong Y, Kaffenberger B, et al. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J Clin Invest (2014) 124:4867–76. doi: 10.1172/JCI75328
40. Young JS, Wu T, Chen Y, Zhao D, Liu H, Yi T, et al. Donor B cells in transplants augment clonal expansion and survival of pathogenic CD4+ T cells that mediate autoimmune-like chronic graft-versus-host disease. J Immunol (2012) 189:222–33. doi: 10.4049/jimmunol.1200677
41. Forcade E, Paz K, Flynn R, Griesenauer B, Amet T, Li W, et al. An activated Th17-prone T cell subset involved in chronic graft-versus-host disease sensitive to pharmacological inhibition. JCI Insight (2017) 2(12):e92111. doi: 10.1172/jci.insight.92111
42. Allen JL, Tata PV, Fore MS, Wooten J, Rudra S, Deal AM, et al. Increased BCR responsiveness in B cells from patients with chronic GVHD. Blood (2014) 123:2108–15. doi: 10.1182/blood-2013-10-533562
43. Flynn R, Allen JL, Luznik L, MacDonald KP, Paz K, Alexander KA, et al. Targeting Syk-activated B cells in murine and human chronic graft-versus-host disease. Blood (2015) 125:4085–94. doi: 10.1182/blood-2014-08-595470
44. Socié G, Blazar BR. Acute graft-versus-host disease: from the bench to the bedside. Blood (2009) 114:4327–36. doi: 10.1182/blood-2009-06-204669
45. Zhang C, Todorov I, Zhang Z, Liu Y, Kandeel F, Forman S, et al. Donor CD4+ T and B cells in transplants induce chronic graft-versus-host disease with autoimmune manifestations. Blood (2006) 107:2993–3001. doi: 10.1182/blood-2005-09-3623
46. Zorn E, Kim HT, Lee SJ, Floyd BH, Litsa D, Arumugarajah S, et al. Reduced frequency of FOXP3+ CD4+CD25+ regulatory T cells in patients with chronic graft-versus-host disease. Blood (2005) 106:2903–11. doi: 10.1182/blood-2005-03-1257
47. Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med (2013) 5:179ra43. doi: 10.1126/scitranslmed.3005265
48. Xiaomei C, Weng J, Lai P, Huang X, Yulian W, Geng S, et al. Artesunate Attenuate Chronic Graft-Versus-Host Disease By Regulating Th17/Treg Balance. Blood (2018) 132:5680–0. doi: 10.1182/blood-2018-99-114556
49. Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, et al. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood (2016) 127:2144–54. doi: 10.1182/blood-2015-10-678706
50. Ramos T, Rodríguez-Gil A, Nufer M, Barbado MV, Guerrero EG, Caracuel-García R, et al. Pre-Clinical Trial to Evaluate the Efficacy of Delayed Administration of Ixazomib in the Prophylaxis of Chronic Graft-Versus-Host Disease. Blood (2018) 132:4521–1. doi: 10.1182/blood-2018-99-118098
51. Pai C-CS, Chen M, Mirsoian A, Grossenbacher SK, Tellez J, Ames E, et al. Treatment of chronic graft-versus-host disease with bortezomib. Blood (2014) 124:1677–88. doi: 10.1182/blood-2014-02-554279
52. Pidala JA, Jaglowski S, Im AP, Chen GL, Onstad L, Kurukulasuriya C, et al. Carfilzomib for Treatment of Refractory Chronic Gvhd: A Chronic GVHD Consortium Pilot Trial. Biol Blood Marrow Transplant (2019) 25:S233. doi: 10.1016/j.bbmt.2018.12.224
53. Schutt SD, Fu J, Nguyen H, Bastian D, Heinrichs J, Wu Y, et al. Inhibition of BTK and ITK with Ibrutinib Is Effective in the Prevention of Chronic Graft-versus-Host Disease in Mice. PloS One (2015) 10:e0137641. doi: 10.1371/journal.pone.0137641
54. Mohamed AJ, Yu L, Bäckesjö C-M, Vargas L, Faryal R, Aints A, et al. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev (2009) 228:58–73. doi: 10.1111/j.1600-065X.2008.00741.x
55. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis (2019) 10:128. doi: 10.1038/s41419-019-1413-8
56. Qi Q, Kannan AK, August A. Structure and function of Tec family kinase Itk. Biomol Concepts (2011) 2:223–32. doi: 10.1515/bmc.2011.020
57. Gevrey J-C, Isaac BM, Cox D. Syk is required for monocyte/macrophage chemotaxis to CX3CL1 (Fractalkine). J Immunol (2005) 175:3737–45. doi: 10.4049/jimmunol.175.6.3737
58. Gomez-Rodriguez J, Kraus ZJ, Schwartzberg PL. Tec family kinases Itk and Rlk / Txk in T lymphocytes: cross-regulation of cytokine production and T-cell fates. FEBS J (2011) 278:1980–9. doi: 10.1111/j.1742-4658.2011.08072.x
59. Henden AS, Hill GR. Cytokines in Graft-versus-Host Disease. J Immunol (2015) 194:4604–12. doi: 10.4049/jimmunol.1500117
60. Gu JJ, Ryu JR, Pendergast AM. Abl tyrosine kinases in T-cell signaling. Immunol Rev (2009) 228:170–83. doi: 10.1111/j.1600-065X.2008.00751.x
61. Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer (2018) 17:57. doi: 10.1186/s12943-018-0779-z
62. Miklos D, Cutler CS, Arora M, Waller EK, Jagasia M, Pusic I, et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood (2017) 130:2243–50. doi: 10.1182/blood-2017-07-793786
63. Morin F, Kavian N, Marut W, Chéreau C, Cerles O, Grange P, et al. Inhibition of EGFR Tyrosine Kinase by Erlotinib Prevents Sclerodermatous Graft-Versus-Host Disease in a Mouse Model. J Invest Dermatol (2015) 135:2385–93. doi: 10.1038/jid.2015.174
64. Shukla S, Chen Z-S, Ambudkar SV. Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance. Drug Resist Update (2012) 15:70–80. doi: 10.1016/j.drup.2012.01.005
65. Crossman LC, Druker BJ, Deininger MWN, Pirmohamed M, Wang L, Clark RE. hOCT 1 and resistance to imatinib. Blood (2005) 106:1133–4. doi: 10.1182/blood-2005-02-0694. author reply 1134.
66. Galimberti S, Bucelli C, Arrigoni E, Baratè C, Grassi S, Ricci F, et al. The hOCT1 and ABCB1 polymorphisms do not influence the pharmacodynamics of nilotinib in chronic myeloid leukemia. Oncotarget (2017) 8:88021–33. doi: 10.18632/oncotarget.21406
67. Ma Y, Zhang Q, Kong P, Xiong J, Zhang X, Zhang C. Treatment Selection for Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia in the Era of Tyrosine Kinase Inhibitors. Chemotherapy (2019) 64:1–13. doi: 10.1159/000501061
68. Robak T, Robak E. Tyrosine kinase inhibitors as potential drugs for B-cell lymphoid malignancies and autoimmune disorders. Expert Opin Invest Drugs (2012) 21:921–47. doi: 10.1517/13543784.2012.685650
69. García-Gutiérrez V, Hernández-Boluda JC. Tyrosine Kinase Inhibitors Available for Chronic Myeloid Leukemia: Efficacy and Safety. Front Oncol (2019) 9:603:603. doi: 10.3389/fonc.2019.00603
70. Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med (2010) 362:2260–70. doi: 10.1056/NEJMoa1002315
71. Akhmetshina A, Venalis P, Dees C, Busch N, Zwerina J, Schett G, et al. Treatment with imatinib prevents fibrosis in different preclinical models of systemic sclerosis and induces regression of established fibrosis. Arthritis Rheum (2009) 60:219–24. doi: 10.1002/art.24186
72. Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol (2011) 6:425–56. doi: 10.1146/annurev-pathol-011110-130246
73. Niemann CU, Herman SEM, Maric I, Gomez-Rodriguez J, Biancotto A, Chang BY, et al. Disruption of in vivo Chronic Lymphocytic Leukemia Tumor-Microenvironment Interactions by Ibrutinib–Findings from an Investigator-Initiated Phase II Study. Clin Cancer Res (2016) 22:1572–82. doi: 10.1158/1078-0432.CCR-15-1965
74. Poe JC, Jia W, Di Paolo JA, Reyes NJ, Kim JY, Su H, et al. SYK inhibitor entospletinib prevents ocular and skin GVHD in mice. JCI Insight (2018) 3(19):e122430. doi: 10.1172/jci.insight.122430
75. Zeiser R, Burchert A, Lengerke C, Verbeek M, Maas-Bauer K, Metzelder SK, et al. Ruxolitinib in corticosteroid-refractory graft-versus-host disease after allogeneic stem cell transplantation: a multicenter survey. Leukemia (2015) 29:2062–8. doi: 10.1038/leu.2015.212
76. González Vicent M, Molina B, González de Pablo J, Castillo A, Díaz MÁ. Ruxolitinib treatment for steroid refractory acute and chronic graft vs host disease in children: Clinical and immunological results. Am J Hematol (2019) 94:319–26. doi: 10.1002/ajh.25376
77. Belle L, Fransolet G, Somja J, Binsfeld M, Delvenne P, Drion P, et al. Limited Impact of Imatinib in a Murine Model of Sclerodermatous Chronic Graft-versus-Host Disease. PloS One (2016) 11:e0167997. doi: 10.1371/journal.pone.0167997
78. Spriewald BM, Zerr P, Akhmetshina A, Mackensen A, Schett G, Distler JHW. Novel Anti-Fibrotic Therapies for Experimental Chronic Graft-Versus-Host Disease. Blood (2009) 114:4490–0. doi: 10.1182/blood.V114.22.4490.4490
79. Olivieri A, Locatelli F, Zecca M, Sanna A, Cimminiello M, Raimondi R, et al. Imatinib for refractory chronic graft-versus-host disease with fibrotic features. Blood (2009) 114:709–18. doi: 10.1182/blood-2009-02-204156
80. Baird K, Comis LE, Joe GO, Steinberg SM, Hakim FT, Rose JJ, et al. Imatinib mesylate for the treatment of steroid-refractory sclerotic-type cutaneous chronic graft-versus-host disease. Biol Blood Marrow Transplant (2015) 21:1083–90. doi: 10.1016/j.bbmt.2015.03.006
81. Olivieri A, Cimminiello M, Corradini P, Mordini N, Fedele R, Selleri C, et al. Long-term outcome and prospective validation of NIH response criteria in 39 patients receiving imatinib for steroid-refractory chronic GVHD. Blood (2013) 122:4111–8. doi: 10.1182/blood-2013-05-494278
82. Zerr P, Distler A, Palumbo-Zerr K, Tomcik M, Vollath S, Dees C, et al. Combined inhibition of c-Abl and PDGF receptors for prevention and treatment of murine sclerodermatous chronic graft-versus-host disease. Am J Pathol (2012) 181:1672–80. doi: 10.1016/j.ajpath.2012.07.017
83. Khanjarsim V, Karimi J, Khodadadi I, Mohammadalipour A, Goodarzi MT, Solgi G, et al. Ameliorative Effects of Nilotinib on CCl4 Induced Liver Fibrosis Via Attenuation of RAGE/HMGB1 Gene Expression and Oxidative Stress in Rat. Chonnam Med J (2017) 53:118–26. doi: 10.4068/cmj.2017.53.2.118
84. Chen GL, Carpenter PA, Broady R, Gregory TK, Johnston LJ, Storer BE, et al. Anti-Platelet-Derived Growth Factor Receptor Alpha Chain Antibodies Predict for Response to Nilotinib in Steroid-Refractory or -Dependent Chronic Graft-Versus-Host Disease. Biol Blood Marrow Transplant (2018) 24:373–80. doi: 10.1016/j.bbmt.2017.10.021
85. Al-Homsi AS, Goodyke A, Cole K, Muilenburg M, McLane M, Abdel-Mageed S, et al. Ixazomib suppresses human dendritic cell and modulates murine graft-versus-host disease in a schedule-dependent fashion. Exp Hematol (2017) 48:50–7. doi: 10.1016/j.exphem.2016.12.002
86. Choi J, Cooper ML, Staser K, Ashami K, Vij KR, Wang B, et al. Baricitinib-induced blockade of interferon gamma receptor and interleukin-6 receptor for the prevention and treatment of graft-versus-host disease. Leukemia (2018) 32:2483–94. doi: 10.1038/s41375-018-0123-z
87. Gupta R, Albabtain A, Arai S, Kwong B, Weng W-K. Hedgehog Pathway Inhibitor for Treatment of Steroid-Refractory Sclerodermatous Chronic Graft-Versus-Host Disease (GVHD). Blood (2018) 132:3409–9. doi: 10.1182/blood-2018-99-113915
88. Zerr P, Palumbo-Zerr K, Distler A, Tomcik M, Vollath S, Munoz LE, et al. Inhibition of hedgehog signaling for the treatment of murine sclerodermatous chronic graft-versus-host disease. Blood (2012) 120:2909–17. doi: 10.1182/blood-2012-01-403428
89. DeFilipp Z, Nazarian RM, El-Jawahri A, Li S, Brown J, Del Rio C, et al. Phase 1 study of the Hedgehog pathway inhibitor sonidegib for steroid-refractory chronic graft-versus-host disease. Blood Adv (2017) 1:1919–22. doi: 10.1182/bloodadvances.2017011239
90. Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med (2005) 353:1412–3. doi: 10.1056/NEJMc051946
91. Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev (2004) 15:255–73. doi: 10.1016/j.cytogfr.2004.03.006
92. Distler JHW, Jüngel A, Huber LC, Schulze-Horsel U, Zwerina J, Gay RE, et al. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum (2007) 56:311–22. doi: 10.1002/art.22314
93. Aono Y, Nishioka Y, Inayama M, Ugai M, Kishi J, Uehara H, et al. Imatinib as a novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med (2005) 171:1279–85. doi: 10.1164/rccm.200404-531OC
94. Westra IM, Oosterhuis D, Groothuis GMM, Olinga P. Precision-cut liver slices as a model for the early onset of liver fibrosis to test antifibrotic drugs. Toxicol Appl Pharmacol (2014) 274:328–38. doi: 10.1016/j.taap.2013.11.017
95. Qu K, Huang Z, Lin T, Liu S, Chang H, Yan Z, et al. New Insight into the Anti-liver Fibrosis Effect of Multitargeted Tyrosine Kinase Inhibitors: From Molecular Target to Clinical Trials. Front Pharmacol (2015) 6:300. doi: 10.3389/fphar.2015.00300
96. Galimberti S, Baldini C, Baratè C, Ricci F, Balducci S, Grassi S, et al. The CoV-2 outbreak: how hematologists could help to fight Covid-19. Pharmacol Res (2020) 157:104866. doi: 10.1016/j.phrs.2020.104866
97. Beham-Schmid C, Apfelbeck U, Sill H, Tsybrovsky O, Höfler G, Haas OA, et al. Treatment of chronic myelogenous leukemia with the tyrosine kinase inhibitor STI571 results in marked regression of bone marrow fibrosis. Blood (2002) 99:381–3. doi: 10.1182/blood.v99.1.381
98. Bueso-Ramos CE, Cortes J, Talpaz M, O’Brien S, Giles F, Rios MB, et al. Imatinib mesylate therapy reduces bone marrow fibrosis in patients with chronic myelogenous leukemia. Cancer (2004) 101:332–6. doi: 10.1002/cncr.20380
99. Chung L, Fiorentino DF, Benbarak MJ, Adler AS, Mariano MM, Paniagua RT, et al. Molecular framework for response to imatinib mesylate in systemic sclerosis. Arthritis Rheum (2009) 60:584–91. doi: 10.1002/art.24221
100. Magro L, Catteau B, Coiteux V, Bruno B, Jouet J-P, Yakoub-Agha I. Efficacy of imatinib mesylate in the treatment of refractory sclerodermatous chronic GVHD. Bone Marrow Transplant (2008) 42:757–60. doi: 10.1038/bmt.2008.252
101. Magro L, Mohty M, Catteau B, Coiteux V, Chevallier P, Terriou L, et al. Imatinib mesylate as salvage therapy for refractory sclerotic chronic graft-versus-host disease. Blood (2009) 114:719–22. doi: 10.1182/blood-2009-02-204750
102. Osumi T, Miharu M, Tanaka R, Du W, Takahashi T, Shimada H. Imatinib is effective for prevention and improvement of fibrotic fasciitis as a manifestation of chronic GVHD. Bone Marrow Transplant (2012) 47:139–40. doi: 10.1038/bmt.2011.10
103. Galeotti L, Ceccherini F, Domingo D, Laurino M, Polillo M, Di Paolo A, et al. Association of the hOCT1/ABCB1 genotype with efficacy and tolerability of imatinib in patients affected by chronic myeloid leukemia. Cancer Chemother Pharmacol (2017) 79:767–73. doi: 10.1007/s00280-017-3271-3
104. Harrach S, Barz V, Pap T, Pavenstädt H, Schlatter E, Edemir B, et al. Notch Signaling Activity Determines Uptake and Biological Effect of Imatinib in Systemic Sclerosis Dermal Fibroblasts. J Invest Dermatol (2019) 139:439–47. doi: 10.1016/j.jid.2018.08.021
105. Gomez-Puerta JA, Mócsai A. Tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Curr Top Med Chem (2013) 13:760–73. doi: 10.2174/15680266113139990094
106. Du FH, Mills EA, Mao-Draayer Y. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Auto Immun Highlights (2017) 8:12. doi: 10.1007/s13317-017-0100-y
107. Lundin J, Kimby E, Björkholm M, Broliden P-A, Celsing F, Hjalmar V, et al. Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood (2002) 100:768–73. doi: 10.1182/blood-2002-01-0159
108. Shapira MY, Abdul-Hai A, Resnick IB, Bitan M, Tsirigotis P, Aker M, et al. Alefacept treatment for refractory chronic extensive GVHD. Bone Marrow Transplant (2009) 43:339–43. doi: 10.1038/bmt.2008.324
109. Marinelli Busilacchi E, Costantini A, Viola N, Costantini B, Olivieri J, Butini L, et al. Immunomodulatory Effects of Tyrosine Kinase Inhibitor In Vitro and In Vivo Study. Biol Blood Marrow Transplant (2018) 24:267–75. doi: 10.1016/j.bbmt.2017.10.039
110. Olivieri A, Mancini G, Olivieri J, Marinelli Busilacchi E, Cimminiello M, Pascale SP, et al. Nilotinib in steroid-refractory cGVHD: prospective parallel evaluation of response, according to NIH criteria and exploratory response criteria (GITMO criteria). Bone Marrow Transplant (2020). doi: 10.1038/s41409-020-0902-9
111. King-Kallimanis BL, Wroblewski T, Kwitkowski V, De Claro RA, Gwise T, Bhatnagar V, et al. FDA review summary of patient-reported outcome results for ibrutinib in the treatment of chronic graft versus host disease. Qual Life Res (2020) 29:1903–11. doi: 10.1007/s11136-020-02448-y
112. Flynn R, Allen JL, Luznik L, MacDonald KP, Paz K, Alexander KA, et al. Targeting Syk-activated B cells in murine and human chronic graft-versus-host disease. Blood (2015) 125:4085–94. doi: 10.1182/blood-2014-08-595470
113. Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood (2014) 123:3832–42. doi: 10.1182/blood-2013-12-543736
114. Schoettler M, Subramaniam M, Margossian SP. Ruxolitinib and Steroid Refractory/Dependent Bronchiolitis Obliterans after Hematopoietic Cell Transplantation: A Steroid Sparing Agent That Also Resulted in Improved Lung Function in Children. Blood (2018) 132:3407–7. doi: 10.1182/blood-2018-99-114971
115. Hui L, Qi L, Guoyu H, Xuliang S, Meiao T. Ruxolitinib for treatment of steroid-refractory graft-versus-host disease in adults: a systematic review and meta-analysis. Expert Rev Hematol (2020) 13:565–75. doi: 10.1080/17474086.2020.1738214
116. You H, Xu D, Zhao J, Li J, Wang Q, Tian X, et al. JAK Inhibitors: Prospects in Connective Tissue Diseases. Clinic Rev Allerg Immunol (2020). doi: 10.1007/s12016-020-08786-6
117. Schroeder MA, Khoury HJ, Jagasia M, Ali H, Schiller GJ, Staser K, et al. A phase 1 trial of itacitinib, a selective JAK1 inhibitor, in patients with acute graft-versus-host disease. Blood Adv (2020) 4:1656–69. doi: 10.1182/bloodadvances.2019001043
118. Herrera AF, Kim HT, Bindra B, Jones KT, Alyea EP, Armand P, et al. A phase II study of bortezomib plus prednisone for initial therapy of chronic graft-versus-host disease. Biol Blood Marrow Transplant (2014) 20:1737–43. doi: 10.1016/j.bbmt.2014.06.040
119. Jain M, Budinger G, Jovanovic B, Dematte J, Duffey S, Mehta J. Bortezomib is safe in and stabilizes pulmonary function in patients with allo-HSCT-associated pulmonary CGVHD. Bone Marrow Transplant (2018) 53:1124–30. doi: 10.1038/s41409-018-0134-4
120. Al-Salama ZT, Garnock-Jones KP, Scott LJ. Ixazomib: A Review in Relapsed and/or Refractory Multiple Myeloma. Target Oncol (2017) 12:535–42. doi: 10.1007/s11523-017-0504-7
121. Pidala JA, Bhatt VR, Hamilton BK, Pusic I, Wood WA, Onstad L, et al. Ixazomib for Treatment of Refractory Chronic Graft Vs. Host Disease: A Chronic Gvhd Consortium Phase II Trial. Biol Blood Marrow Transplant (2019) 25:S28. doi: 10.1016/j.bbmt.2018.12.099
122. Chhabra S, Hari P, Visotcky A, Zhu F, Cantrall K, Abedin S, et al. A Phase I/2 Study of Ixazomib for Chronic Graft-Versus-Host Disease (cGVHD) Prophylaxis after Allogeneic Transplantation (alloHCT). Biol Blood Marrow Transplant (2019) 25:S224–5. doi: 10.1016/j.bbmt.2018.12.213
123. Nahas MR, Soiffer RJ, Kim HT, Alyea EP, Arnason J, Joyce R, et al. Phase 1 clinical trial evaluating abatacept in patients with steroid-refractory chronic graft-versus-host disease. Blood (2018) 131:2836–45. doi: 10.1182/blood-2017-05-780239
124. Taylor RP, Lindorfer MA. Immunotherapeutic mechanisms of anti-CD20 monoclonal antibodies. Curr Opin Immunol (2008) 20:444–9. doi: 10.1016/j.coi.2008.05.011
125. Bologna L, Gotti E, Da Roit F, Intermesoli T, Rambaldi A, Introna M, et al. Ofatumumab is more efficient than rituximab in lysing B chronic lymphocytic leukemia cells in whole blood and in combination with chemotherapy. J Immunol (2013) 190:231–9. doi: 10.4049/jimmunol.1202645
126. Kharfan-Dabaja MA, Mhaskar AR, Djulbegovic B, Cutler C, Mohty M, Kumar A. Efficacy of rituximab in the setting of steroid-refractory chronic graft-versus-host disease: a systematic review and meta-analysis. Biol Blood Marrow Transplant (2009) 15:1005–13. doi: 10.1016/j.bbmt.2009.04.003
127. Cutler C, Kim HT, Bindra B, Sarantopoulos S, Ho VT, Chen Y-B, et al. Rituximab prophylaxis prevents corticosteroid-requiring chronic GVHD after allogeneic peripheral blood stem cell transplantation: results of a phase 2 trial. Blood (2013) 122:1510–7. doi: 10.1182/blood-2013-04-495895
128. Cutler C, Miklos D, Kim HT, Treister N, Woo S-B, Bienfang D, et al. Rituximab for steroid-refractory chronic graft-versus-host disease. Blood (2006) 108:756–62. doi: 10.1182/blood-2006-01-0233
129. van der Wagen L, Te Boome L, Schiffler M, Nijhof I, Schoordijk M, van Dorp S, et al. Prospective evaluation of sequential treatment of sclerotic chronic graft versus host disease with rituximab and nilotinib. Bone Marrow Transplant (2018) 53:1255–62. doi: 10.1038/s41409-018-0158-9
130. Sarantopoulos S, Stevenson KE, Kim HT, Washel WS, Bhuiya NS, Cutler CS, et al. Recovery of B-cell homeostasis after rituximab in chronic graft-versus-host disease. Blood (2011) 117:2275–83. doi: 10.1182/blood-2010-10-307819
131. Ji S-M, Bao X-B, Lu J, Ma X, Tao T, Sun A-N, et al. Protective Effect of Rituximab in Chronic Graft-Versus-Host Disease Occurrence in Allogeneic Transplant patients with Epstein Barr Virus Viremia. Indian J Hematol Blood Transfus (2017) 33:525–33. doi: 10.1007/s12288-017-0783-2
132. Solomon SR, Sizemore CA, Ridgeway M, Zhang X, Brown S, Holland HK, et al. Safety and efficacy of rituximab-based first line treatment of chronic GVHD. Bone Marrow Transplant (2019) 54:1218–26. doi: 10.1038/s41409-018-0399-7
133. Soe ZN, Allsup D. The use of ofatumumab in the treatment of B-cell malignancies. Future Oncol (2017) 13:2611–28. doi: 10.2217/fon-2017-0275
134. Urian L, Renaudineau Y, Berthou C, Zdrenghea M, Petrov L, Tempescul A. Ofatumumab in a chronic lymphocytic leukemia patient with graft versus host disease and relapse after mini-allo BMT. J BUON (2015) 20:668–9.
135. Pidala J, Kim J, Betts BC, Alsina M, Ayala E, Fernandez HF, et al. Ofatumumab in combination with glucocorticoids for primary therapy of chronic graft-versus-host disease: phase I trial results. Biol Blood Marrow Transplant (2015) 21:1074–82. doi: 10.1016/j.bbmt.2015.03.014
136. Bacigalupo A, Deeg HJ, Caballero D, Gualandi F, Raiola AM, Varaldo R, et al. Treatment of Patients with Steroid Refractory Acute Graft Vs Host Disease (SR-GvHD): A Matched Paired Analysis of Anti-CD26 (Begelomab) Compared to Other Treatment. Blood (2016) 128:671–1. doi: 10.1182/blood.V128.22.671.671
137. Ohnuma K, Hatano R, Aune TM, Otsuka H, Iwata S, Dang NH, et al. Regulation of pulmonary graft-versus-host disease by IL-26+CD26+CD4 T lymphocytes. J Immunol (2015) 194:3697–712. doi: 10.4049/jimmunol.1402785
138. Yang J, Cheuk DKL, Ha SY, Chiang AKS, Lee TL, Ho MHK, et al. Infliximab for steroid refractory or dependent gastrointestinal acute graft-versus-host disease in children after allogeneic hematopoietic stem cell transplantation. Pediatr Transplant (2012) 16:771–8. doi: 10.1111/j.1399-3046.2012.01756.x
139. Rubio PM, Labrandero C, Riesco S, Plaza D, González B, Muñoz GM, et al. Successful response to infliximab of recurrent pericardial graft versus host disease in a pediatric patient. Bone Marrow Transplant (2013) 48:144–5. doi: 10.1038/bmt.2012.91
140. Busca A, Locatelli F, Marmont F, Ceretto C, Falda M. Recombinant human soluble tumor necrosis factor receptor fusion protein as treatment for steroid refractory graft-versus-host disease following allogeneic hematopoietic stem cell transplantation. Am J Hematol (2007) 82:45–52. doi: 10.1002/ajh.20752
141. Akgün K, Blankenburg J, Marggraf M, Haase R, Ziemssen T. Event-Driven Immunoprofiling Predicts Return of Disease Activity in Alemtuzumab-Treated Multiple Sclerosis. Front Immunol (2020) 11:56. doi: 10.3389/fimmu.2020.00056
142. Hu Y, Turner MJ, Shields J, Gale MS, Hutto E, Roberts BL, et al. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology (2009) 128:260–70. doi: 10.1111/j.1365-2567.2009.03115.x
143. Ruiz-Argüelles GJ, Gil-Beristain J, Magaña M, Ruiz-Delgado GJ. Alemtuzumab-induced resolution of refractory cutaneous chronic graft-versus-host disease. Biol Blood Marrow Transplant (2008) 14:7–9. doi: 10.1016/j.bbmt.2007.09.013
144. Green K, Pearce K, Sellar RS, Jardine L, Nicolson PLR, Nagra S, et al. Impact of Alemtuzumab Scheduling on Graft-versus-Host Disease after Unrelated Donor Fludarabine and Melphalan Allografts. Biol Blood Marrow Transplant (2017) 23:805–12. doi: 10.1016/j.bbmt.2017.02.007
145. Marsh JC, Gupta V, Lim Z, Ho AY, Ireland RM, Hayden J, et al. Alemtuzumab with fludarabine and cyclophosphamide reduces chronic graft-versus-host disease after allogeneic stem cell transplantation for acquired aplastic anemia. Blood (2011) 118:2351–7. doi: 10.1182/blood-2010-12-327536
146. Scott LJ. Brentuximab Vedotin: A Review in CD30-Positive Hodgkin Lymphoma. Drugs (2017) 77:435–45. doi: 10.1007/s40265-017-0705-5
147. Prince HM, Kim YH, Horwitz SM, Dummer R, Scarisbrick J, Quaglino P, et al. Brentuximab vedotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): an international, open-label, randomised, phase 3, multicentre trial. Lancet (2017) 390:555–66. doi: 10.1016/S0140-6736(17)31266-7
148. DeFilipp Z, Li S, Kempner ME, Brown J, Del Rio C, Valles B, et al. Phase I Trial of Brentuximab Vedotin for Steroid-Refractory Chronic Graft-versus-Host Disease after Allogeneic Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant (2018) 24:1836–40. doi: 10.1016/j.bbmt.2018.05.012
149. Alexander KA, Flynn R, Lineburg KE, Kuns RD, Teal BE, Olver SD, et al. CSF-1-dependant donor-derived macrophages mediate chronic graft-versus-host disease. J Clin Invest (2014) 124:4266–80. doi: 10.1172/JCI75935
150. Miller GT, Hochman PS, Meier W, Tizard R, Bixler SA, Rosa MD, et al. Specific interaction of lymphocyte function-associated antigen 3 with CD2 can inhibit T cell responses. J Exp Med (1993) 178:211–22. doi: 10.1084/jem.178.1.211
151. Majeau GR, Meier W, Jimmo B, Kioussis D, Hochman PS. Mechanism of lymphocyte function-associated molecule 3-Ig fusion proteins inhibition of T cell responses. Structure/function analysis in vitro and in human CD2 transgenic mice. J Immunol (1994) 152:2753–67.
152. Langley RG, Cherman AM, Gupta AK. Alefacept: an expert review concerning the treatment of psoriasis. Expert Opin Pharmacother (2005) 6:2327–33. doi: 10.1517/14656566.6.13.2327
153. Gartlan KH, Bommiasamy H, Paz K, Wilkinson AN, Owen M, Reichenbach DK, et al. A critical role for donor-derived IL-22 in cutaneous chronic GVHD. Am J Transplant (2018) 18:810–20. doi: 10.1111/ajt.14513
154. Forcade E, Paz K, Flynn R, Griesenauer B, Amet T, Li W, et al. An activated Th17-prone T cell subset involved in chronic graft-versus-host disease sensitive to pharmacological inhibition. JCI Insight (2017) 2:e92111. doi: 10.1172/jci.insight.92111
155. Curtis LM, Pavletic SZ. IL-2, the next best thing in chronic GVHD therapy? Blood (2016) 128:13–5. doi: 10.1182/blood-2016-05-711796
156. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
157. Chen W, Nyuydzefe MS, Weiss JM, Zhang J, Waksal SD, Zanin-Zhorov A. ROCK2, but not ROCK1 interacts with phosphorylated STAT3 and co-occupies TH17/TFH gene promoters in TH17-activated human T cells. Sci Rep (2018) 8:16636. doi: 10.1038/s41598-018-35109-9
158. Jagasia M, Salhotra A, Bachier CR, Essell J, Weisdorf DJ, Zoghi B, et al. KD025-208: A Phase 2a Study of KD025 for Patients with Chronic Graft Versus Host Disease (cGVHD) — Pharmacodynamics and Updated Results. Blood (2018) 132:602–2. doi: 10.1182/blood-2018-99-111896
159. Wu J, Gu J, Zhou S, Lu H, Lu Y, Lu L, et al. Anti-IL-22 Antibody Attenuates Acute Graft-versus-Host Disease via Increasing Foxp3+ T Cell through Modulation of CD11b+ Cell Function. J Immunol Res (2018) 2018:1605341. doi: 10.1155/2018/1605341
160. Zhu ZX, Fan LY, Wang Q. Simultaneous blockade of costimulatory signals CD28-CD80 and CD40-CD154 combined with monoclonal antibody against CD25 induced a stable chimerism and tolerance without graft-versus-host disease in rat. Eur Surg Res (2011) 46:109–17. doi: 10.1159/000323011
161. Beier UH, Wang L, Bhatti TR, Liu Y, Han R, Ge G, et al. Sirtuin-1 targeting promotes Foxp3+ T-regulatory cell function and prolongs allograft survival. Mol Cell Biol (2011) 31:1022–9. doi: 10.1128/MCB.01206-10
162. Akimova T, Xiao H, Liu Y, Bhatti TR, Jiao J, Eruslanov E, et al. Targeting sirtuin-1 alleviates experimental autoimmune colitis by induction of Foxp3+ T-regulatory cells. Mucosal Immunol (2014) 7:1209–20. doi: 10.1038/mi.2014.10
163. Lim HW, Kang SG, Ryu JK, Schilling B, Fei M, Lee IS, et al. SIRT1 deacetylates RORγt and enhances Th17 cell generation. J Exp Med (2015) 212:607–17. doi: 10.1084/jem.20132378
164. Zhang J, Lee S-M, Shannon S, Gao B, Chen W, Chen A, et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J Clin Invest (2009) 119:3048–58. doi: 10.1172/JCI38902
165. Daenthanasanmak A, Iamsawat S, Chakraborty P, Nguyen HD, Bastian D, Liu C, et al. Targeting Sirt-1 controls GVHD by inhibiting T-cell allo-response and promoting Treg stability in mice. Blood (2019) 133:266–79. doi: 10.1182/blood-2018-07-863233
166. Ramalho-Santos M, Melton DA, McMahon AP. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development (2000) 127:2763–72.
167. Jia Y, Wang Y, Xie J. The Hedgehog pathway: role in cell differentiation, polarity and proliferation. Arch Toxicol (2015) 89:179–91. doi: 10.1007/s00204-014-1433-1
168. Radojcic V, Paz K, Chung J, Du J, Perkey ET, Flynn R, et al. Notch signaling mediated by Delta-like ligands 1 and 4 controls the pathogenesis of chronic GVHD in mice. Blood (2018) 132:2188–200. doi: 10.1182/blood-2018-03-841155
169. Shindo T, Kim TK, Benjamin CL, Wieder ED, Levy RB, Komanduri KV. MEK inhibitors selectively suppress alloreactivity and graft-versus-host disease in a memory stage-dependent manner. Blood (2013) 121:4617–26. doi: 10.1182/blood-2012-12-476218
170. Itamura H, Shindo T, Tawara I, Kubota Y, Kariya R, Okada S, et al. The MEK inhibitor trametinib separates murine graft-versus-host disease from graft-versus-tumor effects. JCI Insight (2016) 1:e86331. doi: 10.1172/jci.insight.86331
171. Inamoto Y, Storer BE, Petersdorf EW, Nelson JL, Lee SJ, Carpenter PA, et al. Incidence, risk factors, and outcomes of sclerosis in patients with chronic graft-versus-host disease. Blood (2013) 121:5098–103. doi: 10.1182/blood-2012-10-464198
172. Čeović R, Desnica L, Pulanić D, Serventi Seiwerth R, Ilić I, Grce M, et al. High frequency of cutaneous manifestations including vitiligo and alopecia areata in a prospective cohort of patients with chronic graft-vs-host disease. Croat Med J (2016) 57:229–38. doi: 10.3325/cmj.2016.57.229
173. Herrero-Sánchez MC, Rodríguez-Serrano C, Almeida J, San Segundo L, Inogés S, Santos-Briz Á, et al. Targeting of PI3K/AKT/mTOR pathway to inhibit T cell activation and prevent graft-versus-host disease development. J Hematol Oncol (2016) 9:113. doi: 10.1186/s13045-016-0343-5
174. Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol (2013) 31:675–704. doi: 10.1146/annurev-immunol-032712-095946
175. Paz K, Flynn R, Du J, Tannheimer S, Johnson AJ, Dong S, et al. Targeting PI3Kδ function for amelioration of murine chronic graft-versus-host disease. Am J Transplant (2019) 19:1820–30. doi: 10.1111/ajt.15305
176. Zirlik K, Veelken H. Idelalisib. Recent Results Cancer Res (2018) 212:243–64. doi: 10.1007/978-3-319-91439-8_12
177. Dreger P, Koster L, Passweg JR, Collin M, Scheid C, Charbonnier A, et al. Safety and Efficacy of Idelalisib Treatment of Chronic Lymphocytic Leukemia (CLL) or Lymphoma Relapsing after Allogeneic Hematopoietic Cell Transplantation (alloHCT): A Survey By the EBMT Chronic Malignancies and Lymphoma Working Parties. Blood (2017) 130:3273–3.
178. Pouillon V, Maréchal Y, Frippiat C, Erneux C, Schurmans S. Inositol 1,4,5-trisphosphate 3-kinase B (Itpkb) controls survival, proliferation and cytokine production in mouse peripheral T cells. Adv Biol Regul (2013) 53:39–50. doi: 10.1016/j.jbior.2012.08.001
Keywords: chronic graft-versus-host disease, tyrosine kinase inhibitors, immunotherapy, Janus kinase 1/2, hematopoietic stem cell transplantation
Citation: Saidu NEB, Bonini C, Dickinson A, Grce M, Inngjerdingen M, Koehl U, Toubert A, Zeiser R and Galimberti S (2020) New Approaches for the Treatment of Chronic Graft-Versus-Host Disease: Current Status and Future Directions. Front. Immunol. 11:578314. doi: 10.3389/fimmu.2020.578314
Received: 30 June 2020; Accepted: 18 September 2020;
Published: 09 October 2020.
Edited by:
Aurore Saudemont, GlaxoSmithKline, United KingdomReviewed by:
Sergio Querol, Banc de Sang i Teixits, SpainCopyright © 2020 Saidu, Bonini, Dickinson, Grce, Inngjerdingen, Koehl, Toubert, Zeiser and Galimberti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathaniel Edward Bennett Saidu, bi5lLmIuc2FpZHVAb3VzLXJlc2VhcmNoLm5v; Sara Galimberti, c2FyYS5nYWxpbWJlcnRpQG1lZC51bmlwaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.