 Charles Feldman
Charles Feldman Ronald Anderson
Ronald Anderson- 1Department of Internal Medicine, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
- 2Department of Immunology, Faculty of Health Sciences, Institute of Cellular and Molecular Medicine, University of Pretoria, Pretoria, South Africa
Community-acquired pneumonia (CAP) remains an important cause of morbidity and mortality throughout the world with much recent and ongoing research focused on the occurrence of cardiovascular events (CVEs) during the infection, which are associated with adverse short-term and long-term survival. Much of the research directed at unraveling the pathogenesis of these events has been undertaken in the settings of experimental and clinical CAP caused by the dangerous, bacterial respiratory pathogen, Streptococcus pneumoniae (pneumococcus), which remains the most common bacterial cause of CAP. Studies of this type have revealed that although platelets play an important role in host defense against infection, there is also increasing recognition that hyperactivation of these cells contributes to a pro-inflammatory, prothrombotic systemic milieu that contributes to the etiology of CVEs. In the case of the pneumococcus, platelet-driven myocardial damage and dysfunction is exacerbated by the direct cardiotoxic actions of pneumolysin, a major pore-forming toxin of this pathogen, which also acts as potent activator of platelets. This review is focused on the role of platelets in host defense against infection, including pneumococcal infection in particular, and reviews the current literature describing the potential mechanisms by which platelet activation contributes to cardiovascular complications in CAP. This is preceded by an evaluation of the burden of pneumococcal infection in CAP, the clinical features and putative pathogenic mechanisms of the CVE, and concludes with an evaluation of the potential utility of the anti-platelet activity of macrolides and various adjunctive therapies.
Introduction
A number of recent reviews, published by authors from different regions of the world, including North America, Latin America, Europe and Africa, attests to the fact that, globally, community-acquired pneumonia (CAP) remains a common cause of hospitalization, morbidity, mortality, and health-care costs (1–5). There are, however, notable variations in the burden and epidemiology of CAP in different regions of the world. These result from a number of factors, such as differences in population aging, increasing incidence of comorbidities, prevalence of smoking, antibiotic resistance, the introduction of pneumococcal conjugate vaccination (PCV) in children, and the use of pneumococcal and influenza vaccination in adults. Moreover, regional differences in the reported microbial etiology of CAP exist that are related, at least in part, to variations in the diagnostic methods used to identify the causative pathogens (1–5). Irrespective of these regional differences in prevalence and reported etiology, it is abundantly evident from most publications on this important public health issue that the clinical and economic burden of CAP “…is staggering, far-reaching, and expected to increase in the future…” (1).
The main aim of the current review, which fits with the theme of this special issue of “Frontiers in Immunology,” is to describe the emerging role of platelets in the pathogenesis of cardiovascular (CV) complications in patients with severe CAP, a major cause of morbidity and mortality in this condition. This encompasses not only CV complications in the setting of acute disease, particularly that caused by Streptococcus pneumoniae (the pneumococcus), but also the long-term sequelae of severe CAP and its apparent associations with persistent antigenemia and inflammation.
Burden of CAP
The Global Burden of Disease (GBD) Study, which evaluated the impact of lower respiratory tract infections (LRTIs; pneumonia and bronchiolitis) in 195 countries (6), documented in 2015 that these infections accounted for some 2.74 million cases (95% uncertainty interval [UI] of 2.5 million to 2.86 million). While the burden of LRTIs decreased dramatically between 2005 and 2015 in children younger than 5 years, the burden in adults >70 years has however, increased in many regions. With regard to etiology, some authors have noted that the prevalence of pneumococcal CAP appears to be decreasing in North America, but not in other regions of the world, such as Europe, and have attributed this to differences in vaccination rates and smoking habits (7). In the 2015 GBD study, the pneumococcus was the most common cause of LRTIs on a global scale, causing more deaths than all of the other studied respiratory pathogens combined, including Haemophilus influenzae, influenza virus and rhinovirus (6). Additional regional and national studies, such as those from the Eastern Mediterranean region (8) and Brazil (2, 9), supported by investigations from Europe (10), confirm the predominance of the pneumococcus as a cause of CAP.
Importantly, mortality rates of patients with CAP remain very high (11). Indeed, when reviewing the literature, it becomes apparent that mortality in CAP may occur early, described as occurring within the first 72 h to 7 days after hospital admission, or over the short term, commonly measured as the first 28–30 days after diagnosis or hospital admission (11). Importantly, even over the long term, as measured in months or years after hospital discharge, the mortality of CAP patients remains very high, being greater than that of patients admitted to the medical ward for conditions other than CAP, even when adjusted for age and comorbidity (11). While underlying co-morbidities may be important risk factors, there is increasing awareness that the occurrence of cardiac complications, even those occurring while the patient is in hospital, may impact not only on immediate and short-term mortality, but also on long-term mortality in patients who have survived an episode of CAP. In this context, a number of studies have investigated the utility of various biomarkers in predicting both the early and long-term mortality of CAP. Although elevated levels of several biomarkers of cardiovascular disease (CVD) or coagulation, measured either in hospital, or on hospital discharge of patients apparently recovered from acute CAP, were significantly associated with poorer outcomes, the significance of these findings is not entirely understood (11, 12). Do these biomarkers simply indicate the presence of underlying cardiac comorbidity, or are they non-specific markers of inflammation in CAP, or could they be indicators of acute cardiac injury due to CAP (11, 13)?
Cardiac Complications in All-Cause CAP
The occurrence of CV complications as a consequence of both acute bacterial and viral infections, has been known for some time (14, 15); however, it is now well-recognized that cases admitted with infections such as pneumonia and sepsis have an increased incidence of CV conditions such as acute myocardial infarction (AMI) or stroke, not only during hospital admission, but also occurring after hospital discharge. In one study comparing hospitalized patients with and without sepsis, the former patients had an increased incidence of AMI or stroke [(adjusted odds ratio 1.72; 95% confidence interval (CI) 1.60–1.85)], which occurred within 70 days of hospital discharge and was associated with an increased 180 day and 1 year mortality (15). Following adjustment for age, sex and comorbidity, these CV events (CVEs) were found to be independent risk factors for 180 day mortality.
Some studies have shown that both S. pneumoniae and influenza virus were particularly important in triggering a CVE (16, 17). However, one recently published abstract suggested that the frequency of major CVEs, referred to as MACE, and encompassing all-cause mortality, non-fatal AMI, stroke and heart failure, occurring within the first 90 days was 60% higher in CAP patients diagnosed as having a bacterial infection compared to those with viral pneumonia (17). The occurrence of these CAP-related complications is not surprising, since severe sepsis occurs early in the course of this infection in more than 30% of cases, and while it may involve multiple organ systems, it commonly involves the heart (18). In fact, some reviews have labeled pneumonia as a CVD (19). Clearly, there is no doubt that the occurrence of CVEs, is common, and after adjusting for confounders, is a significant risk factor for in-hospital mortality, as well as increased short-term mortality (20, 21).
Besides an increase in mortality, patients with CAP who develop CV dysfunction in hospital also have a significantly higher requirement for mechanical ventilation, need for inotropes and vasopressors, a higher rate of development of acute renal failure, and a more prolonged hospital stay, compared to CAP patients without a cardiac event (22). While the risk of AMI is greatest early in the course of infection and is related to the severity of infection, importantly it persists beyond the short-term infection period, exceeding the baseline risk even up to 10 years after infection (14).
Clearly, the major long-term negative outcome consequences in patients who have survived an episode of CAP are the occurrence of CVEs and mortality, which, as indicated above, are interrelated. Accordingly, prediction of these events early in the course of infection would represent a major advance that could enable early intervention. To this end, various investigators have assessed the utility of biomarkers, both systemic and cardiac, and risk stratification tools, such as severity of illness scores, as well as a combination of these as strategies to optimize outcome prediction in CAP patients (12, 23, 24). Given the absence of an ideal, single prognostic biomarker, current evidence suggests that measurement of a combination of biomarkers in CAP patients may offer the best predictive strategy. This involves combining measurement of the systemic inflammatory biomarkers, procalcitonin (PCT) and C-reactive protein (CRP), as indices of disease severity and short-term outcome assessment, with biomarkers of CV risk such as N-terminal pro-brain natriuretic peptide (proBNP), mid-regional pro-adrenomedullin (proADM), endothelin and troponins, together with severity of illness scores, such as the CURB-65 score (12, 23–25). In this setting, it is noteworthy that the CHA2DS2-VASc score has been found to be a good predictor of new-onset atrial fibrillation in patients hospitalized for CAP (25).
Cardiac Complications in Pneumococcal CAP
Musher et al. were the first to describe the occurrence of acute cardiac events in patients with pneumococcal CAP (26). They described 170 patients hospitalized with pneumococcal CAP, of whom 33 (19.4%) had one or more of the major cardiac events that have been described in patients with CAP; 12 patients had AMI [of whom two also had arrhythmia and five had new-onset or worsening congestive heart failure (CHF)]; eight had new-onset atrial fibrillation or ventricular tachycardia (of whom six also had new-onset CHF); and 13 had newly-diagnosed or worsening CHF on its own. While these are the classical cardiac changes described in patients with pneumococcal CAP, Chiong et al. have, more recently, described a teenager, with bacteremic pneumococcal pneumonia, who presented with features suggestive of an ST-elevation acute MI (STEMI) and cardiogenic shock that was due to myocarditis and rhabdomyolysis (27).
Several other recent studies have shown that (CVEs) occur relatively frequently in patients with pneumococcal CAP, which is the cause of CAP that is best studied with regard to pathogenic mechanisms (13, 28, 29). This contention is underscored by the findings of a recent secondary analysis of data from an international, multicenter, observational, cohort study involving 18 hospitals in 7 countries, encompassing 2,088 patients, 921 (44%) of whom had bacteremic pneumococcal pneumonia and 1,167 (55.9%) non-bacteremic pneumococcal pneumonia (30). Cardiovascular events were noted in 275 (13%) patients with a significantly higher incidence in bacteremic vs. non-bacteremic cases (15% vs. 12%; p = 0.02). There were 316 CVEs in total, of which 25 (7.9%) were AMI, 150 (47.5%) new and 51 (16.1%) worsening arrhythmia, and 51 (16%) new and 39 (12.4%) worsening heart failure. The rate of new-onset cardiac arrhythmias was also significantly higher in the bacteremic group.
With respect to the pathogenesis of CVEs in severe pneumococcal CAP, a number of very recent studies, mostly involving different animal models of experimental pneumococcal infection, have documented that the pneumococcus is able to translocate across the vascular endothelium and invade the myocardium (31–33). Once in the heart, the microorganism kills myocytes, as well as resident and infiltrating macrophages, via activation of necroptosis, causing significant damage to the myocardium that results in tissue remodeling (31–33). In their study, Shenoy et al. using a murine model of invasive pneumococcal disease, demonstrated that cardiac damage only occurred in the setting of infection with those pneumococcal serotypes that caused high-grade bacteremia (31). Cardiac damage was assessed according to elevated serum cardiac troponin-I levels, as well as cardiac histology that varied according to pneumococcal serotype (31). These findings in the experimental setting confirm those of the clinical study reported by Borsa et al. in patients with bacteremic and non-bacteremic pneumococcal pneumonia (30).
Cardiac damage in the setting of invasive experimental disease appears to be caused by pro-inflammatory mechanisms triggered by pneumococcal cell wall components, as well as by pneumolysin, the thiol-activated, pore-forming toxin of the pathogen (13, 28, 31, 34). Pneumolysin-mediated cardiac damage may result from the direct cardiotoxic/immunosuppressive activities of the toxin, as well as from its indirect pro-inflammatory/prothrombotic activities (28). In addition, work from our laboratory, and that reported by other investigators, has suggested that platelet activation and platelet-driven neutrophil activation may also contribute to the pathogenesis of these acute cardiac events (13, 35).
The following section of this review encompasses the role of platelets, both protective and harmful, in anti-bacterial host defense in the setting of CAP, most prominently in relation to the pneumococcus.
Platelets and Host Defense
Platelets originate during thrombopoiesis in the bone marrow, a process regulated by the hormone, thrombopoietin, produced in the liver and kidney (36). During thrombopoiesis, small, anuclear pro-platelets originate from marrow-derived megakaryocytes. Adult humans produce platelets at a steady state of 1011 cells/daily and maintain circulating levels of these cells at between 150 and 450 × 109 platelets per liter (L) of blood, although the tempo of production may increase in times of heightened demand by 20-fold or even more (36). In the circulation, platelets have a lifespan of 7–10 days that is determined by the balance between the cellular levels of the anti-apoptotic and pro-apopototic factors, Bcl-x(L) (B cell lymphoma-extra large) and Bak [protein encoded by the BAK1 gene (BCL2 antagonist/killer1)], respectively (37). Although circulating platelet counts decline with age, somewhat paradoxically, this decrease in the numbers of these cells is associated with acquisition of a pro-inflammatory phenotype (38). The age-related transition of platelets to a more reactive state is associated with increased generation of reactive oxygen species (ROS), mitochondrial dysfunction and activation of the mammalian target of rapamycin (mTOR) pathway (38). In addition, the serine esterase, granzyme A, has recently been detected in non-α-granule, sub-membranous areas of human platelets, attaining levels that are up to 9-fold higher in platelets from older as opposed to younger adults (39). Platelet-derived granzyme A also appears to contribute to the pro-inflammatory phenotype of aging platelets via activation of Toll-like receptor (TLR) 4- and caspase-1-dependent mechanisms that potentiate the synthesis of the monocyte-derived chemokines, interleukin (IL)-8 (CXCL8) and monocyte chemoattractant protein-1 (MCP-1, CCL2) (39).
These age-related alterations in the numbers and reactivity of circulating platelets are likely to reflect the chronic, low-grade, systemic inflammation that is associated with age-related co-morbidities (39). This type of sub-clinical, intravascular instability may predispose elderly adult humans for development of severe organ dysfunction, or even failure, during episodes of serious microbial or viral infection.
Putative mechanisms underpinning these threats, specifically in the context of serious CVEs in patients with invasive CAP, are covered in a later section of this review.
Platelets and Infection
Although the primary function of the circulating platelet is to maintain homeostasis through continuous monitoring of vascular endothelial integrity (36), platelets also play a key role in the systemic recognition and control of invasive microbial and viral pathogens. As highlighted in a series of recent reviews, platelets trap, sequester and, in some cases, eliminate invasive pathogens (40–47). Importantly, platelets also enlist assistance from more powerful cellular components of host defense. In this context, by trapping, weakening and immobilizing invasive pathogens, facilitating their access to neutrophils and monocytes, platelets coordinate and amplify the protective activities of the cellular and humoral elements of both the innate and adaptive immune systems (40–47).
Mechanisms utilized by platelets to counter the threat posed by pathogens that breach local host defenses at sites of primary infection have recently been covered in detail elsewhere (43–47). Accordingly, only direct mechanisms of antimicrobial activity mediated by platelet-derived anti-infective peptides/polypeptides, as well as indirect mechanisms involving high mobility group box 1 (HMGB1) protein, have been included here. Thereafter, the remaining sections of this part of the review focus on mechanisms and adverse consequences of systemic over-reactivity of platelets in the setting of CAP caused by highly invasive bacterial pathogens, particularly the pneumococcus.
Mechanisms Utilized by Platelets to Trap Pathogens During Invasive CAP
Despite their small size (2–3 μm diameter), platelets are endowed with abundant cytoplasmic granules containing an array of pre-formed mediators with a range of biological activities. Alpha-granules predominate in the platelet cytosol, numbering 50–80/cell, while dense granules that number 3–5/cell and small numbers of T granules (contain TLR9 and protein disulfide isomerase) and lysozomes (contain acid hydrolases) are also present (48). Platelet activation occurs most prominently following exposure to thrombin, adenosine 5′-diphosphate (ADP), thromboxane A2, fibronectin and collagen; these platelet activators interact with proteinase-activated receptors 1 and 4, P2Y1 and P2Y12 purinergic receptors, the TP2 prostanoid receptor, the GPIIb/IIIa integrin and the α2β1 integrin/GPVI, respectively. Following activation, platelets undergo degranulation that results in the release of various cytokines, chemokines, pro-coagulants, pro-angiogenic mediators and growth factors that either act extracellularly, or replenish membrane receptors (45, 47). These various mediators initiate key platelet pro-thrombotic/pro-inflammatory activities, including intercellular adhesion (endothelial cells, neutrophils and monocytes), homotypic aggregation, autocrine activation, coagulation, migration, and, in the context of this review, pathogen capture and antimicrobial activity.
Platelet Receptors Involved in Trapping of CAP Pathogens
Platelets possess a range of mechanisms that enable trapping of invasive CAP pathogens, as well as other types of bacterial pathogens (40, 47, 49). Some are expressed constitutively while others are triggered during platelet activation. These mechanisms include:
• expression of the FcγIIA receptor, which interacts with pathogen-bound antibodies of the IgG2 subclass (49), a mechanism that may be of particular relevance in the context of capsular polysaccharide-expressing pathogens such as the pneumococcus and Haemophilus influenzae;
• expression of complement receptors, specifically cC1qR and gC1qR that interact with pathogen-bound, activated complement component C1q, as well as receptors for C3a and C5a, which may be potentiated by discharge of platelet α-granule-derived C3 (50);
• binding of Staphylococcus aureus and various streptococcal species to platelet glycoprotein 1b (GP1b) via surface protein adhesins, or, in the case of S. aureus, by an additional, albeit indirect, mechanism involving von Willebrand factor (40);
• expression of a range of Toll-like receptors (TLRs 1, 2, 3, 4, 6, 7, and 9) that have the potential to trap invasive bacterial and viral pathogens (51); TLR2, via interactions with lipoteichoic acids and peptidoglycan, as well as TLR4 interactions with bacterial lipopolysaccharide, have the potential to entrap Gram-positive and Gram-negative bacteria, respectively. In the case of viral pathogens, platelets capture influenza virus particles via a TLR7-dependent mechanism (52). Although this mechanism may be protective, it also poses the risk of vascular occlusion due to release of C3 from platelets and excessive neutrophil activation (52).
With respect to the pneumococcus, “selective” interaction of this pathogen with platelets may be mediated by several putative mechanisms. These involve interaction of: (i) cell-wall phosphorylcholine with the platelet-activating factor receptor (PAFR) (53); (ii) the pneumococcal adhesins, PavB and PavC, with the GPIIb/IIIa(α2b/β3a)/fibrin/thrombospondin-1 complex (54); (iii) the phage-encoded pneumococcal adhesins, pblA and pblB, with platelet membrane gangliosides (55); and (iv) activation of TLR4 by pneumolysin, as well as by toxin devoid of cytolytic activity (56). Moreover, it is noteworthy that platelets are activated by hydrogen peroxide (57, 58), which is produced in large amounts by the pneumococcus, representing a putative, albeit unproven, additional mechanism of activation of these cells by the pathogen.
These various mechanisms of pathogen/platelet interactions, several of which result in platelet activation, expose the pathogens to the anti-infective mechanisms of the platelet, mostly antimicrobial peptides/polypeptides located predominantly, but not exclusively, in α-granules.
Granule-Derived Mechanisms Utilized by Platelets to Neutralize Microbial Pathogens
Platelet granule-derived, anti-infective peptides/polypeptides are mostly cationic amphiphiles with broad-spectrum activity against microbial and viral pathogens. They include α- and β-defensins, kinocidins, thrombocidins, and thymosin β-4.
Platelet Defensins
Human platelets and megakaryocytes have recently been reported to express defensin α-1 (DEFA1), also known as human neutrophil peptide-1 (HNP1), which co-localizes with platelet α-granules and has also been found in the cytoplasm of a megakaryoblastic leukemia cell line (59). On exposure to activators such as thrombin, ADP or lipopolysaccharide, DEFA1 relocates to the platelet outer surface where it exhibits anti-bacterial activity against the Gram-negative bacterial pathogen, Escherichia coli, confirming its involvement in platelet-mediated antimicrobial activity (59).
Platelets apparently also contain two human β-defensins (HBDs), namely HBD1 and HBD3, usually found in epithelial cells (60). In the case of HBD1, mRNA encoding this antimicrobial peptide, as well as the intact protein, are located in platelet extra-granular cytoplasmic compartments (60). In keeping with a non-granule intracellular location, extracellular discharge of HBD1 did not occur following exposure of platelets to receptor-mediated mobilization of cytoplasmic granules (60). Exposure of platelets to the pore-forming α-toxin of S. aureus, however, did induce release of HBD1, which was associated with inhibition of the growth of various clinical strains of this pathogen (60). In addition, HBD1 induced the formation of neutrophil extracellular traps (NETs) as an auxiliary mechanism of antimicrobial activity (60). Interestingly, proteolysis of the reduced, active form of HBD1 resulted in the formation of an octapeptide with significantly increased antimicrobial potency and a broader spectrum of activity, encompassing antibiotic resistant organisms including E. coli, Pseudomonas aeruginosa, and Candida albicans (61).
In the case of HBD3, the evidence in support of an intracellular localization of this antimicrobial peptide is somewhat less convincing, as it is based on the presence of this anti-infective agent in human platelet concentrates, as opposed to isolated cells, with no evidence provided in support of intracellular detection and location (62). Although interesting, further exploration is necessary to establish or refute the role of HBD3 in platelet anti-infective activity.
Kinocidins
These are primarily chemokines, specifically CXCL4 [also known as platelet factor 4 (PF4)], CXCL7 [also known as platelet basic protein (PBP)] and CCL5 (also known as Regulated on Activation, Normal T Cell Expressed and Secreted (RANTES)] that possess secondary antimicrobial activities, with CXCL4 appearing to be the most potent of these (63–66). Platelet kinocidins are vulnerable to proteolytic cleavage by thrombin, resulting in the generation of smaller antimicrobial peptides known as thrombocidins that are C-terminal deletion products of CXC chemokines (64, 65). In this context, CXCL7 undergoes proteolytic modification to generate two antimicrobial peptides known as thrombocidin-1 (TC-1) and thrombocidin-2 (TC-2) (67). Kinocidins and thrombicidins are present in platelet α-granules and, like defensins, they are amphipathic, cationic antimicrobial agents that promote membrane disruption in target pathogens (68).
Thymosin Beta-4
Platelet α-granules also contain high concentrations of the ubiquitous 43-amino acid reparative, actin-binding polypeptide, thymosin β-4 (TB4), which also possesses antimicrobial properties (65, 69, 70).
High Mobility Group Box 1 (HMGB1) Protein and Platelets
HMGB1 serves a critical role in nucleated cells by maintaining genomic stability and structural integrity (71). Somewhat surprisingly, however, anucleate platelets represent a major source of HMGB1, underscoring the existence of roles for this protein beyond maintaining nuclear stability (72, 73). In this context, megakaryocytes synthesize and deliver both HMGB1 and its encoding mRNA to platelets, where both have a cytosolic location (73). Following receptor-mediated activation of platelets by recognized agonists, mobilization of α-granules results in upregulation of expression the adhesion molecule, CD62P (P-selectin). This event, in turn, promotes the formation of platelet/leucocyte (neutrophils and monocytes) heterotypic aggregates, as well as binding of platelets and platelet aggregates to vascular endothelial cells. Cellular adhesion is achieved via expression of the counter receptor for CD62P, namely P-selectin glycoprotein ligand 1 (PSGL-1). These platelet-driven cellular interactions result in the translocation of HMGB1 from the cytosol of these cells to the plasma membrane where it interacts with several types of pro-inflammatory receptors expressed on adherent neutrophils, monocytes and endothelial cells, as well as with platelets per se, causing secondary autocrine activation of these cells. These receptors are TLR4 and RAGE (receptor for advanced glycation end products) (73, 74). In addition to interacting with these receptors, HMGB1 also functions as a ligand for TREM-1 (triggering receptor expressed on myeloid cells 1), a receptor expressed on neutrophils and monocytes that amplifies TLR4- and RAGE-mediated intracellular signaling (75).
Importantly, HMGB1 is a composite protein that exists as different isoforms determined by the redox status of the three cysteine residues, namely C23, C45, and C106. The fully oxidized isoform is seemingly biologically inactive. However, the remaining two isoforms possess distinct pro-inflammatory activities. These include firstly, an isoform of HMGB1 that results from selective oxidation of the proximal cysteines, C23 and C45, resulting in formation of an intramolecular disulfide bond, while C106 remains in the reduced state (76). These redox modifications enable this variant of HMGB1 to function as a ligand for TLR4 (77). However, the predominant isoform of HMGB1with respect to functioning as an effective ligand for RAGE and TREM-1 has not been identified (78, 79).
Secondly, an isoform in which all three cysteine residues are unmodified, this being the fully reduced form (76, 80). This isoform of HMGB1 forms a heterocomplex with the chemokine, CXCL12 (stromal-derived factor, SDF-1) (81), that is present in platelet α-granules (82). Interaction of CXCL12 with the fully reduced isoform of HMGB1 augments the affinity of the chemokine for its receptor, CXCR4, thereby amplifying recruitment of immune and inflammatory cells (81).
In the case of neutrophils, engagement of platelet-derived HMGB1 with TLR4 or RAGE has been reported to promote the formation of NETs (83, 84), which, if well controlled, may also contribute to intravascular host defenses.
Platelets, HMGB1, Neutrophils, and Reactive Oxygen Species
Like neutrophils, platelets possess the NOX2 isoform of NADPH oxidase. However, the levels of superoxide and H2O2 produced by platelets following activation with thrombin are apparently 1,500- and 4,000-fold lower than those generated by activated neutrophils (85). Nevertheless, as described in several earlier studies, this low level of production of ROS does appear to be important in augmenting activation of the fibrinogen/fibronectin-binding integrin, GPIIb/IIIa, following activation of platelets with thrombin (86, 87), a contention that has, however, been challenged in a more recent study (88). Accordingly, ROS derived from platelet-activated neutrophils may predominate with respect to augmentation of platelet activation. In this context, platelet-derived HMGB1 activates neutrophil NOX2, both in vitro and in vivo, by a TLR4-dependent mechanism (89, 90). These neutrophil-targeted, pro-oxidative activities of HMGB1 may further amplify neutrophil/platelet heterotypic interactions as a result of ROS-mediated, Ca2+-dependent activation of the neutrophil integrin, αMβ2 (Mac-1; CR3) (91), via interaction with its recently identified alternate ligand, PF4 (CXCL4), expressed on the platelet membrane (92).
Table 1 summarizes the mechanisms of direct and indirect platelet-mediated anti-infective activity involving antimicrobial peptides/polypeptides and HMGB1, respectively.
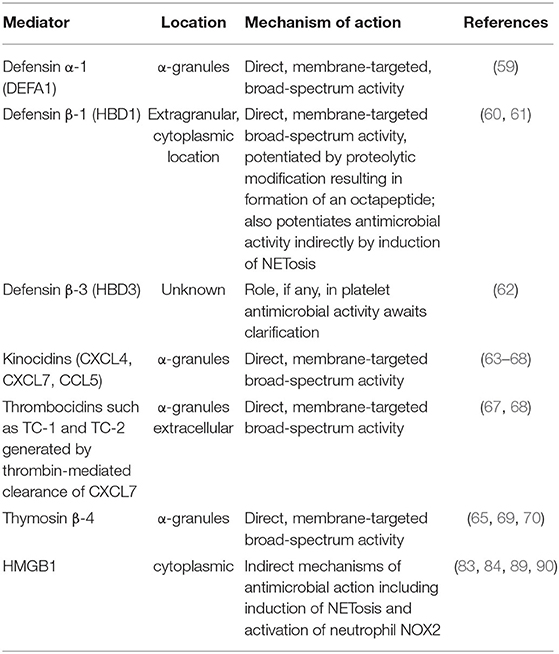
Table 1. Platelet-derived mediators of antimicrobial activity.
Platelets, HMGB1, and Protection Against Bacterial Infection in the Experimental Setting
Zhou et al. using a murine model of intra-abdominal sepsis induced by cecal ligation and puncture have recently described the involvement of platelet-derived HMGB1 in countering the threat posed by invasive bacterial infection (90). To explore, more definitively, the role of platelet HMGB1 in the pathogenesis of experimental sepsis, specifically clearance of bacteria from the peritoneum and circulation, these authors generated gene knockout mice with selective deletion of the genes encoding either HMGB1 individually, or the combination of HMGB1/PF4, in megakaryocytes (90). Using their model of polymicrobial sepsis, the authors observed that mice with HMGB1/PF4-depleted platelets demonstrated significantly greater mortality than wild-type mice that was associated with increased bacterial loads in the peritoneum and blood, as well as an intense systemic inflammatory response (90). Mice harboring platelets selectively depleted of HMGB1 demonstrated decreased levels of the platelet-derived chemokines, RANTES and PF4 in the peritoneal cavity, as well as decreased platelet/neutrophil interaction in the airways. With respect to in vitro studies, exposure of neutrophils to platelets genetically depleted of HMGB1 resulted in significant impairment of formation of neutrophil/platelet aggregates and production of ROS by neutrophils (90).
The authors concluded that these observations revealed the previously unrecognized involvement of platelet-derived HMGB1 in the regulation of neutrophil recruitment and activation by modulating platelet activation during sepsis (90), presumably augmented via the participation of PF4.
Clinical Studies on the Role of Platelets in Protection Against Invasive Bacterial Infections
Claushuis et al. have described clinical studies supporting the involvement of platelets in protecting against invasive bacterial infection. In 2016, these authors reported a retrospective analysis of a study undertaken between 2011 and 2013 that was focused on the relationship between circulating platelet counts and mortality in critically ill sepsis patients (n = 929) (93). Thrombocytopenia, particularly very low circulating platelet counts of <50 × 109/L blood, detected in patients with severe sepsis on admission to a hospital intensive care unit, was associated with intense systemic inflammation and significantly increased mortality relative to those patients with intermediate and normal platelet counts (93). More recently, the same authors reported a similar type of retrospective analysis undertaken in Thailand between 2002 and 2006, in which they investigated relationships between circulating platelet counts and mortality in a cohort of patients (n = 1,160) hospitalized with culture-proven melioidosis (94). This infective condition presents as a severe pneumosepsis caused by the Gram-negative bacterial pathogen, Burkholderia pseudomallei, which is common in Southeast Asia and has a mortality rate of up to 40% (94). The authors again reported that the presence of thrombocytopenia measured at the time of hospital admission was predictive of mortality (94).
To explore mechanisms underpinning the involvement of platelets in disease pathogenesis, the same authors developed a murine model of experimental melioidosis (94). This model revealed the following: i) experimental infection with B. pseudomallei resulted in decreased circulating platelet counts and increased mortality that was enhanced by administration of an anti-platelet antibody targeted against the von Willebrand factor-binding glycoprotein, GPIbα; and ii) early lung bleeding that was associated with altered vascular integrity (94). This latter observation is consistent with the role of platelets in maintaining vascular endothelial integrity, especially in preventing vascular leakage during neutrophil extravasation (95).
Kirby et al. confirmed the findings of the clinical component of the aforementioned study (96). These Australian investigators also conducted a retrospective analysis covering the period 1999–2017, encompassing patients (n = 758) with proven melioidosis. These authors reported that patients who experienced bacteremia or septic shock had significantly lower platelet counts (227 × 109/L and 217 × 109/L, respectively) than those who did not (corresponding values of 323 × 109/L and 266 × 109/L; p < 0,001 and p = 0,002, respectively). Although found to be an independent predictor of mortality, the authors concluded that the platelet count is not a useful biomarker to predict severity of melioidosis as most patients with severe disease still had counts that were within the normal range (96).
Pathogen-Mediated Chronic and Hyperacute Activation of Platelets
The evidence presented in the preceding sections of this review is clearly supportive of a protective role for tightly controlled activation of platelets in anti-infective host defense in the setting of maintenance of vascular integrity. This contention must, however, be balanced against evidence to the contrary, which has implicated platelets in the pathogenesis of organ dysfunction in both chronic and acute inflammatory disorders of infective origin, including severe invasive CAP.
Platelets in the Pathogenesis of Organ Dysfunction Associated With Chronic Bacterial Infection
In the case of chronic infection, Entezari et al. in an earlier study reported that levels of HMGB1 were significantly elevated in sputa and bronchoalveolar lavage fluid (BALF) from patients with cystic fibrosis (CF) chronically infected with P. aeruginosa (97). Although the cellular source of HMGB1 was not identified, it is nevertheless noteworthy that platelets appear to be intimately involved in the pathogenesis of CF (98, 99). In an extension of their clinical study, Entezari et al. developed a murine model of CF in which the gene encoding the CF transmembrane conductance regulator had been deleted (CFTR −/− mice). This murine model of CF enabled the authors to explore possible associations between elevated HMGB1 and impairment of pulmonary anti-bacterial host defenses in the airways of mice experimentally infected with P. aeruginosa via oropharyngeal aspiration (97). A monoclonal antibody (mAb) targeted against rat rHMGB1, or a dummy Ab, were administered to animals in the experimental and control groups, respectively. The authors observed that administration of the HMGB1-neutralizing antibody either prior to, or at the time of infection, protected both CFTR −/− and wild-type mice against influx of neutrophils into the airways and pulmonary damage in the setting of a decreased bacterial load (97). In addition, they also observed that: (i) BALF from CF patients suppressed the phagocytic activity of both murine peritoneal macrophages and a macrophage cell line (RAW264.7); and (ii) murine macrophages in which the TLR4 gene had been inactivated were resistant to the anti-phagocytic action of HMGB1 (97).
Platelets in the Pathogenesis of Hyperacute Systemic Inflammation and Associated Organ Dysfunction in Severe, Invasive CAP
Evidence in support of this contention is derived in large part from studies that have investigated relationships between the numbers, dimensions and activation status of circulating platelets with prognosis in patients with severe, all-cause, invasive CAP.
With respect to alterations in the numbers of circulating platelets in patients hospitalized with severe CAP, both thrombocytopenia (100–106) and thrombocytosis (104–108), measured in most cases at the time of hospital admission, are associated with significantly increased mortality, either in-hospital and/or post-discharge. With respect to alterations in platelet turnover and reactivity, an increase in the mean platelet volume (MPV) measured as an increment over the first 4 days of hospital admission, is, according to one study, a significant predictor of mortality among adult patients admitted to an intensive care unit with severe pneumonia (109). In another recent study, the MPV/platelet count (MPV/PC) ratio, which interestingly is a marker of CVD, significantly predicted 30 day mortality in patients with pneumonia and acute ischemic stroke relative to those with acute ischemic stroke only (110).
With respect to systemic activation of platelets in severe pneumococcal CAP, Tunjungputri et al. described a strong association between expression of the platelet-binding pneumococcal adhesin, pblB, and 30 day mortality in hospitalized patients (n = 349) with proven (molecular/microbiological confirmation) pneumococcal bacteremia (111). In vitro experiments revealed a mechanistic interaction between expression of pblB on the pathogen and platelet activation. In this context, exposure of a pblB-expressing strain of the pneumococcus, but not a paired pblB gene-knockout strain, to isolated platelets resulted in increased expression of platelet CD62P, activation of GPIIb/IIIa, and formation of platelet/monocyte aggregates (111).
In the case of patients hospitalized with all-cause CAP, prolonged platelet activation detected at the time of hospital admission that persisted for up to 30 days has been described (112). In these studies, sustained platelet activation was measured according to prolonged upregulation of expression of CD62P and formation of platelet/leukocyte and platelet/monocyte heterotypic aggregates (112).
As mentioned above, approximately 30% of patients with severe CAP develop sepsis. In this context platelet gene expression (mRNA transcripts) has recently been measured in patients hospitalized with severe sepsis (n = 118, mean platelet counts 186 ± 92 vs. 287 ± 59 for 52 matched healthy subjects, P < 0.0001; MPV values comparable; sites of primary infection and causative pathogens not mentioned) (113). Within 24 h of onset of sepsis, both in humans and in mice, it was revealed that megakaryocytes initiate trafficking of mRNA encoding the ITGA2B gene (encodes integrin α-chain 2b) to platelets (113). Subsequent translation resulted in de novo synthesis of the αIIb component of integrin GPIIb/IIIa, as well as integrin activation, which were associated with mortality in both humans and mice (113).
Table 2 summarizes these various abnormalities of platelet numbers and reactivity in patients with severe CAP.

Table 2. Abnormalities of platelet numbers and reactivity in patients with severe CAP.
Platelets and the Pneumococcus
As alluded to above, the pneumococcus possesses several types of adhesin that promote binding of the pathogen to human platelets, which in the case of the FcGIIAR, TLR2, and pblB, are likely to trigger platelet activation and mobilization of antimicrobial peptides. Interestingly, however, it has recently been reported that the pneumococcus, unlike E. coli and S. aureus, is resistant to killing by TRAP-6 (thrombin mimetic)-activated human platelets or by exposure to platelet releasate (cell-free supernatant of TRAP-6-activated platelets); however, the anti-pneumococcal effects of platelet lysate were not reported (115). Although the mechanism of resistance of the pneumococcus to platelet-mediated killing was not established, it is noteworthy that platelets did not appear to survive exposure to the pathogen (115).
With respect to HMGB1/pneumococcus interactions, invasive pneumococcal disease is associated with elevated levels of this protein in sputum (116). Increased sputum levels of HMGB1, albeit of unknown cellular origin, are significantly associated with bacteremia, but not disease severity, indicative of a possible, albeit unproven, role for HMGB1 in promoting dissemination of the pneumococcus that possibly involves platelets (116).
Taken together with the various strategies utilized by the pneumococcus to evade NETosis (114, 117–120), a process triggered and amplified by platelet-dependent mechanisms, the aforementioned evidence suggests that the pneumococcus may be particularly adept at evading and even hijacking platelet-driven host defenses. In the setting of invasive pneumococcal CAP, the resultant misdirected, systemic pro-inflammatory/pro-thrombotic activities of platelets may contribute to the pathogenesis of the high prevalence of CV complications associated with this condition.
Pathogenic Mechanisms of Cardiovascular Complications in Patients With CAP
Multiple mechanisms are likely to contribute to the pathogenesis of acute cardiac events in CAP, including pneumococcal CAP. These include the pro-thrombotic, pro-coagulant state associated with acute infection that encompasses platelet activation and production of pro-coagulants, NETosis and impaired anti-coagulant activity of the endothelium (14). Although it is clear that invasive CAP is associated with increased systemic reactivity of platelets, and with the occurrence of AMI, a mechanistic association between these events, while plausible, has nevertheless not been conclusively established. Given the therapeutic potential of adjunctive platelet-targeted therapies in severe CAP, this represents a topical and compelling theme of current clinical and laboratory infectious diseases research.
Evidence From Clinical Studies Implicating the Involvement of Platelets in the Pathogenesis of Cardiovascular Events in Patients With All-Cause CAP
Cangemi et al. were among the first clinical researchers to identify a potential link between platelet activation and the development of CVEs in the setting of acute, albeit all-cause, CAP (114). The investigators studied 278 consecutive patients hospitalized for CAP, who were followed to discharge (114). They determined systemic levels of several platelet activation markers, including soluble P-selectin and CD40 ligand, thromboxane B2 (TxB2, a surrogate for TxA2) and high-sensitivity cardiac troponin T, together with measurement of electrocardiograms at 12 and 24 h intervals. Among 144 patients with elevated high-sensitivity troponin, 31 had signs of AMI and 113 did not, while baseline levels of the platelet markers were elevated in all patients with signs of AMI. Logistic regression analysis revealed that elevated soluble P-selectin (p < 0.001), soluble CD40 ligand (p < 0.001), TxB2 (p = 0.030), mean platelet volume (p = 0.037), the Pneumonia Severity Index (PSI; p = 0.030) and cardiac ejection fraction (p = 0.001) were independent predictors of MI (114). Interestingly, no significant differences in the occurrence of MI were detected between patients (n = 123) on aspirin prophylaxis [100 mg/day; inhibitor of cyclooxygenase 1 (COX-1)] and those that were not and aspirin-treated patients with MI had higher serum TxB2 levels than those without MI (p = 0.005).
The accompanying editorial raised several questions. Firstly, is platelet activation the cause of MI, given that aspirin therapy did not reduce the risk of MI, and recognizing that there are many pathways to platelet activation and simply interrupting one pathway may not be sufficient (120)? Secondly, is platelet activation simply a marker of disease severity, noting that the studies, including the current one, document that severe CAP is an independent risk factor for MI (120)? In addition, a vigorous debate ensued in the literature as to whether this study had in fact demonstrated a causal link between platelet activation and MI in patients with CAP. This contention was based on a number of issues, such as studying platelet activation rather than aggregation, the suitability of the platelet markers used, the possibility of underlying comorbidities being associated with elevated platelet reactivity, and the presence of renal failure, among others (121–123), to which the authors of the study and the editorial responded (124–127). Interestingly, one of the letters raised the issue of the use of statins as adjunctive therapy in severe CAP, a suggestion with which the authors of the initial study agreed and this is discussed more fully below (128).
A later study undertaken by the same authors involved consecutive patients with CAP admitted to a University Hospital in Rome who were followed up prospectively to discharge or death (129). The primary endpoint was death up to 30 days after admission, while the secondary endpoint was intra-hospital occurrence of non-fatal MI and ischemic stroke. One thousand and five patients (mean age 74.7 ± 15.1 years) were recruited of whom 390 were receiving aspirin (100 mg/day), while 615 were aspirin free. Overall, 16.2% of patients died [19 (4.9%]) among the aspirin users and 144 (23.4%; p < 0.001) among the non-users]. Non-fatal CVEs occurred in 4.9% of aspirin users and 8.3% of non-users (odds ratio 1.77; 95%CI 1.03–3.04; p = 0.040). Compared with patients taking aspirin the propensity score adjusted analysis confirmed that those not taking aspirin had a hazard ratio of 2.07 (95% CI 1.08–3.98; p = 0.029) for total mortality. An additional study by the same investigators was a post hoc analysis of patients presenting with septic shock in association with CAP and healthcare-associated pneumonia (130). The findings of this study suggested that use of both low-dose aspirin (100 mg/day) and macrolide antibiotics was associated with a lower mortality, possibly because of the anti-inflammatory effects of both the study drugs, together with the reduction in CVEs with aspirin (130).
This apparent link between excessive systemic activation of platelets activation and development of CVEs in CAP is supported by a more recent retrospective study of 351 hospitalized CAP patients, in which platelet counts were measured on admission and after 3–5 days (100). The authors noted that initial platelet counts were significantly lower in those patients who suffered MACE, which was defined as mortality, acute myocardial infarction or stroke, during the 1 year follow-up, compared with those that did not (225 × 109/L vs. 261 × 109/L; p = 0.036) (100). Those patients with a normal initial platelet count on hospital admission who subsequently developed thrombocytopenia (<150 × 109/L) had a worse outcome, not only for all-cause mortality (HR 7.75; p = 0.002), but also for MACE (HR 7.4; p = 0.002) regardless of age, severity of pneumonia or comorbidity (100).
Evidence Linking Platelet Activation to the Pathogenesis of Cardiovascular Events in Severe Pneumococcal CAP
Although the pneumococcus remains the most common and threatening bacterial cause of CAP, clinical studies linking platelet activation to outcome have invariably focused on mortality rates in all-cause CAP, rather than associations between platelets, pneumococcal infection and the prevalence of CVEs. Nevertheless, the study reported by Musher et al. has clearly identified that invasive pneumococcal disease carries a very high risk for development of serious CVEs (26), while that reported by Tunjungputri et al. has identified the association of severe pneumococcal infection with platelet activation and mortality (111).
Several experimental studies have also suggested a possible role for platelets in the pathogenesis of CVEs associated with severe pneumococcal disease. In this context, platelets, which capture, but do not kill, viable pneumococci, may act as vehicles that transport the pathogen to distant anatomical sites (47, 115).
Following translocation to the myocardium, the pneumococcus may intensify systemic platelet activation via interaction of these cells with various adhesins and other surface components of the pathogen, including complement activation products (40, 47–55).
In addition, release of pro-thrombotic/pro-inflammatory pneumolysin from disintegrating organisms also contributes to platelet activation. At sub-lytic concentrations, this pore-forming toxin induces Ca2+-dependent upregulation of expression of platelet CD62P, promoting homotypic platelet aggregation, heterotypic platelet/neutrophil aggregation and NET formation (131–133) that is exacerbated by mobilization of platelet HMGB1 (83, 84).
These events increase the risk of cardiac microvascular occlusion and myocardial dysfunction, a contention that is supported by findings that various biomarkers of platelet activation and neutrophil:platelet aggregates, as well as NETs and their constituents, such as histones, are major components of intravascular thrombi (134–136). In this context it is noteworthy that systemic histones, presumably derived from NETs, are associated with thrombocytopenia in critically ill patients, an association that has also been observed following experimental administration of histones to mice (137, 138). Although again focused on all-cause CAP and mortality (among other clinical end-points), the threat posed by intravascular NETosis in this condition has recently been underscored by the findings of a study communicated by Ebrahimi et al. (139). These authors observed that increased systemic levels of cell-free nucleosomes, a surrogate biomarker of NETosis, measured at the time of hospital admission, were associated with a 3.8-fold increased adjusted odds ratio of 30 day mortality (139).
Although the aforementioned mechanisms, together with the direct cytotoxic effects of pneumolysin on cardiomyocytes and resident cardiac macrophages (31–33), may underpin the pathogenesis of CVEs in acute, invasive pneumococcal infection, they do not explain the long-term cardiac sequelae in those who have seemingly recovered from this condition. Although unproven, persistent antigenemia has been proposed as a putative risk factor in this clinical setting, particularly in the case of the elderly, many of whom may have age-related platelet hyperreactivity and associated, persistent, low-grade systemic inflammation (38, 39).
Indeed, persistent antigenemia, particularly of pneumococcal capsular polysaccharides, for a duration up to 6 months post-infection, and possibly longer, has been detected in those who have recovered from an episode of pneumococcal CAP [reviewed in (13)]. Although the cellular reservoirs of pneumococcal antigen remain to be established, it is noteworthy that pneumolysin has been reported to interact with the mannose receptor C type 1 (MRC-1) expressed predominantly on M2-like anti-inflammatory macrophages and dendritic cells (140). Interaction of surface-expressed pneumolysin enables the pathogen to interact with MRC-1, resulting in uptake into non-lysozomal intracellular compartments that provide a safe haven for the pneumococcus (140).
Figure 1 summarizes the prominent role of pneumolysin in the pathogenesis of CVEs in pneumococcal CAP, encompassing platelet activation, as well as direct cardiotoxicity.
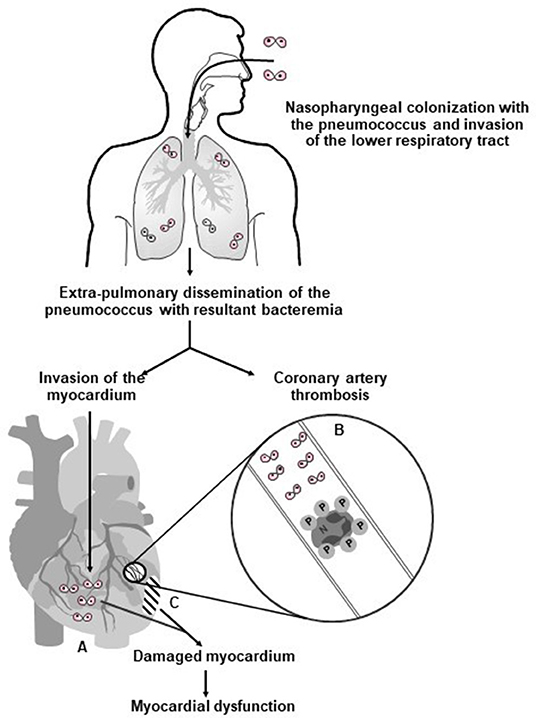
Figure 1. Proposed mechanisms involved in the pathogenesis of pneumolysin-mediated myocardial injury during invasive pneumococcal disease in humans. Following nasopharyngeal colonization, invasion of the lower respiratory tract, extra-pulmonary dissemination, and cardiac invasion by the pneumococcus (⊙⊙–symbol represents diplococci), intra-myocardial and intravascular release of pneumolysin (PLY) by the pathogen results in (A) PLY-mediated death and dysfunction of cardiomyocytes; (B) intravascular activation of platelets and neutrophils with resultant formation of pro-thrombotic/pro-NETotic neutrophil (N):platelet (P) aggregates (as illustrated in the magnification of an affected coronary arteriole/artery); and (C) development of myocardial damage and dysfunction. Reproduced with permission of Anderson et al. (28) under a Creative Commons License (https://creativecommons.org/licenses/by/4.0/).
Treatment of CAP
A detailed description of appropriate antibiotic, adjunctive, and supportive therapy in CAP is clearly beyond the brief of the current manuscript. However, it would seem appropriate to mention host- and pathogen-related risk factors that appear to predict the likelihood of a patient with CAP having a CVE, who could then be prioritized for personalized management with routine cardiac monitoring in the acute phase and targeted control of cardiovascular parameters regularly, both during and after hospital discharge (141, 142). This is followed by a section describing those aspects of antibiotic and adjunctive therapy that may relate to effects that these agents may have on platelet activity.
While CVEs have been documented to occur even in young, otherwise healthy, individuals, studies have indicated that older age, nursing home residence, pre-existing CVD, hyperlipidemia, hypoalbuminemia and higher pneumonia severity, such as assessed by the Pneumonia Severity Index (PSI), but also other markers of severity, appear to be important host risk factors (141, 143, 144). Viasus et al. (141) derived a simple rule based on demographic and clinical features that was helpful in identifying patients with CAP at higher risk of acute cardiac events. With regard to bacterial factors of importance, the latter study indicated that those patients with pneumococcal pneumonia appeared to be at greater risk of CVEs and we have highlighted previously in this manuscript additional studies that have noted the importance of pneumococcal bacteremia, and/or infection with pneumococcal serotypes that cause high-grade bacteremia as being most important in respect of this pathogen (30, 31). Other authors have suggested that patients with CAP due to Staphylococcus aureus and Klebsiella pneumoniae may also be at increased risk of CVEs (144). Lastly, investigators from Spain have noted that measurement of a range of cardiac biomarkers in the blood may be useful in identifying patients with CAP, at increased risk not only of early, but also long-term occurrence of CVE (12).
Platelet-Targeted Adjunctive Strategies in CAP
Notwithstanding the potentially beneficial primary and secondary inhibitory effects of macrolide antibiotics on platelets, adjunctive strategies to antibiotics based on the use of recognized platelet-targeted therapies clearly have the potential, albeit relatively unexplored, to attenuate the development of both acute and delayed-onset, life-threatening CVEs in severe CAP. These include pharmacological strategies that directly target platelet activation and their mediators of thrombosis and inflammation, while direct targeting of platelet-activating bacterial toxins represents an additional, albeit indirect, approach. In the case of the former, potential options include the use of: (i) inhibitors of thromboxane synthesis such as aspirin and corticosteroids; (ii) antagonists of ADP-responsive P2Y12 purinergic receptors; and (iii) antagonists of thrombin-responsive proteinase-activated receptors 1 and 4 (PAR1/PAR4). Future, potential strategies include targeting of P2Y1 purinergic receptors, HMGB1 and necroptosis.
Macrolide Antibiotics
Antibiotics are the mainstay of therapy in patients with CAP and must be administered as soon as the diagnosis is made to minimize the risk of mortality, while adjunctive therapies are used in those patients with more severe infection [reviewed in (145–147)]. In the case of severe infection, the antibiotic choice most commonly used is dual therapy, with the combination of a beta-lactam and a macrolide, usually azithromycin or clarithromycin (146). The rationale underpinning the use of macrolides in the combination therapy of all-cause CAP was initially based on the premise of expanded antibiotic coverage to include Legionella pneumophila, Mycoplasma pneumoniae, and Chlamydia pneumoniae. It is now well-recognized, however, that this latter class of antimicrobial agents possesses numerous additional activities that are beneficial in the treatment of CAP, including both antibiotic (148, 149) and non-antibiotic, neutrophil-targeted anti-inflammatory properties (148–151).
With respect to the former, macrolides as predominantly bacteriostatic agents that inhibit protein synthesis, appear to counteract the pro-inflammatory, platelet-activating activity of cell wall structures and toxins, such as pneumolysin, released following disintegration of bacterial pathogens by beta-lactam agents. This may be the mechanism by which most benefit is derived from combination therapy, where the macrolide is administered prior to the beta-lactam (152). In addition, macrolide antibiotics accumulate to extremely high levels in eukaryotic cells, including cells of the innate immune system and structural cells, countering both intracellular bacterial pathogens and development of antibiotic resistance (149).
With respect to non-antibiotic, anti-inflammatory effects of macrolides, we are aware of only one study that has investigated the effects of macrolides on platelet function. In this study reported by Tsoupras et al. in vitro exposure of washed rabbit platelets to therapeutically attainable concentrations of clarithromycin in particular, as well as azithromycin, resulted in significant attenuation of platelet-activating factor (PAF)-mediated aggregation of these cells, while the corresponding responses activated by thrombin were relatively unaffected (153). These findings await confirmation in the clinical setting.
The aforementioned beneficial effects of macrolides on reducing mortality in severe CAP imply that a similar effect may exist with respect to the occurrence of CVEs. To date, however, we are, unaware of stringently controlled clinical trials that have investigated the potential of macrolide-containing therapeutic regimens to ameliorate the development of CVEs in patients with severe CAP.
Aspirin Therapy
One of the earliest studies undertaken to investigate the utility of aspirin in preventing the development of acute coronary syndrome (ACS) in patients with CAP was reported by Oz et al. (154). Using a multicenter, prospective, randomized trial design, these authors allocated patients who were hospitalized with CAP, and who had more than one risk factor for CVEs, to a control group (n = 94) or to a group that received treatment with aspirin (300 mg/daily, n = 91 patients) for 1 month. The primary endpoint was development of ACS within 1 month, while high-sensitivity cardiac troponin T (hs-cTnT) as a biomarker of cardiac dysfunction was measured on admission and after 48 h (154). The frequencies of development of ACS were 1.1% (n = 1) and 10.6% (n = 10) in the aspirin-treated and control groups, respectively (relative risk = 0.103; 95% CI = 0.005–0.746; p = 0.015). In addition, a significantly higher proportion of patients in the control group had elevated levels of hs-cTnT at 48 h post-hospital admission (55.3% vs. 35.2% for the control and aspirin-treated groups, respectively, p = 0.006). Concurrent diabetes mellitus and a Framingham cardiac disease risk score of >20 were significant risk factors for development of ACS. Although of interest, perceived imitations of the study included small numbers of patients, lack of pathogen identification and absence of data on circulating platelet counts and activation status (154).
As mentioned above, the study reported by Cangemi et al. indicated that patients with CAP who received low-dose aspirin prophylaxis prior to and during hospitalization experienced no significant in-hospital benefit with respect to either mortality or occurrence of non-fatal CVEs (114). However, in a follow-up study, to which a larger number of hospitalized patients with CAP was recruited, both 30 day mortality and in-hospital occurrence of non-fatal CVEs as primary and secondary endpoints, respectively, were significantly decreased in patients receiving low-dose aspirin prophylaxis (129). Two subsequent studies by the same authors, one in a small cohort of patients (n = 188) with CAP-associated septic shock (130) and a much larger multicenter study spanning four countries, these being Italy, USA, China and Japan, encompassing 1,295 patients with severe CAP, revealed that the combination of low-dose aspirin prophylaxis (≥100 mg/daily) and treatment with a macrolide antibiotic significantly improved the 30 day survival rate (155). In the latter study, the 30 day survival rate in patients treated with the aspirin/macrolide combination (n = 148) was significantly better (p = 0.002) than the corresponding rates observed in the subgroups of patients treated with aspirin without a macrolide (n = 237), macrolide without aspirin (n = 294) and aspirin/macrolide untreated (n = 616) (155).
The authors of the latter study, while acknowledging the limitation of its observational design, proposed that clinical benefit of the macrolide/aspirin combination in severe CAP results from a dual anti-inflammatory (macrolide) and anti-platelet (aspirin) mechanism of action (155).
On a cautionary note, however, a recently reported retrospective study from Denmark is noteworthy. The aim of this study, authored by Basille et al. was to determine the possible association between the use of non-steroidal anti-inflammatory drugs (NSAIDs) and clinical outcome in hospitalized patients with CAP (156). Of the 59,250 patients aged >15 years recruited to the study, which was conducted from 1997 to 2011, 27,714 had a history of usage of NSAIDs (current, new, long-term and former). The authors reported that users of NSAIDs hospitalized with CAP were at higher risk than non-users for development of pleuropulmonary complications, with younger patients and those with no comorbidities seemingly at highest risk (156). The authors conclude that this risk may result from masking of symptoms of serious disease and/or interference with host pulmonary defenses (156).
Adjunctive Corticosteroid Therapy
Several recent meta-analyses have noted that adjunctive administration of corticosteroids (CS) to adult patients with severe CAP was associated with significant outcome benefits (157–160). In the latter study, there were differences documented in the mortality rates of the different corticosteroids, with prednisolone and methylprednisolone reducing total mortality, but use of hydrocortisone did not (160). With respect to the effects of CS on platelet function, older investigations noted differing results, which they attributed to different types and concentrations of CS used; however, in one study prednisolone was shown to have exquisite inhibitory properties on platelet function, which appeared to be related to selective regulation of ADP-activated P2Y12 receptor signaling, with the potential to attenuate vascular thrombotic episodes (161). In addition, Cangemi et al. who had previously reported an increased occurrence of AMI that was associated with elevated systemic levels of TxB2 in CAP patients (114), subsequently investigated the effects of CS on platelet activation in vitro and in vivo. In the first of two studies, these authors reported that betamethasone and methylprednisolone at concentrations of 150–600 nanograms/mL caused statistically significant inhibition ADP-activated aggregation of human platelets in vitro, in the setting of decreased activity of cytosolic phospholipase A2, and production of TxB2, seemingly consistent with an inhibitory effect on signaling mediated via activation of platelet P2Y12 and/or P2Y1 receptors (162). This was associated with levels of urinary 11-dehydro-TxB2 that were significantly higher (p < 0.001) in hospitalized CAP patients (n = 300) relative to those of control subjects (outpatients, n = 150) and independently predicted the occurrence of MI (162). These same investigators confirmed that CS use was associated with a lower incidence of MI in patients hospitalized with CAP (n = 758) in a retrospective record review of consecutively recruited patients followed prospectively to hospital discharge (163).
In the study alluded to above in which Ebrahimi et al. described an association between elevated biomarkers of intravascular NETosis and 30 day mortality in patients with severe CAP, it is noteworthy that these authors also noted that patients treated with prednisone demonstrated decreased NETosis and a beneficial outcome (139). However, others have recommended caution with regard to the routine use of CS other than perhaps for patients with severe CAP requiring ICU admission, citing the fact that many of the studies of CS use in CAP are relatively small, have some methodological issues, and the potential harmful effects need to be more fully elucidated; hence, while they concede that CS may have benefit in a small subset of cases with severe CAP, these cases have not yet been adequately identified (164). Other study results suggest that it is not only important to demonstrate benefits in stringently controlled randomized controlled trials, but also in the real world situation (165). It is hoped that current and ongoing studies of CS use in patients with CAP will clarify some of these issues (164, 166).
P2Y12 Receptor Antagonists
Platelets possess two receptors for ADP, namely P2Y1 and P2Y12 receptors. Because ADP-mediated stimulation of platelets is dependent on co-activation of these receptors, blockade of only one receptor type may be adequate to attenuate responses initiated by exposure to ADP (167). Of the three pharmacological P2Y12 receptor antagonists currently in clinical use, namely clopidogrel, prasugrel and ticagrelor, which vary with respect to onset of action and efficacy (168), only clopidogrel and ticagrelor, to our knowledge, have been evaluated in the setting of CAP
In the first of these, Gross et al. in a retrospective study spanning the period 2001–2005, investigated firstly the frequency of development of CAP in subjects who had a history of having been prescribed clopidogrel on at least six occasions (n = 5,166) in comparison with those who had not (n = 52,809) (169). Secondly, assessment of the requirement for hospital admission following development of CAP in the clopidogrel-treated and untreated groups, as well as indices of disease severity, including mortality, intensive care unit (ICU) admission, need for mechanical ventilation, development of sepsis, acute respiratory distress syndrome/acute lung injury, but not, somewhat surprisingly, development of CVEs (169). Surprisingly, the authors reported that both the incidence of CAP, as well as the frequency of hospital admission, were significantly higher in the clopidogrel-treated group, with trends, albeit not attaining statistical significance, indicative of less severe disease (169).
The Platelet inhibition and patient Outcomes (PLATO) study was a multicenter, double-blind, randomized trial designed to compare the effects of ticagrelor (180 mg loading dose, 90 mg twice daily thereafter) and clopidogrel (300–600 mg loading dose, 75 mg daily thereafter) on the prevention of CVEs in patients admitted to hospital with an ACS (170). The study recruited a total of 18,624 patients allocated in equal numbers to receive treatment with either ticagrelor or clopidogrel for 1 year. The findings of the study revealed that treatment with ticagrelor, as opposed to clopidogrel, significantly decreased the rate of death from MI or stroke (170). A subsequent secondary analysis of the data emanating from this trial was focused on the prevalence of pulmonary adverse events, mostly pneumonia, as well as associated mortality, in both treatment arms (171). This analysis revealed significantly lower prevalence rates of pulmonary adverse events and sepsis, as well as associated mortality, in the ticagrelor-treated group. The authors concluded that the mortality risk following pulmonary adverse events and sepsis in ACS patients appears to be lower in those receiving ticagrelor (171). An important caveat, however, is that all patients in both treatment groups also received aspirin (175 mg/daily), indicating that the combination of ticagrelor and aspirin may in fact be most effective, as opposed to ticagrelor alone.
In a more recent study known as the XANTHIPPE study (Examining the effect of Ticagrelor on Platelet Activation, Platelet-Leukocyte Aggregates and Acute Lung Injury in Pneumonia), Sexton et al. investigated the effects of ticagrelor on systemic biomarkers of platelet activation, as well as pulmonary function in hospitalized patients with pneumonia [CAP and hospital-acquired pneumonia (HAP)] (172). The study design was a randomized, placebo-controlled trial with 30 patients in each of the placebo-treated (21 CAP and 9 HAP) and ticagrelor-treated (17 CAP and 13 HAP) groups. Patients who were recruited within 48 h of diagnosis, were treated for 7 days, or until discharge (172). Biomarker and lung function measurements were performed at baseline and after 1, 2, and 7 days of treatment or at the time of discharge and, finally, after 30 days. Briefly, administration of ticagrelor was associated with significant decreases in systemic levels of IL-6, as well as in ADP-activated formation of platelet-leukocyte aggregates ex vivo that was evident from day 1. In addition, trends were evident toward improved pulmonary function and need for supplemental oxygen (172). Similar effects of ticagrelor were observed in a murine model of experimental sepsis, including significantly decreased mortality (172). Limitations of this study identified by the authors include the small numbers of patients, all with moderate disease (172).
Dual Anti-platelet Therapy
To our knowledge there are no stringently controlled studies in the current literature that have described the effects of dual anti-platelet therapy, usually aspirin in combination with a P2Y12 receptor antagonist, in the clinical setting of severe CAP. This may relate to concerns about the risk of bleeding complications.
Proteinase-Activated Receptor 1 (PAR-1) Antagonists
Vorapaxor remains the only selective antagonist of thrombin-activated PAR-1 receptors. To our knowledge, however, this agent has not been used in the treatment of severe CAP, either alone or in combination with aspirin and/or a P2Y12 receptor antagonist.
Statins
These cholesterol-lowering agents, which target the enzyme 3-hydroxy-3-methyl-glutaryl CoA reductase, suppress platelet reactivity by several mechanisms that have been reviewed by us elsewhere (173). These include: (i) inhibition of the formation of oxLDL-C, which triggers platelet activation via interaction with the scavenger receptors, LOX-1 and CD36, both of which are expressed on platelets (174, 175); (ii) inhibition of PLA2, resulting in decreased release of TxA2 (176); (iii) decreased expression of the adhesion molecule CD40L (177); and (iv) although largely unexplored, by lowering membrane cholesterol concentrations, statins may attenuate the pro-inflammatory/pro-thrombotic activities of cholesterol-binding toxins such as pneumolysin. In this context administration of simvastatin at a dose of 20 mg/daily for 6 weeks to hypercholesterolemic patients resulted in significantly decreased concentrations of cholesterol in the plasma membranes of isolated blood neutrophils (178). These anti-platelet activities of statins have also been described in the clinical setting, albeit in newly-diagnosed patients with primary hypercholesterolemia treated with simvastatin (179). In this study, Barale et al. observed that aside from improved lipid profiles, administration of simvastatin was associated with significant reduction in ADP- and collagen-activated platelet aggregation ex vivo, as well as reductions in systemic biomarkers of platelet activation, including sCD62P, sCD40L, platelet-derived growth factor BB and RANTES. With respect to clinical studies in patients hospitalized with CAP, these have been mostly of a retrospective design in patient cohorts that had received statins prior to hospital admission, the broad consensus finding being that of a modest-to-moderate beneficial effect on mortality (173).
More recently, Sapey et al. reported the findings of a pilot, randomized, controlled clinical trial designed primarily to investigate the effects of short-term administration of simvastatin on neutrophil function and clinical outcomes in elderly patients (n = 62, aged > 55 years) admitted to a secondary care facility for “milder” CAP (180). Patients received either simvastatin (n = 32 patients, 80 mg daily for 7 days or until discharge) or a placebo (n = 30 patients), together with a macrolide antibiotic. Neutrophil functions, measured using isolated peripheral blood neutrophils, were performed on treatment day 4 and included, most importantly, measurement of chemoattractant-stimulated NETosis, as well as IL-8-induced chemotaxis, apoptosis and systemic neutrophil elastase activity, while clinical assessment was based on changes in the sequential organ failure assessment (SOFA) score. Treatment with simvastatin was associated with statistically significant decreases in NETosis (p = 0.0034) and systemic elastase activity (p = 0,001), while chemotactic responsiveness increased significantly and apoptosis was unaffected (180). Although, as expected, SOFA scores were relatively low in this cohort of CAP patients, they nevertheless decreased significantly (p < 0.026) in simvastatin-treated patients. With respect to long-term benefit, analysis of readmission and survival data as a composite endpoint revealed significant increases in hospital-free survival at both 180 and 365 days (p = 0.03 for both).
The authors, while conceding that the findings of this small proof-of-concept study require confirmation in large, stringently controlled multicenter trials, contend that elderly persons with mild CAP may be the principal beneficiaries of statin-based adjuvant therapy. They propose that statins may induce the transition of systemic neutrophils from a harmful, possibly platelet-driven, phenotype to a protective anti-inflammatory phenotype (180).
Liposomes That Target CAP Pathogen-Derived, Cholesterol-Binding, Pore-Forming Toxins
Liposomes that contain a 1:1 mixture of cholesterol:sphingomyelin have been developed as a therapeutic strategy to counter the damaging activities of bacterial cholesterol-binding, pore-forming toxins such as pneumolysin (181). When administered to mice with severe, experimental invasive pneumococcal disease, these liposome preparations significantly attenuated the development of myocardial injury (34). Following from these promising earlier studies, a Swiss Biotechnology company, COMBIOXIN SA, developed a therapeutic liposome preparation known as CAL02 (182). As described by the developers “CAL02 consists of a mixture of liposomes that create artificially large and stable liquid-ordered lipid microdomains that function as docking sites for a large range of bacterial toxins” (181, 182).
CAL02 has recently been evaluated in a double-blind, placebo-controlled, randomized phase I clinical trial, encompassing six intensive care units in France and Belgium and a total of 19 patients with severe pneumococcal pneumonia 14 and 5 of whom received CAL02 or placebo, respectively (182). Although primarily focused on safety and tolerability issues, both of which were “promising”, it is noteworthy that the mean SOFA scores at 8 days decreased by 69.0 and 29.2% in the CAL02- and placebo-treated groups, respectively, while the corresponding figures for the occurrence of atrial fibrillation were 14.3% (2/14) and 40% (2/5) (182). These findings provide a basis for further clinical evaluation of CAL02 in larger clinical trials.
Future Platelet-Targeted Strategies
These include pharmacological and biological agents that target ADP-activated P2Y1 receptors on platelets as well as platelet-derived, pro-inflammatory HMGB1. In addition, inhibitors of necroptosis may indirectly attenuate the harmful effects of platelet activation in the setting of severe pneumococcal disease.
P2Y1 Receptors
Although P2Y12 and P2Y1 receptors appear to harmonize with respect to ADP-mediated platelet activation, Amison et al. have recently described a novel mechanism involving only P2Y1 receptors that promotes platelet-dependent leukocyte recruitment at sites of inflammation (183). A P2Y1 receptor-mediated signaling pathway that is distinct from that involving phospholipase C and initiation of platelet aggregation drives this novel mechanism of platelet-dependent neutrophil chemotaxis. Although selective pharmacological inhibitors of P2Y1 receptors are available for use in experimental settings, none is yet available for clinical application.
HMGB1
Strategies targeting this pro-inflammatory protein have been the subject of a very recent review (184). Amongst others, these include pharmacological agents such as metformin that inhibit the translocation of HMGB1 from the nucleus to the cytosol that is likely to be most relevant to megakaryocytes, as well as direct targeting of the protein by monoclonal antibodies, such as m2G7 that target the interaction of HMGB1 with TLR4 and RAGE (184).
Inhibitors of Necroptosis
Very recently, Beno et al. using high-resolution echocardiography, observed that administration of ponatinib, a novel multi-tyrosine kinase inhibitor that attenuates necroptosis, to mice with experimentally induced severe pneumococcal disease, resulted in significant attenuation of long-term cardiac damage (185, 186). Although these cardio-protective effects of ponatinib were attributed to attenuation of pneumolysin-mediated cardiomyocyte necroptosis, it is also noteworthy that toxin-induced platelet activation and resultant microvascular occlusion represents an additional cause of cardiac necroptosis.
These various anti-platelet strategies are summarized in Table 3.

Table 3. Potential pharmacological and biological strategies to counter platelet activation in severe community-acquired pneumonia.
Conclusions
It is quite clear from this review, that platelets play an important role not only in host defense against infections, including, but not limited to CAP, but that platelet activation may also contribute to some of the potentially life-threatening complications that occur in patients with CAP, such as the acute CVEs. The involvement of platelets in the pathogenesis of these acute CVEs, exacerbated by bacterial invasion of the myocardium and release of cell wall components and toxins, such as pneumolysin, in the case of the pneumococcus, is relatively well-established. However, the events that are associated with the pathogenesis of the long-term cardiac events are less clear. Nevertheless, evidence is emerging that persistent antigenemia predisposes to a persistent, systemic pro-inflammatory/prothrombotic phenotype that is associated with an ongoing risk of future CVE. Although various, mainly pharmacological, platelet-targeted adjunctive therapies have been identified, evaluation of these in the clinical setting has been somewhat fragmented, necessitating future stringent assessment in well-controlled, definitive clinical trials.
Author Contributions
Both authors contributed equally to the conceptualization and compilation of the manuscript and approved its submission.
Funding
The National Research Foundation of South Africa supports CF.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Peyrani P, Mandell L, Torres A, Tillotson G. The burden of community-acquired bacterial pneumonia in the era of antibiotic resistance. Expert Rev Respir Med. (2019) 13:139–52. doi: 10.1080/17476348.2019.1562339
2. Michelin L, Weber FM, Scolari BW, Menezes BK, Gullo MC. Mortality and costs of pneumococcal pneumonia in adults: a cross-sectional study. J Bras Pneumol. (2019) 45:e20180374. doi: 10.1590/1806-3713/e20180374
3. Cillóniz C, Cardozo C, García-Vidal C. Epidemiology, pathophysiology, and microbiology of community-acquired pneumonia. Ann Res Hosp. (2018) 2:1. doi: 10.21037/arh.2017.12.03
4. Ferreira-Coimbra J, Sarda C, Rello J. Burden of community-acquired pneumonia and unmet clinical needs. Adv Ther. (2020) 37:1302–18. doi: 10.1007/s12325-020-01248-7
5. Feldman C, Shaddock E. Epidemiology of lower respiratory tract infections in adults. Expert Rev Respir Med. (2019) 13:63–77. doi: 10.1080/17476348.2019.1555040
6. GBD 2015 LRI Collaborators. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory tract infections in 195 countries: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect Dis. (2017) 17:1133–61. doi: 10.1016/S1473-3099(17)30396-1
7. Musher DM, Abers MS, Bartlett JG. Evolving understanding of the causes of pneumonia in adults, with special attention to the role of pneumococcus. Clin Infect Dis. (2017) 65:1736–44. doi: 10.1093/cid/cix549
8. GBD 2015 Eastern Mediterranean Region Lower Respiratory Infections Collaborators. Burden of lower respiratory infections in the Eastern Mediterranean Region between 1990 and 2015: findings from the Global Burden of Disease 2015 study. Int J Public Health. (2018) 63:97–108. doi: 10.1007/s00038-017-1007-0
9. Corrêa RA, José BPS, Malta DC, Passos VMA, França FB, Teixeira RA, et al. Burden of disease by lower respiratory tract infections in Brazil, 1990 to 2015: estimates of the Global Burden of Disease 2015 study. Rev Bras Epidemiol. (2017) 20(Suppl. 1):171–81. doi: 10.1590/1980-5497201700050014
10. Torres A, Cillóniz C, Blasi F, Chalmers JD, Gaillat J, Dartois N, et al. Burden of pneumococcal community-acquired pneumonia in adults across Europe: a literature review. Respir Med. (2018) 137:6–13. doi: 10.1016/j.rmed.2018.02.007
11. Vìasus D, Cillóniz C, Cardozo CG, Puerta P, Garavito A, Torres A, et al. Early, short and long-term mortality in community-acquired pneumonia. Ann Res Hosp. (2018) 2:5. doi: 10.21037/arh.2018.04.02
12. Menéndez R, Méndez R, Aldás I, Reyes S, Gonzalez-Jimernez P, Pablo España P, et al. Community-acquired pneumonia patients at risk for early and long-term cardiovascular events are identified by cardiac biomarkers. Chest. (2019) 156:1080–91. doi: 10.1016/j.chest.2019.06.040
13. Feldman C, Normark S, Henriques-Normark B, Anderson R. Pathogenesis and prevention of risk of cardiovascular events in patients with pneumococcal community-acquired pneumonia. J Intern Med. (2019) 285:635–52. doi: 10.1111/joim.12875
14. Musher DM, Abers MS, Corrales-Medina VF. Acute infection and myocardial infarction. N Engl J Med. (2019) 380:171–6. doi: 10.1056/NEJMra1808137
15. Wu MH, Tsou PY, Wang YH, Lee MG, Chao CCT, Lee WC, et al. Impact of post-sepsis cardiovascular complications on mortality in sepsis survivors: a population-based study. Crit Care. (2019) 23:293. doi: 10.1186/s13054-019-2579-2
16. Warren-Gash C, Blackburn R, Whitaker H, McMenamin J, Hayward AC. Laboratory-confirmed respiratory infections as triggers for acute myocardial infarction and stroke: a self-controlled case series analysis of national linked datasets from Scotland. Eur Respir J. (2018) 51:1701794. doi: 10.1183/13993003.01794-2017
17. Steiner GS, Knight S, Miller RR, Bair TL, Horne BD, Lopansri BK, et al. Abstract 11296: bacterial pneumonia compared to viral pneumonia is associated with a higher risk of future major adverse cardiovascular events. Circulation. (2018) 138:A11296. Available online at: https://www.ahajournals.org/doi/10.1161/circ.138.suppl_1.11296
18. Feldman C, Anderson R. Pneumonia as a systemic illness. Curr Opin Pulm Med. (2018) 24:237–43. doi: 10.1097/MCP.0000000000000466
19. Restrepo MI, Reyes LF. Pneumonia as a cardiovascular disease. Respirology. (2018) 23:250–9. doi: 10.1111/resp.13233
20. Cìlli A, Cakin O, Aksoy E, Kargin F, Adiguzel N, Karakurt Z, et al. Acute cardiac events in severe community-acquired pneumonia: a multicenter study. Clin Respir J. (2018) 12:2212–9. doi: 10.1111/crj.12791
21. Tralhão A, Póvoa P. Cardiovascular events after community-acquired pneumonia: a global perspective with systematic review and meta-analysis of observational studies. J Clin Med. (2020) 9:414. doi: 10.3390/jcm9020414
22. Shaheen ES, Elmelegy E, Hossam El Din W. Cardiac dysfunction in patients with community-acquired pneumonia. J Med Sci Res. (2019) 2:209–13. doi: 10.4103/JMISR.JMISR_4_19
23. Méndez R, Aldás I, Menéndez R. Biomarkers in community-acquired pneumonia (cardiac and non-cardiac). J Clin Med. (2020) 9:549. doi: 10.3390/jcm9020549
24. Özlek E, Biteker FS, Çil C, Çelik O, Özlek B, Dogan V, et al. The risk stratification in community-acquired pneumonia. Am J Emerg Med. (2019) 37:151–72. doi: 10.1016/j.ajem.2018.05.061
25. Pieralli F, Biondo B, Vannucchi V, Falcone M, Antonielli E, De Marzi G, et al. Performance of the CHA2DS2−VASc score in predicting new onset atrial fibrillation during hospitalization for community-acquired pneumonia. Eur J Intern Med. (2019) 62:24–8. doi: 10.1016/j.ejim.2019.01.012
26. Musher DM, Rueda AM, Kaka AS, Mapara SM. The association between pneumococcal pneumonia and acute cardiac events. Clin Infect Dis. (2007) 45:158–65. doi: 10.1086/518849
27. Chiong YK, Tan I, Lim TC, Thong WY, Chan MY, Chan SM. Pneumococcal pneumonia resembling acute myocardial infarction in an adolescent male. Pediatr Infect Dis J. (2020) 39:81–4. doi: 10.1097/INF.0000000000002527
28. Anderson R, Nel JG, Feldman C. Multifaceted role of pneumolysin in the pathogenesis of myocardial injury in community-acquired pneumonia. Int J Mol Sci. (2018) 19:E1147. doi: 10.3390/ijms19041147
29. Bartlett B, Ludewick HP, Lee S, Dwivedi G. Cardiovascular complications following pneumonia: focus on pneumococcus and heart failure. Curr Opin Cardiol. (2019) 34:233–9. doi: 10.1097/HCO.0000000000000604
30. Borsa N, Iturriaga LAR, Fernandez LS, España PP, Aliberti S, Ramirez JA, et al. Bacteremic pneumococcal pneumonia is associated with an increased rate of cardiovascular events. Eur Respir J. (2019) 54:OA3306. doi: 10.1183/13993003.congress-2019.OA3306
31. Shenoy AT, Beno SM, Brissac T, Bell JW, Novak L, Orihuela CJ. Severity and properties of cardiac damage caused by Streptococcus pneumoniae are strain dependent. PLoS ONE. (2018) 13:e0204032. doi: 10.1371/journal.pone.0204032
32. Reyes LF, Hinojosa CA, Soni NJ, Noda J, Winter VT, de la Garza MA, et al. Cardiac injury due to Streptococcus pneumoniae invasion during severe pneumococcal pneumonia in a novel nonhuman primate model. J Clin Transl Sci. (2018) 2:6. doi: 10.1017/cts.2018.51
33. Borsa N, Pasquale MD, Restrepo MI. Animal models of Pneumococcal pneumonia. Int J Mol Sci. (2019) 20:4220. doi: 10.3390/ijms20174220
34. Alhamdi Y, Neill DR, Abrams ST, Malak HA, Yahya R, Barrett-Jolley R, et al. Circulating pneumolysin is a potent inducer of cardiac injury during pneumococcal infection. PLoS Pathog. (2015) 11:e1004836. doi: 10.1371/journal.ppat.1004836
35. Feldman C, Anderson R. Pneumococcal virulence factors in community-acquired pneumonia. Curr Opin Pulm Med. (2020) 26:222–31. doi: 10.1097/MCP.0000000000000674
36. Kaushansky K. Historical review: megakaryopoiesis and thrombopoiesis. Blood. (2008) 111:981–6. doi: 10.1182/blood-2007-05-088500
37. Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell. (2007) 128:1173–86. doi: 10.1016/j.cell.2007.01.037
38. Montenont E, Rondina MT, Campbell RA. Altered functions of platelets during aging. Curr Opin Hematol. (2019) 26:336–42. doi: 10.1097/MOH.0000000000000526
39. Campbell RA, Franks Z, Bhatnagar A, Rowley JW, Manne BK, Supiano MA, et al. Granzyme A in human platelets regulates the synthesis of proinflammatory cytokines by monocytes in aging. J Immunol. (2018) 200:295–304. doi: 10.4049/jimmunol.1700885
40. Hamzeh-Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, Garraud O. Platelets and infections - complex interactions with bacteria. Front Immunol. (2015) 6:82. doi: 10.3389/fimmu.2015.00082
41. Chabert A, Hamzeh-Cognasse H, Pozzetto B, Cognasse F, Schattner M, Gomez RM, et al. Human platelets and their capacity of binding viruses: meaning and challenges? BMC Immunol. (2015) 16:26. doi: 10.1186/s12865-015-0092-1
42. Ali RA, Wuescher LM, Worth RG. Platelets: essential components of the immune system. Curr Trends Immunol. (2015) 16:65–78.
43. Deppermann C, Kubes P. Start a fire, kill the bug: the role of platelets in inflammation and infection. Innate Immun. (2018) 24:335–48. doi: 10.1177/1753425918789255
44. Guo L, Rondina MT. The era of thromboinflammation: platelets are dynamic sensors and effector cells during infectious diseases. Front Immunol. (2019) 10:2204. doi: 10.3389/fimmu.2019.02204
45. Gomez-Casado C, Villaseñor A, Rodriguez-Nogales A, Bueno JL, Barber D, Escribese MM. Understanding platelets in infectious and allergic lung diseases. Int J Mol Sci. (2019) 20:E1730. doi: 10.3390/ijms20071730
46. Gaertner F, Massberg S. Patrolling the vascular borders: platelets in immunity to infection and cancer. Nat Rev Immunol. (2019) 19:747–60. doi: 10.1038/s41577-019-0202-z
47. Page MJ, Pretorius E. A champion of host defense: a generic large-scale cause for platelet dysfunction and depletion in infection. Semin Thromb Hemost. (2020) 46:302–19. doi: 10.1055/s-0040-1708827
48. Heijnen H, van der Sluijs P. Platelet secretory behaviour: as diverse as the granules or not? J Thromb Haemost. (2015) 13:2141–51. doi: 10.1111/jth.13147
49. Riaz AH, Tasma BE, Woodman ME, Wooten RM, Worth RG. Human platelets efficiently kill IgG-opsonized E. coli. FEMS Immunol Med Microbiol. (2012) 65:78–83. doi: 10.1111/j.1574-695X.2012.00945.x
50. Arbesu I, Bucsaiova M, Fischer MB, Mannhalter C. Platelet-borne complement proteins and their role in platelet-bacteria interactions. J Thromb Haemost. (2016) 14:2241–52. doi: 10.1111/jth.13495
51. Mancuso ME, Santagostino E. Platelets: much more than bricks in a breached wall. Br J Haematol. (2017) 178:209–19. doi: 10.1111/bjh.14653
52. Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. (2019) 10:1780. doi: 10.1038/s41467-019-09607-x
53. Anderson R, Feldman C. Review manuscript: mechanisms of platelet activation by the pneumococcus and the role of platelets in community-acquired pneumonia. J Infect. (2017) 75:473–85. doi: 10.1016/j.jinf.2017.09.013
54. Binsker U, Kohler TP, Krauel K, Kohler S, Schwertz H, Hammerschmidt S. Pneumococcal adhesins PavB and PspC are important for the interplay with human thrombospondin-1. J Biol Chem. (2015) 290:14542–55. doi: 10.1074/jbc.M114.623876
55. Mitchell J, Sullam PM. Streptococcus mitis phage-encoded adhesins mediate attachment to α2-8-linked sialic acid residues on platelet membrane gangliosides. Infect Immun. (2009) 77:3485–90. doi: 10.1128/IAI.01573-08
56. Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, et al. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci USA. (2003) 100:1966–71. doi: 10.1073/pnas.0435928100
57. Loiko EN, Samal AB, Shulyakovskaya SM. H2O2-induced platelet aggregation and increase in intracellular Ca2+ concentration are blocked by inhibitors of intracellular signaling. Biochemistry. (2003) 68:1210–6. doi: 10.1023/B:BIRY.0000009135.85883.65
58. Redondo PC, Salido GM, Rosado JA, Pariente JA. Effect of hydrogen peroxide on Ca2+ mobilisation in human platelets through sulphydryl oxidation dependent and independent mechanisms. Biochem Pharmacol. (2004) 67:491–502. doi: 10.1016/j.bcp.2003.09.031
59. Valle-Jiménez X, Ramírez-Cosmes A, Aquino-Domínguez AS, Sánchez-Peña F, Bustos-Arriaga J, Romero-Tlalolini MLÁ, et al. Human platelets and megakaryocytes express defensin alpha 1. Platelets. (2020) 31:344–54. doi: 10.1080/09537104.2019.1615612
60. Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, et al. Novel anti-bacterial activities of β-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog. (2011) 7:e1002355. doi: 10.1371/journal.ppat.1002355
61. Wendler J, Schroeder BO, Ehmann D, Koeninger L, Mailänder-Sánchez D, Lemberg C, et al. Proteolytic degradation of reduced human beta defensin 1 generates a novel antibiotic octapeptide. Sci Rep. (2019) 9:3640. doi: 10.1038/s41598-019-40216-2
62. Tohidnezhad M, Varoga D, Podschun R, Wruck CJ, Seekamp A, Brandenburg LO, et al. Thrombocytes are effectors of the innate immune system releasing human beta defensin-3. Injury. (2011) 42:682–6. doi: 10.1016/j.injury.2010.12.010
63. Yung SC, Murphy PM. Antimicrobial chemokines. Front Immunol. (2012) 3:276. doi: 10.3389/fimmu.2012.00276
64. Stoldt VR, Scharf RE. Determination of the microbicidal capacity of platelets by a biomonitoring system. Blood. (2014) 124:4993. doi: 10.1182/blood.V124.21.4993.4993
65. Lieff J. Platelet Intelligence. (2014) Available online at: http://jonlieffmd.com/blog/platelet-intelligence (accessed April 24, 2020).
66. Yue L, Pang Z, Li H, Yang T, Guo L, Liu L, et al. CXCL4 contributes to host defense against acute Pseudomonas aeruginosa lung infection. PLoS ONE. (2018) 13:e0205521. doi: 10.1371/journal.pone.0205521
67. Wiesner J, Vilcinskas A. Antimicrobial peptides: the ancient arm of the human immune system. Virulence. (2010) 1:440–64. doi: 10.4161/viru.1.5.12983
68. Omardien S, Drijfhout JW, Vaz FM, Wenzel M, Hamoen LW, Zaat SAJ, et al. Bactericidal activity of amphipathic cationic antimicrobial peptides involves altering the membrane fluidity when interacting with the phospholipid bilayer. Biochim Biophys Acta Biomembr. (2018) 1860:2404–15. doi: 10.1016/j.bbamem.2018.06.004
69. Tan WKY, Purnamawati K, Pakkiri LS, Tan SH, Yang X, Chan MY, et al. Sources of variability in quantifying circulating thymosin beta-4: literature review and recommendations. Expert Opin Biol Ther. (2018) 18:141–7. doi: 10.1080/14712598.2018.1448382
70. Carion TW, Ebrahim AS, Kracht D, Agrawal A, Strand E, Kaddurah O, et al. Thymosin beta-4 and ciprofloxacin adjunctive therapy improves Pseudomonas aeruginosa-Induced keratitis. Cells. (2018) 7:E145. doi: 10.3390/cells7100145
71. Mandke P, Vasquez KM. Interactions of high mobility group box protein 1 (HMGB1) with nucleic acids: implications in DNA repair and immune responses. DNA Repair. (2019) 83:102701. doi: 10.1016/j.dnarep.2019.102701
72. Rouhiainen A, Imai S, Rauvala H, Parkkinen J. Occurrence of amphoterin (HMG1) as an endogenous protein of human platelets that is exported to the cell surface upon platelet activation. Thromb Haemost. (2000) 84:1087–94. doi: 10.1055/s-0037-1614175
73. Mardente S, Mari E, Massimi I, Tafani M, Guerriero R, Morsilli O, et al. From human megakaryocytes to platelets: effects of aspirin on high-mobility group box 1/receptor for advanced glycation end products axis. Front Immunol. (2018) 8:1946. doi: 10.3389/fimmu.2017.01946
74. Maugeri N, Rovere-Querini P, Manfredi AA. Disruption of a regulatory network consisting of neutrophils and platelets fosters persisting inflammation in rheumatic diseases. Front Immunol. (2016) 7:182. doi: 10.3389/fimmu.2016.00182
75. Subramanian S, Pallati PK, Sharma P, Agrawal DK, Nandipati KC. Significant association of TREM-1 with HMGB1, TLRs and RAGE in the pathogenesis of insulin resistance in obese diabetic populations. Am J Transl Res. (2017) 9:3224–44.
76. Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. (2013) 93:865–73. doi: 10.1189/jlb.1212662
77. Yang H, Lundbäck P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med. (2012) 18:250–9. doi: 10.2119/molmed.2011.00389
78. Allette YM, Due MR, Wilson SM, Feldman P, Ripsch MS, Khanna R, et al. Identification of a functional interaction of HMGB1 with receptor for advanced glycation end-products in a model of neuropathic pain. Brain Behav Immun. (2014) 42:169–77. doi: 10.1016/j.bbi.2014.06.199
79. Paudel YN, Semple BD, Jones NC, Othman I, Shaikh MF. High mobility group box 1 (HMGB1) as a novel frontier in epileptogenesis: from pathogenesis to therapeutic approaches. J Neurochem. (2019) 151:542–57. doi: 10.1111/jnc.14663
80. Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. (2012) 209:1519–28. doi: 10.1084/jem.20120189
81. Schiraldi M, Raucci A, Muñoz LM, Livoti E, Celona B, Venereau E, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. (2012) 209:551–63. doi: 10.1084/jem.20111739
82. Chatterjee M, von Ungern-Sternberg SN, Seizer P, Schlegel F, Büttcher M, Sindhu NA, et al. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis. (2015) 6:e1989. doi: 10.1038/cddis.2015.233
83. Tadie JM, Bae HB, Jiang S, Park DW, Bell CP, Yang H, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol. (2013) 304:L342–9. doi: 10.1152/ajplung.00151.2012
84. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. (2014) 12:2074–88. doi: 10.1111/jth.12710
85. Zander DM, Klinger M. The blood platelets contribution to innate host defense - what they have learned from their big brothers. Biotechnol J. (2009) 4:914–26. doi: 10.1002/biot.200800362
86. Wachowicz B, Olas B, Zbikowska HM, Buczyński A. Generation of reactive oxygen species in blood platelets. Platelets. (2002) 13:175–82. doi: 10.1080/09533710022149395
87. Begonja AJ, Gambaryan S, Geiger J, Aktas B, Pozgajova M, Nieswandt B, et al. Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIb/beta3-integrin activation independent of the NO/cGMP pathway. Blood. (2005) 106:2757–60. doi: 10.1182/blood-2005-03-1047
88. Sonkar VK, Kumar R, Jensen M, Wagner BA, Sharathkumar AA, Miller FJ Jr, et al. Nox2 NADPH oxidase is dispensable for platelet activation or arterial thrombosis in mice. Blood Adv. (2019) 3:1272–84. doi: 10.1182/bloodadvances.2018025569
89. Fan J, Li Y, Levy RM, Fan JJ, Hackam DJ, Vodovotz Y, et al. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1-TLR4 signaling. J Immunol. (2007) 178:6573–80. doi: 10.4049/jimmunol.178.10.6573
90. Zhou H, Deng M, Liu Y, Yang C, Hoffman R, Zhou J, et al. Platelet HMGB1 is required for efficient bacterial clearance in intra-abdominal bacterial sepsis in mice. Blood Adv. (2018) 2:638–48. doi: 10.1182/bloodadvances.2017011817
91. Kim K, Li J, Tseng A, Andrews RK, Cho J. NOX2 is critical for heterotypic neutrophil-platelet interactions during vascular inflammation. Blood. (2015) 126:1952–64. doi: 10.1182/blood-2014-10-605261
92. Lishko VK, Yakubenko VP, Ugarova TP, Podolnikova NP. Leukocyte integrin Mac-1 (CD11b/CD18, αMβ2, CR3) acts as a functional receptor for platelet factor 4. J Biol Chem. (2018) 293:6869–82. doi: 10.1074/jbc.RA117.000515
93. Claushuis TA, van Vught LA, Scicluna BP, Wiewel MA, Klein Klouwenberg PM, Hoogendijk AJ, et al. Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood. (2016) 127:3062–72. doi: 10.1182/blood-2015-11-680744
94. Birnie E, Claushuis TAM, Koh GCKW, Limmathurotsakul D, Day NPJ, Roelofs JJTH, et al. Thrombocytopenia impairs host defense against Burkholderia pseudomallei (melioidosis). J Infect Dis. (2019) 219:648–59. doi: 10.1093/infdis/jiy541
95. Braun LJ, Stegmeyer R, Schäfer K, Volkery S, Currie SM, Kempe B, et al. Platelets docking to VWF prevent leaks during leukocyte extravasation by stimulating Tie-2. Blood. (2020) 136:627–39. doi: 10.1182/blood.2019003442
96. Kirby P, Smith S, Ward L, Hanson J, Currie BJ. Clinical utility of platelet count as a prognostic marker for melioidosis. Am J Trop Med Hyg. (2019) 100:1085–7. doi: 10.4269/ajtmh.18-0698
97. Entezari M, Weiss DJ, Sitapara R, Whittaker L, Wargo MJ, Li J, et al. Inhibition of high-mobility group box 1 protein (HMGB1) enhances bacterial clearance and protects against Pseudomonas aeruginosa pneumonia in cystic fibrosis. Mol Med. (2012) 18:477–85. doi: 10.2119/molmed.2012.00024
98. Ortiz-Muñoz G, Yu MA, Lefrançais E, Mallavia B, Valet C, Tian JJ, et al. Cystic fibrosis transmembrane conductance regulator dysfunction in platelets drives lung hyperinflammation. J Clin Invest. (2020) 130:2041–53. doi: 10.1172/JCI129635
99. Hovold G, Lindberg U, Ljungberg J, Shannon O, Påhlman LI. BPI-ANCA is expressed in the airways of cystic fibrosis patients and correlates to platelet numbers and Pseudomonas aeruginosa colonization. Respir Med. (2020) 170:105994. doi: 10.1016/j.rmed.2020.105994
100. Luquero-Bueno S, Galván-Román JM, Curbelo J, Lancho-Sánchez A, Roy-Vallejo E, Ortego-Gómez M, et al. Platelet count as an evolution marker of late mortality and cardiovascular events after an episode of community-acquired pneumonia. Eur Respir J. (2019) 54:PA4532. doi: 10.1183/13993003.congress-2019.PA4532
101. Feldman C, Kallenbach JM, Levy H, Reinach SG, Hurwitz MD, Thorburn JR, et al. Community-acquired pneumonia of diverse aetiology: prognostic features in patients admitted to an intensive care unit and a “severity of illness” core. Intensive Care Med. (1989) 15:302–7. doi: 10.1007/BF00263865
102. Feldman C, Kallenbach JM, Levy H, Thorburn JR, Hurwitz MD, Koornhof HJ. Comparison of bacteraemic community-acquired lobar pneumonia due to Streptococcus pneumoniae and Klebsiella pneumoniae in an intensive care unit. Respiration. (1991) 58:265–70. doi: 10.1159/000195943
103. Brogly N, Devos P, Boussekey N, Georges H, Chiche A, Leroy O. Impact of thrombocytopenia on outcome of patients admitted to ICU for severe community-acquired pneumonia. J Infect. (2007) 55:136–40. doi: 10.1016/j.jinf.2007.01.011
104. Mirsaeidi M, Peyrani P, Aliberti S, Filardo G, Bordon J, Blasi F, et al. Thrombocytopenia and thrombocytosis at time of hospitalization predict mortality in patients with community-acquired pneumonia. Chest. (2010) 137:416–20. doi: 10.1378/chest.09-0998
105. ElMaraghy AA, AbdelFattah EB, Ahmed MS. Platelet count: Is it a possible marker for severity and outcome of community acquired pneumonia? Egypt J Chest Dis Tuberc. (2016) 65:499–504. doi: 10.1016/j.ejcdt.2015.09.001
106. Gorelik O, Izhakian S, Barchel D, Almoznino-Sarafian D, Tzur I, Swarka M, et al. Prognostic significance of platelet count changes during hospitalization for community-acquired pneumonia. Platelets. (2017) 28:380–6. doi: 10.1080/09537104.2016.1219032
107. Prina E, Ferrer M, Ranzani OT, Polverino E, Cillóniz C, Moreno E, et al. Thrombocytosis is a marker of poor outcome in community-acquired pneumonia. Chest. (2013) 143:767–75. doi: 10.1378/chest.12-1235
108. Hesham AA, Heba HA. Thrombocytosis at time of hospitalization is a reliable indicator for severity of CAP patients in ICU. Egypt J Chest Dis Tuberc. (2012) 61:145–9. doi: 10.1016/j.ejcdt.2012.10.030
109. Lee JH, Park M, Han S, Hwang JJ, Park SH, Park SY. An increase in mean platelet volume during admission can predict the prognoses of patients with pneumonia in the intensive care unit: a retrospective study. PLoS ONE. (2018) 13:e0208715. doi: 10.1371/journal.pone.0208715
110. Nageeb RS, Abozaid MMN, Nageeb GS, Omran AA. Mean platelet volume to platelet count ratio as a laboratory indicator of mortality in pneumonia following ischemic stroke. Egypt J Neurol Psychiatr Neurosurg. (2018) 54:27. doi: 10.1186/s41983-018-0028-9
111. Tunjungputri RN, Mobegi FM, Cremers AJ, van der Gaast-de Jongh CE, Ferwerda G, Meis JF, et al. Phage-derived protein induces increased platelet activation and is associated with mortality in patients with invasive pneumococcal disease. MBio. (2017) 8:e01984–16. doi: 10.1128/mBio.01984-16
112. Méndez R, Moscardó A, Latorre A, Piró A, Amara-Elori I, Gimeno A, et al. Platelet activation is boosted early in community-acquired pneumonia and sustained until 30 days later. Eur Respir J. (2018) 52(Suppl. 62):PA2633. doi: 10.1183/13993003.congress-2018.PA2633
113. Middleton EA, Rowley JW, Campbell RA, Grissom CK, Brown SM, Beesley SJ, et al. Sepsis alters the transcriptional and translational landscape of human and murine platelets. Blood. (2019) 134:911–23. doi: 10.1182/blood.2019000067
114. Cangemi R, Casciaro M, Rossi E, Calvieri C, Bucci T, Calabrese CM, et al. Platelet activation is associated with myocardial infarction in patients with pneumonia. J Am Coll Cardiol. (2014) 64:1917–25. doi: 10.1016/j.jacc.2014.07.985
115. Wolff M, Handtke S, Palankar R, Wesche J, Kohler TP, Kohler C, et al. Activated platelets kill Staphylococcus aureus, but not Streptococcus pneumoniae - the role of FcγRIIa and platelet factor 4/heparin antibodies. J Thromb Haemost. (2020) 18:1459–68. doi: 10.1111/jth.14814
116. Alpkvist H, Athlin S, Mölling P, Norrby-Teglund A, Strålin K. High HMGB1 levels in sputum are related to pneumococcal bacteraemia but not to disease severity in community-acquired pneumonia. Sci Rep. (2018) 8:13428. doi: 10.1038/s41598-018-31504-4
117. Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol. (2006) 16:401–7. doi: 10.1016/j.cub.2006.01.056
118. Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, et al. Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol. (2007) 9:1162–71. doi: 10.1111/j.1462-5822.2006.00857.x
119. Martinez PJ, Farhan A, Mustafa M, Javaid N, Darkoh C, Garrido-Sanabria E, et al. PspA facilitates evasion of pneumococci from bactericidal activity of neutrophil extracellular traps (NETs). Microb Pathog. (2019) 136:103653. doi: 10.1016/j.micpath.2019.103653
120. Santos-Gallego CG, Badimon JJ. The sum of two evils: pneumonia and myocardial infarction: is platelet activation the missing link? J Am Coll Cardiol. (2014) 64:1926–8. doi: 10.1016/j.jacc.2014.08.023
121. Gavriilaki E, Gkaliagkousi E, Douma S. Platelets interplay between pneumonia and cardiovascular events: establishing a link? J Am Coll Cardiol. (2015) 65:1487–88. doi: 10.1016/j.jacc.2014.11.073
122. Savas G, Kalay N. Myocardial infarction in patients with pneumonia. J Am Coll Cardiol. (2015) 65:1491. doi: 10.1016/j.jacc.2014.12.059
123. Gurbel PA, Kreutz RP, Bliden KP, Tantry US. Platelet activation and pneumonia: is soluble P-selectin the right marker? J Am Coll Cardiol. (2015) 65:1492–3. doi: 10.1016/j.jacc.2014.12.061
124. Violi F, Calvieri C, Falcone N, Taliani G, Cangemi R, SIXTUS Study Group. Reply: platelets interplay between pneumonia and cardiovascular events: establishing a link? J Am Coll Cardiol. (2015) 65:1489–90. doi: 10.1016/j.jacc.2014.12.057
125. Violi F, Calvieri C, Falcone N, Taliani G, Cangemi R, SIXTUS Study Group. Reply: myocardial infarction in patients with pneumonia. J Am Coll Cardiol. (2015) 65:1491–2. doi: 10.1016/j.jacc.2014.12.060
126. Violi F, Calvieri C, Falcone N, Taliani G, Cangemi R, SIXTUS Study Group. Reply: platelet activation and pneumonia. Is soluble P-selectin the right marker? J Am Coll Cardiol. (2015) 65:1493–4. doi: 10.1016/j.jacc.2015.02.011
127. Santos-Gallego CG, Badimon J. Reply: platelet interplay between pneumonia and cardiovascular events. Establishing a link? J Am Coll Cardiol. (2015) 65:1490–1. doi: 10.1016/j.jacc.2014.12.058
128. Khan AR, Bin Abdulhak AA, Luni FK, Assaly R. Platelet activation and myocardial infarction in patients with pneumonia: are statins the answer? J Am Coll Cardiol. (2015) 65:1488. doi: 10.1016/j.jacc.2014.11.074
129. Falcone M, Russo A, Cangemi R, Farcomeni A, Calvieri C, Barillà F, et al. Lower mortality rate in elderly patients with community-onset pneumonia on treatment with aspirin. J Am Heart Assoc. (2015) 4:e001595. doi: 10.1161/JAHA.114.001595
130. Falcone M, Russo A, Farcomeni A, Pieralli F, Vannucchi V, Vullo V, et al. Septic shock from community-onset pneumonia: is there a role for aspirin plus macrolides combination? Intensive Care Med. (2016) 42:301–2. doi: 10.1007/s00134-015-4139-9
131. Nel JG, Durandt C, Mitchell TJ, Feldman C, Anderson R, Tintinger GR. Pneumolysin mediates platelet activation in vitro. Lung. (2016) 194:589–93. doi: 10.1007/s00408-016-9900-5
132. Nel JG, Theron AJ, Durandt C, Tintinger GR, Pool R, Mitchell TJ, et al. Pneumolysin activates neutrophil extracellular trap formation. Clin Exp Immunol. (2016) 184:358–67. doi: 10.1111/cei.12766
133. Nel JG, Durandt C, Theron AJ, Tintinger GR, Pool R, Richards GA, et al. Pneumolysin mediates heterotypic aggregation of neutrophils and platelets in vitro. J Infect. (2017) 74:599–608. doi: 10.1016/j.jinf.2017.02.010
134. Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. (2014) 123:2768–76. doi: 10.1182/blood-2013-10-463646
135. Kimball AS, Obi AT, Diaz JA, Henke PK. The emerging role of NETs in venous thrombosis and immunothrombosis. Front Immunol. (2016) 7:236. doi: 10.3389/fimmu.2016.00236
136. Calvieri C, Tanzilli G, Bartimoccia S, Cangemi R, Arrivi A, Dominici M, et al. Interplay between oxidative stress and platelet activation in coronary thrombus of STEMI patients. Antioxidants. (2018) 7:83. doi: 10.3390/antiox7070083
137. Alhamdi Y, Abrams ST, Lane S, Wang G, Toh CH. Histone-associated thrombocytopenia in patients who are critically ill. JAMA. (2016) 315:817–9. doi: 10.1001/jama.2016.0136
138. Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood. (2011) 118:3708–14. doi: 10.1182/blood-2011-01-332676
139. Ebrahimi F, Giaglis S, Hahn S, Blum CA, Baumgartner C, Kutz A, et al. Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: secondary analysis of a randomised controlled trial. Eur Respir J. (2018) 51:1701389. doi: 10.1183/13993003.01389-2017
140. Subramanian K, Neill DR, Malak HA, Spelmink L, Khandaker S, Marchiori GDL, et al. Pneumolysin binds to the Mannose receptor C type 1 (MRC-1) leading to anti-inflammatory responses and enhanced pneumococcal survival. Nat Microbiol. (2019) 4:62–70. doi: 10.1038/s41564-018-0280-x
141. Viasus D, Garcia-Vidal C, Manresa F, Dorca J, Gudiol F, Carratalà J. Risk stratification and prognosis of acute cardiac events in hospitalized adults with community-acquired pneumonia. J Infect. (2013) 66:27–33. doi: 10.1016/j.jinf.2012.09.003
142. Welte T, Pletz M. Pneumonia and the risk of cardiovascular death. Time to change our strategy. Am J Respir Crit Care Med. (2017) 196:541–3. doi: 10.1164/rccm.201707-1421ED
143. Corrales-Medina VF, Musher DM, Wells GA, Chirinos JA, Chen L, Fine MJ. Cardiac complications in patients with community-acquired pneumonia: incidence, timing, risk factors, and association with short-term mortality. Circulation. (2012) 125:773–81. doi: 10.1161/CIRCULATIONAHA.111.040766
144. Griffin AT, Wiemken TL, Arnold FW. Risk factors for cardiovascular events in hospitalized patients with community-acquired pneumonia. Int J Infect Dis. (2013) 17:e1125–9. doi: 10.1016/j.ijid.2013.07.005
145. Feldman C, Richards G. Appropriate antibiotic management of bacterial lower respiratory tract infections. F1000Res. (2018) 7:F1000 Faculty Rev−1121. doi: 10.12688/f1000research.14226.1
146. Garnacho-Montero J, Barrero-García I, Gómez-Prieto MG, Martin-Loeches I. Severe community-acquired pneumonia: current management and future therapeutic alternatives. Expert Rev Anti Infect Ther. (2018) 16:667–77. doi: 10.1080/14787210.2018.1512403
147. Chalmers S, Khawaja A, Wieruszewski FM, Gajic O, Odeyemi Y. Diagnosis and treatment of acute pulmonary inflammation in critically ill patients: the role of inflammatory biomarkers. World J Crit Care Med. (2019) 8:59–71. doi: 10.5492/wjccm.v8.i5.74
148. Anderson R, Tintinger G, Cockeran R, Potjo M, Feldman C. Beneficial and harmful interactions of antibiotics with microbial pathogens and the host innate immune system. Pharmaceuticals. (2010) 3:1694–710. doi: 10.3390/ph3051694
149. Steel HC, Theron AJ, Cockeran R, Anderson R, Feldman C. Pathogen- and host-directed anti-inflammatory activities of macrolide antibiotics. Mediat Inflamm. (2012) 2012:584262. doi: 10.1155/2012/584262
150. Zimmermann P, Ziesenitz VC, Curtis N, Ritz N. The immunomodulatory effects of macrolides - a systematic review of the underlying mechanism. Front Immunol. (2018) 9:302. doi: 10.3389/fimmu.2018.00302
151. Reijnders TDY, Saris A, Schultz MJ, van der Poll T. Immunomodulation by macrolides: therapeutic potential for critical care. Lancet Respir Med. (2020) 8:619–30. doi: 10.1016/S2213-2600(20)30080-1
152. Peyrani P, Wiemken TL, Metersky ML, Arnold FW, Mattingly WA, Feldman C, et al. The order of administration of macrolides and beta-lactams may impact the outcomes of hospitalized patients with community-acquired pneumonia: results from the community-acquired pneumonia organization. Infect Dis. (2018) 50:13–20. doi: 10.1080/23744235.2017.1350881
153. Tsoupras AB, Chini M, Tsogas N, Lioni A, Tsekes G, Demopoulos CA, et al. In vitro anti-inflammatory and anti-coagulant effects of antibiotics towards platelet activating factor and thrombin. J Inflamm. (2011) 8:17. doi: 10.1186/1476-9255-8-17
154. Oz F, Gul S, Kaya MG, Yazici M, Bulut I, Elitok A, et al. Does aspirin use prevent acute coronary syndrome in patients with pneumonia: multicenter prospective randomized trial. Coron Artery Dis. (2013) 24:231–7. doi: 10.1097/MCA.0b013e32835d7610
155. Falcone M, Russo A, Shindo Y, Farcomeni A, Pieralli F, Cangemi R, et al. A hypothesis-generating study of the combination of aspirin plus macrolides in patients with severe community-acquired pneumonia. Antimicrob Agents Chemother. (2019) 63:e01556–18. doi: 10.1128/AAC.01556-18
156. Basille D, Thomsen RW, Madsen M, Duhaut P, Andrejak C, Jounieaux V, et al. Nonsteroidal antiinflammatory drug use and clinical outcomes of community-acquired pneumonia. Am J Respir Crit Care Med. (2018) 198:128–31. doi: 10.1164/rccm.201802-0229LE
157. Stern A, Skalsky K, Avni T, Carrara E, Leibovici L, Paul M. Corticosteroids for pneumonia. Cochrane Database Syst Rev. (2017) 12:CD007720. doi: 10.1002/14651858.CD007720.pub3
158. Wu WF, Fang Q, He GJ. Efficacy of corticosteroid treatment for severe community-acquired pneumonia: a meta-analysis. Am J Emerg Med. (2018) 36:179–84. doi: 10.1016/j.ajem.2017.07.050
159. Jiang S, Liu T, Hu Y, Li R, Di X, Jin X, et al. Efficacy and safety of glucocorticoids in the treatment of severe community-acquired pneumonia: a meta-analysis. Medicine. (2019) 98:e16239. doi: 10.1097/MD.0000000000016239
160. Huang J, Guo J, Li H, Huang W, Zhang T. Efficacy and safety of adjunctive corticosteroids therapy for patients with severe community-acquired pneumonia: a systematic review and meta-analysis. Medicine. (2019) 98:e14636. doi: 10.1097/MD.0000000000014636
161. Liverani E, Banerjee S, Roberts W, Naseem KM, Perretti M. Prednisolone exerts exquisite inhibitory properties on platelet functions. Biochem Pharmacol. (2012) 83:1364–73. doi: 10.1016/j.bcp.2012.02.006
162. Cangemi R, Carnevale R, Nocella C, Calvieri C, Cammisotto V, Novo M, et al. Glucocorticoids impair platelet thromboxane biosynthesis in community-acquired pneumonia. Pharmacol Res. (2018) 131:66–74. doi: 10.1016/j.phrs.2018.03.014
163. Cangemi R, Falcone M, Taliani G, Calvieri C, Tiseo G, Romiti GF, et al. Corticosteroids use and incident myocardial infarction in adults hospitalized for community-acquired pneumonia. Ann Am Thorac Soc. (2019) 16:91–8. doi: 10.1513/AnnalsATS.201806-419OC
164. Waterer G, Metersky ML. Corticosteroids for community-acquired pneumonia: overstated benefits and understated risks. Chest. (2019) 156:1049–53. doi: 10.1016/j.chest.2019.06.017
165. Lloyd M, Karahailos A, Janus E, Skinner EH, Haines T, De Silva A, et al. Effectiveness of the bundled intervention including adjunctive corticosteroids on outcomes of hospitalized patients with community-acquired pneumonia. A stepped-wedged randomized clinical trial. JAMA Intern Med. (2019) 179:1052–60. doi: 10.1001/jamainternmed.2019.1438
166. Violi F, Calvieri C, Cangemi R. Effect of corticosteroids on myocardial injury among patients hospitalized for community-acquired pneumonia: rationale and study design. The colosseum trial. Intern Emerg Med. (2020) 15:79–86. doi: 10.1007/s11739-019-02117-0
167. Jin J, Kunapuli SP. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc Natl Acad Sci USA. (1998) 95:8070–4. doi: 10.1073/pnas.95.14.8070
168. Schilling U, Dingemanse J, Ufer M. Pharmacokinetics and pharmacodynamics of approved and investigational P2Y12 receptor antagonists. Clin Pharmacokinet. (2020) 59:545–66. doi: 10.1007/s40262-020-00864-4
169. Gross AK, Dunn SP, Feola DJ, Martin CA, Charnigo R, Li Z, et al. Clopidogrel treatment and the incidence and severity of community acquired pneumonia in a cohort study and meta-analysis of antiplatelet therapy in pneumonia and critical illness. J Thromb Thrombolysis. (2013) 35:147–54. doi: 10.1007/s11239-012-0833-4
170. Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. (2009) 361:1045–57. doi: 10.1056/NEJMoa0904327
171. Storey RF, James SK, Siegbahn A, Varenhorst C, Held C, Ycas J, et al. Lower mortality following pulmonary adverse events and sepsis with ticagrelor compared to clopidogrel in the PLATO study. Platelets. (2014) 25:517–25. doi: 10.3109/09537104.2013.842965
172. Sexton TR, Zhang G, Macaulay TE, Callahan LA, Charnigo R, Vsevolozhskaya OA, et al. Ticagrelor reduces thromboinflammatory markers in patients with pneumonia. JACC Basic Transl Sci. (2018) 3:435–49. doi: 10.1016/j.jacbts.2018.05.005
173. Feldman C, Anderson R. Community-acquired pneumonia: pathogenesis of acute cardiac events and potential adjunctive therapies. Chest. (2015) 148:523–32. doi: 10.1378/chest.15-0484
174. Marwali MR, Hu CP, Mohandas B, Dandapat A, Deonikar P, Chen J, et al. Modulation of ADP-induced platelet activation by aspirin and pravastatin: role of lectin-like oxidized low-density lipoprotein receptor-1, nitric oxide, oxidative stress, and inside-out integrin signaling. J Pharmacol Exp Ther. (2007) 322:1324–32. doi: 10.1124/jpet.107.122853
175. Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. (2007) 13:1086–95. doi: 10.1038/nm1626
176. Fontana P, Zufferey A, Daali Y, Reny JL. Antiplatelet therapy: targeting the TxA2 pathway. J Cardiovasc Transl Res. (2014) 7:29–38. doi: 10.1007/s12265-013-9529-1
177. Owens AP III, Mackman N. The antithrombotic effects of statins. Annu Rev Med. (2014) 65:433–45. doi: 10.1146/annurev-med-051812-145304
178. Day AP, Bellavia S, Jones OT, Stansbie D. Effect of simvastatin therapy on cell membrane cholesterol content and membrane function as assessed by polymorphonuclear cell NADPH oxidase activity. Ann Clin Biochem. (1997) 34:269–75. doi: 10.1177/000456329703400308
179. Barale C, Frascaroli C, Senkeev R, Cavalot F, Russo I. Simvastatin effects on inflammation and platelet activation markers in hypercholesterolemia. Biomed Res Int. (2018) 2018:6508709. doi: 10.1155/2018/6508709
180. Sapey E, Patel JM, Greenwood H, Walton GM, Grudzinska F, Parekh D, et al. Simvastatin improves neutrophil function and clinical outcomes in pneumonia. A pilot randomized controlled clinical trial. Am J Respir Crit Care Med. (2019) 200:1282–93. doi: 10.1164/rccm.201812-2328OC
181. Henry BD, Neill DR, Becker KA, Gore S, Bricio-Moreno L, Ziobro R, et al. Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat Biotechnol. (2015) 33:81–8. doi: 10.1038/nbt.3037
182. Laterre PF, Colin G, Dequin PF, Dugernier T, Boulain T, da Silveira SA, et al. CAL02, a novel antitoxin liposomal agent, in severe pneumococcal pneumonia: a first-in-human, double-blind, placebo-controlled, randomised trial. Lancet Infect Dis. (2019) 19:620–30. doi: 10.1016/S1473-3099(18)30805-3
183. Amison RT, Jamshidi S, Rahman KM, Page CP, Pitchford SC. Diverse signalling of the platelet P2Y1 receptor leads to a dichotomy in platelet function. Eur J Pharmacol. (2018) 827:58–70. doi: 10.1016/j.ejphar.2018.03.014
184. Yang H, Wang H, Andersson U. Targeting inflammation driven by HMGB1. Front Immunol. (2020) 11:484. doi: 10.3389/fimmu.2020.00484
185. Beno SM, Riegler AN, Gilley RP, Brissac T, Wang Y, Kruckow KL, et al. Necroptosis inhibition prevents long-term cardiac damage during pneumococcal pneumonia and invasive disease. J Infect Dis. (2020). doi: 10.1093/infdis/jiaa295. [Epub ahead of print].
Keywords: anti-platelet agents, community-acquired pneumonia, pneumococcus, platelets, pneumolysin, thrombocytopenia, high mobility group box 1 protein, cardiovascular events
Citation: Feldman C and Anderson R (2020) Platelets and Their Role in the Pathogenesis of Cardiovascular Events in Patients With Community-Acquired Pneumonia. Front. Immunol. 11:577303. doi: 10.3389/fimmu.2020.577303
Received: 29 June 2020; Accepted: 24 August 2020;
Published: 17 September 2020.
Edited by:
Laurel L. Lenz, University of Colorado, United StatesReviewed by:
Grant Waterer, Royal Perth Hospital, AustraliaDaniel Neill, The University of Liverpool, United Kingdom
Copyright © 2020 Feldman and Anderson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles Feldman, Y2hhcmxlcy5mZWxkbWFuQHdpdHMuYWMuemE=
†ORCID: Charles Feldman orcid.org/0000-0002-6881-8314
Ronald Anderson orcid.org/0000-0002-5925-6452