 Chang Yang
Chang Yang Bai-Rong Xia
Bai-Rong Xia Zhao-Cong Zhang1
Zhao-Cong Zhang1 Ge Lou
Ge Lou Wei-Lin Jin
Wei-Lin Jin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 October 2020
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.577869
Ovarian cancer is the most lethal gynecologic malignancy. Surgery and chemotherapy are the primary treatments for ovarian cancer; however, patients often succumb to recurrence with chemotherapeutic resistance within several years after the initial treatment. In the past two decades, immunotherapy has rapidly developed, and has revolutionized the treatment of various types of cancer. Despite the fact that immunotherapy response rates among ovarian cancer patients remain modest, treatment with immune checkpoint inhibitors (ICIs), chimeric antigen receptor (CAR)- and TCR-engineered T cells is rapidly developing. Therapeutic efficiency could be improved significantly if immunotherapy is included as an adjuvant therapy, in combination with chemotherapy, radiation therapy, and the use of anti-angiogenesis drugs, and poly ADP ribose polymerase inhibitors (PARPi). Newly developed technologies that identify therapeutic targets, predict treatment efficacy, rapidly screen potential immunotherapy drugs, provide neoadjuvant immunotherapy, and utilize nanomedicine technology provide new opportunities for the treatment of ovarian cancer, and have the potential to prolong patient survival. However, important issues that may hinder the efficacy of such approaches, including hyperprogressive disease (HPD), immunotherapy-resistance, and toxicity of the treatments, including neurotoxicity, must be taken into account and addressed for these therapies to be effective.
According to the World Health Organization, it is estimated that the global incidence of ovarian cancer will be 308,069 patients and the total mortality from ovarian cancer will be 193,811 patients, in 2020 (1). The standard treatment for ovarian cancer includes surgery followed by platinum-based chemotherapy. Currently, the five-year survival rate of ovarian cancer is approximately 47%, predominately due to relapse and chemoresistance (2). In recent years, poly-ADP-ribose polymerase (PARP) inhibitors (PARPi) have seemed to be a promising option to treat cases that have BRCA mutations and BRCA wild-type EOC tumors (3). Despite an increase in the number of clinical trials, and a growing number of approved drugs, the therapeutic effects of PARPi are limited, as treatment only extends survival by a few months, and does not provide any long-term benefit (4). Resistance to PARPi in BRCA1/2-mutated tumors suggests that this therapy may not be as effective as previously thought (5–7). The most frequently reported mechanism of resistance is (partial) restoration of homology directed DNA repair. Understanding the mechanisms underlying this resistance and combining therapies may be useful to counteract PARPi resistance. A combination of Bevacizumab and a platinum/taxane-based chemotherapy has been recommended as a first-line therapy for ovarian cancer. Results suggest that progression free survival is prolonged by 3.5-months, whereas no significant differences in overall survival were observed. Thus, a more effective treatment regime is needed to prolong the overall survival of patients.
In the past two decades, immunotherapy has developed rapidly, revolutionizing the treatment of various types of cancer. Recently, immune checkpoint inhibitors, including CTLA-4 and programmed cell death protein 1 (PD-1)/programmed cell death 1 ligand 1 (PD-L1) inhibitors, which reverse the signals from the immunosuppressive tumor microenvironment (TME), are being investigated as potential treatment modalities (8, 9). The application of oncolytic viruses, cancer vaccines, and adoptive cell therapies is advancing rapidly (10–12). Immunotherapies have also attracted significant attention in ovarian cancer therapy (Figure 1).
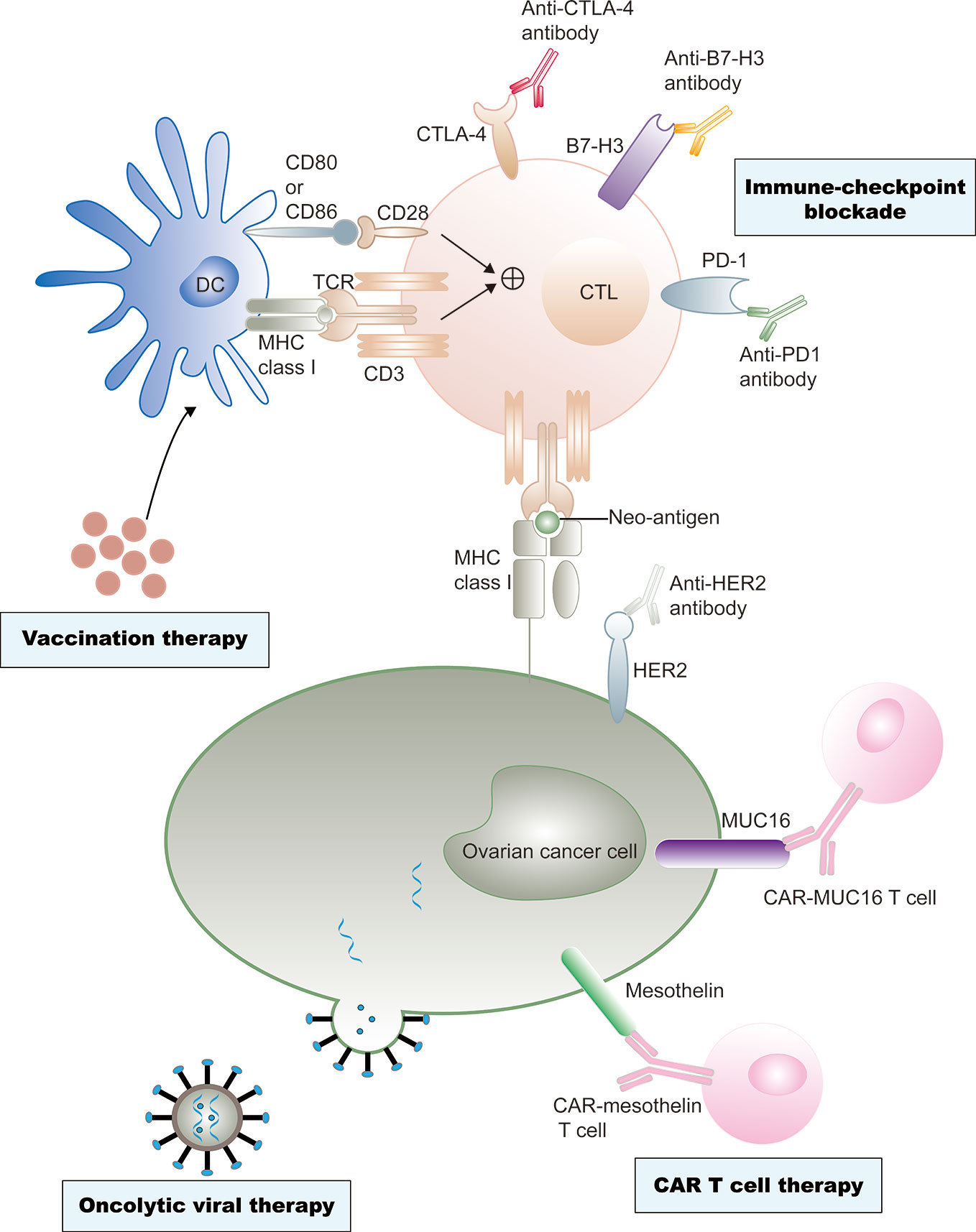
Figure 1 Current immunotherapy treatments of ovarian cancer. Ovarian cancer vaccine therapy is based on dendritic cell (DC)-mediated presentation of neo-antigens derived from malignant ovarian cancer cells to T cells through MHC class I–T cell receptors (TCRs) and a co-stimulation signal of CD80 and/or CD86-CD28 interactions. CTLs are subsequently activated to destroy tumor cells. However, tumor cells often escape immune destruction by CTLs through upregulation of immune checkpoint ligands, such as programmed cell death 1 ligand 1 (PD- L1), that can bind the programmed cell death protein 1 (PD-1) on the CTLs. Immune checkpoint inhibitors could effectively prevent this effect. Antibody-mediated blockade of cytotoxic T lymphocyte protein 4 (CTL A-4), an inhibitory immune- checkpoint molecule that binds CD80 and CD86 and prevents their interaction with CD28, can promote T cell priming by DCs. Similarly, anti–B7-H3 antibody-mediated blockade could neutralize T cell exhaustion in patients with ovarian cancer. Neo-antigens, including human epidermal growth factor receptor 2 (Her2), cancer antigen 125 (MUC16), and mesothelin, are also presented on tumor cell surfaces independent of MHC class I receptors, and these neo-antigens are specific targets of chimeric antigen receptor (CAR) T cell therapies. Genetic engineering is also applied to produce viruses that selectively infect or replicate in tumor cells, and finally destruct tumor cells. Cell destruction can also promote immunogenic tumor cell death, which can active antigen presentation and an adaptive antitumor immune response.
Epithelial ovarian cancers (EOCs) have been considered “immunogenic tumors”, non-spontaneous antitumor immune responses could be detected in the tumors, peripheral blood, and ascites of patients with EOCs (13). Immune cells in the tumors and ascites, including T and B lymphocytes, regulatory T cells (Tregs), natural killer (NK) cells, tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs) play key roles in ovarian cancer. Fibroblasts and adipocytes in the tumor microenvironment may also affect the efficacy of immunotherapeutic and chemotherapeutic drugs (14).
In this review, we summarize recent advances in immunotherapy for ovarian cancer, including cancer vaccines, adoptive cell therapy (ACT), immune checkpoint inhibitors (ICIs), oncolytic viruses, and the immunosuppressive tumor microenvironment. We also introduce diverse treatment modalities that can be used in combination with immunotherapy. In addition, we briefly discuss the future directions of these therapies.
The immunosuppressive immune cells include myeloid-derived suppressor cells (MDSCs), Tregs, TAMs, cancer-associated fibroblasts (CAFs), and adipocytes (Figure 2). In order to develop effective immunotherapies against ovarian cancer, immunosuppressive networks within primary ovarian tumors, ascites, metastatic tumors, and related mechanisms must be considered.
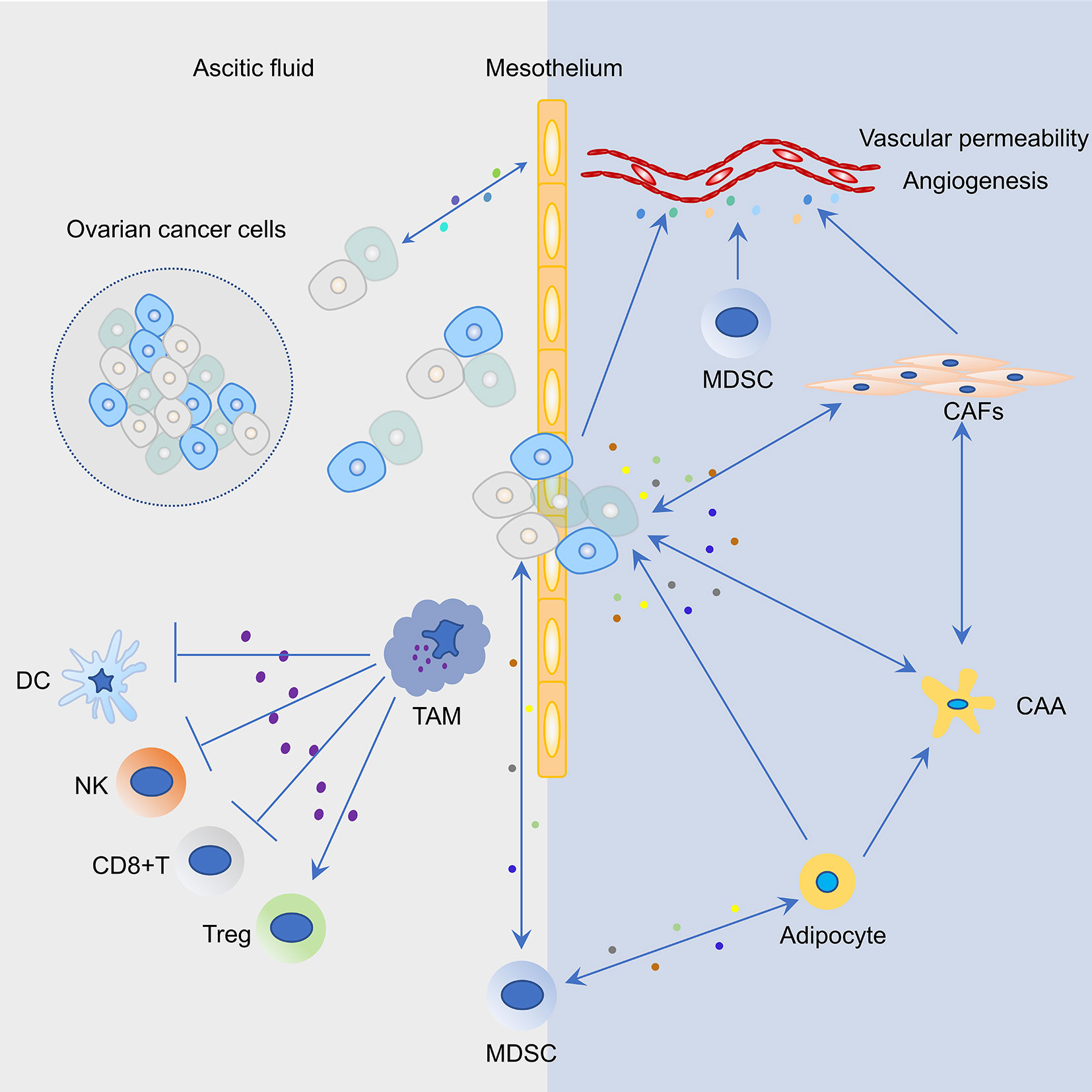
Figure 2 Representative cell types and their interactions in the tumor microenvironment (TME) of ovarian cancer. Cancer cells and mesothelium cells secrete pro-inflammatory cytokines and chemokines, including macrophage chemoattractant protein-1 (MCP1/CCL2), which recruits tumor associated macrophages (TAM) to the peritoneum. Increased infiltration of TAMs in the peritoneal TME not only promotes ovarian cancer cell invasion but also induces an immunosuppressive environment that suppresses the function of T cells, dendritic cells (DCs), and natural killer (NK) cells and activates the function of regulatory T cells (Tregs). Mesothelial cells are the first barrier to this process; however, the bidirectional cross-talk between cancer and mesothelial cells results in mesothelial clearance and invasion of the sub-mesothelial layers. Myeloid-derived suppressor cells (MDSCs) are attracted to the tumor site in response to growth factors and inflammatory cytokines/chemokines that are secreted by ovarian cancer cells. MDSCs could suppress T cell function, maintain the ovarian cancer stem cell pool, and interact with adipocytes. In the ovarian TME, blood vessel structure is modulated by a plethora of factors that are secreted by ovarian cancer cells, MDSCs, and cancer associated fibroblasts (CAFs). Molecular cross-talk between cancer cells and CAFs in the ovarian TME produce a pro-inflammatory TME and promote tumor progression. The bidirectional interaction between omental adipocytes and cancer cells promote dedifferentiation and reprogramming of adipocytes into cancer-associated adipocytes (CAAs). In turn, cancer cells absorbing fat rapidly promote tumor proliferation.
MDSCs consist of multiple immature myeloid cells that are elevated in pathological situations, and which weaken the efficacy of the T cell response (15, 16). In 2017, Horikawa et al. demonstrated that the number of MDSCs is increased by VEGF expression in ovarian cancer, which suppresses local immunity (17). Taki et al. have identified that Snail, a major transcription factor, could increase the expression of CXCR2 ligands and recruit MDSCs, thus resulting in poor prognoses (18). In 2019, a study using mouse EOC cells identified MDSCs as a driving factor for immunosuppression in a ID8-fLuc ovarian cancer mouse model (19).
Tregs directly or indirectly inhibit antitumoral responses, suggesting that Tregs are a primary means of tumor immune escape (20). As early as 2005, Sato et al. concluded that the number of intraepithelial CD8+ TILs and a high ratio of CD8+/Treg are associated with a positive prognosis in EOC (21). TGFβ, IL-10, and tumor-derived exosomes, which contain IL-10, TGFβ, IDO1, and PDL1/L2, are secreted from tumor cells and potentiate the differentiation of inhibitory Tregs, which is characterized by increased expression of FoxP3 and CTLA4 (22–24).
TAMs that infiltrate tumors and differentiate into macrophages are derived from the bone marrow. TAMs consist of both M1 and M2 type macrophages (25, 26). In EOCs, M2 type macrophages in the abdominal cavity and ascites are cancer promoting. Zhang et al. demonstrated that patients with upregulated overall or intra-islet M1/M2 TAMs ratios have an advanced five-year survival rate (27). Yin et al. identified that TAMs play a prominent role in promoting early peritoneal cavity metastasis of ovarian cancer via EGF secretion (28).
However, the regulation between cancer cells and TAMs is bidirectional. Ovarian cancer cells play a critical role in promoting M2 polarization of TAMs, and TAMs can result in cisplatin resistance via the enhancement of cellular stemness in cisplatin-sensitive cells (29).
Cancer-associated fibroblasts (CAFs) are the primary type of stromal cell, and express α-smooth muscle actin and fibroblast activation protein in ovarian cancer (30). CAFs are enhanced in ovarian cancer tumor cells, and therefore secrete high levels of hepatocyte growth factor, which facilitates tumor cell proliferation, chemoresistance, invasion, and migration though activation of the cMet/PI3K/Akt pathways and glucose-regulated protein 78 (31). CAFs produce pro-inflammatory cytokines, such as COX-2, CXCL1, CCL5, CXCL11, and IL-6, which increase tumor cell proliferation and EMT (32–36).
Many studies have reported that obesity is associated with increased incidence and poor prognosis of ovarian cancer. Adipocytes in the omentum secret cytokines and chemokines, such as IL-6, IL-8, MCP-1, tissue inhibitor of metalloproteinases-1, and adiponectin, to promote transcoelomic metastasis and tumor progression (37, 38). Lipid metabolism has also been shown to shape the tumor microenvironment (TME), which could affect the treatment efficacy of immunotherapies. Strategies based on this knowledge have the potential to increase immunotherapeutic response and patient survival (39).
Since the production of immunosuppressive cytokines and chemokines is likely to be shaped by the intrinsic biologic properties of the tumor, therapeutic combinations that can minimize toxicity and maximize benefits, to eliminate EOC, are needed.
Due to the immunosuppressive microenvironment of ovarian cancer, tumor-specific T cells generated by immunotherapy could not destroy tumors in EOC patients. Communication between tumor cells and other cells in the TME occurs via contact-dependent and -independent mechanisms. Tumor cells are in direct contact with cells in the extracellular matrix for contact-dependent mechanisms, whereas communication is achieved via soluble molecules such as cytokines, lipid mediators, and growth factors in contact-independent mechanisms (40). Ovarian tumors have been reported to recruit Tregs and myeloid-derived suppressor cells, which can inhibit the activation and efficacy of CD8+ effector cells (41, 42). Moreover, the stromal cells in the TME, such as MDSCs, Tregs, TAMs, CAFs, and adipocytes could be “educated” to facilitate and sustain cancer cells (43). Soluble factors in the TME function as a limiting factor for the maturation of local antigen-presenting cells, rendering them unable to generate costimulatory signals to effector cells, and consequently inducing the failure of T cell efficacy.
To conclude, the mechanisms of immunosuppressive networks of ovarian cancer include inhibition of CD8+ effector cells by Tregs, suppression of receptor PD-1 engaging by the ligand PD-L1; myeloid-derived suppressor cells and inhibitory cytokines (44).The immunosuppressive network is a significant obstacle in immunotherapy, which must be overcome to ensure the implementation of effective immunotherapeutic strategies.
Most types of ovarian cancer immunotherapy treatment modalities are currently being tested in clinical trials (Table 1). Adaptive immunity in ovarian cancer is rapidly expanding to enhance dendritic cell (DC)-mediated presentation of ovarian cancer, predominantly by vaccination (45, 46) (Figure 1). Cytotoxic T lymphocytes (CTLs) are activated after recognizing tumor-associated antigens (TAAs), and particularly the neoantigens (47). Ovarian cancer tumor cells often evade destruction by CTLs through inhibitory signals in the tumor (48). Mucin 16, α-folate receptor, and mesothelin are being investigated as specific targets of genetic modification of chimeric antigen receptor T (CAR-T) cell therapies for ovarian cancer (49). Oncolytic viral therapy that utilizes genetic engineering is also being used to create viruses that selectively infect tumor cells and lead to tumor cell lysis (11). In this section, we have discussed the evolution of immunotherapies for ovarian cancer.
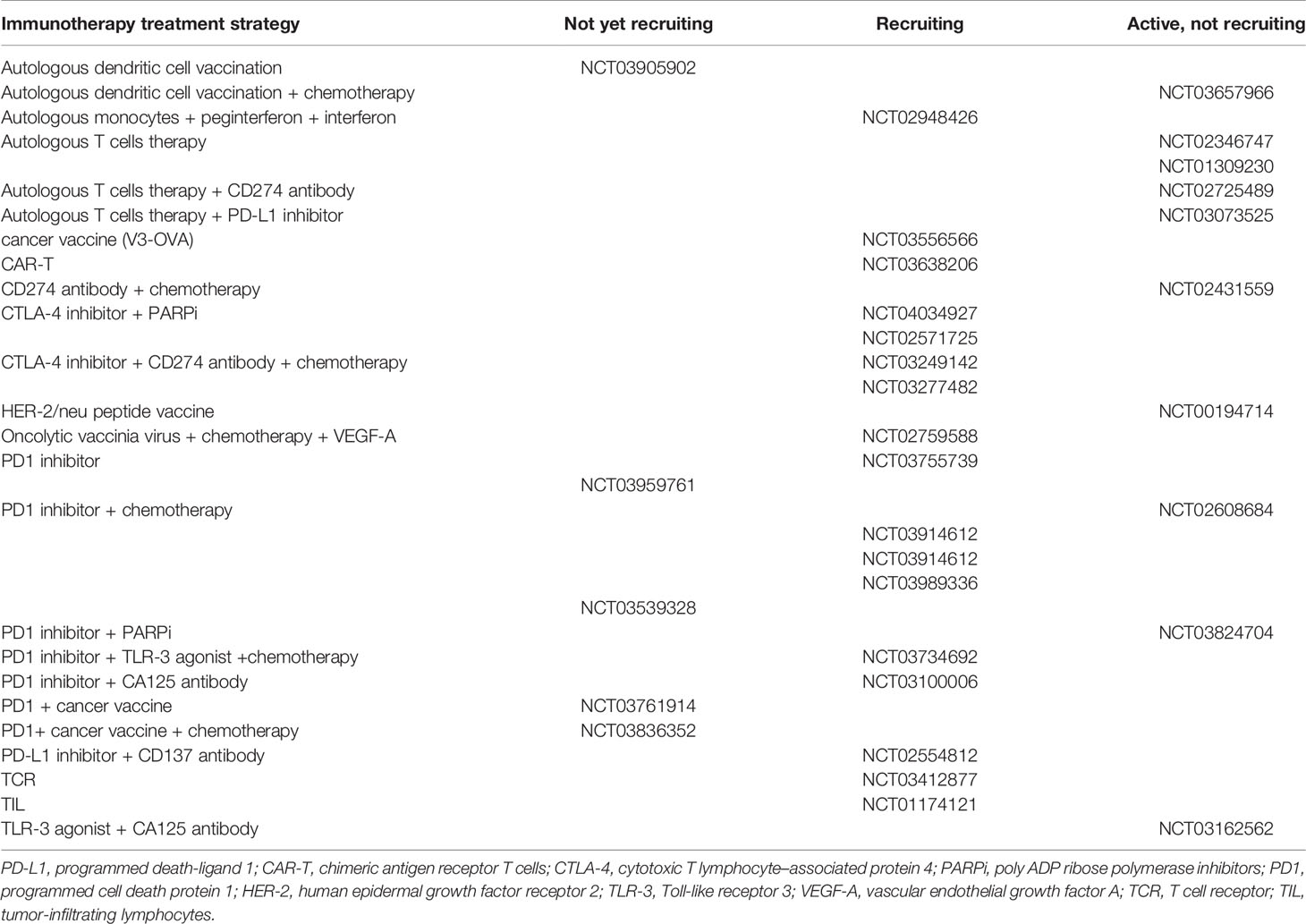
Table 1 Clinical trials of immunotherapy for ovarian cancer.
Successful immunotherapy for ovarian cancer relies on the stimulation of antigen-presenting cells, attenuating the immunosuppressive microenvironment, and bolstering effector T cell activity. The T cell–mediated immune response is regulated by inhibitory and stimulatory signals. Immune checkpoint receptors negatively regulate T cell activation and are critical to prevent over-activation. However, various types of tumors express immune checkpoints, leading to immune escape. Thus, immune-checkpoint blockade inhibitors play an important role in immunotherapy. To date, the most promising immune checkpoint inhibitors have been antibodies that block cytotoxic T lymphocyte antigen 4 (CTLA4) and PD1, which are expressed on T cells, or PD L1, which is expressed on certain immune cell subsets and is aberrantly expressed on tumor cells. The Food and Drug Administration–approved immune checkpoint inhibitors consist of a CTLA-4 antibody (Ipilimumab), PD-1 antibodies (Pembrolizumab and Nivolumab), and PD-L1 antibodies (Avelumab, Atezolizumab, and Durvalumab) (50). These ICIs have shown significant clinical benefits in multiple types of tumors, particularly melanoma (51). Little success has been found in the clinical use of checkpoint inhibitors in ovarian cancer, whose single-agent objective response rates in clinical trials are approximately 6–15% (52, 53).
CTLA-4 is a receptor on T cells that has the same set of ligands as costimulatory receptor CD28, but with a higher binding affinity, leading to competitive inhibition (54). Anti-CTLA-4 could block the binding of CTLA-4 and its ligands and prevent further inhibitory signal transduction, resulting in increased CD28-mediated co-stimulation.
The most commonly investigated immune checkpoint target in ovarian cancer is the PD-1/PD-L1 pathway. PD-1 is also expressed on T cells, and regulates the activation of effector T cells, mainly in the effector phase in peripheral tissue and the tumor microenvironment (55). After binding with its ligands, PD-L1 or PD-L2, PD-1 is phosphorylated and recruits the inhibitory phosphatase that can rapidly dephosphorylate CD28 and inhibit the co-stimulatory signaling pathway. Thus, an antibody targeting PD-1 could counteract its inhibitory effects. The efficacy of dual inhibition of PD-L1 and PD-L2 is more effective than anti–PD-1 or PD-L1 alone in various types of cancers that express both PD-L2 and PD-L1, including ovarian cancer (56). Exosomes expressing PD-L1 can repress anti-tumor immune responses (57). Developing a better understanding the mechanisms of exosomal PD-L1 in immune oncology is important, because inhibition of exosome production may be exploited as a potential new therapy.
Other potential targets of immune checkpoint blockage are listed in Table 2. B7-H3 is an immunosuppressive molecule that is expressed on tumor cells, but not host cells. The antitumor effects of B7-H3 and PD-1 blockade have been studied. Findings suggest that B7-H3, rather than PD-1 blockade, extends the median survival time of ID8 tumor-bearing mice with ovarian cancer. Collectively, B7-H3 may be utilized as a novel target in ovarian cancer patients that are not responsive to PD-L1/PD-1 inhibition (58). B and T lymphocyte attenuator is a novel inhibitory receptor, whose structure and function are similar to those of CTLA-4 and PD-1 (63). LAG-3 (CD223) is a cell surface molecule that is expressed on several immune cells, including activated T cells. LAG-3 has been shown to be involved in CD8+ T cell exhaustion (64). TIM-3 is a co-inhibitory receptor on IFN-γ-producing T cells, FoxP3+ Treg cells, and innate immune cells (macrophages and DCs). By binding with its ligands, TIM3 can suppress the immune response of these cells (65). The V-domain Ig suppressor of T cell activation, which is expressed on tumor cells, has been identified as a novel inhibitory immune-checkpoint protein in ovarian cancer (62). These novel immunosuppressive factors within the tumor microenvironment are promising targets for immunotherapy.
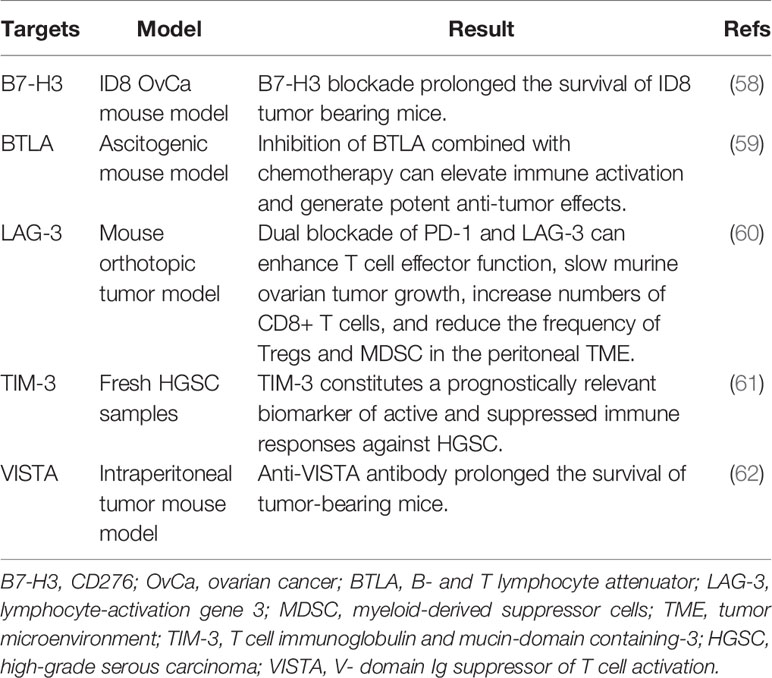
Table 2 Selective targets that block immune checkpoints in ovarian cancer.
Currently, in order to test the clinical response to ICI therapy, there is a need to identify patients who would be expected to benefit from this immunotherapy (66, 67). 5-methylcytosine and 5-hydroxymethylcytosine may act as prognostic and predictive biomarkers of ICI-sensitive cancers (68). Thus, exploration of multimodal predictive models of target assessment, tumor-intrinsic features, independent host features, and the tumor microenvironment are needed to optimize the treatment of ICIs in the future (66).
Adoptive cell therapy (ACT) is a promising strategy for the treatment of cancer, that utilizes the cells of the immune system to eliminate cancer. Currently, ACT can be classified into adoptive T cell therapy and other immune cell types, such as NK cells (69), cytokine-induced killer (CIK) cells (70), and macrophages (71). In this review, we primarily discuss adoptive T cell therapy. Adoptive T cell therapy that infuses the ex vivo–expanded tumor-specific T cells has shown promise as an immunotherapy treatment for cancer patients (72). Adoptive therapy with tumor-specific T cells consists of two major forms: genetic modification of T cells for expression of a specific T cell receptor (TCR) and CAR (73, 74). Both approaches have been tested in preclinical ovarian cancer models and have also been evaluated in early-phase clinical trials.
Therapy with autologous T cells that have been genetically modified to express a cloned TCR directed toward a specific antigen has considerable potential for clinical application in cancer patients. Similar to endogenous T cells, these engineered cells are primed by recognition of the antigen in the context of the major histocompatibility complex (MHC), and can be negatively regulated by immunosuppressive signals in the TME.
TCR-engineered T cells have induced significant objective responses in the majority of treated patients. In ovarian cancer, two clinical trials to evaluate the efficacy of TILs (NCT02482090 and NCT01883297) are ongoing. Recently, Matsuda et al. have built a rapid and efficient process for the production of neoantigen-specific TCR-engineered T cells exploiting blood from an HLA-matched healthy donor. They successfully identified three neoantigen-specific TCRαβ pairs from 14 estimated neoantigen candidates, and also revealed the importance of careful validation for the specificity of TCRs against neoantigens (75).
CAR-T cell therapy is another strategy for antitumor treatment that provides recognition specificity to engineered T cells. The CAR molecule consists of an antigen-binding domain and a cytoplasmic signaling motif. The targets of CAR-T therapy in ovarian cancer are listed in Table 3. Phase I/II clinical trials are currently in progress in order to investigate CAR-T cells targeting MUC16 (NCT02498912), mesothelin (NCT01583686), and NY-ESO-1 (NCT01567891 and NCT02457650) for ovarian cancer. Recently, Garcia et al. demonstrated that T cells expressing the Müllerian inhibiting substance type 2 receptor (MISIIR)–specific CAR exhibited significant antigen specification reactivity and eliminated MISIIR overexpression in tumors in vivo. This group also conducted in vitro experiments and confirmed that without cytotoxicity to normal primary human cells, MISIIR CAR-T recognized various types of human ovarian and endometrial cancer cell lines (76).
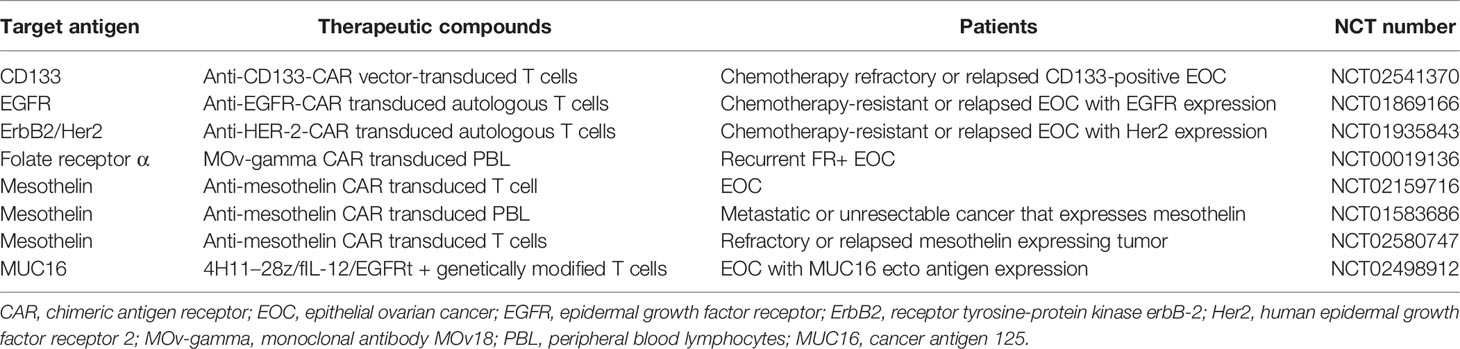
Table 3 Clinical trials of CAR-T therapy in epithelial ovarian cancer.
Despite their promising results in hematological malignancies, application of CAR-T cells in solid tumors has been limited by physical barriers and tumor heterogeneity (77, 78). Compared with liquid tumors, physical barriers between CAR-T cells and ovarian cancer cells inhibit solid tumor accessibility by CAR-T cells. To address this issue, local administration of CAR-T cells will improve T cell therapies (79).
With the development of synthetic biology techniques, more robust generation of TCR or CAR modified T cells is possible. Next generation TCR or CAR-modified T cells may increase the number of decoy receptors for inhibitory molecules (80, 81) or and it is possible that further clinical research and application of next generation approaches can be used to treat ovarian cancer.
Cancer vaccines are used to strengthen tumor-associated antigen (TAA) presentation by APCs and spark TAA-specific CD8+ T cells to kill tumor cells. Vaccine-induced immune responses provide long-term immunologic memory. Cancer vaccines may be classified into cell-based vaccines, peptide/protein vaccines, epigenetic vaccines, and genetic vaccines (82). Among these, peptide/protein vaccines and cell-based vaccines are usually based on well-defined TAAs (51). TAAs in ovarian cancer can be classified into two categories: shared common TAAs and individually mutated neo-antigens. The shared antigens contain three types of antigens: overexpressed antigens, such as mesothelin, tissue-specific TAAs, and TAAs whose expression is generally restricted to male germline cells, such as NY-ESO-1 (83). The TP53 gene, which encodes the tumor suppressor p53, is commonly mutated in ovarian cancer. Malekzadeh et al. systematically studied intertumoral T cell responses to the eight most commonly mutated positions of P53. The findings suggest that TCRs were identified in mutated TP53 and may be candidates with which to evaluate targeted immune cancer therapies (84).
The immunogenicity of ovarian cancer is still ambiguous. Schumacher et al. demonstrated that ovarian tumors have a very heterogeneous and comparatively low mutational load, thus making immune recognition of neo-antigens uncertain (85). In the future, combinations of multiple immunotherapies will be necessary to effectively provoke immune responses and take advantage of the immunogenicity of ovarian cancer (83).
The use of oncolytic virus anticancer therapies has been considered as an independent treatment strategy that is separate from immunotherapy. Viruses can replicate in cancer cells, leading to subsequent destruction of the cells. However, oncolytic viral infections could generate antitumor immune responses. Viruses can stimulate the immune system through pathogen-associated molecular patterns and pattern recognition receptors, and viruses often activate macrophages through receptors (86).
Santos et al. established patient-derived ex vivo tumor cultures in order to investigate the possibility of using TNFα and IL-2 encoding oncolytic adenovirus to restore and enhance the tumor reactivity of TILs in the context of immunosuppressive human ovarian cancer (87). Oncolytic adenovirus Ad5/3-E2FD24-hTNFa-IRES-hIL2 was able to rewire the ovarian tumor microenvironment to heighten antitumor TIL reactivity.
In the future, it will be important to combine oncolytic viral therapies with other immunotherapy strategies to establish prolonged anticancer immune responses initiated by viral infection.
In 2010, Weinberg summarized ten hallmarks of cancer, including proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis, reprogramming energy metabolism, evading immune destruction, promoting genome instability and mutation, and activating tumor-promoting inflammation (88, 89). Almost all cancers have acquired similar capabilities during tumor progress via different mechanistic strategies. The hallmarks of cancers, especially deregulating cellular metabolism and evading immunological destruction, are an increasingly common problem faced by immunotherapies; therefore, the combination of immunotherapy with chemotherapy, radiation therapy, anti-angiogenesis drugs, and PARP inhibitors is essential.
Chemotherapy is the cornerstone of the treatment of EOC, and can lead to the destruction of cancer cells and the release of immunogenic molecules (90). Platinum-based chemotherapy can interfere with the STAT6-mediated immunosuppression in the TME via downregulation of the expression of PD-L2 on human DCs and tumor cells and increasing tumor T cell recognition (91, 92). Paclitaxel treatment may also be beneficial for immunotherapy in ovarian cancer, as it has been shown to elevate the levels of CD8+ T cell infiltration in an ovarian cancer mouse model through increasing expression of both MHC-I and PD-L1 (93). Mkrtichyan et al. have claimed that a combination of cyclophosphamide and immune checkpoint inhibitors, such as anti–PD-1, could collaboratively decrease Treg infiltration and stimulate the yield of CD8+ TILs (94). Combining gemcitabine chemotherapy drugs with a CTLA-4 blockade could induce a potent CD4+ and CD8+ T cell–dependent antitumor immune response (95). Chemotherapy in combination with immunotherapy may be mutually beneficial, as chemotherapy can generate antigenic molecules as an in situ vaccine, and immunotherapy can counterbalance the acute immunosuppression induced by chemotherapy (96).
EOC is sensitive to radiation therapy, but abdominal radiotherapy-induced side effects, such as intestinal obstruction and ureteral stenosis or fistulae can occur, suggesting the need for close attention to dosimetry (97). Radiation therapy can enhance immunotherapy efficacy via inducing in-situ vaccination and immune reprogramming (98). Radiation therapy-induced double-strand break DNA fragments are recognized by cyclic GMP-AMP synthase, a pattern-recognition receptor that promotes the accumulation of type I interferon (99). Interferon is essential to DC recruitment and cross-activation of T effector cells, which is necessary in order to convert a tumor into an in-situ vaccine (100). Besides providing chemoattractants to attract T cells, radiotherapy also accelerates their homing into the tumor bed by increasing the expression of adhesion molecules, such as ICAM-1, on the tumor vasculature endothelium, promoting leucocyte endothelial transmigration (101). Tumor cells damaged by radiation also release damage-associated molecular pattern molecules, including high-mobility group box 1, which stimulate APCs. High-mobility group box 1 is a chromatin nuclear protein that is released mainly after necrotic cell death, and serves as a toll-like receptor 4 ligand on APCs (102). A preclinical study demonstrated that mice treated with radiotherapy in combination with CTLA-4 had increased survival compared to those treated only with radiotherapy (103). In conclusion, immunotherapy efficacy was enhanced by radiotherapy, as radiotherapy can function as an in-situ vaccination and accelerate T cell arrival to the tumor site.
Bevacizumab has been shown to elevate the antitumor efficacy of cisplatin in a xenograft ovarian cancer model (104), and has been used as a first-line treatment in advanced EOC, in combination with carboplatin and paclitaxel, according to positive phase III data (105). Previous studies have shown that VEGF increases the levels of PD-1 expression in intratumoral CD8+ T cells, which could be impaired by anti-VEGF drugs (106). The combination of anti–PD-1 and VEGF-A blockade strengthened the collaborative antitumor effect in patients with high expression of VEGF, as compared with those treated with a monotherapy (107). VEGF antibodies, in combination with a tumor vaccine that facilitates granulocyte-macrophage colony stimulating factor secretion, have been demonstrated to downregulate the number of CD4+ CTLs and improve vaccine efficacy (108). However, the severe side effects of this combination therapy need to be fully evaluated. A clinical trial has reported the PD-1 inhibitor, durvalumab, in combination with endothelial growth factor receptor 1-3 inhibitor, cediranib, induced high incidence of drug-associated treatment-emergent adverse events (109). Therefore, the safety of combination therapies for ovarian cancer treatment should be given priority in further clinical trials.
PARPs are a family of 17 nucleoproteins that are characterized by a common catalytic site that is involved in DNA damage repair (3, 4, 7). Germline mutations in BRCA1 and BRCA2 are harmful in ovarian cancer and other malignant tumors, making these tumors particularly sensitive to PARP inhibitors (PARPis) (110, 111). PARP inhibitors enhance tumor antigenicity, which sensitizes cancers to checkpoint blockade therapies. PARPis have also been thought to improve the response of homologous recombination-deficient ovarian cancers to immunotherapy through the generation of a higher mutation burden, which elevates the level of neoantigen expression. The stimulator of interferon genes pathway is activated by DNA damage and neoantigen expression, and plays an indispensable role in innate immunity (112). PD-L1 inhibitors have been shown to strengthen the antitumor activity of PARPis by restoring antitumor immunity (113), and showed modest clinical activity in recurrent ovarian cancer (114). Therefore, evaluating the efficacy of immune checkpoint blockade in combination with PARPis in ovarian cancer clinical trials is a potential treatment strategy. In a phase I/II TOPACIO trial, combination therapy with niraparib and pembrolizumab was evaluated for the treatment of platinum-resistant ovarian cancer (115). In the cohort with BRCA1/2 mutations, ORR and DCR were 45 and 73%, respectively. The phase I/II basket MEDIOLA trial demonstrated that olaparib in combination with an anti–PD-L1 antibody, durvalumab, for the treatment of germline BRCA 1/2 mutations that were platinum-sensitive resulted in relapse of ovarian cancer (116). At 12 weeks, DCR was 81% and ORR was 63%.
Hyperprogressive disease (HPD) is a side effect of immune checkpoint inhibitors in various types of tumors, and is associated with shorter progression-free and overall survival (117). A number of studies have reported that the incidence of HPD ranges from 4% to 29% (118–121). For ovarian cancer, retrospective analysis of data from a clinical trial with a cohort of 89 patients that received ICB showed that over half of the patients (N = 46, 51.6%) experienced early treatment discontinuation (≤12 weeks after treatment initiation) due to radiographic or clinical disease progression (122). The biological basis and mechanisms underlying HPD, such as the Fc region of antibodies (123), EGFR and MDM2/MDM4 amplification (123), and senescent CD4+ T cells (124), are being clarified. This phenomenon has polarized oncologists, who debate whether this effect could still reflect the natural history of the disease. Therefore, it is important to identify these underlying mechanisms to predict which patients are susceptible to HPD, so that it can be prevented.
Although the overall survival rate of ovarian cancer benefits from spontaneous anti-tumor immune responses and cytotoxic T cell infiltration, the presence of clinical disease indicates that immune effector cells are insufficient to inhibit tumor growth (125, 126). Since the interactions between the immune system and cancer cells are durable, active, and progressive, the development of an initial cancer cell into metastatic disease depends on immune evasion. There are two factors that can drive immune escape: recruitment of suppressive cells mediated by tumor cells, and iatrogenic factors such as treatment programs that include lymphotoxic drugs (126).
Immunotherapy resistance mechanisms contain primary resistance, adaptive immune resistance, and acquired resistance (127). Primary resistance is a clinical condition in which a cancer does not respond to an immunotherapeutic agent (128–130). Adaptive immune resistance occurs when a tumor has been recognized by the immune system, but is able to adapt to immune attack (131, 132). With acquired resistance, the cancer is sensitive to immunotherapy at first, but then relapses and progresses after a period of time (133–135). In the clinic, immunotherapy has been applied to solid tumors for a long time, but there are still issues that limit the development of cancer immunotherapies. First, the effect of the immunotherapies must be validated in preclinical animal models, prior to being administered to actual cancer patients in clinical trials. In most cases, immunotherapy strategies have been shown to suppress tumor development in animal models, but have often been ineffective in patients. Since the immune reaction is a complex and highly regulated biological process, the animal models used to test cancer immunotherapies do not sufficiently replicate the complex phenomenon of tumor immunity in humans. Mestas et al. have summarized the differences between mouse and human immunology in terms of both innate and adaptive immunity, including: balance of leukocyte subsets, defensins, Toll receptors, inducible NO synthase, the NK inhibitory receptor families, the B cell and T cell signaling pathway components, cytokines and cytokine receptors, T helper cells 1/T helper cells 2 differentiation, costimulatory molecule expression and function, and chemokine and chemokine receptor expression (136). Thus, discrepancies should be taken into account when using mice as preclinical models to investigate the mechanisms and efficacy of cancer immunotherapies. Since the anti-tumor efficacy of immunotherapies is long term, it is difficult to access long-term clinical efficacy in preclinical animal models. For immunotherapies, an initial antitumor effect in animal models is not synonymous with final clinical efficacy.
Collectively, a better understanding of the mechanisms of an effective antitumor response and the different intrinsic and extrinsic factors acting on the tumor cells that result in primary, adaptive, and acquired resistance to immunotherapy is needed (127).
Immune-related adverse events (irAEs) are autoimmune manifestations induced by the alteration of the immune system via immunotherapy treatment, such as checkpoint inhibitors and adoptive cell therapy (137). IrAEs have a high incidence in multiple types of cancers, with anti-CTLA-4 therapy, ipilimumab, and anti–PD-1 or anti–PD-L1 therapies, at 90 and 70%, respectively. Studies suggest that immune-related adverse events (irAEs) may develop through an integrated pathway, including autoreactive T cells, autoantibodies, and cytokines (138). For instance, T cell infiltration in tumor tissue induces T cell activation and leads to the production of inflammatory cytokines, which promote the development of irAEs (138). Immune-related adverse events often affect a range of organs, including skin, colon, endocrine glands, lungs, and liver. Mild effects that can be managed via transitory immunosuppression therapy with corticosteroids account for the majority of adverse events; however, severe events often lead to hospitalization and require specialized treatment. Patients, nurses, and other collaborative staff must be educated on these adverse events when considering the use of these drugs for the treatment of cancer (138).
Single-cell genomics is an advanced technology that could revolutionize the way we evaluate complex immune cell assemblies and explore their spatial organization, dynamics, clonal distribution, pathways, function, and crosstalk (139). Krieg et al. utilized high-dimensional single-cell mass cytometry and a bioinformatics pipeline to qualify the immune cell subsets in the peripheral blood of patients with melanoma, before and after anti–PD-1 immunotherapy. They found that CD14+CD16−HLA-DRhi monocytes are a strong predictor of progression-free and overall survival in response to anti–PD-1 immunotherapy (140). Multi-omic datasets at single-cell resolution, in combination with advanced computational methods, will improve the determination of immune cell identity. Current datasets, integrated with ‘big data’ methodologies, can serve as a platform to sustain future immunology research. Thus, in the future, these methods may apply to functional studies of immune cell populations and precision medicine (141). Azizi and his colleagues have demonstrated that combining single-cell analysis of the tumor immune microenvironment in breast cancer with computational analysis can result in the production of an immune map of breast cancer (BC) that points to continuous T cell activation and differentiation states (142). Single cell analysis was also used to reveal the association between CAF clusters (CAF-S1) and immunotherapy resistance. These findings indicated that there is a positive feedback loop between specific CAF-S1 clusters and Tregs that plays a vital role in immunotherapy resistance (143). Recently, single-cell RNA sequencing (scRNAseq) computational analyses were applied to melanoma tumors to investigate the tumor cell states that promote immune evasion. Using this novel technology, a resistance program that is expressed by tumor cells has been identified, which is involved in T cell exclusion and immune evasion (144). With the development of single-cell technology, it is possible that immunotherapy will have fewer adverse events and a more effective response, based on modified patient stratifications, identification of novel biomarkers, and identification of novel cell targets and pathways (145).
Nanotechnology is critically important for immunotherapy for several reasons, including its potential to enhance efficacy, it can be translated, and it can improve novel therapeutic strategies that are based on current cancer immunotherapies (146). In the near future, we envision that nanotechnology will be a key driver of cancer immunotherapy success. Jin et al. have identified that more adaptive and durable responses and more robust antitumor effects will enhance the effects of immunotherapeutic cells through nano-immunoengineering via regulation of the immune-network and identification of precise cancer-targeted theranostics (147). Nanomedicines can also regulate myeloid and lymphoid cell behavior, consequently strengthening anticancer immunity and immunotherapy efficacy (148, 149). Nanoparticles that are used as advanced biomaterials could enhance the efficacy of immunotherapies and reduce harmful side effects (150). Nanoparticles can be applied to reprograming the immunosuppressive tumor microenvironment and triggering systemic antitumor immunity, coupled with immunotherapy agents against advanced cancer (151, 152).
In ovarian cancer, nanotechnology-based immune checkpoint inhibitor delivery systems have the potential to overcome the immunosuppressive environment and transport barriers. This have already been utilized to improve the distribution and targeting-capabilities of drugs against tumor-associated immune cells, such as DCs and macrophages (153). Radiotherapy is frequently applied for the treatment of various kinds of cancer. However, the limitations of radiotherapy are resistance induced by tumor tissue hypoxia and uncontrollable metastases. To address these issues, scientists have designed core–shell nanoparticles that are composed of an enzyme that can decompose H2O2 to generate O2 and a toll-like-receptor-7 agonist that can regulate the immune suppressive tumor microenvironment.(154).
Organoids that simulate the structure and function of their in vivo counterpart organs are grown from stem cells in vitro. This technique has been used as a novel human cancer treatment, as described in our previous studies (155–158). Tumor organoids, in combination with immune cells and fibroblasts, can be utilized for immune-oncology applications (159). Recently, an air-liquid interface (ALI) method has been used to fabricate patient-derived organoids (PDOs) with native embedded immune cells (T cells, B cells, NK cells, and macrophages) that enables investigation of the tumor microenvironment and personalized immunotherapy testing (160).
Hill et al. have used organoid models that are defective in homologous recombination (HR) and replication fork protection for drug sensitivity screening. They found that HR deficiency is related to PARP inhibitor sensitivity, and that replication fork protection deficiency is correlated with carboplatin and CHK1 and ATR inhibitor sensitivity (161). Schnalzger et al. have developed a three-dimensional (3D) patient-derived colon organoid to evaluate CAR efficacy and tumor specificity in a personalized manner. This new preclinical model allowed testing of CAR-mediated cytotoxicity in a tissue-like environment, but further basic and clinical trials are needed to confirm these findings (162). Recently, a novel organoid composed of cells derived from lymph node and tumor tissue from the same patient was developed to evaluate the efficacy of immunotherapy and to assess the relationship between the clinical response of the patient to therapy (163). Lymph node stromal cells (LNSCs) have been reported to be involved in the inhibition of early activation of autoreactive immune cells and peripheral tolerance (164). Mechanisms can be clarified using this advanced organoid model.
Cancer immunotherapies can be grouped into two categories based on the presence or absence of a suppressed immune response to each patient’s tumor. If immunotherapy can trigger a prior immune response, patients are eligible to choose checkpoint inhibitor drugs to trigger the prior immune response and kill tumor cells. Whereas, if patients do not have a prior immune response, then a checkpoint inhibitor will have no effect on the activity of the immune response. Strategies that stimulate a new immune response are more appropriate in these cases.
Immunohistochemistry (IHC) testing to detect the expression of PD-L1 has been one of the first predicted biomarkers for pembrolizumab efficacy (165). However, about 15% of patients that showed PD-L1 negativity exhibit a response to the treatment with PD-1 or PD-L1. The IHC test was also used to evaluate the treatment efficacy of cancer vaccines or with bispecific T cell redirection therapies. Detection of expression of the antigen in the patient for the cancer vaccine or the tumor-specific antigen on the bispecific molecule is necessary.
Interleukin-8 has been reported as a poor predictor of outcome of immune checkpoint blockade in urothelial carcinoma (mUC) and metastatic renal cell carcinoma (166). Anti–PD-1 monoclonal antibody therapy (atezolizumab) showed poor therapeutic effect with high levels of IL-8 in plasma, peripheral blood mononuclear cells, and tumors. A large-scale retrospective analysis reported that the poor prognoses of advanced cancer patients treated with nivolumab and/or ipilimumab, everolimus, or docetaxel in phase III clinical trials were related to upregulated baseline serum IL-8 (167). The effect of IL-8 in ovarian cancer remains unclear, and further investigation is warranted. Molecular analysis of the T cell repertoire (168) could digitize the immune response. Patients with high T cell infiltration in solid tumors and high clonality had a significantly increased response to PD-1; however, patients with low T cell infiltration and no evidence of clonal T cell expansion had a poor response.
Next-generation high-throughput DNA (NGS) sequencing techniques have provided new opportunities for immunotherapy. The high-throughput and deep coverage of NGS techniques could help with whole exome sequencing or RNA sequencing to identify somatic mutations that may encode neoantigens (169). These neoantigens may be used to customize cancer vaccines and predict the response to immunotherapy. Recent studies have reported that recognition of neoantigens is a crucial factor in the activity of clinical immunotherapies, and cutting-edge technologies enable the dissection of the immune response to patient-specific neoantigens. These findings indicate that neoantigens may form a biomarker in cancer immunotherapy and provide novel insights into the development of enhanced T cell efficacy against this class of antigens (85).
Epithelial ovarian cancer (EOC) has early metastasis, peritoneal dissemination, and omentum infiltration. The omentum is an organ rich in lipids, which play a vital role in EOC. Lipid metabolic disorder of ovarian cancer cells, characterized by the alteration of lipid uptake and lipogenesis, are involved in EOC metastasis, alterations of ovarian cancer stem cells, chemotherapy resistance, and immunotherapy (170). Interestingly, for the immune system, alteration of lipid metabolism also has impact on T cells, TAMs, regulatory T cells, and MDSCs. Veglia et al. have reported that the fatty acid transport proteins that are upregulated in MDSCs, result in immunosuppression in tumors. Deletion of fatty acid transport proteins ablated the suppressive activity of MDSCs. Combining an inhibitor of fatty acid transport proteins with checkpoint inhibitors suppressed tumor progression in mice (171). Thus, with omentum infiltration in ovarian cancer, immunotherapy strategies, in combination with agents that target lipid metabolism, is a new direction that can be explored in cancer treatment.
Ovarian cancer is a solid cancer which is still a clinical challenge for CAR-T therapy. Challenges in CAR-T cell therapy are mainly due to antigen heterogeneity, physical barriers, and the complex network of the tumor microenvironment (78). To address the issue of heterogeneity of antigen expression, next-generation CAR-T cells that target more than one antigen, such as EGFR, HER2, and IL13Rα2, have been examined in pre-clinical models (172, 173). Combining CAR-T cell therapy with epigenetic drugs that can promote the expression of target antigens is also an option to deal with antigen heterogeneity (174). In order to overcome the physical barrier, there are three strategies as reported by Fucá (78): local delivery, overcoming the aberrant tumor vasculature, and enhancing trafficking. The TME is a complex network composed of extracellular matrix and stromal cells that are associated with reduced efficacy of CAR-T therapy. Overexpression of the FasL ligand (CD95L) in the TME interacts with the Fas (CD95) receptor that is expressed in CAR-T cells and can lead to T cell apoptotic death (72). Immunosuppressive catabolites, such as adenosine soluble catabolites (175), inhibitory factors rich in TME (176), and cytokines, such as IL15 (177), have direct or indirect effects on survival, expansion, and the anti-tumor function of CAR-T cells. In order to solve the problem of solid tumors and immune surveillance, translational, cooperative, and interdisciplinary efforts are required.
Neoadjuvant chemotherapy (NACT) has been applied in patients with stage IIIC or IV ovarian cancer, who are not suitable for primary debulking surgery (PDS). Research suggests that NACT, followed by interval debulking surgery (IDS) and adjuvant chemotherapy, was not inferior to primary debulking surgery followed by chemotherapy (178). NACT may exert multiple influences on the immune system, including induction of “immunogenic” cell death, presentation of neoantigens, and an increase in acute inflammatory and tumor-destructive responses (179–181) (Figure 3A). In 2017, Balkwill et al. evaluated the effect of NACT on immune activation in ovarian cancer, and found that NACT may enhance the host immune response; however, the response was weakened by upregulation of PD-1 (182). NACT can amplify the TIL responses but fails to turn TIL-negative cases into TIL-positive cases in ovarian cancer (183). Thus, a personalized treatment protocol of NACT and immunotherapy should be evaluated according to the baseline features of the tumor microenvironment.
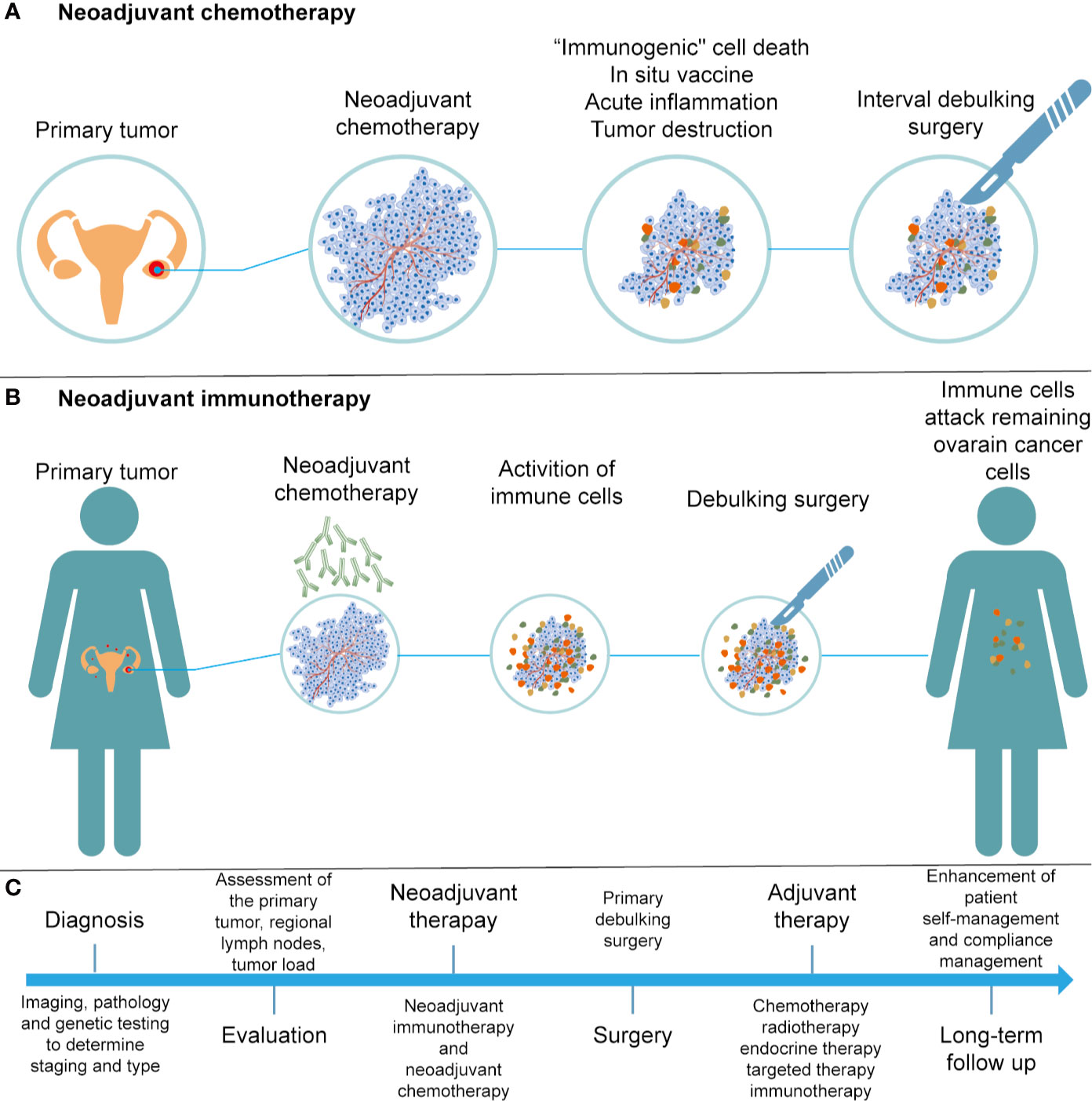
Figure 3 Development of treatments for ovarian cancer. (A) Illustration of NACT. (B) Illustration of neoadjuvant immunotherapy. (C) Flow chart of diagnosis and treatment for ovarian cancer.
Neoadjuvant immunotherapy has demonstrated remarkable efficacy in cancer patients as compared to surgery followed by adjuvant immunotherapy, as the adverse effects of surgery-induced metastasis are associated with innate and adaptive immunity damage (184) (Figure 3B). Early preclinical trials in melanoma (185), non–small cell lung carcinoma (186), and glioblastoma (187) have shown that neoadjuvant immunotherapy was better than adjuvant immunotherapy in promoting clinical efficacy. Neoadjuvant immunotherapy could enhance systemic immunity against tumor antigens and eliminate micrometastases that can be considered as the source of recurrence (188). With a comprehensive understanding of efficacy and safety, neoadjuvant therapies are expected to bring substantial benefits to patients suffering from cancer.
Immunotherapy is a revolution in ovarian cancer management. We present a flow chart that describes the treatment of patients with EOCs (Figure 3C). Despite the promising treatments that have been developed for cancer immunotherapy, such as immune checkpoint inhibitors and CAR-T therapies, there is still a need to overcome the immunosuppressive tumor microenvironment in order to improve the efficacy of cancer immunotherapy. The tumor immune microenvironment is an important regulator of immune suppression and immune tolerance, and can destroy the number and activity of TILs. A better understanding of the relationship between the tumor and stromal environment in EOCs is crucial to identify effective treatment methods and reliable predictive biomarkers. In conclusion, with an increased understanding and advanced technology, such as 3D-organoid models and single-cell technologies, more sophisticated and personal immunotherapy treatments based on tumor biology and TME characteristics can be applied in clinical practice. Thus, these efforts will enhance the benefits of immunotherapy to more patients with EOC and allow them to benefit from the long-lasting responses of immunotherapy.
W-LJ and GL designed the manuscript. CY, Y-JZ, and B-RX wrote the manuscript. CY and Z-CZ drew the figures and tables. W-LJ mainly revised the manuscript. GL made some revisions of the review. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (nos. 81872507 and 81872430), the Harbin Medical University (CN) Research and Innovation Project (YJSKYCX2019-55HYD), and the Special Fund in China Postdoctoral Science Foundation (2019T120281).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to those colleagues whose important work could not be cited due to space constraints.
ACT, Adoptive cell therapy; CAF, Cancer-associated fibroblast; CAR, Chimeric antigen receptor; CIK, Cytokine-induced killer; CTL, Cytotoxic T lymphocyte; CTLA4, Cytotoxic T lymphocyte antigen 4; DC, Dendritic cell; EMT, Epithelial-mesenchymal transition; EOC, Epithelial ovarian cancer; HPD, Hyperprogressive disease; ICI, Immune checkpoint inhibitors; irAEs, Immune-related adverse events; MDSC, Myeloid derived suppressor cells; MHC, Major histocompatibility complex; MISIIR, Müllerian inhibiting substance type 2 receptor; NK, Natural killer; OC, Ovarian cancer; PARP, Poly-ADP-ribose polymerase; TAA, Tumor-associated antigens; TAM, Tumor-associated macrophages; TCR, T cell receptors; TME, Tumor microenvironment; Treg, Regulatory T cell.
1. International Agency for Research on Cancer. Estimated number of incident cases from 2018 to 2040, ovary, females, all ages. (2020). Available at: https://gco.iarc.fr/tomorrow/graphic-isotype (last accessed 2020.Sep 30).
2. Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin (2018) 68:284–96. doi: 10.3322/caac.21456
3. Xie H, Wang W, Xia B, Jin W, Lou G. Therapeutic applications of PARP inhibitors in ovarian cancer. Biomed Pharmacother (2020) 127:110204. doi: 10.1016/j.biopha.2020.110204
4. Franzese E, Centonze S, Diana A, Carlino F, Guerrera LP, Di Napoli M, et al. PARP inhibitors in ovarian cancer. Cancer Treat Rev (2019) 73:1–9. doi: 10.1016/j.ctrv.2018.12.002
5. Francica P, Rottenberg S. Mechanisms of PARP inhibitor resistance in cancer and insights into the DNA damage response. Genome Med (2018) 10:101. doi: 10.1186/s13073-018-0612-8
6. Gogola E, Rottenberg S, Jonkers J. Resistance to PARP Inhibitors: Lessons from Preclinical Models of BRCA-Associated Cancer. Annu Rev Cancer Biol (2019) 3:235–54. doi: 10.1146/annurev-cancerbio-030617-050232
7. D’Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst) (2018) 71:172–6. doi: 10.1016/j.dnarep.2018.08.021
8. Dai L, Li K, Li M, Zhao X, Luo Z, Lu L, et al. Size/Charge Changeable Acidity-Responsive Micelleplex for Photodynamic-Improved PD-L1 Immunotherapy with Enhanced Tumor Penetration. Adv Funct Mater (2018) 28:1707249. doi: 10.1002/adfm.201707249
9. Zhang X, Wang C, Wang J, Hu Q, Langworthy B, Ye Y, et al. PD-1 Blockade Cellular Vesicles for Cancer Immunotherapy. Adv Mater (2018) 30:e1707112. doi: 10.1002/adma.201707112
10. Guo ZS, Lu B, Guo Z, Giehl E, Feist M, Dai E, et al. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J Immunother Cancer (2019) 7:6. doi: 10.1186/s40425-018-0495-7
11. Matuszewska K, Santry LA, van Vloten JP, AuYeung AWK, Major PP, Lawler J, et al. Combining Vascular Normalization with an Oncolytic Virus Enhances Immunotherapy in a Preclinical Model of Advanced-Stage Ovarian Cancer. Clin Cancer Res (2019) 25:1624–38. doi: 10.1158/1078-0432.Ccr-18-0220
12. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer (2016) 16:566–81. doi: 10.1038/nrc.2016.97
13. Santoiemma PP, Reyes C, Wang LP, McLane MW, Feldman MD, Tanyi JL, et al. Systematic evaluation of multiple immune markers reveals prognostic factors in ovarian cancer. Gynecol Oncol (2016) 143:120–7. doi: 10.1016/j.ygyno.2016.07.105
14. Rodriguez GM, Galpin KJC, McCloskey CW, Vanderhyden BC. The Tumor Microenvironment of Epithelial Ovarian Cancer and Its Influence on Response to Immunotherapy. Cancers (Basel) (2018) 10:242. doi: 10.3390/cancers10080242
15. Mi Y, Guo N, Luan J, Cheng J, Hu Z, Jiang P, et al. The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front Immunol (2020) 11:737. doi: 10.3389/fimmu.2020.00737
16. Jin MZ, Jin WL. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther (2020) 5:166. doi: 10.1038/s41392-020-00280-x
17. Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, et al. Expression of Vascular Endothelial Growth Factor in Ovarian Cancer Inhibits Tumor Immunity through the Accumulation of Myeloid-Derived Suppressor Cells. Clin Cancer Res (2017) 23:587–99. doi: 10.1158/1078-0432.ccr-16-0387
18. Taki M, Abiko K, Baba T, Hamanishi J, Yamaguchi K, Murakami R, et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat Commun (2018) 9:1685. doi: 10.1038/s41467-018-03966-7
19. Baert T, Vankerckhoven A, Riva M, Van Hoylandt A, Thirion G, Holger G, et al. Myeloid Derived Suppressor Cells: Key Drivers of Immunosuppression in Ovarian Cancer. Front Immunol (2019) 10:1273. doi: 10.3389/fimmu.2019.01273
20. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
21. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci (2005) 102:18538–43. doi: 10.1073/pnas.0509182102
22. Hanahan D, Coussens LM. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
23. Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, et al. Targeting the TGFβ pathway for cancer therapy. Pharmacol Ther (2015) 147:22–31. doi: 10.1016/j.pharmthera.2014.11.001
24. Neel JC, Humbert L, Lebrun JJ. The Dual Role of TGFβ in Human Cancer: From Tumor Suppression to Cancer Metastasis. ISRN Mol Biol (2012) 2012:1–28. doi: 10.5402/2012/381428
25. Yuan X, Zhang J, Li D, Mao Y, Mo F, Du W, et al. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol Oncol (2017) 147:181–7. doi: 10.1016/j.ygyno.2017.07.007
26. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell (2015) 27:462–72. doi: 10.1016/j.ccell.2015.02.015
27. Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A, et al. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res (2014) 7:19. doi: 10.1186/1757-2215-7-19
28. Yin M, Li X, Tan S, Zhou HJ, Ji W, Bellone S, et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J Clin Invest (2016) 126:4157–73. doi: 10.1172/jci87252
29. Mlynska A, Povilaityte E, Zemleckaite I, Zilionyte K, Strioga M, Krasko J, et al. Platinum sensitivity of ovarian cancer cells does not influence their ability to induce M2-type macrophage polarization. Am J Reprod Immunol (2018) 80:e12996. doi: 10.1111/aji.12996
30. Labiche A, Heutte N, Herlin P, Chasle J, Gauduchon P, Elie N. Stromal compartment as a survival prognostic factor in advanced ovarian carcinoma. Int J Gynecol Cancer (2010) 20:28–33. doi: 10.1111/IGC.0b013e3181bda1cb
31. Kwon Y, Smith BD, Zhou Y, Kaufman MD, Godwin AK. Effective inhibition of c-MET-mediated signaling, growth and migration of ovarian cancer cells is influenced by the ovarian tissue microenvironment. Oncogene (2015) 34:144–53. doi: 10.1038/onc.2013.539
32. Erez N, Glanz S, Raz Y, Avivi C, Barshack I. Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem Biophys Res Commun (2013) 437:397–402. doi: 10.1016/j.bbrc.2013.06.089
33. Lau TS, Chung TKH, Cheung TH, Chan LKY, Cheung LW-H, Yim S-F, et al. Cancer cell-derived lymphotoxin mediates reciprocal tumour-stromal interactions in human ovarian cancer by inducing CXCL11 in fibroblasts. J Pathol (2014) 232:43–56. doi: 10.1002/path.4258
34. Wang L, Zhang F, Cui JY, Chen L, Chen YT, Liu BW. CAFs enhance paclitaxel resistance by inducing EMT through the IL−6/JAK2/STAT3 pathway. Oncol Rep (2018) 39:2081–90. doi: 10.3892/or.2018.6311
35. Givel A-M, Kieffer Y, Scholer-Dahirel A, Sirven P, Cardon M, Pelon F, et al. miR200-regulated CXCL12β promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat Commun (2018) 9:1056. doi: 10.1038/s41467-018-03348-z
36. Zhou B, Sun C, Li N, Shan W, Lu H, Guo L, et al. Cisplatin-induced CCL5 secretion from CAFs promotes cisplatin-resistance in ovarian cancer via regulation of the STAT3 and PI3K/Akt signaling pathways. Int J Oncol (2016) 48:2087–97. doi: 10.3892/ijo.2016.3442
37. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med (2011) 17:1498–503. doi: 10.1038/nm.2492
38. Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene (2018) 37:2285–301. doi: 10.1038/s41388-017-0093-z
39. Peck B, Schulze A. Lipid Metabolism at the Nexus of Diet and Tumor Microenvironment. Trends Cancer (2019) 5:693–703. doi: 10.1016/j.trecan.2019.09.007
40. Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol (2016) 27:1482–92. doi: 10.1093/annonc/mdw168
41. Gujar S, Dielschneider R, Clements D, Helson E, Shmulevitz M, Marcato P, et al. Multifaceted therapeutic targeting of ovarian peritoneal carcinomatosis through virus-induced immunomodulation. Mol Ther (2013) 21:338–47. doi: 10.1038/mt.2012.228
42. Conrad C, Gilliet M. Plasmacytoid dendritic cells and regulatory T cells in the tumor microenvironment: A dangerous liaison. Oncoimmunology (2013) 2:e23887. doi: 10.4161/onci.23887
43. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science (2013) 339:286–91. doi: 10.1126/science.1232227
44. Odunsi K. Immunotherapy in ovarian cancer. Ann Oncol (2017) 28:viii1–7. doi: 10.1093/annonc/mdx444
45. Mantia-Smaldone GM, Corr B, Chu CS. Immunotherapy in ovarian cancer. Hum Vaccin Immunother (2012) 8:1179–91. doi: 10.4161/hv.20738
46. Tanyi JL, Bobisse S, Ophir E, Tuyaerts S, Roberti A, Genolet R, et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med (2018) 10:eaao5931. doi: 10.1126/scitranslmed.aao5931
47. Kiyotani K, Chan HT, Nakamura Y. Immunopharmacogenomics towards personalized cancer immunotherapy targeting neoantigens. Cancer Sci (2018) 109:542–9. doi: 10.1111/cas.13498
48. Ojalvo LS, Thompson ED, Wang TL, Meeker AK, Shih IM, Fader AN, et al. Tumor-associated macrophages and the tumor immune microenvironment of primary and recurrent epithelial ovarian cancer. Hum Pathol (2018) 74:135–47. doi: 10.1016/j.humpath.2017.12.010
49. Zhu X, Cai H, Zhao L, Ning L, Lang J. CAR-T cell therapy in ovarian cancer: from the bench to the bedside. Oncotarget (2017) 8:64607–21. doi: 10.18632/oncotarget.19929
50. Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol (2015) 33:1974–82. doi: 10.1200/jco.2014.59.4358
51. Wang W, Liu JR, Zou W. Immunotherapy in Ovarian Cancer. Surg Oncol Clin N Am (2019) 28:447–64. doi: 10.1016/j.soc.2019.02.002
52. Hinchcliff E, Hong D, Le H, Chisholm G, Iyer R, Naing A, et al. Characteristics and outcomes of patients with recurrent ovarian cancer undergoing early phase immune checkpoint inhibitor clinical trials. Gynecol Oncol (2018) 151:407–13. doi: 10.1016/j.ygyno.2018.10.008
53. Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol (2015) 33:4015–22. doi: 10.1200/JCO.2015.62.3397
54. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
55. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi: 10.1038/nm730
56. Miao YR, Thakkar KN, Qian J, Kariolis MS, Wei H, Nandagopal S, et al. Neutralizing PD-L1 and PD-L2 Enhances the Efficacy of Immune Checkpoint Inhibitors in Ovarian Cancer. bioRxiv (2020). doi: 10.1101/2020.01.19.911941
57. Daassi D, Mahoney KM, Freeman GJ. The importance of exosomal PDL1 in tumour immune evasion. Nat Rev Immunol (2020) 20:209–15. doi: 10.1038/s41577-019-0264-y
58. Cai D, Li J, Liu D, Hong S, Qiao Q, Sun Q, et al. Tumor-expressed B7-H3 mediates the inhibition of antitumor T-cell functions in ovarian cancer insensitive to PD-1 blockade therapy. Cell Mol Immunol (2019) 17:227–36. doi: 10.1038/s41423-019-0305-2
59. Chen YL, Lin HW, Chien CL, Lai YL, Sun WZ, Chen CA, et al. BTLA blockade enhances Cancer therapy by inhibiting IL-6/IL-10-induced CD19high B lymphocytes. J Immuno Ther Cancer (2019) 7:313. doi: 10.1186/s40425-019-0744-4
60. Huang RY, Francois A, McGray AJR, Miliotto A, Odunsi K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. OncoImmunology (2017) 6:e1249561. doi: 10.1080/2162402X.2016.1249561
61. Fucikova J, Rakova J, Hensler M, Kasikova L, Belicová L, Hladikova K, et al. TIM-3 dictates functional orientation of the immune infiltrate in ovarian cancer. J Clin Cancer Res (2019) 276:97–111. doi: 10.1158/1078-0432.CCR-18-4175
62. Mulati K, Hamanishi J, Matsumura N, Chamoto K, Mise N, Abiko K, et al. VISTA expressed in tumour cells regulates T cell function. Br J Cancer (2019) 120:115–27. doi: 10.1038/s41416-018-0313-5
63. Derré L, Rivals J-P, Jandus C, Pastor S, Rimoldi D, Romero P, et al. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest (2010) 120:157–67. doi: 10.1172/JCI40070
64. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol (2009) 10:29–37. doi: 10.1038/ni.1679
65. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev (2017) 276:97–111. doi: 10.1111/imr.12520
66. Ganesan S, Mehnert J. Biomarkers for Response to Immune Checkpoint Blockade. Annu Rev Cancer Biol (2019) 4:331–51. doi: 10.1146/annurev-cancerbio-030419-033604
67. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer (2019) 19:133–50. doi: 10.1038/s41568-019-0116-x
68. Xiao Q, Nobre A, Pineiro P, Berciano-Guerrero MA, Alba E, Cobo M, et al. Genetic and Epigenetic Biomarkers of Immune Checkpoint Blockade Response. J Clin Med (2020) 9:286. doi: 10.3390/jcm9010286
69. Nersesian S, Glazebrook H, Toulany J, Grantham SR, Boudreau JE. Naturally Killing the Silent Killer: NK Cell-Based Immunotherapy for Ovarian Cancer. Front Immunol (2019) 10:1782. doi: 10.3389/fimmu.2019.01782
70. Gao X, Mi Y, Guo N, Xu H, Xu L, Gou X, et al. Cytokine-Induced Killer Cells As Pharmacological Tools for Cancer Immunotherapy. Front Immunol (2017) 8:774. doi: 10.3389/fimmu.2017.00774
71. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
72. Yamamoto TN, Lee PH, Vodnala SK, Gurusamy D, Kishton RJ, Yu Z, et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J Clin Invest (2019) 129:1551–65. doi: 10.1172/jci121491
73. Galluzzi L, Vacchelli E, Pedro JMBS, Buqué A, Senovilla L, Baracco EE, et al. Classification of current anticancer immunotherapies. Oncotarget (2014) 5:12472–508. doi: 10.18632/oncotarget.2998
74. Perica K, Varela JC, Oelke M, Schneck J. Adoptive T Cell Immunotherapy For Cancer. Rambam Maimonides Med J (2015) 6:e0004. doi: 10.5041/rmmj.10179
75. Matsuda T, Leisegang M, Park JH, Ren L, Kato T, Ikeda Y, et al. Induction of Neoantigen-Specific Cytotoxic T Cells and Construction of T-cell Receptor-Engineered T Cells for Ovarian Cancer. Clin Cancer Res (2018) 24:5357–67. doi: 10.1158/1078-0432.CCR-18-0142
76. Rodriguez-Garcia A, Sharma P, Poussin M, Boesteanu AC, Minutolo NG, Gitto SB, et al. CAR T Cells Targeting MISIIR for the Treatment of Ovarian Cancer and Other Gynecologic Malignancies. Mol Ther (2020) 28:548–60. doi: 10.1016/j.ymthe.2019.11.028
77. Filley AC, Henriquez M, Dey M. CART Immunotherapy: Development, Success, and Translation to Malignant Gliomas and Other Solid Tumors. Front Oncol (2018) 8:453. doi: 10.3389/fonc.2018.00453
78. Fucá G, Reppel L, Landoni E, Savoldo B, Dotti G. Enhancing Chimeric Antigen Receptor T cell Efficacy in Solid Tumors. Clin Cancer Res (2020) 26:2444–51. doi: 10.1158/1078-0432.Ccr-19-1835
79. Nizzero S, Shen H, Ferrari M, Corradetti B. Immunotherapeutic Transport Oncophysics: Space, Time, and Immune Activation in Cancer. Trends Cancer (2020) 6:40–8. doi: 10.1016/j.trecan.2019.11.008
80. Jaspers JE, Brentjens RJ. Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacol Ther (2017) 178:83–91. doi: 10.1016/j.pharmthera.2017.03.012
81. Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol (2016) 13:370–83. doi: 10.1038/nrclinonc.2016.36
82. Martin Lluesma S, Wolfer A, Harari A, Kandalaft L. Cancer Vaccines in Ovarian Cancer: How Can We Improve? Biomedicines (2016) 4:10. doi: 10.3390/biomedicines4020010
83. Tanyi JL, George E. Personalized vaccination against ovarian cancer: what are the possibilities? Expert Rev Vaccines (2018) 17:955–8. doi: 10.1080/14760584.2018.1541743
84. Malekzadeh P, Pasetto A, Robbins PF, Parkhurst MR, Paria BC, Jia L, et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J Clin Invest (2019) 129:1109–14. doi: 10.1172/JCI123791
85. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Sci (New York NY) (2015) 348:69–74. doi: 10.1126/science.aaa4971
86. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol (2018) 15:422–42. doi: 10.1038/s41571-018-0003-5
87. Santos JM, Heinio C, Cervera-Carrascon V, Quixabeira DCA, Siurala M, Havunen R, et al. Oncolytic adenovirus shapes the ovarian tumor microenvironment for potent tumor-infiltrating lymphocyte tumor reactivity. J Immunother Cancer (2020) 8:e000188. doi: 10.1136/jitc-2019-000188
88. Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell (2000) 100:57–70. doi: 10.1016/s0092-8674(00)81683-9
89. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
90. Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol (2015) 6:187. doi: 10.3389/fimmu.2015.00187
91. Lesterhuis WJ, Punt CJA, Hato SV, Eleveld-Trancikova D, Jansen BJH, Nierkens S, et al. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J Clin Invest (2011) 121:3100–8. doi: 10.1172/JCI43656
92. Hato SV, Khong A, de Vries IJ, Lesterhuis WJ. Molecular pathways: the immunogenic effects of platinum-based chemotherapeutics. Clin Cancer Res (2014) 20:2831–7. doi: 10.1158/1078-0432.CCR-13-3141
93. Peng J, Hamanishi J, Matsumura N, Abiko K, Murat K, Baba T, et al. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-κB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res (2015) 75:5034–45. doi: 10.1158/0008-5472.CAN-14-3098
94. Mkrtichyan M, Najjar YG, Raulfs EC, Abdalla MY, Samara R, Rotem-Yehudar R, et al. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur J Immunol (2011) 41:2977–86. doi: 10.1002/eji.201141639
95. Lesterhuis WJ, Salmons J, Nowak AK, Rozali EN, Khong A, Dick IM, et al. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS One (2013) 8:e61895. doi: 10.1371/journal.pone.0061895
96. Hartl CA, Bertschi A, Puerto RB, Andresen C, Cheney EM, Mittendorf EA, et al. Combination therapy targeting both innate and adaptive immunity improves survival in a pre-clinical model of ovarian cancer. J Immunother Cancer (2019) 7:199. doi: 10.1186/s40425-019-0654-5
97. Kandalaft LE, Odunsi K, Coukos G. Immunotherapy in Ovarian Cancer: Are We There Yet? J Clin Oncol (2019) 37:2460–71. doi: 10.1200/JCO.19.00508
98. Herrera FG, Irving M, Kandalaft LE, Coukos G. Rational combinations of immunotherapy with radiotherapy in ovarian cancer. Lancet Oncol (2019) 20:e417–33. doi: 10.1016/s1470-2045(19)30401-2
99. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity (2014) 41:843–52. doi: 10.1016/j.immuni.2014.10.019
100. Herrera FG, Bourhis J, Coukos G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA: Cancer J Clinicians (2017) 67:65–85. doi: 10.3322/caac.21358
101. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell (2013) 24:589–602. doi: 10.1016/j.ccr.2013.09.014
102. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13:1050–9. doi: 10.1038/nm1622
103. Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Polychemotherapy for early breast cancer: an overview of the randomised trials. Lancet (1998) 352:930–42. doi: 10.1016/s0140-6736(98)03301-7
104. Mabuchi S, Terai Y, Morishige K, Tanabe-Kimura A, Sasaki H, Kanemura M, et al. Maintenance treatment with bevacizumab prolongs survival in an in vivo ovarian cancer model. Clin Cancer Res (2008) 14:7781–9. doi: 10.1158/1078-0432.CCR-08-0243
105. Rossi L, Verrico M, Zaccarelli E, Papa A, Colonna M, Strudel M, et al. Bevacizumab in ovarian cancer: A critical review of phase III studies. Oncotarget (2017) 8:12389–405. doi: 10.18632/oncotarget.13310
106. Tada Y, Togashi Y, Kotani D, Kuwata T, Sato E, Kawazoe A, et al. Targeting VEGFR2 with Ramucirumab strongly impacts effector/ activated regulatory T cells and CD8 T cells in the tumor microenvironment. J Immunother Cancer (2018) 6:106. doi: 10.1186/s40425-018-0403-1
107. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun (2016) 7:12624. doi: 10.1038/ncomms12624
108. Li B, Lalani AS, Harding TC, Luan B, Koprivnikar K, Huan Tu G, et al. Vascular Endothelial Growth Factor Blockade Reduces Intratumoral Regulatory T Cells and Enhances the Efficacy of a GM-CSF-Secreting Cancer Immunotherapy. Clin Cancer Res (2006) 12:6808–16. doi: 10.1158/1078-0432.ccr-06-1558
109. Lee JM, Cimino-Mathews A, Peer CJ, Zimmer A, Lipkowitz S, Annunziata CM, et al. Safety and Clinical Activity of the Programmed Death-Ligand 1 Inhibitor Durvalumab in Combination With Poly (ADP-Ribose) Polymerase Inhibitor Olaparib or Vascular Endothelial Growth Factor Receptor 1-3 Inhibitor Cediranib in Women’s Cancers: A Dose-Escalation, Phase I Study. J Clin Oncol (2017) 35:2193–202. doi: 10.1200/JCO.2016.72.1340
110. Scott CL, Swisher EM, Kaufmann SH. Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J Clin Oncol (2015) 33:1397–406. doi: 10.1200/JCO.2014.58.8848
111. Papa A, Caruso D, Strudel M, Tomao S, Tomao F. Update on Poly-ADP-ribose polymerase inhibition for ovarian cancer treatment. J Transl Med (2016) 14:267. doi: 10.1186/s12967-016-1027-1
112. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol (2015) 15:760–70. doi: 10.1038/nri3921
113. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu J-M, et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res (2017) 23:3711–20. doi: 10.1158/1078-0432.CCR-16-3215
114. Lampert EJ, Zimmer AS, Padget MR, Cimino-Mathews A, Nair JR, Liu Y, et al. Combination of PARP inhibitor olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: a proof-of-concept phase 2 study. Clin Cancer Res (2020) 26:4268–79. doi: 10.1158/1078-0432.CCR-20-0056
115. Konstantinopoulos PA, Waggoner SE, Vidal GA, Mita MM, Fleming GF, Holloway RW, et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. J Clin Oncol (2018) 36:106–6. doi: 10.1200/JCO.2018.36.15_suppl.106
116. Drew Y, de Jonge M, Hong SH, Park YH, Wolfer A, Brown J, et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in germline BRCA-mutated (gBRCAm) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecol Oncol (2018) 149:246–7. doi: 10.1016/j.ygyno.2018.04.555
117. Adashek JJ, Subbiah IM, Matos I, Garralda E, Menta AK, Ganeshan DM, et al. Hyperprogression and Immunotherapy: Fact, Fiction, or Alternative Fact? Trends Cancer (2020) 6:181–91. doi: 10.1016/j.trecan.2020.01.005
118. Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin Cancer Res (2017) 23:4242–50. doi: 10.1158/1078-0432.CCR-16-3133
119. Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, et al. Hyperprogressive Disease in Patients With Advanced Non-Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. JAMA Oncol (2018) 4:1543–52. doi: 10.1001/jamaoncol.2018.3676
120. Frelaut M, Le Tourneau C, Borcoman E. Hyperprogression under Immunotherapy. Int J Mol Sci (2019) 20:2674. doi: 10.3390/ijms20112674
121. Saâda-Bouzid E, Defaucheux C, Karabajakian A, Coloma VP, Servois V, Paoletti X, et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol (2017) 28:1605–11. doi: 10.1093/annonc/mdx178
122. Boland JL, Zhou Q, Martin M, Callahan MK, Konner J, O’Cearbhaill RE, et al. Early disease progression and treatment discontinuation in patients with advanced ovarian cancer receiving immune checkpoint blockade. Gynecol Oncol (2019) 152:251–8. doi: 10.1016/j.ygyno.2018.11.025
123. Lo Russo G, Moro M, Sommariva M, Cancila V, Boeri M, Centonze G, et al. Antibody-Fc/FcR Interaction on Macrophages as a Mechanism for Hyperprogressive Disease in Non-small Cell Lung Cancer Subsequent to PD-1/PD-L1 Blockade. Clin Cancer Res (2019) 25:989–99. doi: 10.1158/1078-0432.CCR-18-1390
124. Arasanz H, Zuazo M, Bocanegra A, Gato M, Martinez-Aguillo M, Morilla I, Fernandez G, Hernandez B, Lopez P, Alberdi N, et al Early Detection of Hyperprogressive Disease in Non-Small Cell Lung Cancer by Monitoring of Systemic T Cell Dynamics. Cancers (Basel) (2020) 12(2):334.
125. Adams SF, Benencia F. Immunotherapy for ovarian cancer: what are the targets of the future? Future Oncol (London Engl) (2015) 11:1293–6. doi: 10.2217/fon.15.44
126. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med (2003) 348:203–13. doi: 10.1056/NEJMoa020177
127. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
128. Chae YK, Oh MS, Giles FJ. Molecular Biomarkers of Primary and Acquired Resistance to T-Cell-Mediated Immunotherapy in Cancer: Landscape, Clinical Implications, and Future Directions. Oncologist (2018) 23:410–21. doi: 10.1634/theoncologist.2017-0354
129. Nowicki TS, Hu-Lieskovan S, Ribas A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J (2018) 24:47–53. doi: 10.1097/PPO.0000000000000303
130. Balachandran VP, Beatty GL, Dougan SK. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology (2019) 156:2056–72. doi: 10.1053/j.gastro.2018.12.038
131. McGray AJR, Bramson J. Adaptive Resistance to Cancer Immunotherapy. Adv Exp Med Biol (2017) 1036:213–27. doi: 10.1007/978-3-319-67577-0_14
132. Miao Y, Yang H, Levorse J, Yuan S, Polak L, Sribour M, et al. Adaptive Immune Resistance Emerges from Tumor-Initiating Stem Cells. Cell (2019) 177:1172–86.e14. doi: 10.1016/j.cell.2019.03.025
133. O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
134. Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer (2016) 16:121–6. doi: 10.1038/nrc.2016.2
135. O’Donnell JS, Long GV, Scolyer RA, Teng MWL, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev (2017) 52:71–81. doi: 10.1016/j.ctrv.2016.11.007
136. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol (Baltimore Md 1950) (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
137. Sosa A, Lopez Cadena E, Simon Olive C, Karachaliou N, Rosell R. Clinical assessment of immune-related adverse events. Ther Adv Med Oncol (2018) 10:175883591876462. doi: 10.1177/1758835918764628
138. Postow MA, Sidlow R, Hellmann MD. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N Engl J Med (2018) 378:158–68. doi: 10.1056/NEJMra1703481
139. Giladi A, Amit I. Single-Cell Genomics: A Stepping Stone for Future Immunology Discoveries. Cell (2018) 172:14–21. doi: 10.1016/j.cell.2017.11.011
140. Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med (2018) 24:144–53. doi: 10.1038/nm.4466
141. Gomes T, Teichmann SA, Talavera-Lopez C. Immunology Driven by Large-Scale Single-Cell Sequencing. Trends Immunol (2019) 40:1011–21. doi: 10.1016/j.it.2019.09.004
142. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell (2018) 174:1293–1308.e1236. doi: 10.1016/j.cell.2018.05.060
143. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov (2020) 10:1330–51. doi: 10.1158/2159-8290.CD-19-1384
144. Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell (2018) 175:984–997.e924. doi: 10.1016/j.cell.2018.09.006
145. Yofe I, Dahan R, Amit I. Single-cell genomic approaches for developing the next generation of immunotherapies. Nat Med (2020) 26:171–7. doi: 10.1038/s41591-019-0736-4
146. Song W, Das M, Chen X. Nanotherapeutics for Immuno-Oncology: A Crossroad for New Paradigms. Trends Cancer (2020) 6:288–98. doi: 10.1016/j.trecan.2020.01.011
147. Jin W, Cui D. Cancer Nano-Immunoengineering: The Marriage of Immunoengineering and Nanotechnology for Cancer Therapy. Nano Biomed Eng (2016) 8:105–7. doi: 10.5101/nbe.v8i2.p105-107
148. van der Meel R, Sulheim E, Shi Y, Kiessling F, Mulder WJM, Lammers T. Smart cancer nanomedicine. Nat Nanotechnol (2019) 14:1007–17. doi: 10.1038/s41565-019-0567-y
149. Irvine DJ, Dane EL. Enhancing cancer immunotherapy with nanomedicine. Nat Rev Immunol (2020) 20:321–34. doi: 10.1038/s41577-019-0269-6
150. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov (2019) 18:175–96. doi: 10.1038/s41573-018-0006-z
151. Nam J, Son S, Park KS, Zou W, Shea LD, Moon JJ. Cancer nanomedicine for combination cancer immunotherapy. Nat Rev Mater (2019) 4:398–414. doi: 10.1038/s41578-019-0108-1
152. Duan X, Chan C, Lin W. Nanoparticle-Mediated Immunogenic Cell Death Enables and Potentiates Cancer Immunotherapy. Angewandte Chemie Int Edition (2019) 58:670–80. doi: 10.1002/anie.201804882
153. Corradetti B, Pisano S, Conlan RS, Ferrari M. Nanotechnology and Immunotherapy in Ovarian Cancer: Tracing New Landscapes. J Pharmacol Exp Ther (2019) 370:636–46. doi: 10.1124/jpet.118.254979
154. Chen Q, Chen J, Yang Z, Xu J, Xu L, Liang C, et al. Nanoparticle-Enhanced Radiotherapy to Trigger Robust Cancer Immunotherapy. Adv Mater (Deerfield Beach Fla) (2019) 31:e1802228. doi: 10.1002/adma.201802228
155. Jin WL, Jin MZ, Tu YY. Organoids: A Platform Ready for Glioblastoma Precision Medicine? Trends Cancer (2020) 6:265–7. doi: 10.1016/j.trecan.2020.01.016
156. Yang C, Xia BR, Jin WL, Lou G. Circulating tumor cells in precision oncology: clinical applications in liquid biopsy and 3D organoid model. Cancer Cell Int (2019) 19:341. doi: 10.1186/s12935-019-1067-8
157. Jin MZ, Han RR, Qiu GZ, Ju XC, Lou G, Jin WL. Organoids: An intermediate modeling platform in precision oncology. Cancer Lett (2018) 414:174–80. doi: 10.1016/j.canlet.2017.11.021
158. Liu HD, Xia BR, Jin MZ, Lou G. Organoid of ovarian cancer: genomic analysis and drug screening. Clin Transl Oncol (2020) 22:1240–51. doi: 10.1007/s12094-019-02276-8
159. Tuveson D, Clevers H. Cancer modeling meets human organoid technology. Sci (New York NY) (2019) 364:952–5. doi: 10.1126/science.aaw6985
160. Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell (2018) 175:1972–88.e1916. doi: 10.1016/j.cell.2018.11.021
161. Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ Jr., et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov (2018) 8:1404–21. doi: 10.1158/2159-8290.CD-18-0474
162. Schnalzger TE, de Groot MH, Zhang C, Mosa MH, Michels BE, Roder J, et al. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J (2019) 38:e100928. doi: 10.15252/embj.2018100928
163. Votanopoulos KI, Skardal A. ASO Author Reflections: Co-cultured Lymph Node and Tumor Organoids as a Platform for the Creation of Adaptive Immunity and Predict Response to Immunotherapy. Ann Surg Oncol (2020) 27:1968–9. doi: 10.1245/s10434-020-08351-7
164. Nadafi R, Gago de Graca C, Keuning ED, Koning JJ, de Kivit S, Konijn T, et al. Lymph Node Stromal Cells Generate Antigen-Specific Regulatory T Cells and Control Autoreactive T and B Cell Responses. Cell Rep (2020) 30:4110–23.e4114. doi: 10.1016/j.celrep.2020.03.007
165. Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515:563–7. doi: 10.1038/nature14011
166. Yuen KC, Liu LF, Gupta V, Madireddi S, Keerthivasan S, Li C, et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat Med (2020) 26:693–8. doi: 10.1038/s41591-020-0860-1
167. Schalper KA, Carleton M, Zhou M, Chen T, Feng Y, Huang S-P, et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat Med (2020) 26:688–92. doi: 10.1038/s41591-020-0856-x
168. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515:568–71. doi: 10.1038/nature13954
169. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500:415–21. doi: 10.1038/nature12477
170. Zhao G, Cardenas H, Matei D. Ovarian Cancer—Why Lipids Matter. Cancers (2019) 11:69–74. doi: 10.3390/cancers11121870
171. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature (2019) 569:73–8. doi: 10.1038/s41586-019-1118-2
172. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov (2015) 5:1282–95. doi: 10.1158/2159-8290.CD-15-1020
173. Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids (2013) 2:e105. doi: 10.1038/mtna.2013.32
174. Kailayangiri S, Altvater B, Lesch S, Balbach S, Göttlich C, Kühnemundt J, et al. EZH2 Inhibition in Ewing Sarcoma Upregulates G Expression for Targeting with Gene-Modified T Cells. Mol Ther J Am Soc Gene Ther (2019) 27:933–46. doi: 10.1016/j.ymthe.2019.02.014
175. Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest (2017) 127:929–41. doi: 10.1172/JCI89455
176. Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, et al. Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients With Relapsed Hodgkin Lymphoma. J Clin Oncol (2018) 36:1128–39. doi: 10.1200/JCO.2017.74.3179
177. Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clin Cancer Res (2019) 25:2915–24. doi: 10.1158/1078-0432.CCR-18-1811
178. Vergote I, Tropé CG, Amant F, Kristensen GB, Ehlen T, Johnson N, et al. Neoadjuvant Chemotherapy or Primary Surgery in Stage IIIC or IV Ovarian Cancer. N Engl J Med (2010) 363:943–53. doi: 10.1056/nejmoa0908806
179. Vacchelli E, Aranda F, Eggermont A, Galon J, Sautès-Fridman C, Cremer I, et al. Trial Watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology (2014) 3:e27878. doi: 10.1056/NEJMoa0908806
180. Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med (2012) 4:143ra199. doi: 10.1126/scitranslmed.3003807
181. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Sci (New York NY) (2011) 334:1573–7. doi: 10.1126/science.1208347
182. Bohm S, Montfort A, Pearce OM, Topping J, Chakravarty P, Everitt GL, et al. Neoadjuvant Chemotherapy Modulates the Immune Microenvironment in Metastases of Tubo-Ovarian High-Grade Serous Carcinoma. Clin Cancer Res (2016) 22:3025–36. doi: 10.1158/1078-0432.CCR-15-2657
183. Lo CS, Sanii S, Kroeger DR, Milne K, Talhouk A, Chiu DS, et al. Neoadjuvant Chemotherapy of Ovarian Cancer Results in Three Patterns of Tumor-Infiltrating Lymphocyte Response with Distinct Implications for Immunotherapy. Clin Cancer Res (2017) 23:925–34. doi: 10.1158/1078-0432.CCR-16-1433
184. Bakos O, Lawson C, Rouleau S, Tai LH. Combining surgery and immunotherapy: turning an immunosuppressive effect into a therapeutic opportunity. J Immunother Cancer (2018) 6:86. doi: 10.1186/s40425-018-0398-7
185. Amaria RN, Reddy SM, Tawbi HA, Davies MA, Ross MI, Glitza IC, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med (2018) 24:1649–54. doi: 10.1038/s41591-018-0197-1
186. Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. New Engl J Med (2018) 378:1976–86. doi: 10.1056/nejmoa1716078
187. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med (2019) 25:470–6. doi: 10.1038/s41591-018-0339-5
Keywords: ovarian cancer, immunotherapy, tumor microenvironment, combination therapy, immune checkpoint inhibitors (ICI)
Citation: Yang C, Xia B-R, Zhang Z-C, Zhang Y-J, Lou G and Jin W-L (2020) Immunotherapy for Ovarian Cancer: Adjuvant, Combination, and Neoadjuvant. Front. Immunol. 11:577869. doi: 10.3389/fimmu.2020.577869
Received: 30 June 2020; Accepted: 16 September 2020;
Published: 06 October 2020.
Edited by:
Nurit Hollander, Tel Aviv University, IsraelReviewed by:
Gian Franco Zannoni, Catholic University of the Sacred Heart, ItalyCopyright © 2020 Yang, Xia, Zhang, Zhang, Lou and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ge Lou, bG91Z2VAZW1zLmhyYm11LmVkdS5jbg==; Wei-Lin Jin, d2VpbGluamluQHlhaG9vLmNvbQ==; d2VpbGluamluQHNqdHUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.